 Jiaquan Huang1,2
Jiaquan Huang1,2 Iobana Alanís-Martínez3
Iobana Alanís-Martínez3 Lucita Kumagai4
Lucita Kumagai4 Zehan Dai1
Zehan Dai1 Zheng Zheng1
Zheng Zheng1 Adalberto A. Perez de Leon5
Adalberto A. Perez de Leon5 Jianchi Chen5*
Jianchi Chen5* Xiaoling Deng1*
Xiaoling Deng1*- 1Department of Plant Pathology, South China Agricultural University, Guangzhou, Guangdong, China
- 2Center for Biological Science and Technology, Advanced Institute of Natural Sciences, Beijing Normal University at Zhuhai, Zhuhai, Guangdong, China
- 3National Station of Plant Epidemiology, Quarantine and Sanitation, SENASICA, Queretaro, Mexico
- 4Plant Pest Diagnostic Center, California Department of Food and Agriculture, Sacramento, CA, United States
- 5United States Department of Agriculture-Agricultural Research Service (USDA-ARS), San Joaquín Valley Agricultural Sciences Center, Parlier, CA, United States
Background: Huanglongbing (HLB, yellow shoot disease) is a highly destructive citrus disease associated with a nonculturable bacterium, “Candidatus Liberibacter asiaticus” (CLas), which is transmitted by Asian citrus psyllid (ACP, Diaphorina citri). In Mexico, HLB was first reported in Tizimin, Yucatán, in 2009 and is now endemic in 351 municipalities of 25 states. Understanding the population diversity of CLas is critical for HLB management. Current CLas diversity research is exclusively based on analysis of the bacterial genome, which composed two regions, chromosome (> 1,000 genes) and prophage (about 40 genes).
Methods and results: In this study, 40 CLas-infected ACP samples from 20 states in Mexico were collected. CLas was detected and confirmed by PCR assays. A prophage gene(terL)-based typing system (TTS) divided the Mexican CLas strains into two groups: Term-G including four strains from Yucatán and Chiapas, as well as strain psy62 from Florida, USA, and Term-A included all other 36 Mexican strains, as well as strain AHCA1 from California, USA. CLas diversity was further evaluated to include all chromosomal and prophage genes assisted by using machine learning (ML) tools to resolve multidimensional data handling issues. A Term-G strain (YTMX) and a Term-A strain (BCSMX) were sequenced and analyzed. The two Mexican genome sequences along with the CLas genome sequences available in GenBank were studied. An unsupervised ML was implemented through principal component analysis (PCA) on average nucleotide identities (ANIs) of CLas whole genome sequences; And a supervised ML was implemented through sparse partial least squares discriminant analysis (sPLS-DA) on single nucleotide polymorphisms (SNPs) of coding genes of CLas guided by the TTS. Two CLas Geno-groups, Geno-group 1 that extended Term-A and Geno-group 2 that extended Term-G, were established.
Conclusions: This study concluded that: 1) there were at least two different introductions of CLas into Mexico; 2) CLas strains between Mexico and USA are closely related; and 3) The two Geno-groups provide the basis for future CLas subspecies research.
Introduction
Huanglongbing (HLB, yellow shoot disease, also called citrus greening disease) is a highly destructive disease threatening the world citrus industry. HLB is associated with a nonculturable proteobacterium bacterium, “Candidatus Liberibacter asiaticus” (CLas). Under field conditions, CLas is transmitted by Asian citrus psyllid (ACP, Diaphorina citri). HLB was observed for over a hundred years in Asia (Bove, 2006; Lin, 1956), but was first reported in the United States in Florida in 2005 (Halbert, 2005), then in California in 2012 (Kumagai et al., 2013) and in Texas in 2012 (Kunta et al., 2012). In Mexico, the disease was first reported in Tizimin, Yucatán, in 2009 (Trujillo-Arriaga, 2011) and is now endemic in 351 municipalities of 25 states (SENASICA, 2022).
For effective HLB management, it is important to understand the population diversity of CLas strains and relationships among strains from different geographical sources. Since CLas cannot be cultured in vitro, strain characterizations are mostly derived from genomic sequence analyses. A CLas genome is about 1.2 Mbp and composed of a chromosomal region that hosts core genes, and a prophage region that contains dispensable genes (Duan et al., 2009; Zheng et al., 2014), and prophage could be absent (Katoh et al., 2014). The CLas prophage region is highly variable and has been the target in CLas diversity studies (Chen et al., 2010; Liu et al., 2011; Deng et al., 2014; Zheng et al., 2017; Alanís-Martínez et al., 2018; Dai et al., 2019; Fu et al., 2020; Zheng et al., 2021). Based on single nucleotide polymorphisms (SNPs) in a 370-bp region in terL, a gene encoding a phage terminase large subunit, the terL typing system (TTS) was proposed (Deng et al., 2014). In contrast, due to the highly conserved nature of the CLas genome, few chromosomal loci have been found to be effective for CLas diversity research (Bastianel et al., 2005; Deng et al., 2008). The exception was a tandem repeat locus located in chromosomal region annotated as a gene encoding for a bacteriophage repressor protein (Chen et al., 2010; Katoh et al., 2011; Ma et al., 2014; Ghosh et al., 2015; Singh et al., 2019). Analyses on both prophage and chromosomal regions are needed for a complete evaluation of CLas genomic diversity. Since TTS is prophage-based, use of chromosomal gene(s) that support TTS will strengthen CLas diversity research.
Thanks to the development of next-generation sequencing (NGS) technology, there are currently 39 whole genome sequences of CLas with various completion status (either complete with a single contig or draft with multiple contigs) in GenBank database (version 249). Eighteen CLas genome sequences were from the United States (9 from California, 5 from Florida, and 6 from Texas) (Supplementary Table 1). In contrast, there is only one Mexican CLas genome sequence (Mex8, related to Texas and Florida strains) published (Thapa et al., 2020). More CLas genome sequences from Mexico are in need for CLas diversity research.
Genome sequence analyses involve data sets of Mbp or Gbp that generate multi-dimensional data outputs, i.e., data matrices with a large number of columns and rows. Handling and interpreting such complex data sets are highly challenging through conventional approaches. Machine learning (ML) is a technology that uses computational statistics to resolve complex data problems. In general, there are two types of ML. Unsupervised ML takes a high-dimensional set of data to find or predict structures such as clusters, e.g., principal component analysis (PCA) (Abdi and Williams, 2010) Supervised ML builds a mathematical model of a set of data that contains guidance to generate desired outputs, e.g., sparse partial least-squares discriminant analysis (sPLS-DA) (Lê Cao et al., 2011; Lee et al., 2018).
The goal of this study was to characterize the genomic diversity of CLas strains from Mexico. The information was used to evaluate strain evolutionary paths and to reveal relationships of CLas strains within Mexico and between Mexico and the neighboring USA. CLas strains were collected from 20 states in Mexico and typed by TTS. Representative strains were sequenced. CLas diversity were evaluated using ML approaches to resolve research issues associated with data processing and result interpretations. Two Geno-groups of CLas were established. Potential impacts of the Geno-groups on HLB epidemiology and CLas biological research were discussed.
Materials and methods
CLas strain DNA collection
The ACP samples from 20 states in Mexico were collected in the period from September, 2017 to August, 2018 from commercial citrus orchards and backyard trees in urban areas (Table 1). The trees where the psyllids were collected were Citrus aurantiifolia, C. sinensis, C. latifolia, C. reticulata, as well as the ornamental species Murraya paniculata. Each ACP sample contained 1 to 50 insects preserved in 70% ethanol. DNA was obtained using the E.Z.N.A.® Tissue DNA Kit (Omega Bio-Tek, D3396-02) according to the manufacturer’s instructions.
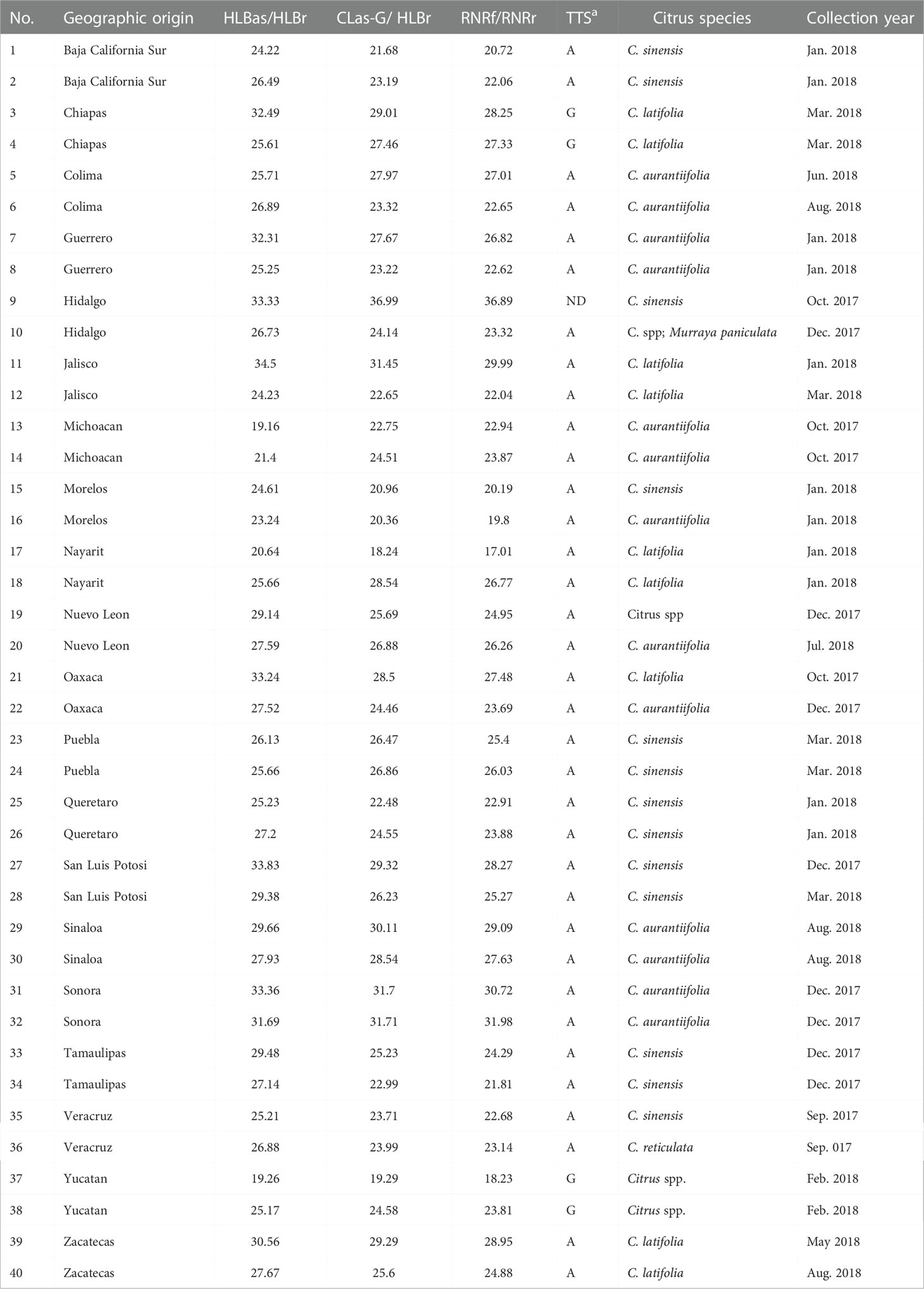
Table 1 General information of Asian citrus psyllid (ACP, Diaphorina citri) samples collected from 20 states in Mexico and the result of PCR detections and typing with terL typing system (TTS).
CLas PCR
CLas in a sample was determined by three quantitative PCR assays. 1) TaqMan qPCR with primer set HLBas/HLBr and probe HLBp (HLBas-PCR, Li et al., 2006) performed in Mexico; 2) TaqMan PCR with primer set CLas-4G/HLBr and probe HLBp (CLas-4G-PCR, Bao et al., 2020); and TaqMan PCR with primer set RNRf-RNRr and probe RNRp (RNRf-PCR, Zheng et al., 2016) performed in California, USA.
HLBas-PCR (Li et al., 2006) was used as a primary detection. PCR was carried out in the CFX96 thermocycler (Bio-Rad Laboratories) in a volume of 25 µl with the following reagents: 1U Platinum Taq DNA polymerase (Invitrogen, USA), 1× Buffer PCR, 6 mM MgCl2, 0.2 mM dNTPs, 240 nM of each primer, 120 nM of target probe, and 2 µl DNA template. PCR amplification started at 95°C for 20 seconds, followed by 40 cycles of 95°C for 1 second and 58°C for 40 seconds. The fluorescence signal was captured at the end of each 58°C step. The data were generated from CFX96 Manager software with default parameters.
CLas-4G-PCR and RNRf-PCR were carried out as confirmatory tests. PCR was performed in Applied Biosystems (ABI) StepOnePlus Real-time PCR system. A reaction mixture (20μl) contained 10 μl of TaqMan® Fast Universal PCR Master Mix (2X) (Applied Biosystems, ABI), 1 μl of DNA template (25 ng), 0.2 μl of TaqMan® probe (5 μM), 0.4 μl of each forward and reverse primer (10 μM). PCR amplification started at 95°C for 20 seconds, followed by 40 cycles of 95°C for 10 seconds and 60°C for 20 seconds. The fluorescence signal was captured at the end of each 60°C step. StepOnePlusTM Software v2.3 (Applied Biosystems) was used to analyze the data, with automated baseline settings and a manually set threshold of 0.1.
Strain typing using TTS
For PCR typing, the procedure described previously (Deng et al., 2014) was followed. Primer set CT3f/CT3r (5’ CCAGAAACGA TTAGC GTTCT GAT 3’/5’ GTCAATAAGA CCTGGTATGT CTA 3’) was used for Term-A type, and primer set FC3f/FC3r (5’ CCAGAAACGA TTAGCGTTCC ACT 3’/5’ CGACCAGGGA TATCGG 3’) was used for Term-G typing.
For sequencing typing, CLas DNA samples were amplified by conventional PCR using primer set 766-f/766-r (5’ CAATAACAGG ACCTCTACGC 3’/5’ ATTGGAAAGA CGACGTTAAA 3’) (Liu et al., 2011) with the following steps: initial denaturation for 3 min at 95°C, followed by 35 cycles of denaturation for 45 seconds at 95°C, annealing for 30 seconds at 55°C, elongation for 2 min 30 seconds at 72°C, and a final extension step of 72°C for 10 min. The amplified DNA fragments were collected from NucleoSpin® Gel and purified by PCR Clean-up kit (QIAGEN, Valencia, USA) following the manufacturer’s protocol, and sequenced using ABI BigDye Terminator Chemistry (Life Technologies) in an ABI 3730 sequencer (Life Technologies) with 766-f/766-r. The terL-370 bp segment, including the CT3f/CT3r and FC3f/FC3r regions, was identified and extracted from each amplicon sequence. All sequences were aligned using CLUSTAL program (Thompson et al., 1994). Single nucleotide polymorphisms (SNPs) were identified manually for assignment of Term-A or Term-G group.
Whole genome sequencing and assembling
Two samples, YTMX (Term-G) from Yucatán (No. 1 in Table 1) and BCSMX (Term-A) from Baja California Sur (No. 37 in Table 1) were selected for whole genome sequencing following the procedure described previously (Zheng et al., 2014; Huang et al., 2021). Briefly, 2–4 µl of sample DNA (mixture of bacteria + ACP) was enlarged (i.e., increased all DNA simultaneously) with GenomiPhiTM V2 DNA Amplification kit (GE Healthcare, Sigma-Aldrich Corp., St. Louis, MO, USA) following manufacturer’s instructions. The enlarged DNAs were sequenced by Illumina HiSeq (2x100) formats through commercial sources. Only sequence reads with Q score > 30 were collected. Quality check of the Illumina reads was performed using FastQC (Andrews, 2010). The HiSeq reads were assembled using reference-mapping with the whole genome sequences of psy62 (NC_012985.3), A4 (NZ_CP010804.2) and AHCA1 (NZ_CP029348.1) using CLC Genomic Workbench 10.0 with the parameters: Length Fraction = 1 and Similarity Fraction = 0.9. de novo assembly was also conducted using default parameters. A final complete genome sequence was obtained by combining the assemblies of reference-mapping and de novo assembly. The genome sequence annotations were performed using Prokka v1.14.6 (Seemann, 2014).
Unsupervised ML
All 39 CLas genome sequences deposited in GenBank database (version 249) were downloaded (Supplementary Table 1). One representative sequence each from “Ca. L. africanus” (CLaf), “Ca. L. americanus” (CLam), and “Ca. L. solanacearum” (CLso) were also downloaded. Strain Tabriz.3 showed average nucleotide identity (ANI) value lower than 95 and (Supplementary Table 2), therefore not considered as CLas in this study. The genome sequences HHCA (JMIL02), SGpsy (QFZJ01), and SGCA1 (QFZT01) that had few contigs longer than 1,000 bp were excluded for ANI calculation. The 35 (39 – 4) CLas strain sequences along with the BCSMX and YTMX genome sequences, adding to a total of 37 CLas genome sequences, and the 3 non-CLas genome sequences (1 CLaf, 1 CLam, and 1 CLso) were used for pair-wise ANI calculation using FastANI v1.33 (Jain et al., 2018) with fragment size set to 1,000 bp. Relationships among CLas strains embedded in the ANI matrix were revealed through PCA with mixOmics R package (v6.10.8) (Rohart et al., 2017). The TTS typing of each genome was implemented through BLAST search with the terL-370-bp sequence in reference to SNPs matching CT3f/CT3r or FC3f/FC3r. If no SNP match, the genome was labeled as non-typeable.
Supervised ML
The label of Term-A or Term-G type of a genome was used as a guide to identify genes with supportive SNPs. Details procedures were listed in Table 2. Briefly, 1) With the exception of the non-TTS typeable strain Ishi-1 (NZ_AP014595.1), 10 CLas whole genome sequences with a single contig (“complete” status), and the two Mexican CLas genome sequences from this study were selected (Supplementary Table 1). The genome sequences were reannotated to generate uniform feature format (.gff) file; 2) Single-copy core genes were identified using Panaroo v1.2.10 software (Tonkin-Hill et al., 2020); 3) SNPs in the core gene were identified using MAFFT v7.490 software (Katoh and Standley, 2013) and SNP-sites v2.5.1 (Page et al., 2016); 4). Based on TTS, each genome was labeled as either Term-A or Term-G for parameters tuning; and 5) sPLS-DA was performed with the mixOmics R package (v6.10.8) to identify genes with supportive SNPs for TTS. The features with contribution values to each component >= |0.2| were selected. Each SNPs in the two Mexican genomes were confirmed by strict read mapping performed with CLC Genomic Workbench 10.0 (Length Fraction = 1; Similarity Fraction = 0.97).
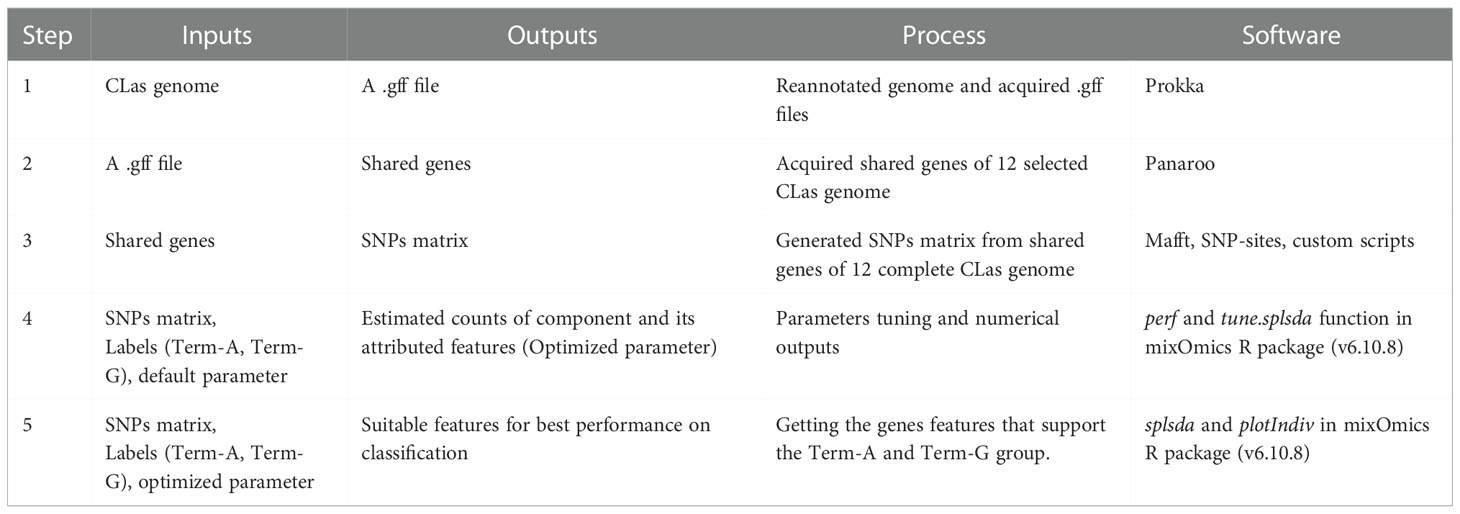
Table 2 A workflow of sparse partial least-squares discriminant analysis (sPLS-DA) in this study.
Using the genome sequence of strain AHCA1 (NZ_CP029348.1) as a reference, all genes with TTS supportive SNPs were identified. For each TTS SNP, a 51-bp sequence with the SNP in the center (nucleotide position 21) was copied. The 51-bp sequence was used as a query to BLAST search against the 40 CLas genome sequences (38 sequences in GenBank excluding strain Tabriz.3 and two Mexican strain genomes in this study, Supplementary Table 1). TTS SNPs in each genome were identified manually. TTS SNPs in each CLas genome were concatenated for cluster analysis using ggdendro and ggplot2 (Wickham, 2016).
Results
A total of 40 Asian citrus psyllid (ACP) samples were collected from 20 states in Mexico (Table 1 and Figure 1). The HLBas-PCR Ct values ranged from 19.16 to 34.50. CLas was further confirmed by CLas4G-PCR despite of the variations of Ct value ranges in some samples. In all samples, Ct values of RNRf-PCR were lower than those of CLas4G-PCR, a further validation of CLas detection (Bao et al., 2020). Of particular interest was sample #9 from Hidalgo with a Ct of 36.99 by CLas4G-PCR. Such a high Ct value could have been attributed to the presence of non-CLas bacteria (Huang et al., 2021). However, when sample #9 was cross-checked using the more sensitive RNRf-PCR, a lower Ct value confirmed the presence of CLas (Bao et al., 2020).
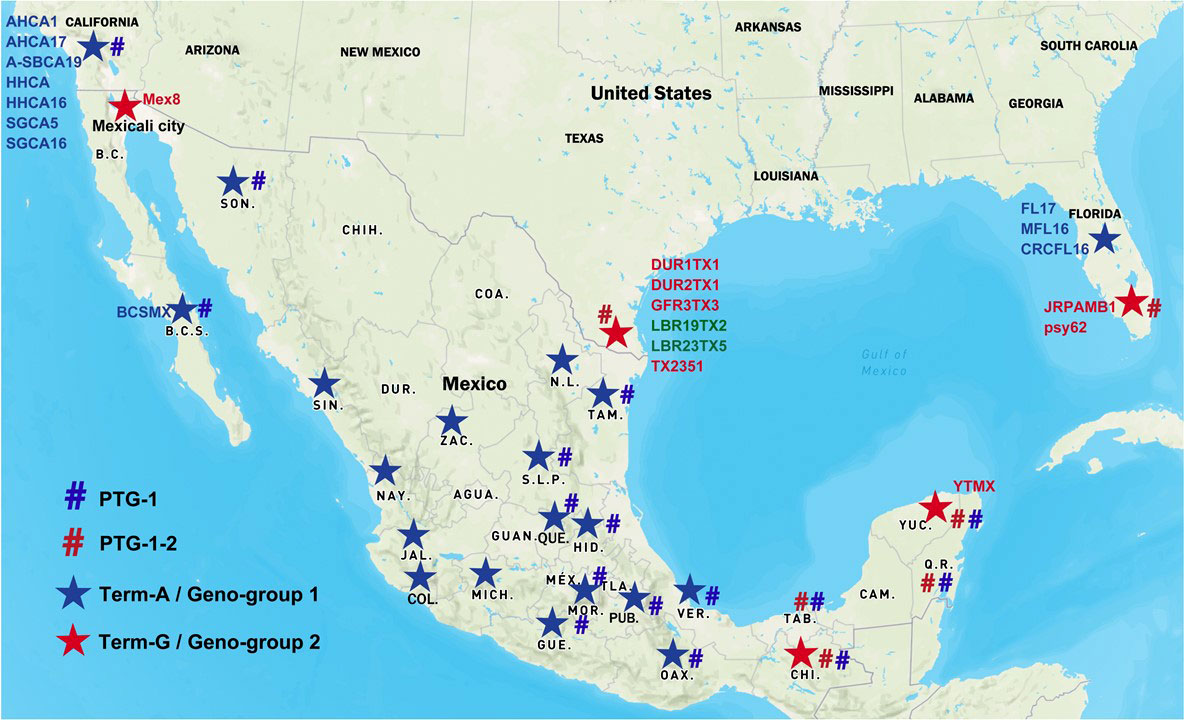
Figure 1 Distribution of “Candidatus Liberibacter asiaticus” (CLas) strains in Mexico and the United States. Blue star signs represent Term-A group/Geno-group 1; Red star signs represent Term-G group/Geno-group 2. Blue number signs represent PTG-1 prophage group; Red number signs represent PTG-1-2 prophage group. Two CLas strains in green were non-typeable for neither Term-A or Term-G, but determined to be in Geno-group 2.
TTS by PCR grouped the Mexican CLas strains into Term-A and Term-G groups (Table 1 and Figure 1). Term-G included strains in the states of Yucatán and Chiapas in Peninsula and southern Mexico, and strain psy62, the major CLas strain in Florida, USA. Term-A included the other 36 strains from central and northern Mexico, and strain AHCA1 reported in California, USA (Dai et al., 2019). Analyses of amplicon sequences confirmed the TTS typing results with the exception of sample #9, where no amplicon was available for sequencing. Interestingly, the Mexicali strain in the central and northern Mexico was in Term-G group (Figure 1).
Selected metrics of whole genome sequences of CLas strains YTMX and BCSMX are summarized in Table 3. HiSeq sequencing generated a total of 492,619,792 reads (49,754,598,992 bp, ~50 G bp) for YTMX. HiSeq reads mapped the best to psy62 with the largest number of 625,260 reads, as comparing to A4 (623,691 reads) and AHCA1 (622,913 reads). Therefore, psy62 was used as the main reference for YTMX genome assembling. The CLas strain YTMX genome was 1,229,040 bp in 4 contigs with three prophages. The circular form of all three prophages was detected. Similarly, a total of 571,293,176 reads (57,700,610,776 bp, ~58 G bp) for strain BCSMX. HiSeq reads mapped the best to AHCA1 (141,652 reads), comparing to psy62 (140,531 reads) and A4 (137,757 reads). Therefore, AHCA1 was used as the main reference for BCSMX genome assembling. The BCSMX genome was 1,230,223 bp in 4 contigs with one prophage and the circular form was detected.
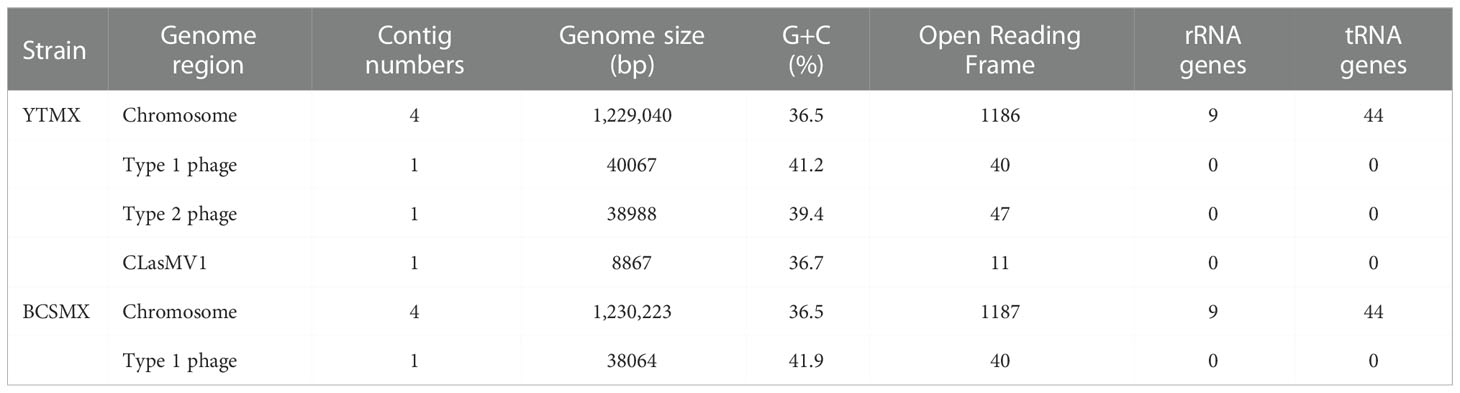
Table 3 Selected metrics of two whole genome sequences of “Candidatus Liberibacter asiaticus” strains from Mexico.
In the unsupervised machine learning (ML) study, pair-wise average nucleotide identity (ANI) values of the 37 CLas strains ranged from 99.67 to 100.00 (Supplementary Table 2), indicating the high level of genome sequence congruence of CLas strains. The ANIs of all CLas strains to the non-CLas strains were significantly lower with CLaf at 76.21, CLam at 75.91, and CLso at 76.72. Results of PCA analysis on the CLas strain matrix (Supplementary Table 2) are presented in Figure 2. The three PCAs accounted for 72% of the total variance. Term-G strains (in red) are separated from Term-A strains (in blue). For the five non-typeable strains (in green), strains LBR19TX2 (#34) and LBR23TX5 (#35) were grouped with Term-G, so was strain Ishi-1 (#33). Strain PA19 (#36) and PA20 (#37) (green dash-line circle) appeared to be distant from Term-A and Term-G groups, along with the Term-A strain Myan16 (#17). It is also interested that strains circled by blue dash-line, TaiYZ2 (#21), YNXP-1(#24), A4 (#2), HHCA16 (#13), GDHZ11D(#11), harbored Type 2 prophage only.
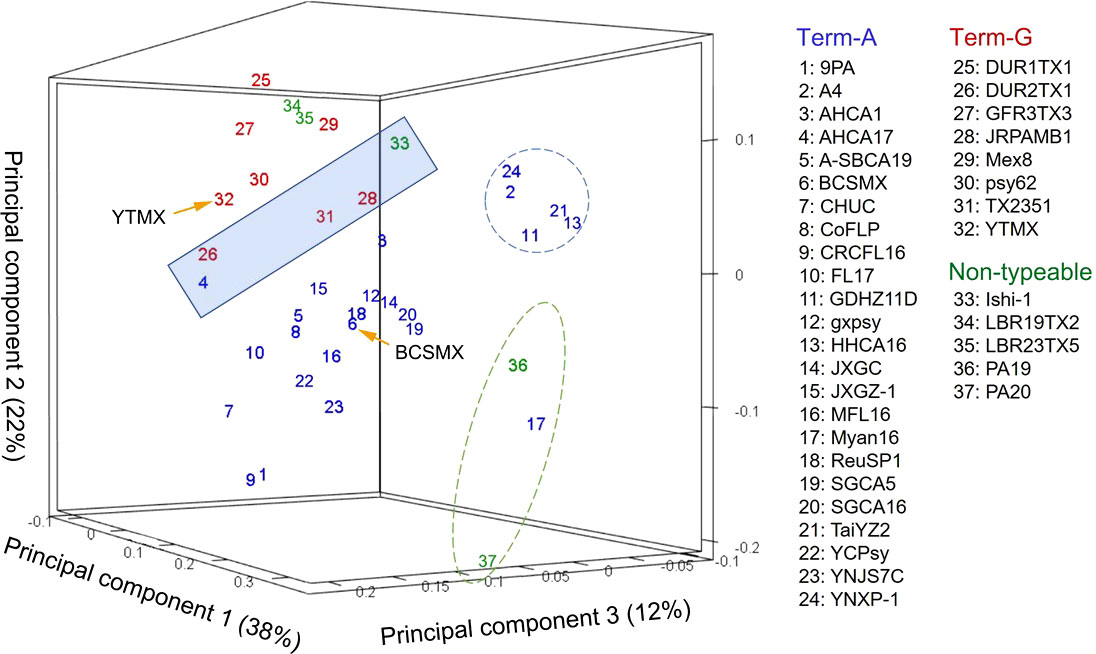
Figure 2 Relationships of “Candidatus Liberibacter asiaticus” (CLas) strains revealed by an unsupervised marchine learning (ML) tool (principal component analysis) on average nucleotide identity (ANI) values. Each strain is represented by a number with the correspondent names listed on the right. Blue numbers represent Term-A group; Red numbers represents Term-G group; and green numbers represents non-typable for Term-A or Term-G. Note that strains in the shaded box borderline between Term-A and Term-G groups. The grouping status of the borderline strains as well as the non-typeable strains was resolved/confirmed in a supervised ML approach (Figure 3). Although not the objective of this study, it was interesting to note that strains in blue circle harbored Type 2 prophage only, and strains in green circle were of South Asia origins (#17 from Myanmar; #36 and #37 from Pakistan).
In the supervised ML study, analysis of the 12 selected CLas genomes identified a total of 1,044 single-copy core genes. Sparse partial least squares discriminant analysis (sPLS-DA) identified 15 terL typing system (TTS) supportive genes, each having 1 to 5 SNPs. A closer examination found that some SNPs could subgroup within Term-A and Term-G groups. SNPs supporting TTS are shown in Table 4, along with their correspondent positions in the reference genomes (AHCA1, NZ_CP029348.1 and psy62, NZ_CP010804.2). Gene 1 to 14 were from the chromosomal region and Gene 15 was from the prophage region (Table 4). The length of these genes varied from 183 to 5,487 bp according to the annotation of CLas strain AHCA1 and psy62 (Table 4).
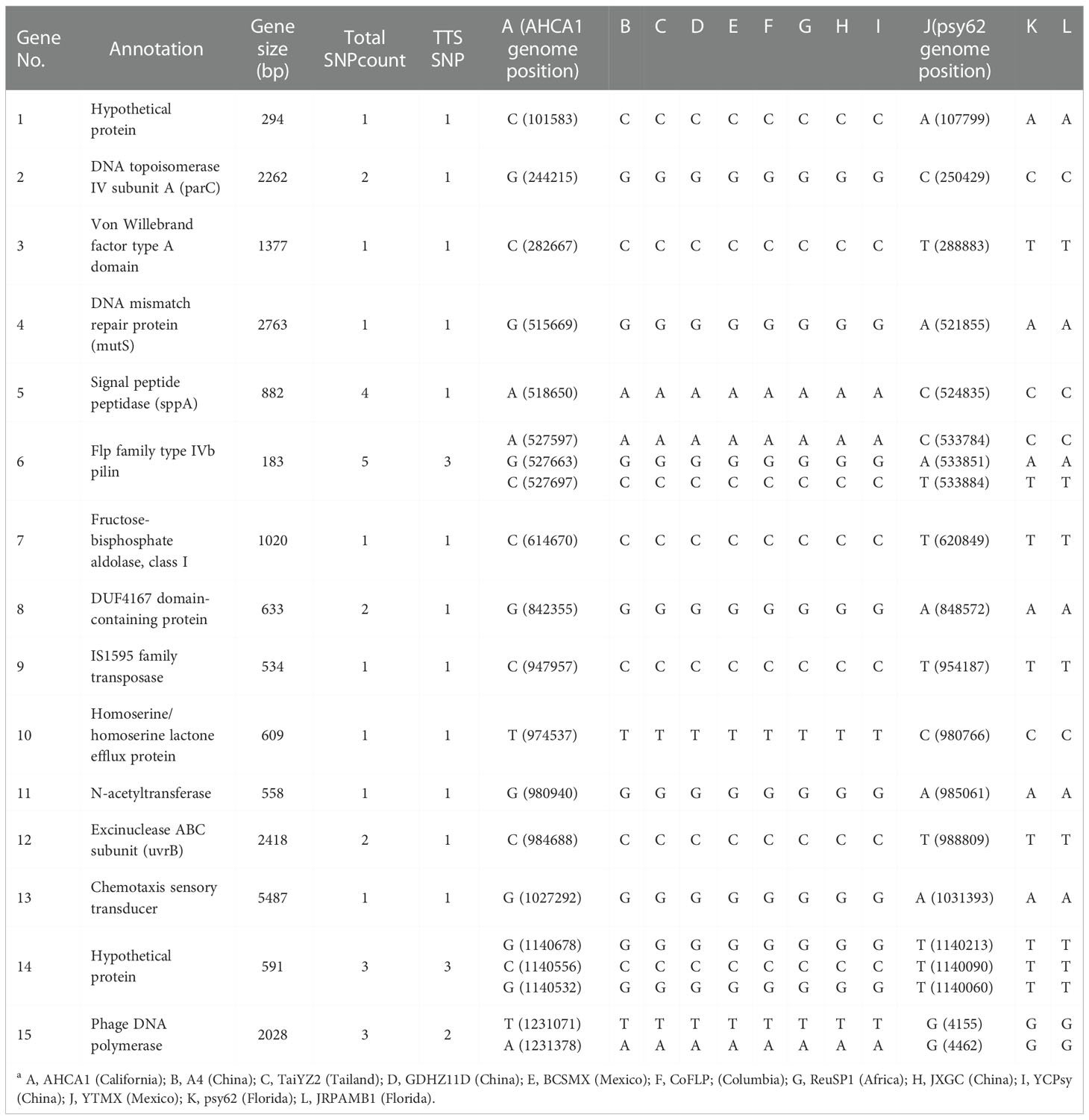
Table 4 List of genes with single nucleotide polymorphisms (SNPs) supporting the terL typing system (TTS) based on partial least squares discriminant analysis (sPLS-DA) on 12 selected genomes sequences of “Candidatus Liberibacter asiaticus” strains (column A to La).
The TTS SNPs identified through BLAST search against the 40 CLas genomes (12 complete and 28 draft) sequences are summarized in Supplementary Table 3. Because of the incomplete status, not all TSS SNPs were found in each draft genomes. A dendrogram based on the concatenated SNPs (Supplementary Table 3) is shown in Figure 3. The Term-A and Term-G clusters are clearly defined. For the non-typeable strains, LBR19TX2 and LBR23TX5 fall into the Term-G group; strains PA19 and PA20 formed a unique cluster within Term-A group, and strain Ishi-1 is within the Term-A group.
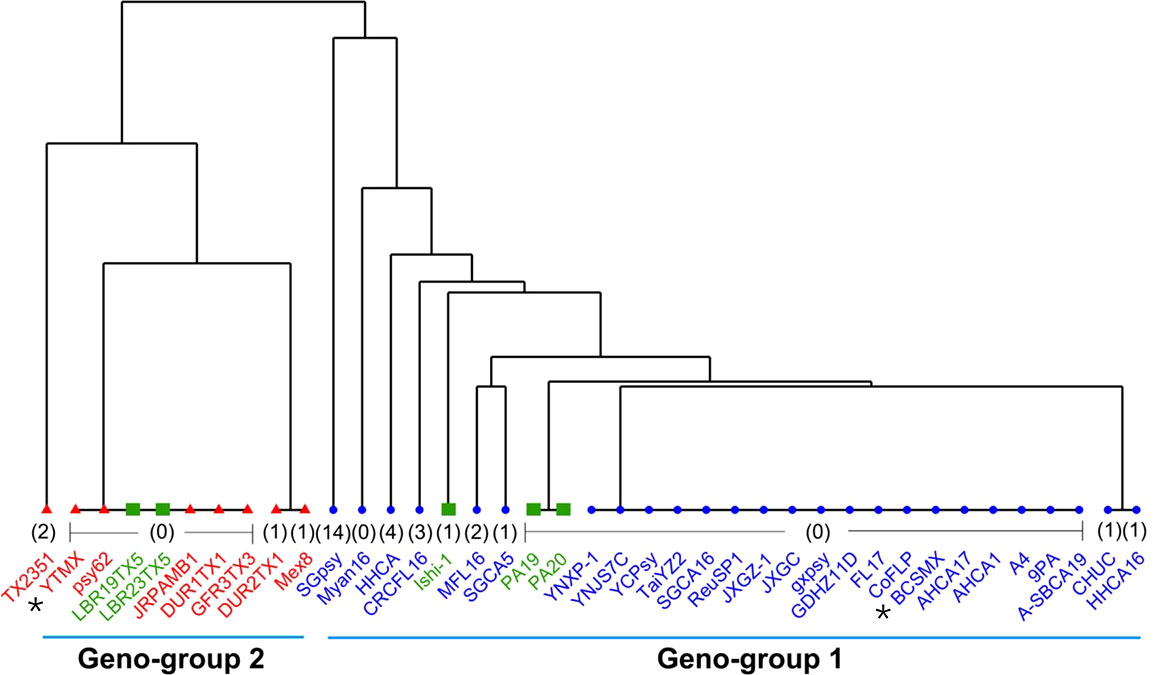
Figure 3 A dendrogram showing the Geno-group status of “Candidatus Liberibacter asiaticus” (CLas) strains with whole genome sequences available in GenBank based on concatenated SNPs from 15 TTS (terL typing system) supportive genes. Strains in blue are in Term-A group; Strain in red are in Term-G group; and strains in green are non-typeable for Term-A or Term-G. The number in bracket indicates absent SNPs of that strain due to the incomplete nature of the draft genome sequence. The two Mexican CLas strains in this study are indicated by *.
Discussions
An important contribution of this study was the identification of chromosomal core genes with SNPs for typing CLas strains from Mexico, as well as from other geographical regions (Table 4 and Supplementary Table 3). This was achieved by the analyses of CLas whole genome sequences using ML approaches. While the prophage-based TTS (terL typing system) has shown its capacity for CLas strain typing (Deng et al., 2014), there was no support from chromosomal genes until this study. Theoretically, each chromosomal gene can be analyzed one by one to identify TTS SNPs. This is apparently not practical for >1,000 genes in a CLas genome. The unsupervised ML (PCA) served as a first step to show the pattern of CLas strain clusters at the whole genome sequence level (Figure 2). Some strains fall in the border region between Term-A and Term-G groups and some strains were not typeable by TSS. The supervised ML (sPLS-DA) defined the CLas groups with SNPs in 14 core genes along with a prophage gene and resolved the CLas strain typing issue (Figure 3). Putting together all the evidence, two CLas genomic groups, designated as Geno-groups, are proposed: Geno-group 1 that extends the Term-A group, and Geno-group 2 that extends the Term-G group.
The two distinct Geno-groups in Mexico set the foundation for epidemiological analysis. First, it suggests that there were at least two different introductions of CLas in Mexico. The exact timelines and routes of introduction could not be determined with the current information. It is, however, clear that CLas strains in Mexico were related to those in the US (Figure 1). It is generally believed that long distant CLas spread is through infected propagative materials along with ACPs. The original source of CLas and ACP in Mexico is currently not clear (Fuentes et al., 2018). Mexico allows importation of citrus propagative material (buds) from the US and Spain. Although strict quarantine procedures were enforced through the Mexico Citrus Propagation Material Certification Program, CLas was not in the scope of a regulation until 2005 when HLB was detected in Florida. Furthermore, one cannot rule out illicit host material brought or shipped in as a source of introduction. However, it should be noted that for a better understanding of CLas introduction and spread, much more information, including ACP diversity and records of citrus material movements are needed.
With the detection of CLas (now confirmed as Geno-group 2) in Yucatan in 2009, the Mexico federal government implemented various phytosanitary actions to limit the spread of the disease. Through regulatory mechanisms, the movement of citrus plants within the country was restricted. The efforts appeared to be effective since Geno-group 2 is still restricted in the Peninsula states (Figure 1). However, there was evidence that Geno-group 1 strains spread quickly from west to east in the central and north of Mexico (Figure 1). CLas was recorded in the states of the Pacific Ocean coastal zone between December, 2009 and July, 2010, in the central states from April, 2011 to September, 2013, and finally in the states located in the Gulf of Mexico in December, 2015. The rapid dispersion of CLas from the Pacific coast to other areas has probably been favored by the prevalence of lemon plantations, whose almost continuous vegetative sprouting for most of the year favors the reproduction of the ACP (Robles-González et al., 2018).
The whole genome sequencing efforts reveal that both strains BCSMX and YTMX harbor prophages/phages (Table 3). BCSMX has only one Type 1 phage, and therefore belongs in the prophage typing group (PTG-1) that was also found in California (Dai et al., 2019). Strain YTMX contained Type 1 and Type 2 phages (PTG-1-2) like those from Florida (Zhang et al., 2011) and Texas (Kunta et al., 2012). This supports the finding of two different prophage types of CLas, PTG-1-2 in Peninsula and South of Mexico and PTG1 in Central and Northern Mexico (Figure 1) (Alanís-Martínez et al., 2018; Martínez-Carrillo et al., 2019).
In addition, CLasMV1, a single-stranded DNA phage recently reported in China (Zhang et al., 2021), was found in strain YTMX. This is the first report of CLasMV1 outside China, although fractions of CLasMV1 sequences (but not the complete phage genome) could be found in some published CLas genomes (Zhang et al., 2021). The CLasMV1 sequence from YTMX was highly similar to those described in the Chinese CLas strains (34 gaps, 11 SNPs, data not shown). No CLasMV1 type of phage was found in strain BCSMX. The different status of CLasMV1 between strains YTMX and BCSMX adds further evidence to support the two CLas Geno-groups in Mexico.
The establishment of two CLas Geno-groups encourages CLas subspecies research. Intra-specific diversity information is important for both CLas biology research and HLB management. There have not been reports on subspecies in CLas, despite the many efforts of strain diversity investigations. Multiple subspecies have been proposed for CLaf, the pathogen of the African form HLB (Garnier et al., 2000; Roberts et al., 2015; Roberts and Pietersen, 2017). Along this line, it is also noted that, based on PCA results (Figure 2) and core gene SNPs (Figure 3), the non-TTS typeable strains, PA19 and PA20, from Pakistan (Cui et al., 2021) could represent a new Geno-group although more studies are needed in the future.
Conclusion
This study characterized the genomic diversity of CLas strains from Mexico using ML assisted genomic approaches. Two CLas Geno-groups were established. Research results suggested that 1) there were at least two different introductions of CLas into Mexico; 2) CLas strains between Mexico and USA are closely related; and 3) The two Geno-groups provides a base for future CLas subspecies study.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
JC, XD, and JH designed the project, conducted the data analysis, and wrote the manuscript. IA-M, LK and AP performed the samples collection and facilitate sample and data processing. ZD and ZZ assisted in data analyses. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by USDA-ARS base fund 2034-22000-012-00D, California Citrus Research Board (5300-188), Key-Area Research and Development Program of Guangdong Province (2019B020217003).
Acknowledgments
We thank Y. Andrade for technical assistance. Any mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture (USDA). USDA is an equal-opportunity employer. In-silico analysis was supported by Interdisciplinary Intelligence SuperComputer Center of Beijing Normal University at Zhuhai.
Conflict of interest
The handling editor WW declared a shared parent affiliation with the author(s) JC, APL at the time of review.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1052680/full#supplementary-material
Supplementary Table 1 | General information of “Candidatus Liberibacter” genome sequences available in GenBank database (version 249)
Supplementary Table 3 | List of single nucleotide polymorphisms (SNPs) in genes supporting the terL typing system (TTS) from 26 draft genomes sequences of “Candidatus Liberibacter asiaticus” strains with Strain AHCA1 and Psy62 as references highlighted in yellow.
References
Abdi, H., Williams, L. J. (2010). Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat. 2, 433–459. doi: 10.1002/wics.101
Alanís-Martínez, E. I., López-Arroyo, J. I., Cora-Valencia, E., Torres-Martinez, G., López-Buenfil, A. (2018). Characterization of Candidatus liberibacter asiaticus populations in Mexico, based on presence of prophages. Rev. Mexicana Fitopatología 36, S83.
Andrews, S. (2010) FastQC: A quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Bao, M., Zheng, Z. Z., Sun, X., Chen, J., Deng, X. (2020). Enhancing PCR capacity to detect “Candidatus liberibacter asiaticus” utilizing whole genome sequence information. Plant Dis. 104, 527–532. doi: 10.1094/PDIS-05-19-0931-RE
Bastianel, C., Garnier-Semancik, M., Renaudin, J., Bové, J. M., Eveillard, S. (2005). Diversity of “Candidatus liberibacter asiaticus” based on the omp gene sequence. Appl. Environ. Microbiol. 71, 6473–6478. doi: 10.1128/AEM.71.11.6473-6478.2005
Bové, J. M. (2006). Huanglongbing: A destructive, newly-emerging, century-old disease of citrus. J. Plant Pathol. 88, 7–37. doi: 10.4454/jpp.v88i1.828
Chen, J., Deng, X., Sun, X., Jones, D., Irey, M., Civerolo, E. (2010). Guangdong and florida populations of “Candidatus liberibacter asiaticus” distinguished by a genomic locus with short tandem repeats. Phytopathology 100, 567–572. doi: 10.1094/PHYTO-100-6-0567
Cui, X., Liu, K., Atta, S., Zeng, C., Zhou, C., Wang, X. (2021). Two unique prophages of “Candidatus liberibacter asiaticus” strains Pakistan. Phytopathology 111, 784–788. doi: 10.1094/PHYTO-10-20-0454-SC
Dai, Z., Wu, F., Zheng, Z., Yokomi, R., Kumagai, L., Cai, W., et al. (2019). Prophage diversity of ‘Candidatus liberibacter asiaticus’ strains in California. Phytopathology 109, 551–559. doi: 10.1094/PHYTO-06-18-0185-R
Deng, X., Chen, J., Feng, Z., Shan, Z., Guo, H., Zhu, J., et al. (2008). Identification and characterization of the huanglongbing bacterium in pummelo from multiple locations in guangdong, p. r. China. Plant Dis. 92, 513–518. doi: 10.1094/PDIS-92-4-0513
Deng, X., Lopes, S., Wang, X., Sun, X., Jones, D., Irey, M., et al. (2014). Characterization of “Candidatus liberibacter asiaticus” populations by double-locus analyses. Curr. Microbiol. 69, 554–560. doi: 10.1007/s00284-014-0621-9
Duan, Y., Zhou, L., Hall, D. G., Li, W., Doddapaneni, H., Lin, H., et al. (2009). Complete genome sequence of citrus huanglongbing bacterium, “Candidatus liberibacter asiaticus” obtained through metagenomics. Mol. Plant-Microbe Interact. 22, 1011–1020. doi: 10.1094/MPMI-22-8-1011
Fu, S., Bai, Z., Su, H., Liu, J., Hartung, J. S., Zhou, C., et al. (2020). Occurrence of prophage and historical perspectives associated with the dissemination of huanglongbing in mainland China. Plant Pathol. 69, 132–138. doi: 10.1111/ppa.13100
Fuentes, A., Braswell, W. E., Ruiz-Arce, R., Racelis, A. (2018). Genetic variation and population structure of diaphorina citri using cytochrome oxidase I sequencing. PloS One 13 (6), 1–17. doi: 10.1371/journal.pone.0198399
Garnier, M., Jagoueix-Eveillard, S., Cronje, P. R., Le Roux, H. F., Bové, J. M. (2000). Genomic characterization of a liberibacter present in an ornamental rutaceous tree, calodendrum capense, in the Western cape province of south africa. proposal of “Candidatus liberibacter africanus subsp. capensis.” Int. J. Syst. Evol. Microbiol. 50, 2119–2125. doi: 10.1099/00207713-50-6-2119
Ghosh, D. K., Bhose, S., Motghare, M., Warghane, A., Mukherjee, K., Ghosh, D. K., et al. (2015). Genetic diversity of the Indian populations of “Candidatus liberibacter asiaticus” based on the tandem repeat variability in a genomic locus. Phytopathology 105, 1043–1049. doi: 10.1094/PHYTO-09-14-0253-R
Halbert, S. (2005). “The discovery of huanglongbing in Florida,” in Proc. 2nd int. citrus canker and huanglongbing research, workshop, vol. H-3. Available at: https://swfrec.ifas.ufl.edu/hlb/database/pdf/22_CankerHuang_05.pdf.
Huang, J., Dai, Z., Zheng, Z., da Silvia, P. A., Kumagai, L., Xiang, Q., et al. (2021). Bacteriomic analyses of Asian citrus psyllid and citrus samples infected with “Candidatus liberibacter asiaticus” in southern California and huanglongbing management implications. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.683481
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 1–8. doi: 10.1038/s41467-018-07641-9
Katoh, H., Miyata, S., Inoue, H., Iwanami, T. (2014). Unique features of a Japanese 'Candidatus liberibacter asiaticus' strain revealed by whole genome sequencing. PloS One 9, e106109. doi: 10.1371/journal.pone.0106109
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Katoh, H., Subandiyah, S., Tomimura, K., Okuda, M., Su, H. J., Iwanami, T. (2011). Differentiation of “Candidatus liberibacter asiaticus” isolates by variable-number tandem-repeat analysis. Appl. Environ. Microbiol. 77, 1910–1917. doi: 10.1128/AEM.01571-10
Kumagai, L. B., LeVesque, C. S., Blomquist, C. L., Madishetty, K., Guo, Y., Woods, P. W., et al. (2013). First report of “Candidatus liberibacter asiaticus” associated with citrus huanglongbing in California. Plant Dis. 97 (2), 282–283. doi: 10.1094/PDIS-09-12-0845-PDN
Kunta, M., Sétamou, M., Skaria, M., Li, W., Nakhla, M. K., da Graça, J. V. (2012). First report of citrus huanglongbing in Texas. Phytopathology 102, S4. 66.
Lê Cao, K. A., Boitard, S., Besse, P. (2011). Sparse PLS discriminant analysis: Biologically relevant feature selection and graphical displays for multiclass problems. BMC Bioinf. 12. doi: 10.1186/1471-2105-12-253
Lee, L. C., Liong, C. Y., Jemain, A. A. (2018). Partial least squares-discriminant analysis (PLS-DA) for classification of high-dimensional (HD) data: A review of contemporary practice strategies and knowledge gaps. Analyst 143, 3526–3539. doi: 10.1039/c8an00599k
Li, W., Hartung, J. S., Levy, L. (2006). Quantitative real-time PCR for detection and identification of Candidatus liberibacter species associated with citrus huanglongbing. J. Microbiol. Methods 66, 104–115. doi: 10.1016/j.mimet.2005.10.018
Liu, R., Zhang, P., Pu, X., Xing, X., Chen, J., Deng, X. (2011). Analysis of a prophage gene frequency revealed population variation of “Candidatus liberibacter asiaticus” from two citrus-growing provinces in China. Plant Dis. 95, 431–435. doi: 10.1094/PDIS-04-10-0300
Ma, W., Liang, M., Guan, L., Xu, M., Wen, X., Deng, X., et al. (2014). Population structures of “Candidatus liberibacter asiaticus” in southern China. Phytopathology 104, 158–162. doi: 10.1094/PHYTO-04-13-0110-R
Martínez-Carrillo, J. L., Suarez-Beltrán, A., Nava-Camberos, U., Aguilar-Medel, S., Valenzuela-Lagarda, J., Gutiérrez-Coronado, M. A., et al. (2019). Successful area-wide management of the Asian citrus Psyllid1 in southwestern Sonora, méxico. Southwest Entomol. 44, 173. doi: 10.3958/059.044.0119
Page, A. J., Taylor, B., Delaney, A. J., Soares, J., Seemann, T., Keane, J. A., et al. (2016). SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genomics 2, e000056. doi: 10.1099/mgen.0.000056
Roberts, R., Pietersen, G. (2017). A novel subspecies of ‘Candidatus liberibacter africanus’ found on native teclea gerrardii (Family: Rutaceae) from south Africa. Antonie van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 110, 437–444. doi: 10.1007/s10482-016-0799-x
Roberts, R., Steenkamp, E. T., Pietersen, G. (2015). Three novel lineages of ‘Candidatus liberibacter africanus’ associated with native rutaceous hosts of trioza erytreae in south africa. Int. J. Syst. Evol. Microbiol. 65, 723–731. doi: 10.1099/ijs.0.069864-0
Robles-González, M. M., Orozco-Santos, M., Manzanilla-Ramírez, M.Á., Velázquez-Monreal, J. J., Mediana-Urrutia, V. M., Sanches-Stuchi, E. (2018). Experiencias con huanglonbing en limón mexicano en el estado de colima, méxico. Citrus Res. Technol. 39, 1–12. doi: 10.4322/crt.16518
Rohart, F., Gautier, B., Singh, A., Lê Cao, K. A. (2017). mixOmics: An r package for ‘omics feature selection and multiple data integration. PloS Comput. Biol. 13, 1–19. doi: 10.1371/journal.pcbi.1005752
Seemann, T. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
SENASICA (2022). “Acciones contra el huanglongbing y su vector en méxico,” in Dirección general de sanidad vegetal. dirección de protección fitosanitaria. Available at: https://www.gob.mx/cms/uploads/attachment/file/722452/01_Enero_2022_Plagas_de_los_c_-_tricos.pdf.
Singh, Y. H., Sharma, S. K., Sinha, B., Baranwal, V. K., Singh, N. B., Chanu, N. T., et al. (2019). Genetic variability based on tandem repeat numbers in a genomic locus of ‘Candidatus liberibacter asiaticus’ prevalent in north east india. Plant Pathol. J. 35, 644–653. doi: 10.5423/PPJ.OA.03.2019.0061
Thapa, S. P., De Francesco, A., Trinh, J., Gurung, F. B., Pang, Z., Vidalakis, G., et al. (2020). Genome-wide analyses of liberibacter species provides insights into evolution, phylogenetic relationships, and virulence factors. Mol. Plant Pathol. 21, 716–731. doi: 10.1111/mpp.12925
Thompson, J. D., Higgins, D. G., Gibson, T. J. (1994). CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Tonkin-Hill, G., MacAlasdair, N., Ruis, C., Weimann, A., Horesh, G., Lees, J. A., et al. (2020). Producing polished prokaryotic pangenomes with the panaroo pipeline. Genome Biol. 21, 1–21. doi: 10.1186/s13059-020-02090-4
Trujillo-Arriaga, F. J. (2011). HLB en México. Situación actual, regulación y perspectivas de áreas regionales para el manejo del HLB. 3er Encuentro Internacional de Investigación en Cítricos. Septiembre de 2011. (Veracruz, México: Martínez de la Torre).
Zhang, S., Flores-Cruz, Z., Zhou, L., Kang, B. H., Fleites, L. A., Gooch, M. D., et al. (2011). “Ca. liberibacter asiaticus” carries an excision plasmid prophage and a chromosomally integrated prophage that becomes lytic in plant infections. Mol. Plant-Microbe Interact. 24, 458–468. doi: 10.1094/MPMI-11-10-0256
Zhang, L., Li, Z., Bao, M., Li, T., Fang, F., Zheng, Y., et al. (2021). A novel microviridae phage (CLasMV1) from “Candidatus liberibacter asiaticus”. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.754245
Zheng, Z., Deng, X., Chen, J. (2014). Whole-genome sequence of “Candidatus liberibacter asiaticus” from guangdong, China. Genome Announc. 2, 3–4. doi: 10.1128/genomeA.00273-14
Zheng, Y., Huang, H., Huang, Z., Deng, X., Zheng, Z., Xu, M. (2021). Prophage region and short tandem repeats of “Candidatus liberibacter asiaticus” reveal significant population structure in China. Plant Pathol. 70, 959–969. doi: 10.1111/ppa.13332
Zheng, Z., Wu, F., Kumagai, L. B., Polek, M., Deng, X., Chen, J. (2017). Two “Candidatus liberibacter asiaticus” strains recently found in California harbor different prophages. Phytopathology 107, 662–668. doi: 10.1094/PHYTO-10-16-0385-R
Keywords: “Candidatus Liberibacter asiaticus”, citrus HLB, mexico HLB, genomic diversity, machine learning, single nucleotide polymorphisms, sparse partial least squares discriminant analysis (sPLS-DA), principal component analysis (PCA)
Citation: Huang J, Alanís-Martínez I, Kumagai L, Dai Z, Zheng Z, Perez de Leon AA, Chen J and Deng X (2022) Machine learning and analysis of genomic diversity of “Candidatus Liberibacter asiaticus” strains from 20 citrus production states in Mexico. Front. Plant Sci. 13:1052680. doi: 10.3389/fpls.2022.1052680
Received: 24 September 2022; Accepted: 23 November 2022;
Published: 15 December 2022.
Edited by:
Wei Wei, Agricultural Research Service (USDA), United StatesReviewed by:
Bo Min Kim, Oak Ridge Institute for Science and Education (ORISE), United StatesXuefeng Wang, Citrus Research Institute, Chinese Academy of Agricultural Sciences, China
Copyright © 2022 Huang, Alanís-Martínez, Kumagai, Dai, Zheng, Perez de Leon, Chen and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianchi Chen, SmlhbmNoaS5jaGVuQHVzZGEuZ292; Xiaoling Deng, eGxkZW5nQHNjYXUuZWR1LmNu