 Yan-Fang Cui1†
Yan-Fang Cui1† Peng Zhou1†
Peng Zhou1† Kun-Li Xiang2
Kun-Li Xiang2 Qiang Zhang3Hua Yan1
Qiang Zhang3Hua Yan1 Li-Guo Zhang1Bo Pan3Yu-Song Huang3Zhi-You Guo4Zhen-Yu Li2
Li-Guo Zhang1Bo Pan3Yu-Song Huang3Zhi-You Guo4Zhen-Yu Li2 Xiao-Guo Xiang1*
Xiao-Guo Xiang1*- 1Jiangxi Province Key Laboratory of Watershed Ecosystem Change and Biodiversity, Institute of Life Science and School of Life Sciences, Nanchang University, Nanchang, Jiangxi, China
- 2State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, Beijing, China
- 3Guangxi Key Laboratory of Plant Conservation and Restoration Ecology in Karst Terrain, Guangxi Institute of Botany, Guangxi Zhuang Autonomous Region and Chinese Academy of Sciences, Guilin, China
- 4Qiannan Normal College for Nationalities, College of Biological Sciences and Agriculture, Duyun, Guizhou, China
Trichosporeae is the largest and most taxonomically difficult tribe of Gesneriaceae due to its diverse morphology. Previous studies have not clarified the phylogenetic relationships within this tribe on several DNA markers, including the generic relationships within subtribes. Recently, plastid phylogenomics have been successfully employed to resolve the phylogenetic relationships at different taxonomic levels. In this study, plastid phylogenomics were used to explore the relationships within Trichosporeae. Eleven plastomes of Hemiboea were newly reported. Comparative analyses, phylogeny and morphological character evolution within Trichosporeae were conducted on 79 species representing seven subtribes. The Hemiboea plastomes range from 152,742 bp to 153,695 bp in length. Within Trichosporeae, the sampled plastomes range from 152,196 bp to 156,614 bp and GC content from 37.2% to 37.8%. A total of 121–133 genes were annotated in each species, including 80–91 protein-coding genes, 34–37 tRNA genes, and 8 rRNA genes. The contraction and expansion of IR borders were not detected, and gene rearrangements and inversions did not occur. The 13 hypervariable regions were proposed as the potential molecular markers for species identification. A total of 24,299 SNPs and 3,378 indels were inferred, and most of the SNPs were functionally missense and silent variations. There were 1968 SSRs, 2055 tandem repeats and 2802 dispersed repeats. The RSCU and ENC values indicated that the codon usage pattern was conserved in Trichosporeae. Both the phylogenetic frameworks based on the whole plastome and 80 CDSs were basically concordant. The sister relationships between Loxocarpinae and Didymocarpinae were confirmed, and Oreocharis was a sister group of Hemiboea with high support. The morphological characters showed a complex evolutionary pattern of Trichosporeae. Our findings may contribute to future research on genetic diversity, morphological evolutionary patterns, and conservation of the tribe Trichosporeae.
1 Introduction
Gesneriaceae comprises ca. 150 genera and 3000 species and mainly distributes in the tropics and subtropics of the world (Li and Wang, 2005; Möller et al., 2016). These plants are usually perennial herbs with gorgeous flowers and various leaves, which have high ornamental value and great potential for garden and horticultural applications (Li and Wang, 2005). For example, Sinningia speciosa Benth. & Hook., Episcia cupreata (Hook.) Hanst. and Streptocarpus ionanthus (H. Wendl.) Christenh. have become famous ornamental flowers. Furthermore, many Gesneriaceae species, such as Corallodiscus lanuginosus (Wall. ex R. Br.) B.L. Burtt, Dorcoceras hygrometricum Bunge, Hemiboea subcapitata Clarke, Lysionotus pauciflorus Maxim. and Primulina eburnea (Hance) Yin Z. Wang, have been used as folk medicines in China for a long time (Li and Wang, 2005). As the largest tribe of Gesneriaceae, Trichosporeae includes 10 subtribes and distributes in tropical and subtropical Asia, Europe and Africa. Its morphological characters are diverse, and traditional classifications based on morphology have identified genera that belong to different alliances and geographical groups. This makes it the most taxonomically difficult tribe (e.g., Weber et al., 2013; Möller et al., 2016). Phylogenetically, the tribe is subdivided into distinct clades, but their relationships are not fully resolved and not consistently well supported (e.g., Wang et al., 2010; Möller et al., 2011; Li et al., 2022). Based on 16 plastid, 9 nuclear and 1 mitochondrial DNA makers, Roalson and Roberts (2016) showed that Streptocarpinae and Loxocarpinae were sister groups, while Li et al. (2022), based on 11 chloroplast regions and nuclear ITS, indicated that Loxocarpinae was a sister group to Didymocarpinae. Besides, the phylogenetic positions of some genera within Didymocarpinae were also uncertain. For example, both Roalson and Roberts (2016) and Li et al. (2022) indicated that Hemiboea was close to Lysionotus with weak supporting values, while Hsieh et al. (2022) supported that Oreocharis was a sister group of Hemiboea based on plastomes with one IR excluded. Möller et al. (2011) proposed to employ more molecular markers, especially conserved plastome sequences, to improve the phylogenetic relationships within Trichosporeae. Chen et al. (2020) also intensively suggested adopting more DNA sequences to resolve the phylogeny of Gesneriaceae. Therefore, developing more effective molecular markers is crucial for the conservation and utilization of Trichosporeae as well as Gesneriaceae.
With the advancement of high-throughput sequencing technologies, more and more plastomes are being successfully sequenced and annotated (Dobrogojski et al., 2020). Most angiosperms plastomes are circular with four regions: a large single copy (LSC) region, a small single copy (SSC) region and two copies of inverted repeats (IRa/b), with a length ranging from 120–160 kb with 30–40% GC content (Mower and Vickrey, 2018). The high conservatism and slow evolution rate of plastomes make them suitable for inferring phylogenetic relationships (e.g., Lu et al., 2016; Chi et al., 2018; Lee et al., 2019). Rosaceae is one of the taxonomically difficult lineages with hybridization and rapid radiation, and the phylogenetic relationships among subfamilies, tribes and genera have been successfully resolved by employing plastid phylogenomics (Zhang et al., 2017). Wei et al. (2021) clarified the phylogenetic relationships within Euphorbia based on plastomes, which have highly homogenous morphological characters, and revealed that it may have undergone complex plastome evolution. Overall, plastome data has a high power to explore the phylogenetic relationships of different taxonomic levels.
In this study, we newly sequenced, assembled, and annotated plastomes of 11 Hemiboea species, and combined them with 68 previously reported plastomes of Trichosporeae, we conducted comparative analyses, phylogenetic reconstruction and morphological characters appraisal within Trichosporeae. Our objectives are: (1) to investigate general plastome features and sequence divergence in Trichosporeae; (2) to identify the most variable regions as potential DNA barcodes for future species identification within Trichosporeae; (3) to explore the phylogenetic relationships within this tribe; (4) to assess the morphological evolution of Trichosporeae. Our study provides abundant information for the phylogeny, biogeography, and conservation of Trichosporeae.
2 Materials and methods
2.1 Sampling, sequencing, assembly, and annotation
In this study, eleven new plastomes of Hemiboea species were obtained. Sixty-eight published plastomes of Trichosporeae were downloaded from GenBank and updated the annotations (one species of Actinostephanus, two species of Beccarinda, one species of Boeica, one species of Corallodiscus, one species of Dorcoceras, one species of Haberlea, one species of Henckelia, one species of Leptoboea, one species of Litostigma, one species of Lysionotus, five species of Oreocharis, 12 species of Paraboea, four species of Petrocodon, 28 species of Primulina, two species of Rhynchotechum, one species and five varieties of Streptocarpus), representing seven subtribes (Corallodiscinae, Didymocarpinae, Leptobaeinae, Litostigminae, Loxocarpinae, Ramondinae and Streptocarpinae). Additionally, two species of Gesnerioideae (Achimenes cettoana H.E. Moore and Achimenes erecta (Lam.) H.P. Fuchs) were selected as outgroups. The detailed information of samples was listed in Table S1.
Total DNA was extracted from silica gel-dried leaves using the modified CTAB method (Doyle and Doyle, 1987). Library construction was performed with the NEB Next® Ultra DNA Library Prep Kit (NEB, USA) following the manuals. Libraries for paired-end 150 bp sequencing were conducted using an Illumina HiSeq 2000 platform to generate approximately 4 Gb of data for each sample. The library preparation and sequencing were finished at the Kunming Institute of Botany, Chinese Academy of Sciences (Yunnan, China). The quality of raw sequence reads was assessed in FastQC v0.11.9 (Brown et al., 2017) and the adapters and low-quality reads were filtered in Trimmomatic v0.39 (Bolger et al., 2014). Then, the clean reads were assembled using GetOrganelle v1.7.3.2 with default parameters (Jin et al., 2020). The assembled genomes were checked and visualized in Bandage v0.7.1 (Wick et al., 2015). Finally, the plastomes were annotated and manually checked in Geneious v9.05 (Kearse et al., 2012) with Hemiboea ovalifolia (W.T. Wang) A. Weber & Mich. Möller (NC_054358) as a reference. The physical map was drawn using OGDRAW (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html).
2.2 Structure and sequence divergence analysis
The expansion and contraction of IR boundary were visualized in IRscope v3.1 (Amiryousefi et al., 2018). Sequence alignments were performed using MAFFT v7 (Katoh and Standley, 2013) and manually adjusted in Geneious v9.05 (Kearse et al., 2012). Before the collinearity analysis, one copy of the IR region was removed. Possible rearrangements and inversions were detected using the Mauve v1.1.3 (Darling et al., 2010) plugin in Geneious v9.05 (Kearse et al., 2012). To further detect hypervariable regions within Trichosporeae, nucleotide diversity (pi) of protein-coding genes (CDSs) and non-coding regions were evaluated using DnaSP v6.12.03 (Rozas et al., 2017). Functional annotations for the nucleotide variations within Trichosporeae plastomes were performed using snpEff v5.1 (Cingolani, 2012).
2.3 Repetitive sequence analysis
Three types of repeats sequences, including simple sequence repeats (SSRs), tandem repeats, and dispersed repeats, were analyzed. SSRs were detected using MISA v2.1 (Beier et al., 2017) and visualized using R packages ggpubr and ggplot2 (Wickham, 2016; Kassambara, 2023). Tandem repeats were performed using Tandem Repeats Finder v0.9 (Benson, 1999). The identification of dispersed repeats (including forward, reverse, complement, and palindromic) was analyzed in REPuter (Kurtz et al., 2001) following the parameter settings in Cauz-Santos et al. (2020).
2.4 Codon usage analysis
To quantify the degree of the codon usage bias, both the relative synonymous codon usage (RSCU) ratio and the effective number codons (ENC) for 79 Trichosporeae plastomes were estimated using CodonW v1.4.2 (Peden, 1999). The RSCU>1 indicates a preferred codon, while the RSCU<1 indicates the opposite. The ENC values range from 20 (where only one synonymous codon is used to encode amino acids) to 61 (where every synonymous codon is used equally). The ENC<35 indicates that a gene has significant codon bias (Tao and Yao, 2020).
2.5 Phylogenetic analysis
The phylogenetic relationships were inferred from whole plastome sequences (excluding one IR) and concatenation of 80 CDSs. Before performing maximum likelihood (ML) and Bayesian inference (BI), we calculated the best nucleotide substitution model for each dataset using jModelTest2 (Darriba et al., 2012) under the Akaike information criterion. We conducted ML analyses in RAxML v8.2.12 (Stamatakis, 2014) with a rapid bootstrap analysis (1000 replicates) and searching for the best-scoring tree simultaneously. BI analyses were performed in MrBayes v3.2.6 (Ronquist et al., 2012) with four Markov Chain Monte Carlo chains starting with a random tree. We ran 2,000,000 generations and sampled every 1000 generations. The convergence was assessed in Tracer v1.7 (Rambaut et al., 2018). The majority rule (>50%) consensus tree was obtained after removing the “burn-in” samples (the first 20% of the sampled trees).
2.6 Ancestral state reconstruction of morphological characters
Following Li and Wang (2005); Wang et al. (1998), and Weber et al. (2013), nine morphological characters and their states were selected to study their evolution within Trichosporeae. These characters were habit (perennial herb, annual herb, shrub), stem (rhizome, creeping stem, erect stem), leaf margin (dentation, entire, undulate, cleft), inflorescence (cymes, solitary), number of stamens (2, 4), number of staminodes (0, 1, 2, 3), capsule (twisted, not twisted), capsule dehiscing type (valves 2, valves 4, berry), and seeds (appendaged, unappendaged). Ancestral state reconstruction were performed under the ER model (equal-rates model) using R packages phytools (Revell, 2012).
3 Result
3.1 Plastome features and gene contents
The plastomes of the 11 Hemiboea species displayed the typical quadripartite structure (Figure 1). The genome sizes ranged from 152,742 bp (H. purpurea Yan Liu & W.B. Xu) to 153, 695 bp (H. fangii Chun) (Table S2). The plastomes contained a pair of IRs (25,432–25,494 bp), LSC region (83,724–84,670 bp), and SSC region (17,788–18,256 bp). The GC content of the plastomes ranged from 37.5% to 37.6%. The GC content of the IR region ranged from 43.1% to 43.2%, the GC content of the LSC region ranged from 35.5% to 35.6%, while the GC content of the SSC region ranged from 30.9% to 31.2% (Table S2). Each Hemiboea plastome consisted of 132 genes, comprising 87 CDSs, 37 transfer RNA (tRNA) genes, and eight ribosomal RNA (rRNA) genes.
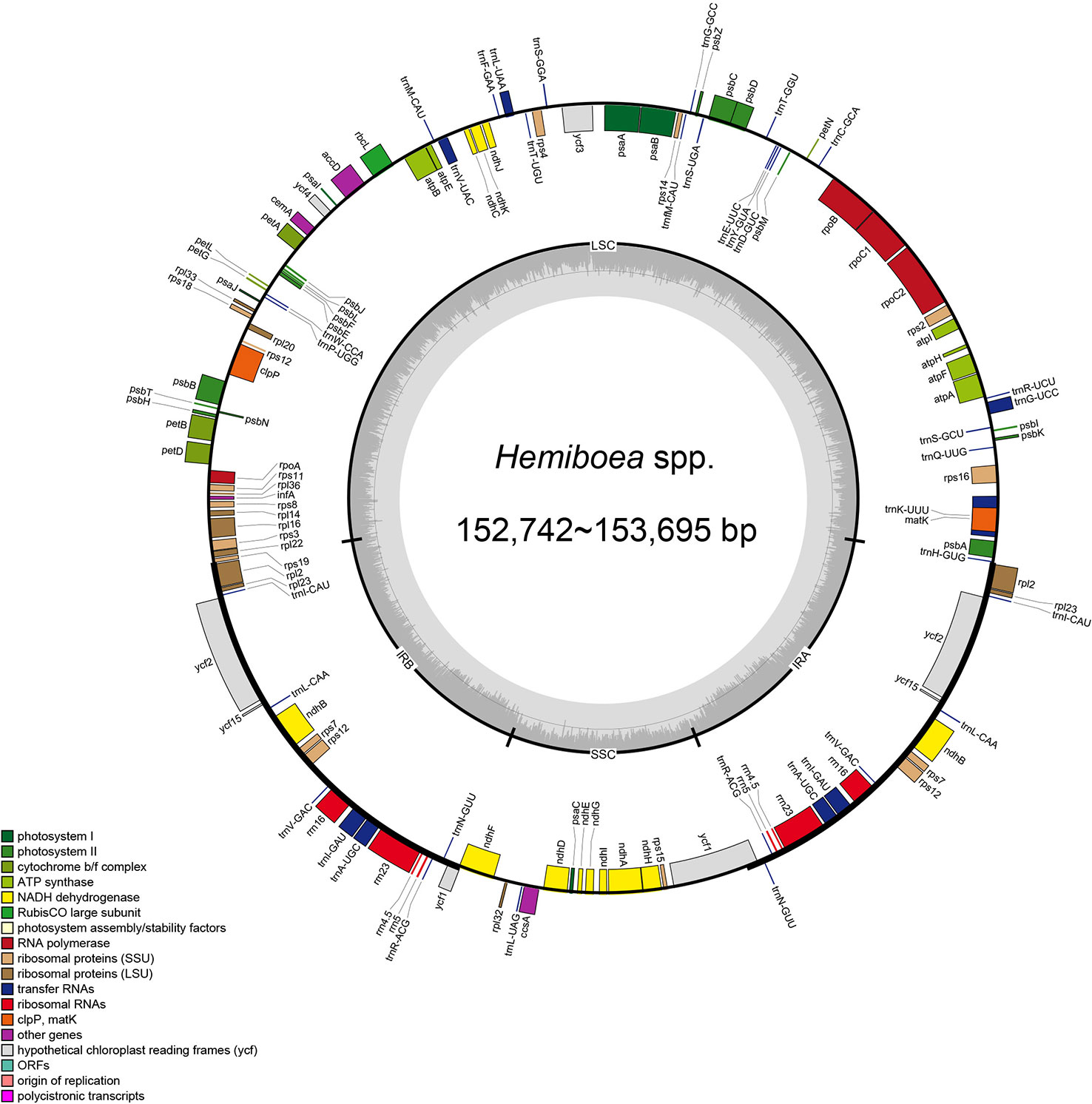
Figure 1 Plastome structure of 11 Hemiboea species. Bars of different colors indicate different functional groups. LSC, large single copy region; SSC, small single copy region; IRa and IRb, two inverted repeat regions.
The total length of the 79 plastomes in the tribe Trichosporeae ranged from 152,196 bp (Primulina suichuanensis X. L. Yu & J. J. Zhou) to 156,614 bp (Corallodiscus lanuginosus (Wallich ex R. Brown) B. L. Burtt) (Table S2). All the plastomes also displayed a typical quadripartite structure, including LSC region (83,063–87,429 bp), SSC region (17,478–18,330 bp) as well as a pair of IRs regions (25,272–25,657 bp) (Table S2). The total GC content ranged from 37.2% to 37.8%. The GC content of the IR region ranged from 43.1% to 43.2%. The GC content of the LSC region ranged from 34.9% to 35.8%, while the GC content of the SSC region ranged from 30.9% to 31.7%. The 79 Trichosporeae plastomes contained 121–133 genes, including 80–91 CDSs, 34–37 tRNAs, and eight rRNAs (Table S2). Among these genes, six CDSs (ndhB, rpl2, rpl23, rps7, ycf15, and ycf2), seven tRNA (trnA-UGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, and trnV-GAC), four rRNA (rrn16, rrn23, rrn4.5 and rrn5) were duplicated in the IR regions. The gene rps12 contained three exons, two of which were duplicated in the IRs. Further, 16 genes (ndhA, ndhB, petB, petD, atpF, rpl16, rpl2, rps12, rps16, rpoC1, trnG-UCC, trnA-UGC, trnI-GAU, trnK-UUU, trnL-UAA, and trnV-UAC) had one intron, and two genes (clpP and ycf3) had two introns (Table S3).
3.2 Plastome structural variations and sequence divergence
The expansion and contraction in the IR/SC boundary regions were shown in Figure 2. Within Trichosporeae, rpl2 and rps19 genes located in the LSC-IRb borders. Although the length of rps19 was 179 bp, its location showed differences. In 62 species, rps19 crossed the border, leading to the appearance of a ψrps19 in IRa-LSC borders. The ndhF located across the IRb-SSC borders, except for Haberlea rhodopensis Friv. The SSC-IRa borders located in ycf1 gene. The ycf1 gene crossed the border in 76 species, leading to the appearance of a ψycf1 in IRa-LSC borders. However, the ycf1 gene in Boeica ferruginea Drake, Leptoboea multiflora (Clarke) Clarke, and Beccarinda cordifolia (J. Anthony) B.L. Burtt was completely located in the SSC region. The IRa-LSC borders were between the ψrps19, rpl2, and trnH.

Figure 2 Visualization of the expansion and contraction in the inverted repeat region (IR) boundary. The topology was the ML tree based on plastomes with one IR excluded.
The collinearity analysis revealed no gene rearrangements or inversions in the Trichosporeae plastomes (Figure S1). The pi of the CDSs ranged from 0.001876 (rps7) to 0.036559 (rps15). Notably, five CDSs (rps15, matK, ndhF, rps16, and rpl32) exhibited high pi values (pi>0.025; Figure 3A). Among the non-coding regions, the pi values ranged from 0 (i.e., rpoC1/rpoB, trnI-GAU/trnA-UGC and ndhA/ndhH) to 0.409580 (rpl16/rps3). Eight non-coding regions (i.e., rpl16/rps3, rpl14/rpl16, petB/petD, trnC-GCA/petN, rps15/ycf1, trnfM-CAU/rps14, petD/rpoA and trnS-GGA/rps4) showed significantly high pi values (pi>0.25; Figure 3B). The aligned matrix of the 79 Trichosporeae plastomes contained 24,299 single nucleotide polymorphisms (SNPs) and 3378 insertion-deletions (indels). The majority of SNPs from coding genes were functionally missense variations, while 6448 SNPs (43.20%) and 104 SNPs (0.70%) were silent (synonymous) and nonsense variations, respectively (Table 1).
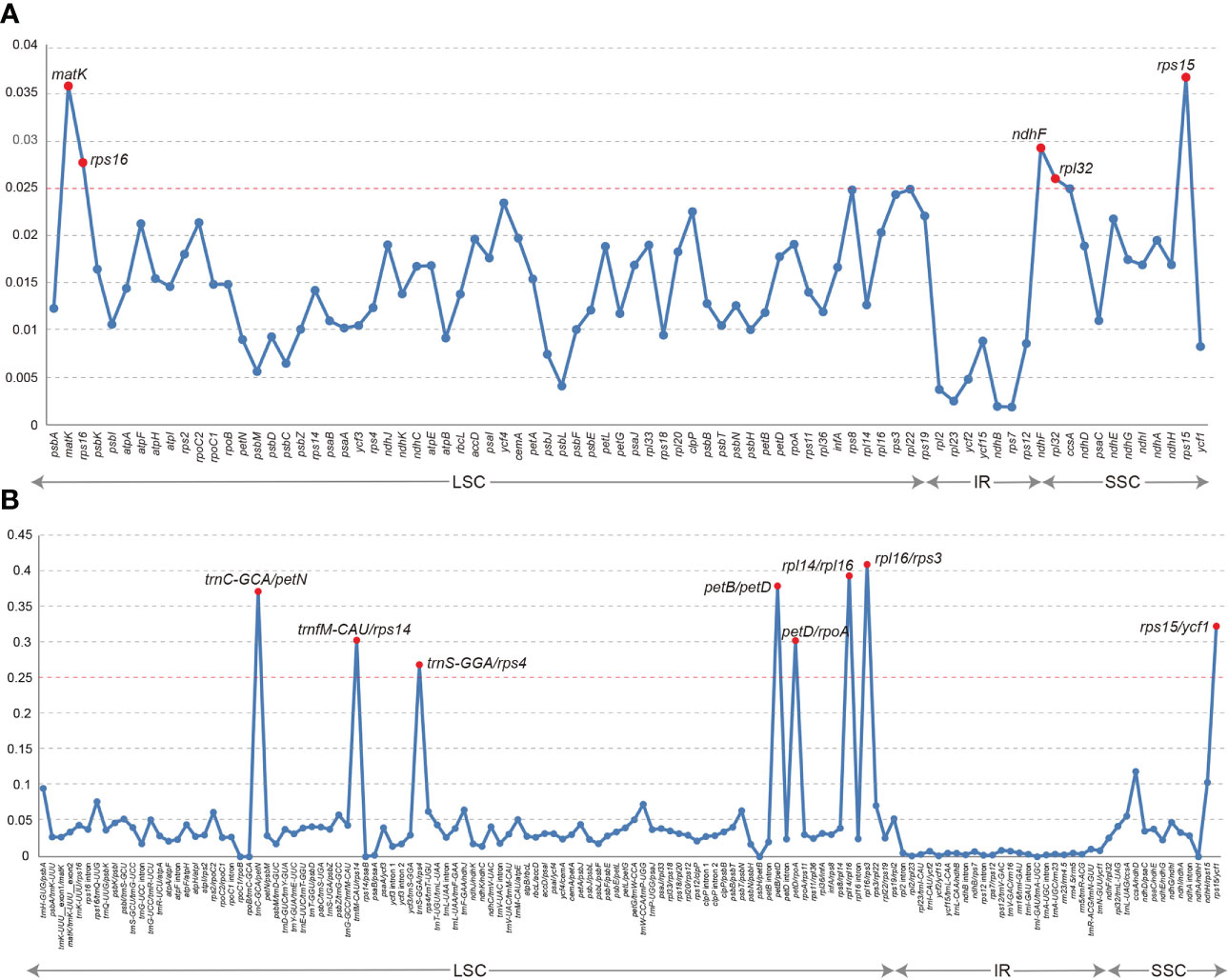
Figure 3 Comparison of nucleotide diversity (Pi) values in Trichosporeae plastomes. (A) Pi values of CDSs, (B) Pi values of non-coding regions.
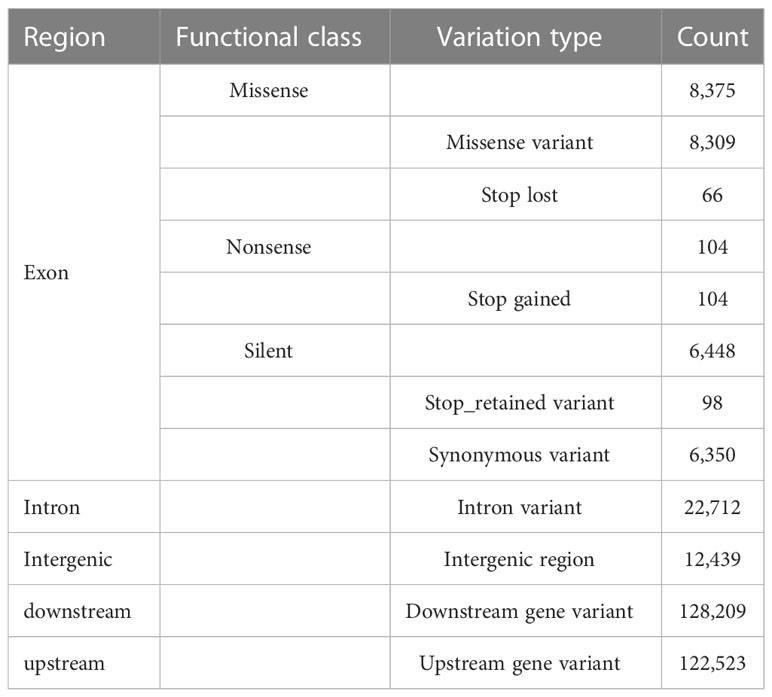
Table 1 Functional annotaitons for the single-nucleotide variants (SNVs) detected in the Trichosporeae plastomes.
The results of SSRs, tandem repeats, and dispersed repeats in the 79 Trichosporeae plastomes were shown in Figure 4 and Table S4. The number of identified SSRs ranged from 11 (Dorcoceras hygrometricum) to 53 (Corallodiscus lanuginosus). A total of 1968 SSRs were detected in the 79 Trichosporeae plastomes, of which 1432 SSRs (62.60%) located in the LSC region, 280 SSRs (14.23%) in the SSC region, and 258 SSRs (13.11%) in the IR regions. Four types of SSRs (mono-nucleotide, di-nucleotide, tri-nucleotide and tetra-nucleotide) were identified, with 1771 SSRs (89.99%) being mono-nucleotide type, particularly A and T repeat motifs. In the di-nucleotide repeat type, 176 SSRs were AT/TA repeat motifs. Only one TC and AG repeat motifs were found in H. ovalifolia, Dorcoceras hygrometricum, Litostigma coriaceifolium Y. G. Wei, F. Wen & Mich. Möller, Primulina lobulata (W.T. Wang) Mich. Möller & A. Weber and Primulina xiziae F. Wen, Yue Wang & G. J. Hua, respectively. In tri-nucleotide repeat type, only TTA, ATA and ATT repeat motifs were found in Paraboea filipes (Hance) Burtt, P. peltifolia D. Fang & L. Zeng, P. clavisepala D. Fang & D. H. Qin, P. dictyoneura (Hance) Burtt, P. dolomitica Z.Y. Li, X.G. Xiang & Z.Y. Guo and Primulina fimbrisepala (Hand.-Mazz.) Yin Z. Wang. Similarly, in the tetra-nucleotide repeat type, only one ATCT repeat motif was found in H. malipoensis Y. H. Tan, H. sinovietnamica, H. subacaulis Hand.-Mazz. Petrocodon jingxiensis (Yan Liu, H. S. Gao & W. B. Xu) A. Weber & Mich. Möller, and Primulina linearifolia (W. T. Wang) Yin Z. Wang. A total of 2055 tandem repeats were detected, ranging from 13 (Petrocodon longitubus Cong R. Li & Yang Luo) to 47 (Corallodiscus lanuginosus). There were 2802 dispersed repeats in the Trichosporeae plastomes, of which 1185 (42.29%) were forward repeats, 1502 (53.60%) were palindromic repeats, 86 (3.07%) were reverse repeats, and 29 (1.03%) were complement repeats.

Figure 4 Plot of each SSR repeat pattern numbers of 79 Trichosporeae plastomes with one IR excluded. The total number of SSRs in each plastome is shown in parentheses.
3.3 Codon usage bias
The codon usage bias of CDSs in Trichosporeae was shown in Figures S2, 3. There were 30 preferred codons (RSCU>1), 2 non-preferred codons (RSCU=1), and 32 less frequently used codons (RSCU<1). The preferred codons typically ended with A or U, except for UUG. Moreover, leucine (Leu, encoded by UUA, UUG, CUU, CUC, CUA and CUG) was the most frequently encoded amino acid, while cysteine (Cys, encoded by UGU and UGC) was the least frequently encoded amino acid. The two non-preferred codons (Met and Trp) only had a unique codon. Within Trichosporeae, the ENC values of CDSs ranged from 25.17 to 61. Remarkably, the ENC values of psbI in 78 species (except Corallodiscus lanuginosus), petN in 74 species (except Corallodiscus lanuginosus, Litostigma coriaceifolium, Paraboea filipes (Hance) Burtt, Petrocodon longitubus and Primulina renifolia (D.Fang & D.H.Qin) J.M.Li & Yin Z.Wang), and psbJ in 77 species (except Corallodiscus lanuginosus and Primulina huaijiensis Z. L. Ning & J. Wang) were less than 35, indicating significant codon bias for these genes.
3.4 Phylogenetic reconstruction
The topologies based on the whole plastome (excluding one IR) and 80 CDs were basically concordant, with both showing strong support for the monophylies of the 7 sampled subtribes and 17 genera (Figure 5). BI and ML analyses yielded nearly identical topologies, with some differences in the supporting values of certain nodes. The whole plastome had a higher overall resolution power compared to the 80 CDSs. The subtribe Corallodiscinae diverged first, followed by Litostigminae. Ramondinae and Leptobaeinae formed a sister group (PPBI=1.00, BSML=100%). Loxocarpinae and Didymocarpinae were strongly supported as a sister group (PPBI=1.00, BSML=100%) (Figure 5A). Within subtribe Didymocarpinae, Henckelia pumila (D. Don) A. Dietr. diverged first (PPBI=1.00, BSML=100%). Oreocharis formed a sister group of Hemiboea with high supporting values (PPBI=1.00, BSML=100%). However, the phylogenetic position of Lysionotus could not be determined.
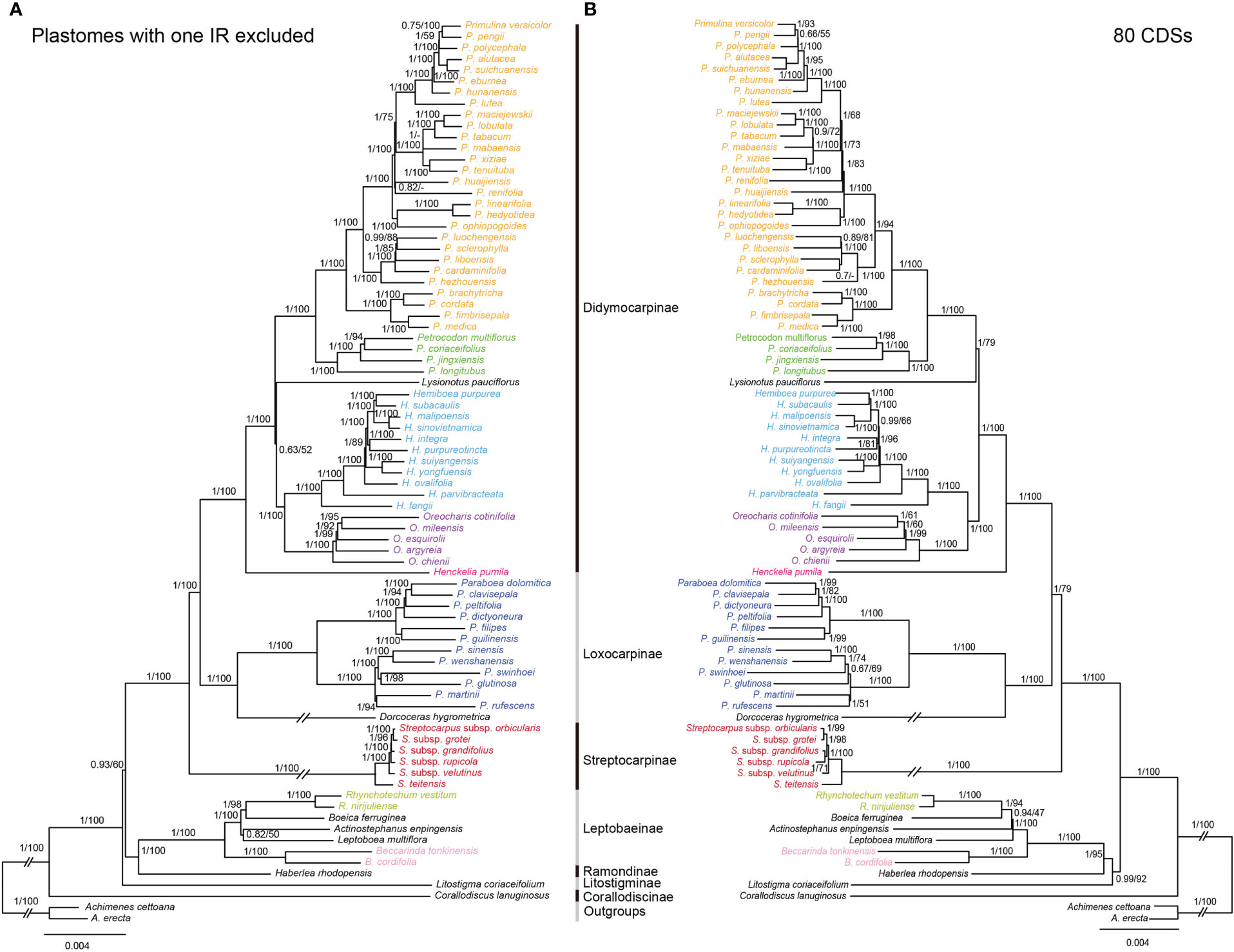
Figure 5 Comparisons of phylogenetic topologies for plastomes with one IR excluded (A) and 80 CDSs (B) based on BI and ML analyses in Trichosporeae species. Numbers above the branches are Bayesian posterior probabilities and ML bootstrap values, respectively. A dash (-) indicates that the supporting values are less than 50%.
3.5 Evolution of morphological characters
The ancestral reconstructions of the nine morphological characters were shown in Figure S4. The results indicated that the evolution patterns of morphological characters in Trichosporeae were complex. Some characters, such as habit, inflorescence and seeds, remain constant or nearly similar within tribe. However, some generic diagnostic morphological characters, including stem, leaf margin, numbers of stamens and staminodes, capsule, and its dehiscing type, have evolved several times independently within tribe. Additionally, the leaf margin has shifted several times among Primulina species.
4 Discussion
4.1 Plastome evolution within Trichosporeae
Plastomes in angiosperms are generally maternal inherited and exhibit a highly conserved structure among different plant lineages (Wicke et al., 2011). In this study, the whole plastome sequences of 11 Hemiboea species were obtained and found to be very similar in size (152,742–153, 695 bp), structure, gene order, gene content, and GC content (Table S2; Figures S1). The high conservatism of plastome has also been observed in other Gesneriaceae genera. For instance, Wang et al. (2022) found that the plastomes length of Paraboea ranged from 153,166–154,245 bp and exhibited similar structure, gene order, and gene content. Additionally, our results showed that the GC content of the IR region (43.1–43.2%) was higher than that of the LSC region (35.5–35.6%) and the SSC region (30.9–31.2%).
Within Trichosporeae, no contraction or expansion of IR borders was detected. However, there was relatively larger plastome variation in gene content (121–133), plastome length (152,196–156,614 bp), and GC content (37.2–37.8%) (Tables S2). Higher GC content is usually related to the stability of the DNA strands, which might be due to the presence of rRNA and tRNA in the IR region (Necşulea and Lobry, 2007; Ding et al., 2021). In this study, 30 preferred codons were found within Trichosporeae, all ending with A or U except for UUG (Figure S2). The codon usage patterns of plastomes were relatively conserved and highly similar over long evolutionary periods, possibly due to natural selection and mutation (Necşulea and Lobry, 2007; Sharp et al., 2010). Among the 20 amino acids, Cys was the least frequently encoded amino acid (Figure S2), possibly due to the gene being highly sensitive to physiological and environmental conditions (Marino and Gladyshev, 2012). Codon bias is crucial for understanding species evolution and predicting gene function and expression levels (Uddin et al., 2015).
SSRs are very often used as genetic molecular markers in phylogenetic studies of closely related species (e.g., Cheng et al., 2005; Angioi et al., 2009; Ebert and Peakall, 2009). In this study, a total of 1968 SSRs were identified in the plastomes of 79 Trichosporeae species, with 89.99% of them being mono-nucleotide repeats (Figure 4; Table S4). These results are consistent with previous studies of other angiosperms (Xiang et al., 2022; Yu et al., 2022). Plastome SSRs usually consist of short polyA or polyT repeats and mono-nucleotide repeats (Provan et al., 2001). The variation in the non-coding region was higher than that in the coding region, with the variation in the SC region being higher than that in the IR region, which is like other angiosperms plastomes (Xiang et al., 2022). Five hypervariable CDSs (rps15, matK, ndhF, rps16 and rpl32) and eight hypervariable non-coding regions (rpl16/rps3, rpl14/rpl16, petB/petD, trnC-GCA/petN, rps15/ycf1, trnfM-CAU/rps14, petD/rpoA and trnS-GGA/rps4) were identified in Trichosporeae (Figure 3). Among these, ndhF, matK and rps16 have been employed in previous phylogenetic studies (Roalson and Roberts, 2016; Li et al., 2022). Other hypervariable regions, such as rps15, rpl32, rpl16/rps3, rpl14/rpl16, petB/petD, trnC-GCA/petN, rps15/ycf1, trnfM-CAU/rps14, petD/rpoA and trnS-GGA/rps4, have great potential to be exploited as DNA barcodes markers, which might be used for further species identification. SNPs and indels play an important role in elucidating genome evolution (Matsuoka et al., 2002; Grover et al., 2008). In Trichosporeae, the majority of SNPs from coding genes were functionally missense variations, indicating a relatively complex evolutionary history.
4.2 Phylogenomics and morphological evolution within Trichosporeae
Previously, the phylogenetic relationships within Trichosporeae were unclear based on trnL-F and ITS (Möller et al., 2011). Although Roalson and Roberts (2016) and Li et al. (2022) obtained more molecular data, the relationships among subtribes and genera have not been resolved. In this study, the most extensive phylogenetic relationships of Trichosporeae were established (Figure 5). Our study strongly supported the monophyly of seven sampled subtribes and 17 genera (Figure 5). Also most relationships among subtribal level and genera were clarified with strong support based on the whole plastomes without one IR (Figure 5). The sister relationships between Loxocarpinae and Didymocarpinae were confirmed here (PPBI=1.00, BSML=100%), which was consistent with Li et al. (2022). Furthermore, Oreocharis was found to be a sister group of Hemiboea with high supporting values (PPBI=1.00, BSML=100%). However, the phylogenetic positions of Lysionotus remain controversial based on whole plastomes (PPBI=0.63, BSML=52%) and 80 CDSs (PPBI=1.00, BSML=79%). This controversial result was also found in previous studies. For example, the phylogenetic relationships among Lysionotus, Hemiboea, Oreocharis, Primulina and Petrocodon were unclear based on whole plastomes (Xu et al., 2022). Gu et al. (2020) showed that Lysionotus was a sister group of Oreocharis based on whole plastomes (PPBI=0.68, BSML=44%). However, Wen et al. (2022) showed that Lysionotus was close related to Primulina and Petrocodon based on 80 CDSs (PPBI=0.87, BSML=88%). These findings might contribute to future research on systematic analysis, genetic diversity and evolutionary patterns of the tribe Trichosporeae, and support the use of plastome to resolve the phylogenetic relationships of different taxonomic levels.
The Trichosporeae is characterized by morphological heterogeneity and is considered the most taxonomically difficult group (Weber et al., 2013). While traditional classifications based on morphological characters recognized genera in different alliances and geographical groups, recent phylogenomic data have almost clarified the relationships among subtribes. Our results showed that, except for conserved vegetative characters, Trichosporeae is greatly divergent with regard to its floral and capsule characters (Figure S4). For instance, the ancestors of this tribe had 2 stamens, which independently evolved into 4 stamens in Oreocharis and Leptobaeniae; the numbers of staminodes have evolved several times and even diverged in genus Primulina. Weber et al. (2013) considered that the advanced Asiatic and Malesian genera of this tribe were divided into a comparatively small group of genera with predominantly twisted fruits and a much larger group with straight fruits. However, our results inferred that the ancestor groups had a straight capsule, and the twisted capsule had independently evolved at least three times in different subtribes. Therefore, the complex morphological character evolution of Trichosporeae would be significant in defining larger groups and understanding their wide distribution.
5 Conclusion
In this study, we reported eleven new Hemiboea plastome sequences. Comparative analysis revealed that Trichosporeae plastomes were relatively conserved in genome structure, gene order, gene content, GC content and codon usage bias. Contraction and expansion of IR borders were not detected, and the 13 hypervariable regions were identified as potential molecular markers for species identification in Trichosporeae. We identified 1968 simple sequence repeats, 2055 tandem repeats and 2802 dispersed repeats. Our results highly confirmed the monophyly of seven sampled subtribes and 17 genera. We clarified the sister relationships between Loxocarpinae and Didymocarpinae, and supported the close relationship between Oreocharis and Hemiboea with highly supported values. Furthermore, the morphological character evolution was complex, and Trichosporeae was greatly divergent in its floral and capsule characters. Future research should employ more samples and molecular markers to investigate the phylogenetic relationships and comprehensively infer the complex evolutionary history of Trichosporeae based on extensive morphological characters.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OP820508-OP820512, OQ799915-OQ799920.
Author contributions
X-GX and Z-YL conceived and designed the study. X-GX, QZ, BP, Y-SH, and Z-YG did fieldwork. Y-FC, PZ, HY, and L-GZ conducted experiments and data analysis. Y-FC, X-GX, K-LX, and PZ wrote the draft. All authors revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Natural Science Foundation of China (32060056, 31670212, 31370227 and 31300181), and the Thousand Talents Program of Jiangxi Province (jxsq2018106032).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1160535/full#supplementary-material
References
Amiryousefi, A., Hyvönen, J., Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Angioi, S. A., Desiderio, F., Rau, D., Bitocchi, E., Attene, G., Papa, R. (2009). Development and use of chloroplast microsatellites in Phaseolus spp. and other legumes. Plant Biol. (Stuttg.) 11, 598–612. doi: 10.1111/j.1438-8677.2008.00143.x
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brown, J., Pirrung, M., McCue, L. A. (2017). FQC dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139. doi: 10.1093/bioinformatics/btx373
Cauz-Santos, L. A., da Costa, Z. P., Callot, C., Cauet, S., Zucchi, M. I., Bergès, H., et al. (2020). A repertory of rearrangements and the loss of an inverted repeat region in Passiflora chloroplast genomes. Genome Biol. Evol. 12, 1841–1857. doi: 10.1093/gbe/evaa155
Chen, W. H., Wen, F., Ren, M. X., Yang, L., Hong, X., Qiu, Z. J., et al. (2020). Gesneriaceae in China and Vietnam: perfection of taxonomy based on comprehensive morphological and molecular evidence. PhytoKeys 157, 1–5. doi: 10.3897/phytokeys.157.56842
Cheng, Y. J., de Vicente, M. C., Meng, H., Guo, W. W., Tao, N., Deng, X. X. (2005). A set of primers for analyzing chloroplast DNA diversity in Citrus and related genera. Tree Physiol. 25, 661–672. doi: 10.1093/treephys/25.6.661
Chi, X. F., Wang, J. L., Gao, Q. B., Zhang, F. Q., Chen, S. L. (2018). The complete chloroplast genomes of two Lancea species with comparative analysis. Molecules 23, 602. doi: 10.3390/molecules23030602
Cingolani, P. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Flying 6, 80–92. doi: 10.4161/fly.19695
Darling, A. E., Mau, B., Perna, N. T. (2010). ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PloS One 5, e11147. doi: 10.1371/journal.pone.0011147
Darriba, D., Taboada, G. L., Doallo, R., Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. doi: 10.1038/nmeth.2109
Ding, S. X., Dong, X., Yang, J. X., Guo, C. C., Cao, B. B., Guo, Y., et al. (2021). Complete chloroplast genome of Clethra fargesii franch., an original sympetalous plant from central China: comparative analysis, adaptive evolution, and phylogenetic relationships. Forests 12, 441. doi: 10.3390/f12040441
Dobrogojski, J., Adamiec, M., Luciński, R. (2020). The chloroplast genome: a review. Acta Physiol. Plant 42, 98. doi: 10.1007/s11738-020-03089-x
Doyle, J. J., Doyle, J. L. (1987). A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Ebert, D., Peakall, R. (2009). A new set of universal de novo sequencing primers for extensive coverage of noncoding chloroplast DNA: new opportunities for phylogenetic studies and cpSSR discovery. Mol. Ecol. Resour. 9, 777–783. doi: 10.1111/j.1755-0998.2008.02320.x
Grover, C. E., Yu, Y., Wing, R. A., Paterson, A. H., Wendel, J. F. (2008). A phylogenetic analysis of indel dynamics in the cotton genus. Mol. Biol. Evol. 25, 1415–1428. doi: 10.1093/molbev/msn085
Gu, L., Su, T., An, M. T., Hu, G. X. (2020). The complete chloroplast genome of the vulnerable Oreocharis esquirolii (Gesneriaceae): structural features, comparative and phylogenetic analysis. Plants 9, 1692. doi: 10.3390/plants9121692
Hsieh, C. L., Xu, W. B., Chung, K. F. (2022). Plastomes of limestone karst gesneriad genera Petrocodon and Primulina, and the comparative plastid phylogenomics of gesneriaceae. Sci. Rep. 12, 15800. doi: 10.1038/s41598-022-19812-2
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Kassambara, A. (2023). ggpubr: “ggplot2” Based publication ready plots. R package version 0.6.0. Available at: https://rpkgs.datanovia.com/ggpubr/.
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lee, S. R., Kim, K., Lee, B. Y., Lim, C. E. (2019). Complete chloroplast genomes of all six Hosta species occurring in Korea: molecular structures, comparative, and phylogenetic analyses. BMC Genom. 20, 833. doi: 10.1186/s12864-019-6215-y
Li, Z. Y., Wang, Y. Z. (2005). Plants of gesneriaceae in China (Zhengzhou: Henan Science and Technology Publishing House).
Li, X. Q., Xiang, X. G., Zhang, Q., Jabbour, F., Ortiz, R.d. C., Erst, A. S., et al. (2022). Immigration dynamics of tropical and subtropical southeast Asian limestone karst floras. Proc. R. Soc B 289, 20211308. doi: 10.1098/rspb.2021.1308
Lu, R. S., Li, P., Qiu, Y. X. (2016). The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: comparative genomic and phylogenetic analyses. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.02054
Marino, S. M., Gladyshev, V. N. (2012). Analysis and functional prediction of reactive cysteine residues. J. Biol. Chem. 287, 4419–4425. doi: 10.1074/jbc.R111.275578
Matsuoka, Y., Yamazaki, Y., Ogihara, Y., Tsunewaki, K. (2002). Whole chloroplast genome comparison of rice, maize, and wheat: implications for chloroplast gene diversification and phylogeny of cereals. Mol. Biol. Evol. 19, 2084–2091. doi: 10.1093/oxfordjournals.molbev.a004033
Möller, M., Forrest, A., Wei, Y. G., Weber, A. (2011). A molecular phylogenetic assessment of the advanced Asiatic and malesian didymocarpoid gesneriaceae with focus on non-monophyletic and monotypic genera. Plant Syst. Evol. 292, 223–248. doi: 10.1007/s00606-010-0413-z
Möller, M., Wei, Y. G., Wen, F., Clark, J. L., Weber, A. (2016). You win some you lose some: updated generic delineations and classification of gesneriaceae implications for the family in China. Guihaia 36, 44–60. doi: 10.11931/guihaia.gxzw201512015
Mower, J. P., Vickrey, T. L. (2018). Structural diversity among plastid genomes of land plants. Adv. Bot. Res. 85, 263–292. doi: 10.1016/bs.abr.2017.11.013
Necşulea, A., Lobry, J. R. (2007). A new method for assessing the effect of replication on DNA base composition asymmetry. Mol. Biol. Evol. 24, 2169–2179. doi: 10.1093/molbev/msm148
Provan, J., Powell, W., Hollingsworth, P. M. (2001). Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol. Evol. 16, 142–147. doi: 10.1016/s0169-5347(00)02097-8
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Revell, L. J. (2012). Phytools: an r package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Roalson, E. H., Roberts, W. R. (2016). Distinct processes drive diversification in different clades of gesneriaceae. Syst. Biol. 65, 662–684. doi: 10.1093/sysbio/syw012
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sharp, P. M., Emery, L. R., Zeng, K. (2010). Forces that influence the evolution of codon bias. Philos. Trans. R. Soc Lond. B Biol. Sci. 365, 1203–1212. doi: 10.1098/rstb.2009.0305
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tao, J., Yao, H. P. (2020). Comprehensive analysis of the codon usage patterns of polyprotein of zika virus. Prog. Biophys. Mol. Biol. 150, 43–49. doi: 10.1016/j.pbiomolbio.2019.05.001
Uddin, A., Mazumder, T. H., Choudhury, M. N., Chakraborty, S. (2015). Codon bias and gene expression of mitochondrial ND2 gene in chordates. Bioinformatics 11, 407–412. doi: 10.6026/97320630011407
Wang, Y. Z., Liang, R. H., Wang, B. H., Li, J. M., Qiu, Z. J., Li, Z. Y., et al. (2010). Origin and phylogenetic relationships of the old world gesneriaceae with actinomorphic flowers inferred from ITS and trnL-trnF sequences. Taxon 59, 1044–1052. doi: 10.1002/tax.594005
Wang, W. T., Pan, K. Y., Li, Z. Y., Weitzman, A. L., Skog, L. E. (1998). “Gesneriaceae,” in Flora of China. Eds. Wu, Z. Y., Raven, P. H. (Beijing and St. Louis: Science Press and Missouri Botanical Garden Press).
Wang, Y. F., Wen, F., Hong, X., Li, Z. L., Mi, Y. L., Zhao, B. (2022). Comparative chloroplast genome analyses of Paraboea (Gesneriaceae): insights into adaptive evolution and phylogenetic analysis. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1019831
Weber, A., Clark, J. L., Möller, M. (2013). A new formal classification of gesneriaceae. Selbyana 31, 68–94.
Wei, N., Pérez-Escobar, O. A., Musili, P. M., Huang, W., Yang, J., Hu, A. Q., et al. (2021). Plastome evolution in the hyperdiverse genus Euphorbia (Euphorbiaceae) using phylogenomic and comparative analyses: large-scale expansion and contraction of the inverted repeat region. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.712064
Wen, F., Xin, Z. B., Hong, X., Cai, L., Chen, X. Y., Liang, J. J., et al. (2022). Actinostephanus (Gesneriaceae), a new genus and species from guangdong, south China. PhytoKeys 193, 89–106. doi: 10.3897/phytokeys.193.80715
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wicke, S., Schneeweiss, G. M., de Pamphilis, C. W., Muller, K. F., Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Xiang, K. L., Mao, W., Peng, H. W., Erst, A. S., Yang, Y. X., He, W. C., et al. (2022). Organization, phylogenetic marker exploitation, and gene evolution in the plastome of Thalictrum (Ranunculaceae). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.897843
Xu, X. L., Shi, M. M., Shi, L. C., Zhang, W. J., Liu, J. X., Duan, X. L. (2022). The first complete chloroplast genome of Briggsia chienii w. y. chun and its phylogenetic position within gesneriaceae. Mitochondrial DNA B Resour. 7, 1120–1122. doi: 10.1080/23802359.2022.2087551
Yu, J. J., Fu, J., Fang, Y. P., Xiang, J., Dong, H. J. (2022). Complete chloroplast genomes of Rubus species (Rosaceae) and comparative analysis within the genus. BMC Genom. 23, 32. doi: 10.1186/s12864-021-08225-6
Keywords: Gesneriaceae, Hemiboea, molecular markers, phylogenomics, plastome evolution, Trichosporeae
Citation: Cui Y-F, Zhou P, Xiang K-L, Zhang Q, Yan H, Zhang L-G, Pan B, Huang Y-S, Guo Z-Y, Li Z-Y and Xiang X-G (2023) Plastome evolution and phylogenomics of Trichosporeae (Gesneriaceae) with its morphological characters appraisal. Front. Plant Sci. 14:1160535. doi: 10.3389/fpls.2023.1160535
Received: 07 February 2023; Accepted: 21 April 2023;
Published: 09 May 2023.
Edited by:
Gang Yao, South China Agricultural University, ChinaReviewed by:
Xiao-Jian Qu, Shandong Normal University, ChinaCornelius Mulili Kyalo, University of Chinese Academy of Sciences, China
Fei Zhao, Chinese Academy of Sciences (CAS), China
Copyright © 2023 Cui, Zhou, Xiang, Zhang, Yan, Zhang, Pan, Huang, Guo, Li and Xiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao-Guo Xiang, eGlhbmd4ZzIwMTBAMTYzLmNvbQ==
†These authors have contributed equally to this work