 Jiahui Sun1,2†
Jiahui Sun1,2† Dan Zhao3†
Dan Zhao3† Ping Qiao2†
Ping Qiao2† Yiheng Wang1,2
Yiheng Wang1,2 Ping Wu4
Ping Wu4 Keren Wang5
Keren Wang5 Lanping Guo1*
Lanping Guo1* Luqi Huang1*Shiliang Zhou4*
Luqi Huang1*Shiliang Zhou4*- 1State Key Laboratory for Quality Ensurance and Sustainable Use of Dao-di Herbs, National Resource Center for Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, China
- 2Dexing Research and Training Center of Chinese Medical Sciences, China Academy of Chinese Medical Sciences, Dexing, China
- 3College of Pharmacy, Guizhou University of Traditional Chinese Medicine, Guiyang, China
- 4State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, Beijing, China
- 5Institute of Highland Forest Science, Chinese Academy of Forestry, Kunming, China
In Rosaceae, the replacement of the traditional four-subfamily division (Amygdaloideae or Prunoideae, Maloideae, Rosoideae, and Spiraeoideae) by the three-subfamily division (Dryadoideae, Rosoideae, and Amygdaloideae), the circumscription, systematic position, and phylogeny of genera in Maleae need to be reconsidered. The study aimed to circumscribe Maleae, pinpoint its systematic position, and evaluate the status of all generally accepted genera in the tribe using complete chloroplast genome data. Results indicated that Maleae consisted of pome-bearing genera that belonged to Maloideae as well as four genera (Gillenia, Kageneckia, Lindleya, and Vauquelinia) that were formerly considered to be outside Maloideae. The tribe could be subdivided into four subtribes: Gilleniinae (Gillenia), Lindleyinae (Kageneckia and Lindleya), Vaugueliniinae (Vauquelinia), and Malinae (all other genera; the core Maleae). Among the 36 recognized genera, Aria, Docyniopsis, Chamaemespilus, and Mespilus were not considered distinct and more research is needed to determine the taxonomic status of Rhaphiolepis from Eriobotrya. Within the core Maleae, five groups were revealed, whereas Sorbus L. was split as its members belonged to different groups.
1 Introduction
Molecular systematics has deeply changed the classification of the Rosaceae family from the subdivision of subfamilies to species. This family includes approximately 2,950 species in 91 genera, of which many are economically important such as apples, pears, peaches, strawberries, roses, and cherries (Christenhusz and Byng, 2016). The conventional four-subfamily division (Amygdaloideae, Maloideae, Rosoideae, and Spiraeoideae) was replaced by a three-subfamily division [Amygdaloideae (including Maloideae and Spiraeoideae), Dryadoideae (divided from Rosoideae), and Rosoideae] that was based on molecular data (Potter et al., 2002, 2007; Xiang et al., 2016; Zhang et al., 2017). In the three-subfamily division, subfamily Maloideae (2n = 34) with inferior ovaries was merged with the subfamily Amygdaloideae (2n = 16, 18) with superior ovaries, whereas the species of Maloideae were grouped into Maleae (Pyrinae; Potter et al., 2007). Besides, the four capsule-producing genera Gillenia Moench (= Porteranthus Britton, 2n = 18), Kageneckia Ruiz & Pav. (2n = 34), Lindleya Kunth (2n = 34), and Vauquelinia Corrêa ex Bonpl. (2n = 30) that formerly included in Spiraeoideae showed a closer genetic relationships with the pome-bearing Maleae (Evans & Campbell, 2002; Campbell et al., 2007; Potter et al., 2007; Li et al., 2012; Lo & Donoghue, 2012; Xiang et al., 2016; Zhang et al., 2017; Sun et al., 2018). Sun et al. (2018) showed that the four capsule-producing genera and the pome-bearing genera comprise the tribe Maleae and that the latter forms the core Maleae.
Taxonomic classification within the core Maleae has been a subject of considerable debate, primarily due to the complex interplay of polyploidy, hybridization, and apomixis. Robertson et al. (1991) identified 28 genera of pome-bearing species, while Gu & Spongberg (2003) delineated 16, underscoring the variability in genus circumscriptions. The pome-bearing species with 2n = 34 (x = 17) were once believed to be originated by a genome merge between species of Amygdaloideae (x = 8) and Spiraeoideae (x = 9) (Sax, 1932; Stebbins, 1950; Phipps et al., 1991). However, such an inter-subfamilial origin was not supported by morphological data (Phipps et al., 1991). Evans & Campbell (2002) proposed a hypothesis of aneuploidy (x = 17 from x = 18) and Gillenia as a possible ancestor based on the nuclear gene GBSSI (granule-bound starch synthase I). Recent studies based on genome information supported aneuploidization events that occurred approximately 50 million years ago (Velasco et al., 2010; Considine et al., 2012). Nonetheless, the presence of two copies of the GBSSI locus in Amelanchier Medik (Evans et al., 2000). may indicate that the allotetraploid origin of genera is one of the pathways in the evolution of the core Maleae, which obscures the distinction among genera and inevitably complicates taxonomic studies. Intergeneric hybridization is rather common in the core Maleae (Robertson et al., 1991, Figure 1), owing to the nature of multiple copies of genes in polyploids and apomixis for the survival of hybrids as well as the close genetic relationships among the genera.
The pome-bearing core Maleae were subdivided into one group that included the connate endocarps and another that included the polypyrenous drupes (Hutchinson, 1964; Schulze-Menz and Melchior, 1964; Phipps et al., 1990; Kalkman and Kubitzki, 2004). In fact, the subdivision of the core Maleae beyond the generic level based on morphological features is considered unreliable due to their complexity (Phipps et al., 1991).
Previous the attempts for the circumscription of genera were of diverse opinions due to their understanding of morphological variations. In any case, there were 36 genera have been recognized previously (Phipps et al., 1990; Robertson et al., 1991; Sun et al., 2018, Supplementary Table S1). Crataegus L., Eriobotrya Lindl., Mespilus L., and Rhaphiolepis Lindl. were taxonomically uncontroversial; however, the genetic divergence of each Mespilus from Crataegus and Rhaphiolepis from Eriobotrya were too small for generic ranking based on molecular evidence (Lo and Donoghue, 2012; Sun et al., 2018). Morphological data did not help to effectively address any taxonomic problems based on phylogeny, especially in the Sorbus-related, Malus-related, and Photinia-related genera. Therefore, molecular tools might help to reconstruct the phylogeny of Maleae (Xiang et al., 2016; Zhang et al., 2017; Sun et al., 2018). Xiang et al. (2016) built a phylogeny of Rosaceae with sequences of 113 genes in which 16 genera of the core Maleae were sampled. Zhang et al. (2017) sampled 25 genera of the core Maleae in their study of Rosaceae. Sun et al. (2018) used 15 chloroplast regions to reconstruct the phylogeny of Maleae and concluded that “it is still premature to make a formal taxonomic treatment for these genera” in the tribe.
In this study, we used the complete chloroplast genome of 35 genera of Maleae except Chamaemeles Lindl. to reconstruct a well-presented phylogeny of the tribe Maleae. Based on our chloroplast genome data and molecular information provided by previously published studies (Xiang et al., 2016; Zhang et al., 2017; Sun et al., 2018), a validated taxonomy of the genera in Maleae will be presented after considering all the morphological differences and genetic divergences.
2 Materials and methods
2.1 Taxon sampling and data collection
A total of 49 species representing nine tribes in three subfamilies of Rosaceae was sampled (Table 1), including one genus in Dryadoideae, two genera in Rosoideae, and 46 genera in Amygdaloideae. The tribes in Amygdaloideae were well represented with three genera in Exochordeae, two genera in Kerrieae, two genera in Neillieae, one genus in Sorbarieae, three genera in Spiraeeae, and 35 genera in Maleae. DNA was deposited in the Plant DNA Bank of China and associated specimens in the PE National Specimen Resources Bank.
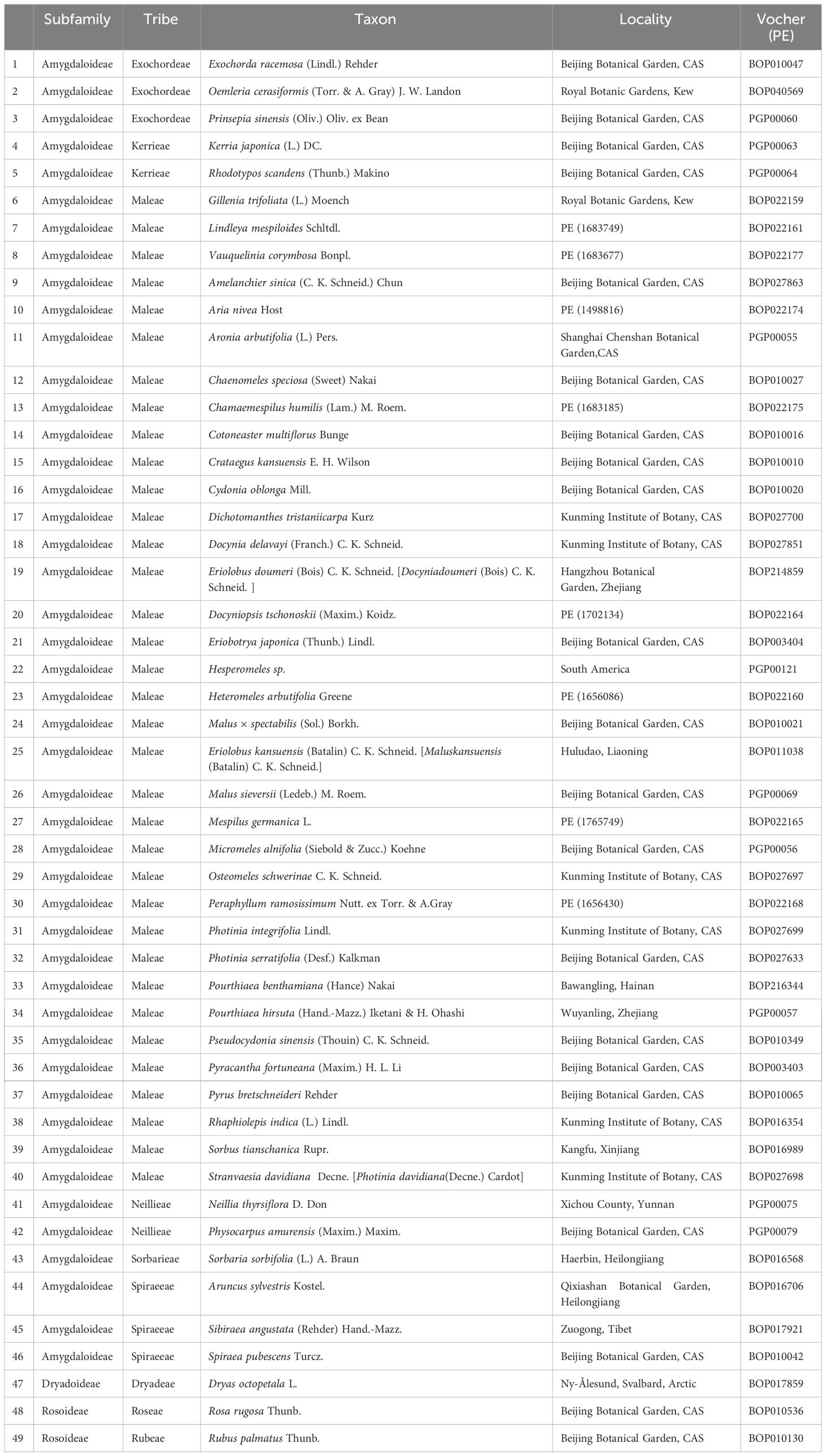
Table 1 Taxa in Rosaceae that sampled for chloroplast genome determinations with voucher information.
A total of 81 chloroplast genomes from an equal number of species, representing 69 genera in all three subfamilies (three genera of Dryadeae in Dryadoideae; 12 genera of Agrimonieae, two genera of Colurieae, and 10 genera of Potentilleae in Rosoideae; and one genus of Amygdaleae, two genera of Kerrieae, one genus of Lyonothamneae, 30 genera of Maleae, two genera of Sorbarieae, and five genera of Spiraeeae in Amygdaloideae), was downloaded from the GenBank (Supplementary Table S2).
In total, the newly determined chloroplast genomes (Table 1) along with those available in the GenBank (Supplementary Table S2) represented most tribes in Rosaceae, all 8 tribes in Amygdaloideae, and 35 out of 36 genera in Maleae were included in this study. However, we were unable to obtain material from Chamaemeles, a monotypic genus of South America, as only three chloroplast fragments are deposited in GenBank, which are insufficient for identifying its taxonomic position.
2.2 DNA extraction and chloroplast genome sequencing
Total genomic DNA was extracted from silica-gel dried leaf materials using the mCTAB method (Li et al., 2013) and purified using the Wizard DNA Clean-Up System (Promega, Madison, WI, USA). Chloroplast genomes were amplified using the primers listed in Supplementary Table S3. PCR amplification was performed in a final volume of 20 μl, containing 1× Taq buffer (1 mol L-1 KCl; 20 mmol Tris-HCl, pH 9.0; and 1% Triton X-100), 2.0 μl dNTPs (2 mmol L-1), 1.0 μl of each primer (5 μmol L-1), 20 ng of genomic DNA, and 1 unit of Taq polymerase. PCR was carried out in a C1000 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA) as follows: initial denaturation at 94°C for 3 min, followed by 35 cycles denaturation at 94°C for 30 s, annealing at 50°C for 30 s, and elongation at 72°C for 10 min, and a final elongation step at 72°C for 5 min. The PCR products were purified with a 1:1 mixture of 40% PEG 8000 and 5 mol L-1 NaCl, followed by a washing step with 80% ethanol. The library construction and sequencing with the pair-end Hiseq PE 150 on the Illumina Xten platform were performed by Novogene (Chaoyang, Beijing).
2.3 Data preparation
Reads were cleaned by removing all low-quality paired-end reads. Clean reads were de novo assembled using SPAdes 3.9 (Bankevich et al., 2012), and the generated contigs were mapped to the closest references using BLASTn 2.8.10 (Altschul et al., 1990). The mapped contigs were assembled using Sequencher 5.4 (Gene Codes, Ann Arbor, MI, USA), and gaps were filled by Sanger sequencing in Sangon Biotech.
2.4 Genome splitting and homologous fragment alignment
The newly determined chloroplast genomes were annotated by Geseq (Tillich et al., 2017). The option “annotate plastid IR” was used to determine the boundary of invert-repeat regions. After a manual check, the annotated genomes along with those retrieved from the GenBank were split into fragments (coding vs. non-coding regions) using BarcodeFinder (https://github.com/wpwupingwp/BarcodeFinder). Homologous regions were grouped, and then, each one was aligned using MAFFT 7.408 (Katoh and Standley, 2013) and adjusted manually with Se-Al 2.0 (Rambaut, 1996).
2.5 Dataset preparation
The datasets were separated into three taxonomic categories to increase accuracy. At the family level, the dataset contained 88 genomes (all the representative members of tribes and one genome from each genus in Maleae); at the Maleae level, the dataset contained 65 genomes (one or two representative members from all genera); and at the core Maleae level, the dataset contained 59 genomes (one or two representative members from all genera except Gillenia, Kageneckia, Lindleya, and Vauquelinia). Genome data were concatenated according to coding and non-coding regions using SequenceMatrix (Vaidya et al., 2011). Sequences in the two inverted regions were only used once. The nucleotide substitution models for the coding and non-coding datasets were selected by MrModelTest (Nylander, 2004) using the Bayesian information criteria (BIC).
2.6 Phylogenetic analyses
All datasets were analyzed with PAUP 4.0b10 (Swofford, 2003) for maximum parsimony (MP), with RaxML-HPC2 on XSEDE 8.2.12 (Stamatakis, 2014). for maximum likelihood (ML), and with MrBayes on XSEDE 3.2.6 (Ronquist and Huelsenbeck, 2003) for Bayesian inference (BI). ML and BI analyses were performed on CIPRES (Miller et al., 2010).
MP analyses employed a heuristic search strategy of 10,000 replicates that treated all characters as equally weighted and unordered, obtaining the starting trees with stepwise addition, random stepwise addition of 100 replicates, tree-bisection-reconnection (TBR), and MulTrees enabled. Branch support for MP trees was assessed with 1,000 bootstrap replicates, and all trees were saved at each replicate.
The nucleotide substitution models were selected by ModelFinder using BIC. Candidate models were restricted to RaxML supported by the “-mset raxml” option for ML analysis or by the “-mset mrbayes” option for BI analysis. ML analysis with 1,000 nonparametric bootstrap (BP) replicates was performed using the best-fit model.
Default settings were used for MrBayes; 2× four chains were run for 100,000,000 generations and sampled every 1,000 generations. Posterior probabilities (PP) were calculated from almost all the sampled trees when the standard deviation of the split frequencies permanently fell below 0.01. The trees sampled during the burn-in phase were discarded.
3 Results
3.1 Chloroplast genome features
The dataset used in this study contained 49 newly determined chloroplast genomes (Table 1) as well as 81 chloroplast genomes downloaded from GenBank (Supplementary Table S2), representing 117 species from 88 genera in Rosaceae. The general genome features of the 49 newly determined chloroplast genomes were presented in Table 2.
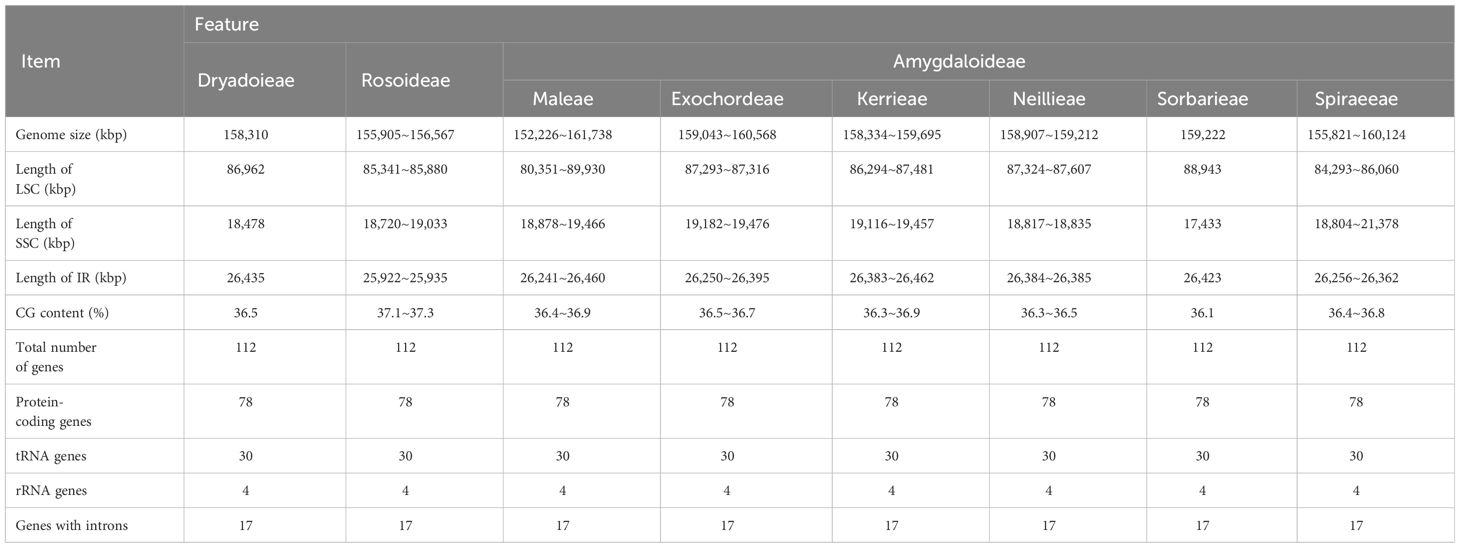
Table 2 Features of 49 chloroplast genomes of Rosaceous species.
The chloroplast genome of Maleae showed a typical structure (Supplementary Figure S1): a circular double-stranded structure with two short inverted repeat (IRa and IRb) regions separated by a large single copy (LSC) region and a small single-copy (SSC) region. The genome size ranged from 155,367 bp to 159,695 bp, whereas the overall GC content was 36.35–37.23%. No significant differences were found in the chloroplast genome size, the overall GC content, or the size of each region within the Maleae but only among the subfamilies in Rosaceae.
A total of 113 coding genes was found in the chloroplast genome of Maleae, of which 79 were protein-coding genes, 30 were distinct tRNA genes, and four were rRNA genes (16S, 23S, 5S, and 4.5S). Based on their functions, the genes were divided into three categories: I) 60 genes related to transcription and translation, including subunits of RNA polymerase, rRNA, and ribosomal proteins (most of which were tRNA genes); II) 47 genes related to photosynthesis, including Rubisco large subunit genes, genes of various components in the photosynthetic electron transport chain, and genes presumed to be NAD(P)H dehydrogenase subunits; and III) six genes related to the biosynthesis of amino acids, fatty acids, and other substances, as well as some genes with unknown functions. Among the 113 genes, 19 genes contained introns: 17 contained one intron, whereas ycf3 and clpP contained two introns. The gene rps12 was a special trans-splicing gene with the 5’-terminal exon in LSC and the 3’-terminal exon in the IR region.
3.2 Variability of chloroplast genomes
A total of 83 coding regions and 267 noncoding regions (introns and intergenic spacers) were identified. Variability in 162 regions (>100 bp) of the 65 core Maleae chloroplast genomes was parameterized in percentages of variable sites [p, ratio of number of variable sites (S) to net length (nL); p = S/nL] and nucleotide diversity (π) (Supplementary Table S4). The top 10 variable regions were ndhC-trnVuac, ndhF-rpl32, ndhG-ndhI, psbZ-rps14, rpl33-rps18, trnGgcc-trnRucu, trnHgug-psbA, trnRucu-atpA, trnTugu-trnLuaa, and trnWcca-trnPugg. No variable sites were observed in the regions of rpl23-ycf2, trnAugc, trnNguu-ycf1, and trnVgac-rrn16.
3.3 Systematic position of Maleae in Rosaceae
To reduce the computational burden, only one representative genome from each genus was selected for reconstructing the phylogeny of Rosaceae. All genera in the family were well represented, and the branch length of clades was a good indicator of the tribe rank. The pome-bearing genera formed a highly supported (bs = 100) clade and along with Gillenia, Kageneckia, Lindleya, and Vauquelinia composed the tribe Maleae (Supplementary Figure S2). The branch length of the clade was much longer than those within the clade. Six additional monophyletic clades in the subfamily Amygdaloideae corresponded to Lyonothamneae, Neillieae, Exochordeae, Kerrieae, Amygdaleae, Sorbarieae, and Spiraeeae. The latter was considered the sister tribe of Maleae.
3.4 Phylogenetic relationships within the tribe Maleae
Phylogenetic relationships among the major lineages within the tribe Maleae were fully determined (Supplementary Figures S3, S4). Four highly supported major lineages with relatively long branches were identified: (1) Gillenia, (2) Lindleya & Kageneckia, (3) Vauquelinia, and (4) the core Maleae. Thus, the monophyly of the former four genera as shown in Supplementary Figure S2 was polished, whereas that of the core Maleae was highly supported by Pyracantha M. Roem. at the base. However, the genera in the core Maleae were very closely related, and in some cases, the systematic relationships were sensitive to data alignment.
3.5 Phylogenetic relationships within the core Maleae
The sequence data alignment of the core Maleae was carefully adjusted. The unreliable microsatellite regions were excluded from the analysis, resulting in a phylogenetic tree with well-supported clades (Figure 1). Five groups of lineages were indicated: Group I that consisted of Pyracantha; Group II that consisted of Amelanchier, Crataegus, Hesperomeles Lindl., Malacomeles (Decne.) Engl., Mespilus, and Peraphyllum Nutt.; Group III that consisted of Cormus Spach, Cotoneaster Medik., Eriobotrya, Heteromeles M.Roem., Micromeles Decne., Rhaphiolepis, Photinia Lindl., Pyrus L., Sorbus L., and Stranvaesia Lindl.; Group IV that consisted of Osteomeles Lindl.; and Group V that consisted of Aria (Pers.) Host, Aronia Medik., Chaenomeles Lindl., Chamaemespilus Medik., Cydonia Mill., Dichotomanthes Kurz, Docynia Decne., Docyniopsis (C. K. Schneid.) Koidz, Eriolobus (DC.) M. Roem., Malus Mill., Pourthiaea Decne., Pseudocydonia C. K. Schneid., and Torminalis Medik.
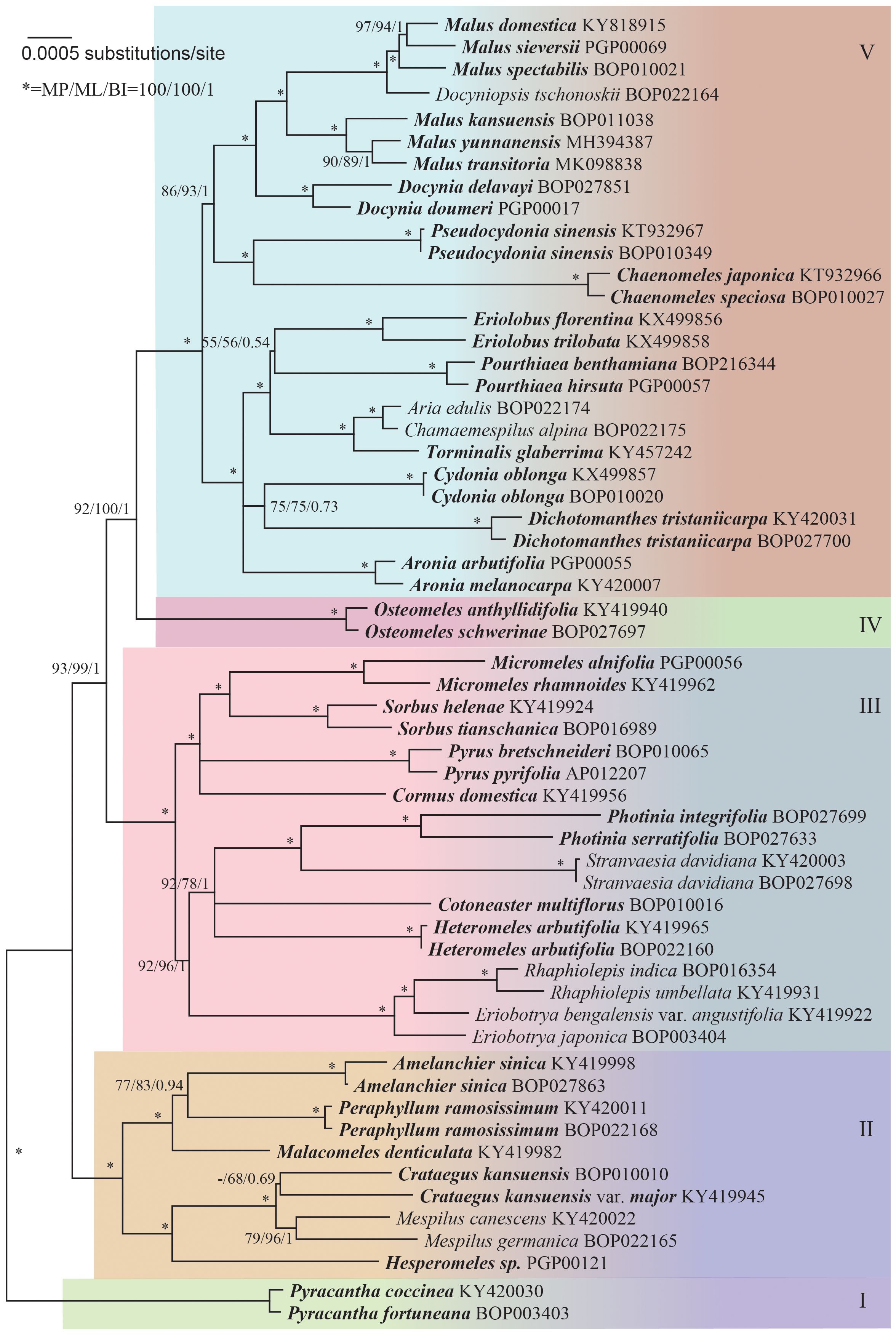
Figure 1 Maximum likelihood tree based on 59 complete chloroplast genomes, representing the phylogenetic relationships within the core Maleae. Supports to the branches are provided in the order of maximum parsimony bootstrap, maximum likelihood bootstrap, and Bayesian posterior probabilities. “-” indicates a branch collapse in the maximum parsimony and maximum likelihood trees. “*” indicates a branch with fully supports in MP/ML/BI. The tree was rooted using Pyracantha genomes as outgroups according to Supplementary Figures S2, S3.
4 Discussion
4.1 Systematic position of Gillenia
After the merge of Maloideae with Amygdaloideae, it seemed reasonable to also merge the pome-bearing taxa with the non-pome-bearing taxa. The inclusion of Kageneckia, Lindleya, and Vauquelinia in the Maleae cannot be considered controversial as they are all tetraploids with the same basal chromosome number (x = 15 or 17), similarly as the core Maleae. The diploid Gillenia (x = 9) has been either placed in the distinct tribe Gillenieae (i.e., Angiosperm Phylogeny Website; https://www.mobot.org) or at an uncertain systematic position (i.e., National Center for Biotechnology Information; https://www.ncbi.nlm.nih.gov/Taxonomy). Although Gillenia diverged earlier than Kageneckia, Lindleya, Vauquelinia, and the core Maleae, the branch length of the clade was long enough to include all of them in the same tribe (Xiang et al., 2016, Figure 2; Zhang et al., 2017, Figure 1). It would be trivial to create a new tribe that included only two species from the same genus. In Amygdaloideae, some tribes and genera diverged in the Cretaceous but the divergence of Gillenia from the other members of Maleae probably occurred in the Eocene (Xiang et al., 2016, Figure 4). The inclusion of Gillenia in Maleae explained the origin of the tetraploid taxa from ancestral paleo-allotetraploid maternal parents.
4.2 Subdivision of Maleae
A hypothetical subdivision of Maleae would create four natural groups: Group A that would include Gillenia, a genus with the most basal position and ancestors of all other members; Group B that would include Kageneckia and Lindleya, two genera that form a well-supported clade and share some apomorphic morphological characters such as dry and dehiscent fruits; Group C that would include Vauquelinia, a genus of two North American species that differs from other members in the basal chromosome number (x = 15 instead of x = 9 or 17); and Group D that would include the pome-bearing genera or the core Maleae. Thus, the tribe could be subdivided into four subtribes: Gilleniinae (Gillenia), Lindleyinae (Kageneckia and Lindleya), Vauqueliniinae (Vauquelinia), and Malinae (the core Maleae).
4.3 Merging or splitting intractable genera in core Maleae
The phylogeny of the core Maleae remained unclear owing to the low resolution and incongruence of used molecular markers as well as to paralog problems (Campbell et al., 2007). Despite the controversies, especially at the generic level (Robertson et al., 1991), we supported the existence of five groups (Figure 1). Since there was only one genus each in Group I and IV, disputes were focused on Groups II, III and V.
In Group II, Crataegus and Mespilus were monophyletic but also extremely closely related. Besides, Mespilus included only two species, M. canescens (triploid) and M. germanica (diploid). Eugenia et al. (2007); Lo and Donoghue (2012) showed that Mespilus was nested within a clade that mostly consisted of Crataegus species and consequently, merged the two genera.
In Group III, Rhaphiolepis was nested within Eriobotrya, making the latter paraphyletic. Eriobotrya and Rhaphiolepis have a similar morphology, which indicates their close relationship (Robertson et al., 1991). Besides, Rhaphiolepis is not reproductively isolated from Eriobotrya species since hybrids have been reported between the two genera (Aldasoro et al., 2005; Li et al., 2016). Our result supported the merge of Eriobotrya with Rhaphiolepis, which was consistent with Liu et al. (2020).
Species in Stranvaesia are morphologically similar to Photinia. Besides, S. davidiana is considered a member of Photinia (Liu et al., 2019). However, the close relationship between Stranvaesia and Cotoneaster that previously suggested by Campbell et al. (2007) and Sun et al. (2018) was not confirmed in the present study.
The phylogenetic relationships among the members of Sorbus s. l. have been previously reviewed by Sennikov and Kurtto (2017). Here, the splitting of Sorbus s. l. was necessary because some members belonged to Group III while others to Group V. The pinnately compound-leaved Cormus, Sorbus s. str., and the simple-leaved Micromeles remained in Group III.
In Group V, the close phylogenetic relationship among Aria, Chamaemespilus, and Torminalis that was previously suggested by Campbell et al. (2007) and Lo and Donoghue (2012) was also confirmed in the present study. In contrast to previous studies (Campbell et al., 2007; Potter et al., 2007), our clades were highly supported and had short branches, indicating low phylogenetic divergence. Only the stem branch was long, revealing a relatively long evolutionary history. It was reasonable to merge them into one genus.
The genus Malosorbus was first proposed by Browicz in 1970 to justify the hybrid origin of Malosorbus florentina (or Malus florentina), which was later defined as a true species that occurs in many European countries (Schneider, 1906; Huckins, 1972; Qian et al., 2008). In the present study, Malosorbus florentina and Malus trilobata (or correctly Eriolobus trilobata) formed a clade that had a close relationship with Pourthiaea but not with Torminalis or Malus. Considering the unique systematic positions of Malosorbus florentina and Malus trilobata, Eriolobus was adopted to host the two species; however, the former should be renamed to Eriolobus florentina (Zuccagni) Stapf.
Phipps et al. (1990) reported that the genus Malus consists of sect. Malus, sect. Sorbomalus, sect. Chloromeles (North American species), sect. Eriolobus, and sect. Docyniopsis (East Asian species). The present study supported the inclusion of Docyniopsis but not of Eriolobus. Originally, sect. Eriolobus included only M. trilobata (= Eriolobus trilobata) for the eastern Mediterranean; however, our data indicated that the North American species Malus florentina (= Eriolobus florentina) was closely related to E. trilobatus.
4.4 Systematic position of Chamaemeles
The systematic position of Chamaemeles remains uncertain owing to the lack of chloroplast genome data. The sequences of three available genes suggested that it might belong to Group III; however, its exact position remains to be confirmed.
4.5 Taxonomic implications
All genera presented in the current study are widely accepted, and the clades are well-supported, considering the complexity of their origins. Our objective was to provide additional data that would shed light on the taxonomy of Maleae and especially on the core Maleae. We suggested that the tribe could be subdivided into four subtribes with all pome-bearing species in Malinae (the core Maleae) and also that the five groups in the core Maleae could be given a subtribal rank. However, additional research is needed to confirm the systematic position of Chamaemeles.
Chloroplast genome sequences are very helpful to clarify the maternal origin of species and identify their systematic position. However, two-copy nuclear genes are necessary for revealing the bi-parental origin of Maleae species and confirm their taxonomy.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (https://www.ncbi.nlm.nih.gov) with accession numbers: OK375413 to OK375461.
Author contributions
JS: Writing – original draft, Writing – review & editing. DZ: Writing – review & editing. PQ: Writing – review & editing. YW: Investigation, Methodology, Resources, Writing – review & editing. PW: Methodology, Software, Writing – review & editing. KW: Investigation, Resources, Writing – review & editing. LG: Project administration, Supervision, Visualization, Writing – review & editing. LH: Funding acquisition, Supervision, Visualization, Writing – review & editing. SZ: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was partly supported by NSFC 32170226, 31872679 and the Fundamental Research Funds for the Central Public Welfare Research Institutes (Grand No. ZZXT201802; No. ZZXT202003; No. ZZXT202104), CACMS Innovation Fund (CI2021A03909&CI2023E002) and China Agriculture Research System (CARS-21).
Acknowledgments
The authors would like to thank Bing Liu and Jianfei Ye for providing plant materials.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1367645/full#supplementary-material
Supplementary Figure 1 | Chloroplast genome map of Crataegus kansuensis. Genes inside and outside the circle are transcribed clockwise and counterclockwise, respectively. Genes of different functional groups are shown in different colors. Thick lines indicate the extent of inverted repeats (IRa and IRb) that separate the genomes into small single copy (SSC) and large single copy (LSC) regions.
Supplementary Figure 2 | Maximum parsimonious tree of 88 chloroplast genomes, showing the phylogenetic relationships within the Rosaceae and the systematic position of Maleae. The tree was rooted using Dryadoideae as an outgroup. Bootstrap support is 100% unless otherwise indicated. “*” indicates the branch of core Maleae.
Supplementary Figure 3 | Maximum likelihood tree of 88 chloroplast genomes, showing the phylogenetic relationships within the Rosaceae and the systematic position of Maleae. The tree was rooted using Dryadoideae as an outgroup. Bootstrap support value is 100% unless otherwise indicated, and the branch support value below 50 were collapsed. “*” indicates the branch of core Maleae.
Supplementary Figure 4 | Maximum likelihood tree of 65 chloroplast genomes, showing the phylogenetic relationships within the Maleae. The tree was rooted using Gillenia genome as an outgroup.
Supplementary Figure 5 | Maximum parsimonious tree of 65 chloroplast genomes, showing the phylogenetic relationships within the Maleae. The tree was rooted using Gillenia genome as an outgroup.
Supplementary Table 1 | Thirty-six generally accepted genera in Maleae.
Supplementary Table 2 | Taxa of chloroplast genomes downloaded from the GenBank and used in phylogenetic analysis.
Supplementary Table 3 | Primers used to amplify the chloroplast genomes of Rosaceae.
Supplementary Table 4 | Variability of 162 regions of 59 chloroplast genomes of species in the core Maleae.
References
Aldasoro, J. J., Aedo, C., Navarro, C. (2005). Phylogenetic and phytogeographical relationships in Maloideae (Rosaceae) based on morphological and anatomical characters. Blumea-biodiversity, evolution biogeography of plants 50 (1), 3–32.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Campbell, C. S., Evans, R. C., Morgan, D. R., Dickinson, T. A., Arsenault, M. P. (2007). Phylogeny of subtribe Pyrinae (formerly the Maloideae, Rosaceae): Limited resolution of a complex evolutionary history. Plant Systematics Evol. 266, 119–145. doi: 10.1007/s00606-007-0545-y
Christenhusz, M. J. M., Byng, J. W. (2016). The number of known plants species in the world and its annual increase. Phytotaxa 261, 201–217. doi: 10.11646/phytotaxa.261.3.1
Considine, M. J., Wan, Y., D’Antuono, M. F., Zhou, Q., Han, M., Gao, H., et al. (2012). Molecular genetic features of polyploidization and aneuploidization reveal unique patterns for genome duplication in diploid Malus. PloS One 7, 1–9. doi: 10.1371/journal.pone.0029449
Eugenia, Y., Stefanović, S., Dickinson, T. A. (2000). Molecular reappraisal of relationships between Crataegus and Mespilus (Rosaceae, Pyreae)—Two genera or one? Systematic Botany 32 (3), :596–616.
Evans, R. C., Alice, L. A., Campbell, C. S., Kellogg, E. A., Dickinson, T. A. (2000). The granule-bound starch synthase (GBSSI) gene in the Rosaceae: multiple loci and phylogenetic utility. Mol. Phylogenet. Evol. 17, 388–400. doi: 10.1006/mpev.2000.0828
Evans, R. C., Campbell, C. S. (2002). The origin of the apple subfamily (Maloideae; Rosaceae) is clarified by DNA sequence data from duplicated GBSSI genes. Am. J. Bot. 89, 1478–1484. doi: 10.3732/ajb.89.9.1478
Gu, C., Spongberg, S. (2003). “Chaenomeles” in Flora of China, vol. 9 Eds. Wu, Z., Raven, P. (Science Press; St. Louis: Missouri Botanical Garden Press, Beijing), 46–434.
Huckins, C. A. (1972). A revision of the sections of the genus Malus Miller (Ithaca, New York: Cornell University). Ph.D. thesis.
Kalkman, C., Kubitzki, K. (2004). “Rosaceae,” in The families and genera of vascular plants (Springer, Berlin), 343–386.
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Li, Q. Y., Guo, W., Liao, W. B., Macklin, J. A., Li, J. H. (2012). Generic limits of Pyrinae: Insights from nuclear ribosomal DNA sequences. Botanical Stud. 53, 151–164.
Li, J., Wang, S., Yu, J., Wang, L., Zhou, S. (2013). A modified CTAB protocol for plant DNA extraction. Chin. Bull. Bot. 48, 72–78. doi: 10.3724/SP.J.1259.2013.00072
Li, G. F., Zhang, Z. K., Yang, X. H., Qiao, Y. C., He, X. L., Gao, Y. S., et al. (2016). Inter-specific and inter-generic hybridization compatibility of eriobotrya species (Loquat) and related genera. Hortic. Plant J. 2, 315–322. doi: 10.1016/j.hpj.2017.02.001
Liu, B. B., Hong, D. Y., Zhou, S. L., Xu, C., Dong, W. P., Johnson, G., et al. (2019). Phylogenomic analyses support the recognition of a new genus Phippsiomeles and the resurrection of a redefined Stranvaesia in Maleae (Rosaceae). J. Systematics Evol. In press. 57 (6), 678–694. doi: 10.1111/jse.12542
Liu, B. B., Liu, G. N., Hong, D. Y., Wen, J. (2020). Eriobotrya belongs to Rhaphiolepis (Maleae, Rosaceae): Evidence from chloroplast genome and nuclear ribosomal DNA data. Front. Plant Sci. 10, 1731. doi: 10.3389/fpls.2019.01731
Lo, E. Y. Y., Donoghue, M. J. (2012). Expanded phylogenetic and dating analyses of the apples and their relatives (Pyreae, Rosaceae). Mol. Phylogenet. Evol. 63, 230–243. doi: 10.1016/j.ympev.2011.10.005
Miller, M. A., Pfeiffer, W., Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in 2010 gateway computing environments workshop (GCE), (New Orleans, LA, USA: IEEE, Conference) 1–8. doi: 10.1109/GCE.2010.5676129
Nylander, J. A. A. (2004). MrModeltest v2. Program distributed by the author (Uppsala, Sweden: Evolutionary Biology Centre, Uppsala University).
Phipps, J. B., Robertson, K. R., Rohrer, J. R., Smith, P. G. (1991). Origins and evolution of subfam. Maloideae (Rosaceae). Systematic Bot. 16, 303–332. doi: 10.2307/2419283
Phipps, J. B., Robertson, K. R., Smith, P. G., Rohrer, J. R. (1990). A checklist of the subfamily Maloideae (Rosaceae). Botany 68, 2209–2269. doi: 10.1139/b90-288
Potter, D., Eriksson, T., Evans, R. C., Oh, S., Smedmark, J. E. E., Morgan, D. R., et al. (2007). Phylogeny and classification of rosaceae. Plant Systematics Evol. 266, 5–43. doi: 10.1007/s00606-007-0539-9
Potter, D., Gao, F., Bortiri, P. E., Oh, S.-H., Baggett, S. (2002). Phylogenetic relationships in Rosaceae inferred from chloroplast matK and trnL-trnF nucleotide sequence data. Plant Systematics Evol. 231, 77–89. doi: 10.1007/s006060200012
Qian, G. Z., Liu, L. F., Hong, D. Y., Tang, G. G. (2008). Taxonomic study of Malus section Florentinae (Rosaceae). Botanical J. Linn. Soc. 158, 223–227. doi: 10.1111/j.1095-8339.2008.00841.x
Rambaut, A. (1996) Se-Al: Sequence alignment editor. ver. 2.0 Available online at: http://tree.bio.ed.ac.uk/software/seal/.
Robertson, K. R., Phipps, J. B., Rohrer, J. R., Smith, P. G. (1991). A synopsis of genera in Maloideae (Rosaceae). Systematic Bot. 16, 376–394. doi: 10.2307/2419287
Ronquist, F., Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Sax, K. (1932). Chromosome relationships in the pomoideae. J. Arnold Arboretum 13, 363–367. doi: 10.5962/p.185263
Schneider, C. K. (1906). Illustrietes handbuch der Lanbholzukunde Vol. 1 (Jena: Verlag yon Gustav Fischer).
Schulze-Menz, G. K., Melchior, H. (1964). “Rosaceae,” in Engler’s syllabus der pflanzenfamilien. II (Gebrüder Borntraeger, Berlin), 209–218.
Sennikov, A. N., Kurtto, A. (2017). “A phylogenetic checklist of Sorbus s.l. (Rosaceae) in Europe,” in Memoranda Societatis pro Fauna et Flora Fennica, (Davis, California, USA: University of California Davis) vol. 93, 1–78.
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stebbins, G. L. (1950). Variation and evolution in plants (New York: Columbia University Press). doi: 10.7312/steb94536
Sun, Jh, Shi, S., Li, JL, Yu, J., Wang, L., XY, Y., et al. (2018). Phylogeny of maleae (Rosaceae) based on multiple chloroplast regions: implications to genera circumscription. BioMed. Res. Int. 2018, 1–10. doi: 10.1155/2018/7627191
Swofford, D. L. (2003). PAUP*: phylogenetic analysis using parsimony (and other methods). Version 4.0b10 (Sunderland: Sinauer Associates).
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Vaidya, G., Lohman, D. J., Meier, R. (2011). SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. doi: 10.1111/cla.2011.27.issue-2
Velasco, R., Zharkikh, A., Affourtit, J., Dhingra, A., Cestaro, A., Kalyanaraman, A., et al. (2010). The genome of the domesticated apple (Malus ×domestica Borkh.). Nat. Genet. 42, 833–839. doi: 10.1038/ng.654
Xiang, Y., Huang, C.-H., Hu, Y., Wen, J., Li, S., Yi, T., et al. (2016). Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 34 (2), 262–281. doi: 10.1093/molbev/msw242
Keywords: chloroplast genome, generic classification, Maleae, phylogeny, Rosaceae
Citation: Sun J, Zhao D, Qiao P, Wang Y, Wu P, Wang K, Guo L, Huang L and Zhou S (2024) Phylogeny of genera in Maleae (Rosaceae) based on chloroplast genome analysis. Front. Plant Sci. 15:1367645. doi: 10.3389/fpls.2024.1367645
Received: 09 January 2024; Accepted: 14 March 2024;
Published: 26 March 2024.
Edited by:
Robert Philipp Wagensommer, Free University of Bozen-Bolzano, ItalyReviewed by:
Enrico Vito Perrino, International Centre for Advanced Mediterranean Agronomic Studies, ItalyChristiane M. Ritz, Senckenberg Museum of Natural History Görlitz, Germany
Copyright © 2024 Sun, Zhao, Qiao, Wang, Wu, Wang, Guo, Huang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shiliang Zhou, c2x6aG91QGliY2FzLmFjLmNu; Luqi Huang, aHVhbmdsdXFpMDFAMTI2LmNvbQ==; Lanping Guo, Z2xwMDFAMTI2LmNvbQ==
†These authors have contributed equally to this work