 Ke Hu
Ke Hu Xiao-Qin Sun
Xiao-Qin Sun Min Chen1,2,3
Min Chen1,2,3 Rui-Sen Lu
Rui-Sen Lu- 1Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing, China
- 2Jiangsu Key Laboratory for the Research and Utilization of Plant Resources, Nanjing, China
- 3Jiangsu Provincial Science and Technology Resources Coordination Platform (Agricultural Germplasm Resources) Germplasm Resources Nursery of Medicinal Plants, Nanjing, China
Dioscorea sect. Stenophora (Dioscoreaceae) comprises about 30 species that are distributed in the temperate and subtropical regions of the Northern Hemisphere. Despite being evolutionarily “primitive” and medically valuable, genomic resources and molecular studies of this section are still scarce. Here, we conducted low-coverage whole genome sequencing of 11 Stenophora species/subspecies to retrieve their plastome information (whole plastome characteristics, plastome-divergent hotspots, plastome-derived SSRs, etc.) and polymorphic nuclear SSRs, as well as performed comparative plastome and phylogenetic analyses within this section. The plastomes of Stenophora species/subspecies ranged from 153,691 bp (D. zingiberensis) to 154,149 bp (D. biformifolia) in length, and they all contained the same 114 unique genes. All these plastomes were highly conserved in gene structure, gene order and GC content, although variations at the IR/SC borders contributed to the whole length differences among them. The number of plastome-derived SSRs among Stenophora species/subspecies varied from 74 (D. futschauensis) to 93 (D. zingiberensis), with A/T found to be the most frequent one. Seven highly variable regions and 12 polymorphic nuclear SSRs were identified in this section, thereby providing important information for further taxonomical, phylogenetic and population genetic studies. Phylogenomic analyses based on whole plastome sequences and 80 common protein coding genes strongly supported D. biformifolia and D. banzhuana constituted the successive sister species to the remaining sampled species, which could be furtherly divided into three clades. Overall, this study provided a new perspective for plastome evolution of Stenophora, and proved the role of plastome phylogenomic in improving the phylogenetic resolution in this section. These results also provided an important reference for the protection and utilization of this economically important section.
1 Introduction
Dioscorea is the largest genus in the family Dioscoreaceae with over 600 species, which contains about ten major clades: Stenophora, New World I, New World II, African, Mediterranean, New World III, Compound Leaved, Malagasy, Shannicorea and Enantiophyllum (Viruel et al., 2016; Couto et al., 2018; Viruel et al., 2018). Among them, Stenophora, the subject of our study, coincides with the section Stenophora Uline circumscribed by Burkill (1960), and contains about 30 species disjunctively distributed in the Northern Hemisphere (Gao et al., 2008; Vinogradova et al., 2022). The sect. Stenophora likely originated in Himalayas-Hengduan Mountains, China, and is the most basal clade of Dioscorea, differing from the rest of this genus by having rhizomes, monosulcate pollen and a diploid chromosome number (x = 10) (Wilkin et al., 2005; Gao et al., 2008; Hsu et al., 2013; Viruel et al., 2016; Couto et al., 2018; Noda et al., 2020). Plant species in this section are reported to have great medicinal values. In particular, the rhizomes of D. nipponica and D. zingiberensis are extensively used to extract diosgenin, which is an important precursor for the synthesis of steroid drugs in the pharmaceutical industry (Gong et al., 2011; Cheng et al., 2021). The immense evolutionary and medicinal value of Stenophora species has also brought new challenges to their conservation and sustainable use. One major concern is that the increasing demands for naturally growing plants has threaten their wild populations and genetic variations (Das et al., 2013; Sun et al., 2017). For example, D. nipponica has been listed as a secondary-level endangered plant species in China, as its wild resource is facing extinction (Fu, 1992; Chen et al., 2007). Another concern is the misidentification and misuse of Stenophora species, as they are similar in morphological characteristics and local names. Therefore, accurate and rapid identification of Stenophora species (e.g., molecular markers) is urgently required.
Previous studies of Stenophora have concentrated on external morphology, cytology, pollen morphology and phytochemistry (Pei et al., 1979; Huang et al., 2010). Morphological and embryological features have been shown to be important for systematics and species identification of Stenophora, and could divide this section into several subclades (e.g., Titova and Torshilova, 2015; Vinogradova et al., 2022), however, it is still difficult to find clear gaps of morphological variations among closely related species (Noda et al., 2020). Moreover, although previous molecular-based studies have accelerated species identification and phylogenetic inference of Stenophora, the plastid loci used (e.g., atpB, matK, rbcL) always showed low discriminatory power (Gao et al., 2008; Noda et al., 2020). For example, Gao et al. (2008) revealed that D. nipponica was sister to D. althaeoides, and D. nipponica ssp. rosthornii was not related to these two species, but their interspecific relationships receive weak bootstrap support. Evidently more effective molecular markers are needed to solve the remaining phylogenetic dilemma.
Plastomes of land plants generally have a quadripartite circular structure, with a pair of inverted repeats (IRs) separated by a large single-copy (LSC) region and a small single-copy (SSC) region, ranging from 100 to 200 kb in length (Raubeson and Jansen, 2005; Jansen and Ruhlman, 2012; Olejniczak et al., 2016; Lu et al., 2023). Due to many advantages such as highly conserved structure, usually uniparental inheritance, absence of recombination, and large copy numbers, plastome sequences have been widely used for accurate species identification and phylogenetic inferences, especially at low taxonomic levels (Gitzendanner et al., 2018; Yang et al., 2022). Furthermore, comparative plastome genomics could provide essential information for plastome evolution, such as gene loss and IR boundary shifts, and can develop mutational hotspots, which may contribute to species discrimination, phylogenetic, and population genetic studies (Lu et al., 2021; Yang et al., 2022). In sect. Stenophora, although some representative plastomes have been sporadically released, previous studies mainly focused on the plastome characteristics of single species (e.g., Wu et al., 2016; Zhou et al., 2016), or performed comparative and phylogenetic analyses only based on a small number of plastomes (e.g., Zhao et al., 2018; Xia et al., 2019). Thus, it is necessary to provide more genomic resources for further understanding the plastome evolution and phylogeny of Stenophora.
With the rapid development of next generation sequencing (NGS) technologies, it is cheap and fast to obtain low-coverage (~0.1–10×) of the whole genome sequencing data (or called genome skimming data), which could provide sufficient data for complete plastome assemblies (Straub et al., 2012; Twyford and Ness, 2017; Jin et al., 2020). Besides, the assembled nuclear scaffolds from low-coverage whole genome sequencing data could be used for mining polymorphic nuclear SSRs (nSSRs) (e.g., Liu et al., 2018; Xia et al., 2018; Lu et al., 2022). Here, we performed low-coverage whole genome shotgun sequencing for 11 Stenophora species/subspecies (i.e., D. banzhuana Pei & Ting, D. biformifolia Pei & Ting, D. collettii Hook.f., D. deltoidea Wall., D. futschauensis Uline ex R.Knuth, D. gracillima Miq., D. nipponica Makino, D. nipponica subsp. rosthorni (Prain & Burkill) C.T.Ting, D. spongiosa J.Q.Xi, M.Mizuno & W.L.Zhao, D. tokoro Makino, D. zingiberensis C.H.Wright). Using this data, we aimed to i) present the complete and annotated plastome sequences of these 11 Stenophora species/subspecies, and assess plastome structural evolution of them; ii) identify plastomic SSRs and mutational hotspot regions (plastome-derived markers); iii) develop polymorphic nSSRs based on assembled nuclear scaffolds of Stenophora species/subspecies; and iv) conduct phylogenetic analyses of these species/subspecies using plastome data. Overall, this study will not only provide a valuable resource for species identification and phylogenetic studies of Stenophora, but also be useful for conservation and utilization of this economically important section.
2 Materials and methods
2.1 Plant materials, DNA extraction and genomic sequencing
Fresh leaves of 11 Stenophora species/subspecies, i.e., D. banzhuana, D. biformifolia, D. collettii, D. deltoidea, D. futschauensis, D. gracillima, D. nipponica, D. nipponica subsp. rosthorni, D. spongiosa, D. tokoro, D. zingiberensis, were field-collected and dried with silica-gel. The voucher specimens were deposited at Herbarium of Institute of Botany, Jiangsu Province and Chinese Academy of Sciences (NAS) [details about sampling information can be found in Gao et al. (2008)]. For each species/subspecies, genomic DNA was extracted from silica gel-dried leaves using DNAsecure Plant Kit (Tiangen Biotech, Beijing, China), following the manufacturer’s protocol. DNA concentration and integrity were measured by Agilent 2100 BioAnalyzer and agarose gel electrophoresis. Paired-end library with insert size of 350 bp was constructed for each species/subspecies by using the Genomic DNA Sample Prep, and then sequenced on the Illumina HiSeqTM 4000 platform (Illumina, San Diego, California, USA) according to the paired-end 2 × 150 bp protocol. Library construction, genome sequencing and raw data filtering were conducted by Novogene Bioinformatics Technology Co., Ltd., Beijing, China.
2.2 Plastome assembly and annotation
After removing library barcodes and filtering low-quality data, the clean reads (about 4 Gb per sample) were used for de novo assembly of whole plastome sequences using GetOrganelle v.1.7.6 (Jin et al., 2020), with the default parameters as suggested by its authors. All the assembly graphs were subsequently visually inspected using Bandage v.0.8.1 (Wick et al., 2015). Initial annotations of all newly assembled plastomes were performed with MAFFT v.7 plugin (Katoh and Standley, 2013) in Geneious Prime® 2022.0.1 (https://www.geneious.com) by aligning them to two closely related and previously published plastomes, i.e., D. aspersa (NC_039807) and D. collettii (NC_037717), and transferring reference annotations to these newly assembled plastomes. Then, the initial annotations were checked and adjusted manually to confirm the accuracy of exon/intron boundaries and start/stop codon locations. All newly generated plastome sequences were deposited in GenBank (accession numbers: OQ525992-OQ526002). High-resolution circular plastome maps of these 11 Stenophora species/subspecies were generated using the web-based tool OrganellarGenomeDRAW (OGDRAW) v.1.3.1 (Greiner et al., 2019).
2.3 Whole plastome comparison within sect. Stenophora
To visualize sequence similarity of plastomes within sect. Stenophora, all 11 newly sequenced plastomes and one plastome of D. villosa (NC_034686) were aligned using the global alignment program LAGAN (Brudno et al., 2003), and visualized in VISTA browser (Frazer et al., 2004), taking annotations of D. villosa plastome as reference. To further illustrate the IR expansions and contractions among Stenophora plastomes, the four junctions between two invert repeats (IRs) and large/small single copy (LSC/SSC) regions were identified and compared by Repeat Finder plugin as implemented in Geneious Prime® 2022.0.1 (https://www.geneious.com/plugins/repeat-finder/).
2.4 Identification of mutational hotspots and plastome-derived SSRs
To identify mutational hotspot regions for PCR-based identification of Stenophora species and subspecies, a total of 12 plastomes (one plastome per species/subspecies, Table 1) were first aligned using the MAFFT v.7 plugin (Katoh and Standley, 2013) in Geneious Prime® 2022.0.1. Then, protein coding sequences (CDS), intergenic spacer regions (IGS), introns and tRNAs, with aligned length > 200 bp and the total number of mutations > 0 were extracted from the alignment matrix of these 12 plastome sequences. Finally, the nucleotide diversity (π) of these regions was calculated in DnaSP v.6.12.03 (Rozas et al., 2017). In addition, the MISA-web application (Beier et al., 2017) was employed to screen SSRs across the 12 Stenophora plastomes, with thresholds (minimum numbers) of 10, 5, 4, 3, 3, and 3 repeat units for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide SSRs, respectively.
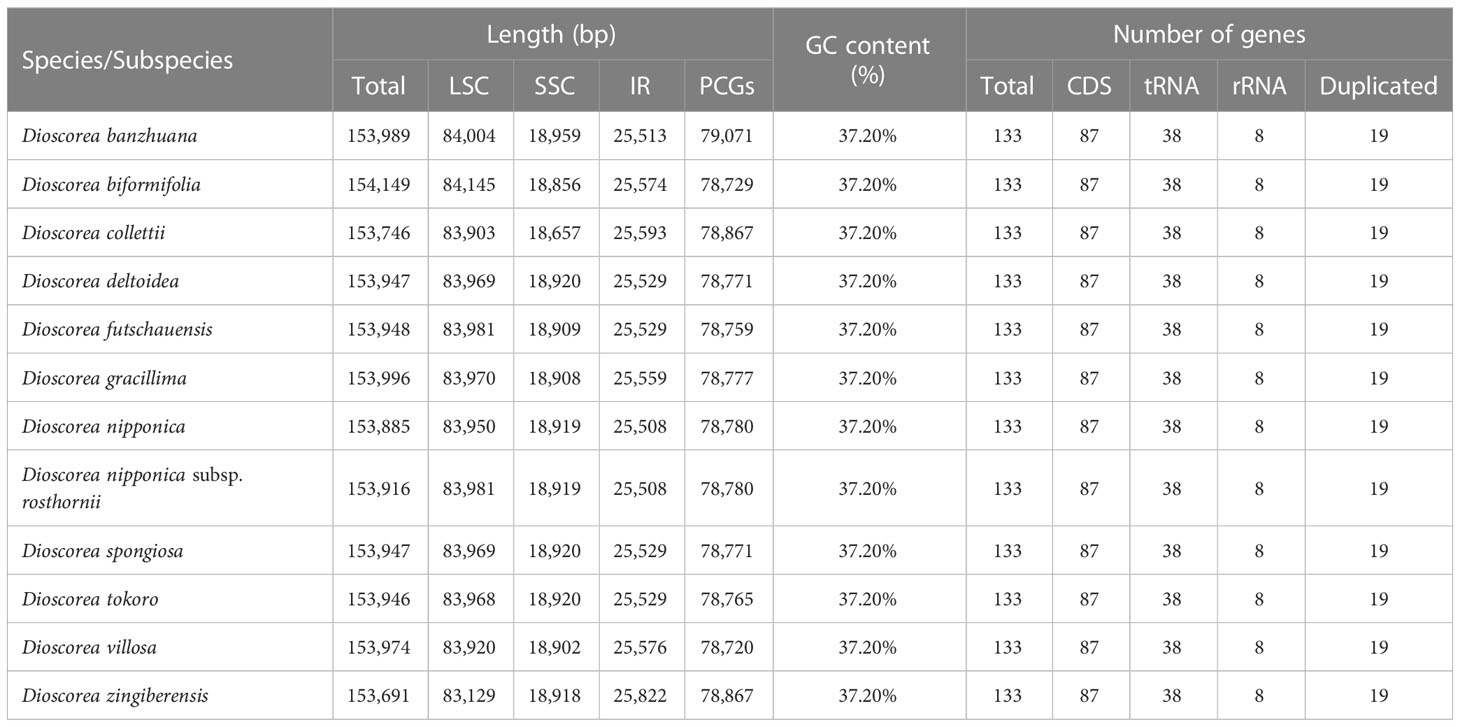
Table 1 The basic features of 11 Stenophora plastomes newly generated in this study.
2.5 Development of polymorphic nuclear SSRs
To develop polymorphic nuclear SSRs in sect. Stenophora, low-coverage whole genome sequence reads of these 11 species/subspecies were aligned to the reference genome sequence of D. zingiberensis (Cheng et al., 2021) to remove mitochondria and plastome reads, using BWA-MEM v.0.7.17 (Li, 2013). Aligned files were then sorted using SAMtools v.1.9 (Li et al., 2009). The resultant Binary Alignment/Map (BAM) data (only containing nuclear reads) were de novo assembled into scaffolds using a de Bruijn graph-based assembly program, SOAPdenovo v.1.0.4 (Xie et al., 2014). Based on these nuclear scaffolds, the potential polymorphic nuclear SSRs were identified using CandiSSR (Xia et al., 2016), with default parameters.
2.6 Phylogenetic analyses
Phylogenetic relationships among the 12 species/subspecies of sect. Stenophora (Table 1) were inferred based on two datasets: (1) whole plastome sequences and (2) 80 shared protein coding genes, taking D. aspersa (NC_039807) and D. alata (NC_039707) as outgroups. For the latter dataset, three partitioning scenarios: (1) unpartitioned scenarios; (2) partitioned by each gene and intergenic region; and (3) partitioned by codon position were employed. Both whole plastome sequences and protein coding sequences were aligned using the MAFFT v.7 plugin (Katoh and Standley, 2013) in Geneious Prime® 2022.0.1. The best nucleotide substitution model was determined by the Akaike Information Criterion (AIC) in jModelTest v2.1.4 (Darriba et al., 2012), and the GTR + G substitution model was selected for both datasets. Maximum likelihood (ML) analyses were performed using RAxML v.8.2.12 (Stamatakis, 2014) available in the CIPRES Science Gateway v.3.3 (http://www.phylo.org/portal2/). Clade support values were estimated by 1000 bootstrap replicates. Bayesian inference (BI) analyses were conducted on MrBayes v.3.2.7 (Ronquist et al., 2012), which consists of two independent runs of 1 × 106 generations, with four independent Markov chain Monte Carlo (MCMC) chains (i.e., one cold and three heated) each, and a sampling frequency of 1000 generations. The first 200 trees were discarded as ‘burn-in’, and the remaining trees were used to construct a majority-rule consensus tree and estimate posterior probabilities (PPs).
3 Results and discussion
3.1 Plastome characteristics
The whole length of these Stenophora plastomes ranged from 153,691 bp (D. zingiberensis) to 154,149 bp (D. biformifolia) (Figure 1; Table 1). All these plastomes shared the typical quadripartite structure of angiosperm plastomes, with a pair of IR regions (25,508–25,822 bp) separated by the LSC (83,129–84,145 bp) and SSC (18,657–18,959 bp) regions. The length variation of Dioscorea plastomes is a very common phenomenon (Zhao et al., 2018; Xia et al., 2019), which is often caused by the expansion and contraction of the IR regions (see details below). These Stenophora plastomes have the same overall GC content (37.20%), higher than that in LSC (35.00–35.10%) and SSC (31.2%) regions, but lower than that in IR regions (43.29–43.0%) (Table 1), possibly influenced by the high GC content (55.3%) of the four ribosomal RNA (rRNA) sequences.
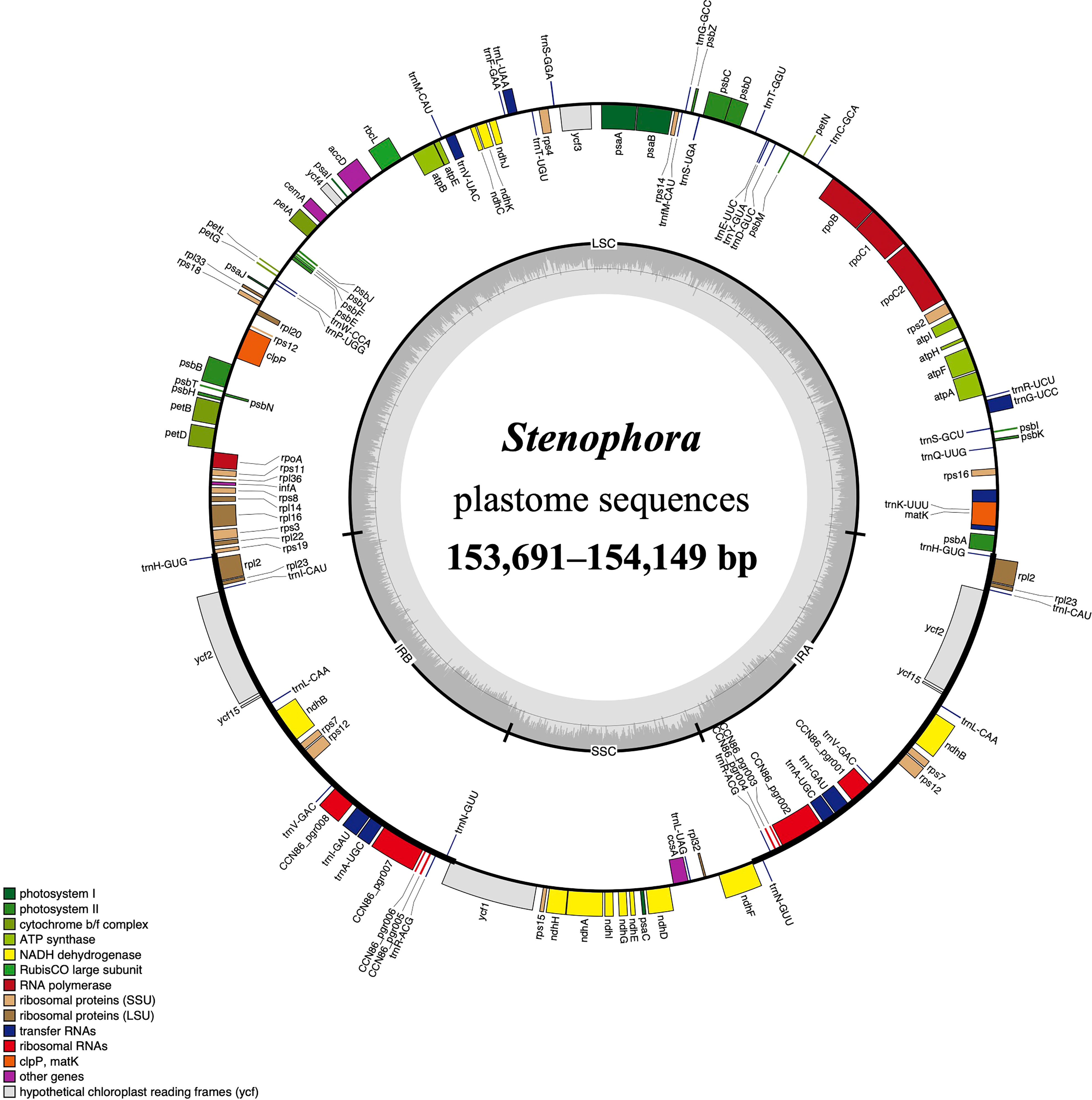
Figure 1 The plastome map of Stenophora species/subspecies. Thick lines on the outer complete circle identify the inverted repeat regions (IRa and IRb). Genes shown on the outside of the circle are transcribed clockwise, while genes inside are transcribed counter-clockwise. Genes are color coded according to their functional groups. GC/AT content is displayed by darker/lighter grey bars in the inner ring.
All these Stenophora plastomes encoded the same 114 unique genes, including 80 protein-coding genes (PCGs), 30 transfer RNA (tRNA) genes, and four rRNA genes. Nineteen unique genes (seven PCGs, eight tRNA genes, and all four rRNA) were duplicated in the IRs, giving a total of 133 genes (Figure 1; Table S1). Among these unique genes, nine PCGs (i.e., atpF, petB, petD, ndhA, ndhB, rpoC1, rpl2, rpl16, and rps16) and six tRNAs (trnK-UUU, trnG-UCC, trnL-UAA, trnV-UAC, trnI-GAU and trnA-UGC) possessed a single intron, while three PCGs (ycf3, rps12 and clpP) contained two introns (Figure 1; Table S1). The rps12 gene consists of three exons that were trans-spliced together: exon 1 was located in the LSC region, while exons 2 and 3 were proximal and located in the IR regions (Figure 1; Table S1). Furthermore, all Stenophora plastomes reported in this study harbored the complete rps16 gene, contrary to previous studies indicating the entire loss of rps16 gene in several clades of Dioscorea (Jansen et al., 2007; Lu et al., 2023). To further improve our understanding of rps16 gene evolution in Dioscorea, a ML phylogenetic tree was reconstructed (with the same method above) based on whole plastome sequences of 42 Dioscorea species, using Trichopus zeylanicus and Tacca leontopetaloides as outgroups. Phylogenetic result showed that rps16 gene was lost in all other Dioscorea clades except Stenophora, suggesting a single loss of this gene within Dioscorea (Figure S1). Since Stenophora and the rest of the genus diverged about 48.3 (47.6–49.1) million years ago (Mya) (Viruel et al., 2016), the gene loss mentioned above may have occurred in sync with this divergence event, implying that the loss of rps16 gene may have occurred about 48.3 Mya.
3.2 Comparative plastome analyses of Stenophora
Comprehensive comparison of 12 Stenophora plastomes revealed a high degree of overall sequence similarity and collinearity within this section (Figure 2). Similar to previous monocot plastome studies (e.g., Asaf et al., 2017; Lu et al., 2017; Lu et al., 2021; Lu et al., 2022), our mVISTA analysis demonstrated that IRs exhibited a lower level of sequence divergence compared with LSC and SSC regions (Figure 2). This could be attributed to copy correction between IR sequences by gene conversion, and the abundance of conserved rRNA genes in the IRs (Khakhlova and Bock, 2006). In addition, the protein-coding regions were found to be more conserved than non-coding regions (including intergenic spacers and introns), which were likely to be subject to natural selection (Shaw et al., 2007; Lu et al., 2022).
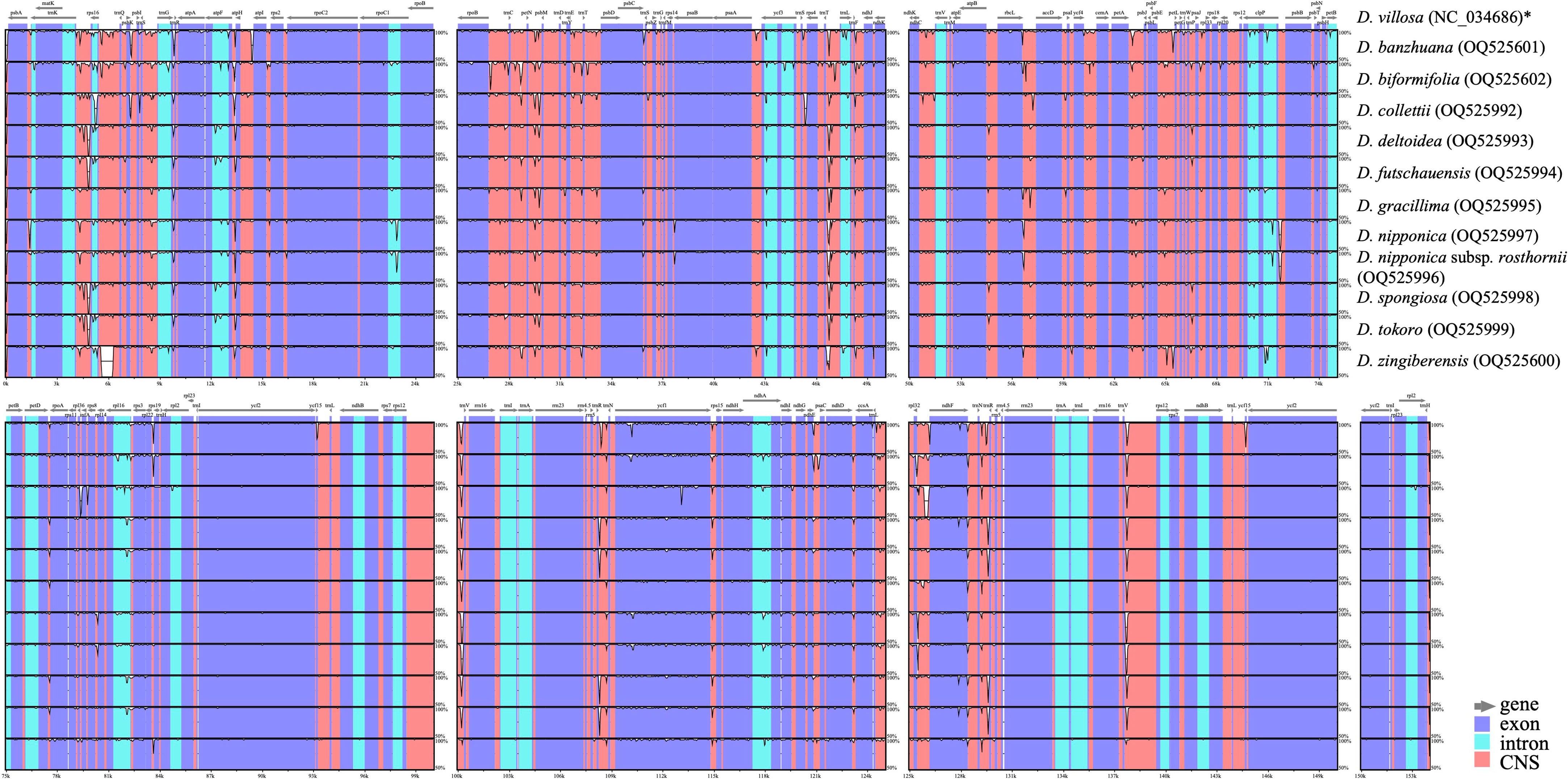
Figure 2 Sequence identity plots among Stenophora plastomes, with D. villosa (NC_034686) as a reference. Annotated genes are shown along the top. Gray arrows above the alignment indicates genes with their orientation. The vertical scale represents the percent identity between 50% and 100%. Genome regions are color coded as exon, intron, and conserved non-coding sequences (CNS). * previously published plastome sequence.
Despite the similarity of plastome sequences, and the conservation of gene content and linear order of genes, the 12 Stenophora plastomes exhibited obvious differences at the IR/SC borders (Figure 3). For example, the ndhF gene crossed the SSC/IRa border in D. collettii and D. zingiberensis, while it was completely included in the SSC region in the other 10 species (Figure 3). The IRb region extended 238 bp into the rps19 gene in D. zingiberensis, much deeper than those extended into all other species (2–22 bp), and further extended 280–296 bp into the ycf1 gene. IR expansion into rps19 gene has also been observed in other sections in the genus Dioscorea, e.g., Opsophyton, Testudinaria, Enantiophyllum (Zhao et al., 2018; Lu et al., 2023), suggesting that this phenomenon may be an ancestral symplesiomorphy of the genus Dioscorea. In addition, the trnH gene was totally located within the IR region and duplicated in all these species, 142–378 bp away from its proximal IR/SC border.

Figure 3 Comparison of IR/SC junctions among 12 Stenophora plastomes. * previously published plastome sequence.
3.3 Plastome-derived hotspot regions and SSRs for Stenophora
Morphology-based species identification in sect. Stenophora has always been difficult, because it is challenging to find clear gaps of morphological variations among closely related species (Kawabe et al., 1997; Wilkin et al., 2005; Gao et al., 2008). In this case, barcoding has been performed for this section using nuclear gene phosphoglucose isomerase (PGI) and plastid DNA (matK, rbcL and trnL-F) regions (Kawabe et al., 1997; Gao et al., 2008). However, these markers are today considered intermediately variable regions (Shaw et al., 2014), and always showed low species discriminatory power and poor phylogenetic resolution (Gao et al., 2008; Noda et al., 2020). Therefore, we here used these plastome sequences to develop novel genetic markers (hypervariable regions) for taxonomic and phylogenetic analysis of Stenophora. A total of 130 regions (58 CDS, 53 IGS, 13 introns, five tRNAs and one rRNA) was eventually extracted to calculate the nucleotide diversity, and the π values ranged from 0.01% (rrn16) to 3.36% (ndhD–ccsA) (Figure 4). Six IGS regions (i.e., ndhD-ccsA, petA-psbJ, trnL-rpl32, psbZ-trnG, trnD-trnY and rpl32-ndhF), and rps16 intron sequence were the top seven highly variable regions (π > 1.00%) (Figure 4), which could be served as section-specific molecular markers for future identification, conservation and utilization of Stenophora species.
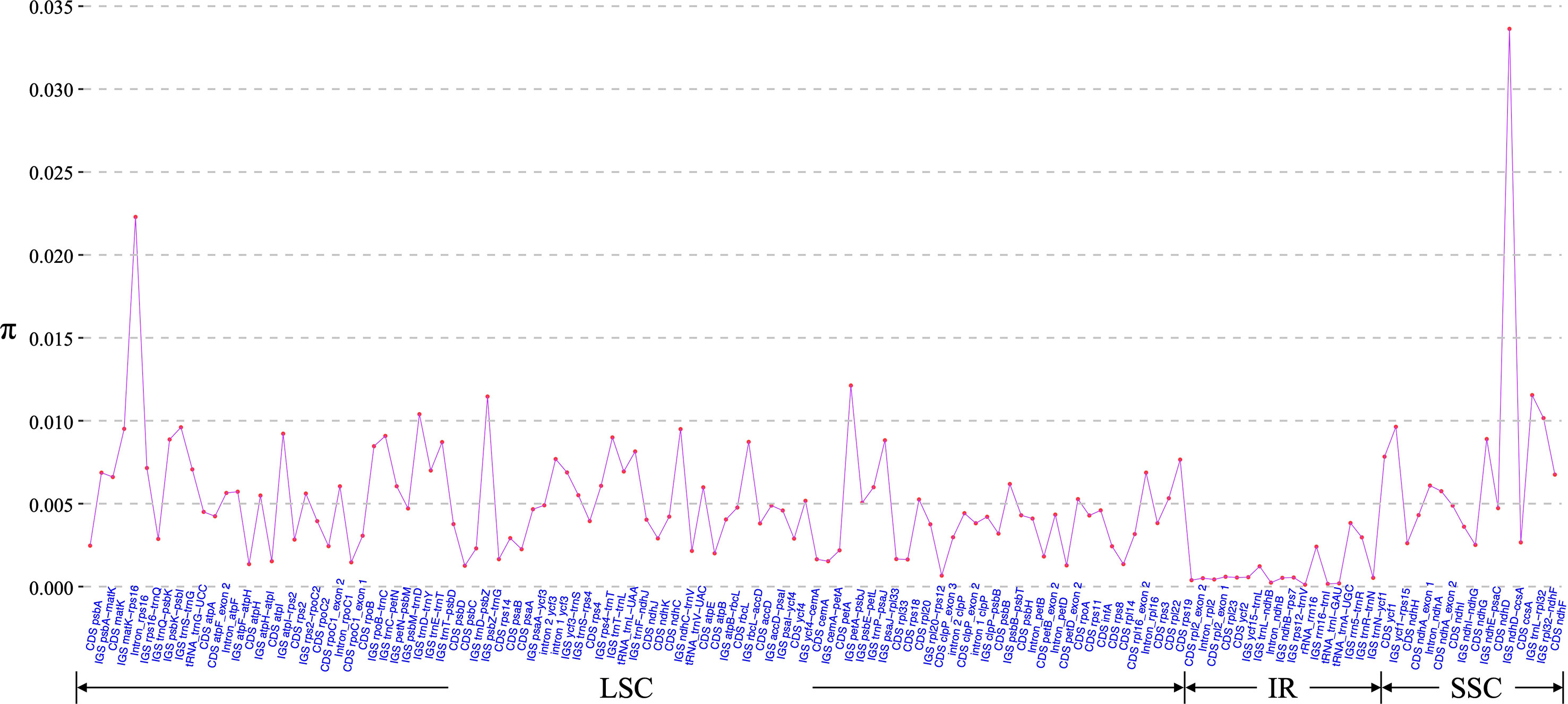
Figure 4 Nucleotide variability (π) values of 130 regions (58 CDS, 53 IGS, 13 introns, five tRNAs and one rRNA) extracted from the alignment matrix of 12 Stenophora plastome sequences.
Plastome-derived SSRs (chloroplast SSRs, cpSSRs) are scattered in the plastomes across different plant taxa, and have been widely used in population genetic studies and breeding programs (Jiménez, 2010; Chmielewski et al., 2015; Hazra et al., 2021; Ping et al., 2021). In this study, the MISA analysis identified a total of 960 SSRs across the 12 Stenophora plastomes. The number of SSRs for each plastome ranged from 74 (D. futschauensis) to 93 (D. zingiberensis). Mononucleotide repeats were predominant, with numbers ranging from 35 (D. collettii) to 50 (D. biformifolia), followed by dinucleotides ranging from 14 (D. banzhuana) to 18 (D. villosa), and tetranucleotides (10 in D. collettii and 9 in the other 11 plastomes), while trinucleotides (4–6 per plastome), pentanucleotides (0–5 per plastome) and hexanucleotides (3 per plastome) were relatively few in Stenophora plastomes (Figure 5; Table S2). The most common motifs were A/T and AT/TA for mono- and dinucleotides, accounting for 46.75%–68.76% and 17.20%–22.97% of the total SSRs in Stenophora plastomes, respectively, which may lead to the AT richness of the Stenophora plastomes (Figure 5; Table S2). These results were also consistent with the previous findings that plastome-based SSRs are largely composed of short polyadenine (polyA) and polythymine (polyT) repeats, while rarely contained tandem guanine (G) and cytosine (C) repeats (Kuang et al., 2011; Lu et al., 2022). In addition, several potential species-specific SSRs were identified in the present study. For example, AGC/CTG and AAGTAT/ACTTAT were only observed in D. collettii and D. biformifolia, respectively, but not appeared in the other 10 species/subspecies. Both AATAG/ATTCT and AATAT/ATATT were only presented in D. nipponica and its subspecies, while absence in other 10 species (Figure 5; Table S2). Clearly, these SSRs could be developed as effective molecular markers for species identification.

Figure 5 Plastome-derived SSRs in the 12 Stenophora species/subspecies.
3.4 Polymorphic nuclear SSRs of Stenophora
Unlike plastome-derived SSRs with a certain degree of conservatism and usually uniparental inheritance, nuclear SSR markers are co-dominant and generally highly polymorphic, thus can complement plastome-derived SSR analysis in plants (Aecyo et al., 2021). In this study, based on the multiple assembled nuclear scaffolds of 11 newly sequenced Stenophora species/subspecies, a total of 12 polymorphic nSSRs (including six dinucleotides and six trinucleotides) were determined within this section by using CandiSSR (Table S3). Among these polymorphic nSSRs, nSSR_7 could divide these Stenophora species/subspecies into five groups, while four nSSRs (i.e., nSSR_1, nSSR_2, nSSR_5, nSSR_6) and seven nSSRs (i.e., nSSR_3, nSSR_4, nSSR_8, nSSR_9, nSSR_10, nSSR_11, nSSR_12) could divide them into four and three groups, respectively (Table S3). Apparently, these polymorphic nSSRs would be useful for species identification and conservation of this section, especially in the population genetic context.
3.5 Phylogenetic relationships within Stenophora
Previous phylogenetic studies have laid an important foundation for the phylogeny and classification of Stenophora species, however the selected loci (e.g., atpB, matK, rbcL and trnL-F) unfortunately could not provide sufficient information for elucidating the phylogenetic and evolutionary relationships among them (Gao et al., 2008; Viruel et al., 2016; Noda et al., 2020). Recently, plastome sequences have been extensively used for phylogenetic analyses, especially in addressing unresolved relationship at low taxonomic levels (Carbonell-Caballero et al., 2015; Li et al., 2017). In this study, two datasets including the complete plastome sequences and 80 commonly present protein-coding genes of 12 Stenophora species/subspecies were used to perform phylogenetic analyses, with D. aspersa and D. alata as outgroups. Both ML and BI analyses of these two datasets (including different partitioning scenarios on protein-coding genes) yielded the same topology, with moderate to high bootstrap support values (BS = 65–100) and maximal posterior probability support values (PP = 1.0) at each node (Figure 6). The topology strongly supported D. biformifolia and D. banzhuana constituted the successive sister species to the rest. The remaining 10 sampled species/subspecies within this section could be further divided into three clades. Clade I contained four species, in which D. futschauensis was sister to the clade of D. tokoro + (D. deltoidea + D. spongiosa). Clade II, i.e., (D. nipponica + D. nipponica subsp. rosthornii) + D. zingiberensis, and clade III, i.e., (D. gracillima + D. villosa) + D. collettii were sister to each other, and jointly sister to Clade I (Figure 6). Contrary to previous hypothesis that D. nipponica ssp. rosthornii was not related to D. nipponica (Gao et al., 2008), our study strongly supported the monophyly of D. nipponica and D. nipponica ssp. rosthornii, which was consistent to the taxonomic treatments of this species in Flora of China (Ting et al., 2000).
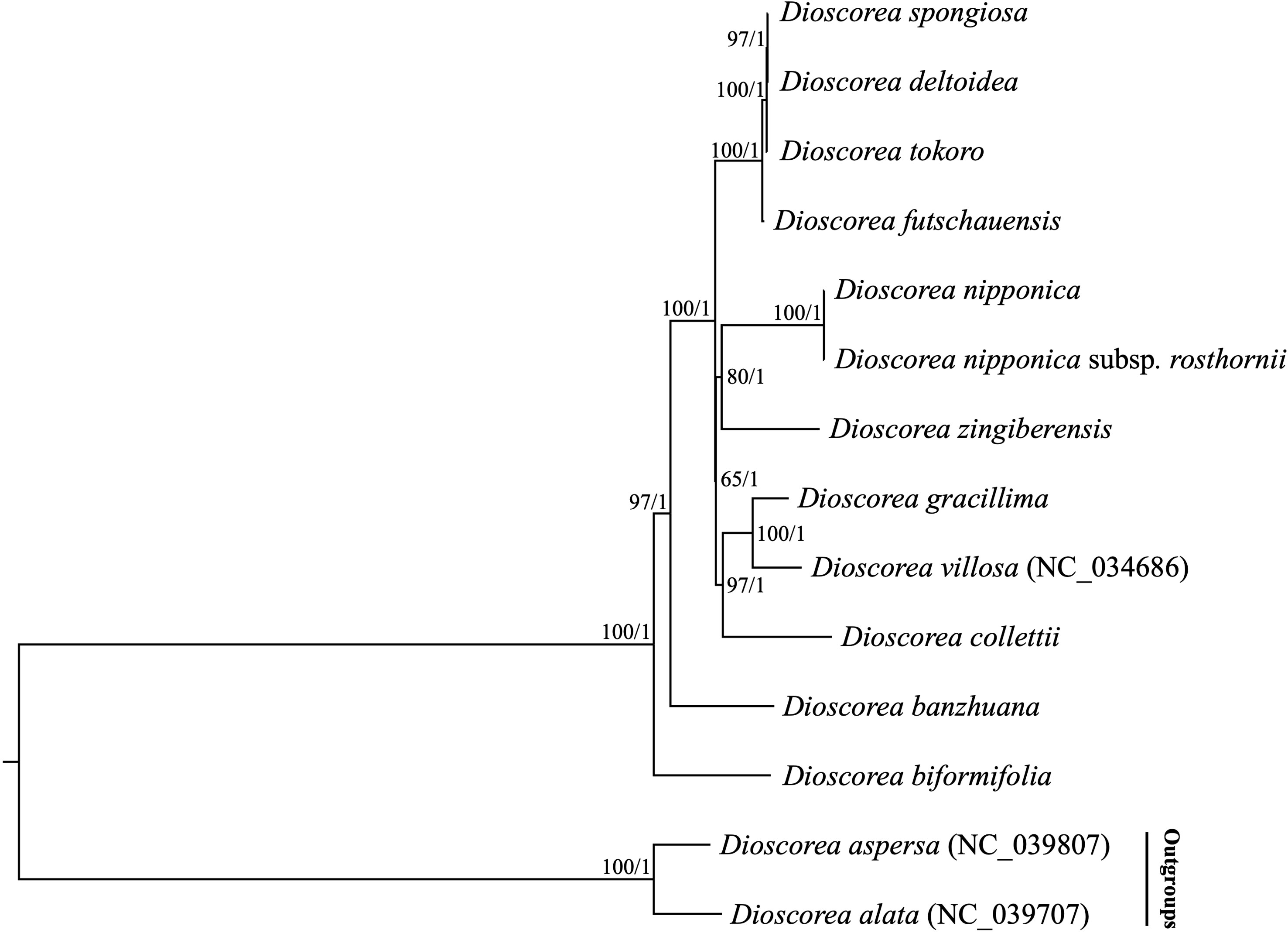
Figure 6 Phylogenetic relationships of 12 Stenophora species/subspecies inferred from Maximum likelihood (ML) and Bayesian inference (BI) methods, according to complete plastome sequences. Numbers above the lines represent ML bootstrap values and BI posterior probabilities. Phylogenetic trees based on 80 commonly present protein-coding genes with different partitioning scenarios are completely consistent with this topology.
Although our analyses have demonstrated the power of plastome phylogenetics to improve the resolutions of phylogenetic relationships in sect. Stenophora, this study was conducted based on insufficient taxa sampling, thus could not establish a complete picture of phylogenetic relationships within this section. Also considering that hybridization and polyploidization has been reported within this section (Qin and Zhang, 1985), plastome data could not accurately capture hybridization and polyploidization events, as plastome is usually uniparentally inherited, and acts as a linked single locus (Birky, 1995; Stull et al., 2015). Thus, moving beyond the plastomes and analyzing multilocus nuclear DNA sequence data with more extensive sampling is necessary in the future, to explore the phylogenetic and biogeographic hypotheses of sect. Stenophora.
4 Conclusions
In this study, we first assembled and annotated the complete plastomes of 11 D. sect. Stenophora species/subspecies, based on low-coverage whole genome sequencing data. Together with previously published plastome sequence of D. villosa, we then provided comparative plastome analyses within this section. All sampled Stenophora plastomes (153,691–154,149 bp) shared the same gene content, gene order and GC content. The rps16 gene was lost in all other Dioscorea clades except Stenophora, which may have occurred about 48.3 Mya. A total of 960 plastome-derived SSRs and seven plastomic mutational hotspots were identified in Stenophora. Besides, we also successfully developed 12 polymorphic nuclear SSRs within this section, based on multiple assembled nuclear scaffolds. Phylogenetic analyses strongly supported that D. biformifolia and D. banzhuana constituted the successive sister species to the rest, which can be furtherly divided into three clades. Overall, the data obtained here will not only contribute to our understanding of plastome evolution of Stenophora, but also aid in the conservation and utilization of their genetic resources.
Data availability statement
All data generated in this study has been publicly available.
Author contributions
R-SL conceived the ideas. KH, X-QS and MC contributed to the sampling. X-QS and MC performed the experiment and analyzed the data. The manuscript was written by KH and R-SL. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the National Natural Science Foundation of China (32200192), the Jiangsu Key Laboratory for the Research and Utilization of Plant Resources (JSPKLB202206), the Jiangsu Forestry Science and Technology Innovation and Promotion Project (LYKJ[2020]08), the Jiangsu Institute of Botany Talent Fund (JIBTF202102), and the Jiangsu Innovative and Enterpreneurial Talent Programme (JSSCBS20211311).
Acknowledgments
The authors gratefully acknowledge helpful comments from reviewers on earlier versions of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1196176/full#supplementary-material
Supplementary Figure 1 | Phylogenetic relationships among Dioscorea based on complete plastome sequences. The loss of rps16 gene is indicated on the branches of the tree.
References
Aecyo, P., Marques, A., Huettel, B., Silva, A., Esposito, T., Ribeiro, E., et al. (2021). Plastome evolution in the caesalpinia group (Leguminosae) and its application in phylogenomics and populations genetics. Planta 254, 1–19. doi: 10.1007/s00425-021-03655-8
Asaf, S., Waqas, M., Khan, A. L., Khan, M. A., Kang, S. M., Imran, Q. M., et al. (2017). The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00304
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Birky, J. C.W. (1995). Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc. Natl. Acad. Sci. 92, 11331–11338. doi: 10.1073/pnas.92.25.11331
Brudno, M., Do, C. B., Cooper, G. M., Kim, M. F., Davydov, E., Green, E. D., et al. (2003). LAGAN and multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13, 721–731. doi: 10.1101/gr.926603
Burkill, I. H. (1960). The organography and the evolution of dioscoreaceae, the family of the yams. J. Linn. Soc London Bot. 56, 319–412. doi: 10.1111/j.1095-8339.1960.tb02508.x
Carbonell-Caballero, J., Alonso, R., Ibañez, V., Terol, J., Talon, M., Dopazo, J. (2015). A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 32, 2015–2035. doi: 10.1093/molbev/msv082
Chen, F. Q., Fu, Y., Wang, D. L., Gao, X., Wang, L. (2007). The effect of plant growth regulators and sucrose on the micropropagation and microtuberization of Dioscorea nipponica makino. J. Plant Growth Regul. 26, 38–45. doi: 10.1007/s00344-005-0147-2
Cheng, J., Chen, J., Liu, X., Li, X., Zhang, W., Dai, Z., et al. (2021). The origin and evolution of the diosgenin biosynthetic pathway in yam. Plant Commun. 2, 100079. doi: 10.1016/j.xplc.2020.100079
Chmielewski, M., Meyza, K., Chybicki, I. J., Dzialuk, A., Litkowiec, M., Burczyk, J. (2015). Chloroplast microsatellites as a tool for phylogeographic studies: the case of white oaks in Poland. iforest-Biogeosciences Forestry 8, 765–771. doi: 10.3832/ifor1597-008
Couto, R. S., Martins, A. C., Bolson, M., Lopes, R. C., Smidt, E. C., Braga, J. M. A. (2018). Time calibrated tree of Dioscorea (Dioscoreaceae) indicates four origins of yams in the neotropics since the Eocene. Bot. J. Linn. Soc 188, 144–160. doi: 10.1093/botlinnean/boy052
Darriba, D., Taboada, G. L., Doallo, R., Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–772. doi: 10.1038/nmeth.2109
Das, S., Choudhury, M. D., Mazumdar, P. B. (2013). Micropropagation of Dioscorea alata l. through nodal segments. Afr. J. Biotechnol. 12, 6611–6617. doi: 10.5897/AJB2013.12191
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Gao, X., Zhu, Y. P., Wu, B. C., Zhao, Y. M., Chen, J. Q., Hang, Y. Y. (2008). Phylogeny of Dioscorea sect. Stenophora based on chloroplast matK, rbcL and trnL-F sequences. J. Syst. Evol. 46, 315–321. doi: 10.3724/sp.J.1002.2008.08007
Gitzendanner, M. A., Soltis, P. S., Yi, T. S., Li, D. Z., Soltis, D. E. (2018). “Plastome phylogenetics: 30 years of inferences into plant evolution,” in Advances in botanical research (New York: Elsevier), 293–313.
Gong, G., Qin, Y., Huang, W. (2011). Anti-thrombosis effect of diosgenin extract from Dioscorea zingiberensis C.H. Wright in vitro and in vivo. Phytomedicine 18, 458–463. doi: 10.1016/j.phymed.2010.08.015
Greiner, S., Lehwark, P., Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Hazra, A., Das, S., Bhattacharya, S., Sur, S., Sengupta, C., Das, S. (2021). Phylogenetic inference of ericales based on plastid genomes and implication of cp-SSRs. BioTechnologia. J. Biotechnol. Comput. Biol. Bionanotechnology 102, 277–283. doi: 10.5114/bta.2021.108723
Hsu, K. M., Tsai, J. L., Chen, M. Y., Ku, H. M., Liu, S. C. (2013). Molecular phylogeny of Dioscorea (Dioscoreaceae) in East and southeast Asia. Blumea - Biodiversity Evol. Biogeography Plants 58, 21–27. doi: 10.3767/000651913x669022
Huang, H. P., Gao, S. L., Huang, P., Zhou, L. (2010). Resources of medicinal classification and geographical distribution on plants of sect. Stenophora uline (Dioscorea l.) in China. Strait Pharm. J. 22, 64–66.
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Depamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Jansen, R. K., Ruhlman, T. A. (2012). Plastid genomes of seed plants. Genomics chloroplasts mitochondria 35, 103–126. doi: 10.1007/978-94-007-2920-9_5
Jiménez, A. (2010). “Microsatellites: a powerful genetic marker for fern research,” in Working with ferns: issues and applications (New York: Springer), 207–220.
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., DePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 1–31. doi: 10.1186/s13059-020-02154-5
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kawabe, A., Miyashita, N. T., Terauchi, R. (1997). Phylogenetic relationship among the section Stenophora in the genus Dioscorea based on the analysis of nucleotide sequence variation in the phosphoglucose isomerase (Pgi) locus. Genes Genet. Sys. 72, 253–262. doi: 10.1266/ggs.72.253
Khakhlova, O., Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j.1365-313X.2006.02673.x
Kuang, D. Y., Wu, H., Wang, Y. L., Gao, L. M., Zhang, S. Z., Lu, L. (2011). Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome 54, 663–673. doi: 10.1139/g11-026
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM (arXiv preprint). doi: 10.48550/arXiv.1303.3997.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, P., Lu, R. S., Xu, W. Q., Ohi-Toma, T., Cai, M. Q., Qiu, Y. X., et al. (2017). Comparative genomics and phylogenomics of East Asian tulips (Amana, liliaceae). Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00451
Liu, L., Wang, Y., He, P., Li, P., Lee, J., Soltis, D. E., et al. (2018). Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genomics 19, 1–17. doi: 10.1186/s12864-018-4633-x
Lu, R., Chen, M., Feng, Y., Yuan, N., Zhang, Y., Cao, M., et al. (2022). Comparative plastome analyses and genomic resource development in wild rice (Zizania spp., poaceae) using genome skimming data. Ind. Crops Prod. 186, 115224. doi: 10.1016/j.indcrop.2022.115244
Lu, R. S., Hu, K., Zhang, F. J., Sun, X. Q., Chen, M., Zhang, Y. M. (2023). Pan-plastome of greater yam (Dioscorea alata) in China: intraspecific genetic variation, comparative genomics, and phylogenetic analyses. Int. J. Mol. Sci. 24, 3341. doi: 10.3390/ijms24043341
Lu, R. S., Li, P., Qiu, Y. X. (2017). The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: comparative genomic and phylogenetic analyses. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.02054
Lu, R. S., Yang, T., Chen, Y., Wang, S. Y., Cai, M. Q., Cameron, K. M., et al. (2021). Comparative plastome genomics and phylogenetic analyses of liliaceae. Bot. J. Linn. Soc 196, 279–293. doi: 10.1093/botlinnean/boaa109
Noda, H., Yamashita, J., Fuse, S., Pooma, R., Poopath, M., Tobe, H., et al. (2020). A large-scale phylogenetic analysis of Dioscorea (Dioscoreaceae), with reference to character evolution and subgeneric recognition. Acta Phytotax. Geobot. 71, 103–128. doi: 10.18942/apg.201923
Olejniczak, S. A., Łojewska, E., Kowalczyk, T., Sakowicz, T. (2016). Chloroplasts: state of research and practical applications of plastome sequencing. Planta 244, 517–527. doi: 10.1007/s00425-016-2551-1
Pei, C., Ting, C. T., Chin, H. C., Su, P., Tang, S. Y., Chang, H. C. (1979). A preliminary systematic study of Dioscorea l. sect. Stenophora uline. Acta Phytotaxon. Sin. 17, 61–72.
Ping, J., Feng, P., Li, J., Zhang, R., Su, Y., Wang, T. (2021). Molecular evolution and SSRs analysis based on the chloroplast genome of Callitropsis funebris. Ecol. Evol. 11, 4786–4802. doi: 10.1002/ece3.7381
Qin, H. Z., Zhang, M. Z. (1985). A study on the classification of Dioscorea cells in China: chromosome number, the origin and evolution of the genus. Acta Phytotaxon. Sin. 23, 11–18.
Raubeson, L. A., Jansen, R. K. (2005). “Chloroplast genomes of plants,” in Plant diversity and evolution: genotypic and phenotypic variation in higher plants (Wallingford: CABI publishing), 45–68.
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Shaw, J., Lickey, E. B., Schilling, E. E., Small, R. L. (2007). Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am. J. Bot. 94, 275–288. doi: 10.3732/ajb.94.3.275
Shaw, J., Shafer, H. L., Leonard, O. R., Kovach, M. J., Schorr, M., Morris, A. B. (2014). Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. Am. J. Bot. 101, 1987–2004. doi: 10.3732/ajb.1400398
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Straub, S. C., Parks, M., Weitemier, K., Fishbein, M., Cronn, R. C., Liston, A. (2012). Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am. J. Bot. 99, 349–364. doi: 10.3732/ajb.1100335
Stull, G. W., Duno de Stefano, R., Soltis, D. E., Soltis, P. S. (2015). Resolving basal lamiid phylogeny and the circumscription of icacinaceae with a plastome-scale data set. Am. J. Bot. 102, 1794–1813. doi: 10.3732/ajb.1500298
Sun, W., Wang, B., Yang, J., Wang, W., Liu, A., Leng, L., et al. (2017). Weighted gene co-expression network analysis of the dioscin rich medicinal plant Dioscorea nipponica. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00789
Ting, C., Michael, G., Wu, Z., Raven, P. (2000). “Dioscorea linnaeus,” in Flora in china. Eds. Wu, Z. Y., Raven, P. H. (St. Louis: Missouri Botanical Garden Press and Beijing: Science Press).
Titova, G. E., Torshilova, A. A. (2015). To the evolution of seed morphological type in the Dioscorea section Stenophora (Dioscoreaceae). Botanicheskiĭ Zhurnal 100, 761–786.
Twyford, A. D., Ness, R. W. (2017). Strategies for complete plastid genome sequencing. Mol. Ecol. Resour. 17, 858–868. doi: 10.1111/1755-0998.12626
Vinogradova, G., Torshilova, A., Machs, E. (2022). Flower morphology and phylogenetic analysis of some Dioscorea species of the section Stenophora (Dioscoreaceae). Plant Syst. Evol. 308, 42. doi: 10.1007/s00606-022-01834-y
Viruel, J., Forest, F., Paun, O., Chase, M. W., Devey, D., Couto, R. S., et al. (2018). A nuclear Xdh phylogenetic analysis of yams (Dioscorea: dioscoreaceae) congruent with plastid trees reveals a new Neotropical lineage. Bot. J. Linn. Soc 187, 232–246. doi: 10.1093/botlinnean/boy013
Viruel, J., Segarra-Moragues, J. G., Raz, L., Forest, F., Wilkin, P., Sanmartín, I., et al. (2016). Late Cretaceous–early Eocene origin of yams (Dioscorea, dioscoreaceae) in the laurasian palaearctic and their subsequent oligocene–Miocene diversification. J. Biogeogr. 43, 750–762. doi: 10.1111/jbi.12678
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wilkin, P., Schols, P., Chase, M. W., Chayamarit, K., Furness, C. A., Huysmans, S., et al. (2005). A plastid gene phylogeny of the yam genus, Dioscorea: roots, fruits and Madagascar. Syst. Bot. 30, 736–749. doi: 10.1600/036364405775097879
Wu, L., Wang, B., Yang, J., Song, C., Wang, P., Chen, S., et al. (2016). The chloroplast genome sequence of an important medicinal plant Dioscorea nipponica. Mitochondrial DNA Part A 27, 2559–2560. doi: 10.3109/19401736.2015.1038803
Xia, Y., Luo, W., Yuan, S., Zheng, Y., Zeng, X. (2018). Microsatellite development from genome skimming and transcriptome sequencing: comparison of strategies and lessons from frog species. BMC Genom. 19, 1–13. doi: 10.1186/s12864-018-5329-y
Xia, E. H., Yao, Q. Y., Zhang, H. B., Jiang, J. J., Zhang, L. P., Gao, L. Z. (2016). CandiSSR: an efficient pipeline used for identifying candidate polymorphic SSRs based on multiple assembled sequences. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.01171
Xia, W., Zhang, B., Xing, D., Li, Y., Wu, W., Xiao, Y., et al. (2019). Development of high-resolution DNA barcodes for Dioscorea species discrimination and phylogenetic analysis. Ecol. Evol. 9, 10843–10853. doi: 10.1002/ece3.5605
Xie, Y., Wu, G., Tang, J., Luo, R., Patterson, J., Liu, S., et al. (2014). SOAPdenovo-trans: de novo transcriptome assembly with short RNA-seq reads. Bioinformatics 30, 1660–1666. doi: 10.1093/bioinformatics/btu077
Yang, Z., Ma, W., Yang, X., Wang, L., Zhao, T., Liang, L., et al. (2022). Plastome phylogenomics provide new perspective into the phylogeny and evolution of betulaceae (Fagales). BMC Plant Biol. 22, 611. doi: 10.1186/s12870-022-03991-1
Zhao, Z., Wang, X., Yu, Y., Yuan, S., Jiang, D., Zhang, Y., et al. (2018). Complete chloroplast genome sequences of Dioscorea: characterization, genomic resources, and phylogenetic analyses. PeerJ 6, e6032. doi: 10.7717/peerj.6032
Keywords: Dioscorea sect. Stenophora, plastome, comparative analyses, hotspots, phylogeny
Citation: Hu K, Sun X-Q, Chen M and Lu R-S (2023) Low-coverage whole genome sequencing of eleven species/subspecies in Dioscorea sect. Stenophora (Dioscoreaceae): comparative plastome analyses, molecular markers development and phylogenetic inference. Front. Plant Sci. 14:1196176. doi: 10.3389/fpls.2023.1196176
Received: 29 March 2023; Accepted: 26 April 2023;
Published: 06 June 2023.
Edited by:
Yunpeng Cao, CAS, ChinaReviewed by:
Aiping Song, Nanjing Agricultural University, ChinaZhitao Niu, Nanjing Normal University, China
Li Feng, Xi’an Jiaotong University, China
Copyright © 2023 Hu, Sun, Chen and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui-Sen Lu, bHVyc0BjbmJnLm5ldA==