 Shixi Chen
Shixi Chen Fardous Mohammad Safiul Azam
Fardous Mohammad Safiul Azam Mst. Lovely Akter3
Mst. Lovely Akter3- 1College of Life Science, Neijiang Normal University, Neijiang, Sichuan, China
- 2Fishes Conservation and Utilization in the Upper Reaches of the Yangtze River, Key Laboratory of Sichuan Province, Neijiang Normal University, Sichuan, China
- 3Department of Biotechnology and Genetic Engineering, Faculty of Life Sciences, University of Development Alternative, Dhaka, Bangladesh
- 4Key Laboratory of Regional Characteristic Agricultural Resources, College of Life Sciences, Neijiang Normal University, Neijiang, Sichuan, China
- 5Branch of The First Affiliated Hospital of Xinjiang Medical University, Changji, Xinjiang, China
Introduction: Thalictrum fargesii is a medicinal plant belonging to the genus Thalictrum of the Ranunculaceae family and has been used in herbal medicine in the Himalayan regions of China and India. This species is taxonomically challenging because of its morphological similarities to other species within the genus. Thus, herbal drugs from this species are frequently adulterated, substituted, or mixed with other species, thereby endangering consumer safety.
Methods: The present study aimed to sequence and assemble the entire chloroplast (cp) genome of T. fargesii using the Illumina HiSeq 2500 platform to better understand the genomic architecture, gene composition, and phylogenetic relationships within the Thalictrum.
Results and discussion: The cp genome was 155,929 bp long and contained large single-copy (85,395 bp) and small single-copy (17,576 bp) regions that were segregated by a pair of inverted repeat regions (26,479 bp) to form a quadripartite structure. The cp genome contains 133 genes, including 88 protein-coding genes (PCGs), 37 tRNA genes, and 8 rRNA genes. Additionally, this genome contains 64 codons that encode 20 amino acids, the most preferred of which are alanine and leucine. We identified 68 SSRs, 27 long repeats, and 242 high-confidence C-to-U RNA-editing sites in the cp genome. Moreover, we discovered seven divergent hotspot regions in the cp genome of T. fargesii, among which ndhD-psaC and rpl16-rps3 may be useful for developing molecular markers for identifying ethnodrug species and their contaminants. A comparative study with eight other species in the genus revealed that pafI and rps19 had highly variable sites in the cp genome of T. fargesii. Additionally, two special features, (i) the shortest length of the ycf1 gene at the IRA-SSC boundary and (ii) the distance between the rps19 fragment and trnH at the IRA-LSC junction, distinguish the cp genome of T. fargesii from those of other species within the genus. Furthermore, phylogenetic analysis revealed that T. fargesii was closely related to T. tenue and T. petaloidium.
Conclusion: Considering all these lines of evidence, our findings offer crucial molecular and evolutionary information that could play a significant role in further species identification, evolution, and phylogenetic studies on T. fargesii.
1 Introduction
The genus Thalictrum belongs to the subfamily Thalictroideae of Ranunculaceae, with the subgenera Thalictrum and Lecoyerium, which includes approximately 200 species and is distributed in Asia, Europe, Africa, North America, and South America (Zhu and Xiao, 1989). At least 43 species of this genus have been used medicinally (Chen et al., 2003), 26 of which have been used as medicinal herbs in China and have been found to contain bioactive compounds, such as thalidomide, thalicarpine, lithospermoside, and dasycarponin (Wu et al., 1979; Huang and Williams, 1998). Among them, Thalictrum fargesii Franch. ex Finet & Gagnep. is distributed throughout Asia and has been reported to have ethnomedicinal and traditional uses in the Tibetan region of China (Zhang et al., 2021) and the Himalayan region of India (Singh et al., 2016). In China, T. fargesii is geographically distributed in the Anhui, Chongqing, Fujian, Gansu, Guizhou, Henan, Hubei, Hunan, Jiangxi, Shaanxi, Sichuan, and Zhejiang Provinces (Wang and Wang, 1979; Fu and Zhu, 2001; Xie, 2016; Wang et al., 2018). Thalictroideae is a monophyletic group that is well supported by systematic molecular studies of Ranunculaceae (Johansson and Jansen, 1993; Hoot, 1995; Johansson, 1995; Ro et al., 1997; Wang and Chen, 2007). Ren et al. (2011) first examined detailed floral development in this subfamily. They classified this species based on chloroplast rbcL, matK, trnL-F, and nuclear ITS primers (Wang and Chen, 2007), and a taxonomic report on this species was provided by Zeng et al. (2021).
The plant of T. fargesii is termed “CHENG KOU TANG SONG CAO” in China for its thalidasine roots (Zhou et al., 2011). The roots of this species are also rich in the naturally active compounds 5-methoxy salicylic acid, thalfoetidine, thaligasinine, and thalisopidine (Wu and Yi, 1991; NCBI, 2022). Alkaloids from this species have been reported to exhibit various pharmacological activities including antitumor, antimicrobial, antitussive, antiamoebic, antiparasitic, antiplatelet aggregation, antisilicosis, hypotensive, and HIV antiviral activities (Gao, 1999; Singh et al., 2023). This species is used in traditional Chinese medicine to treat dysentery, diarrhea, viral hepatitis, influenza, measles, carbuncles, boils, and eye congestion (Chen et al., 2003). A pharmacophylogenic study showed that aporphine, protopine, pavine, phenanthrene, and bisbenzylisoquinoline are prominent in the Thalictrum genus (Da-Cheng et al., 2015). Additionally, metabolites related to their structural features and roles have been studied in T. fargesii (Khamidullina et al., 2006).
Because of its immense potential in herbal medicine, this species is collected from the wild or grown by rural farmers in China. Consequently, in local herbal markets, the species is often confused by morphological similarities with other related species in the genus, and adulterated, altered, or mixed in the herbal formulation with or without intention (Bainsal et al., 2022). No doubt, it poses a serious threat to consumer safety. Thus, proper identification of T. fargesii is of great importance in herbal formulations, which are often misunderstood, confused, or incorrectly recorded during plant identification. For example, during the course of herbarium specimens, T. fortune was found to be mixed with T. fargesii Franchet ex Finet & Gagnepain (1903: 608), resulting in wrong records of the geographical distribution of these species (Zeng et al., 2022). Moreover, Li et al. (2016) clarified the variations in T. fargesii and proposed a synonym for T. xingshanicum, revealing that the name of T. fargesii was previously misunderstood. In addition, herbarium collections of T. fargesii from Neijiang, Sichuan Province were misled by misidentification of the collections of T. ramosum in Gansu Province (Zeng et al., 2023). These misidentifications can be explained by the morphological variation related to environmental factors (e.g., weather, soil properties, elevation level, etc.) where the plants have grown (Hernández-Nicolás et al., 2017). It is important to eliminate such dilemmas using molecular approaches rather than morphological observations (Duminil and Di Michele, 2009).
Identifying member species within the Ranunculaceae family, especially within the Thalictrum, is taxonomically challenging because of the inability to distinguish these species through universal molecular markers and similar morphological traits (Li et al., 2020). Previous studies have revealed that Thalictrum species form a highly supported clade, indicating that they are monophyletic (Cai et al., 2022; Michimoto et al., 2022). Thalictrum species exhibit small chromosomes compared with those of Rununculus, and this genus is challenging and diverse in terms of taxonomy and phylogeny based on variations in morphological traits (Langlet, 1927; Tamura, 1995; Xiang et al., 2022). As a critical solution, chloroplast (cp) genomic resources have facilitated the classification of members of the Ranunculaceae family, providing data and insight into evolutionary relationships (Zhai et al., 2019), phylogenetic resolution (bootstrap support and tree certainty) (Morales-Briones et al., 2019), and marker exploration (Xiang et al., 2022).
In the photosynthesis of green plants, chloroplasts play the most significant role in coordinating the expression of different genes between these organelles and the nuclear genome in response to environmental stress, including cellular signaling (Daniell et al., 2016; Serrano et al., 2016; Yu et al., 2019; Tano et al., 2023). Interestingly, because of the presence of inherited conserved genes and their organization and low recombination and substitution rates, compared with nuclear genomes, cp genomes serve as resources for phylogenetic analysis and evolutionary studies (Wolfe et al., 1987; Corriveau and Coleman, 1988; Birky, 2001; Provan et al., 2001; Zhang et al., 2003; Ravi et al., 2008). Moreover, DNA barcoding and molecular breeding have been studied using cp genomes (Daniell et al., 2016). Several strategies have been adopted to sequence the organelle genome, including amplification of the whole cp genome using a universal set of primers, sequencing (Cronn et al., 2008; Dong et al., 2013), and using total genomic DNA to extract the cp genome (Velasco et al., 2007; McPherson et al., 2013). Considering the available taxonomic evidence, the importance of cp genome studies of folk medicinal plants has been demonstrated by several studies (He et al., 2021; Lin et al., 2021; Pu et al., 2022).
From several previous studies, significant molecular cross-talks have been reported on the characteristics of cp genomes within Thalictrum, such as T. cirrhosum (Zhe et al., 2023), T. coreanum (Park et al., 2015), T. foliolosum (Pu et al., 2022), and Thalictrum aquilegiifolium var. sibiricum (Michimoto et al., 2022). Current research has focused predominantly on T. fargesii, emphasizing the need to construct the whole cp genome using next-generation sequencing, characterization, long and simple sequence repeats (SSRs), phylogenetic analysis, nucleotide diversity, and selection pressure of genes within the Thalictrum. This study aimed to generate molecular data for the cp genome of T. fargesii to provide taxonomic and molecular information to biologists, practitioners, and professionals in herbal medicine to develop molecular markers to avoid misidentification.
2 Materials and methods
2.1 DNA extraction and next-generation sequencing
The T. fargesii plant sample used for cp genome sequencing was identified and artificially reproduced in the Wenchuan Botanical Garden of the Aba Tibetan and Qiang Autonomous Prefecture in Sichuan Province, China. The voucher specimen (Accession No. CP00002) was identified and deposited at the Herbarium of Neijiang Normal University (Neijiang City, China; Shixi Chen,c2FpaGVpQGZveG1haWwuY29t). After collecting leaf specimens, they were stored at room temperature and packaged in 0.2 g of silicon dioxide. We used a modified cetyltrimethylammonium bromide (CTAB) method to extract total genomic DNA from fresh leaves (Porebski et al., 1997). Short reads of the T. fargesii cp genome were sequenced using a genomic library with an insert size of 260 bp. The products were prepared and sequenced on an Illumina HiSeq 2500 platform with approximately 436× coverage.
2.2 Chloroplast genome assembly and annotation
The sequenced reads were filtered using the Trimmomatic program (version 0.39) (Bolger et al., 2014). The filtered reads were assembled into the cp genome using NOVOPlasty (version 4.3.1) (Dierckxsens et al., 2017), and annotation was conducted using GeSeq with 3rd Party Stand-Alone Annotators from Chloë (version 0.1.0) and tRNA annotation from tRNAscan-SE (version 2.0.7) (Tillich et al., 2017). The annotation was followed by a manual check against the information from NCBI. Later, the sequences were deposited in the NCBI database (GenBank accession No. ON868919.1). The T. fargesii cp genome was visualized and plotted using Chloroplot (web-based tool: https://irscope.shinyapps.io/Chloroplot/) (Zheng et al., 2020).
2.3 Comparison of the chloroplast genome within Thalictrum
Eight relevant Thalictrum cp genome sequences were downloaded from the GenBank database. Nine sequences, including T. fargesii, were analyzed to determine the degree of variation and sequence conservation within the same genus (Supplementary Table 1). We compared the IR regions of all species using the web-based tool IRplus (https://irscope.shinyapps.io/IRplus/) to visualize the IR-SC boundaries and their gene orientations (Díez Menéndez et al., 2023). Sequence alignment was performed to identify the variation in sites between the nine Thalictrum cp genome sequences on mVISTA using the alignment program LAGAN (Mayor et al., 2000; Frazer et al., 2004). The sites were checked manually using BioEdit version 7 (Hall, 1999). The sites were subsequently analyzed, and the results are presented in Supplementary Table 2.
2.4 Relative synonymous codon usage analysis
Each amino acid has a minimum of one codon and a maximum of six codons owing to the simplicity of the codons. MEGA11 software was used to calculate the relative synonymous codon usage (RSCU) in the cpDNA of T. fargesii (Tamura et al., 2021).
2.5 Repeat analysis
SSR locus analysis was performed on the assembled T. fargesii cp genome sequence using the MicroSarellite identification tool (MISA) (Beier et al., 2017). The parameters were set as follows: 1–10, 2–5, 3–4, 4–3, 5–3, and 6–3, and the minimum distance between the two SSRs was set to 0 bp. The software package REPuter was used to conduct a long-repeat analysis and a minimum repeat size of 30 bp was assigned along a Hamming distance of 3 bp (Kurtz et al., 2001). The tandem repeat finder is an online program that detects tandem repeats using the default parameter settings (Benson, 1999).
2.6 Predicting RNA editing sites
We predicted RNA editing sites in the cp genome of T. fargesii using a convolutional neural network (CNN) model-based tool called Deepred-mt (Edera et al., 2021). We extracted the PCGs of this cp genome and inputted them into the Deepred-mt tool for prediction, considering threshold probability values greater than 0.9 as reliable results.
2.7 Synonymous substitution and selective pressure
The natural selection that drives molecular evolution was analyzed for the Thalictrum chloroplast genome; thus, we calculated the ratio of nonsynonymous (dn) to synonymous (ds) substitutions called ω (dn/ds). The ω value is an indicator of natural selection of PCGs. Values ω > 1, ω = 1, and ω < 1 indicate positive, neutral, and negative/purifying selection, respectively. The stop codons of all 73 PCGs from these species were deleted and subsequently aligned, and the dn, ds, and ω values were calculated using MEGA 11 (Tamura et al., 2021).
2.8 Phylogenetic analysis
To ascertain the phylogenetic position of T. fargesii within Thalictrum, nine species were analyzed, and Aconitum delavayi (NC_038097) was chosen as the outgroup. Sequence (full-length) alignment was performed using the MAFFT v7 plugin integrated into PhyloSuite v1.1.15 (Katoh and Standley, 2013). The dataset was refined using the Gblocks program with stringent parameter settings and then subjected to maximum likelihood (ML) analyses. Substitutional saturation was assessed using DAMBE version 7.0.68 (Xia, 2018) and phylogenetic trees were constructed using ML algorithms. ML phylogenies were conducted using RAxML v8.2.12, with 1,000 bootstrap replicates, and the GTRGAMMA model (Stamatakis, 2014).
3 Results
3.1 Features of the chloroplast genome
After removing the adapter and low-quality reads, we found 9.4 Gb data for the T. fargesii cp genome. The cp genome was 155,929 bp in length, displayed a circular structure, and was divided into typical quadripartite regions, similar to most land plants (Figure 1). The cp genome includes two 26,479-bp inverted repeats (IRA and IRB), and they separated the genome into a large single-copy (LSC) region of 85,395 bp and a small single-copy (SSC) region of 17,576 bp (Supplementary Table 1). The overall GC content of the cp genome was 38%.
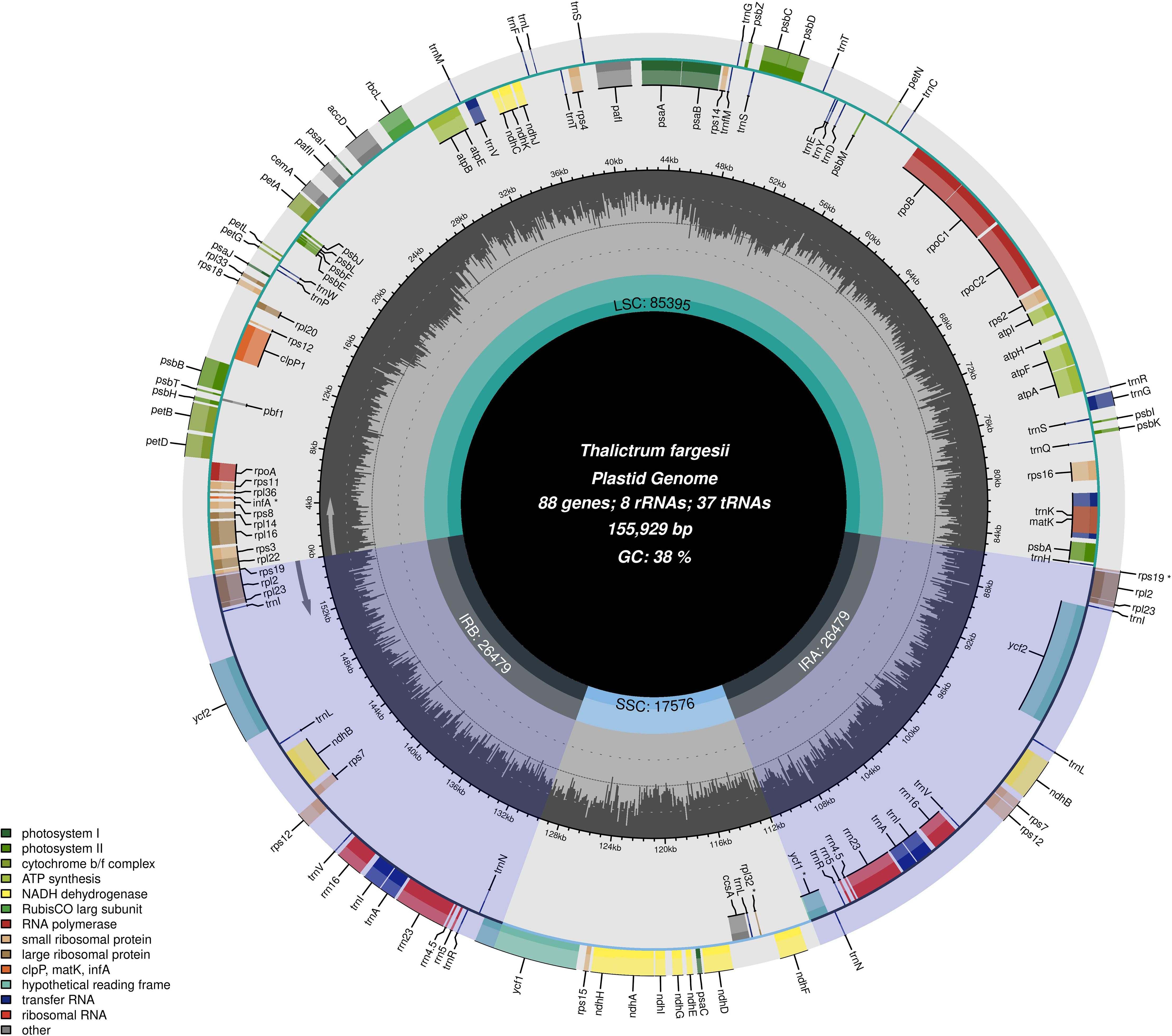
Figure 1 Graphical representation of the features identified in the cp genome of T. fargesii. The species name, genome length, GC content, and number of genes are represented in the center of the plot. Represented by arrows, transcripts for genes inside and outside the circle are generated in opposite directions of the tetrad structure of the cp genome. Different colors are used to distinguish between genes that belong to specific functional categories. At the lower left, the legend for the classification of the cp genes according to their function is shown. The darker shaded region inside the inner circle indicates the GC content, while the light color indicates the AT content of the cp genome. The gradient GC content of the cp genome is represented by the second circle, with a zero level based on the outer circle. The GC content of each gene is displayed as the proportion of shaded areas. The boundaries of the small single-copy (SSC), large single-copy (LSC), and inverted repeat (IRA, IRB) regions are denoted in the inner circle.
The size of the cp genomes of the Thalictrum ranged from 154,889 bp (T. thalictroides) to 156,103 bp (T. tenue). The cp genome of T. fargesii (155,929 bp) was the fourth largest and most similar to those of other species within the genus (Supplementary Table 1). Moreover, we found the most extended LSC region (85,395 bp) in the T. fargesii cp genome, and the shortest LSC region (84,733 bp) in T. coreanum. In terms of count, the total number of genes found ranged from 128 to 134, and for PCGs, it ranged from 83 to 89. Nonetheless, except for T. viscosum (38 tRNAs), most species, including T. fargesii, possess 37 tRNA genes.
3.2 Gene composition of the chloroplast genome
Annotation of the cp genome revealed 133 genes, including 88 PCGs (including six duplicated genes), 37 tRNA genes (including seven duplicated genes), and 8 ribosomal RNA genes. The identified genes were categorized into four groups: photosynthesis-related genes (n = 45), self-replicating genes (n = 58), other genes (n = 6), and unknown genes (n = 6) (Table 1). Four pseudogenes have been identified in the genome. The IR regions carried two copies of ndhB, rps7, rps12, rpl2, rpl23, ycf2, trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnR-ACG, trnN-GUU, rrn16, rrn23, rrn4.5, and rrn5. Of the genes identified, 21 contained introns and 16 (trnK-UUU, trnG-UCC, petB, petD, rpl16, rpl2, ndhB, rps12, trnI-GAU, trnA-UGC, rrn23, trnA-UGC, trnI-GAU, rps12, ndhB, and rpl2) contained one intron. The other five genes (two copies of rps16, trnV-UAC, and atpF) contained two introns (Supplementary Table 3). Moreover, 10 genes were located in the LSC region, 10 in the two IR regions, and 1 in the SSC region. The largest intron (2,532 bp) was observed in trnK-UUU, and the smallest intron (199 bp) was found in rrn23. tRNA genes are distributed throughout the genome and are encoded by 61 possible codons (excluding the stop codon).
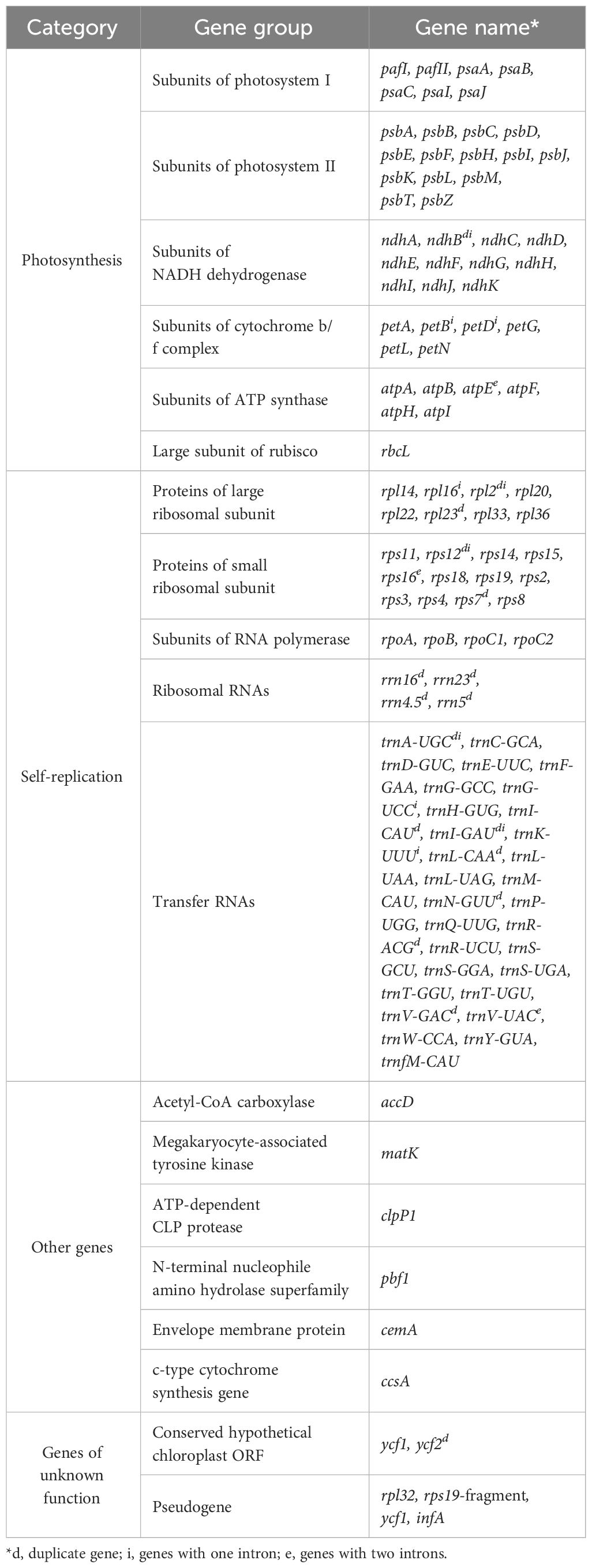
Table 1 Genes according to the categories within the T. fargesii chloroplast genome.
As shown in Table 1, the PCG genes have different functions, including one acetyl-CoA carboxylase, six ATP synthases, one ATP-dependent CLP protease, five cytochrome b6, one cytochrome c biogenesis protein, one cytochrome b/f, one envelope membrane protein, two hypothetical chloroplast RF2, one megakaryocyte-associated tyrosine kinase, 12 NADH dehydrogenases, one N-terminal nucleophile amino hydrolase superfamily, seven photosystem I, 14 photosystem II, 25 ribosomal proteins, one ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit, and four RNA polymerases. Each RNA had two copies and was equally distributed within the IR region. Duplicated tRNAs were not present in the LSC region. Moreover, rpl32, the rps19 fragment, one copy of ycf1, and infA were pseudogenes.
3.3 The base composition of the chloroplast genome
Base compositions of the LSC, SSC, and IR regions and codons at various locations were examined in the cp genome. The percentages of GC content in LSC, SSC, and IRs were 40.57%, 32.53%, and 43.23%, respectively, indicating that the percentage in the IR region was greater than that in the other regions. Moreover, the frequencies of adenine (A), thymine (T), guanine (G), and cytosine (C) contents were 30.46%, 31.12%, 18.84%, and 19.58%, respectively. The single-copy regions, LSC (59.43%) and SSC (67.48), had higher AT content than the repeat regions (56.77%) (Table 2). The reduced AT content in the IR regions may be related to the presence of rRNA, which contains fewer AT nucleotides, contributing to genome stability and sequence complexity. Similarly, the CSD (protein-coding region) was rich in AT (61.24%), whereas the GC content was 38.73%. At the first, second, and third codon positions, each had a length of 25,038 bp and contained a range of 29.10%–31.44% A, 23.44%–37.25% T, 16.69%–27.35% G, and 14.61%–20.67% C contents, respectively. These findings disclosed that the cp genome had a higher AT than the GC content and that the codons preferred bases A and T.
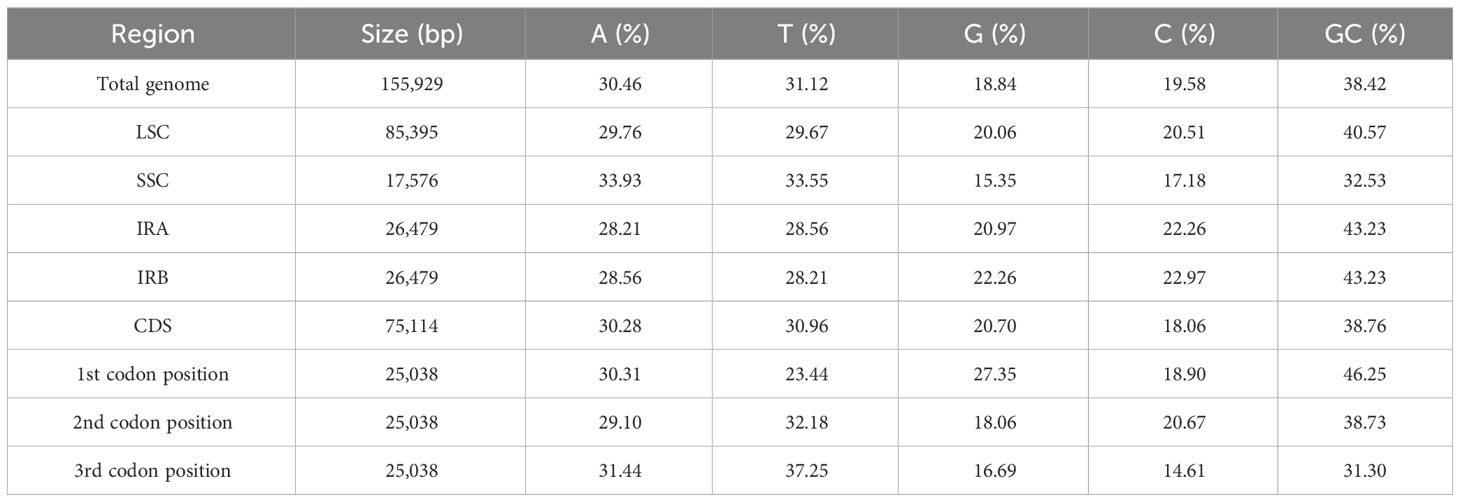
Table 2 Nucleotide composition in different regions of the chloroplast genome of T. fargesii.
3.4 Codon usage
The ratio of the frequency of a codon observed to that expected, termed “relative synonymous codon usage” (RSCU), was determined, which indicates codon usage bias in coding sequences. The protein-coding sequences (CDSs) of the T. fargesii cp genome were used to calculate RSCU via MEGA 11, and 64 codons were found. Three of these were stop codons and 61 encoded 20 amino acids. A single codon encodes methionine (Met) and tryptophan (Try), whereas the other amino acids are encoded by two to six codons (Figure 2). Among the 64 codons, 31 had RSCU > 1 in the CDS of the cp genome, indicating that they were high-frequency and optimal codons. A or U was preferred as the third base of the high-frequency codon, as evidenced by the fact that 16 of these codons ended in U, 13 in A, 1 in G, and 1 in C. The most preferred codon was GCU, which encodes alanine (Ala) and has an RSCU value of 2. UUA, which encodes leucine (Leu) and has an RSCU value of 1.86, was the second-most preferred codon.
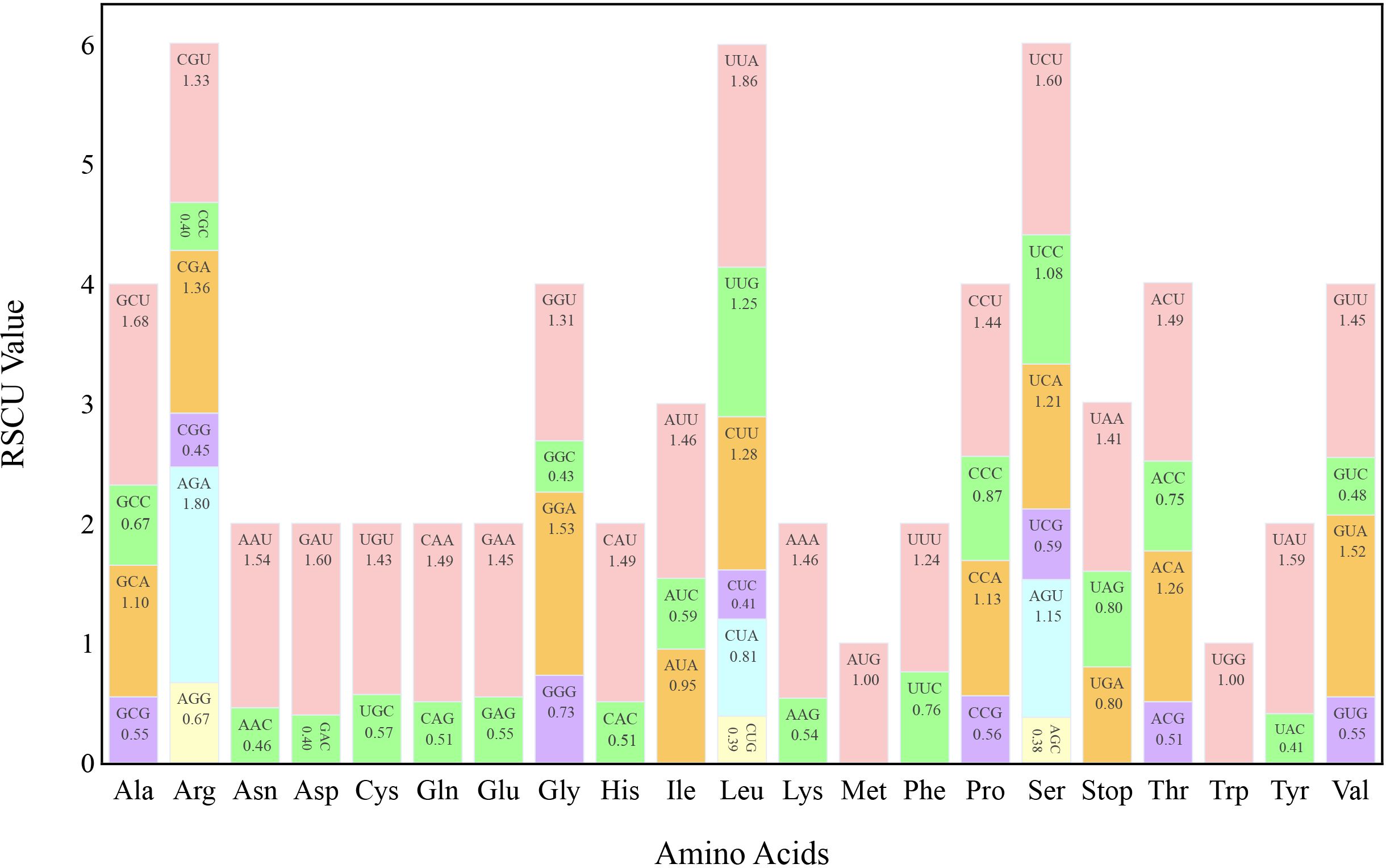
Figure 2 Relative synonymous codon usage (RSCU) of 20 amino acids and stop codons in all protein-coding genes of the chloroplast genome of T. fargesii. The colors of the histograms correspond to the colors of the codons.
3.5 Simple sequence repeats
SSRs from cp genomes can be used to explore evolutionary relationships and population genetics, owing to their high polymorphism rates and consistent repetition (Xia et al., 2022). Most SSRs are A or T units that contribute to the AT richness of the chloroplast genome (Chew et al., 2023). Analysis of SSRs using the MISA web-based tool (Beier et al., 2017) revealed that the T. fargesii cp genome comprises 68 SSR loci. The most abundant were mononucleotides (35, 51%), followed by dinucleotides (20, 30%), trinucleotides (2, 3%), tetranucleotides (9, 13%), and pentanucleotides (2, 3%) (Supplementary Table 4) (Figure 3A). Hexanucleotide repeats were absent. Among these repeats, 43.33%, 23.33%, and 25% were present in the LSC, SSC, and IR region, respectively (Figure 3B). In the cp genome of T. fargesii, the single-base repeat unit A/T (51%) is the most prevalent, followed by AT/TA (29%), AAAT/ATTT (7%), AATAT/ATATT (3%), and AAT/ATT (2%). The forenamed repeat units accounted for 92% of the total SSRs, whereas G/C repeat units accounted for only 8% of the total SSRs (Figure 3C). Among the SSRs, 21 were located in the intergenic region, 45 were within the CDS region, and the remaining 2 were miscellaneous.
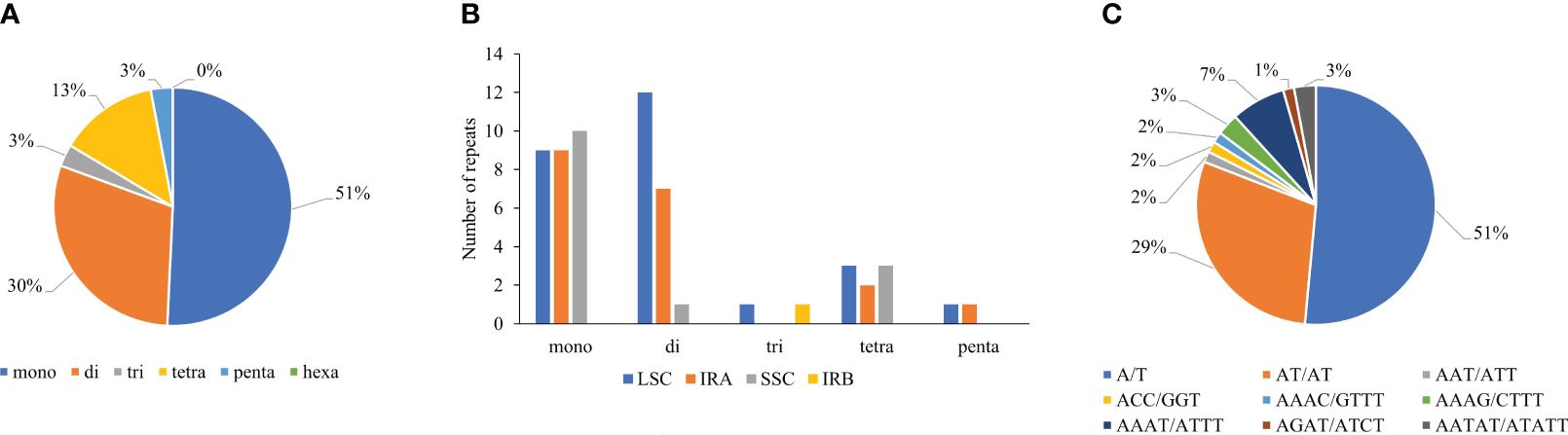
Figure 3 Repeat type and number of analyzed SSRs in the cp genome of T. fargesii. (A) SSR-type distribution and their frequency in the complete cp genome. (B) Distribution of SSRs in the LSC, SSC, and IR regions. (C) Type and frequency of each identified SSR.
3.6 Long repeats
Long repetitive sequences have proven to be valuable markers for comparative genomics, phylogenetics, genomic recombination studies, and plant evolution (Ivanova et al., 2017). According to our analysis, T. fargesii consisted of major repetitive sequences between 30 and 40 bp in length, including 30–32 bp, 33–35 bp, 36–38 bp, 38–40 bp, and ≥40 bp, accounting for 15, 4, 2, 4, and 3 repeats, respectively (Figure 4A). In total, the cp genome contained 27 long repeats, namely, 17 palindromic repeats (P), 9 forward repeats (F), 1 reverse repeat (R), and no complementary repeats (Supplementary Table 5) (Figure 4B). Among the total repeats, 66.66% were in the LSC region, 29.62% were in the IR region, and only 3.70% were in the SSC region (Figure 4C). Only six genes (pafI, psaB, trnS-GCU, rpl16, ccsA, and ycf2) possessed long repeat elements, and ycf2 contained the highest number of repeat sites (n = 8, 29.62%). The remaining repeats were located in intergenic regions.
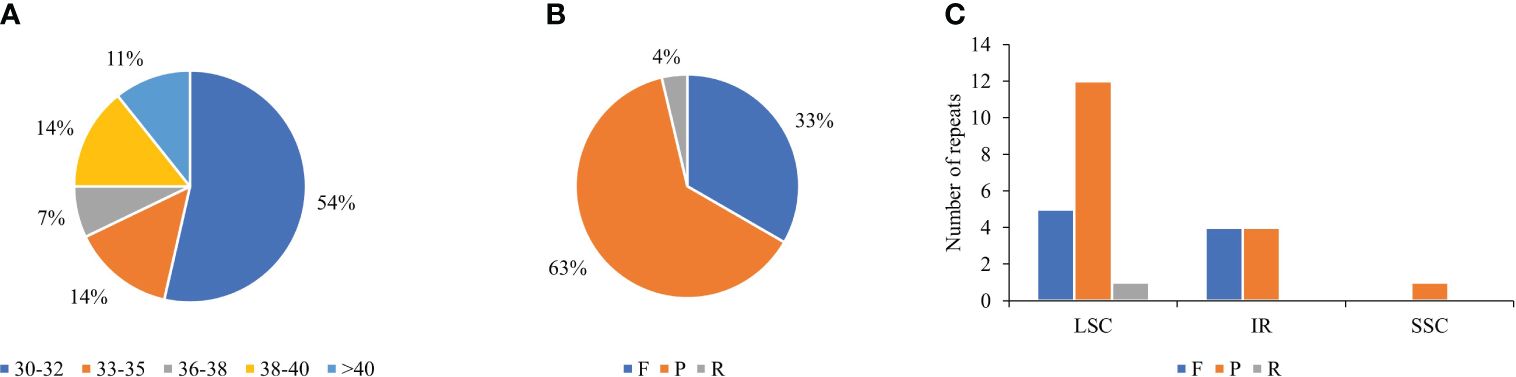
Figure 4 Distribution types and number of long repeats in the cp genome of T. fargesii. (A) Frequencies of different groups of repeats according to their length in the cp genome. Different colors indicate repeats of various sizes in bp. (B) Frequencies of different types of repeats: forward (F), palindrome (P), and reverse (R). (C) Distribution of the long repeats in the LSC, SSC, and IR regions. Different colors indicate the types of repeats.
3.7 Tandem repeat analysis
Centromeres are essential for chromosomal segregation and contain megabase-scale arrays of tandem repeats. These tandem repeats share common properties among species across different phyla (Melters et al., 2013). In total, 21 tandem repeats were identified in the cp genome of T. fargesii, 12 of which were dispersed in the gene spacer regions (rpoB/trnC-GCA, petN/psbM, petN/psbM, petN/psbM, psbZ/trnG-GCC, psbZ/trnG-GCC, atpB/rbcL, trnW-CCA/trnP-UGG, trnP-UGG/psaJ, trnP-UGG/psaJ, rps12/trnV-GAC, and trnV-GAC/rps1), and the remaining nine repeats were spread in the CDS region. One tandem repeat was found in rpl16 and each ycf2 gene had four repeats (Supplementary Table 6).
3.8 Prediction of RNA-editing sites
Using Deepred-Mt, 242 high-confidence C-to-U RNA editing sites were successfully identified across 60 PCGs (Supplementary Figure 1; Supplementary Table 7). Among these, ycf2 had the highest number of RNA editing sites (24), followed by ycf1 and rpoC2 (17 and 13, respectively), making them the top three genes for RNA editing. Additionally, both ndhB and psaB contained 11 spots, and accD, atpA, matK, ndhF, psaA, and psbA carried seven editing sites. In descending order, fewer editing sites were present in the PCGs, including six (psbC and psbD), five (ndhA and rpoB), four (atpI, ccsA, ndhD, ndhG, and ndhK), three (atpE, cemA, pafII, petB, and psbB), and two (atpB, atpF, ndhC, ndhH, rbcL, rpl16, rpl2, rpl36, and rpoC1). One editable site was detected in genes atpH, clpP1, ndhI, pafI, petA, petD, petL, psaI, psdJ, psbK, psbL, psbT, psbZ, rpl20, rpl23, rpoA, rps14, rps16, rps18, and rps7.
3.9 Comparison of IR boundaries within Thalictrum chloroplast genomes
Significant contributions to variations within cp genomes among different species were observed, owing to the contraction and expansion of IR regions, which resulted in gene duplication, deletion, and pseudogene generation (Song et al., 2022). The locations of LSC/IR and SSC/IR junctions are regarded as indices of cp genome evolution (Zhang et al., 2013a). Figure 5 shows that the size of the cp genome within the Thalictrum was 154,889–156,103 bp; the lengths of the LSC and SSC were 84,733–85,507 bp and 17,470–17,657 bp, respectively; and the lengths of the IR regions were 26,272–26,521 bp. The LSC/IRB border is located within rps19 and extends 53–119 bp into the IRB region. Notably, the rps19 gene spanned 119 bp from the LSC to IRB in the cp genome of T. coreanum, whereas it stretched by only 100 bp in the same direction in the T. fargesii plastome. In T. thalictroides, the LSC/IRB border is flanked by an intergenic region of 28 bp between rps19 and the border, and the IRB/SSC border is between the partial ycf1 and ndhF genes.
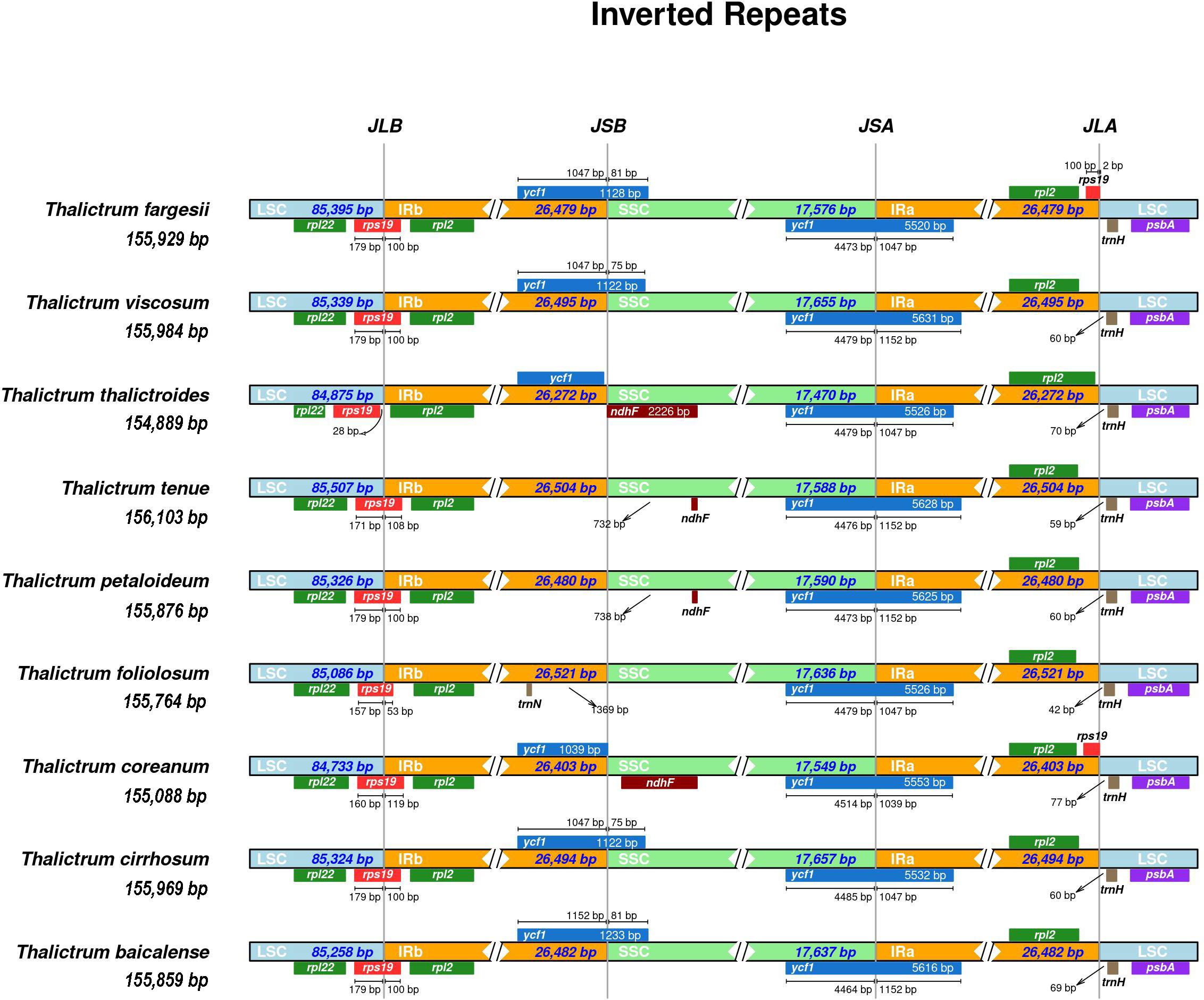
Figure 5 Comparison of IR boundaries of chloroplast genomes within Thalictrum. The junctions are the LSC-IRB, IRB-SSC, SSC-IRA, and IRA-LSC. The length inside the boxes indicates the distance between the end of the gene and the border sites. Arrows indicate the distance from the junction.
In the SSC region, the ndhF gene was larger in T. thalictroides than in the other species. Moreover, T. foliolosum lacks ndhF. The ycf1 fragment gene crosses the IRB/SSC border as a pseudogene in T. viscosum, T. cirrhosum, T. balcalense, and T. fargesii. In contrast, the SSC/IRA junction was positioned on the full-length ycf1 gene in all the species. However, the size of the overlapping ycf1 gene at the SSC/IRA junction varies among the Thalictrum species. Remarkably, this gene enveloped 1,039–1,152 bp of the IRA region in all species. In addition, the IRA/LSC border was pointed between the rpl2 and trnH genes and 42–77 bp from the trnH region toward the LSC region. Notably, exceptions were observed in T. fargesii and T. coreanum, where the rps19 fragment was present at the edge of the IRA/LSC. The rps19 fragment ended at the termination of the IRA region in T. coreanum, whereas it was 2 bp extended toward the LSC in T. fargesii. These results demonstrate that the IR and SC borders of the cp genomes within the Thalictrum are primarily similar in terms of organization, gene content, and gene order.
3.10 Divergence hotspot regions
Highly variable chloroplast genome sequences can be used to ascertain phylogenetic relationships between species and genera (Liu et al., 2018; Wu et al., 2018). To assess the degree of sequence divergence, we computed nucleotide diversity values (pi) to locate hotspots; hence, the results are presented for the entire T. fargesii cp genome in a plot of pi values ranging from 0 to 0.033 (Figure 6). In the plot, the IR regions were shown to have a lower variability than the SSC and LSC regions. Remarkably, seven highly variable regions (pi > 0.02) were detected: three in the LSC (psbM-trnD-GUC, trnF-GAA-ndhj, and atpB), two in the SSC (ycf1 and trnR-ACG-rrn4.5), and two in the IRA (ndhD-psaC and rpl16-rps3). These regions may also be employed as high-potential fragments to distinguish between different Thalictrum species and may help in the development of molecular markers.
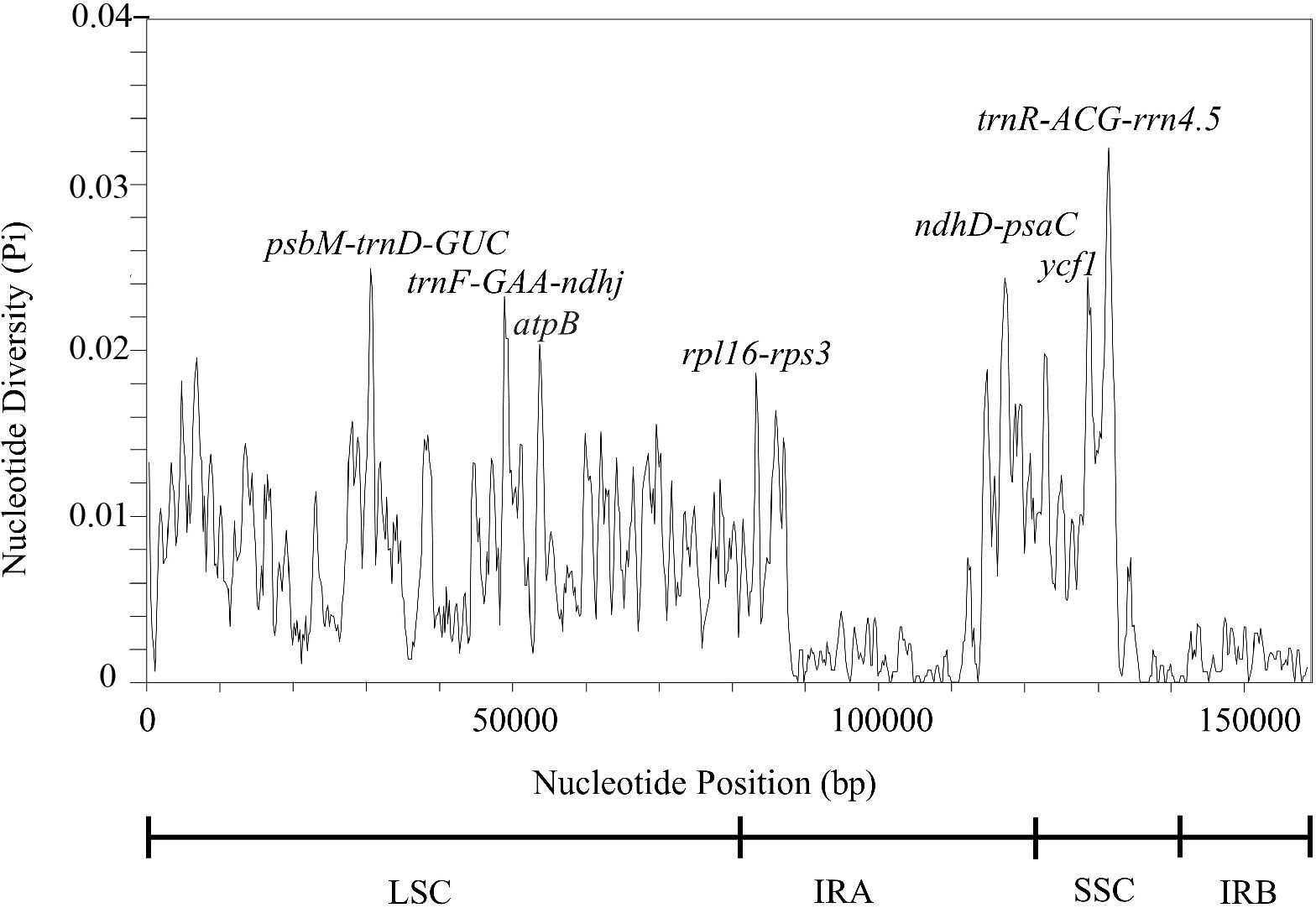
Figure 6 Sliding window analysis of the complete T. fargesii cp genome. Nucleotide diversity analysis (π value) of the complete chloroplast genome of T. fargesii. Window length: 600 bp; step size: 200 bp. X-axis: Position of the midpoint of the window. Y-axis: Nucleotide diversity of each window.
3.11 Selection pressure on genes
In this study, we analyzed the rates of nonsynonymous (dn), synonymous (ds), and dn/ds (ω) ratios among 77 PCGs common to Thalictrum, and the results are shown in Supplementary Table 8. Among the 77 PCGs, the results of the statistical neutrality test indicated that the genes were relatively stable during the evolutionary process, and only 12 genes were found to have ω values less than 1 (Supplementary Figure 2). To manifest, seven genes (atpF, matK, petG, rpl22, rps2, ycf1, and pafI) were under neutral selection (ω = 1), and four genes (atpE, ccsA, ndhG, and rpl20) had ω values of precisely 0.5, indicating that the genes were under purifying selection. The remaining gene, ndhF, had a ω = 0.33, suggesting that it was also under purifying selection.
3.12 Phylogenetic analysis
The appearance of a complete T. fargesii cp genome provided us with access to sequencing data that could be used to investigate the phylogeny and, therefore, contribute to our understanding of evolutionary relationships within the Thalictrum. In the present study, nine species were analyzed to ascertain their phylogenetic position, and Aconitum delavayi (NC_038097) was chosen as an outgroup. ML analysis revealed a single phylogenetic tree with higher bootstrap values, which resulted in constructing one cluster from the complete cp genome of Thalictrum species divided into two clades (Figure 7). Hence, Thalictrum was strongly supported as a monophyletic group [bootstrap support (bs) = 100%]. This result indicates that T. fargesii is closely related to T. tenue and T. petaloideum.
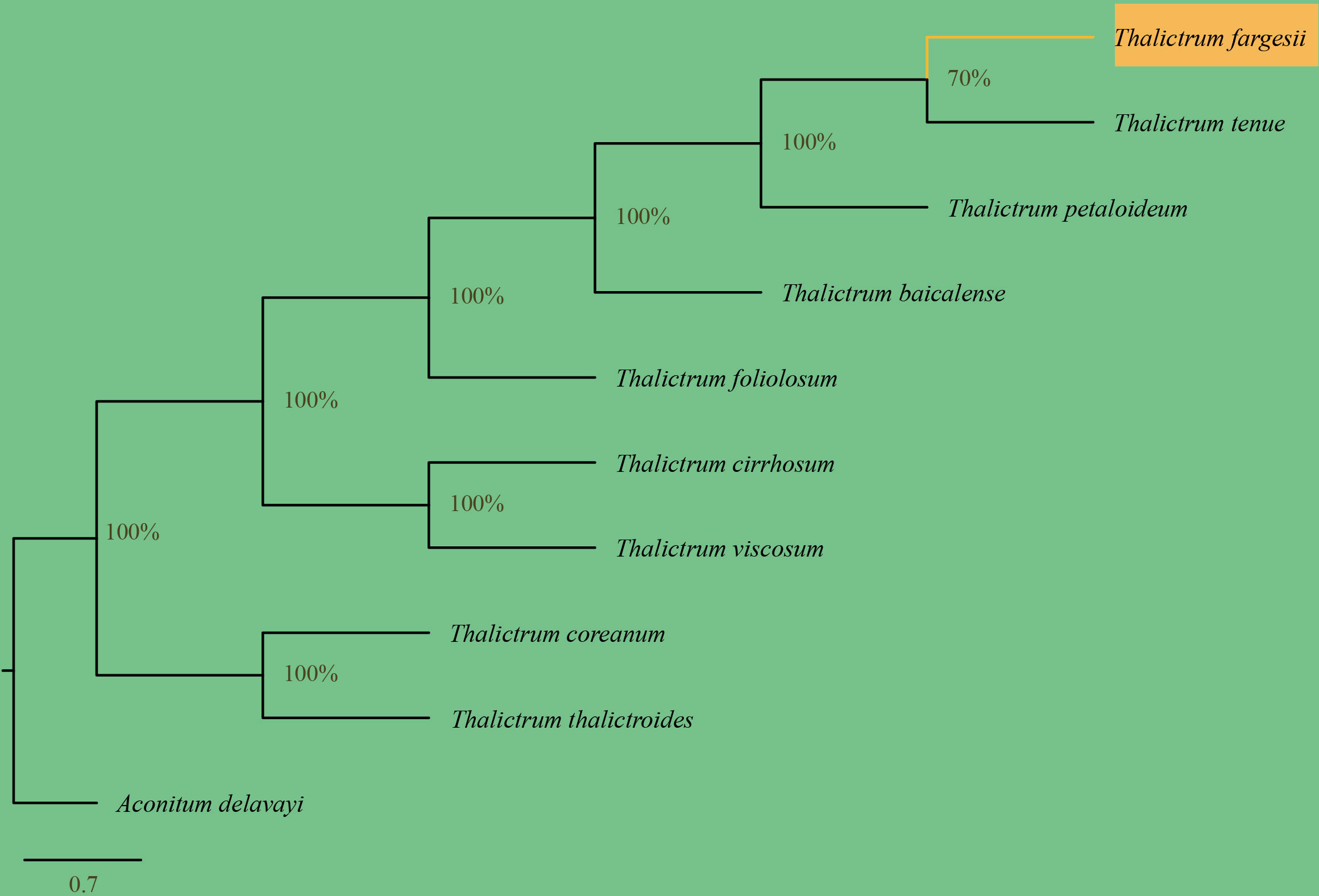
Figure 7 Phylogenetic relationships of Thalictrum inferred from maximum likelihood (ML) analysis based on whole chloroplast genomes. The numbers in each node indicate ML bootstrap values. The scale bar shows 0.7 changes.
3.13 Variations in gene content within Thalictrum
Regarding variation in gene content, we observed that pafI and pafII were found only in the cp genomes of T. fargesii and T. cirrhosum (Supplementary Table 9). Moreover, in the case of pseudogenes, ycf1, infA, and rpl32 were identified in the T. baicalense, T. coreanum, T. thalictroides, and T. fragesii cp genomes, whereas the rps19 fragment (as a pseudogene) existed only in T. coreanum and T. fargesii, indicating significant variations. The remaining species in the genus showed a relative absence of pseudogenes in their cp genomes.
3.14 Hotspots of variant sites in the chloroplast genome
Variant sites are the most useful for identifying genes with significant variation and can facilitate the development of molecular markers (Li and Cullis, 2023). The whole cp genomes of the nine Thalictrum species were compared with those of T. fargesii, which served as a reference to investigate variant sites. Our results showed that 3.60% of the variant sites were present, and the conserved sites accounted for 96.40%, suggesting that these sequences were highly conserved within one genus (Supplementary Table 2; Figure 8). Of the variant sites, 5,609 (3.60%) included 1.51% extragenic variant sites and 2.08% intragenic variant sites, indicating that the noncoding regions were highly conserved (Figure 8A). Within the intragenic sites, there were 1,500 (48%), 951 (30%), 550 (18%), 111 (4%), and 11 (0%) variant loci in the genes related to photosynthesis, self-replication, other, t-RNA, and unknown, respectively (Figure 8B). Furthermore, we calculated the frequency of variant sites within each category. Genes that contained more than 5% of the variant sites within each category included photosynthesis (ndhD, ndhA, pafI, petA, psbB, and ndhK; Figure 8C), self-replication (rpl32, rpl33, rps18, rps19, rpl16, rps16, rpl20, and rpl2; Figure 8D), t-RNAs (trnQ-UUG, trnC-GCA, trnK-UUU, and trnG-UCC; Figure 8E), unknown (ycf1 and yfc15; Figure 8F), and others (cemA, matK, accD, clpP1, and ycf1; Figure 8G).
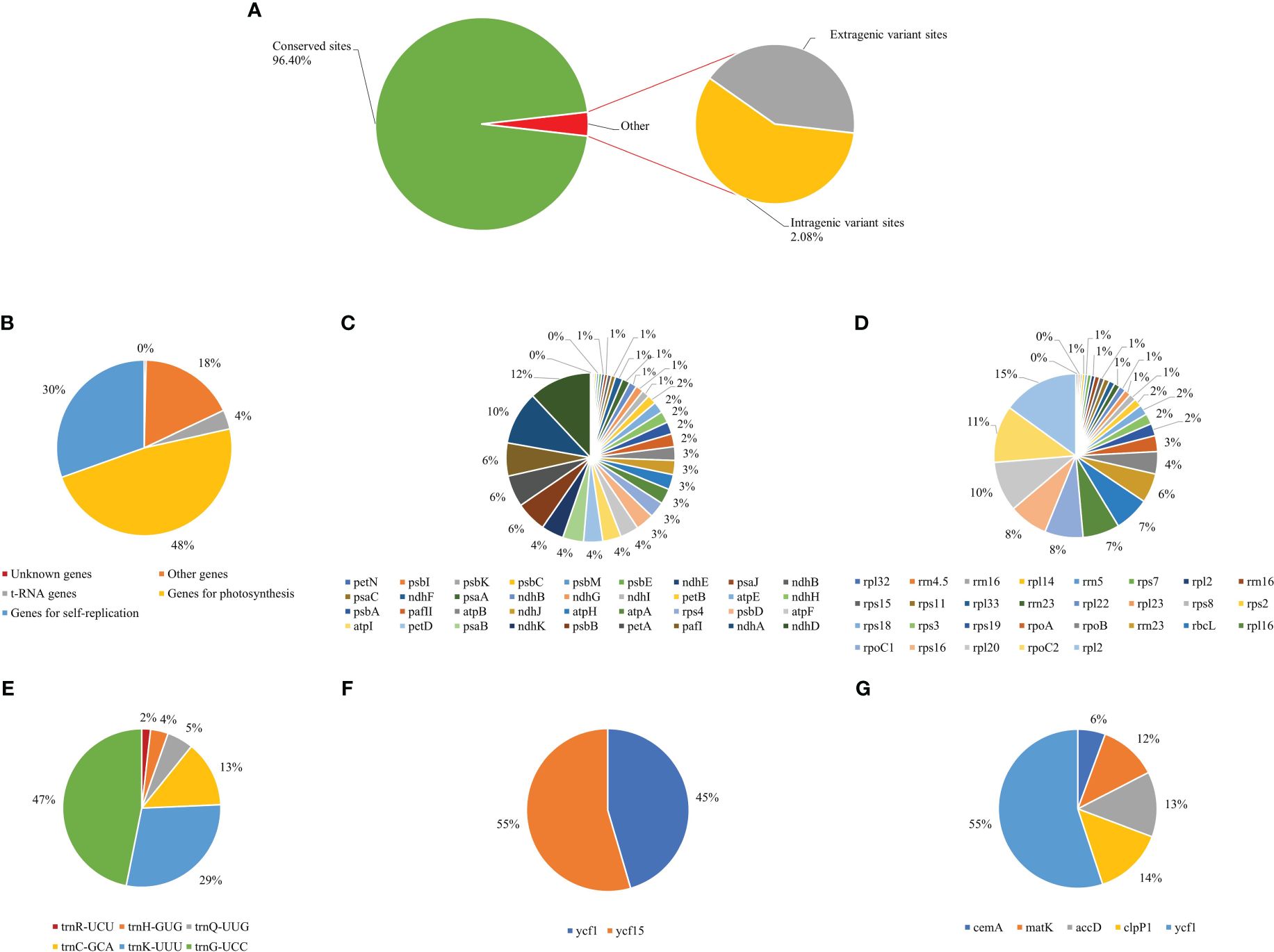
Figure 8 The ratio of chloroplast genome variant sites in Thalictrum species. (A) Distribution of the different variant sites. (B) The frequency of intragenic variant loci in different gene categories. (C) Frequency of variant loci among photosynthesis genes. (D) Frequency of variant loci among genes involved in self-replication. (E) Frequency of variant loci in the t-RNA genes. (F) Frequency of variant loci in the unknown genes. (G) Frequency of variant loci in the other genes.
4 Discussion
4.1 The conserved genome of Thalictrum
T. fargesii is of considerable importance in herbal medicine in Asian countries; however, taxonomic controversies need to be resolved, which may be possible from a detailed study of its plastome genome (Li et al., 2020). Such a systemic investigation and evolutionary analysis of T. fargesii has not been published previously. Therefore, our study reports the first whole cp genome composition of T. fargesii, which provides potential evidence via phylogenomics. Previous studies on other angiosperm groups have shown that chloroplast genomes are conserved or highly polymorphic (Abdullah et al., 2020, Abdullah et al., 2021). In the present study, the cp genome was highly conserved in terms of gene order, gene content, and intron number, following the findings of many other taxa, including Thalictrum (Tang et al., 2015; Yu et al., 2018; Xiang et al., 2022). However, in some taxa, such as Amorphophallus of Araceae, some genes were lost (Liu et al., 2019), and in others, such as Pelargonium (Chumley et al., 2006), the structure and gene order diverged from those reported here and in most other angiosperms.
Overall, the structure and organization of the nine observed cp genomes in Thalictrum, including T. fargesii, follow the typical structure commonly found in angiosperms, except for a difference in tRNA content in T. viscosum, which contains 38 tRNAs instead of 37 (Park et al., 2015; Daniell et al., 2016; He et al., 2019; Morales-Briones et al., 2019; He et al., 2021; Cai et al., 2022; Zhe et al., 2023). The total length, GC content, and gene composition were almost identical in the cp genomes of all studied species, and these highly conserved features might be due to a typical evolutionary passage in Thalictrum (Khan et al., 2020; Villanueva-Corrales et al., 2021; Feng et al., 2022). Compared to the LSC and SSC regions, the IR region was more conserved among the cp genomes.
In addition, the ratio of nucleotide substitutions (dn/ds) and their occurrence are frequently utilized as powerful tools to elucidate the evolution of protein-encoding genes and species adaptations (Kimura, 1989; Fay and Wu, 2003). It is noteworthy that the chloroplast genome of T. fargesii only contains neutral selection sites and purifying sites, suggesting that these genes have conserved functions throughout their evolutionary history (Khakhlova and Bock, 2006). These results offered further evidence that the cp genomes of the Thalictrum are conserved.
4.2 Spotlights on significant molecular variations within Thalictrum
Next-generation sequencing was used to sequence the cp genomes of the ethnodrug species T. fargesii. The plastome of this species varies in terms of genomic structure, nucleotide diversity, structural alterations, and phylogeny with the plastomes of Thalictrum species (Park et al., 2015; He et al., 2019; Morales-Briones et al., 2019; He et al., 2021; Cai et al., 2022; Zhe et al., 2023). For instance, there is an apparent difference between Thalictrum plastomes in the IR-SC boundary areas, which causes variations in genome size (Xiang et al., 2022). IR-SC contractions or expansions into or out of neighboring single-copy regions are typically observed in angiosperm plastomes, and this process is a primary mechanism and an example of length variation in cp genomes (Kim and Lee, 2004; Yang et al., 2016; Zhang et al., 2016; Ye et al., 2018). Indeed, in our investigation, several genes, including ycf1, rps19, ndhF, ycf15, and ψrpl32, were worth examining more closely. For example, ycf1 is duplicated in the cp genomes of several members of the Thalictrum, with a larger copy (ycf1, 5,616–5,658 bp) positioned at the SSC-IRA boundary and a fragmented copy (ψycf1, 1,144–1,152 bp) located at the SSC-IRB boundaries, including T. fargesii (Xiang et al., 2022). Other taxonomic groups also contain the pseudogene ycf1 at the same junction (Henriquez et al., 2020; Mehmood et al., 2020). According to our findings, the shortest ycf1 gene was present in the cp genome of T. fargesii (5,520 bp), with more than 5% of the variant sites within the genus. Thus, ycf1 has enormous potential for use in developing molecular markers and has also been reported in Fritillaria and Pulsatilla (Bi et al., 2018; Li et al., 2018, Li et al., 2020).
Among such variations in cp genomes, molecular evolutionary phenomena including mutations, substitutions, selections, and random genetic drifts play a significant role. One of the molecular features known as “codon use bias” also helps unveil chloroplast and nuclear genome relationships and has been influenced by the GC content at codon locations in various organisms (Yang et al., 2018). Moreover, it is a critical feature for studying the functions of genes and contributes to its evolutionary relationship (Wang et al., 2023; Yang et al., 2023). We disclosed the presence of AT-rich intergenic regions in our present analysis, which is harmonized with the phenomenon that the overall GC content in the cp genomes of terrestrial plants is often low (Yang et al., 2017). Codon usage has a strong relationship with GC content in the codon position, and it is considered an important parameter in understanding evolutionary processes and selection pressure on genes (Zhang et al., 2013b). Our investigation revealed that of the 31 codons with RSCU values higher than one, 16 ended in U and 13 ended in A, indicating natural selection and mutation (Necşulea and Lobry, 2007). This finding suggests that T. fargesii prefers to employ synonymous codons with a third base, A or U, which is comparable to that of other Thalictrum species (Xiang et al., 2022; Zhe et al., 2023). This may be attributed to the abundance of A/T nucleotides in the angiosperm cp genomes (Abdullah et al., 2020). For instance, alanine and leucine were the most common codons in the T. fargesii chloroplast, which might be related to the abundance of A or T in the IR region (Chen et al., 2015), and is consistent with previous studies on the cp genomes of angiosperms (Orešič and Shalloway, 1998). Lauraceae exhibited a similar trend (Cao et al., 2023); however, the cp genome of Lespedeza showed comparable results (Somaratne et al., 2019).
Repeat sequences, particularly SSRs, are widely distributed in cp genomes, as microsatellite repeats play an essential role in gene expression, transcription control, and chromosomal construction, and display a high level of polymorphism (Qi et al., 2018; Huang et al., 2022; Xia et al., 2022). Our results support the hypothesis that cp-genome SSRs have substantial A/T bias, and this trend frequently occurs in many plants (Vieira et al., 2014; Wang et al., 2018). Similar to T. fargesii, mononucleotide repeats are the most abundant in Fritillaria, Lilium, and Epimedium (Rønsted et al., 2005; Zhang et al., 2016). Xiang et al. (2022) have reported 39 to 60 SSRs among 11 Thalictrum species; however, we have identified a total of 68 such repeats. In future genetic studies, we may leverage that SSRs could be used as a source of molecular markers and genomic barcodes for the T. fargesii cp genome. Previous studies have demonstrated that improperly combined repeating or sliding sequences can cause genomic rearrangements (Lu et al., 2017). Long repeat sequences promote cp genome rearrangement and increase the genetic diversity of a population (Guo et al., 2017; Cui et al., 2019). For example, the ycf2 gene provides a suitable resource for genomic studies in the cp genome of T. fargesii, similar to those in other plant species (Cauz-Santos et al., 2017; Shen et al., 2017; Liu et al., 2018). Owing to the presence of different repeats in genes, such as ycf1, ycf2, and rpl32, resulting in a pseudogenized gene or loss of functions, become a significant molecular feature of cp genome in plant evolution and identification (Santos and Pereira, 2018; Park et al., 2019; Miao et al., 2022).
In higher plants, RNA editing occurs as a codon-specific event for converting cysteine (C) to uridine (U) during posttranslational modification. Therefore, this molecular process is crucial for RNA maturation to prevent unwanted mutations and to act as a signal for adaptation (Chu and Wei, 2019; Somaratne et al., 2019). Numerous plants, including Arabidopsis, tobacco, pea, and tomato, have RNA editing sites (Wang et al., 2016). Thus, our findings on the significant C-to-U editing mechanism in the plastid genome of T. fargesii can aid in detecting missense mutations in its genes (Chen et al., 2018; Small et al., 2020).
Divergent regions in cp genomes can help to identify closely related species and reveal phylogenetic information (Dong et al., 2012; Du et al., 2017). These regions have been adopted to generate barcodes for plant identification in other families of plants, including woody plants (Tan et al., 2018; Yan et al., 2023). According to our findings, ndhD-psaC and rpl16-rps3 were found to be divergent hotspots in the cp genome of T. fargesii, and have also been reported in other studies on the plastome of Thalictrum (Xiang et al., 2022). The same phenomenon has been witnessed for numerous other taxa such as Lagerstroemia and Adrinandra (Cui et al., 2019; Dong et al., 2021; Nguyen et al., 2021; Ren et al., 2022; Yang et al., 2022). In addition, these mutational hotspots might aid researchers in studying population genetics and identifying Thalictrum species.
To evaluate the selection pressure on genes and genomes, dn and ds values are important markers in the cp genome. Selection pressure plays a vital role in the adaptation to different environments (Yang and Nielsen, 2000; Gao et al., 2019). Several genes in T. fargesii were found to have a lower dn/ds ratio, suggesting that they are likely to undergo disadvantageous non-synonymous substitutions and purifying selections (Sheikh-Assadi et al., 2022). In T. coreanum, T. foeniculaceum, T. foliolosum, and T. thalictroides, the ndhG gene was significant under positive selection, with a lower codon bias index, suggesting a lower expression level in vivo (Xiang et al., 2022). Moreover, they reported that atpF, petG, rpl20, and rpl22 were under positive selection. In contrast, in the present study, the ndhG and rpl20 were observed under purifying selection, and atpF, petG, and rpl22 were supported by neutral selection in T. fargessii. Thus, purifying selection might be a result of an evolutionary process that preserves the adaptive behavior of this species.
Interestingly, the pafI and rps19 genes in the plastome of T. fargesii exhibited more than 5% variation within the Thalictrum, which was consistent with the findings of Li et al. (2014). In addition, the pafI gene contains two mono-type (10 bp each) SSR repeats and a 39-bp forward repeat at one of its introns, similar to the findings elucidated by Kurt et al. (2023). With the meaningful characteristic architecture of the IRA-LSC, the distance between the rpl2 and trnH genes was markedly different from that of other species in the genus. Moreover, similar to the phenomenon observed in Fritillaria, the presence of the rps19 fragment at the IRA-LSC junction in the T. fargesii cp genome and its distance from trnH make this region a probable string for marker design (Li et al., 2014). Cumulatively, these findings provide molecular evidence for differentiating the ethnodrug plant T. fargesii from other species of Thalictrum.
4.3 Insights into the phylogeny of Thalictrum
The phylogeny and classification of Thalictrum have long been debated, and distinguishing T. fargesii plants from plants with similar morphological traits remains unclear (Xiang et al., 2022). Plastid genome sequences have proven useful for phylogenetic investigations of angiosperms (Jansen et al., 2007; Huang et al., 2014; Kim et al., 2015; Li et al., 2019). This approach is valuable for classifying organisms at lower taxonomic levels (He et al., 2012; Zhang et al., 2016). Previous phylogenetic studies of the genus Thalictrum have used traditional approaches to classify this genus that includes rpl16 intron and ndhC-trnV-UAC by Soza et al., (2012, 2013), and ndhA intron, trnLUAA-trnF-GAA, rpl32-trnL-UAG, and rbcL by Wang et al. (2019). Moreover, Xiang et al. (2022) found that 116 IGS regions had stronger support than 114 gene sequences and revealed that the closest phylogenetic relationship existed within T. minus and T. aquilegiifolium, and T. coreanum and T. thalictroides. Several studies have uncovered that close relationships were present between T. minus and T. tenue (He et al., 2019; Lin et al., 2021; Pu et al., 2022), T. tenue and T. petaloideum (He et al., 2019; Pu et al., 2022), T. minus and T. petaloideum (He et al., 2019; Pu et al., 2022), T. minus and T. aquilegiifolium (Cai et al., 2022; Michimoto et al., 2022; Xiang et al., 2022), T. viscosum and T. cirrhosum (Cai et al., 2022; Michimoto et al., 2022), and T. coreanum and T. thalictroides (He et al., 2021; Lin et al., 2021; Michimoto et al., 2022; Xiang et al., 2022) based on all sequences and whole cp genome. However, none of their circumscribed relationships have uncovered the T. fargesii status within this genus. The availability of complete cp genomes of T. fargesii, as investigated by the present authors, provides sufficient information to establish phylogenetic relationships among the same taxa. According to their results, T. fargesii is closely related to T. tenue and T. petaloideum, supporting the conclusion of an earlier study that T. baicalense is closely related to T. tenue, T. minus, and T. petaloideum (He et al., 2021). Interestingly, T. viscosum has been reported to be closely related to T. cirrhosum and T. foeniculaceum (Cai et al., 2022). Conferring with more Thalictrum taxon samples, our phylogenetic studies of cp genome sequences, reported for the first time, revealed significantly enriched phylogenetic resolution and provided robust inferences of intraspecific relationships. This finding expands the scope of future research on the correlation between these mutations in the cp genome in terms of genetic evolution and speciation in this species. Further studies should entail selecting more samples from wild populations based on their ecotypes and collecting more extensive data from both the nuclear and cp genomes to establish more detailed evidence of phylogenetics and species evolution.
5 Conclusion
Altogether, our findings revealed the complete cp genome of T. fargesii using the Illumina HiSeq platform and a comparative analysis with those of other Thalictrum species provides a fundamental reference for phylogenetic studies. The chloroplast genomes of Thalictrum were compared, and although they were found to be highly conserved in terms of structure, organization, gene order, IR boundaries, and gene content, we have reported distinguishable features of T. fargesii. In particular, two features have clearly demarcated the cp genome of T. fargesii within the genus, which is the length of pseudogenes and their distance from IR/SC borders. Moreover, pafI and rps19 had highly variable sites in the cp genome of T. fargesii compared with other species in the genus. In addition, the divergent regions identified in the present study via nucleotide divergence analysis could help design molecular markers to identify this ethnodrug and its contaminants. Phylogenetic analysis revealed close relationships among T. fargesii, T. tenue, and T. petaloidium. The aforementioned analytical findings significantly contribute to the understanding of the cp genome of T. fargesii and provide genomic information to aid taxonomic identification and phylogenetic relationships within the Thalictrum species. In conclusion, our study provides powerful resources and valuable scientific references for identifying T. fargesii plants for the safety and effectiveness of ethnodrug use and contributes to the bioprospecting and conservation of this species.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
SC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing, Funding acquisition, Project administration. FS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. LAk: Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. LAo: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Writing – review & editing. YZ: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – review & editing. YQ: Methodology, Validation, Resources, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the Natural Science Foundation of Sichuan Province (http://202.61.89.120/) (No. 2022NSFSC1721 to SC, 2021YFN0033 to YZ); Science and Technology Program Projects of Sichuan Provincial Science and Technology Department (2021YFN0028 to SC), and the Neijiang Normal University School-level Research project (http://www.njtc.edu.cn) 448 (2023YB12 to LAo).
Acknowledgments
The authors acknowledge the support received from Neijiang Normal University in conducting this study. Additionally, we extend our thanks to Farhana Tasnim for her assistance in visualizing and necessary editing of figures in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1356912/full#supplementary-material
Supplementary Figure 1 | Distribution of RNA editing sites among the genes in the cp genome of T. fargesii.
Supplementary Figure 2 | Genes under selective pressure in Thalictrum.
References
Abdullah, Mehmood, F., Shahzadi, I., Ali, Z., Islam, M., Naeem, M., et al. (2021). Correlations among oligonucleotide repeats, nucleotide substitutions, and insertion–deletion mutations in chloroplast genomes of plant family Malvaceae. J. Systematics Evol. 59, 388–402. doi: 10.1111/jse.12585
Abdullah, M. F., Shahzadi, I., Waseem, S., Mirza, B., Ahmed, I., Waheed, M. T. (2020). Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): comparative analyses and identification of mutational hotspots. Genomics 112, 581–591. doi: 10.1016/j.ygeno.2019.04.010
Bainsal, N., Bora, K. S., Singh, J. (2022). Pharmacognostic evaluation and phytochemical screening of an unexplored herb: Thalictrum foliolosum DC. Biomed. Pharmacol. J. 15, 985–991. doi: 10.13005/bpj/2434
Beier, S., Thiel, T., Münch, T., Scholz, U., Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, Y., Zhang, M. F., Xue, J., Dong, R., Du, Y. P., Zhang, X. H. (2018). Chloroplast genomic resources for phylogeny and DNA barcoding: a case study on Fritillaria. Sci. Rep. 8, 1184. doi: 10.1038/s41598-018-19591-9
Birky, C. W., Jr. (2001). The inheritance of genes in mitochondria and chloroplasts: laws, mechanisms, and models. Annu. Rev. Genet. 35, 125–148. doi: 10.1146/annurev.genet.35.102401.090231
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Cai, H., Shi, W., Weicai, S., Han, W., Wang, S. (2022). Complete chloroplast genome sequence of Thalictrum viscosum WT Wang & SH Wang 1979(Ranunculaceae). Mitochondrial DNA Part B 7, 1586–1588. doi: 10.1080/23802359.2022.2113751
Cao, Z., Yang, L., Xin, Y., Xu, W., Li, Q., Zhang, H., et al. (2023). Comparative and phylogenetic analysis of complete chloroplast genomes from seven Neocinnamomum taxa (Lauraceae). Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1205051
Cauz-Santos, L. A., Munhoz, C. F., Rodde, N., Cauet, S., Santos, A. A., Penha, H. A., et al. (2017). The Chloroplast Genome of Passiflora edulis (Passifloraceae) Assembled from long sequence reads: structural organization and phylogenomic studies in Malpighiales. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00334
Chen, S. B., Chen, S. L., Xiao, P. G. (2003). Ethnopharmacological investigations on Thalictrum plants in China. J. Asian Natural Products Res. 5, 263–271. doi: 10.1080/1028602031000111941
Chen, J., Hao, Z., Xu, H., Yang, L., Liu, G., Sheng, Y., et al. (2015). The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.00447
Chen, X., Zhou, J., Cui, Y., Wang, Y., Duan, B., Yao, H. (2018). Identification of Ligularia herbs using the complete chloroplast genome as a superbarcode. Front. Pharmacol. 9. doi: 10.3389/fphar.2018.00695
Chew, I. Y. Y., Chung, H. H., Lim, L. W. K., Lau, M. M. L., Gan, H. M., Wee, B. S., et al. (2023). Complete chloroplast genome data of Shorea macrophylla (Engkabang): Structural features, comparative and phylogenetic analysis. Data Brief 47, 109029. doi: 10.1016/j.dib.2023.109029
Chu, D., Wei, L. (2019). The chloroplast and mitochondrial C-to-U RNA editing in Arabidopsis thaliana shows signals of adaptation. Plant Direct 3, e00169. doi: 10.1002/pld3.169
Chumley, T. W., Palmer, J. D., Mower, J. P., Fourcade, H. M., Calie, P. J., Boore, J. L., et al. (2006). The complete chloroplast genome sequence of Pelargonium× hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. doi: 10.1093/molbev/msl089
Corriveau, J. L., Coleman, A. W. (1988). Rapid screening method to detect potential biparental inheritance of plastid DNA and results for over 200 angiosperm species. Am. J. Bot. 75, 1443–1458. doi: 10.1002/j.1537-2197.1988.tb11219.x
Cronn, R., Liston, A., Parks, M., Gernandt, D. S., Shen, R., Mockler, T. (2008). Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 36, e122–e122. doi: 10.1093/nar/gkn502
Cui, Y., Chen, X., Nie, L., Sun, W., Hu, H., Lin, Y., et al. (2019). Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible Amomum species. Int. J. Mol. Sci. 20, 4040. doi: 10.3390/ijms20164040
Da-Cheng, H., Pei-Gen, X., Hong-Ying, M., Yong, P., Chun-Nian, H. (2015). Mining chemo diversity from biodiversity: pharmacophylogeny of medicinal plants of Ranunculaceae. Chin. J. Natural Med. 13, 507–520. doi: 10.1016/s1875-5364(15)30045-5
Daniell, H., Lin, C. S., Yu, M., Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 1–29. doi: 10.1186/s13059-016-1004-2
Dierckxsens, N., Mardulyn, P., Smits, G. (2017). NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18. doi: 10.1093/nar/gkw955
Díez Menéndez, C., Poczai, P., Williams, B., Myllys, L., Amiryousefi, A. (2023). IRplus: An augmented tool to detect inverted repeats in plastid genomes. Genome Biol. Evol. 15, evad177. doi: 10.1093/gbe/evad177
Dong, W., Liu, J., Yu, J., Wang, L., Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One 7, e35071. doi: 10.1371/journal.pone.0035071
Dong, W., Xu, C., Cheng, T., Lin, K., Zhou, S. (2013). Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 5, 989–997. doi: 10.1093/gbe/evt063
Dong, W., Xu, C., Liu, Y., Shi, J., Li, W., Suo, Z. (2021). Chloroplast phylogenomics and divergence times of Lagerstroemia (Lythraceae). BMC Genomics 22, 434. doi: 10.1186/s12864-021-07769-x
Du, Y. P., Bi, Y., Yang, F. P., Zhang, M. F., Chen, X. Q., Xue, J., et al. (2017). Complete chloroplast genome sequences of Lilium: insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 7, 5751. doi: 10.1038/s41598-017-06210-2
Duminil, J., Di Michele, M. (2009). Plant species delimitation: a comparison of morphological and molecular markers. Plant Biosyst. 143 (3), 528–542. doi: 10.1080/11263500902722964
Edera, A. A., Small, I., Milone, D. H., Sanchez-Puerta, M. V. (2021). Deepred-Mt: Deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 136, 104682. doi: 10.1016/j.compbiomed.2021.104682
Fay, J. C., Wu, C. I. (2003). Sequence divergence, functional constraint, and selection in protein evolution. Annu. Rev. Genomics Hum. Genet. 4, 213–235. doi: 10.1146/annurev.genom.4.020303.162528
Feng, Y., Gao, X. F., Zhang, J. Y., Jiang, L. S., Li, X., Deng, H. N., et al. (2022). Complete chloroplast genomes provide insights into evolution and phylogeny of Campylotropis (Fabaceae). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.895543
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., Dubchak, I. (2004). VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Fu, D. Z., Zhu, G. (2001). Thalictrum Linnaeus. Vol. 6. Eds. Wu, Z. Y., Raven, P. H., Hong, D. Y. (St. Louis: Flora of China, Science Press: Beijing & Missouri Botanical Garden Press), 282–302.
Gao, G. Y. (1999). Study on chemical constituents of Thalictrum atriplex and its systematic significance (Beijing: Peking Union Medical College).
Gao, L. Z., Liu, Y. L., Zhang, D., Li, W., Gao, J., Liu, Y., et al. (2019). Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun. Biol. 2, 278. doi: 10.1038/s42003-019-0531-2
Guo, H., Liu, J., Luo, L. I., Wei, X., Zhang, J., Qi, Y., et al. (2017). Complete chloroplast genome sequences of Schisandra chinensis: Genome structure, comparative analysis, and phylogenetic relationship of basal angiosperms. Sci. China Life Sci. 60, 1286–1290. doi: 10.1007/s11427-017-9098-5
Hall, T. A. (1999). BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Ser. 41, 95–98. doi: 10.1007/978-1-4757-0905-6_31
He, Y., Wang, R., Gai, X., Lin, P., Li, Y., Wang, J. (2021). The complete chloroplast genome of Thalictrum baicalense Turcz. ex Ledeb. Mitochondrial DNA Part B 6, 437–438. doi: 10.1080/23802359.2020.1870896
He, S., Wang, Y., Volis, S., Li, D., Yi, T. (2012). Genetic diversity and population structure: implications for conservation of wild soybean (Glycine soja Sieb. et Zucc) based on nuclear and chloroplast microsatellite variation. Int. J. Mol. Sci. 13, 12608–12628. doi: 10.3390/ijms131012608
He, J., Yao, M., Lyu, R. D., Lin, L. L., Liu, H. J., Pei, L. Y., et al. (2019). Structural variation of the complete chloroplast genome and plastid phylogenomics of the genus Asteropyrum (Ranunculaceae). Sci. Rep. 9, 15285. doi: 10.1038/s41598-019-51601-2
Henriquez, C. L., Ahmed, I., Carlsen, M. M., Zuluaga, A., Croat, T. B., McKain, M. R. (2020). Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae). Genomics 112, 2349–2360. doi: 10.1016/j.ygeno.2020.01.006
Hernández-Nicolás, N. Y., Córdova-Téllez, L., Luna-Cavazos, M., Romero-Manzanares, A., Jiménez-Ramírez, J. (2017). Morphological variation related with environmental factors in endemic and threatened jatropha species of tehuacán-cuicatlan, méxico. Genet. Resour. Crop Evol. 64, 557–568. doi: 10.1007/s10722-016-0383-2
Hoot, S. B. (1995). “Phylogeny of the Ranunculaceae based on preliminary atpB, rbcL and 18S nuclear ribosomal DNA sequence data,” in Systematics and evolution of the Ranunculiflorae., vol. 9 . Eds. Jensen, U., Kadereit, J. W. (Springer, Vienna). Supplement 9. doi: 10.1007/978-3-7091-6612-3_24
Huang, Y., Li, J., Yang, Z., An, W., Xie, C., Liu, S., et al. (2022). Comprehensive analysis of complete chloroplast genome and phylogenetic aspects of ten Ficus species. BMC Plant Biol. 22, 1–15. doi: 10.1186/s12870-022-03643-4
Huang, H., Shi, C., Liu, Y., Mao, S. Y., Gao, L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evolutionary Biol. 14, 1–17. doi: 10.1186/1471-2148-14-151
Huang, K. C., Williams, W. M. (1998). The Pharmacology of Chinese herbs. 2nd Edition (Boca Raton: CRC Press). doi: 10.4324/9780367801892
Ivanova, Z., Sablok, G., Daskalova, E., Zahmanova, G., Apostolova, E., Yahubyan, G., et al. (2017). Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.00204
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Depamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Johansson, J. T. (1995). “A revised chloroplast DNA phylogeny of the Ranunculaceae,” in Systematics and Evolution of the Ranunculiflorae (Springer, Vienna), 253–261. doi: 10.1007/978-3-7091-6612-3_25
Johansson, J. T., Jansen, R. K. (1993). Chloroplast DNA variation and phylogeny of the Ranunculaceae. Plant Systemic Evol. 187, 29–49. doi: 10.1007/BF00994090
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Khakhlova, O., Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j.1365-313X.2006.02673.x
Khamidullina, E. A., Gromova, A. S., Lutsky, V. I., Owen, N. L. (2006). Natural products from medicinal plants: non-alkaloidal natural constituents of the Thalictrum species. Natural Product Rep. 23, 117–129. doi: 10.1039/B504014K
Khan, A., Asaf, S., Khan, A. L., Shehzad, T., Al-Rawahi, A., Al-Harrasi, A. (2020). Comparative chloroplast genomics of endangered euphorbia species: insights into hotspot divergence, repetitive sequence variation, and phylogeny. Plants 9, 199. doi: 10.3390/plants9020199
Kim, K. J., Lee, H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kim, K., Lee, S. C., Lee, J., Yu, Y., Yang, K., Choi, B. S., et al. (2015). Complete chloroplast and ribosomal sequences for 30 accessions elucidate the evolution of Oryza AA genome species. Sci. Rep. 5, 15655. doi: 10.1038/srep15655
Kimura, M. (1989). The neutral theory of molecular evolution and the world view of the neutralists. Genome 31, 24–31. doi: 10.1139/g89-009
Kurt, S., Kaymaz, Y., Ateş, D., Tanyolaç, M. B. (2023). Complete chloroplast genome of Lens lamottei reveals intraspecies variation among with Lens culinaris. Sci. Rep. 13, 14959. doi: 10.1038/s41598-023-41287-y
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Langlet, O. F. (1927). Beitrage zur zytologie der Ranunculazeen. Svensk Botanisk Tidskrift 21, 1–17.
Li, J., Cullis, C. (2023). Comparative analysis of 84 chloroplast genomes of Tylosema esculentum reveals two distinct cytotypes. Front. Plant Sci. 13, 1025408. doi: 10.3389/fpls.2022.1025408
Li, Q., Li, Y., Song, J., Xu, H., Xu, J., Zhu, Y., et al. (2014). High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol. 204, 1041–1049. doi: 10.1111/nph.12966
Li, J. Y., Liu, H. J., Wang, Y. Z., Xie, L. (2016). A new synonymy in thalictrum (Ranunculaceae). Phytotaxa 286 (1), 57–58. doi: 10.11646/phytotaxa.286.1.8
Li, Q. J., Su, N., Zhang, L., Tong, R. C., Zhang, X. H., Wang, J. R., et al. (2020). Chloroplast genomes elucidate diversity, phylogeny, and taxonomy of Pulsatilla (Ranunculaceae). Sci. Rep. 10, 19781. doi: 10.1038/s41598-020-76699-7
Li, H. T., Yi, T. S., Gao, L. M., Ma, P. F., Zhang, T., Yang, J. B., et al. (2019). Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 5, 461–470. doi: 10.1038/s41477-019-0421-0
Li, Y., Zhang, Z., Yang, J., Lv, G. (2018). Complete chloroplast genome of seven Fritillaria species, variable DNA markers identification and phylogenetic relationships within the genus. PLoS One 13, e0194613. doi: 10.1371/journal.pone.0194613
Lin, L., He, J., Lyu, R., Yao, M., Luo, Y., Xie, L. (2021). The complete chloroplast genome of a distinctive meadow-rue, Thalictrum foeniculaceum (Ranunculaceae). Mitochondrial DNA Part B 6, 404–405. doi: 10.1080/23802359.2020.1870880
Liu, M. L., Fan, W. B., Wang, N., Dong, P. B., Zhang, T. T., Yue, M., et al. (2018). Evolutionary analysis of plastid genomes of seven Lonicera L. species: implications for sequence divergence and phylogenetic relationships. Int. J. Mol. Sci. 19, 4039. doi: 10.3390/ijms19124039
Liu, E., Yang, C., Liu, J., Jin, S., Harijati, N., Hu, Z., et al. (2019). Comparative analysis of complete chloroplast genome sequences of four major Amorphophallus species. Sci. Rep. 9, 809. doi: 10.1038/s41598-018-37456-z
Lu, R. S., Li, P., Qiu, Y. X. (2017). The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: comparative genomic and phylogenetic analyses. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.02054
Mayor, C., Brudno, M., Schwartz, J. R., Poliakov, A., Rubin, E. M., Frazer, K. A., et al. (2000). VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16, 1046–1047. doi: 10.1093/bioinformatics/16.11.1046
McPherson, H., van der Merwe, M., Delaney, S. K., Edwards, M. A., Henry, R. J., McIntosh, E., et al. (2013). Capturing chloroplast variation for molecular ecology studies: a simple next-generation sequencing approach applied to a rainforest tree. BMC Ecol. 13, 1–11. doi: 10.1186/1472-6785-13-8
Mehmood, F., Shahzadi, I., Waseem, S., Mirza, B., Ahmed, I., Waheed, M. T. (2020). Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): comparative analyses and identification of mutational hotspots. Genomics 112, 581–591. doi: 10.1016/j.ygeno.2019.04.010
Melters, D. P., Bradnam, K. R., Young, H. A., Telis, N., May, M. R., Ruby, J. G., et al. (2013). Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 14, 1–20. doi: 10.1186/gb-2013-14-1-r10
Miao, Y., Chen, H., Xu, W., Yang, Q., Liu, C., Huang, L. (2022). Structural mutations of small single copy (SSC) region in the plastid genomes of five Cistanche species and inter-species identification. BMC Plant Biol. 22, 412. doi: 10.1186/s12870-022-03682-x
Michimoto, K., Ito, T., Maki, M. (2022). The complete chloroplast genome sequence of Thalictrum aquilegiifolium var. sibiricum (Ranunculaceae). Mitochondrial DNA Part B 7, 1171–1173. doi: 10.1080/23802359.2022.2088309
Morales-Briones, D. F., Arias, T., Di Stilio, V. S., Tank, D. C. (2019). Chloroplast primers for clade-wide phylogenetic studies of Thalictrum. Appl. Plant Sci. 7, e11294. doi: 10.1002/aps3.11294
NCBI. (2022). PubChem Taxonomy Summary for Taxonomy 1277774, Thalictrum fargesii. Available at: https://pubchem.ncbi.nlm.nih.gov/taxonomy/Thalictrumfargesii.
Necşulea, A., Lobry, J. R. (2007). A new method for assessing the effect of replication on DNA base composition asymmetry. Mol. Biol. Evol. 24, 2169–2179. doi: 10.1093/molbev/msm148
Nguyen, H. Q., Nguyen, T. N. L., Doan, T. N., Nguyen, T. T. N., Phạm, M. H., Le, T. L., et al. (2021). Complete chloroplast genome of novel Adinandra megaphylla Hu species: molecular structure, comparative and phylogenetic analysis. Sci. Rep. 11, 11731. doi: 10.1038/s41598-021-91071-z
Orešič, M., Shalloway, D. (1998). Specific correlations between relative synonymous codon usage and protein secondary structure. J. Mol. Biol. 281, 31–48. doi: 10.1006/jmbi.1998.1921
Park, S., Jansen, R. K., Park, S. (2015). Complete plastome sequence of Thalictrum coreanum (Ranunculaceae) and transfer of the rpl32 gene to the nucleus in the ancestor of the subfamily Thalictroideae. BMC Plant Biol. 15, 1–12. doi: 10.1186/s12870-015-0432-6
Park, I., Yang, S., Kim, W. J., Song, J. H., Lee, H. S., Lee, H. O., et al. (2019). Sequencing and comparative analysis of the chloroplast genome of angelica polymorpha and the development of a novel indel marker for species identification. Molecules 24, 1038. doi: 10.3390/molecules24061038
Porebski, S., Bailey, L. G., Baum, B. R. (1997). Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15, 8–15. doi: 10.1007/BF02772108
Provan, J., Powell, W., Hollingsworth, P. M. (2001). Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol. Evol. 16, 142–147. doi: 10.1016/S0169-5347(00)02097-8
Pu, T., Zhou, Z., Zhang, Z., Zhang, M. (2022). Characterization of the complete chloroplast genome of Thalictrum foliolosum DC. 1817, a folk medicine plant in China. Mitochondrial DNA Part B 7, 622–623. doi: 10.1080/23802359.2022.2055981
Qi, W. H., Jiang, X. M., Yan, C. C., Zhang, W. Q., Xiao, G. S., Yue, B. S., et al. (2018). Distribution patterns and variation analysis of simple sequence repeats in different genomic regions of bovid genomes. Sci. Rep. 8, 14407. doi: 10.1038/s41598-018-32286-5
Ravi, V., Khurana, J. P., Tyagi, A. K., Khurana, P. J. P. S. (2008). An update on chloroplast genomes. Plant Systematics Evol. 271, 101–122. doi: 10.1007/s00606-007-0608-0
Ren, Y., Gu, T. Q., Chang, H. L. (2011). Floral development of dichocarpum, thalictrum, and aquilegia (Thalictroideae, ranunculaceae). Plant Systematics Evol. 292, 203–213. doi: 10.1007/s00606-010-0399-6
Ren, J., Tian, J., Jiang, H., Zhu, X. X., Mutie, F. M., Wanga, V. O., et al. (2022). Comparative and phylogenetic analysis based on the chloroplast genome of Coleanthus subtilis (Tratt.) Seidel, a protected rare species of monotypic genus. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.828467
Ro, K. E., Keener, C. S., McPheron, B. A. (1997). Molecular phylogenetic study of the Ranunculaceae: utility of the nuclear 26S ribosomal DNA in inferring intrafamilial relationships. Mol. Phylogenet. Evol. 8, 117–127. doi: 10.1006/mpev.1997.0413
Rønsted, N., Law, S., Thornton, H., Fay, M. F., Chase, M. W. (2005). Molecular phylogenetic evidence for the monophyly of Fritillaria and Lilium (Liliaceae; Liliales) and the infrageneric classification of Fritillaria. Mol. Phylogenet. Evol. 35, 509–527. doi: 10.1016/j.ympev.2004.12.023
Santos, C., Pereira, F. (2018). Identification of plant species using variable length chloroplast DNA sequences. Forensic Sci. International: Genet. 36, 1–12. doi: 10.1016/j.fsigen.2018.05.009
Serrano, I., Audran, C., Rivas, S. (2016). Chloroplasts at work during plant innate immunity. J. Exp. Bot. 67, 3845–3854. doi: 10.1093/jxb/erw088
Sheikh-Assadi, M., Naderi, R., Kafi, M., Fatahi, R., Salami, S. A., Shariati, V. (2022). Complete chloroplast genome of Lilium ledebourii (Baker) Boiss and its comparative analysis: lights into selective pressure and adaptive evolution. Sci. Rep. 12, 9375. doi: 10.1038/s41598-022-13449-x
Shen, X., Wu, M., Liao, B., Liu, Z., Bai, R., Xiao, S., et al. (2017). Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 22, 1330. doi: 10.3390/molecules22081330
Singh, H., Singh, D., Lekhak, M. M. (2023). Ethnobotany, botany, phytochemistry and ethnopharmacology of the genus Thalictrum L.(Ranunculaceae): A review. J. Ethnopharmacology 305, 115950. doi: 10.1016/j.jep.2022.115950
Singh, H., Srivastava, A., Husain, T. (2016). Notes on Thalictrum L. (Ranunculaceae) from Indian Himalayas with some ethnobotanical uses. J. Biodiversity Manage. Forestry 05, 2–7. doi: 10.4172/2327-4417.1000162
Small, I. D., Schallenberg-Rüdinger, M., Takenaka, M., Mireau, H., Ostersetzer-Biran, O. (2020). Plant organellar RNA editing: what 30 years of research has revealed. Plant J. 101, 1040–1056. doi: 10.1111/tpj.14578
Somaratne, Y., Guan, D. L., Wang, W. Q., Zhao, L., Xu, S. Q. (2019). The complete chloroplast genomes of two Lespedeza species: Insights into codon usage bias, RNA editing sites, and phylogenetic relationships in Desmodieae (Fabaceae: Papilionoideae). Plants 9, 51. doi: 10.3390/plants9010051
Song, W., Chen, Z., Shi, W., Han, W., Feng, Q., Shi, C., et al. (2022). Comparative analysis of complete chloroplast genomes of nine species of Litsea (Lauraceae): Hypervariable regions, positive selection, and phylogenetic relationships. Genes 13, 1550. doi: 10.3390/genes13091550
Soza, V. L., Brunet, J., Liston, A., Smith, P. S., Di Stilio, V. S. (2012). Phylogenetic insights into the correlates of dioecy in meadow-rues (Thalictrum, ranunculaceae). Mol. Phylogenet. Evol. 63, 180–192. doi: 10.1016/j.ympev.2012.01.009
Soza, V. L., Haworth, K. L., Di Stilio, V. S. (2013). Timing and consequences of recurrent polyploidy in meadow-rues (Thalictrum, ranunculaceae). Mol. Biol. Evol. 30, 1940–1954. doi: 10.1093/molbev/mst101
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tamura, M. (1995). “Ranunculaceae,” in Die Naturlichen Pflanzenfamilien, vol. 17a . Ed. Hiepko, P. (Duncker & Humblot, Berlin, Germany), pp.223–pp.497.
Tamura, K., Stecher, G., Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tan, S. L., Luo, Y. H., Hollingsworth, P. M., Burgess, K. S., Xu, K., Li, D. Z., et al. (2018). DNA barcoding herbaceous and woody plant species at a subalpine forest dynamics plot in Southwest China. Ecol. Evol. 8, 7195–7205. doi: 10.1002/ece3.4254
Tang, Y., Yukawa, T., Bateman, R. M., Jiang, H., Peng, H. (2015). Phylogeny and classification of the East Asian Amitostigma alliance (Orchidaceae: Orchideae) based on six DNA markers. BMC Evolutionary Biol. 15, 1–32. doi: 10.1186/s12862-015-0376-3
Tano, D. W., Kozlowska, M. A., Easter, R. A., Woodson, J. D. (2023). Multiple pathways mediate chloroplast singlet oxygen stress signaling. Plant Mol. Biol. 111, 167–187. doi: 10.1007/s11103-022-01319-z
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Velasco, R., Zharkikh, A., Troggio, M., Cartwright, D. A., Cestaro, A., Pruss, D., et al. (2007). A high-quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One 2, e1326. doi: 10.1371/journal.pone.0001326
Vieira, L. D. N., Faoro, H., Rogalski, M., Fraga, H. P. D. F., Cardoso, R. L. A., de Souza, E. M., et al. (2014). The complete chloroplast genome sequence of Podocarpus lambertii: genome structure, evolutionary aspects, gene content and SSR detection. PLoS One 9, e90618. doi: 10.1371/journal.pone.0090618
Villanueva-Corrales, S., Garcıía-Botero, C., Garceís-Cardona, F., Ramıírez-Rıíos, V., Villanueva-Mejıía, D. F., Aílvarez, J. C. (2021). The complete chloroplast genome of Plukenetia volubilis provides insights into the organelle inheritance. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.667060
Wang, W., Chen, Z. D. (2007). Generic level phylogeny of Thalictroideae (Ranunculaceae)—implications for the taxonomic status of Paropyrum and petal evolution. Taxon 56, 811–821. doi: 10.2307/25065863
Wang, W., Chen, S., Zhang, X. (2018). Whole-genome comparison reveals divergent IR borders and mutation hotspots in chloroplast genomes of herbaceous bamboos (Bambusoideae: Olyreae). Molecules 23, 1537. doi: 10.3390/molecules23071537
Wang, T. N., Clifford, M. R., Martínez-Gómez, J., Johnson, J. C., Riffell, J. A., Di Stilio, V. S. (2019). Scent matters: differential contribution of scent to insect response in flowers with insect vs. wind pollination traits. Ann. Bot. 123, 289–301. doi: 10.1093/aob/mcy131
Wang, M., Liu, H., Ge, L., Xing, G., Wang, M., Weining, S., et al. (2016). Identification and analysis of RNA editing sites in the chloroplast transcripts of Aegilops tauschii L. Genes 8, 13. doi: 10.3390/genes8010013
Wang, Z. K., Liu, Y., Zheng, H. Y., Tang, M. Q., Xie, S. Q. (2023). Comparative analysis of codon usage patterns in nuclear and chloroplast genome of dalbergia (Fabaceae). Genes 14, 1110. doi: 10.3390/genes14051110
Wang, W. T., Wang, S. H. (1979). “Thalictrum L,” in Flora Reipublicae Popularis Sinicae, vol. 27. (Science Press, Beijing), 502–592. Anonymous.
Wolfe, K. H., Li, W. H., Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. 84, 9054–9058. doi: 10.1073/pnas.84.24.9054
Wu, J., Fairchild, E. H., Beal, J. L., Tomimatsu, T., Doskotch, R. W. (1979). Lithospermoside and dasycarponin, cyanoglucosides from Thalictrum. J. Natural Products 42, 500–511. doi: 10.1021/np50005a011
Wu, Y., Liu, F., Yang, D. G., Li, W., Zhou, X. J., Pei, X. Y., et al. (2018). Comparative chloroplast genomics of Gossypium species: insights into repeat sequence variations and phylogeny. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00376
Wu, Z. X., Yi, Y. J. (1991). Studies on chemical constituents of Thalictrum fargesii. J. China Pharm. Univ. 03, 177–178.
Xia, X. (2018). DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 35, 1550–1552. doi: 10.1093/molbev/msy073
Xia, C., Wang, M., Guan, Y., Li, Y., Li, J. (2022). Comparative analysis of complete chloroplast genome of ethnodrug Aconitum episcopale and insight into its phylogenetic relationships. Sci. Rep. 12, 9439. doi: 10.1038/s41598-022-13524-3
Xiang, K. L., Mao, W., Peng, H. W., Erst, A. S., Yang, Y. X., He, W. C., et al. (2022). Organization, phylogenetic marker exploitation, and gene evolution in the plastome of Thalictrum (Ranunculaceae). Front. Plant Sci. 13. doi: 10.3389/fpls.2022.897843
Xie, L. (2016). “Thalictrum L,” in Medical Flora of China, vol. 3 . Ed. Wei, F. N. (Peking University Medical Press, Beijing), 231–287.
Yan, M., Dong, S., Gong, Q., Xu, Q., Ge, Y. (2023). Comparative chloroplast genome analysis of four Polygonatum species insights into DNA barcoding, evolution, and phylogeny. Sci. Rep. 13, 16495. doi: 10.1038/s41598-023-43638-1
Yang, J., Hu, G., Hu, G. (2022). Comparative genomics and phylogenetic relationships of two endemic and endangered species (Handeliodendron bodinieri and Eurycorymbus cavaleriei) of two monotypic genera within Sapindales. BMC Genomics 23, 27. doi: 10.1186/s12864-021-08259-w
Yang, M., Liu, J., Yang, W., Li, Z., Hai, Y., Duan, B., et al. (2023). Analysis of codon usage patterns in 48 Aconitum species. BMC Genomics 24, 703. doi: 10.1186/s12864-023-09650-5
Yang, Z., Nielsen, R. (2000). Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43. doi: 10.1093/oxfordjournals.molbev.a026236
Yang, J., Yue, M., Niu, C., Ma, X. F., Li, Z. H. (2017). Comparative analysis of the complete chloroplast genome of four endangered herbals of Notopterygium. Genes 8, 124. doi: 10.3390/genes8040124
Yang, Y., Zhou, T., Duan, D., Yang, J., Feng, L., Zhao, G. (2016). Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00959
Yang, Y., Zhu, J., Feng, L., Zhou, T., Bai, G., Yang, J., et al. (2018). Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: Highlighting the effect of codon composition bias in phylogenetic inference. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00082
Ye, W. Q., Yap, Z. Y., Li, P., Comes, H. P., Qiu, Y. X. (2018). Plastome organization, genome-based phylogeny and evolution of plastid genes in Podophylloideae (Berberidaceae). Mol. Phylogenet. Evol. 127, 978–987. doi: 10.1016/j.ympev.2018.07.001
Yu, X., Yang, D., Guo, C., Gao, L. (2018). Plant phylogenomics based on genome-partitioning strategies: Progress and prospects. Plant Diversity 40, 158–164. doi: 10.1016/j.pld.2018.06.005
Yu, X., Zuo, L., Lu, D., Lu, B., Yang, M., Wang, J. (2019). Comparative analysis of chloroplast genomes of five Robinia species: Genome comparative and evolution analysis. Gene 689, 141–151. doi: 10.1016/j.gene.2018.12.023
Zeng, Y., Yuan, Q., Yang, Q. E. (2021). Thalictrum nanhuaense (Ranunculaceae), a new species from central Yunnan, China. Phytotaxa 479, 121–128. doi: 10.11646/phytotaxa.479.1.10
Zeng, Y., Yuan, Q., Yang, Q. (2022). Clarification of morphology and distribution of thalictrum fortunei (Ranunculaceae) from china, with reduction of t. fortunei var. bulbiliferum to its synonymy. Phytotaxa 561 (2), 121–137. doi: 10.11646/phytotaxa.561.2.1
Zeng, Y., Yuan, Q., Yang, Q. (2023). Thalictrum minutiflorum (Ranunculaceae), described from guizhou province, china, is merged with t. ramosum. Phytotaxa 609 (3), 165–179. doi: 10.11646/phytotaxa.609.3.1
Zhai, W., Duan, X., Zhang, R., Guo, C., Li, L., Xu, G., et al. (2019). Chloroplast genomic data provide new and robust insights into the phylogeny and evolution of the Ranunculaceae. Mol. Phylogenet. Evol. 135, 12–21. doi: 10.1016/j.ympev.2019.02.024
Zhang, Z., Dai, W., Wang, Y., Lu, C., Fan, H. (2013b). Analysis of synonymous codon usage patterns in torquTFe teno sus virus 1 (TTSuV1). Arch. Virol. 158, 145–154. doi: 10.1007/s00705-012-1480-y
Zhang, Y., Du, L., Liu, A., Chen, J., Wu, L., Hu, W., et al. (2016). The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00306
Zhang, H., Li, C., Miao, H., Xiong, S. (2013a). Insights from the complete chloroplast genome into the evolution of Sesamum indicum L. PLoS One 8, e80508. doi: 10.1371/journal.pone.0080508
Zhang, Q., Liu, Y., Sodmergen (2003). Examination of the cytoplasmic DNA in male reproductive cells to determine the potential for cytoplasmic inheritance in 295 angiosperm species. Plant Cell Physiol. 44, 941–951. doi: 10.1093/pcp/pcg121
Zhang, H., Wang, X., Guo, Y., Liu, X., Zhao, X., Teka, T., et al. (2021). Thirteen bisbenzylisoquinoline alkaloids in five Chinese medicinal plants: botany, traditional uses, phytochemistry, pharmacokinetic and toxicity studies. J. Ethnopharmacology 268, 113566. doi: 10.1016/j.jep.2020.113566
Zhe, H., Xiaoming, L., Ang, L., Changbing, H. (2023). Characteristics of Thalictrum cirrhosum chloroplast genome and its analysis on codon usage bias. J. Southwest Forestry Univ. 43, 47–56. doi: 10.11929/j.swfu.202205026
Zheng, S., Poczai, P., Hyvönen, J., Tang, J., Amiryousefi, A. (2020). Chloroplot: an online program for the versatile plotting of organelle genomes. Front. Genet. 11. doi: 10.3389/fgene.2020.576124
Zhou, J., Xie, G., Yan, X. (2011). Encyclopedia of traditional Chinese medicines - molecular structures, pharmacological activities, natural sources and applications. Eds. Zhou, J., Xie, G., Yan, X. (Heidelberg: Springer Berlin), 1–730. doi: 10.1007/978-3-642-16744-7_1
Keywords: chloroplast genome, Thalictrum fargesii, thalidasine, repeat sequences, RSCU, nucleotide diversity, variant sites, phylogeny
Citation: Chen S, Safiul Azam FM, Akter ML, Ao L, Zou Y and Qian Y (2024) The first complete chloroplast genome of Thalictrum fargesii: insights into phylogeny and species identification. Front. Plant Sci. 15:1356912. doi: 10.3389/fpls.2024.1356912
Received: 16 December 2023; Accepted: 08 April 2024;
Published: 29 April 2024.
Edited by:
Leonardo Miguel Galindo-González, Canadian Food Inspection Agency (CFIA), CanadaReviewed by:
Guanghui Xiao, Shaanxi Normal University, ChinaShirin Aktar, Chinese Academy of Sciences (CAS), China
Copyright © 2024 Chen, Safiul Azam, Akter, Ao, Zou and Qian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fardous Mohammad Safiul Azam, c2hvamliYmlvdGVjaEB5YWhvby5jb20=
†These authors have contributed equally to this work and share first authorship