 Min Tang
Min Tang Jialin Huang2†
Jialin Huang2† Peiwen Guo
Peiwen Guo Lei Wang
Lei Wang- 1College of Landscape and Horticulture, Yunnan Agricultural University, Kunming, China
- 2School of Chemical Biology and Environment, Yuxi Normal University, Yuxi, China
- 3College of Animal Science and Technology, Yunnan Agricultural University, Kunming, China
- 4School of Mathematical Sciences, Xiamen University, Xiamen, China
- 5Scientific Research Department, Kunming Novo Medical Laboratory Co., Ltd., Kunming, China
This study presents a chromosome-level, near-complete genome assembly of Thalia dealbata (Marantaceae), a typical emergent wetland plant with high ornamental and environmental value. Based on 36.99 Gb PacBio HiFi reads and 39.44 Gb Hi-C reads, we obtained a 255.05 Mb assembly, of which 251.92 Mb (98.77%) were anchored into eight pseudo-chromosomes. Five pseudo-chromosomes were completely assembled, and the other three had one to two gaps. The final assembly had a high contig N50 value (29.80 Mb) and benchmarking universal single-copy orthologs (BUSCO) recovery score (97.52%). The T. dealbata genome had 100.35 Mb repeat sequences, 24,780 protein-coding genes, and 13,679 non-coding RNAs. Phylogenetic analysis revealed that T. dealbata was closest to Zingiber officinale, whose divergence time was approximately 55.41 million years ago. In addition, 48 and 52 significantly expanded and contracted gene families were identified within the T. dealbata genome. Moreover, 309 gene families were specific to T. dealbata, and 1,017 genes were positively selected. The T. dealbata genome reported in this study provides a valuable genomic resource for further research on wetland plant adaptation and the genome evolution dynamics. This genome is also beneficial for the comparative genomics of Zingiberales species and flowering plants.
1 Introduction
Wetlands, also known as the “kidneys of the earth”, are of great ecological importance because they have played important roles in biodiversity conservation, carbon management, flood reduction, and water purification (Zedler and Kercher, 2005). Although wetlands cover less than 9% of the land area, they are vital habitats to many aquatic plants and animals (Gray et al., 2013). As key components of wetland ecosystems, wetland plants function as primary producers, habitats for other taxonomic groups, and nutrient removers (Cronk and Fennessy, 2016). Almost all wetland plants are angiosperms, with a few ferns and gymnosperms. These plants are categorized into emergent, submergent, floating-leaved, and floating plants based on their growth types and morphologies (Cronk and Fennessy, 2016). Although wetland plants have developed adaptation strategies to survive periodic soil saturation and the accompanying changes in soil chemistry (Pezeshki, 2001), the underlying genetic mechanisms in survival strategies are rarely studied. With the rapid development of sequencing technologies, the characterization of more wetland plant genomes can provide deeper insights into the adaptive evolution and morphological characteristics of wetland plants.
Thalia dealbata Fraser (Marantaceae), commonly known as powdery alligator flag, is a typical emergent wetland plant native to swamps and ponds in the Southern United States of America and Mexico. It has high ornamental value, given its long-stalked canna-like foliage and violet-blue flowers. This plant is usually covered with a white and water-repellent powdery coating, which enhances its performance. T. dealbata is also widely used in man-made wetlands to improve water quality by breaking down or removing excess pollutants from eutrophic water (Wang et al., 2020). A recent study has presented the complete chloroplast genome of T. dealbata (Deng et al., 2021). However, the T. dealbata nuclear genome has not been sequenced or reported.
Therefore, this study aimed to reconstruct a reference genome sequence of T. dealbata for further genomic and genetic studies. We performed a chromosome-level assembly of the T. dealbata, the first sequenced genome in the Marantaceae family, by integrating PacBio high-fidelity (HiFi) sequencing and chromosome conformation capture (Hi-C) technology. Subsequently, we performed a comparative genomics analysis of T. dealbata and other publicly available Zingiberales species, including Musa acuminata Colla (D’hont et al., 2012), M. balbisiana Colla (Wang et al., 2019), Zingiber officinale Roscoe (Li et al., 2021), and Ensete glaucum (Roxb.) Cheesman (Wang Z. et al., 2022). The reference-level genome assembly in this study will accelerate evolutionary and morphological studies of wetland plants and further phylogenomic studies of Marantaceae and Zingiberales.
2 Materials and methods
2.1 Sample collection and sequencing
Young and healthy T. dealbata leaves were collected from a mature T. dealbata plant growing on the lakeside of Dongan Lake in Chengdu, Sichuan Province, Southwest China. The leaves were immediately frozen in liquid nitrogen and stored at -80°C, awaiting further analysis.
High-quality total genomic DNA was extracted from the T. dealbata leaves using the CTAB method (Doyle and Doyle, 1987). For genome survey analysis, a paired-end library with an insert size of approximately 400 bp was constructed and sequenced on a NovaSeq 6000 platform. For the de novo assembly of the genome, SMRTbell libraries were prepared using the PacBio 15-kb protocol (Pacific Biosciences, CA, USA) and sequenced using the circular consensus sequencing (CCS) mode on the PacBio Sequel II sequencer. Finally, a Hi-C library was constructed and sequenced for the Hi-C scaffolding analysis on a NovaSeq 6000 platform.
In addition, the T. dealbata total RNA was extracted from the fresh stem, leaf, and flower tissues. Next, the RNA sequencing (RNA-seq) libraries were constructed and sequenced on an Illumina NovaSeq 6000 platform. The obtained raw RNA-seq reads were filtered using Trimmomatic (version 0.36) with default parameters (Bolger et al., 2014) for downstream genome annotation and quality assessment.
2.2 Genome survey
A k-mer (k = 19) analysis of Illumina short reads was performed using the Jellyfish (version 2.2.9, parameters, -k 19, -C) (Marçais and Kingsford, 2011). The low-frequency k-mers (frequency < 4) were removed, and the genome size was calculated by dividing the total k-mer number by the homozygous peak depth in the k-mer distribution curve. In addition, a polyploidy peak around the homozygous peak was examined to determine the ploidy level of the T. dealbata genome.
2.3 Genome assembly and quality assessment
HiFi long reads were pre-processed by CCS (version 4.2.0, parameters, min-passes = 3, min-length = 10, and min-rq = 0.99; http://ccs.how). Next, the filtered HiFi reads were assembled into contigs using hifiasm (version 0.14, default parameters) (Cheng et al., 2021) and mapped using Minimap (version 2.24, default parameters) (Li, 2018). Subsequently, the low-quality contigs with read depth < 10 or GC content > 50% were removed based on the GC-depth distribution. For Hi-C scaffolding analysis, Hi-C reads were mapped using Juicebox (version 1.8.8) with default parameters (Durand et al., 2016). Uniquely mapped Hi-C reads were then used to anchor contigs into chromosomes using 3D-DNA software (Dudchenko et al., 2017). Finally, scaffolding errors were checked and corrected according to the Hi-C contact heat maps generated with Juicebox.
The final assembly quality was evaluated by re-mapping the Illumina reads against the assembly using BWA (version 0.7.17) with default parameters (Li and Durbin, 2009). Subsequently, the benchmarking universal single-copy orthologs (BUSCO) completeness score was calculated by mapping 1,614 conserved genes from Embryophyta odb10 against the assembly using BUSCO (version 3.0.2) with default parameters (Simão et al., 2015). We searched the plant telomeres that are listed in the telomerase database (Podlevsky et al., 2007) against the final assembly using an in-house perl script. In addition, we identified centromeres within the T. dealbata genome using quarTeT (http://www.atcgn.com:8080/quarTeT/home.html) with the similar procedures which were described in Yue et al. (2023).
2.4 Identification of repeats
The repetitive elements in the T. dealbata genome were annotated using RepeatMasker (version v4.07) (Tarailo-Graovac and Chen, 2009) and RepeatModeler (version v1.0.11) (Price et al., 2005). First, a repeat library was de novo predicted based on the final assembly using RepeatModeler with default parameters. Next, a known repeat library of green plants was extracted using the “queryRepeatDatabase.pl” script from RepeatModeler. Finally, the two repeat libraries were combined into one comprehensive library, followed by a genome-wide homology-based identification of repeats using RepeatMasker. In addtion, we annotated intact long terminal repeat (LTR) retrotransposons (LTR-RTs) by integrating the predictions from LTR_Finder (version 1.06) (Xu and Wang, 2007) and LTRharvest (version 1.5.10) (Ellinghaus et al., 2008) using LTR_retriever (Ou and Jiang, 2018) with default parameters.
2.5 Annotation of protein-coding genes
Protein-coding genes were annotated as outlined by Wang et al. (2021). First, E. glaucum, M. acuminata, M. balbisiana, Z. officinale, and Oryza sativa L. protein sequences (Jain et al., 2019) were aligned with the T. dealbata genome using TBLASTN (version 2.2.31+, parameters, E-value < 1e-5) (Camacho et al., 2009), and a homology-based prediction was performed using GeneWise (version 2.4.1) with default parameters (Birney et al., 2004). Second, the de novo and genome-guided RNA-seq assemblies were combined for transcriptome-based prediction using the program to assemble spliced alignment (PASA; version 2.3.3) with default parameters (Haas et al., 2003). Third, a de novo prediction of gene models was performed using AUGUSTUS (version 3.2.3) (Stanke et al., 2006) with high-confidence gene model-trained parameters (exon number > 2 and CDS length > 1200 bp) selected from the PASA results. Finally, all the predicted gene models were integrated into a final gene set using EvidenceModeler (version 1.1.1) with default parameters (Haas et al., 2008). The final protein-coding gene set was functionally annotated using the publicly available protein databases, including Swiss-Prot, TrEMBL (Bairoch and Apweiler, 2000), InterPro (Hunter et al., 2009), and KEGG (Moriya et al., 2007), as described by Wang M. et al., (2022). Gene ontology (GO) terms were then assigned based on InterPro entries.
2.6 Annotation of non-coding RNAs
Non-coding RNAs (ncRNAs), including ribosomal RNAs (rRNAs), transfer RNAs (tRNAs), microRNAs (miRNAs), and small nuclear RNAs (snRNAs) were annotated using the de novo and homology-based methods. The rRNAs were predicted by aligning the assembly against the Arabidopsis thaliana rRNA sequences using BLASTN (version 2.2.31+, parameters, E-value < 1e-5). The tRNAs were predicted using tRNAscan-SE (version 1.4) (Lowe and Eddy, 1997), while snRNAs and miRNAs were predicted using Infernal (version 1.1.3) with default parameters (Nawrocki and Eddy, 2013).
2.7 Phylogenetic analysis and divergence time estimation
A phylogenetic analysis was performed based on the protein sequences of T. dealbata and four Zingiberales species, including E. glaucum, M. acuminata, M. balbisiana, and Z. officinale (haplotype A), with O. sativa as the outgroup species. The gene family clustering was performed using OrthoMCL (version 2.0.9) with default parameters (Li et al., 2003). Single copy genes (SCGs) in the six species were identified based on the clustering results. In addition, the SCGs protein sequences were aligned using MAFFT (version 7.313, parameters, LINSI) (Katoh et al., 2002). Finally, a maximum likelihood phylogenetic tree was reconstructed from the alignments of concatenated SCGs using RAxML (version 8.0.17, parameters, PROTGAMMAILGX, n = 500) (Stamatakis, 2014). The divergence time of T. dealbata was estimated using MCMCTREE in the PAML (version 4.9e, parameters, independent rates, F84 model) (Yang, 2007) based on the divergence between O. sativa and E. glaucum (103.2–117.1 million years ago, MYA) from the TimeTree database (Kumar et al., 2017) as the calibration point. Subsequently, the gene family expansions and contractions per species were detected using CAFE (version 3.1) with default parameters (De Bie et al., 2006).
2.8 Whole genome duplication (WGD) analysis
Recent WGD events within the T. dealbata genome were analyzed by comparing T. dealbata and M. acuminata protein sequences using MCScanX (version 1.1) with default parameters (Wang et al., 2012). Next, the synonymous substitution rate (Ks) was calculated per collinear gene pair within and between the two species using “add_ka_and_ks_to_collinearity.pl”. Synteny blocks shared between T. dealbata and M. acuminata and in T. dealbata were visualized using TBtools (version 1.120) (Chen et al., 2020). Different gene duplication types were detected using DupGen_finder with default parameters (Qiao et al., 2019).
2.9 Selection analysis
Three species, including T. dealbata, M. acuminata, and O. sativa were selected for selection analysis. The coding sequences of SCGs among the three species were aligned using MAFFT and trimmed by Gblocks (version 0.91b) with default parameters (Castresana, 2000). Finally, selection analysis was performed based on the branch site model using Codeml in PAML (version 4.9e). The LRT p-value was calculated using Chi2 in PAML.
3 Results
3.1 Genome sequencing and assembly
A total of 21.30 Gb Illumina reads were generated for genome survey analysis (Table S1). The T. dealbata genome was estimated to be 256.15 Mb in size, with no evidence of polyploidy (Figure S1). In addition, 36.99 Gb (144.40× genome coverage) HiFi reads were generated for de novo genome assembly, which yielded 145 contigs with a total length of 260.54 Mb, which is very close to the estimated genome size. After removing the low-quality contigs with low read depth or high GC content (Figure S2), a Hi-C scaffolding analysis on 39.44 Gb Hi-C reads (153.97× genome coverage), yielded 251.92 Mb sequences that were anchored to eight pseudo-chromosomes (Figure S3).
The final genome assembly (Figure 1) was 255.05 Mb in length, with contig and super-scaffold N50 of 29.80 and 30.83 Mb, respectively (Table 1, Table S2). Five of the eight pseudo-chromosomes were completely assembled without any gap; two had one gap, while one pseudo-chromosome had two gaps (Table S3). Approximately 99.78% of the Illumina reads were mapped back to the T. dealbata genome, with a 10-fold minimum genome coverage of 99.87% (Figure S4). The genome assembly had an overall BUSCO score of 97.52% (Table S4). Approximately 96.34% of RNA-seq reads could be successfully aligned to the genome (Table S5). In addition, we found that all pseudo-chromosomes of the T. dealbata genome contained (TTAGGG)n telomeres at both ends (Table S6) and centromeres in the central region (Table S7). Overall, these data implied the T. dealbata genome was of high quality and completeness.
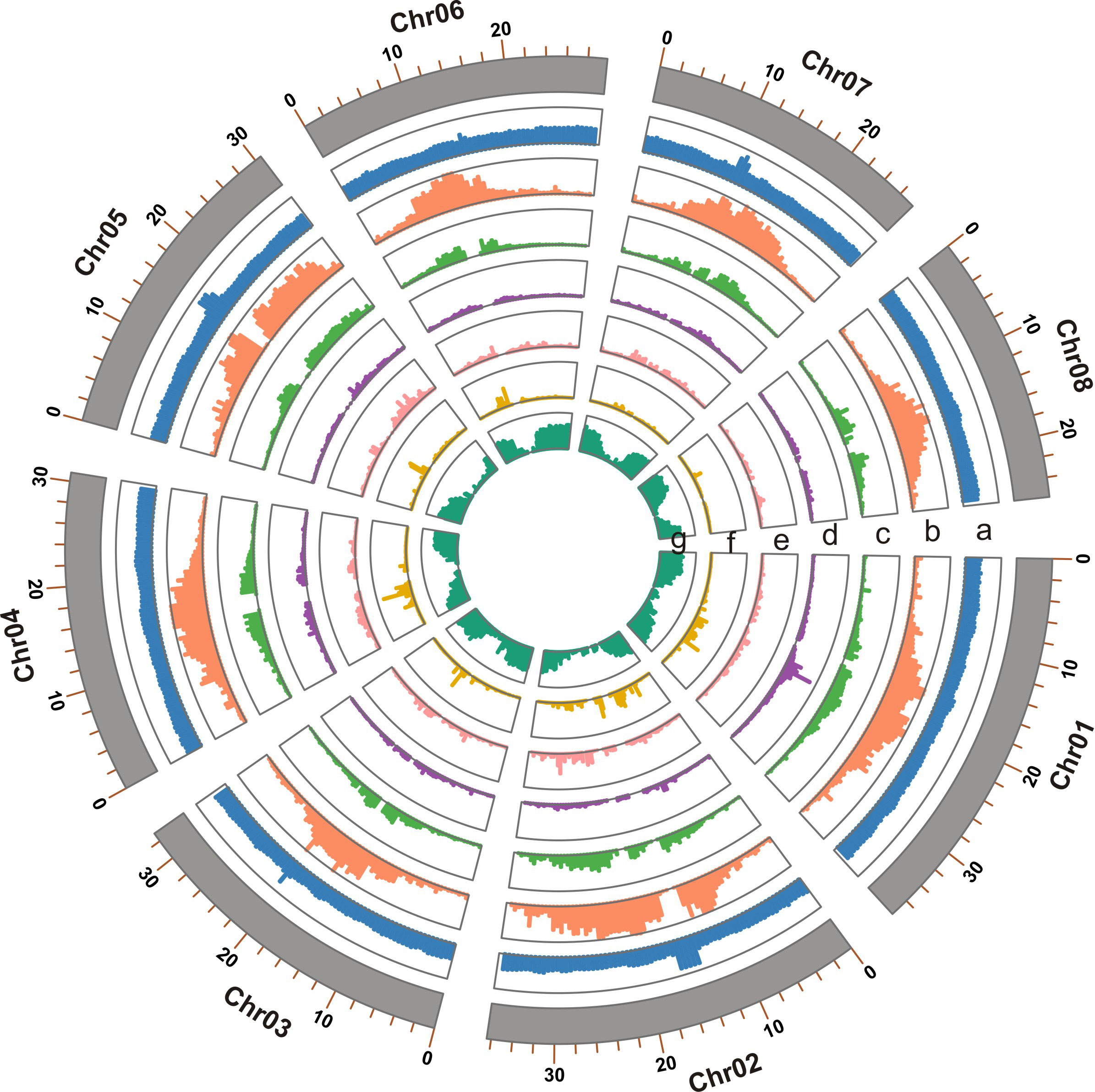
Figure 1 Genome features of the T. dealbata genome indicated in tracks from a to e: (A) GC content; (B) repeat content; (C) LTR density; (D) density of DNA transposons; (E) density of long interspersed nuclear elements; (F) density of short interspersed nuclear elements; and (G) gene density. All features are presented in non-overlapping windows of 500 kb.
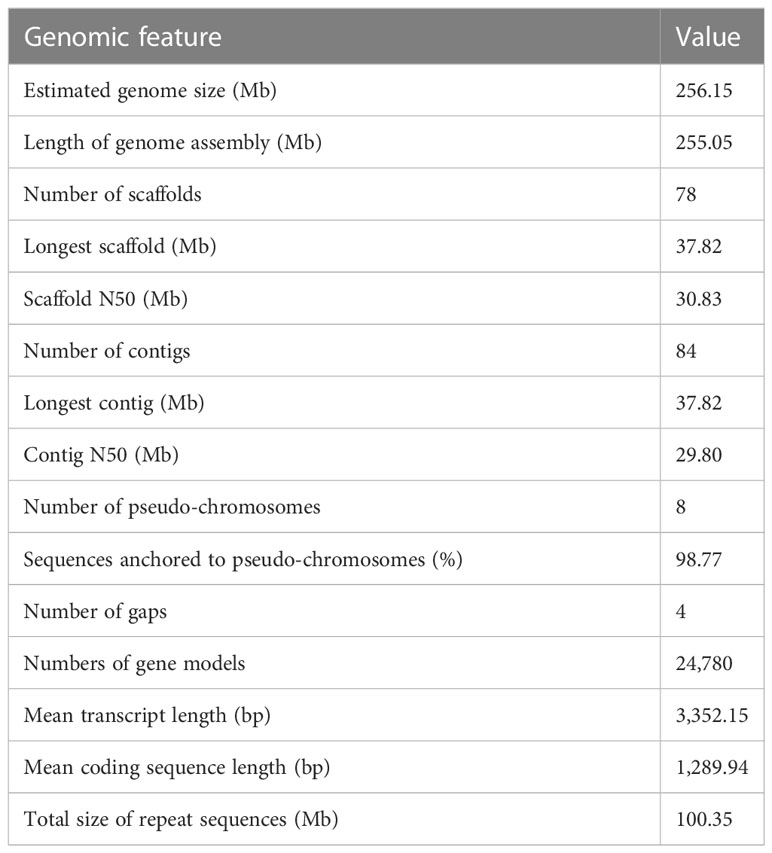
Table 1 Summary statistics for the T. dealbata assembly.
3.2 Genome features
A total of 100.35 Mb of repeat sequences representing 39.34% of the T. dealbata genome were predicted from the high-quality assembly (Table S8). The repeat content of T. dealbata was lower than that of other sequenced Zingiberales species (Figure S5). The LTR-RTs were the most abundant repeat class, with 31.54 Mb (12.36%) of the genome and a Gypsy to Copia ratio of 4.4:1. Within the repeat-masked genome, 24,780 high confidence protein-coding genes covering 92.50% of the complete BUSCO genes were predicted (Table S4).
In addition, 24,020 (96.93%) gene models were assigned to known functions using at least one of the protein databases, with 15,611 (63.00%) assigned to GO terms (Table S9). At the same time, 13,679 ncRNAs with a total length of 2.09 Mb, including 5,358 rRNAs, 7,647 tRNAs, 152 miRNAs, and 522 snRNAs, were identified (Table S10).
3.3 Genome evolution
A total of 21,717 T. dealbata genes were classified into 13,613 families, 2,289 (16.81%) of which were located in the single-copy orthogroups across the six plant species (Table S10). A phylogenetic tree reconstructed based on the SCGs revealed that T. dealbata had the closest genetic relationship with Z. officinale (Figure 2). The divergence between T. dealbata and Z. officinale was estimated to be around 55.41 MYA. In addition, 48 and 52 gene families were significantly (P < 0.01) expanded and contracted in the T. dealbata genome, respectively, and 309 gene families containing 1,161 genes were specific to T. dealbata (Table S11). The 439 genes within the significantly expanded gene families were highly enriched in GO terms related to “glutathione metabolic process”, “response to wounding”, “photosynthesis, light reaction”, and “defense response” (Figure S6), which possibly contributes to T. dealbata adaption to wetland environments. In addition, the T. dealbata specific genes were functionally enriched in “intracellular transport”, “response to hormone”, “cell wall modification”, and “glycerolipid biosynthetic process” (Figure S7).
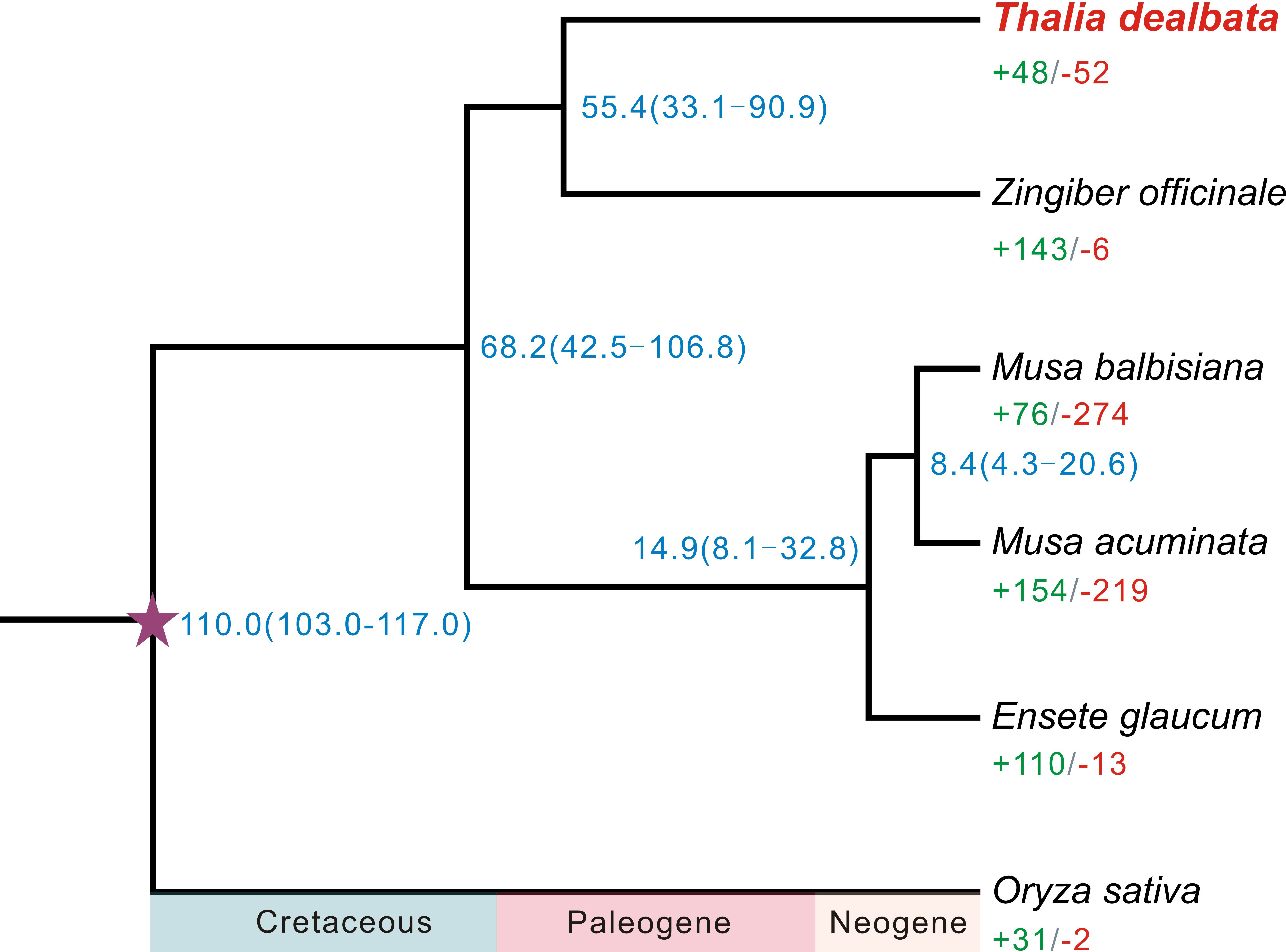
Figure 2 Phylogenetic tree of six plant species and evolution of gene families. The fully supported species tree was reconstructed based on 2,289 SCGs across the six species. Blue number beside each node shows the estimated divergence time of each node with 95% confidence intervals. The purple pentagram represent the calibration point. Green and red numbers indicate the numbers of significantly (P < 0.01) expanded and contracted gene families per species.
Furthermore, the WGD analysis revealed no recent WGD events in the T. dealbata genome (Figure 3A), although most (14,734; 59.46%) of the T. dealbata genes were classified as the WGD-derived genes (Table S12). However, the distribution of synonymous substitution rate (Ks) showed that T. dealbata and M. acuminata shared an WGD event in their common ancestor. This ancient WGD event was also supported by the 2:2 relationship of the synteny blocks between T. dealbata and M. acuminata (Figure S8) and the 1:1 relationship of the synteny blocks within T. dealbata (Figure S9).
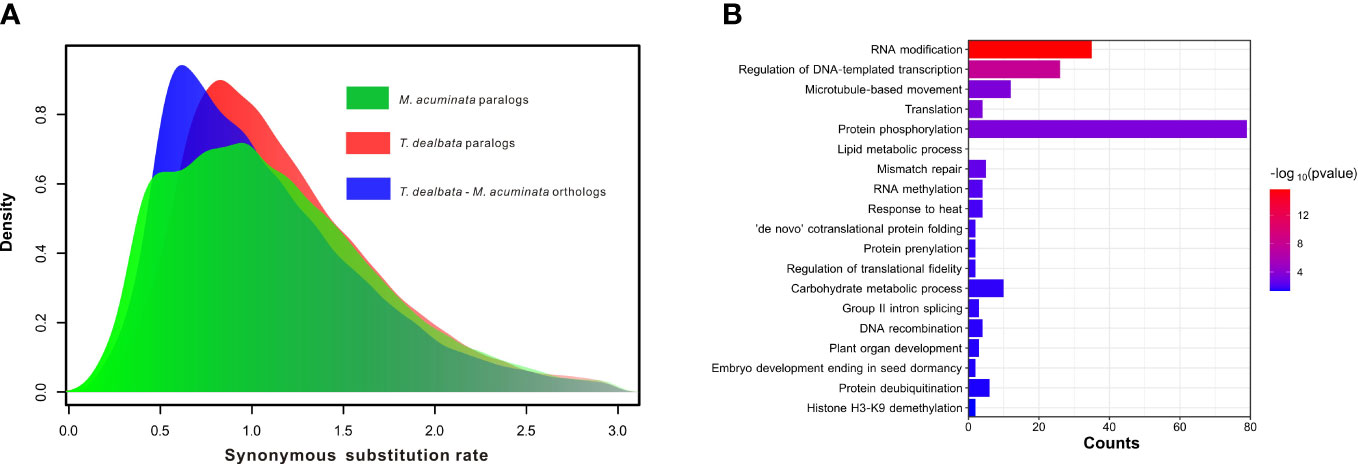
Figure 3 WGD event and positive selection in T. dealbata. (A) Results of recent WGD analysis based on the paralog and ortholog gene pairs within and between T. dealbata and M. acuminata. (B) GO enrichment of the 1,017 positively selected in T. dealbata..
We detected a total of 764 intact LTR-RTs in the T. dealbata genome, all of which were inserted after the split of T. dealbata from M. acuminata (Figure S10). The T. dealbata genome contained significantly more intact LTR-RTs than the M. acuminata (764 vs 278), although T. dealbata had a smaller genome size than M. acuminata (255 Mb vs 473 Mb).
Finally, 3,863 SCGs were identified among T. dealbata, M. acuminata, and O. sativa, of which 1,017 genes were positively selected in T. dealbata. These positively selected genes were mainly related to “RNA modification”, “translation”, “lipid metabolic process”, “RNA methylation”, and “plant organ development” (Figure 3B).
4 Discussion and conclusion
In this study, we performed deep sequencing and chromosome-level genome assembly of T. dealbata, an emergent wetland plant belonging to Marantaceae from the order Zingiberales. Based on high coverage PacBio and Hi-C reads, we assembled a near-complete genome assembly of T. dealbata, the first reported assembly in Marantaceae. This T. dealbata assembly has considerably high completeness and continuity, with most of the pseudo-chromosomes were completely assembled. The high quality of this genome indicated the advantages of PacBio HiFi sequencing in constructing highly continuous genome assemblies with long and accurate reads (Nurk et al., 2020; Wang M. et al., 2022). However, there are still one to two gaps in three pseudo-chromosomes, which might be caused by the species-specific complex repetitive regions in T. dealbata genome. Further integration of ultra-long reads and other high-throughput sequencing data will make it possible to generate a telomere-to-telomere (T2T) genome for T. dealbata.
Using Hi-C scaffolding analysis, we anchored the genome sequences of T. dealbata into eight pseudo-chromosomes, showing the different chromosome number from a previous study (2n = 2x = 12; Suessenguth, 1921). The 3D-DNA software does not require a priori chromosome number as input, and the Hi-C contact map shows a clear pattern of eight chromosome interaction (Figure S3). Thus, we speculated that there was some mistake in the relatively old research of Suessenguth (1921). Future karyotype analysis of T. dealbata can verify the validity of our speculation.
The high-quality T. dealbata genome has lead to accurate structural annotation of pretein-coding genes and ncRNAs, enabling us to gain further insights into the evolutionary history of T. dealbata and related species. We found that T. dealbata had the closest genetic relationship with Z. officinale, which was consistent with the close relationship between Marantaceae and Zingiberaceae that was revealed by two previous studies (Sass et al., 2016; Carlsen et al., 2018). These two species shared an ancient WGD event with M. acuminata in their common ancestor, which was follwed by diploidization events that involved substantial genome reshuffling and gene losses. The early divergence among these species provided a long enough period to allow sufficient divergence in genomic characteristics and adaptation strategies in T. dealbata and Z. officinale. The identified expanded, contracted, and unique gene families together with a number of positively selected genes in T. dealbata genome are possibly responsible to the adaptation of T. dealbata to wetland environment.
Overall, the high-quality T. dealbata genome assembly presented in this study will provide a valuable genomic resource for the study of plant adaptation to wetland environments and the evolutionary analysis of Marantaceae and Zingiberales. We look forward more genetic and genomic analysis and functional studies of this interesting wetland plant in the future.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
MT, JH, LW, and HL designed and supervised the study. MT and JD collected the samples and extracted the genomic DNA and RNA. MT, XM, and PG performed genomic data analysis. MT drafted the manuscript, and YB revised this manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors declare that this study received funding from Kunming Novo Medical Laboratory Co., Ltd. The funder had the following involvement in the study: designed and supervised the study, technical guidance, genome sequencing and assembly, analysis, interpretation of data and the writing of this article. All authors agreed to submit it for publication.
This research was also funded by the research on the collection and identification of forage resources and productive cultivation techniques in Yunnan.
Acknowledgments
We thank the colleagues of College of Landscape Architecture and Horticulture, Yunnan Agricultural University for their generous help, College of Animal Science and Technology, Yunnan Agricultural University for providing a well-founded experimental platform, and Kunming Novo Medical Laboratory Co., Ltd. for financial support.
Conflict of interest
Authors HL and LW were employed by the company Kunming Novo Medical Laboratory Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1183361/full#supplementary-material
References
Bairoch, A., Apweiler, R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi: 10.1093/nar/28.1.45
Birney, E., Clamp, M., Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Carlsen, M. M., Fér, T., Schmickl, R., Leong-Škorničková, J., Newman, M., Kress, W. J. (2018). Resolving the rapid plant radiation of early diverging lineages in the tropical zingiberales: pushing the limits of genomic data. Mol. Phylogenet. Evol. 128, 55–68. doi: 10.1016/j.ympev.2018.07.020
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., Li, H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. doi: 10.1038/s41592-020-01056-5
Cronk, J. K., Fennessy, M. S. (2016). Wetland plants: biology and ecology. CRC press. doi: 10.1201/9781420032925
De Bie, T., Cristianini, N., Demuth, J. P., Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Deng, N., Liu, C., Tian, Y., Song, Q., Niu, Y., Ma, F. (2021). Complete chloroplast genome sequences and codon usage pattern among three wetland plants. Agron. J. 113, 840–851. doi: 10.1002/agj2.20499
D’hont, A., Denoeud, F., Aury, J. M., Baurens, F. C., Carreel, F., Garsmeur, O., et al. (2012). The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217. doi: 10.1038/nature11241
Doyle, J. J., Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytoch. Bull. 19, 11–15.
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the Aedes aegypti genome using Hi-c yields chromosome-length scaffolds. Science 356, 92–95. doi: 10.1126/science.aal3327
Durand, N. C., Shamim, M. S., Machol, I., Rao, S. S., Huntley, M. H., Lander, E. S., et al. (2016). Juicer provides a one-click system for analyzing loop-resolution Hi-c experiments. Cell Syst. 3, 95–98. doi: 10.1016/j.cels.2016.07.002
Ellinghaus, D., Kurtz, S., Willhoeft, U. (2008). LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinf. 9, 1–14. doi: 10.1186/1471-2105-9-18
Gray, M. J., Hagy, H. M., Nyman, J. A., Stafford, J. D. (2013). “Management of wetlands for wildlife,” in Wetland techniques. Eds. Anderson, J., Davis, C. (Dordrecht: Springer). doi: 10.1007/978-94-007-6907-6_4
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, J. R.K., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. doi: 10.1093/nar/gkg770
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7. doi: 10.1186/gb-2008-9-1-r7
Hunter, S., Apweiler, R., Attwood, T. K., Bairoch, A., Bateman, A., Binns, D., et al. (2009). InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215. doi: 10.1093/nar/gkn785
Jain, R., Jenkins, J., Shu, S., Chern, M., Martin, J. A., Copetti, D., et al. (2019). Genome sequence of the model rice variety KitaakeX. BMC Genomics 20, 1–9. doi: 10.1186/s12864-019-6262-4
Katoh, K., Misawa, K., Kuma, K.-I., Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kumar, S., Stecher, G., Suleski, M., Hedges, S. B. (2017). TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. doi: 10.1093/molbev/msx116
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, L., Stoeckert, C. J., Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, H. L., Wu, L., Dong, Z., Jiang, Y., Jiang, S., Xing, H., et al. (2021). Haplotype-resolved genome of diploid ginger (Zingiber officinale) and its unique gingerol biosynthetic pathway. Hortic. Res. 8, 189. doi: 10.1038/s41438-021-00627-7
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Marçais, G., Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Nawrocki, E. P., Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Nurk, S., Walenz, B. P., Rhie, A., Vollger, M. R., Logsdon, G. A., Grothe, R., et al. (2020). HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305. doi: 10.1101/gr.263566.120
Ou, S., Jiang, N. (2018). LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422. doi: 10.1104/pp.17.01310
Pezeshki, S. R. (2001). Wetland plant responses to soil flooding. Environ. Exp. Bot. 46, 299–312. doi: 10.1016/S0098-8472(01)00107-1
Podlevsky, J. D., Bley, C. J., Omana, R. V., Qi, X., Chen, J. J. L. (2007). The telomerase database. Nucleic Acids Res. 36, D339–D343. doi: 10.1093/nar/gkm700
Price, A. L., Jones, N. C., Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358. doi: 10.1093/bioinformatics/bti1018
Qiao, X., Li, Q., Yin, H., Qi, K., Li, L., Wang, R., et al. (2019). Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 20, 38. doi: 10.1186/s13059-019-1650-2
Sass, C., Iles, W. J., Barrett, C. F., Smith, S. Y., Specht, C. D. (2016). Revisiting the zingiberales: using multiplexed exon capture to resolve ancient and recent phylogenetic splits in a charismatic plant lineage. PeerJ 4, e1584. doi: 10.7717/peerj.1584
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., Morgenstern, B. (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Suessenguth, K. (1921). Bemerkungen zur meiotischen und somatischen kernteilung bei einigen monokotylen. Flora oder Allgemeine Botanische Zeitung 114, 313–328. doi: 10.1016/S0367-1615(17)31551-3
Tarailo-Graovac, M., Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. 25, 4–10. doi: 10.1002/0471250953.bi0410s05
Wang, M., Huang, J., Liu, S., Liu, X., Li, R., Luo, J., et al. (2022). Improved assembly and annotation of the sesame genome. DNA Res. 29, dsac041. doi: 10.1093/dnares/dsac041
Wang, J., Lu, X., Zhang, J., Ouyang, Y., Wei, G., Xiong, Y. (2020). Rice intercropping with alligator flag (Thalia dealbata): a novel model to produce safe cereal grains while remediating cadmium contaminated paddy soil. J. Hazard. Mater. 394, 122505. doi: 10.1016/j.jhazmat.2020.122505
Wang, Z., Miao, H., Liu, J., Xu, B., Yao, X., Xu, C., et al. (2019). Musa balbisiana genome reveals subgenome evolution and functional divergence. Nat. Plants 5, 810–821. doi: 10.1038/s41477-019-0452-6
Wang, Z., Rouard, M., Biswas, M. K., Droc, G., Cui, D., Roux, N., et al. (2022). A chromosome-level reference genome of Ensete glaucum gives insight into diversity and chromosomal and repetitive sequence evolution in the musaceae. GigaScience 11, giac027. doi: 10.1093/gigascience/giac027
Wang, Y., Tang, H., DeBarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49–e49. doi: 10.1093/nar/gkr1293
Wang, M., Tong, S., Ma, T., Xi, Z., Liu, J. (2021). Chromosome-level genome assembly of sichuan pepper provides insights into apomixis, drought tolerance, and alkaloid biosynthesis. Mol. Ecol. Resour. 21, 2533–2545. doi: 10.1111/1755-0998.13449
Xu, Z., Wang, H. (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yue, J., Chen, Q., Wang, Y., Zhang, L., Ye, C., Wang, X., et al. (2023). Telomere-to-telomere and gap-free reference genome assembly of the kiwifruit Actinidia chinensis. Hortic. Res. 10, uhac264. doi: 10.1093/hr/uhac264
Keywords: Thalia dealbata, near-complete genome assembly, PacBio HiFi, genome annotation, wetland plant
Citation: Tang M, Huang J, Ma X, Du J, Bi Y, Guo P, Lu H and Wang L (2023) A near-complete genome assembly of Thalia dealbata Fraser (Marantaceae). Front. Plant Sci. 14:1183361. doi: 10.3389/fpls.2023.1183361
Received: 10 March 2023; Accepted: 26 May 2023;
Published: 13 June 2023.
Edited by:
Kai-Hua Jia, Shandong Academy of Agricultural Sciences, ChinaReviewed by:
Hui Liu, Chinese Academy of Sciences (CAS), ChinaJoaquim Martins Jr., Centro Nacional de Pesquisa em Energia e Materiais, Brazil
Copyright © 2023 Tang, Huang, Ma, Du, Bi, Guo, Lu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Wang, Mjg5MDkwMjcwOEBxcS5jb20=; Hao Lu, aW5jYW5hOTZAMTYzLmNvbQ==
†These authors have contributed equally to this work