 Yunli Wang1,2
Yunli Wang1,2 Guichao Wang
Guichao Wang Shuping Qu
Shuping Qu- 1Key Laboratory of Biology and Genetic Improvement of Horticultural Crops (Northeast Region), Ministry of Agriculture and Rural Affairs/Northeast Agricultural University, Harbin, China
- 2College of Horticulture and Landscape Architecture, Northeast Agricultural University, Harbin, China
Trichomes provide an excellent model for studying cell differentiation and proliferation. The aboveground tissues of plants with long dense trichomes (LDTs) can cause skin itching in people working in a zucchini field, in which management, pollination, and fruit harvesting are difficult. In this study, an F2 population was constructed with the LDT inbred line “16” and the sparse micro trichome (SMT) inbred line “63” for QTL analysis of type I and II trichome density. Two QTLs were identified on chromosomes 3 and 15 using the QTL-seq method. Additionally, 191 InDel markers were developed on 20 chromosomes, a genetic map was constructed for QTL mapping, and three QTLs were identified on chromosomes 3, 6, and 15. Two QTLs, CpTD3.1 and CpTD15.1, were identified in both QTL-seq and genetic map-based QTL analyses, and CpTD15.1 was the major-effect QTL. The stability of CpTD3.1 and CpTD15.1 was confirmed using data from F2 plants under different environmental conditions. The major-effect QTL CpTD15.1 was located between markers chr15-4991349 and chr15-5766791, with a physical distance of 775.44 kb, and explained 12.71%–29.37% of the phenotypic variation observed in the three environments. CpTD3.1 was located between markers chr3-218350 and chr3-2891236, in a region with a physical distance of 2,672.89 kb, and explained 5.00%–10.64% of the phenotypic variation observed in the three environments. The functional annotations of the genes within the CpTD15.1 region were predicted, and five genes encoding transcription factors regulating trichome development were selected. Cp4.1LG15g04400 encoded zinc finger protein (ZFP) and harbored nonsynonymous SNPs in the conserved ring finger domain between the two parental lines. There were significant differences in Cp4.1LG15g04400 expression between “16” and “63”, and a similar pattern was found between germplasm resources of LDT lines and SMT lines. It was presumed that Cp4.1LG15g04400 might regulate trichome density in zucchini. These results lay a foundation for better understanding the density of multicellular nonglandular trichomes and the regulatory mechanism of trichome density in zucchini.
Introduction
Cucurbita pepo L. is one of the cultivated Cucurbita spp. and has the most abundant germplasm resources among Cucurbita crops. Zucchini not only shows strong stress resistance and adaptability, but also had high nutritional, medicinal, and ornamental value. Therefore, zucchini is grown widely from tropical to temperate areas. The surfaces of the aboveground tissues of most zucchini germplasm resources are covered with sharp dense trichomes. During the field production of zucchini, the sharp dense trichomes of zucchini plants can cause skin itchiness in people managing or pollinating plants or harvesting their fruit. Furthermore, the dense trichomes of fruit stems easily scratch young zucchini fruits, which is unfavorable for fruit transport. Therefore, it is necessary to identify and clone genes controlling trichome density trait in zucchini.
Trichomes are the first functional barrier in contact with the outside world and play a significant role in plant development and the biotic and abiotic adaptations, and some of them have important commercial value (Amme et al., 2005; Emiko et al., 2010; An et al., 2012). Trichomes are widely distributed on the surfaces of the aboveground parts of land plants, such as leaves, petioles, stems, pericarps, fruits, tendrils, and pedicels (Liu et al., 2016). Different arrangements and numbers of trichome cells develop into different morphologies, and according to trichome morphology, plant trichomes are divided into unicellular or multicellular trichomes (Schnittger et al., 1999). Unicellular trichomes are nonglandular, and some have branches, while multicellular trichomes are divided into glandular or nonglandular trichomes (Schnittger et al., 1999). Common multicellular trichomes had complex structures and consist of three components: head cells, stalk cells, and basal cells (Amme et al., 2005; Chang et al., 2016).
Trichomes in Arabidopsis are typically unicellular branched structures. Numerous genes have been identified as key regulators of trichome initiation. The regulatory network affecting trichome initiation and development was constructed, and approximately 40 genes were involved. In Arabidopsis, the R2R3 MYB transcription factor GLABRA 1 (GL1) (Kirik et al., 2004), the basic helix-loop-helix (bHLH) transcription factor GLABRA 3/ENHANCER OF GLABRA 3 (GL3/EGL3), and the pleiotropic WD40 repeat protein TRANSPARENT TESTA GLABRA 1 (TTG1) (Walker et al., 1999; Zhao et al., 2008) formed the MYB-bHLH-WDR (MBW) complex, which was the key core of the trichome initiation pathway. The MBW complex activates trichome formation by enhancing the expression of downstream homeodomain-leucine zipper (HD-ZIP) IV genes, GL2 and EGL2 (Payne et al., 2000; Ishida et al., 2007). R3-type MYB transcription factors CAPRICE (CPC), ENHANCER OF TRY AND CPC 1 (ETC1), ETC2, ETC3, TRIPTYCHON (TRY), TRICHOMELESS 1 (TCL1), and TCL2 act as negative regulators of trichome formation by competing with GL1 and interacting with GL3/EGL3 to form an inactive complex (Schnittger et al., 1999; Kirik et al., 2004; Wang et al., 2007; Wang et al., 2008; Wester et al., 2009). bHLH transcription factor MYC1 positively regulates trichome number (Zhao et al., 2012). C2H2 type zinc-finger proteins (ZFPs), such as glabrous inflorescence stems (GIS), GIS2, GIS3, ZFP5, ZFP6, and ZFP8, play key roles in the initiation and formation of leaf trichomes (Gan et al., 2007; Zhou et al., 2012; Zhou et al., 2013; Sun et al., 2015; Liu et al., 2017; Kim et al., 2018). The AP2/ERF family transcription factor TARGET OF EARLY ACTIVATION TAGGED 1 (TOE1) regulates trichome initiation of the main-stem inflorescence (Liu et al., 2023). SET Domain Group 26 (SDG26), a histone lysine methyltransferase, affects trichome growth and development (Zeng et al., 2023).
Tomato trichomes have unbranched multicellular structures and are divided into eight types. Types I, IV, VI, and VII are glandular types with secretory capacity, whereas types II, III, V, and VIII are nonglandular types (Amme et al., 2005). The functions of an HD-ZIP IV family gene, Woolly (Wo), are involved in the formation and development of type I and IV trichomes (Yang et al., 2011). A B-type cyclin gene, SlCycB2, participates in the development of type I, III, and V trichomes (Gao et al., 2017). SlMYC1, a bHLH transcription factor, regulates not only the development of type VI trichomes but also mono- and sesquiterpene biosynthesis in tomato (Xu et al., 2018). An R2R3-MYB family gene, SlMIXTA, functions as a positive regulator of tomato trichome formation (Ying et al., 2020). SlHair (SlH), a C2H2 zinc finger protein (ZFP), positively regulates the number and length of type I trichomes in tomato (Chun et al., 2021).
Cucumber fruit is covered with spines (a special type of trichome) and tubercules, which are important agronomic traits that affect commercial value. Fruit spines have branchless multicellular structures. Fruit spines are divided into eight types: types I and VI are glandular and types II, III, IV, V, VII, and VIII are nonglandular (Xue et al., 2019). The HD-ZIP I transcription factor genes Glabrous 1/Tny branched hair/Micro-trichome (CsGL1/TBH/MICT) are key allelic genes involved in spine development and affect spine distribution in cucumber (Chen et al., 2014; Li et al., 2015; Zhao et al., 2015). The HD-ZIP IV transcription factor genes Glabrous 3/Trichome-less/Few spine 1 (CsGL3/TRIL/FS1) are key allelic genes for the positive regulation of cucumber fruit spines’ differentiation, initiation, and development (Liu et al., 2016; Wang et al., 2016; Zhang et al., 2016; Zhang et al., 2019). The C2H2-type zinc finger gene tuberculate (Tu) is a key gene that controls the formation of tuberculate fruit (Yang et al., 2014). The WD-repeat homologues CsTTG1 and MIXTA-LIKE transcription factor CsMYB6 are key factors involved in regulating the differentiation of fruit spines and the tubercule formation (Chen et al., 2014; Chen et al., 2016; Zhao et al., 2020). bHLH transcription factors, such as CsMYC2, CsMYC4, CsMYC5, CsMYC6, CsMYC7, and CsMYC8, play an important role in glandular trichome development (Feng et al., 2023). In addition to transcription factor genes, other genes are also involved in cucumber spine development. NUMEROUS SPINES (NS) acts as a regulatory factor in spine development by modulating the auxin signaling pathway (Xie et al., 2018). The tender spines (TS) gene encodes a C-type lectin receptor-like tyrosine-protein kinase and plays an important role in the formation of cucumber spines (Guo et al., 2018).
Gene mapping studies and analyses of regulatory mechanisms related to trichome/spine initiation and development are mainly conducted in model plants, such as Arabidopsis, tomato, and cucumber. In a previous report, zucchini trichomes were classified into seven types according to the morphology: type I, II, III, and VII trichomes were nonglandular, and type IV, V, and VI trichomes were glandular. Among them, type I and II trichomes were the longest. The zucchini germplasm resources were divided into a long dense trichome (LDT) group and a sparse micro trichome (SMT) group according to the presence of type I and II trichomes. An F2 population derived by crossing the LDT line “16” and SMT line “63” was constructed, and the trichome density exhibited characteristic quantitative inheritance (Lin et al., 2023). Although there have been reports of trichome characteristics, classification, and inheritance, the key genes and regulatory mechanisms related to zucchini trichome development remain to be reported. In our study, quantitative trait loci (QTLs) related to the trichome density were identified using the QTL sequencing (QTL-seq) method and genetic map-based QTL analysis. Additional F2 populations grown under different environmental conditions were used to validate the stability of QTLs, and the candidate genes in the major-effect QTL region associated with trichome development were identified. A C3DHC3-type ZFP gene might regulate trichome density in zucchini. The results lay a foundation for better understanding of multicellular trichome development and improving trichome-related breeding in zucchini.
Materials and methods
Plant material
The high-generation inbred line “16” showed typical LDT, and type I, II, III, and IV trichomes were distributed on the surfaces of these plants. The high-generation inbred line “63” had SMT, and type III, IV, and VII trichomes were distributed on the surfaces of these plants. For the QTL analysis of trichome density of type I and type II, the F2 population was derived from a cross between “16” and “63”. A total of 536 and 238 F2 individuals were grown in the spring (S) of 2019 and 2021, respectively. A total of 886 F2 individuals were grown in the fall (F) of 2020. Ten “16”, “63”, and F1 plants were also investigated under each set of environmental conditions. To investigate the allelic diversity of predicted genes, germplasm resources of five LDT lines (20-23, 21-1, 21-3, 21-6, and 22-4) and five SMT lines (19-7, 19-28, 20-17, 21-9, and 21-15) (15 plants of each line) were planted. Seeds were soaked in water at 55°C for 8 h and then germinated at 28°C. All individuals were planted in a greenhouse at Xiangyang Experimental Agricultural Station of Northeast Agricultural University, Harbin, China (N45°77′, E126°92′). All individuals were subjected to irrigation, hand weeding, and hand pollination.
Classification of zucchini trichomes
During the growth of the F2 population to the flowering stage, the genetic regularity of trichome density was analyzed. Among the aboveground tissues of zucchini, petiole trichomes were the easiest to observe and distinguish. According to the number of trichomes in a 1-mm2 area and the mean distance of type I and II trichomes of petioles on the second young leaf, the trichome density phenotypes of individual F2 plants were categorized into five classes (Lin et al., 2023). The trichomes of line “16” belonged to class 5, the trichomes of line “63” belonged to class 1, and the trichomes of F1 belonged to class 3.
QTL analysis of trichome density with QTL-seq
Young leaves from the parents and F1 and F2 populations were collected for DNA extraction using the cetyltrimethylammonium bromide (CTAB) method (Murray and Thompson, 1980). For QTL-seq, a dense trichome pool (D-pool) and a sparse trichome pool (S-pool) were constructed by mixing equal amounts of DNA from 30 dense trichome individuals (class 5) and 30 sparse trichome individuals (class 1) from F2. A parental dense trichome pool and a parental sparse trichome pool were constructed by mixing an equal amount of DNA from 20 “16” plants and 20 “63” plants. A paired-end sequencing library with DNA fragments of approximately 350 bp DNA was constructed from the four pools on an Illumina Hi-Seq 2000 sequencer using a commercial service at BioMarker (Peking, China). Reads were mapped to the reference genome of zucchini (http://cucurbitgenomics.org/organism/14, Javier et al., 2018). The output of paired-end alignment files was compared with that of the BWA software (Li and Durbin, 2009) to filter clean reads. SNPs and InDels between “16” and “63”, and between the D-pool and S-pool were identified using the variant analysis software GATK (McKenna et al., 2010). To identify candidate regions associated with trichome density of type I and type II, the SNP/InDel index of Euclidean distance (ED) and the Δ(SNP/InDel index) were calculated for all positions (Abe et al., 2012; Hill et al., 2013). According to the null hypothesis, 99% confidence intervals were selected to identify the candidate regions harboring QTLs for trichome density.
Genetic map-based analysis of trichome density QTLs
Ninety-three individuals were randomly selected from the F2 population in Spring 2021 for genetic map construction and QTL analysis of trichome density. InDel markers on 20 chromosomes were developed based on InDels between the two parental lines using Primer Premier 5.0 (Table S1). For InDel markers, PCR was carried out using 10-μl samples containing ~40 ng of genomic DNA, 0.5 μM of each primer, 200 μM dNTPs, 1× reaction buffer, and 0.5 U Taq DNA polymerase (Aidlab Biotechnologies, Beijing, China). PCR amplification was performed with the following PCR program: 94°C for 4 min; 35 cycles of 94°C for 30 s, 57°C for 30 s, and 72°C for 30 s; and 72°C for 10 min. Products were separated on an 8% polyacrylamide gel by electrophoresis (Wang et al., 2020; Wang et al., 2021). After electrophoresis at 220 V for 1.5 h, the gel was stained in 0.2% AgNO3 solution and revealed the silver-stained DNA bands (Ding et al., 2021).
A genetic map was constructed with InDel polymorphism markers from the 20 chromosomes using JoinMap 4.0, and recombination values were converted into map distances in centiMorgans (cM) using the Kosambi mapping function. A, B, and H represent homozygous “16”, homozygous “63”, and heterozygous genotypes, respectively. Spearman’s correlation coefficients for each chromosome were calculated using the Statistical Analysis System (SAS) program (ND Times, Peking, China). The marker information in the genetic map and trichome phenotype data were combined for linkage analysis using RStudio (R/qtl) software with the multiple QTL mapping (MQM) method with a walking speed of 1 cM (Weng et al., 2015). The logarithm of odds (LOD) score for a significant QTL was estimated using 1,000 permutations and a p-value of 0.05. QTLs that could explain more than 15% of the observed phenotypic variation (PVE) were considered major-effect QTLs. The QTLs were named according to the trait name, chromosome number, and QTL number.
Stability analysis of QTLs
To confirm the QTLs for trichome density detected by QTL-seq and genetic map-based QTL mapping, we conducted QTL analysis of 536 F2 individuals from Spring 2019, 886 individuals from Fall 2020, and 238 individuals from Spring 2021 using InDel markers. InDel markers between the two parental lines on the QTL chromosomes were selected (Table S2). The marker information and trichome phenotype data were combined for linkage analysis using RStudio (R/qtl) software via the composite interval mapping (CIM) method with a walking speed of 1 cM (Churchill and Doerge, 1994). The LOD threshold for significant QTL identification was established separately with 1,000 permutation tests (p = 0.05) and ranged from 2.88 to 3 for each environment; therefore, an LOD score of 3 was used for QTL detection.
Sequencing and predictive functions of predicted genes associated with the major-effect QTL
In the major-effect QTL region, the gene IDs, structures, and predicted functions were obtained using online software according to the Cucurbit Genomics Database (Javier et al., 2018). NCBI online software (https://www.ncbi.nlm.nih.gov/) was also used for gene structure identification, conserved domain analysis, and gene function annotation. The coding sequences of full-length candidate genes were sequenced, and primer pairs were designed using Primer 5.0 software.
RNA isolation and gene expression
The trichomes development of the cotyledon base and leaf primordium was observed, and trichomes from the third stage was the key stage for glandular and nonglandular trichome development (Zhang et al., 2021; Dong et al., 2022). Trichomes from the third stage (the key stage for glandular and nonglandular trichome development) were collected from the base of cotyledon at 5 days after germination and leaf primordium at 7 days after germination on “16” and “63” for trichome analysis at the key developmental stage. Leaf primordium of five LDT lines and five SMT lines were also collected to analyze expression differences between LDT and SMT germplasm resources. RNA was extracted using TRIzol reagent (Invitrogen, USA). Dried RNA samples were dissolved in H2O and treated with diethylpyrocarbonate. First-strand cDNA was prepared according to the PrimeScript RT Reagent Kit with the gDNA Eraser (TaKaRa, Kyoto, Japan) protocol. Primer pairs of candidate resistance genes were designed using Primer Premier 5.0 (Table S3). Taq SYBR Green qPCR Premix (Yugong Biolabs, Inc., CN) was used to perform qRT-PCR. The program used in this assay was as follows: 96°C for 1 min, 95°C for 15 s, 58°C for 15 s, and 72°C for 45 s, with 35 cycles. The Actin gene was used as the internal reference. Three technical replicates were performed for each sample, and relative expression levels were quantified using the 2−ΔΔCT method (Livak and Schmittgen, 2001).
Results
QTL analysis of trichome density with QTL-seq
The zucchini trichomes were observed to be unbranched multicellular trichomes and classified into seven types according to their trichome structures. Among these types, type I and II trichomes were long and pointy. In “16”, type I and II trichomes were densely arranged on the surface of aboveground tissues, such as leaves, petioles, tendrils, flower buds, petals, fruits, fruit stalks, and pedicels. In “63”, type I and II trichomes were absent on the surface of aboveground tissues (Figure 1). In previous studies in our lab, an F2 population derived by crossing “16” and “63” was constructed, and the density of type I and type II trichomes exhibited characteristic quantitative inheritance.

Figure 1 Observation of the trichomes on the surface of tissues. The trichomes on the surface of young leaves (A), flower buds and pedicels (B), ovary and fruit stalks (C), petals and sepals (D), and tendrils (E) in “16”. The trichomes on the surface of young leaves (F), flower buds and pedicels (G), ovary and fruit stalks (H), petals and sepals (I), and tendrils (J) in “16”. Scale bars represent 1 cm.
DNA pools of “16” and “63”, the D-pool, and the S-pool were sequenced to obtain the candidate regions for trichome density. A total of 47.29 and 38.48 million clean reads with high average read depths (28- and 23-) were obtained from the D- and S-pools, respectively, and the corresponding Q30 values reached 92.42% and 91.49%. The percentages of clean reads from the D- and S-pools that were mapped to the reference genome of zucchini were 98.07% and 97.96%, respectively (Table S4). These results indicated that the sequencing results were reliable for QTL-seq. A total of 40,404 high-quality InDels and 80,789 high-quality SNPs were obtained on all 20 chromosomes in zucchini.
The ED algorithm was employed to identify significantly differential SNPs between the D-pool and S-pool based on sequencing data, to predict candidate regions of trichome density. A 2.70-Mb region and a 5.00-Mb region were identified on chromosomes 3 and 15, respectively (Figure 2A). The ED algorithm was employed to identify significantly differential InDels between the D-pool and S-pool, and a 3.00-Mb region and a 4.98-Mb region were identified on chromosomes 3 and 15, respectively (Figure 2B). Δ(InDel-index) and Δ(SNP-index) were calculated and plotted by comparing the InDel-index and SNP-index results of the D- and S-pools at the corresponding genomic positions. A 1.11-Mb region of chromosomes 15 was identified based on Δ(InDel-index) values (Figure 2C), and a 1.33-Mb region of chromosomes 15 was identified based on Δ(SNP-index) values higher than the threshold value (Figure 2D). To obtain all the possible regions associated with trichome density, the convergence of these association regions was considered. The candidate region from 0 to 3.00 Mb on chromosome 3 and the candidate region from 3.65 Mb to 8.65 Mb on chromosome 15 were obtained and found to contain a total of 1,116 genes (Table S5).
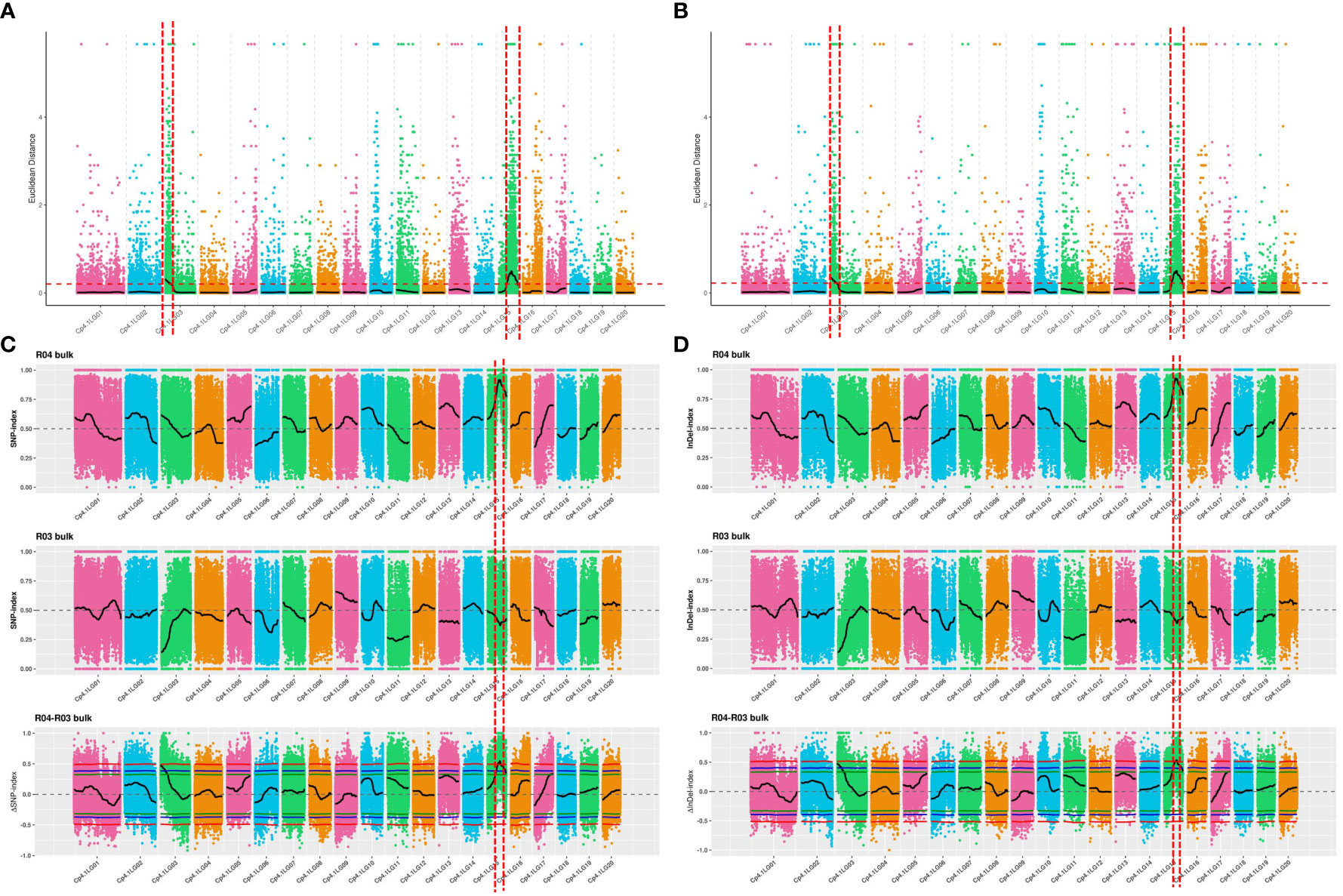
Figure 2 QTL mapping of trichome density by the strategy of QTL-seq combined with linkage analysis. The candidate region from 0 to 3.00 Mb on chromosome 3 and the candidate region from 3.65 Mb to 8.65 Mb on chromosome 15 were obtained. (A) Distribution of ED-based linkage value with SNPs on chromosomes. (B) Distribution of ED-based linkage value with InDels on chromosomes. (C) Distribution of SNP index correlation values on chromosomes. (D) Distribution of InDel index correlation values on chromosomes. The x-axes indicate chromosomes. In (A, B), Median + 3 SD of the fitted value of all sites were defined as linkage threshold and shown by red lines. In (C, D), the top panel is a distribution map of the SNP index (InDel index) values of the D-pool, the middle panel is a distribution map of the SNP index (InDel index) values of the S-pool, and the bottom panel is a distribution map of the ΔSNP index (ΔInDel index) values. The red line represents the confidence threshold of 0.99, the blue line represents a confidence threshold of 0.95, and the green line represents a confidence threshold of 0.90.
QTL analysis of trichome density with genetic mapping
InDel markers on 20 chromosomes were developed based on the InDels identified between the two parental lines, “16” and “63”, for genetic map construction. Finally, a total of 191 polymorphic InDel markers were selected, which were uniformly distributed on 20 chromosomes. The genotypes of the 191 InDel markers in F2 individuals are presented in Table S6. The genetic map spanned a total of 2,765.65 cM, with an average distance of 14.48 cM between adjacent markers (Table S7). The Spearman correlation coefficient of each chromosome ranged from 0.98 to 1 (Table S7), which indicated that the order of the InDel markers was highly consistent with their physical locations in zucchini scaffolds. Therefore, this zucchini genetic map was suitable for QTL mapping.
According to the genotype and phenotype of the F2 population, QTLs for trichome density were identified. Information on the QTLs was provided in Table 1 and Figure 3. The LOD threshold was 3.43, and three QTLs were detected on chromosomes 3, 6, and 15. The major-effect QTL CpTD15.1 was situated in a 1.54-Mb region between two markers, chr15-4229645 and chr15-5766791, and the peak marker was chr15-5766791. The LOD score of CpTD15.1 was 15.38, explaining 36.74% of the observed phenotypic variation. The minor-effect QTL CpTD3.1 was situated in a 3.16-Mb region between two markers, chr3-219017 and chr3-3381239, and the peak marker was chr3-1732969. The LOD score of CpTD3.1 was 3.40, explaining 15% of the phenotypic variation. The minor-effect QTL CpTD6.1 was situated in a 5.64-Mb region between two markers, chr6-2570712 and chr6-8212486, and the peak marker was chr6-5896890. The LOD score of CpTD6.1 was 3.46, explaining 11.29% of the phenotypic variation.
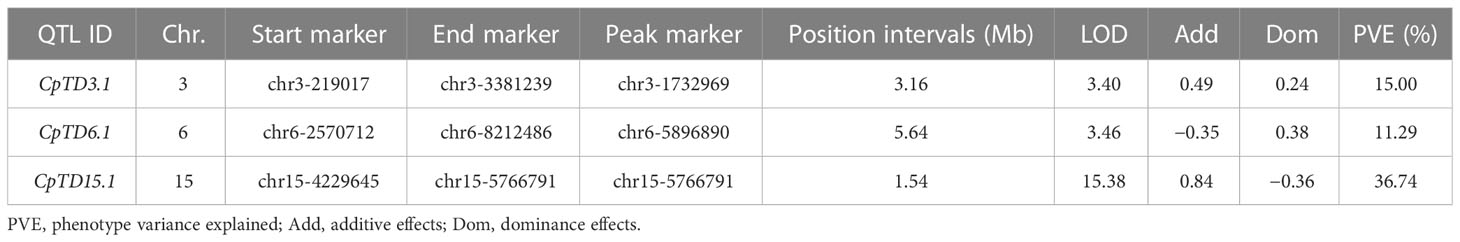
Table 1 Mapping of QTLs for trichome density of type I and type II with genetic mapping.
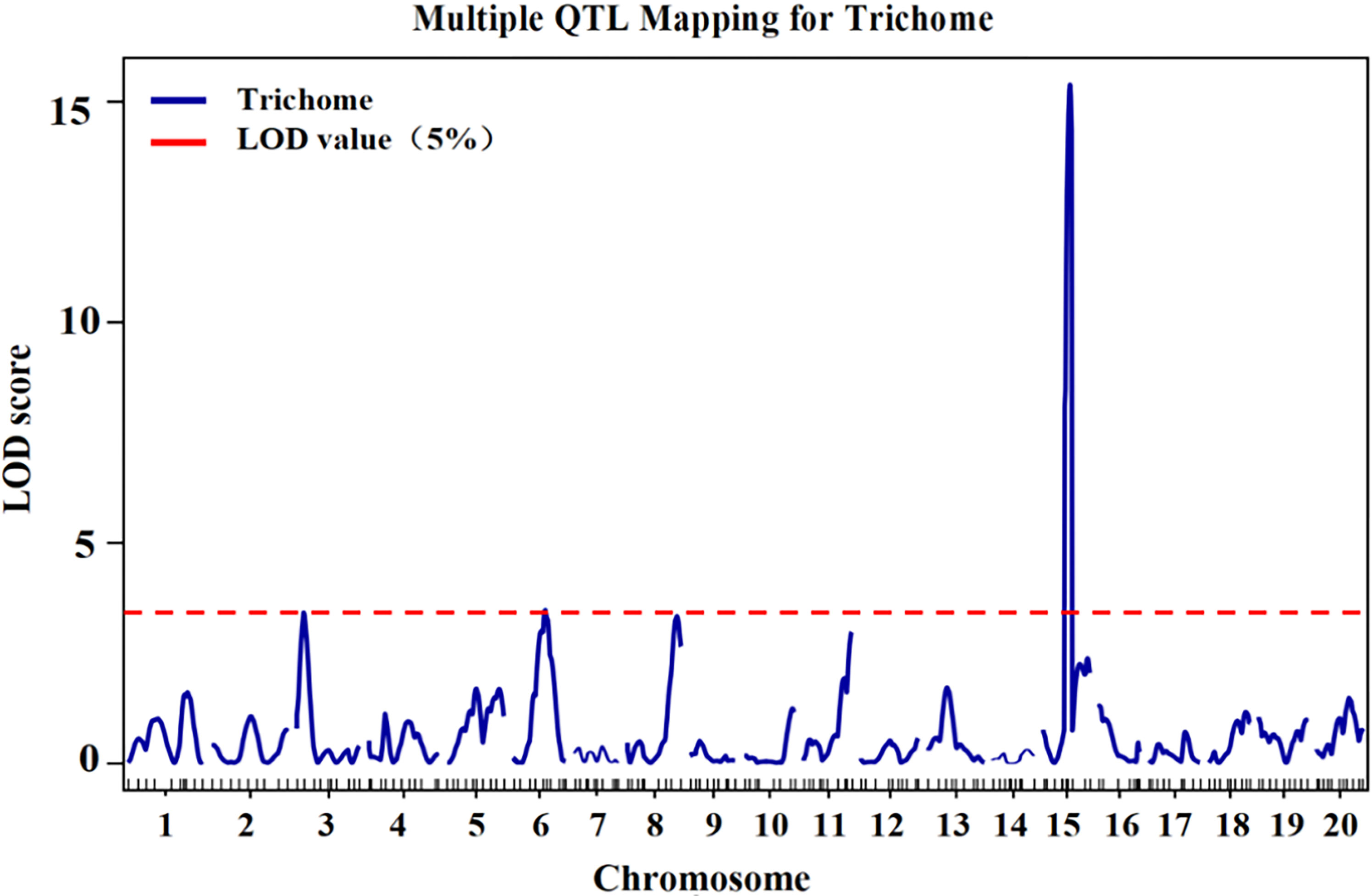
Figure 3 QTL mapping of trichome density by the genetic map in zucchini. According to the genotype and phenotype of the F2 population, three QTLs for trichome density were identified on chromosomes 3, 6, and 15. The x-axes indicate chromosomes, and the y-axes indicate logarithm of odds (LOD) score. The red horizontal lines indicate the LOD threshold value.
Stability analysis of QTLs with InDel markers
Candidate regions associated with trichome density were detected on chromosomes 3 and 15 by both QTL analysis methods. Therefore, polymorphic InDel markers on chromosomes 3 and 15 were developed to evaluate the stability of QTLs for trichome density. Plants of the F2 population grown under different environmental conditions were used for QTL mapping in R/qtl, and the genotype and phenotype of the F2 population are provided in Table S8. QTL starting markers, ending markers, peak markers, position intervals, LODs, PVEs, additive values, and dominance values are shown in Table 2 and Figure 4. In 2019S, two QTLs, CpTD3.1 and CpTD15.1, were identified. CpTD3.1 was delimited by two markers, chr3-764774 and chr3-1732969, with a peak at chr3-1379216. CpTD3.1 explained 10.64% of the observed phenotypic variation, with an LOD value of 12.10. CpTD15.1 was delimited by two markers, chr15-5110786 and chr15-5766791, with a peak at chr15-5311576. CpTD15.1 explained 12.71% of the phenotypic variation, with an LOD value of 11.93. In 2020F, two QTLs, CpTD3.1 and CpTD15.1, were identified. CpTD3.1 was delimited to the same region as CpTD3.1 in 2019S (by two markers, chr3-218350 and chr3-1379216), with the same peak marker as CpTD3.1 in 2019S. CpTD3.1 explained 5.2% of the phenotypic variation, with an LOD value of 19.55. CpTD15.1 was delimited to the same region as CpTD15.1 in 2019S (by two markers, chr15-4991349 and chr15-5461456), with a peak at chr15-5224151. CpTD15.1 explained 29.37% of the phenotypic variation, with an LOD value of 76.37. In 2021S, two QTLs, CpTD3.1 and CpTD15.1, were identified. CpTD3.1 was delimited to the same region as CpTD3.1 in 2019S and 2020F (by two markers, chr3-764774 and chr3-2891236), with the same peak marker as CpTD3.1 in 2019S and 2020F. CpTD3.1 explained 5% of the phenotypic variation, with an LOD value of 3.43. CpTD15.1 was delimited to the same region as CpTD15.1 in 2019S and 2020F (by two markers, chr15-5224151 and chr15-5619256), with the peak marker of CpTD15.1 in 2019S. CpTD15.1 explained 25.22% of the phenotypic variation, with an LOD value of 16.12.
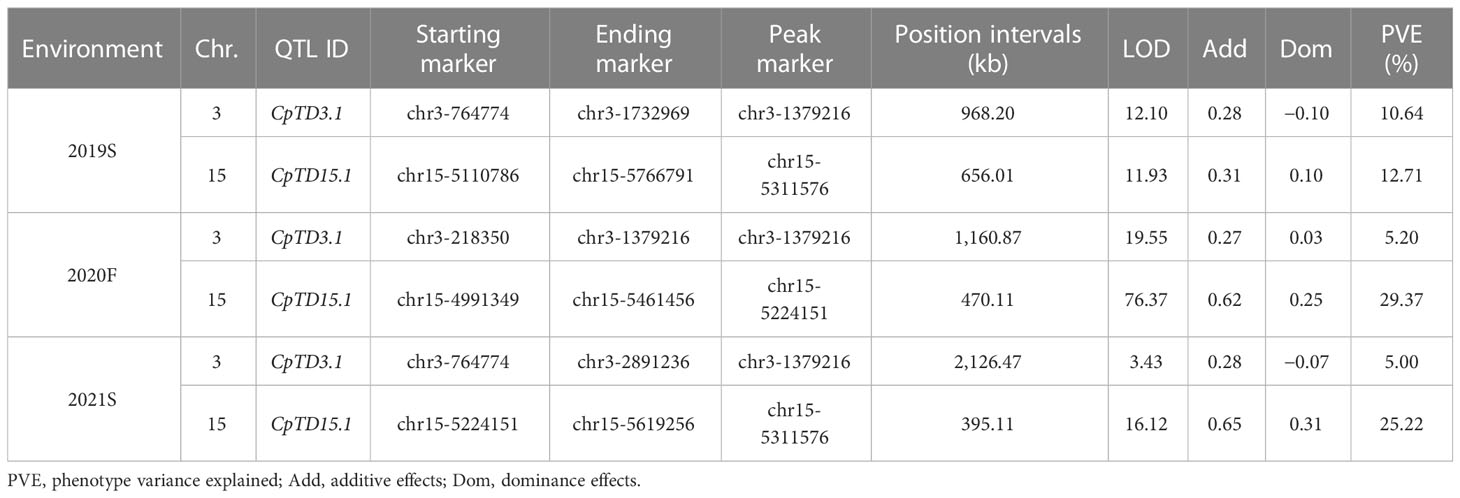
Table 2 QTL analysis of CpTD15.1 and CpTD3.1 in different environmental conditions.
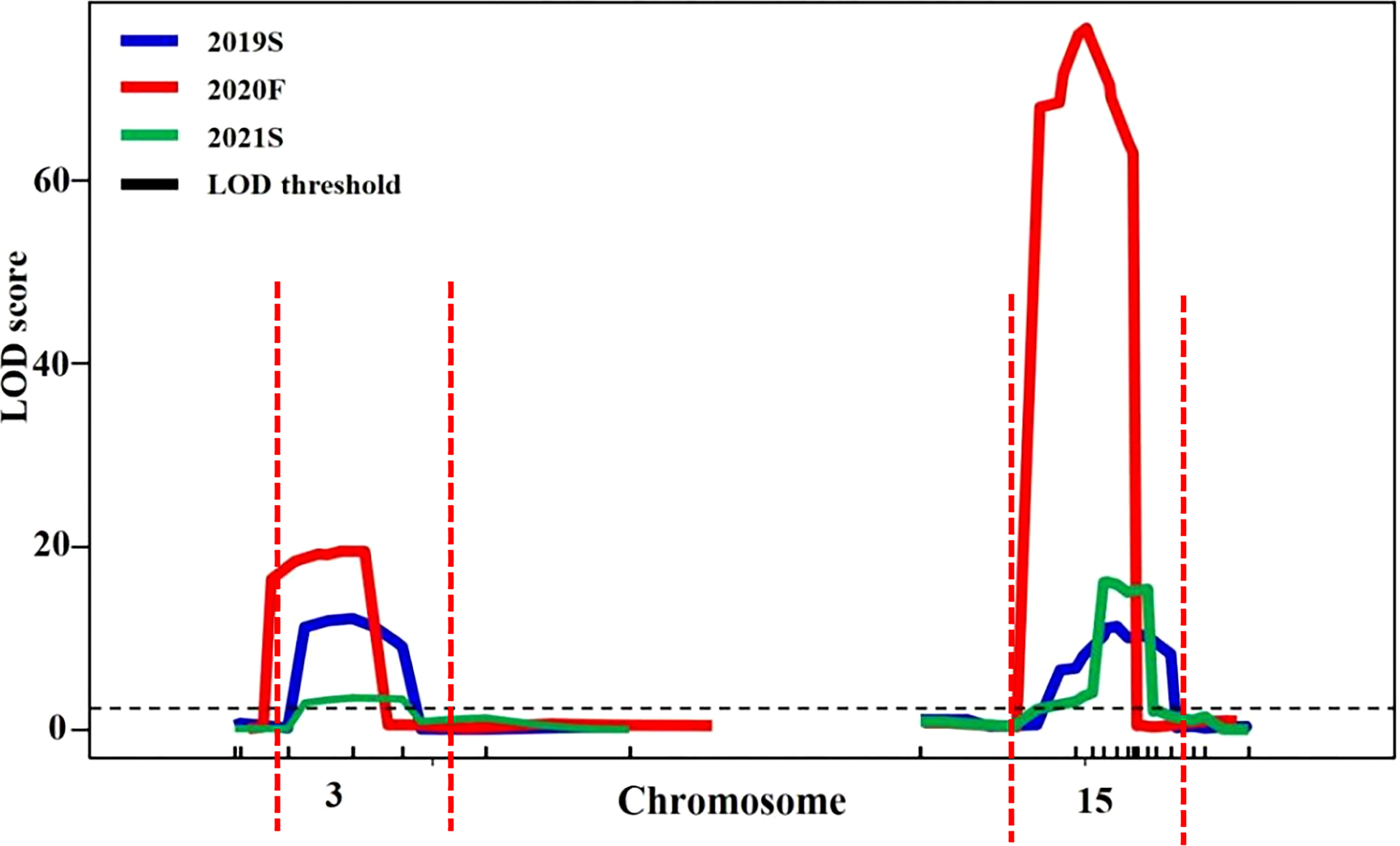
Figure 4 Stability analysis of CpTD3.1 and CpTD15.1 using F2 in three environments. The black horizontal line indicates the LOD = 3.0. According to data of three environments, the CpTD3.1 locus was located in a 2,672.89-kb region by markers chr3-218350 and chr3-2891236, and the CpTD15.1 locus was located in a 775.44-kb region by markers chr15-4991349 and chr15-5766791.
Regarding the intersection of associated regions in different environments, the CpTD3.1 locus was located in a 2,672.89-kb region based on the markers chr3-218350 and chr3-2891236, and the CpTD15.1 locus was located in a 775.44-kb region based on the markers chr15-4991349 and chr15-5766791. CpTD15.1 was the major-effect locus, and the region harbored candidate genes for trichome density. A genetic interaction analysis between CpTD3.1 and CpTD15.1 was performed and is illustrated in Figure 5. Typical parallels were observed across all genotypes indicating that there was no strong epistatic interaction between CpTD3.1 and CpTD15.1. There was a simple additive gene effect between CpTD3.1 and CpTD15.1.
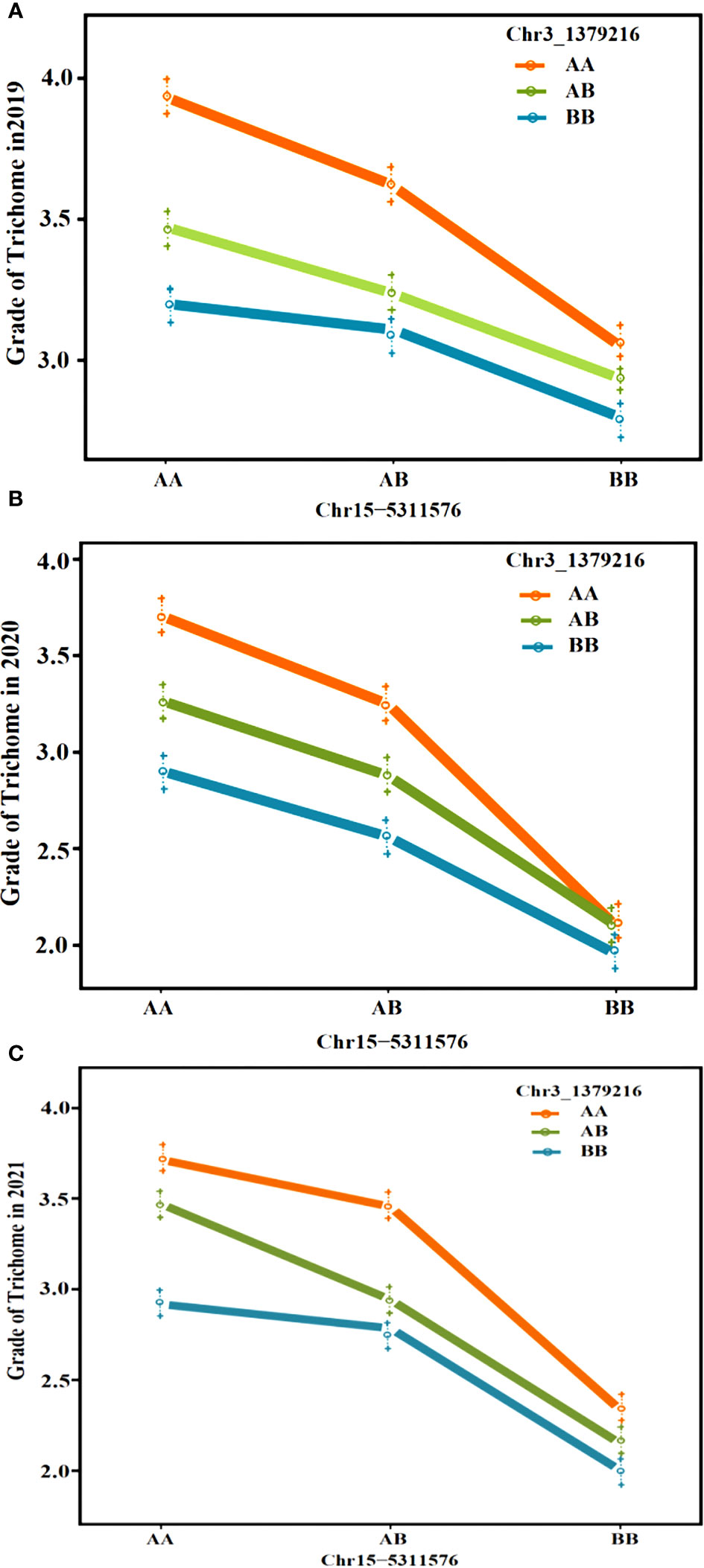
Figure 5 The genetic interaction analysis between CpTD3.1 and CpTD15.1. There was a simple additive gene effect between CpTD3.1 and CpTD15.1. (A) The genetic interaction analysis of two QTLs in 2019S. (B) The genetic interaction analysis of two QTLs in 2020F. (C) The genetic interaction analysis of two QTLs in 2021S.
Candidate genes for trichome density
According to the Cucurbit Genomics Database, a total of 106 coding genes were located in the 775.44-kb region. To select the candidate genes associated with trichome density, gene structures and predicted functions in the region were analyzed. Five genes encoding GATA transcription factors, zinc finger proteins, and basic helix-loop helix (BHLH) transcription factors had the functions related to trichome development (Table 3). Three genes (Cp4.1LG15g04030, Cp4.1LG15g04400, and Cp4.1LG15g04350) showed nonsynonymous SNP/InDels in the CDS regions between “16” and “63” (Figure S1). Two genes (Cp4.1LG15g04040 and Cp4.1LG15g04400) showed SNPs/InDels in the promoter elements between “16” and “63”, and one gene (Cp4.1LG15g03820) showed no SNPs/InDels in the CDS region or the promoter element. Among them, only Cp4.1LG15g04400, encoded ZFP gene, showed nonsynonymous SNPs in the conserved domain of the C3DHC3-type ring finger.
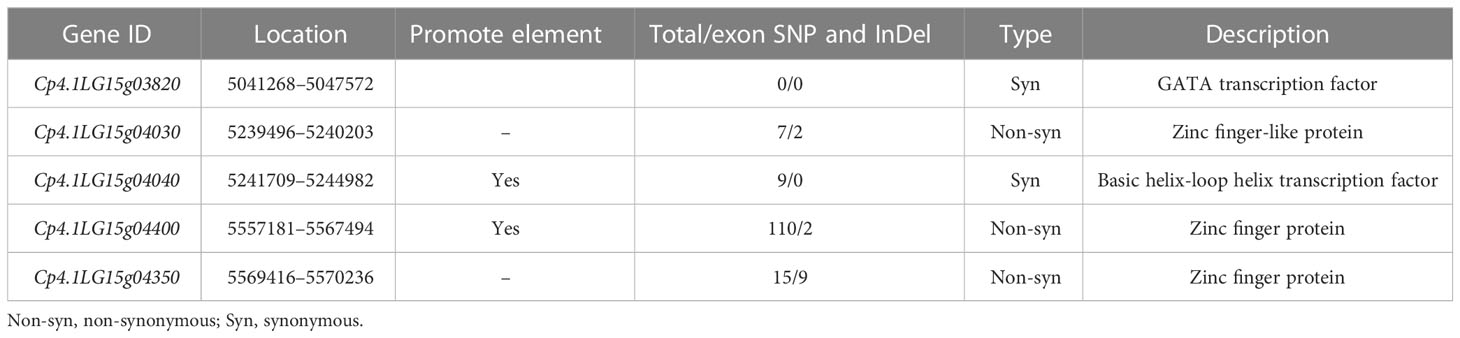
Table 3 Predicted genes in the major-effect QTL CpTD15.1.
Trichomes from the third stage (the key stage for glandular and nonglandular trichome development) were collected from the base of cotyledon at 5 days after germination and leaf primordium at 7 days after germination on “16” and “63” plants. Expression analysis of all five genes was performed. The results showed that the expression levels of Cp4.1LG15g04030 and Cp4.1LG15g04350 in the SMT line “63” were significantly lower than those in the LDT line “16”, and the expression levels of Cp4.1LG15g04040 and Cp4.1LG15g04400 in the SMT line “63” were significantly higher than those in the LDT line “16” (Figure 6). Zucchini germplasm resources consisting of five LDT lines and five SMT lines were selected, and leaf primordium of each line was collected for gene expression analysis. Only the Cp4.1LG15g04400 gene showed significant expression differences between the LDT lines and SMT lines (Figure S2), and there was no significant difference in the expression of other four genes between the LDT lines and SMT lines. These results indicated that Cp4.1LG15g04400 might be a good candidate gene for trichome density.
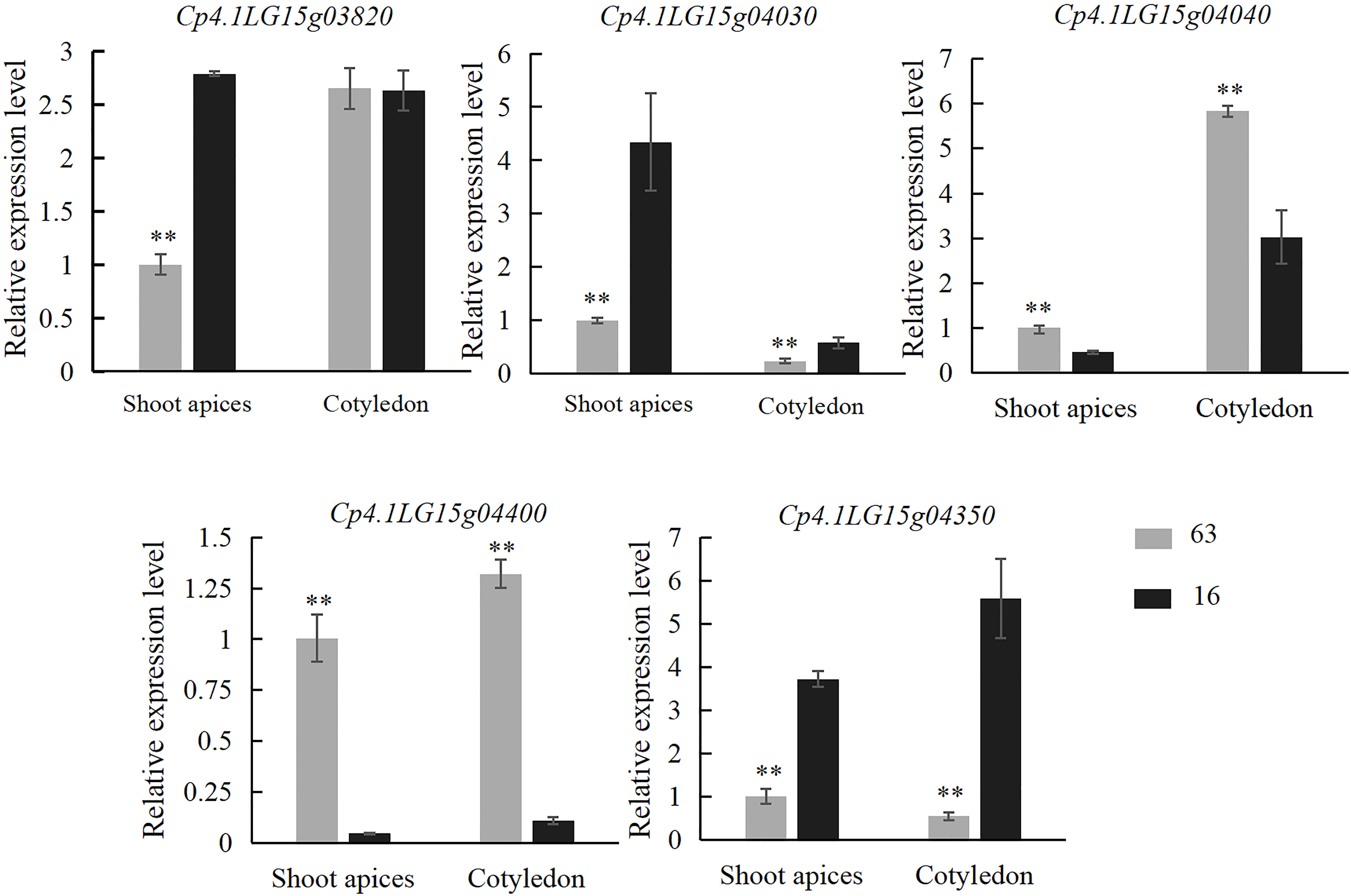
Figure 6 Relative expression levels of five candidate genes (Cp4.1LG15g03820, Cp4.1LG15g04030, Cp4.1LG15g04040, Cp4.1LG15g04400, and Cp4.1LG15g04350) in “16” and “63”. The relative expression levels of five genes were quantified using the 2−ΔΔCT method. The expression level of the respective genes in leaf primordium of “16” was set to a value of 1 and used as a reference, respectively. Each was repeated three times. ** indicates an extremely significant difference, p < 0.01.
Discussion
Trichomes are hairy structures covering aboveground tissues in terrestrial plants and provide an excellent model for studying cell differentiation and proliferation. Trichome development involves trichome density, trichome number, trichome distribution, trichome morphology, the production of secondary metabolic secretions, and so on. The R2R3 MYB, bHLH, HD-ZIP, and C2H2 ZFP families of transcription factors play significant roles in the regulation of trichome development in the model plants Arabidopsis thaliana, tomato, and cucumber. There are few reports on regulatory genes and trichome development mechanisms in zucchini. In our previous study, the multicellular trichome morphology of aboveground tissues of “16” and “63” was observed, and trichomes were classified into seven types: type I, II, III, and VII trichomes were nonglandular, and type IV, V, and VI trichomes were glandular. Type I and II trichomes were longer and harder than the other types, and the presence or absence of type I and II trichomes was an obvious difference between “16” and “63” (Lin et al., 2023). Type I and II trichomes of petioles were larger than those on other tissues and were dispersedly distributed on the surface; therefore, we could more easily observe trichome morphology and density by microscopy. As demonstrated by observing the density of type I and type II trichomes on petioles, the phenotypes of F2 plants were categorized into five classes. F2 individuals of class 1 had the sparsest type I and type II trichomes, and individuals of the class 5 phenotype had the densest type I and type II trichomes (Lin et al., 2023). In cucumber, the developmental processes involved in glandular and nonglandular multicellular trichome formation were observed using cotyledons as a model and divided into five sequential stages (Dong et al., 2022). In our study, zucchini cotyledons were used as a model and the same developmental processes of glandular and nonglandular trichomes were observed. The third stage (tip head formation/glandular head transition) was the key stage for glandular and nonglandular trichome development. The morphological observation and classification of zucchini trichomes laid a foundation for the QTL analysis of trichome density.
In our study, an F2 population derived by crossing “16” and “63” was constructed, and QTL analysis of the density of type I and type II trichomes was performed with QTL-seq and genetic map-based QTL analysis. Two QTLs associated with trichome density, CpTD3.1 and CpTD15.1, were detected on chromosomes 3 and 15 by both QTL analysis methods. The stability of CpTD3.1 and CpTD15.1 was confirmed using data from three environmental conditions. The results showed that CpTD3.1 and CpTD15.1 were detected in similar regions in all environments, which explained 5%–10.64% and 12.71%–29.37% of the observed phenotypic variation, respectively. These two QTLs showed good stability, and the environment seldom affected the major-effect QTL CpTD15.1 or the minor-effect QTL CpTD3.1. The CpTD3.1 locus was within a 2,672.89-kb region between markers chr3-218350 and chr3-2891236, and the CpTD15.1 locus was within a 775.44-kb region between markers chr15-4991349 and chr15-5766791. The genetic interaction analysis indicated that there was a simple additive gene effect between CpTD3.1 and CpTD15.1. F2 individuals with the CpTD15.1 locus derived from parent “16” and the CpTD3.1 locus derived from parent “63” were labeled 15A-3B, individuals with the CpTD15.1 locus derived from parent “63” and the CpTD3.1 locus derived from parent “16” were labeled 15B-3A, and individuals with the CpTD15.1 and CpTD3.1 loci derived from parent “16” were labeled 15A-3A. The phenotypes of 15A-3B, 15B-3A, and 15A-3A in 2020F were collected. The trichome density of 15A-3B ranged from class 2 to class 4 with an average class of 2.86, the trichome density of 15B-3A ranged from class 1 to class 3 with an average class of 2.04, and the trichome density of 15A-3A ranged from 3 to 5 with an average class of 3.74 (Figure S3). The results confirmed that zucchini plants with both loci derived from parent “16” had dense trichomes. Conventional breeding techniques require the selection of phenotypes and evaluation in multiple environments for many years, and this process could require considerable time, labor, and land. Marker-assisted selection (MAS) is a powerful tool for selecting the target trait, which effectively accelerates the breeding process. Markers closely linked to the CpTD15.1 and CpTD3.1 loci (chr3-218350, chr3-2891236, chr15-4991349, and chr15-5766791) could potentially aid in the selection of trichome density at the seedling stage and be successfully used in MAS.
ZFPs are one of the most important transcription factors in plants. Based on the different number and arrangement of Cys and His residues, ZFPs can be categorized into nine types, namely, C2H2, C8, C6, C3HC4, C2HC, C2HC5, C4, C4HC3, and CCCH (Berg and Shi, 1996). ZFPs play vital roles in the regulation of growth, developmental processes, and multiple stress responses in plants (Bernhardt et al., 2003; Davletova et al., 2005; Dinneny et al., 2006; Xiao et al., 2009; Zhang et al., 2012). Many C2H2 ZFP genes have also been reported to be involved in trichome formation in unicellular or multicellular trichome plants (Davletova et al., 2005; Dinneny et al., 2006; Xiao et al., 2009; Zhang et al., 2012). In Arabidopsis, the first-identified C2H2 ZFP, GLABROUS INFLORESCENCE STEMS (GIS), induces trichome initiation in inflorescence organs. It acts upstream of the MYB-bHLH-WD complex and GL1 by gibberellin (GA), and acts downstream of SPINDLY to promote inflorescence trichome initiation (Gan et al., 2006). Two C2H2 ZFPs, GIS2 and ZFP8, are integrated with cytokinin and GA signaling in the regulation of trichome initiation on inflorescence organs and cauline leaves, respectively (Gan et al., 2007). ZFP5, ZFP6, and GIS3 are the key proteins involved in trichome initiation on inflorescence organs, acting upstream of GIS, GIS2, and ZFP8 (Zhou et al., 2012). In tomato, the SlH, SlHair2/SlZFP8L, and SlZFP6 genes are members of the C2H2 family. SlH increases the length of type I, III, and VI trichomes. SlH and its closest homologue, SlZFP8L, physically interact with Wo, and SlH, zinc finger protein-like 8 (SlZFP8L), and Wo function together to regulate long-stalk type I trichome initiation (Chang et al., 2018; Chun et al., 2021). SlZFP6 and SlZFP8L are critical for the density of type I, III/V, VI, and VII trichomes and increase the length of long-stalk type I, III, and VI trichomes. SlH interacts with SlZFP8-like to regulate trichome initiation and elongation by modulating SlZFP6 gene expression. Based on transgenic technology, SlH, SlZFP8L, and SlZFP6 may promote trichome elongation by activating the expression of cell-wall-loosening protein genes (Zheng et al., 2022). In conclusion, C2H2 zinc finger transcription factors play similar roles in the regulation of trichome formation in unicellular trichome plant Arabidopsis and multicellular trichome plant tomato. Cucumber fruits are covered with tubercules and spines. The tuberculate fruit gene Tu, which was identified as a C2H2 ZFP gene, is involved in the CTK biosynthetic pathway (Yang et al., 2014). In our research, genes encoding transcription factors were screened in the region of the major-effect locus CpTD15.1. Among these genes, one (Cp4.1LG15g04400) encoding ZFP harbored nonsynonymous SNPs in the conserved ring finger domain. The ring finger domain of Cp4.1LG15g04400 was of the C3DHC3 type, which is found in ZFPL1 genes. There were significant differences in Cp4.1LG15g04400 expression between the LDT parent “16” and the SMT parent “63”, and a similar pattern was shown between zucchini LDT lines and SMT lines. A phylogenetic analysis of Cp4.1LG15g04400 with known genes was performed, and Cp4.1LG15g04400 was found to be a distant relative of C2H2 ZFP genes (Figure S4). Based on sequence analysis, gene function annotation, and gene expression analysis, it was presumed that Cp4.1LG15g04400 might regulate trichome density as a new type of ZFP gene in zucchini. Near-isogenic lines of CpTD15.1 were constructed to confirm the candidate gene of the major locus. The gene functions and regulatory pathways related to the trichome density of Cp4.1LG15g04400 need to be further validated based on more molecular biological experiments in zucchini.
Conclusion
An F2 population was constructed with the LDT inbred line “16” and the SMT inbred line “63” for QTL analysis of type I and II trichome density. Two QTLs, CpTD3.1 and CpTD15.1, were identified on chromosomes 3 and 15 using the QTL-seq method and genetic map-based QTL analysis, and CpTD15.1 was the major-effect QTL. The stability of CpTD3.1 and CpTD15.1 was confirmed using data from F2 plants under different environmental conditions. The major-effect QTL CpTD15.1 was located between markers chr15-4991349 and chr15-5766791 with a physical distance of 775.44 kb, and explained 12.71%–29.37% of the phenotypic variation observed in the three environments. CpTD3.1 was located between markers chr3-218350 and chr3-2891236, with a physical distance of 2,672.89 kb, and explained 5.00%–10.64% of the phenotypic variation in the three environments. In the CpTD15.1 region, five genes encoding transcription factors regulating trichome development were selected. Cp4.1LG15g04400 harbored nonsynonymous SNPs in the conserved ring finger domain between the two parental lines. There were significant differences in Cp4.1LG15g04400 expression between “16” and “63” and in the germplasm resources of LDT lines and SMT lines. It was inferred that Cp4.1LG15g04400 might regulate trichome density in zucchini. This study lays a foundation for better understanding the density of multicellular nonglandular trichomes and the regulatory mechanism of trichome density in zucchini.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioProject accession number: PRJNA987358.
Author contributions
YW performed data analysis and prepared the manuscript. GW contributed to RNA extraction and qRT-PCR test. DL contributed to collecting phenotypic characteristics and DNA extraction. QL contributed to growing plants. WX and SQ, the corresponding authors, oversaw all activities related to the project implementation and manuscript development. All authors read and approved the final version of the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (32272723 and 32072590), the National Natural Science Foundation of China (32002051), the Academic Backbone Foundation of Northeast Agricultural University (20XG03), the Postdoctoral Science Foundation of China (2019M661244), the Open Project of Key Laboratory of Biology and Genetic Improvement of Horticultural Crops (Northeast Region), and the Ministry of Agriculture and Rural Affairs.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1232154/full#supplementary-material
Supplementary Figure 1 | Alignment of the predicted protein sequences of five genes between “16” and “63”. (A) Alignment acid sequences of Cp4.1LG15g04030. (B) Alignment acid sequences of Cp4.1LG15g04400. (C) Alignment acid sequences of Cp4.1LG15g04350. The red lines indicated ring finger domains.
Supplementary Figure 2 | Alignment of the predicted protein sequences of five genes between germplasm resources of LDT lines and SMT lines. The relative expression levels of five genes were quantified using the 2−ΔΔCT method. The expression level of the respective genes in the first LDT line was set to a value of 1 and used as a reference, respectively. Each was repeated three times. Capital letters indicate an extremely significant difference, p < 0.01.
Supplementary Figure 3 | Trichome density of 15A-3B, 15B-3A, and 15A-3A in F2. Capital letters indicate an extremely significant difference, p < 0.01.
Supplementary Figure 4 | Phylogenetic tree analysis of Cp4.1LG15g04400 and known ZFPs.
References
Abe, A., Kosugi, S., Yoshida, K., Natsume, S., Takagi, H., Kanzaki, H., et al. (2012). Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 30, 174–178. doi: 10.1038/nbt.2095
Amme, S., Rutten, T., Melzer, M., Sonsmann, G., Mock, H. P. (2005). A proteome approach defines protective functions of tobacco leaf trichomes. Proteomics 5, 2508–2518. doi: 10.1002/pmic.200401274
An, L., Zhou, Z., Sun, L., Yan, A., Xi, W., Yu, N., et al. (2012). A zinc finger protein gene ZFP5 integrates phytohormone signaling to control root hair development in Arabidopsis. Plant J. 72, 474–490. doi: 10.1111/j.1365-313X.2012.05094.x
Berg, J. M., Shi, Y. (1996). The galvanization of biology: a growing appreciation for the roles of zinc. Science 271 (5252), 1081–1085. doi: 10.1126/science.271.5252.1081
Bernhardt, C., Lee, M. M., Gonzalez, A., Zhang, F., Lloyd, A., Schiefelbein, J. (2003). The bHLH genes GLABRA3 (GL3) and ENHANCER OF GLABRA3 (EGL3) specify epidermal cell fate in the Arabidopsis root. Development 130 (26), 6431–6439. doi: 10.1242/dev.00880
Chang, J., Yu, T., Gao, S., Xiong, C., Xie, Q., Li, H., et al. (2016). Fine mapping of the dialytic gene that controls multicellular trichome formation and stamen development in tomato. Theor. Appl. Genet. 129 (8), 1531–1539. doi: 10.1093/pcp/pcq118
Chang, J., Yu, T., Yang, Q., Xiong, C., Gao, S., Xie, Q., et al. (2018). Hair, encoding a single C2H2 zincfinger protein, regulates multicellular trichome formation in tomato. Plant J. 96, 90–102. doi: 10.1111/tpj.14018
Chen, C., Liu, M., Jiang, L., Liu, X., Zhao, J., Yan, S., et al. (2014). Transcriptome profiling reveals roles of meristem regulators and polarity genes during fruit trichome development in cucumber (Cucumis sativus L.). J. Exp. Bot. 65 (17), 4943–4958. doi: 10.1093/jxb/eru258
Chen, C. H., Yin, S., Liu, X. W., Liu, B., Yang, S., Xue, S. D., et al. (2016). The WD-repeat protein csttg1 regulates fruit wart formation through interaction with the Homeodomain-Leucine Zipper I Protein Mict. Plant Physiol. 171 (2), 1156–1168. doi: 10.1104/pp.16.00112
Chun, J. I., Kim, S. M., Kim, H., Cho, J. Y., Kwon, H. W., Kim, J. I., et al. (2021). SlHair2 regulates the initiation and elongation of type I trichomes on tomato leaves and stems. Plant Cell Physiol. 62 (9), 1446–1459. doi: 10.1093/pcp/pcab090
Churchill, G. A., Doerge, R. W. (1994). Empirical threshold values for quantitative trait mapping. Genetics 138, 963–971. doi: 10.1093/genetics/138.3.963
Davletova, S., Schlauch, K., Coutu, J., Mittler, R. (2005). The zinc-finger protein Zat12 plays a central role in reactive oxygen and abiotic stress signaling in Arabidopsis. Plant Physiol. 139, 847–856. doi: 10.1104/pp.105.068254
Ding, W., Wang, Y., Qi, C., Luo, Y., Wang, C., Xu, W., et al. (2021). Fine mapping identified the gibberellin 2-oxidase gene CpDw leading to a dwarf phenotype in squash (Cucurbita pepo L.). Plant Sci. 306. doi: 10.1016/j.plantsci.2021.110857
Dinneny, J. R., Weigel, D., Yanofsky, M. F. (2006). NUBBIN and JAGGED define stamen and carpel shape in Arabidopsis. Development 133, 1645–1655. doi: 10.1242/dev.02335
Dong, M., Xue, S., Bartholomew, E. S., Zhai, X., Sun, L., Xu, S., et al. (2022). Transcriptomic and functional analysis provides molecular insights into multicellular trichome development. Plant Physiol. 189, 301–314. doi: 10.1093/plphys/kiac050
Emiko, H., Kim, J. A., Andreas, J. M., Rü digger, H., Stephan, C., Cho, Y. E. (2010). Expression profiling of tobacco leaf trichomes identifies genes for biotic and abiotic stresses. Plant Cell Physiol. 51, 1627–1637. doi: 10.1093/pcp/pcq118
Feng, Z., Sun, L., Dong, M., Fan, S., Shi, K., Qu, Y., et al. (2023). Identification and functional characterization of csMYCs in cucumber glandular trichome development. Int. J. Mol. Sci. 24 (7), 6435. doi: 10.3390/ijms24076435
Gan, Y., Kumimoto, R., Liu, C., Ratcliffe, O., Yu, H., Broun, P. (2006). GLABROUS INFLORESCENCE STEMS modulates the regulation by gibberellins of epidermal differentiation and shoot maturation in Arabidopsis. Plant Cell 18, 1383–1395. doi: 10.1105/tpc.106.041533
Gan, Y., Liu, C., Yu, H., Broun, P. (2007). Integration of cytokinin and gibberellin signalling by Arabidopsis transcription factors GIS, ZFP8 and GIS2 in the regulation of epidermal cell fate. Development 134, 2073–2081. doi: 10.1242/dev.005017
Gao, S. H., Gao, Y. N., Xiong, C., Yu, G., Chang, J., Yang, Q. H., et al. (2017). The tomato B-type cyclin gene, SlCycB2, plays key roles in reproductive organ development, trichome initiation, terpenoids biosynthesis and Prodenia litura defense. Plant Sci. 262, 103–114. doi: 10.1016/j.plantsci.2017.05.006
Guo, C. L., Yang, X. Q., Wang, Y. L., JT, N., Yang, Y., JX, S., et al. (2018). Identification and mapping of ts (tender spines), a gene involved in soft spine development in Cucumis sativus. Theor. Appl. Genet. 131, 1–12. doi: 10.1007/s00122-017-2954-9
Hill, J. T., Demarest, B. L., Bisgrove, B. W., Gorsi, B., Su, Y., Yost, H. J. (2013). MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 23, 687–697. doi: 10.1101/gr.146936.112
Ishida, T., Hattori, S., Sano, R., Inoue, K., Shirano, Y., Hayashi, H., et al. (2007). Arabidopsis TRANSPARENT TESTA GLABRA2 is directly regulated by R2R3 MYB transcription factors and is involved in regulation of GLABRA2 transcription in epidermal differentiation. Plant Cell 19 (8), 2531–2543. doi: 10.1105/tpc.107.052274
Javier, M. P., José, B., Aureliano, B., Peio, Z., Cristina, E., Carlos, M. G., et al. (2018). De-novo assembly of zucchini genome reveals a whole genome duplication associated with the origin of the Cucurbita genus. Plant Biotechnol. J. 16 (6), 1161–1171. doi: 10.1111/pbi.12860
Kim, S. Y., Hyoung, S., So, W. M., Shin, J. S. (2018). The novel transcription factor TRP interacts with ZFP5, a trichome initiation-related transcription factor, and negatively regulates trichome initiation through gibberellic acid signaling. Plant Mol. Biol. 96, 315–326. doi: 10.1007/s11103-018-0697-x
Kirik, V., Simon, M., Huelskamp, M., Schiefelbein, J. (2004). The ENHANCER OF TRY AND CPC1 gene acts redundantly with TRIPTYCHON and CAPRICE in trichome and root hair cell patterning in Arabidopsis. Dev. Biol. 268 (2), 506–513. doi: 10.1016/j.ydbio.2003.12.037
Li, Q., Cao, C. X., Zhang, C. J., Zheng, S. S., Wang, Z. H., Wang, L. N., et al. (2015). The identification of Cucumis sativus Glabrous 1 (CsGL1) required for the formation of trichomes uncovers a novel function for the homeodomain-leucine zipper I gene. J. Exp. Bot. 66 (9), 2515–2526. doi: 10.1093/jxb/erv046
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Lin, D., Li, W., Hou, Y., Ding, W., Wang, C., Zhang, F., et al. (2023). Classification of inheritance of trichomes in different organs in zucchini (Cucurbita pepo L.). Crop Sci. 63, 712–723. doi: 10.1002/csc2.20883
Liu, X., Bartholomew, E., Cai, Y., Ren, H. (2016). Trichome-related mutants provide a new perspective on multicellular trichome initiation and development in cucumber (Cucumis sativus L). Front. Plant Sci. 7, 1187. doi: 10.3389/fpls.2016.01187
Liu, Y., Liu, D., Hu, R., Hua, C., Ali, I., Zhang, A., et al. (2017). AtGIS, a C2H2 zinc-finger transcription factor from Arabidopsis regulates glandular trichome development through GA signaling in tobacco. Biochem. Biophys. Res. Commun. 483, 209–215. doi: 10.1016/j.bbrc.2016.12.164
Liu, Y., Yang, S., Kha, A. R., Gan, Y. (2023). TOE1/TOE2 interacting with GIS to control trichome development in Arabidopsis. Int. J. Mol. Sci. 24 (7), 6698. doi: 10.3390/ijms24076698
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25 (4), 402–408. doi: 10.1006/meth.2001.1262
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Murray, M. G., Thompson, W. F. (1980). Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8 (19), 4321–4325. doi: 10.1093/nar/8.19.4321
Payne, C. T., Zhang, F., Lloyd, A. M. (2000). GL3 encodes a bHLH protein that regulates trichome development in Arabidopsis through interaction with GL1 and TTG1. Genetics 156 (3), 1349–1362. doi: 10.1093/genetics/156.3.1349
Schnittger, A., Folkers, U., Schwab, B., Jürgens, G., Hülskamp, M. (1999). Generation of a spacing pattern: the role of triptychon in trichome patterning in Arabidopsis. Plant Cell 11 (6), 1105–1116. doi: 10.1105/tpc.11.6.1105
Sun, L., Zhang, A., Zhou, Z., Zhao, Y., Yan, A., Bao, S., et al. (2015). GLABROUS INFLORESCENCE STEMS3 (GIS3) regulates trichome initiation and development in Arabidopsis. New Phytol. 206, 220–230. doi: 10.1111/nph.13218
Walker, A. R., Davison, P. A., Bolognesi-Winfield, A. C., James, C. M., Srinivasan, N., Blundell, T. L., et al. (1999). The TRANSPARENT TESTA GLABRA1 locus, which regulates trichome differentiation and anthocyanin biosynthesis in Arabidopsis, encodes a WD40 repeat protein. Plant Cell 11 (7), 1337–1350. doi: 10.1105/tpc.11.7.1337
Wang, S., Hubbard, L., Chang, Y., Guo, J., Schiefelbein, J., Chen, J. G. (2008). Comprehensive analysis of single-repeat R3 MYB proteins in epidermal cell patterning and their transcriptional regulation in Arabidopsis. BMC Plant Biol. 8 (1), 81. doi: 10.1186/1471-2229-8-81
Wang, S., Kwak, S. H., Zeng, Q., BE, E., XY, C., Schiefelbein, J., et al. (2007). TRICHOMELESS1 regulates trichome patterning by suppressing GLABRA1 in Arabidopsis. Development 134, 3873–3882. doi: 10.1242/dev.009597
Wang, Y. L., Nie, J. T., Chen, H. M., Guo, C. L., Pan, J., He, H. L., et al. (2016). Identification and mapping of Tril, a homeodomain−leucine zipper gene involved in multicellular trichome initiation in Cucumis sativus. Theor. Appl. Genet. 129 (2), 305–316. doi: 10.1007/s00122-015-2628-4
Wang, Y., Qi, C., Luo, Y., Zhang, F., Dai, Z., Li, M., et al. (2021). Identification and mapping of CpPM10.1, a major gene involved in powdery mildew (race 2 France of Podosphaera xanthii) resistance in zucchini (Cucurbita pepo L.). Theor. Appl. Genet. 134 (8), 2531–2545. doi: 10.1007/s00122-021-03840-z
Wang, Y., Wang, C., Han, H., Luo, Y., Wang, Z., Yan, C., et al. (2020). Construction of a high-density genetic map and analysis of seed-related traits using specific length amplified fragment sequencing for Cucurbita maxima. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01782
Weng, Y., Colle, M., Wang, Y., Yang, L., Rubinstein, M., Sherman, A., et al. (2015). QTL mapping in multiple populations and development stages reveals dynamic quantitative trait loci for fruit size in cucumbers of different market classes. Theor. Appl. Genet. 128, 1747–1763. doi: 10.1007/s00122-015-2544-7
Wester, K., Digiuni, S., Geier, F., Timmer, J., Fleck, C., Hulskamp, M. (2009). Functional diversity of R3 single-repeat genes in trichome development. Development 136 (9), 1487–1496. doi: 10.1242/dev.021733
Xiao, H., Tang, J., Li, Y., Wang, W. M., Li, X. B., Jin, L., et al. (2009). STAMENLESS 1, encoding a single C2H2 zinc finger protein, regulates floral organ identity in rice. Plant J. 59, 789–801. doi: 10.1111/j.1365-313X.2009.03913.x
Xie, Q., Liu, P. N., Shi, L. X., Miao, H., Bo, K. L., Wang, Y., et al. (2018). Combined fine mapping, genetic diversity, and transcriptome profiling reveals that the auxin transporter gene ns plays an important role in cucumber fruit spine development. Theor. Appl. Genet. 131 (6), 1239–1252. doi: 10.1007/s00122-018-3074-x
Xu, J., van Herwijnen, Z. O., Dra¨ger, D. B., Sui, C., MA, H., Schuurink, R. C. (2018). SlMYC1 regulates type VI glandular trichome formation and terpene biosynthesis in tomato glandular cells. Plant Cell 30, 2988–3005. doi: 10.1105/tpc.18.00571
Xue, S., Dong, M., Liu, X., Xu, S., Pang, J., Zhang, W., et al. (2019). Classifcation of fruit trichomes in cucumber and effects of plant hormones on type II fruit trichome development. Planta 249, 407–416. doi: 10.1007/s00425-018-3004-9
Yang, C. X., Li, H. X., Zhang, J. H., Luo, Z. D., Gong, P. J., Zhang, C. J., et al. (2011). A regulatory gene induces trichome formation and embryo lethality in tomato. Proc. Natl. Acad. Sci. United States America 108 (29), 11836–11841. doi: 10.1073/pnas.1100532108
Yang, X., Zhang, W., He, H., Nie, J., Bie, B., Zhao, J., et al. (2014). Tuberculate fruit gene Tu encodes a C2H2 zinc finger protein that is required for the warty fruit phenotype in cucumber (Cucumis sativus L.). Plant J. 78 (6), 1034–1046. doi: 10.1111/tpj.12531
Ying, S., Su, M., Wu, Y., Zhou, L., Fu, R., Li, Y., et al. (2020). Trichome regulator SlMIXTA-like directly manipulates primary metabolism in tomato fruit. Plant Biotechnol. J. 18 (2), 3541–3563. doi: 10.1111/pbi.13202
Zeng, J., Yang, L., Tian, M., Xie, X., Liu, C., Ruan, Y. (2023). SDG26 is involved in trichome control in Arabidopsis thaliana: affecting phytohormones and adjusting accumulation of H3K27me3 on genes related to trichome growth and development. Plants 12 (8), 1651. doi: 10.3390/plants12081651
Zhang, T., Li, X., Yang, Y., Guo, X., Feng, Q., Dong, X., et al. (2019). Genetic analysis and QTL mapping of fruit length and diameter in a cucumber (Cucumber sativus L.) recombinant inbred line (RIL) population. Scientia Hortic. 250, 214–222. doi: 10.1016/j.scienta.2019.01.062
Zhang, L., Lv, D., Pan, J., Zhang, K., Wen, H., Chen, Y., et al. (2021). A SNP of HD-ZIP I transcription factor leads to distortion of trichome morphology in cucumber (Cucumis sativus L.). BMC Plant Biol. 21 (1), 182. doi: 10.1186/s12870-021-02955-1
Zhang, H., Ni, L., Liu, Y., Wang, Y., Zhang, A., Tan, M., et al. (2012). The C2H2-type zinc finger protein ZFP182 is involved in abscisic acidinduced antioxidant defense in rice. J. Integr. Plant Biol. 54, 500–510. doi: 10.1111/j.1744-7909.2012.01135.x
Zhang, H., Wang, L., Zheng, S., Liu, Z., Wu, X., Gao, Z., et al. (2016). A fragment substitution in the promoter of CsHDZIV11/CsGL3 is responsible for fruit spine density in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 129, 1289–1301. doi: 10.1007/s00122-016-2703-5
Zhao, M., Morohashi, K., Hatlestad, G., Grotewold, E., Lloyd, A. (2008). The TTG1-bHLHMYB complex controls trichome cell fate and patterning through direct targeting of regulatory loci. Development 135 (11), 1991–1999. doi: 10.1242/dev.016873
Zhao, J. L., Pan, J. S., Guan, Y., Zhang, W. W., Bie, B. B., Wang, Y. L., et al. (2015). Micro-trichome as a class I homeodomain-leucine zipper gene regulates multicellular trichome development in Cucumis sativus. J. Integr. Plant Biol. 57, 925–935. doi: 10.1111/jipb.12345
Zhao, H. T., Wang, X. X., Zhu, D. D., Cui, S. J., Li, X., Cao, Y., et al. (2012). A single amino acid substitution in iiif subfamily of basic helix loop-helix transcription factor Atmyc1 leads to trichome and root hair patterning defects by abolishing its interaction with partner proteins in Arabidopsis. J. Biol. Chem. 287 (17), 14109–14121. doi: 10.1074/jbc.M111.280735
Zhao, L., Zhu, H., Zhang, K., Wang, Y., Wu, L., Chen, C., et al. (2020). The MIXTA-LIKE transcription factor CsMYB6 regulates fruit spine and tubercule formation in cucumber. Plant Sci. 300, 110636. doi: 10.1016/j.plantsci.2020.110636
Zheng, F., Cui, L., Li, C., Xie, Q., Ai, G., Wang, J., et al. (2022). Hair interacts with SlZFP8-like to regulate the initiation and elongation of trichomes by modulating SlZFP6 expression in tomato. J. Exp. Bot. 73, 228–244. doi: 10.1093/jxb/erab417
Zhou, Z., An, L., Sun, L., Gan, Y. (2012). ZFP5 encodes a functionally equivalent GIS protein to control trichome initiation. Plant Signaling Behav. 7 (1), 28–30. doi: 10.4161/psb.7.1.18404
Keywords: Cucurbita pepo, trichome density, InDel markers, QTL analysis, zinc finger protein
Citation: Wang Y, Wang G, Lin D, Luo Q, Xu W and Qu S (2023) QTL mapping and stability analysis of trichome density in zucchini (Cucurbita pepo L.). Front. Plant Sci. 14:1232154. doi: 10.3389/fpls.2023.1232154
Received: 31 May 2023; Accepted: 06 July 2023;
Published: 11 August 2023.
Edited by:
Jingtao Nie, Zhejiang Agriculture and Forestry University, ChinaReviewed by:
Yupeng Pan, Northwest A&F University, ChinaXingwang Liu, China Agricultural University, China
Copyright © 2023 Wang, Wang, Lin, Luo, Xu and Qu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenlong Xu, eHdsQG5lYXUuZWR1LmNu; Shuping Qu, c3BxdUBuZWF1LmVkdS5jbg==