 Seema Parveen1
Seema Parveen1 Nutan Singh2
Nutan Singh2 Arjun Adit1
Arjun Adit1 Suman Kumaria2
Suman Kumaria2 Rajesh Tandon1
Rajesh Tandon1 Manu Agarwal1
Manu Agarwal1 Arun Jagannath1
Arun Jagannath1 Shailendra Goel1*
Shailendra Goel1*- 1Department of Botany, University of Delhi, New Delhi, India
- 2Department of Botany, North-Eastern Hill University, Shillong, India
Nymphaea, commonly known as water lily, is the largest and most widely distributed genus in the order Nymphaeales. The importance of Nymphaea in wetland ecosystems and their increased vulnerability make them a great choice for conservation and management. In this work, we studied genetic diversity in a collection of 90 N. micrantha and 92 N. nouchali individuals from six different states of India, i.e., Assam, Manipur, Meghalaya, Maharashtra, Goa, and Kerala, using simple sequence repeat (SSR) markers developed by low throughput Illumina sequencing (10X coverage of genome) of N. micrantha. Nymphaea nouchali is native to India, whereas N. micrantha is suggested to be introduced to the country for its aesthetic and cultural values. The study revealed extensive polymorphism in N. nouchali, while in N. micrantha, no apparent genetic divergence was detected prompting us to investigate the reason(s) by studying the reproductive biology of the two species. The study revealed that N. micrantha predominantly reproduces asexually which has impacted the genetic diversity of the species to a great extent. This observation is of immense importance for a successful re-establishment of Nymphaea species during restoration programs of wetlands. The information generated on reproductive behaviors and their association with genotypic richness can help in strategizing genetic resource conservation, especially for species with limited distribution. The study has also generated 22,268 non-redundant microsatellite loci, out of which, 143 microsatellites were tested for polymorphism and polymorphic markers were tested for transferability in five other Nymphaea species, providing genomic resources for further studies on this important genus.
Introduction
Nymphaea, an aquatic perennial herb commonly known as water lily, is the largest and most widely distributed genus in the order Nymphaeales. The genus is represented by about 45–50 species with a worldwide distribution in the temperate and tropical regions (Borsch et al., 2007). Nymphaea is divided into two main groups, Apocarpae and Syncarpiae, which in turn, are divided into five subgenera, viz., Anecphya, Brachyceras, and Hydrocallis, Lotos, and Nymphaea, respectively (Conard, 1905). It is a small group of flowering plants but generates considerable interest in the context of the origin of angiosperms. It belongs to the ANA-grade (Amborellales, Nymphaeales, and Austrobaileyales) of angiosperms, which forms the first three branches of the phylogenetic tree of angiosperms.
Nymphaea is popular as an ornamental plant, as well as for food and medicine in many parts of the world. Different parts of the plant, such as rhizome, tuber, leaf, fruit, and seeds, have been used as food in different ways by people in various parts of the world, such as West Africa, Madagascar, Australia, Uganda, Poland, and Central America and Asia (Conard, 1905; Irvine and Trickett, 1953). In many religions prevalent in Asia, such as Hindu and Buddhist religions, water lilies are considered sacred (Ansari and Jeeja, 2009; Yakandawala et al., 2017). In India, it is being favored by plant growers and cultivated in gardens and nurseries throughout the country for its much-admired flower colors (Ansari and Jeeja, 2009). In most freshwater wetlands, Nymphaea spp. acts as an important component of plant communities. They provide food and habitat for many aquatic animals and migratory birds, including Siberian cranes and lily-trotters (Jacanas), act to reduce water turbidity and provide in-stream sediment stabilization (Leito et al., 2016; Ranjan and Prakash, 2019; Dalziell et al., 2020). Nymphaea species such as N. mexicana and N. violacea are sensitive to salt stress and are known to occur in freshwater only (Lauer and Ross, 2016; Dalziell et al., 2020). Thus, Nymphaea can also act as a significant wetland health indicator (Dalziell et al., 2020). Therefore, the loss of Nymphaea plants would be detrimental not only nutritionally and culturally, but also ecologically.
There are 10 species of Nymphaea reported from India, of which six (N. nouchali, N. alba, N. candida, N. pubescens, N. rubra, and N. tetragona) are wild and four (N. × khurooi, N. caerulea, N. × marliacea, and N. micrantha) are cultivated (Mitra, 1990). The species N. nouchali, N. rubra, and N. pubescens are distributed throughout the plains of India, whereas others are confined to some areas. Among these, Nymphaea nouchali, a wildly growing species, is well known for its medicinal values and Nymphaea micrantha, a cultivated species, is well known for its cultural and aesthetic values.
Nymphaea nouchali Burm. f. is native to the tropical and subtropical old world to North Australia range, but also found to be distributed in Brazil, Argentina, and South Australia (Plants of the world online; POWO, 2021). It is a well-known medicinal plant, extensively used for the treatment of diabetes, inflammation, liver disorders, urinary disorders, menorrhagia, blennorrhagia, menstruation problem, as an aphrodisiac, and as a bitter tonic (Raja et al., 2010). Nymphaea micrantha Guill. and Perr. is native to West Tropical Africa to Chad range (Wiersema, 1988; POWO, 2021), and is known for its cultural and aesthetic values. In India, the N. nouchali and N. micrantha are the two widely distributed species from the genus Nymphaea. Nymphaea nouchali is most used for its rhizome and in traditional medicine under Ayurveda and Siddha traditional systems, whereas N. micrantha is used for its flowers. In North-East India, the cut flowers of N. micrantha are sold in the market and are offered to gods. Nymphaea nouchali is native to India (Singh and Jain, 2017), whereas N. micrantha is suggested to be introduced as such (Ansari and Jeeja, 2009), although, the source of introduction of N. micrantha is not known. These two species were collected to study genetic diversity patterns using microsatellite markers.
The genetic diversity of a species is important for its long-term survival, adaptability, and evolution with changing environmental conditions (Frankham, 2005; Kahilainen et al., 2014). Reduction in allele diversity or heterozygosity can affect the resilience and adaptive potential of a species (Reed and Frankham, 2003). The genus Nymphaea is phenotypically diverse and exhibits high levels of inter-specific polymorphism (Heslop-Harrison, 1955). However, studies on molecular genetic diversity and structure within and among populations of Nymphaea spp. are limited despite the rich genetic resources worldwide. A limited number of studies have used molecular markers, such as inter simple sequence repeats (ISSR), amplified fragment length polymorphism (AFLP), and random amplified polymorphic DNA (RAPD) for assessing the genetic diversity and resolving relationships among Nymphaea species (Woods et al., 2005b; Volkova et al., 2010; Poczai et al., 2011; Dkhar et al., 2013). Genic and inter-genic spacers viz. ITS, chloroplast trnT-trnF regions, matK have also been used for elucidating phylogenetic relationships among different Nymphaea species (Woods et al., 2005a; Borsch et al., 2007; Löhne et al., 2008). A study with co-dominant molecular markers with a large number of samples representing diverse geographical areas is missing. The simple sequence repeats (SSRs), because of their high degree of polymorphism and co-dominant nature, are useful in conservation genetics. Their wide genome coverage and efficient transfer to closely related species make them a marker of choice to assess genetic variation and structure among and within populations (Kalia et al., 2011), assigning inbred lines to heterotic groups (Ganapathy et al., 2012; Sajib et al., 2012), and fingerprinting of genotypes (Lörz and Wenzel, 2005).
In this work, we collected 90 N. micrantha and 92 N. nouchali var. nouchali individuals from different regions of India and assessed their genetic diversity and population structure using SSR markers. The specific objectives of the study were: (1) to develop and characterize SSR markers for Nymphaea in terms of frequency, genomic distribution, information content, and transferability to related species, and (2) to assess genetic diversity and population structure of the two aforementioned Nymphaea species. We discuss the importance of genetic diversity as a resource used to ensure the species success in varied environments. Emphasis is placed on the significance of genetic diversity in the conservation of biodiversity. This is the first report on the genetic diversity and population structure of Nymphaea species using genomic SSR markers. Therefore, the genetic information obtained in this study will provide useful resources for various genetic studies in Nymphaea and guide strategies to conserve wild species.
Materials and Methods
Survey and Collection of Plant Material
Wetland areas of different states of North-East India (Assam, Meghalaya, and Manipur), Western India (Maharashtra, Goa), and Southern India (Kerala) were explored during the years 2017–2020 to survey and collect biological samples of the genus Nymphaea. Sixteen populations of N. micrantha and 27 populations of N. nouchali consisting of 90 and 92 individuals, respectively, were collected from different locations in Assam, Meghalaya, Manipur, Goa, Maharashtra, and Kerala. The species were identified by Prof. Santhosh Nampy and Dr. A. K. Pradeep, University of Calicut, Kerala, and the voucher specimens were deposited at the herbarium of Department of Botany, University of Delhi (DUH14710-DUH14732) and Botanical Survey of India, Eastern Regional Centre, Meghalaya (ASSAM96585-ASSAM96610). The plant material of 3–5 individuals per population was collected, dried in silica, and brought to the laboratory for DNA extraction. To avoid sampling from the same plant and capture more diversity, the samples were collected from the plants growing at a greater distance than 5 m. Individuals collected from a single wetland were assigned to one population. All the accessions studied in the present study are tabulated in Table 1.
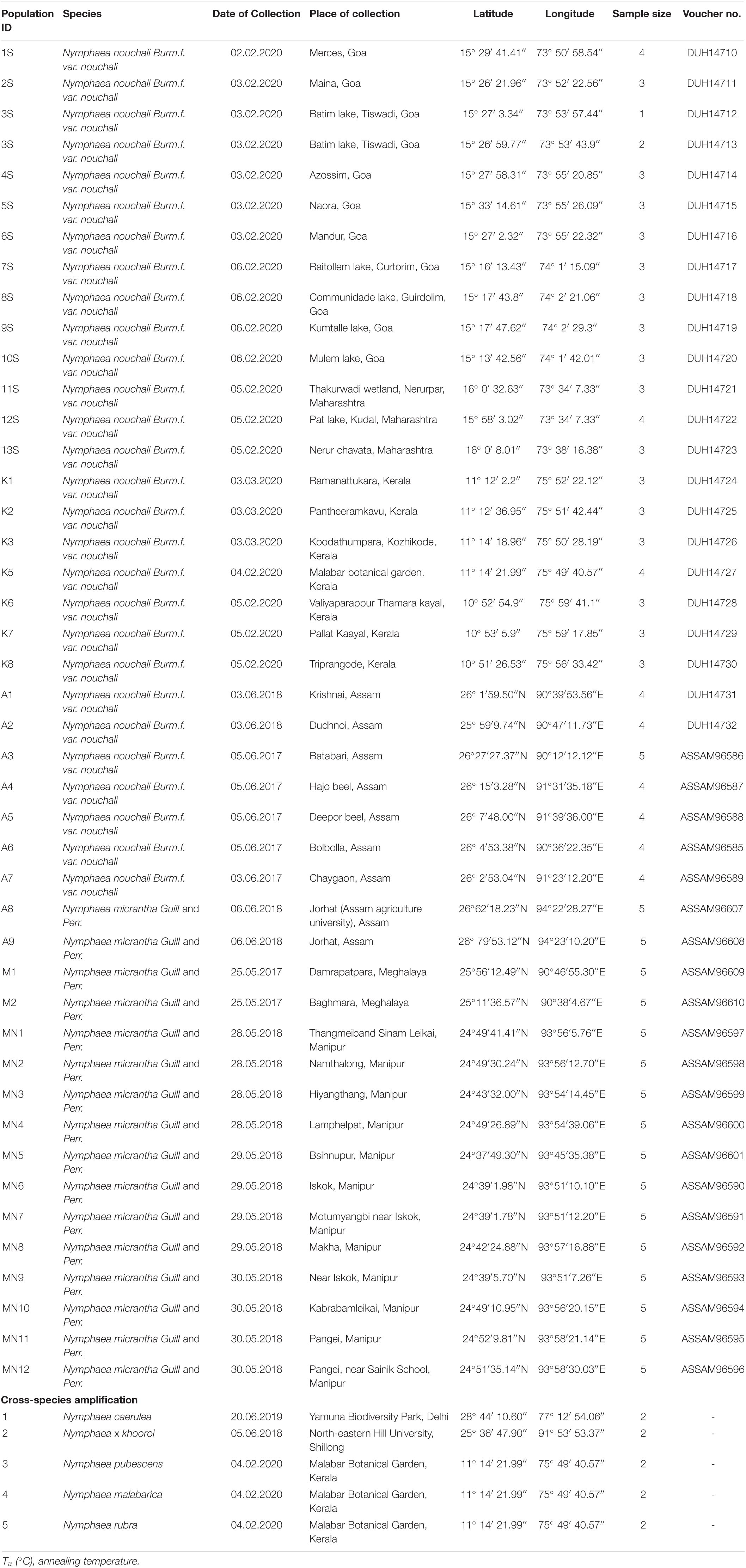
Table 1. Details of plant samples collected and used in this study with their respective geographic locations (latitude and longitude) in six different states of India.
To assess the cross-species transferability of the SSR markers, a few other Nymphaea spp. were also collected (Table 1). Three species, N. Pubescens, N. malabárica, and N. rubra, were collected from the germplasm growing in the wetlands of Malabar botanical garden, Kozhikode, Kerala. Two other species viz. N. × khurooi (earlier named as N. alba var. rubra) and N. caerulea were collected from North-eastern Hill University (NEHU), Shillong, and Yamuna Biodiversity Park, Delhi, respectively.
Genomic DNA Extraction
Genomic DNA was extracted from the silica dried leaf tissues brought from the field using CTAB (hexadecyltrimethylammonium bromide) method described by Doyle and Doyle (1987) with a few modifications. The modifications involved an extended incubation of 1 h in CTAB buffer, an extended RNase treatment of 1 h, a phenol-chloroform extraction (phenol:chloroform: isoamyl alcohol; 25:24:1), and final precipitation by adding 1/10 volume of 3 M sodium acetate and an equal volume of chilled ethanol. The quality and quantity of the extracted DNA were estimated by electrophoresis on a 0.8% agarose gel and using a NanoDrop spectrophotometer (NanoDrop, Wilmington, DE, United States). The final concentration of the DNA was adjusted to 50 ng/μl and the DNA was stored at 4°C.
Genome Size Estimation
The genome size of N. micrantha was estimated by flow cytometry. About 1 cm2 of the young fresh leaf was used for sample preparation using the CyStain® PI Absolute P kit (Sysmex, Germany) following manufacturer instructions. Solanum lycopersicum L. “Stupicke’ polnı’ rane”’ 1C = 0.98 pg (Doležel et al., 1992) was used as an internal standard reference. In brief, the nuclei of the standard and the sample were isolated, stained, and analyzed simultaneously. The nuclear DNA content was determined using the CyFlow® Cube 8 flow cytometer (Sysmex, Germany) for each sample. For both, the standard reference and N. micrantha samples were prepared in replicates, and each was run three times with a minimum of 2,500 nuclei count per sample. The fluorescence values were converted to the DNA content using the following formula:
Genome Sequencing and de novo Assembly
The high-quality genomic DNA of N. micrantha was used for genome sequencing. The sequencing library was prepared using the NEBNext Ultra DNA library prep kit for Illumina (New England BioLabs, United States) following the manufacturer’s instructions. In brief, the genomic DNA was fragmented by sonication, followed by adaptor ligation (TruSeq Index3 adaptor; GATCGGAAGAGCACACGTCTGAACTCCAGTCACTTAGG CATCTCGTATGCCGTCTTCTGCTTG). The adaptor-ligated fragments were then PCR amplified using NEBnext universal (P3; AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTA CACGACGCTCTTCCGATC*T) and Index3 tagged NEBnext indexed primer (P7; CAAGCAGAAGACGGCATACGAGATG CCTAAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC) and gel purified. The concentration of the prepared library was quantified by Qubit 4.0 Fluorometer (Thermo Fisher Scientific, United States) and sequenced on Illumina HiSeq 2000 platform in paired-end sequencing mode (Illumina, United States) by Macrogen Inc., (South Korea). The FastQ files containing the raw reads were submitted to the sequence read archive (SRA) at National Centre for Biotechnology Information (NCBI) under the accession number PRJNA750447. The raw data was filtered for high-quality reads [Reads with ≥ 70% HQ bases (Q ≥ 20)] using the NGS QC toolkit (Patel and Jain, 2012) at default parameters. High-quality reads obtained were used for de novo assembly using Velvet (v1.2.10), with the k-mer length of 55. The assembled sequences were filtered to all contigs greater than or equal to 1,000 bp to remove fragmentation of the assembly.
Identification of Microsatellites and Functional Annotation
The assembled sequences of the N. micrantha genome were mined for perfect microsatellites using MISA (MicroSAtellite identification tool; Thiel et al., 2003). The minimum numbers of repeats used to select the microsatellites were sixteen for mono-, nine for di-, six for tri-, and five for tetra-, penta- and hexanucleotide repeats. For functional annotation, the SSR containing sequences were searched against the NCBI non-redundant (nr) protein sequence database using the BLASTx program with an e-value cutoff 1e–5 or better. Further, Gene Ontology (GO) terms were assigned by adopting the terms that had been assigned to the top hit sequences of the database and mapping them to the higher-level categories using the Functional Annotation Tool of DAVID Bioinformatics Resources 6.8 (Huang et al., 2009).
Primer Design
Sequences harboring SSRs with a minimum repeat length of 20 bases and optimum flanking regions (≥50 nucleotides on both flanks of SSR) were used to design primers. The web-based program, Primer3Plus1, was used to design primer pairs from the SSR flanking regions. The parameters for designing primers were—primer length of 18–25 with an optimum of 22 bases; product size range of 100–500 with an optimum of 250 bases; annealing temperature between 5 and 65°C with an optimum value of 60°C and GC content between 40 and 80%, with an optimum of 60%. Other parameters, such as overall self and 3′ self-complementarity, penalty weight, and sequence quality scores, were kept at the default setting. A total of 217 primer pairs were designed and synthesized at Eurofins Genomics India Pvt. Ltd., India (Supplementary Table 1). Additionally, the 43 genic SSR markers were designed from the transcriptomic data (unpublished data, S. Parveen and S. Goel) (Supplementary Table 1).
Marker Amplification
The 43 genic-SSR, designated as NymTr_1 to NymT_43 and a subset of 100 non-genic SSR markers, designated as Nym_NGS1 to Nym_NGS100 were chosen for experimental validation. These markers were tested for amplification using two N. micrantha DNA samples. The PCR reaction was conducted in a total reaction volume of 15 μl containing 1x PCR buffer, 2 mM of MgCl2, 0.2 mM of each dNTP, 0.4 mM each of forward and reverse primers, 50 ng of template genomic DNA, and 1.25 U of Taq DNA polymerase (iNtRON Biotechnology, South Korea). The DNA amplification was performed in a SimpliAmp™ thermal cycler (Applied Biosystems, United States) with the following cycling parameters: initial denaturation at 94°C for 3 min followed by 30 cycles at 94°C for 20 s, optimized primer annealing temperature (between 5 and 60°C) for 20 s, DNA extension at 72°C for 30 s, and a final extension at 72°C for 7 min. The amplified products were initially resolved on 2% agarose and visualized under UV-light after ethidium bromide staining.
Genotyping and Cross-Species Transferability
Primers producing a clear unambiguous band were selected and further analyzed on 6% polyacrylamide gel for all the accessions of N. micrantha and N. nouchali. Amplicon size was determined by comparing it with a 100 bp DNA ladder (GeneDireX, Inc.). Unambiguously amplified alleles for each SSR locus were scored individually and coded as for their integer size in base pairs (bp). To avoid genotyping errors due to PCR-based amplification failures, the software Micro-Checker 2.2.3 (Van Oosterhout et al., 2004), was used to estimate the errors in allele scoring, presence of null alleles, large allele dropouts, or stuttering during PCR amplification. Null allele frequency (NAF) for each locus was calculated based on the formulas of Chakraborty et al. (1994) and Brookfield (1996). The polymorphic microsatellite markers were further assessed for their cross-species transferability in five Nymphaea species viz. N. pubescence, N. malabarica, N. rubra, N. × khurooi, and N. caerulea (see Supplementary Notes).
Diversity Analysis of Simple Sequence Repeat Markers
Various statistical genetic diversity estimates such as an average number of alleles (Na), overall, an effective number of alleles (Ne), observed (Ho) and expected (He) heterozygosity, Fst = Nei’s differentiation index (Fst), and Gene flow (Nm) were calculated for each locus using GenAlEx 6.5 (Smouse and Peakall, 2012). The polymorphism information content (PI) for each of the primer pairs has been calculated using the package POLYSAT (Clark and Jasieniuk, 2011), with the following formula.
Where n is the number of alleles, and Pi and Pj are the frequencies of alleles i and j, respectively (Botstein et al., 1980). Deviations from Hardy–Weinberg equilibrium were estimated for each locus in each population and across all populations based on Fisher’s exact test using default settings in GENEPOP version 4.7.5 (Rousset et al., 2020). The significance of deviations was adjusted using sequential Bonferroni correction (Rice, 1989). Deviation from hardy-weinberg equilibrium (HWE) for each locus in each population was analyzed using a heat plot (Supplementary Figure 1) generated using the PEGAS package in R (Goss et al., 2014). Pairwise linkage disequilibrium (LD) between SSRs was computed based on log-likelihood ratio statistics (G-test) in GENEPOP.
Genetic Diversity and Population Structure
To estimate within population diversity, the genetic diversity parameters were calculated using GenAlEx 6.5 and poppr R package. Observed mean number of alleles (Ao), the mean number of effective alleles (Ne), percentage of polymorphic loci (PPL), number of private alleles (Ap), and mean observed heterozygosity (Ho) were calculated using Genalex and the Nei’s unbiased gene diversity/expected heterozygosity (He; Nei, 1978), Shannon–Wiener index of diversity (H; Shannon, 2001), Simpson’s index (lambda; Simpson, 1949), and evenness (E5) were calculated using poppr R package. Private alleles were defined as those discovered only in the population considered, discarding those that were found only once as they could reflect genotyping errors.
To evaluate genetic structure in our dataset, we used three different methods: the model-based Bayesian method implemented in the STRUCTURE version 2.3.4 (Pritchard et al., 2010) and maximum likelihood (ML) estimation method implemented in the SNAPCLUST (Beugin et al., 2018); and the model-free DAPC (Discriminant Analysis of Principal Components) (Jombart et al., 2010).
The STRUCTURE uses Markov Chain Monte Carlo (MCMC) approach to estimate every individual’s admixture proportions for a predefined K value (Pritchard et al., 2010). We ran an analysis in the STRUCTURE version 2.3.4 for 150,000 MCMC replications after 50,000 burn-in steps. About 10 replicates each were performed for K values ranging from 1 to 10. The optimum number of populations (K) was estimated using a web-based program, the STRUCTURE HARVESTER (Earl, 2012). The STRUCTURE HARVESTER uses the method outlined by Evanno et al. (2005) and searches for a mode in the ΔK distribution.
The DAPC combines the advantages of principal component analysis (PCA) and discriminant analysis (DA) (Jombart et al., 2010). It first transforms the data using PCA and then performs DA on retained principal components. The number of groups or genetic clusters was defined based on K-means clustering of principal components using find.clusters function in the ADEGENET 2.1.1 package (Jombart, 2008). The optimal K-value was identified by running k-means sequentially for K-value ranging from 1 to 10 and comparing different clustering solutions using BIC (Bayesian Information Criterion). The best-fit K was chosen based on the point at which the elbow in the curve of BIC values as a function of K was observed. The cross-validation function, xvaldapc, was used to determine the minimum number of PCs (Principal Components) to be retained for accurate assignment of every individual to different groups. Finally, the individuals were assigned to clusters based on the posterior membership probabilities of every individual as a measure of the admixture proportion originating in each cluster.
SNAPCLUST allies the advantages of both model-based approaches as used in STRUCTURE and geometric approaches like those used by DAPC and provides a fast maximum-likelihood solution to the specific genetic clustering problem (Beugin et al., 2018). We used Snapclust.choose.k to choose the more adequate number of clusters (K). The most suitable K-value was selected through Bayesian Information Criterion (BIC) by choosing the point at which the BIC value was lowest. After choosing K, we proceeded with the clustering analysis using the function snapclust, implemented in R package ADEGENET.
To further assess the population genetic structure and evaluate the genetic interrelationships among the individuals collected from different locations, unrooted neighbor joining (NJ), and principal coordinate analysis (PCoA) were performed based on pairwise Nei’s genetic distance matrix. The PCoA was generated using dudi. pco function implemented in ade4 R-package (Dray and Dufour, 2007), and the NJ tree was constructed using NJ function from ape R package (Paradis and Schliep, 2019).
Analysis of Molecular Variance
The analysis of molecular variance (AMOVA) analysis was carried out to test the structure of genetic variation within and among genetic clusters inferred based on population structure analysis.
We performed hierarchical AMOVA analysis using the poppr R package (Kamvar et al., 2014) to quantify the partitioning of variance within the individuals, among individuals within the genetic clusters, and among genetic clusters by using pairwise FST as the distance measure. Within individual variance was calculated to quantify variation due to heterozygosity within genotype. The F-statistics was calculated to summarize the degree of differentiation at each hierarchical level. The significance of variance components and F-statistics was tested using 9,999 random permutations of data matrices.
Diversity Analysis Among Genetic Clusters Inferred by Population Structure Analysis
To analyze the dynamics of genetic diversity within each genetic cluster, the measures of genetic diversity were estimated for each cluster obtained during population structure analyses. The genetic diversity indices: observed mean number of alleles (Ao), the mean number of effective alleles (Ne), percentage of polymorphic loci (PPL), number of private alleles (Ap), and mean observed heterozygosity (Ho) were estimated using GenAlEx 6.5 (Smouse and Peakall, 2012), and Nei’s unbiased gene diversity/expected heterozygosity (He), Shannon–Wiener index of diversity (H), Simpson’s index (lambda), and evenness (E5) were calculated using poppr R package. Weir and Cockerham’s (1984) estimator of pairwise Fst between accession groups/clusters were estimated using Hierfstat (Goudet, 2005).
Isolation by Distance
To determine if the genetic variability is structured in geographic space, we performed an isolation by distance (IBD) test (Mantel, 1967) between geographic populations and within each genetic cluster using procedures implemented in the GenAlEx version 6.5 (Peakall and Smouse, 2006). The Mantel test was performed using the Slatkin’s (Slatkin, 1995) pairwise linearized FST {FST [FST/(1 − FST)]} and natural log-transformed geographic distance [ln (1 + geographic distance)] matrix as outlined by Rousset (1997). The statistical significance of the test was conducted by random shuffling (10,000 times) of all individuals among the geographic locations.
The spatial autocorrelation analysis was computed using the multi-locus genetic distance between individuals calculated via the method of Peakall et al. (1995) and was further explained in Smouse and Peakall (1999), and the geographic distance calculated as Euclidean distance using the coordinates of the samples’ collection sites (Peakall and Smouse, 2006). The analysis was performed with an even distance class of 6 km and 10,000 permutations. For testing the statistical significance, a two-tailed 95% confidence interval was constructed around the null hypothesis of no genetic structure (r = 0). A bootstrap of 10,000 trials was used to estimate a 95% confidence interval around the observed r values for each class.
Phenology
Besides making a record of pheno-events covering onset/duration of flowering, fruiting, and production of vegetative propagules through regular field visits (2017–2020), the plantlets/propagules were grown in experimental ponds at the Department of Botany, the University of Delhi to monitor the events. The pollinated flowers/fruits (n = 20) of both species were collected from the natural populations and dissected to observe the seed set.
Reproductive Barriers in Nymphaea micrantha
As N. micrantha exhibited a complete absence of genetic diversity and compromised reproductive output in nature, some of the key aspects of floral biology influencing seed/fruit-set were investigated in the plants growing in experimental ponds at the Department of Botany.
Stigmatic and Ovule Receptivity
Flowers were observed from the bud stage onward until the formation of fruits in nature, and the most receptive stage of stigma was ascertained using the peroxidase test (Galen and Plowright, 1987). Besides the time of onset of anthesis, the timing of dehiscence of anther concerning the duration of the stigma receptivity was also noted down. Receptivity of ovules was ascertained by staining them with toluidine blue O’ (n = 20; Sengupta and Tandon, 2010).
Pollen Biology
Fertility of pollen grains (the ability of the pollen to produce normal male gametes) was determined using 1% acetocarmine (Shivanna and Rangaswamy, 1992), while pollen viability (the ability of the pollen to germinate when conditions are favorable) was determined through fluorochromatic reaction test (Heslop-Harrison and Heslop-Harrison, 1970). For this, fresh pollen grains were extracted from anthers. For each fertility and viability, 20 observations were made using an epifluorescence microscope (Axio plan A.1, Carl Zeiss, Germany) (Supplementary Table 2).
Controlled Pollinations
Flowers were either (i) selfed (n = 15) or (ii) cross-pollinated (n = 15), with fresh pollen on floral stages harboring the most receptive stigma and bagged with a muslin cloth. The pollinated pistils were collected 6 h after pollination (HAP) and fixed in acetic: alcohol (1:3, v/v), cleared with NaOH (1N), and stained with decolorized aniline blue, and visualized under the epifluorescence microscope following Linskens and Esser (1957). A few flowers (n = 20) were left untreated (open-pollination) to determine the fruit-set in the plants under cultivation. The pollinated pistils were collected 15 days after pollination (DAP) and dissected to observe a seed set.
Results
Genome Sequencing and Discovery of Microsatellites
The genome size estimation of N. micrantha, based on the linear relationship of 2C peaks of the sample and the internal standard, indicated an approximate genome size of 1.11 pg per 2C content corresponding to 544 Mbp/1C (Supplementary Figure 2). Illumina paired-end sequencing resulted in 56,645,378 raw reads providing a genome coverage of ∼10 X fold based on the estimated genome size. The raw reads were subjected to quality check to remove sequence artifacts, such as low-quality reads and adapter contamination, using the NGS QC toolkit (Patel and Jain, 2012). A total of 53,511,392 (94.57%) high-quality reads showing a Q value of ≥ 20 were obtained after quality filtration. An assembly of high-quality filtered reads, with Velvet (v1.2.10) at a k-mer value of 55, resulted in 518,464 contigs with an average contig length of 596 bp (Supplementary Table 3). To reduce fragmentation of the assembly, the contigs were further filtered to retain contigs ≥ 1,000 bp resulting in 77,175 contigs with an average contig length of 1,536 bp (Supplementary Figure 3). These contigs were used for the identification of microsatellites. A total of 22,268 non-redundant microsatellite loci were obtained using the Perl script MISA. There were 16,937 sequences containing SSRs of which 4,123 sequences contained more than one SSR. About 2,464 SSRs were present in the compound formation, which were removed from further analysis (see Supplementary Notes and Supplementary Table 4 for details).
Functional Annotation of Simple Sequence Repeat Containing Sequences
To identify the functional significance of 12,556 non-redundant SSR containing sequences, a BLASTx analysis was performed against the NCBI non-redundant (nr) protein database. A total of 3,809 (30.3%) sequences were found to have similarities to the sequences in the NCBI nr protein database, and 1,623 (12.9%) were found to have at least one functional annotation. The remaining 8,747 (69.67%) did not show significant similarity to the known sequences and were not annotated.
Gene Ontology (GO) enrichment analysis, using DAVID Bioinformatics Resources 6.8 (Huang et al., 2009), classified microsatellite sequences into three categories: cellular components (CC), molecular functions (MFs), and biological process (BP). Supplementary Table 5 provides detailed information regarding annotated SSR sequences. Fourteen hundred and thirty-six contigs were mapped to GO terms with 1,009, 1,172, and 892 assignments distributed under the cellular component, molecular function, and biological process ontology, respectively.
Genetic Diversity in Nymphaea micrantha
The 143 SSR primer pairs were analyzed for polymorphism among 90 N. micrantha individuals. Surprisingly, all the markers tested in N. micrantha produced the same DNA profile and were monomorphic.
Genetic Diversity in Nymphaea nouchali
Genetic Diversity Statistics of Microsatellites
In N. nouchali, of 143 SSR markers tested, 50 could not produce clear and scorable bands on PAGE, and 36 were monomorphic and, thus, were excluded from further analysis. The analysis using Micro-Checker software revealed no evidence for stutter error or large allele dropout at any locus for all the populations. The probable presence of null alleles was detected for locus Nym_NGS82 (K1 population) and locus NymTr_41 (K3 population). Accordingly, the adjusted frequencies of amplified alleles for these populations were used for further analysis. Descriptive characterization and genetic diversity analysis among 92 N. nouchali individuals, for each of the remaining 57 polymorphic SSR loci, is given in Table 2. A total of 277 alleles were identified with an average of 4.8 alleles per locus. The effective number of alleles (Ne) ranged between 1.056 (NymTr_13) and 2.339 (NymTr_24), averaging 1.821 alleles per locus. The observed (Ho) and expected (He) heterozygosity ranged from 0.04 (NymTr_13) to 0.975 (NymTr_21), and 0.03 (NymTr_13) to 0.559 (NymTr_24), respectively. The average observed heterozygosity (Ho = 0.578) was higher than the average expected heterozygosity (He = 0.386). Polymorphic information content (PIC) values varied from 0.042 (NymTr_13) to 0.826 (NymTr_5), with a mean PIC of 0.466. Twenty-six SSR markers were considered as highly informative (PIC > 0.5), twenty-five were moderately informative (0.5 > PIC > 0.3) and six markers (Nym_NGS31, Nym_NGS35, Nym_NGS36, NymTr_42, NymTr_17, and NymTr_13) were considered of little polymorphism information having PIC of < 0.3 values (Table 2). Total gene diversity (Ht) was highest for the marker NymTr_5 (Ht = 0.86), followed by NymTr_31 (Ht = 0.751) and Nym_NGS45 (Ht = 0.737), and lowest for the marker NymTr_13 (Ht = 0.04). Null allele frequencies obtained for all loci were close to zero. No significant linkage disequilibrium was found between the locus pairs based on log-likelihood ratio statistics (G-test). Forty-two out of 57 loci significantly deviated from HWE (p < 0.05), of which 35 deviated due to heterozygote excess and 7 loci deviated due to heterozygote deficiency (Table 2). Negative inbreeding coefficient (average Fis = −0.456) and deviations from HWE indicate an excess of heterozygosity among N. nouchali populations.
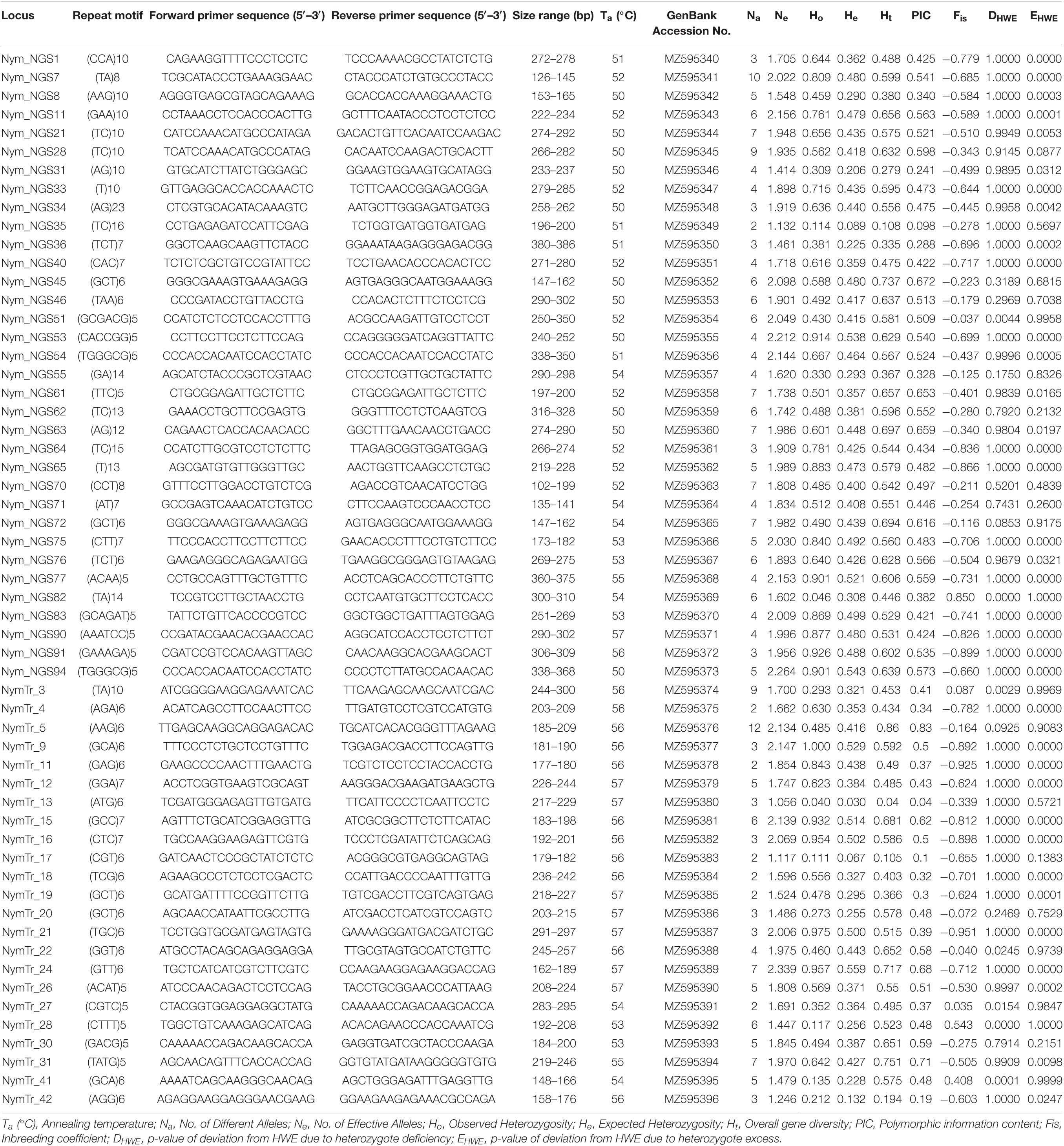
Table 2. Characteristics and genetic diversity indices of 57 polymorphic simple sequence repeat (SSR) markers used for diversity analysis in Nymphaea nouchali Burm.f. var. nouchali.
Genetic Diversity Analysis Within Populations
Genetic diversity indices obtained for each population of N. nouchali are tabulated in Table 3. Average across loci of the observed and effective number of alleles ranged between 1.702 (A2 population) and 2.298 (A1 and A6 populations), and 1.511 (A2 population) to 1.984 (5S population), respectively. The overall mean observed allele number was 2.053 and the effective one was 1.821. The discrepancy between observed and effective allele numbers reflects the uneven distribution of allele frequencies. In total, 277 distinct alleles were identified, of which 32 were private (found only in one population). The population K5 had the maximum number of private alleles (6). The observed heterozygosity (Ho) was higher than the expected heterozygosity (He) for each population except A6. Ten out of 27 populations showed a significant deviation (p < 0.05) from HWE (Table 3 and Supplementary Figure 1). Nei’s unbiased gene diversity ranged between 0.292 (A2) and 0.535 (6S), with an average value of 0.531 across populations. Shannon–Wiener index and Simpson’s Index of genetic diversity suggested the highest diversity in A1 (H = 1.609; lambda = 0.8). The genotype abundance/evenness (E5) for all the populations except one (K2) indicated that the multi-locus genotypes (MLG) in the populations are equally abundant. More than 50% of the studied loci were polymorphic in all the populations with the highest percentage of polymorphic loci in 6S, K3, and A7 (PPL = 87.72%), and lowest in A2 (PPL = 59.65%).
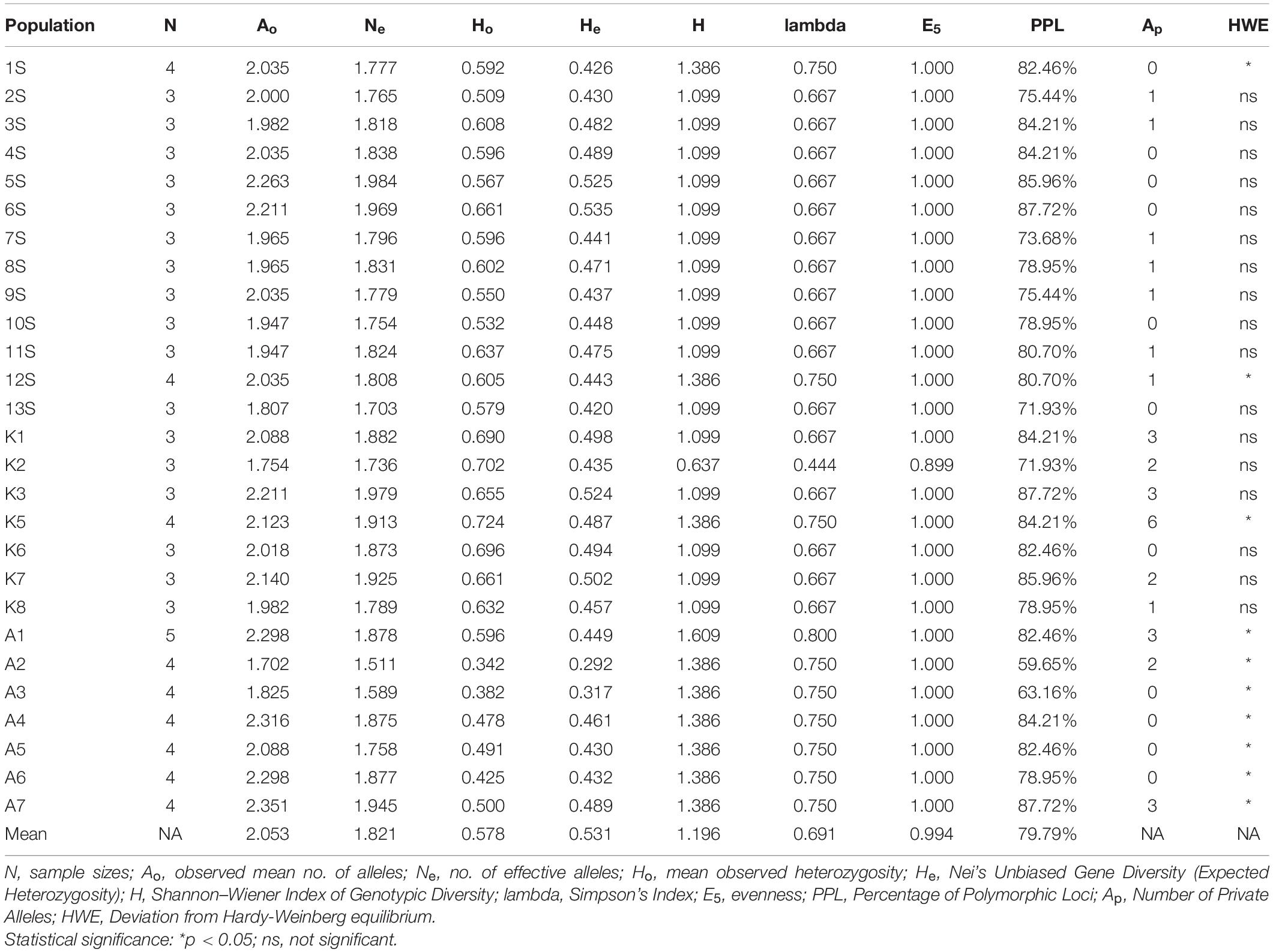
Table 3. Genetic diversity statistics of 27 Nymphaea nouchali populations.
Pairwise FST values ranged between 0.00 (between A4 and A5 populations) and 0.390 (between K2 and A2 populations), showing no genetic differentiation to fairly high genetic differentiation among populations (Table 4).

Table 4. Multi-locus estimates of pairwise FST between all population pairs of Nymphaea nouchali.
Analysis of Population Structure
An analysis using both STRUCTURE and SNAPCLUST recovered three clusters with some degree of correlation between genetic clusters and geographical origin (Figures 1A,C). Although the assignment of some individuals to clusters differed between the two methods, in general, there is a high degree of correspondence between the groups defined by SNAPCLUST and those recovered by STRUCTURE (Figure 1D). Accessions from Goa and Maharashtra, which are less geographically distant, were clustered together (cluster 1). All the accessions from Kerala were assigned to cluster 2. Some accessions of Assam were recovered as admixtures (STRUCTURE) and/or groups with the accessions of Kerala (SNAPCLUST) suggesting movement of genetic material between the two geographical locations. The STRUCTURE failed to classify more individuals (11 were classified as admixed based on the threshold defined) as compared to SNAPCLUST which classified individuals into different clusters. The SNAPCLUST placed all individuals into a single group, except one (1S_3) that could not be assigned to a single group with a group membership score of over 0.75.
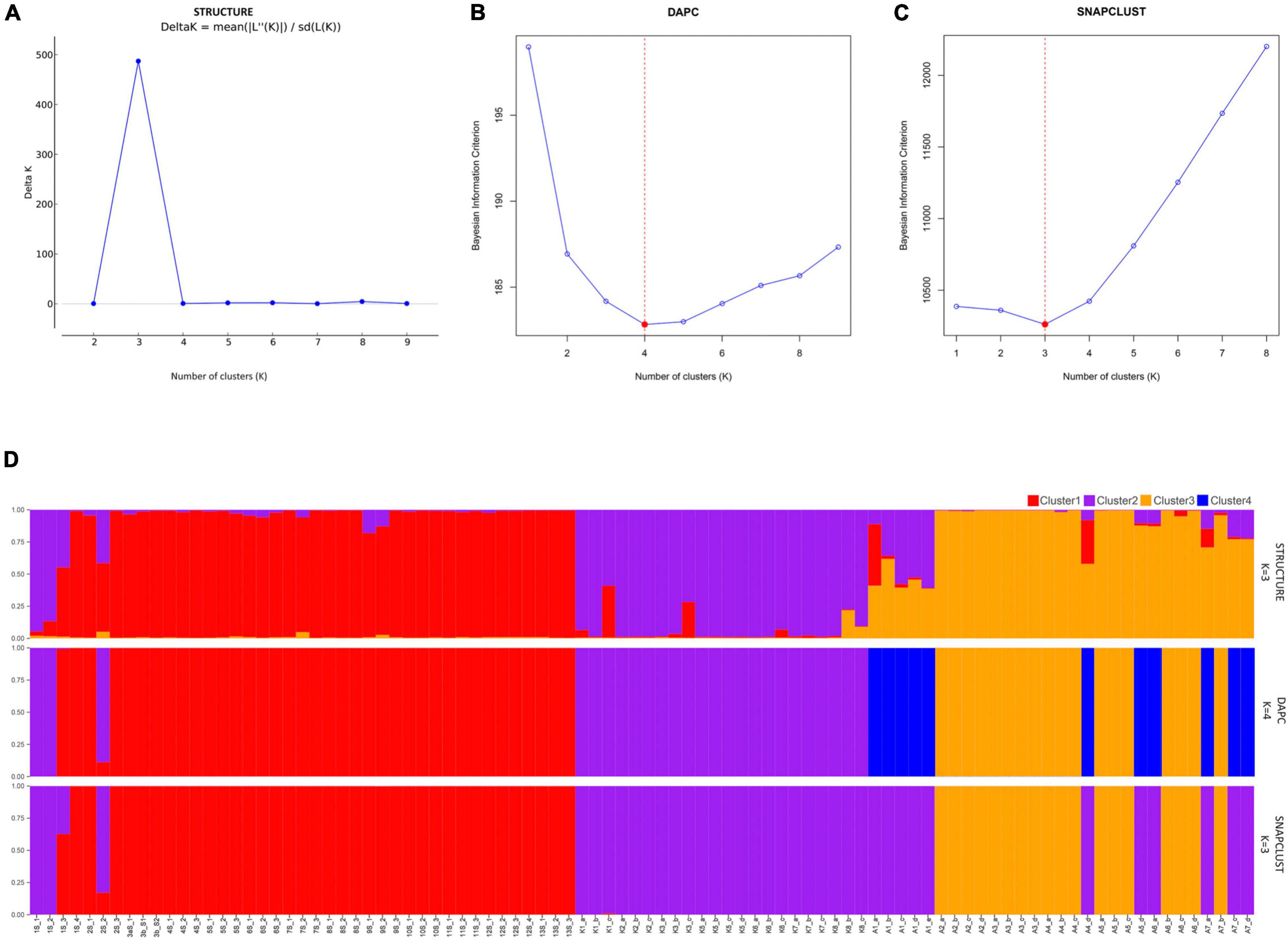
Figure 1. Structure clustering results of population structure analysis. Each inferred cluster is marked with a different color and everyone in the cluster is represented by a vertical bar. Estimation of populations [(K; (A–C)] and distribution of individuals to different clusters at K = 3 and K = 4 (D) using STRUCTURE, Discriminant Analysis of Principal Components (DAPC), and SNAPCLUST.
The DAPC recovered four genetic clusters (Figure 1B), two of which roughly corresponded to the two major groups identified using both STRUCTURE and SNAPCLUST. Unlike STRUCTURE and SNAPCLUST, DAPC classified accessions collected from Assam into two different genetic clusters (cluster 3 and cluster 4; Figure 1D). No individual was classified as admixed based on the threshold defined for this method. All the individuals from Goa and Maharashtra formed a single group except 1S_1, 1S_2, and 2S_2 that were grouped with Kerala.
The PCoA and NJ were conducted to further assess the population’s genetic structure. Both NJ and PCoA showed a grouping of accessions according to their geographical distances (Figure 2). The NJ analysis (Figure 2A) showed that the accessions from 27 natural populations could be divided into three groups, which was consistent with the pattern from the population structure analysis using STRUCTURE and SNAPCLUST software. Accessions from Maharashtra and Goa were clustered together. The accessions from Kerala were less distant to the accessions from Goa and Maharashtra than those from Assam. In addition, the results of PCoA analysis further verified the NJ and the population structure analysis results (Figure 2B). In summary, the populations with less geographic distance are grouped. The first two principal coordinates explained 18.74% and 7.98% individually and explained 26.72% of the total variation (Figure 2B).
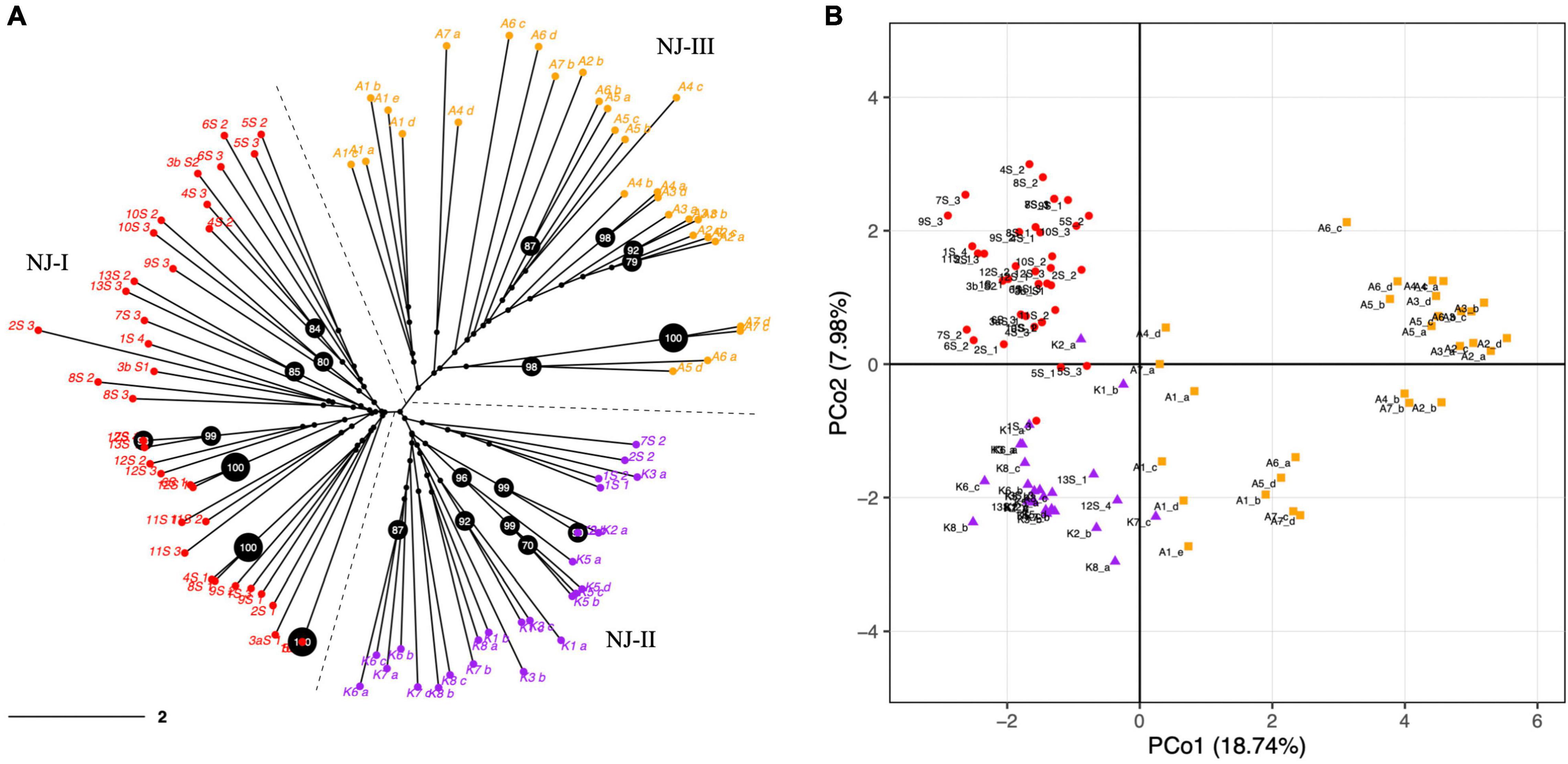
Figure 2. (A) Dendrogram constructed with a neighbor-joining (NJ) clustering algorithm elucidating the genetic relationships among 92 N. nouchali individuals using 57 polymorphic SSR markers. The NJ clusters (NJ-I, NJ-II and NJ-III) are demarcated through dashed lines and different tip colors. Percentage bootstrap values were generated from 1,000 replicates. Bootstrap values ≥ 70% are depicted in black circles. Scale bar represents branch length. (B) Principal Coordinate Analysis (PCoA) exhibiting the distribution of N. nouchali accessions on two main axes. Primary axes 1 and 2 captured 18.74% and 7.98% of the total variance, respectively. Different colors represent accessions from different NJ clusters.
Analysis of Molecular Variance
Considering the clustering of accessions into genetic groups, between K = 3 and K = 4, the AMOVA accounted for more variance for K = 4 than K = 3 (Table 5). The AMOVA results indicated that most of the total genetic variance (>90%) was attributed to the heterozygosity within individuals (Table 5). Variance attributed to individuals within the genetic cluster was zero with no statistical support and negative FIS value (FIS = −0.161, p > 0.05). Significant genetic differentiation (p < 0.001) was detected among genetic clusters for both K = 3 and K = 4 genetic clusters (Table 5).
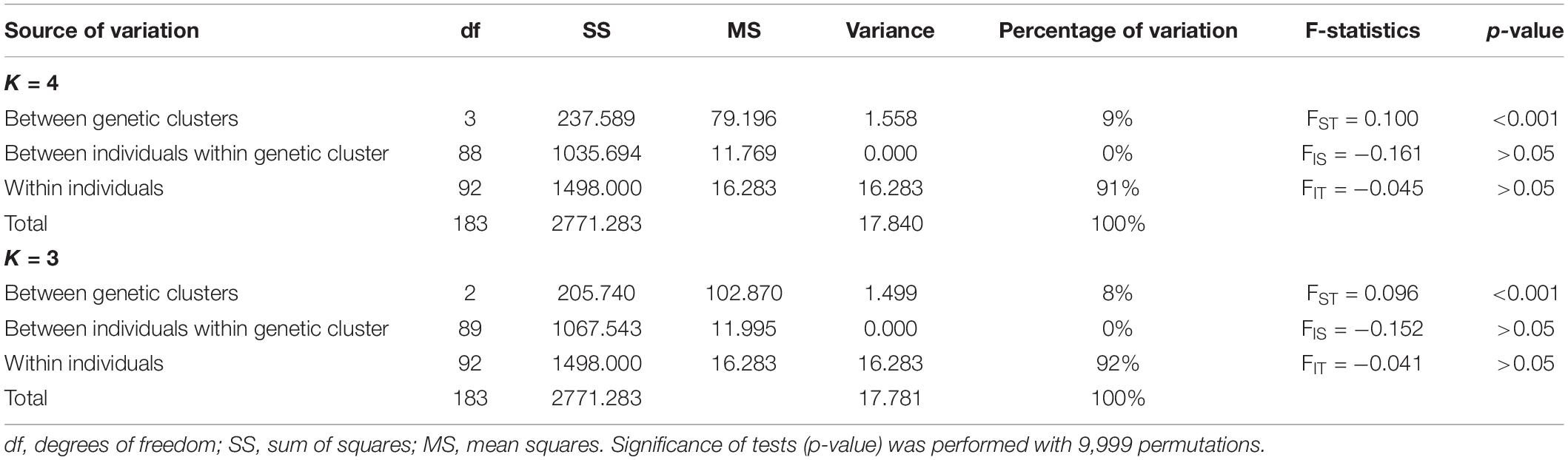
Table 5. Analysis of molecular variance (AMOVA) showing variance percentage explained by four (K = 4) and three (K = 3) genetic clusters inferred by different structure methods.
Diversity Analysis Among Genetic Clusters
Genetic diversity statistics obtained for each genetic cluster are tabulated in Table 6. The observed heterozygosity (Ho) and Nei’s unbiased gene diversity (He) were highest in cluster II (Ho = 0.667; He = 0.520) and lowest in cluster IV (Ho = 0.463; He = 0.383). Simpson’s index (H) and Shannon–Wiener index were highest in cluster I (lambda = 0.973; H = 3.611) and lowest in cluster III (lambda = 0.909; H = 2.398). The genotype abundance/evenness (E5) in each cluster was either 1 or close to 1 which indicates the equal abundance of all multi-locus genotypes (MLGs) in these genetic clusters. More than 90% of the loci studied were polymorphic in all four clusters with the highest percentage of polymorphic loci in cluster II (98.25%). All the accession groups amplified private alleles (Ap) varying between 6 and 30 with the highest number of Ap detected in cluster II.
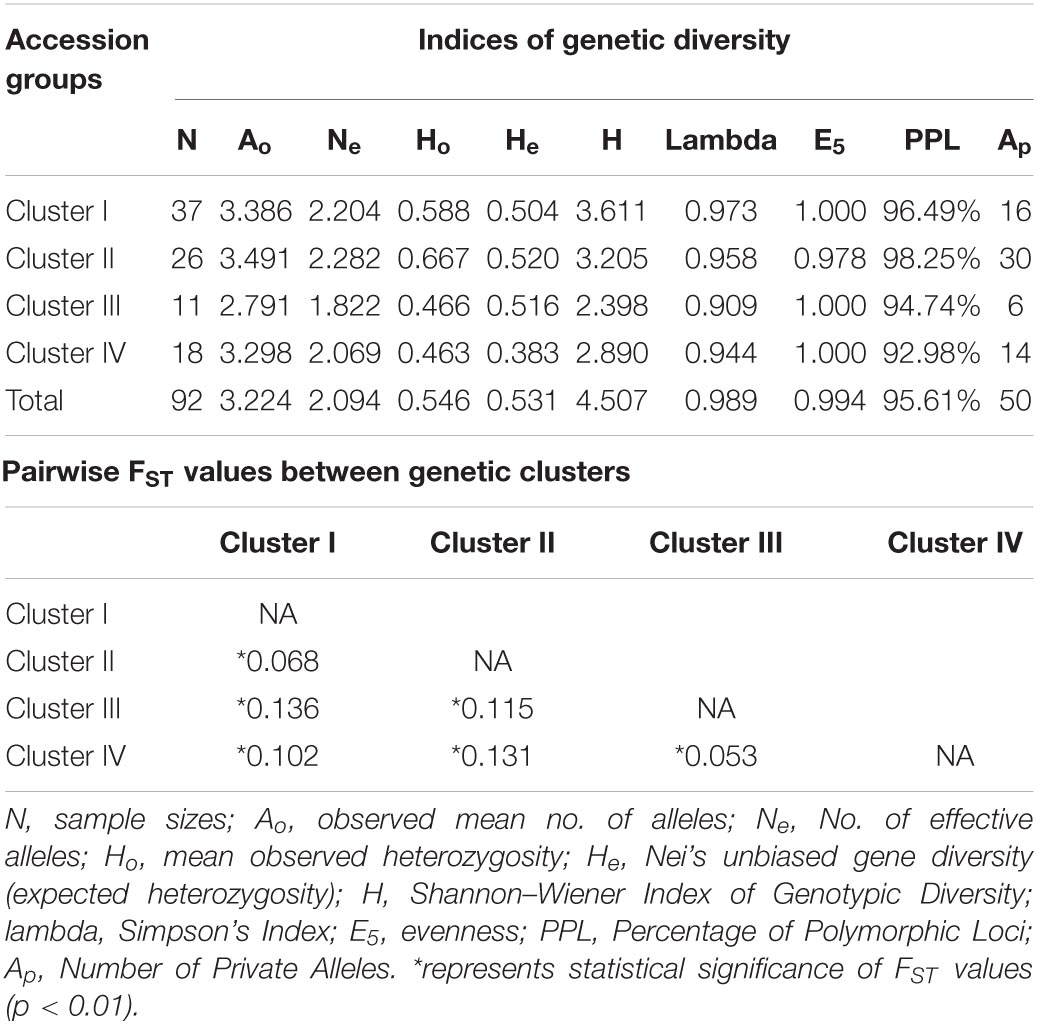
Table 6. Indices of genetic diversity and pairwise Fst among four genetic clusters.
The pairwise Fst values between clusters ranged from 0.053 to 0.136, and all the cluster pairs were significantly differentiated p-value < 0.01 for each pair (Table 6) from each other. Accessions of cluster III and cluster IV were least differentiated (FST = 0.053), followed by accessions from cluster I and cluster II (FST = 0.068). Cluster I and II accessions were more distinct from cluster III and IV accessions than they were from each other, and vice-versa.
Spatial Genetic Analysis
For all geographical locations, as expected under the isolation-by-distance (IBD) model, significant positive correlation was observed between pairwise FST and geographical distance based on Mantel permutation test (Figure 3A; r = 0.654, p < 0.001, permutations = 1,000). The multivariate spatial autocorrelation analysis displayed positive and significant r values up to 12 km (Supplementary Figure 4) illustrating that those individuals, which are less than 12 km apart, had non-random genetic similarities.
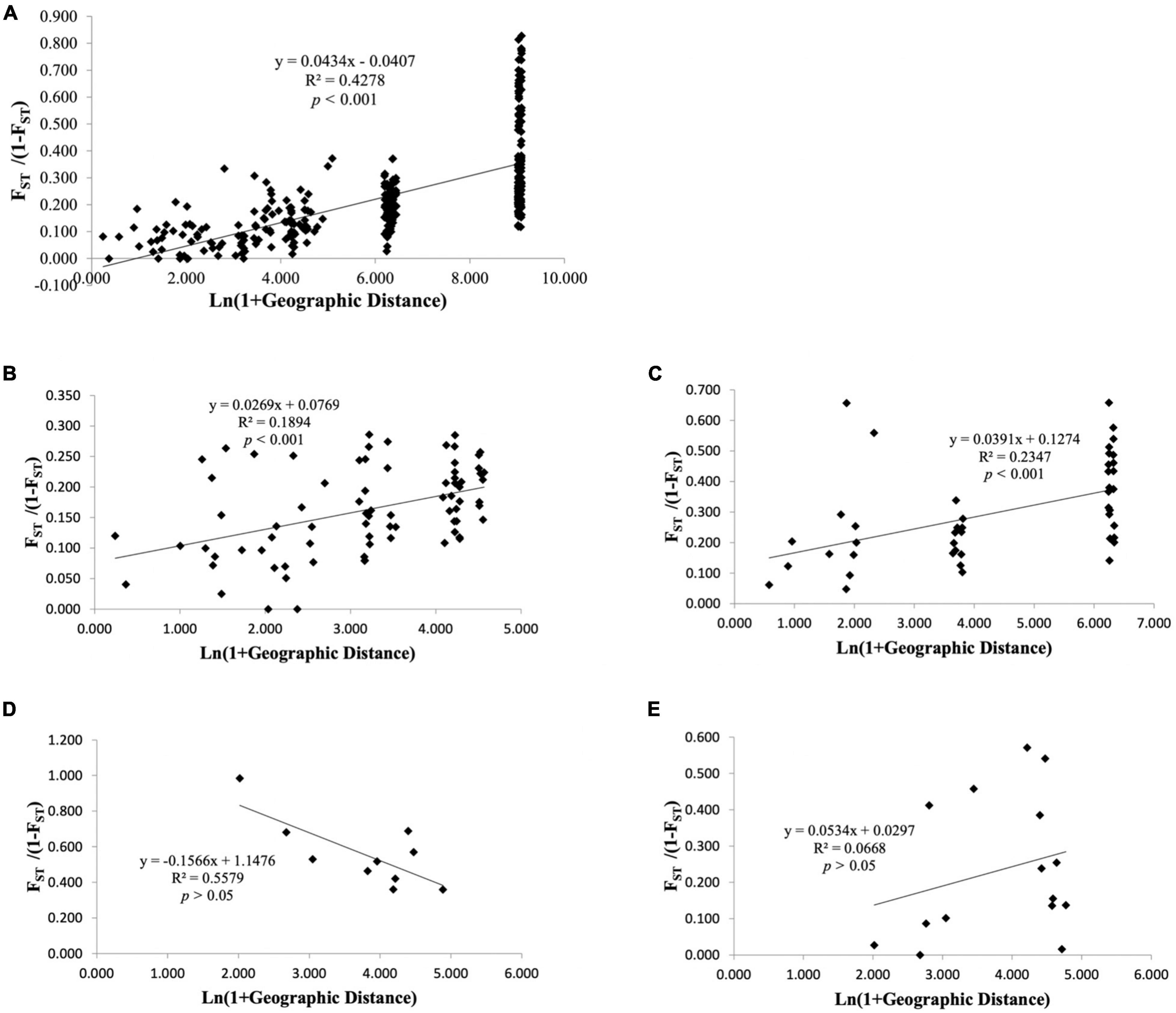
Figure 3. Relationship between Slatkin’s linearized pairwise FST and geographical distance (Isolation-by-distance based on Mantel test) among populations of Nymphaea nouchali (A) and genetic clusters inferred using DAPC: cluster I (B), cluster II (C), cluster III (D), and cluster IV (E). Significance of the test was tested based on 10,000 random permutations of the data.
Among the four genetic clusters inferred with population structure analysis, isolation by distance pattern could be observed within cluster I (Figure 3B; r2 = 0.187, p < 0.001) and cluster II (Figure 3C; r2 = 0.259, p < 0.01), which mostly included the localities from Goa-Maharashtra and Kerala respectively. The multivariate spatial autocorrelation analysis within the genetic groups displayed significant r values for cluster I and suggested a non-random genetic similarity between individuals who are less than 32 km apart. For clusters II, III, and IV, the r values fell within the confidence interval of the null hypothesis (Supplementary Figure 4) suggesting that the individuals within these clusters had random genetic similarities.
Phenology
The flowering and fruiting in N. nouchali were observed from March to October; while the N. micrantha flowers throughout the year, with peak flowering residing between April and September. Vegetative propagules were observed only in the case of N. micrantha arising from the petiolar node (Figures 4b,c). They appeared throughout the year, with copious production during the winters (November–January). Fruits of N. nouchali matured within 25 DAP (Figure 4g,h) and dehisced underwater, with multiple longitudinal splits in the capsule, producing an enormous number of seeds. On the contrary, fruit formation in the naturally growing plants of N. micrantha was not apparent (Figure 4).

Figure 4. Images showing (a) first day flower of Nymphaea micrantha, (b) bulbil arising from the petiolar node of N. micrantha, (c) plantlet produced from the bulbil in N. micrantha, (d) pollinated flower pods of N. micrantha after 5, 15, and 20 days of pollination (DAP), (e) content of a locule extracted from 15 DAP pod of Nymphaea, showing mature seed (blue arrow), aborted seeds (pink arrow) and unfertilized ovules (white arrow), (f) flower of N. nouchali, and (g,h) fruits of N. nouchali, collected 5, 15, and 20 DAP.
Reproductive Barrier in Nymphaea micrantha
The flowers exhibited protogyny, as the stigma became receptive on the day of anthesis, while anthers dehisced a day later. There was a gradual decline in the receptivity after the first day, indicating overlapping dichogamy in the species. Pollen tubes reached the ovules 6 HAP (Hours after pollination). However, only 4.33 ± 1.91% of the ovules showed receptivity. The fertility of pollen grains was significantly greater (87.89 ± 5.18%) than the viability (18.02 ± 4.51%; t = 44.36, df = 38, p < 0.001). Controlled pollinations (both selfed and outcrossed) resulted in the initiation of fruits formation but failed to mature into fruits. The pollinated flowers turned brown and started decomposing after 12–15 days of the anthesis phase (Figure 4d). Such abortive fruits were dissected to ascertain seed formation, showed a 2.8 ± 0.84 % seed set (Figure 4e). No difference in seed set was observed between selfed and cross-pollinated flowers. The results of seed set in the naturally growing populations were also comparable with manual pollination treatment.
Discussion
Aquatic plant species, inhabiting shallow lakes and river ecosystems, are most vulnerable to various anthropogenic disturbances (Guo et al., 2019). The various environmental changes, such as water pollution, habitat destruction, eutrophication, or changed hydrological regime, have led to the global declination and extinction of many aquatic species during the past 50 years (Sand-Jensen et al., 2000; Zhang et al., 2017; Orsenigo, 2018), making them priority targets for conservation. Nymphaea species are the important keystone species within the freshwater wetlands. They provide food and habitat for many herbaceous animals, act to reduce water turbidity and provide in-stream water stabilization (Leito et al., 2016; Ranjan and Prakash, 2019). Fruits and seeds of N. nouchali are eaten by many birds including Red-crested Pochard, Eurasian Wigeon, Spot-billed Duck, and others (Jha, 2013). In India, the wetland ecosystems are under tremendous stress due to rapid increases in industrialization, urbanization, and agricultural intensification (Bassi et al., 2014). Supplementary Figure 5 shows how much human activities have led to the rapid destruction of water bodies and induced rapid decline of many aquatic plants including Nymphaea species.
An assessment of genetic diversity within a species plays an important role in the efficient management and conservation of genetic resources. This study is the first to utilize SSR markers in molecular characterization of two Indian representative species, N. micrantha and N. nouchali, in their natural habitat from different environmental regions. The investigation used 43 genic SSRs and 100 non-genic SSRs to analyze a collection of 90 N. micrantha and 92 N. nouchali individuals from six different states of India, i.e., Assam, Manipur, Meghalaya, Maharashtra, Goa, and Kerala. The N. micrantha, being an introduced species to India, is expected to possess low genetic diversity, although, to our surprise, the SSR markers revealed no apparent genetic divergence in the studied populations. On the contrary, N. nouchali possessed a high level of polymorphism among and within populations.
Genotypic Richness and Genetic Relationships Among and Within Populations of N. nouchali Revealed by Simple Sequence Repeats
High Genetic Diversity Within 27 Geographically Distinct Populations of N. nouchali
A high level of genetic diversity was observed in N. nouchali. The 57 polymorphic SSR markers detected a total of 277 alleles, with an average of 4.8 alleles per locus. The alleles observed for each locus were not equally frequent as the number of alleles observed (Ao) were greater than an effective number of alleles (Ae). The negative inbreeding coefficient (mean Fis = −0.456) values indicate an excess of heterozygosity in the studied populations of N. nouchali. The average total gene diversity (Ht = 0.531) among all polymorphic SSR markers was higher than polymorphic information content (PIC = 0.466) suggesting that it is less likely that the N. nouchali individuals have identical heterozygote genotypes (Table 2).
Various studies (Gupta, 1978; Hossain et al., 2007) have reported polyploidy in the genus Nymphaea. The reported chromosome count in N. nouchali varies between 2n = 28 and 2n = 84 (Hossain et al., 2007). Polyploid populations are expected to exhibit high levels of heterozygosity (Muller, 1914; Haldane, 1930; Soltis and Soltis, 2000). In our study, we observed high levels of heterozygosity, but it is not completely fixed. Moreover, the SSR data did not reveal any sign of polyploidy as a maximum of two alleles were present for each locus within an individual.
The average gene diversity (He) within populations ranged between 0.292 and 0.535 (Table 3). The preservation of this high level of genetic diversity in N. nouchali may be due to prevailing cross-pollination in the species (Stebbins, 1957). Since Nymphaea flowers are protogynous, many Nymphaea spp. utilize xenogamy or geitonogamy as their reproductive strategy (Wiersema, 1988). In 1988, Wiersema reported that N. nouchali predominantly utilizes xenogamy (i.e., the union of genetically unrelated organisms within a species), which might also explain the observed high within-population gene diversity in the species. Several studies on other aquatic plants, however, have reported low genetic variation (Tian et al., 2008; Lambertini et al., 2010; Hu et al., 2012) based on various dominant molecular markers. Han et al. (2007) had reported high genetic diversity in populations of Nelumbo nucifera using ISSR markers. 42 of 57 polymorphic SSR markers was detected in HWE with p ≤ 0.05 (Supplementary Figure 1 and Table 2) in ten populations (Supplementary Figure 1 and Table 3), which included one population each from Goa (1S), Maharashtra (12S), and Kerala (K5), and all the seven populations of Assam. Simpson’s index and Shannon–Wiener index of gene diversity also revealed high levels of genotypic richness within the studied populations (Table 3). The excess of heterozygosity observed in N. nouchali populations may be attributed to: (1) the low effective size of populations studied, (2) the outcrossing breeding system, and (3) selection favoring heterozygotes.
Population Structure and Genetic Clustering
To estimate genetic populations within a group of N. nouchali accessions, we used three different approaches (STRUCTURE, SNAPCLUST, and DAPC). These approaches recovered three or four different clusters with a slightly different partitioning of genotypes into genetic clusters. All the three approaches recovered a high degree of correspondence between genetic clusters and geographical origin, thus, indicating that the individuals with less geographical distances were more genetically similar than those from a greater geographical distance.
Furthermore, the NJ clustering algorithm clustered individuals into three major clusters with correspondence to their geographic origin (Figure 2). Individuals with less geographical distance were clustered together implying that geographical isolation restricted gene exchange among populations (Geraldes et al., 2014; Chen et al., 2020). Estimation of pairwise FST between each population pair revealed that populations collected from Assam were more differentiated from the populations collected from Goa, Maharashtra, and Kerala (Table 4), which again indicates that populations with less geographic distance were more genetically similar.
The Isolation-by-distance (IBD) pattern also suggested restricted dispersal and regional equilibrium between gene flow and genetic drift in controlling the distribution of genetic variation (Hutchison and Templeton, 1999). The detectable similarity among individuals at a distance < 12 km within some regions, again, indicates that gene flow is effective only over very short distances (Figure 3 and Supplementary Figure 4).
Hierarchical AMOVA analysis was used to quantify the partitioning of variance within the individuals, among individuals within the genetic clusters, and among genetic clusters for both K = 4 and K = 3. The highest variance was present within geographical among individuals, followed by among genetic clusters and zero variance was detected among individuals within the genetic cluster (Table 5). High within individual variance suggests that most of the observed variation in N. nouchali was attributed to the heterozygosity within individuals which might have resulted due to prevalent cross-pollination or xenogamy as a mode of reproductive strategy.
Genetic Diversity Measures Within Each Genetic Cluster
The analysis of genetic diversity within each genetic cluster inferred from DAPC based on three indices (Nei’s unbiased genetic diversity, Shannon–Weiner index of genetic diversity, and Simpson’s index) indicated that the populations collected from southern Indian states, Goa, Maharashtra, and Kerala (represented in clusters I and II; Figure 5) were more genotypically rich as compared to Assam (represented in clusters III and IV; Figure 5). Further, the observed mean number of alleles, percentage of polymorphic loci, and private alleles were also high in clusters I and II as compared to clusters III and IV. These observations suggest (1) predominant outcrossing and a great pre-existing genetic variability (Stebbins, 1957), and (2) low anthropogenic influence in Goa, Kerala, and Maharashtra as compared to Assam. The degree of differentiation can be explained by many factors such as habitat destruction, breeding system type, geographic isolation, and gene flow (Shen et al., 2017). Pairwise FST between each cluster pair, significantly differentiated (p < 0.01) the four clusters. The pairwise FST values between the clusters showed greater genetic differentiation with increased geographic distance between the two clusters. Clusters I and II individuals were more genetically differentiated from clusters III and IV individuals than from each other. The cluster III and cluster IV individuals, which includes individuals from Assam, were least differentiated (FST = 0.053), followed by cluster I and cluster II (FST = 0.068), which included individuals from Goa and Maharashtra. The regional population genetic structure and weak or lack of IBD within clusters (Figure 3) indicated higher gene flow between populations that are less geographically distant.
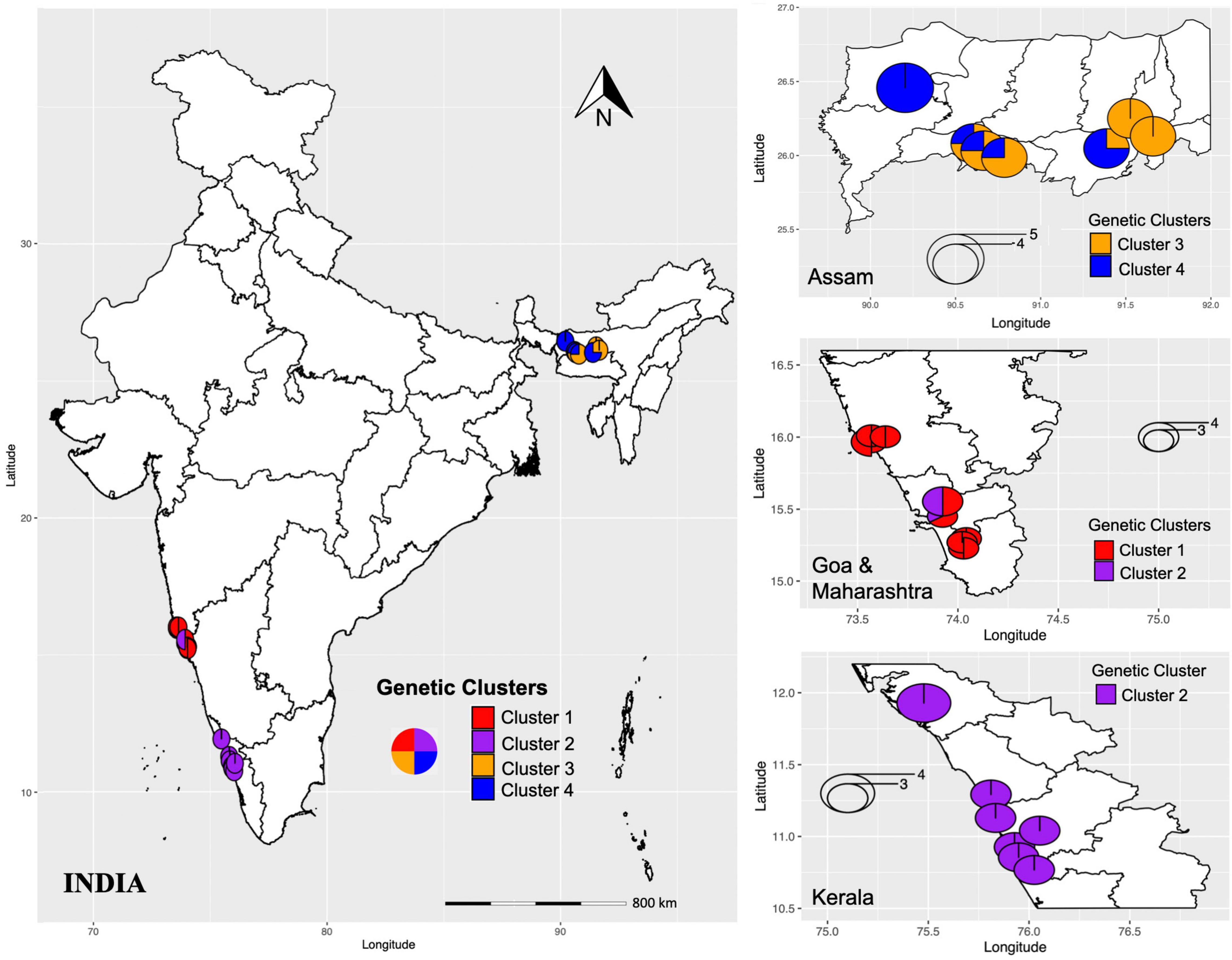
Figure 5. Geographical distribution of genetic clusters (I-IV) inferred by DAPC. The colors represent different genetic clusters and pie charts represent the percentage of individuals assigned to each genetic cluster.
Cross-Species Transferability of Polymorphic Simple Sequence Repeat Markers
The 57 polymorphic SSR markers produced for N. nouchali were also assessed for their cross-species transferability in five other Nymphaea spp. viz. N. malabarica, N. caerulea, N. rubra, N. pubescens, and N. × khooroi (see Supplementary Table 6). The markers with a high transferability rate would be of importance and can be efficiently utilized in conducting basic genetic studies in Nymphaea.
Clonal Reproduction and Lack of Genotypic Diversity in N. micrantha in the Studied Regions
Nymphaea micrantha is non-native to India and, thus, is expected to possess low genetic variation. Surprisingly, however, no genetic variation was found within or between populations investigated as a single multilocus genotype (MLG) was detected in the samples studied.
Clonal reproduction is often considered an evolutionary dead-end (Stebbins, 1957). Lambertini et al. (2010) detected low genetic diversity in three clonal aquatic species, Elodea canadensis, Egeria densa, and Lagarosiphon major. Ren et al. (2005) reported very low genetic differentiation in Eichhornia crassipes (water hyacinth) populations from different localities in China. Ellstrand and Roose (1987) summarized the population genetic structure of 21 clonal plant species and revealed that two species, Taraxacum obliquum, and Gaura triangulata, did not display any apparent intraspecific variability. Pollux et al. (2007) reported that the shifting balance between sexual and asexual reproduction affected the genotypic diversity within populations Sparganium emersum. These studies suggest that the plants reproducing asexually can preserve well-adapted genomes in stable environments, which permits the fixation of well-adapted phenotypes over generations (Niklas and Cobb, 2017).
Various species of Nymphaea are known to be propagated by both seeds and bulbils (Wiersema, 1988; Ansari and Jeeja, 2009). The prevalent mode of reproduction (asexual vs. sexual) in these plants may likely influence the extent of genetic variability of the species. Our results showed that sexual reproduction in N. micrantha is significantly impaired in all the investigated populations. Manually pollinated flowers were aborted at different stages due to low pollen viability (male fitness), and poor ovule receptivity (female fitness). These results were comparable to the flowers that were marked for natural fruit-set (open-pollination). As seen in the present work as well, the species is known to propagate predominantly through leaf propagules (Wiersema, 1988). The extensive clonality accompanied by impaired sexual reproduction could be the major cause for the observed lack of genetic diversity in N. micrantha. Moreover, N. micrantha is not indigenous to India (Ansari and Jeeja, 2009; POWO, 2021), although, the time and source of the introduction are not known. It is predicted that populations in their introduced range are less variable relative to their source or native range, owing to founder effects and genetic drift (Husband and Barrett, 1991; Amsellem et al., 2000; Hagenblad et al., 2015). The successful expansion of its distribution range in India demonstrates its strong adaptability to the local ecological conditions. The non-existence of genetic variation within and between N. micrantha populations, therefore, may be attributed to (1) predominant vegetative propagation, broad tolerance range, and plasticity of genotypes; (2) introduction of the same genotype once or multiple times in the North-eastern parts of India; (3) fixation of well-adapted genomes over generations.
Our results suggest that a single genotype was introduced to India and expanded its distribution range through massive vegetative reproduction. The ability of N. micrantha to conquer water bodies in the North-East regions of India as a silent invader could also be attributed to human interventions. The ornamental and religious values of the species aided by human involvement might have played a role in its spread. Several previous studies have also demonstrated the presence of low genetic diversity in invasive and clonally reproducing plant species supporting our results. Xu et al. (2003) reported extremely low genetic diversity in the invasive alligator weed in China. Li et al. (2006) revealed a lack of genetic variation in the clonally reproducing invasive plant Eichhornia crassipes.
Conclusion
The importance of Nymphaea in wetland ecosystems and their increased vulnerability make them a great choice for conservation and management. This will also be useful for the sustainable use of Nymphaea species. The existing re-establishment practices, which include restoration of degraded habitats and introduction of individuals into locations, may help recover the declining populations of Nymphaea (Guo et al., 2019). However, the successful establishment will require sufficient genetic variability in introduced populations to avoid inbreeding depression and to cope with environmental changes (Forsman and Wennersten, 2016). In this study, we developed a set of novel SSR markers in Nymphaea. These SSR markers were used to study the diversity of two widely distributed species, N. nouchali, and N. micrantha, and had demonstrated contrasting genetic variation patterns in the two species, which was complemented by our results from the reproductive biology of the two species. This contrast in reproductive strategy impacting the existing genetic diversity is an excellent example of varying reasons in nature that can influence the genetic diversity and, hence, the survival of a species. It also underlines the importance of genetic diversity studies in strategizing conservation practices. The information generated in this study is of important consideration for successful wetland restoration programs and genetic resource conservation in Nymphaea species, especially those with limited distribution. High genetic variability detected in N. nouchali makes it the species of choice for the successful restoration of wetlands. The two gene pools, cluster I and cluster II, which detected more genetic diversity, can be chosen to procure plant material during the restoration programs, although, regional adaptation should also be taken into consideration.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material. The raw DNA sequencing data of Nymphaea micrantha has been deposited at NCBI SRA database under the BioProject accession number PRJNA750447.
Author Contributions
SG conceived and designed the experiments, analyzed and interpreted the results, and authored and reviewed drafts of the manuscript. SP collected the plant material, designed and performed lab experiments, performed the statistical analysis, analyzed and interpreted the results, and authored the manuscript. NS collected the plant material and performed lab experiments. AA performed pollen biology and ovule receptivity tests and wrote the manuscript. SK designed the experiments and organized the collection of plant material. RT designed the experiments, analyzed and interpreted the results, and wrote the manuscript. MA and AJ participated in designing lab experiments and reviewed the drafts of the manuscript. All authors have read and approved the manuscript.
Funding
We would like to acknowledge the grant provided by the Department of Biotechnology, Government of India to the University of Delhi, India, and North-Eastern Hill University, India under the Sanction order -BT/PR16385/NER/95/124/2015, through NER Biotechnology Management Cell (NER-BPMC) to support this work. Financial support from the Institution of Eminence, University of Delhi (IoE/FRP/LS/2020/27) is gratefully acknowledged.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
SP acknowledges the fellowship by the Council of Scientific and Industrial Research, Ministry of Science and Technology, Government of India.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.773572/full#supplementary-material
Supplementary Figure 1 | Heat map analysis plot of Fisher’s exact test (goodness-of-fit) for Hardy-Weinberg (HW) equilibrium generated using the hw.test function from the pegas package in R for each locus in each population. All loci in pink are loci suspected to be deviated from HWE with p ≤ 0.05.
Supplementary Figure 2 | Histogram showing relative DNA content of Nymphaea micrantha (peak A, coefficient of variation 4.92 %) and the reference, Solanum lycopersicum L. “Stupicke’polnı’ rane”’ (reference, peak B, coefficient of variation 6.02 %) obtained after analysis of nuclei isolated from young leaves using CyFlow® Cube 8 flow cytometer (Sysmex, Germany). % CV, coefficient of variation of the peaks as percentage (Standard deviation of the peak/mean position of the peak × 100).
Supplementary Figure 3 | Assembly statistics of Nymphaea micrantha genome before and after length filtration.
Supplementary Figure 4 | Spatial autocorrelation correlogram for total population (Omega = 157.275, p = 0.00) and four genetic clusters inferred from DAPC. Solid line indicates autocorrelation (−1 < r < +1) between individuals at each distance; dashed lines indicate 95 % confidence region around the null hypothesis of no genetic structure (r = 0) based on 10,000 permutations. Error bars for each distance class resulted from 10,000 bootstraps of individuals. Values outside the upper (U) and lower (L) 95% confidence interval indicate significant spatial structure.
Supplementary Figure 5 | Decline in the population of Nymphaea species in different parts of Goa, Maharashtra, and Kerala due to construction (top left), habitat destruction by the local people (top right), excessive growth of wild grasses (bottom left) and water pollution (bottom right).
Supplementary Table 1 | Details of 260 SSR primers (217 non-genic + 43 genic) designed in present study.
Supplementary Table 2 | Observations made using an epifluorescence microscope to test pollen fertility and viability in Nymphaea micrantha.
Supplementary Table 3 | Assembly statistics of Nymphaea micrantha genome.
Supplementary Table 4 | Contigs containing simple sequence repeats in Nymphaea genome.
Supplementary Table 5 | Annotation details of 12556 non-redundant SSR containing sequences.
Supplementary Table 6 | Cross species amplification of 57 polymorphic SSR loci in Nymphaea.
Supplementary Notes | Supplementary notes including supplementary findings of microsatellites discovery in Nymphaea micrantha genome and cross-species transferability of polymorphic SSR markers discovered from the study.
Footnotes
References
Amsellem, L., Noyer, J. L., Le Bourgeois, T., and Hossaert-Mckey, M. (2000). Comparison of genetic diversity of the invasive weed Rubus alceifolius Poir.(Rosaceae) in its native range and in areas of introduction, using amplified fragment length polymorphism (AFLP) markers. Mol. Ecol. 9, 443–455. doi: 10.1046/j.1365-294x.2000.00876.x
Ansari, R., and Jeeja, G. (2009). Waterlilies in India: taxonomy and cultivation of the genus Nymphaea L. Nymphaeaceae). Kozhikode: IAAT.
Bassi, N., Kumar, M. D., Sharma, A., and Pardha-Saradhi, P. (2014). Status of wetlands in India: a review of extent, ecosystem benefits, threats and management strategies. J. Hydrol. Reg. Stud. 2, 1–19. doi: 10.1016/j.ejrh.2014.07.001
Beugin, M.-P., Gayet, T., Pontier, D., Devillard, S., and Jombart, T. (2018). A fast likelihood solution to the genetic clustering problem. Methods Ecol. Evol. 9, 1006–1016. doi: 10.1111/2041-210X.12968
Borsch, T., Hilu, K. W., Wiersema, J. H., Löhne, C., Barthlott, W., and Wilde, V. (2007). Phylogeny of Nymphaea (Nymphaeaceae): evidence from substitutions and microstructural changes in the chloroplast trnT-trnF region. Int. J. Plant Sci. 168, 639–671. doi: 10.1086/513476
Botstein, D., White, R. L., Skolnick, M., and Davis, R. W. (1980). Con- struction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 32, 314–331.
Brookfield, J. F. Y. (1996). A simple new method for estimating null allele frequency from heterozygote deficiency. Mol. Ecol. 5, 453–455. doi: 10.1111/j.1365-294x.1996.tb00336.x
Chakraborty, R., Zhong, Y., Jin, L., and Budowle, B. (1994). Nondetectability of restriction fragments and independence of DNA fragment sizes within and between loci in RFLP typing of DNA. Am. J. Hum. Genet. 55:391.
Chen, C., Chu, Y., Ding, C., Su, X., and Huang, Q. (2020). Genetic diversity and population structure of black cottonwood (Populus deltoides) revealed using simple sequence repeat markers. BMC Genet. 21:2. doi: 10.1186/s12863-019-0805-1
Clark, L. V., and Jasieniuk, M. (2011). POLYSAT: an R package for polyploid microsatellite analysis. Mol. Ecol. Resour. 11, 562–566. doi: 10.1111/j.1755-0998.2011.02985.x
Conard, H. S. (1905). The waterlilies: a monograph of the genus Nymphaea (Vol. 4). Washington: Carnegie institution of Washington.
Dalziell, E. L., Lewandrowski, W., and Merritt, D. J. (2020). Increased salinity reduces seed germination and impacts upon seedling development in Nymphaea L.(Nymphaeaceae) from northern Australia’s freshwater wetlands. Aquat. Bot. 165:103235. doi: 10.1016/j.aquabot.2020.103235
Dkhar, J., Kumaria, S., Rao, S. R., and Tandon, P. (2013). New insights into character evolution, hybridization and diversity of Indian Nymphaea (Nymphaeaceae): evidence from molecular and morphological data. Syst. Biodivers. 11, 77–86.
Doležel, J., Sgorbati, S., and Lucretti, S. (1992). Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiol. Plant. 85, 625–631. doi: 10.1111/j.1399-3054.1992.tb04764.x
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Dray, S., and Dufour, A.-B. (2007). The ade4 package: implementing the duality diagram for ecologists. J. Stat. Softw. 22, 1–20.
Earl, D. A. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Ellstrand, N. C., and Roose, M. L. (1987). Patterns of genotypic diversity in clonal plant species. Am. J. Bot. 74, 123–131. doi: 10.2307/2444338
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Forsman, A., and Wennersten, L. (2016). Inter-individual variation promotes ecological success of populations and species: evidence from experimental and comparative studies. Ecography 39, 630–648.
Frankham, R. (2005). Stress and adaptation in conservation genetics. J. Evol. Biol. 18, 750–755. doi: 10.1111/j.1420-9101.2005.00885.x
Galen, C., and Plowright, R. C. (1987). Testing the accuracy of using peroxidase activity to indicate stigma receptivity. Can. J. Bot. 65, 107–111. doi: 10.1139/b87-015
Ganapathy, K. N., Gomashe, S. S., Rakshit, S., Prabhakar, B., Ambekar, S. S., Ghorade, R. B., et al. (2012). Genetic diversity revealed utility of SSR markers in classifying parental lines and elite genotypes of sorghum (‘Sorghum bicolor’L. Moench). Aust. J. Crop Sci. 6, 1486–1493.
Geraldes, A., Farzaneh, N., Grassa, C. J., McKown, A. D., Guy, R. D., Mansfield, S. D., et al. (2014). Landscape genomics of Populus trichocarpa: the role of hybridization, limited gene flow, and natural selection in shaping patterns of population structure. Evolution 68, 3260–3280. doi: 10.1111/evo.12497
Goss, E. M., Tabima, J. F., Cooke, D. E., Restrepo, S., Fry, W. E., Forbes, G. A., et al. (2014). The Irish potato famine pathogen Phytophthora infestans originated in central Mexico rather than the Andes. Proc. Natl. Acad. Sci. U. S. A. 111, 8791–8796. doi: 10.1073/pnas.1401884111
Goudet, J. (2005). Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 5, 184–186. doi: 10.1111/j.1471-8286.2004.00828.x
Guo, J.-L., Yu, Y.-H., Zhang, J.-W., Li, Z.-M., Zhang, Y.-H., and Volis, S. (2019). Conservation strategy for aquatic plants: endangered Ottelia acuminata (Hydrocharitaceae) as a case study. Biodivers. Conserv. 28, 1533–1548.
Gupta, P. P. (1978). Cytogenetics of aquatic ornamentals. II. Cytology of Nymphaeas. Cytologia 43, 477–484.
Hagenblad, J., Hülskötter, J., Acharya, K. P., Brunet, J., Chabrerie, O., Cousins, S. A., et al. (2015). Low genetic diversity despite multiple introductions of the invasive plant species Impatiens glandulifera in Europe. BMC Genet. 16:103. doi: 10.1186/s12863-015-0242-8
Han, Y.-C., Teng, C.-Z., Zhong, S., Zhou, M.-Q., Hu, Z.-L., and Song, Y.-C. (2007). Genetic variation and clonal diversity in populations of Nelumbo nucifera (Nelumbonaceae) in central China detected by ISSR markers. Aquat. Bot. 86, 69–75.
Heslop-Harrison, J., and Heslop-Harrison, Y. (1970). Evaluation of pollen viability by enzymatically induced fluorescence; intracellular hydrolysis of fluorescein diacetate. Stain Technol. 45, 115–120. doi: 10.3109/10520297009085351
Hossain, A., Kabir, G., Ud-deen, M. M., and Alam, A. M. S. (2007). Cytological studies of Nymphaea species available in Bangladesh. J. Bio-Sci. 15, 7–13.
Hu, J., Pan, L., Liu, H., Wang, S., Wu, Z., Ke, W., et al. (2012). Comparative analysis of genetic diversity in sacred lotus (Nelumbo nucifera Gaertn.) using AFLP and SSR markers. Mol. Biol. Rep. 39, 3637–3647. doi: 10.1007/s11033-011-1138-y
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. doi: 10.1093/nar/gkn923
Husband, B. C., and Barrett, S. C. (1991). Colonization history and population genetic structure of Eichhornia paniculata in Jamaica. Heredity 66, 287–296.
Hutchison, D. W., and Templeton, A. R. (1999). Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution 53, 1898–1914. doi: 10.1111/j.1558-5646.1999.tb04571.x
Jha, K. K. (2013). Aquatic food plants and their consumer birds at Sandi Bird Sanctuary, Hardoi, Northern India. Asian J. Conserv. Biol. 2, 30–43.
Jombart, T. (2008). adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Jombart, T., Devillard, S., and Balloux, F. (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11:94. doi: 10.1186/1471-2156-11-94
Kahilainen, A., Puurtinen, M., and Kotiaho, J. S. (2014). Conservation implications of species–genetic diversity correlations. Glob. Ecol. Conserv. 2, 315–323. doi: 10.1016/j.gecco.2014.10.013
Kalia, R. K., Rai, M. K., Kalia, S., Singh, R., and Dhawan, A. K. (2011). Microsatellite markers: an overview of the recent progress in plants. Euphytica 177, 309–334.
Kamvar, Z. N., Tabima, J. F., and Grunwald, N. J. (2014). Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2013, 1–14. doi: 10.7717/peerj.281
Lambertini, C., Riis, T., Olesen, B., Clayton, J. S., Sorrell, B. K., and Brix, H. (2010). Genetic diversity in three invasive clonal aquatic species in New Zealand. BMC Genet. 11:52. doi: 10.1186/1471-2156-11-52
Lauer, N., and Ross, C. (2016). Physiological and oxidative stress responses of baldcypress in response to elevated salinity: linking and identifying biomarkers of stress in a keystone species. Acta Physiol. Plant. 38, 1–15.
Leito, A., Leivits, M., Leivits, A., Raet, J., Ward, R., Ott, I., et al. (2016). Black-headed gull (Larus ridibundus L.) as a keystone species in the lake bird community in primary forest-mire-lake ecosystem. Balt. For. 22, 34–45.
Li, W., Wang, B., and Wang, J. (2006). Lack of genetic variation of an invasive clonal plant Eichhornia crassipes in China revealed by RAPD and ISSR markers. Aquat. Bot. 84, 176–180. doi: 10.1016/j.aquabot.2005.09.008
Linskens, H. F., and Esser, K. L. (1957). Über eine spezifische Anfärbung der Pollenschläuche im Griffel und die Zahl der Kallosepfropfen nach Selbstung und Fremdung. Naturwissenschaften 44, 16–16.
Löhne, C., Borsch, T., Jacobs, S. W., Hellquist, C. B., and Wiersema, J. H. (2008). Nuclear and plastid DNA sequences reveal complex reticulate patterns in Australian water-lilies (Nymphaea subgenus Anecphya, Nymphaeaceae). Aust. Syst. Bot. 21, 229–250. doi: 10.1071/sb07010
Lörz, H., and Wenzel, G. (2005). Molecular marker systems in plant breeding and crop improvement. Heidelberg: Springer.
Mantel, N. (1967). The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220.
Mitra, R. L. (1990). “Nymphaeaceae,” in Fascicles of flora of India, Fascicles 20, eds M. P. Nayar, K. Thothathri, and M. Sanjappa (Howrah: Botanical Survey of India), 11–25.
Muller, H. J. (1914). A new mode of segregation in Gregory’s tetraploid primulas. Am. Nat. 48, 508–512. doi: 10.1086/279426
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590. doi: 10.1093/genetics/89.3.583
Niklas, K. J., and Cobb, E. D. (2017). The evolutionary ecology (evo-eco) of plant asexual reproduction. Evol. Ecol. 31, 317–332. doi: 10.1007/s10682-016-9828-5
Orsenigo, S. (2018). How to halt the extinction of wetland-dependent plant species? The role of translocations and restoration ecology. Aquat. Conserv. Mar. Freshw. Ecosyst. 28, 772–775. doi: 10.1002/aqc.2962
Paradis, E., and Schliep, K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Peakall, R., Smouse, P. E., and Huff, D. R. (1995). Evolutionary implications of allozyme and RAPD variation in diploid populations of dioecious buffalograss Buchloe dactyloides. Mol. Ecol. 4, 135–148.
Peakall, R. O. D., and Smouse, P. E. (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295.
Poczai, P., Mátyás, K. K., Szabó, I., Varga, I., Hyvönen, J., Cernák, I., et al. (2011). Genetic variability of thermal Nymphaea (Nymphaeaceae) populations based on ISSR markers: implications on relationships, hybridization, and conservation. Plant Mol. Biol. Rep. 29, 906–918.
Pollux, B. J. A., Jong, M. D. E., Steegh, A., Verbruggen, E., Van Groenendael, J. M., and Ouborg, N. J. (2007). Reproductive strategy, clonal structure and genetic diversity in populations of the aquatic macrophyte Sparganium emersum in river systems. Mol. Ecol. 16, 313–325. doi: 10.1111/j.1365-294X.2006.03146.x
POWO (2021). Plants of the World Online. Facilitated by the Royal Botanic Gardens, Kew. Available Online at: http://www.plantsoftheworldonline.org/ (accessed 28 November 2021)
Pritchard, J. K., Wen, W., and Falush, D. (2010). Documentation for STRUCTURE software: Version 2. Chicago, IL: University of Chicago.
Raja, M. M. M., Sethiya, N. K., and Mishra, S. H. (2010). A comprehensive review on Nymphaea stellata: a traditionally used bitter. J. Adv. Pharm. Technol. Res. 1:311. doi: 10.4103/0110-5558.72424
Ranjan, R., and Prakash, S. (2019). Seasonal Variation in Primary Productivity and Macrophytes of Baghel Taal. Iconic Res. Eng. J. 3, 210–215.
Reed, D. H., and Frankham, R. (2003). Correlation between fitness and genetic diversity. Conserv. Biol. 17, 230–237. doi: 10.1046/j.1523-1739.2003.01236.x
Ren, M.-X., Zhang, Q.-G., and Zhang, D.-Y. (2005). Random amplified polymorphic DNA markers reveal low genetic variation and a single dominant genotype in Eichhornia crassipes populations throughout China. Weed Res. 45, 236–244.
Rice, W. R. (1989). Analyzing tables of statistical tests. Evolution 43, 223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x
Rousset, F. (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145, 1219–1228. doi: 10.1093/genetics/145.4.1219
Sajib, A. M., Hossain, M., Mosnaz, A., Hossain, H., Islam, M., Ali, M., et al. (2012). SSR marker-based molecular characterization and genetic diversity analysis of aromatic landreces of rice (Oryza sativa L.). J. BioSci. Biotechnol. 1, 107–116. doi: 10.1007/978-81-322-2665-9_5
Sand-Jensen, K., Riis, T., Vestergaard, O., and Larsen, S. E. (2000). Macrophyte decline in Danish lakes and streams over the past 100 years. J. Ecol. 88, 1030–1040.
Sengupta, S., and Tandon, R. (2010). Assessment of ovule receptivity as a function of expected brood size in flowering plants. Int. J. Plant Reprod. Biol. 2, 51–63.
Shannon, C. E. (2001). A mathematical theory of communication. ACM Sigmobile Mob. Comput. Commun. Rev. 5, 3–55.
Shen, Z., Zhang, K., Ma, L., Duan, J., and Ao, Y. (2017). Analysis of the genetic relationships and diversity among 11 populations of Xanthoceras sorbifolia using phenotypic and microsatellite marker data. Electron. J. Biotechnol. 26, 33–39.
Shivanna, K. R., and Rangaswamy, N. S. (1992). “Tests for pollen viability,” in Pollen biology, eds K. R. Shivanna and N. S. Rangaswamy (Berlin: Springer), 33–37. doi: 10.1007/978-3-642-77306-8_5
Singh, M., and Jain, A. P. (2017). A review on genus Nymphaea: multi potential medicinal plant. Asian J. Pharm. Educ. Res. 6, 1–9.
Slatkin, M. (1995). A measure of population subdivision based on microsatellite allele frequencies. Genetics 139, 457–462. doi: 10.1093/genetics/139.1.457
Smouse, P. E., and Peakall, R. O. D. (1999). Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity 82, 561–573. doi: 10.1038/sj.hdy.6885180
Smouse, R. P. P., and Peakall, R. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Soltis, P. S., and Soltis, D. E. (2000). The role of genetic and genomic attributes in the success of polyploids. Proc. Natl. Acad. Sci. U. S. A. 97, 7051–7057. doi: 10.1073/pnas.97.13.7051
Stebbins, G. L. (1957). Self fertilization and population variability in the higher plants. Am. Nat. 91, 337–354. doi: 10.1086/281999
Thiel, T., Michalek, W., Varshney, R., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Tian, H.-L., Xue, J.-H., Wen, J., Mitchell, G., and Zhou, S.-L. (2008). Genetic diversity and relationships of lotus (Nelumbo) cultivars based on allozyme and ISSR markers. Sci. Hortic. 116, 421–429. doi: 10.1016/j.scienta.2008.02.011
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P., and Shipley, P. (2004). MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538.
Volkova, P. A., Trávníček, P., and Brochmann, C. (2010). Evolutionary dynamics across discontinuous freshwater systems: rapid expansions and repeated allopolyploid origins in the Palearctic white water–lilies (Nymphaea). Taxon 59, 483–494.
Weir, B. S., and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 1358–1370.
Wiersema, J. H. (1988). Reproductive biology of Nymphaea (Nymphaeaceae). Ann. Mo. Bot. Gard. 75, 795–804.
Woods, K., Hilu, K. W., Wiersema, J. H., and Borsch, T. (2005b). Pattern of variation and systematics of Nymphaea odorata: I. Evidence from morphology and inter-simple sequence repeats (ISSRs). Syst. Bot. 30, 471–480.
Woods, K., Hilu, K. W., Borsch, T., and Wiersema, J. H. (2005a). Pattern of variation and systematics of Nymphaea odorata: II. Sequence information from ITS and trnL-trnF. Syst. Bot. 30, 481–493. doi: 10.1600/0363644054782143
Xu, C.-Y., Zhang, W.-J., Fu, C.-Z., and Lu, B.-R. (2003). Genetic diversity of alligator weed in China by RAPD analysis. Biodivers. Conserv. 12, 637–645.
Yakandawala, D., Guruge, S., and Yakandawala, K. (2017). The identity of the violet flowered water lily (Nymphaeaceae) and its hybrid origin in the wetland ecosystems of Sri Lanka. J. Natl. Sci. Found. Sri Lanka 45, 381–392. doi: 10.4038/jnsfsr.v45i4.8232
Keywords: conservation, genetic diversity, microsatellite, population structure, propagules, reproduction
Citation: Parveen S, Singh N, Adit A, Kumaria S, Tandon R, Agarwal M, Jagannath A and Goel S (2022) Contrasting Reproductive Strategies of Two Nymphaea Species Affect Existing Natural Genetic Diversity as Assessed by Microsatellite Markers: Implications for Conservation and Wetlands Restoration. Front. Plant Sci. 13:773572. doi: 10.3389/fpls.2022.773572
Received: 10 September 2021; Accepted: 03 February 2022;
Published: 09 March 2022.
Edited by:
Petr Koutecký, University of South Bohemia in České Budějovice, CzechiaCopyright © 2022 Parveen, Singh, Adit, Kumaria, Tandon, Agarwal, Jagannath and Goel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shailendra Goel, c2hhaWxlbmRyYWdvZWxAZ21haWwuY29t