 Qian Xiong
Qian Xiong Luqin Zheng1,4
Luqin Zheng1,4 Lirong Song
Lirong Song- 1Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, China
- 2Center for Environment and Health in Water Source Area of South-to-North Water Diversion, School of Public Health, Hubei University of Medicine, Shiyan, China
- 3National Aquatic Biological Resource Center, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, China
- 4University of Chinese Academy of Sciences, Beijing, China
Sphaeropleales is a diverse group with over one thousand species described, which are found in a wide range of habitats showed strong environmental adaptability. This study presented comprehensive genomic analyses of seven newly sequenced Sphaeropleales strains with BUSCO completeness exceeding 90%, alongside comparative assessments with previously sequenced genomes. The genome sizes of Sphaeropleales species ranged from 39.8 Mb to 151.9 Mb, with most having a GC content around 56%. Orthologous analysis revealed unique gene families in each strain, comprising 2 to 3.5% of all genes. Comparative functional analysis indicated that transporters, genes encoding pyrroline-5-carboxylate reductase and antioxidant enzymes played a crucial role in adaptation to environmental stressors like salinity, cold, heavy metals and varying nutrient conditions. Additionally, Sphaeropleales species were found to be B12 auxotrophy, acquiring this vitamin or its precursors through a symbiotic relationship with bacteria. Phylogenetic studies based on 18S rDNA and the low copy othologues confirmed species identification and the relationships inside core Chlorophyta and between prasinophytes. Evolutionary analyses demonstrated all the species exhibited a large count of gene family expansions and contraction, with rapidly evolving and positive selected genes identified in terrestrial Bracteacoccus species, which contributed to their adaptation to terrestrial habitat. These findings enriched the genomic data for Sphaeropleales, particularly the genus Bracteacoccus, which can help in understanding the ecological adaptations, evolutionary relationships, and biotechnological applications of this group of algae.
1 Introduction
The Chlorophyta are a diverse group of green algae, which, together with the Streptophyta and Prasinodermophyta, belong to the Viridiplantae, an ancient lineage that diverged from a proposed ‘ancestral green flagellate’ (Fang et al., 2017; Leliaert et al., 2012; Li et al., 2020). Within the Chlorophyta, there are two major subdivisions: the core Chlorophyta and the paraphyletic prasinophytes. The monophyletic core Chlorophyta currently comprises five classes: Chlorophyceae, Ulvophyceae, Trebouxiophyceae, Pedinophyceae, and Chlorodendrophyceae (Fucikova et al., 2014; Sun et al., 2016; Turmel et al., 2016, 2017). The order Sphaeropleales belongs to the CS clade (which includes Chlamydomonadales and Sphaeropleales) of Chlorophyceae, a group containing 18 taxonomically recognized families. Over a thousand species have been described in this order, which are found in a wide range of habitats, demonstrating strong environmental adaptability and exhibiting diverse cellular organizations (Krienitz et al., 2011; Wolf et al., 2002).
Members of Sphaeropleales have been explored for bioassays and biofuel production, due to their potential for producing a range of biomolecules such as pigments, lipids, starch, and cellulose (Breuer et al., 2014, 2015, 2012, 2015; Chisti, 2007; De Jaeger et al., 2014; Rawat et al., 2013) Additionally, their rapid growth and strong resistance to environmental stress make them promising candidates for bioproduction. Some species are also valuable in ecological research and applications due to their heightened sensitivity to various substances compared to other algae (Nagai et al., 2016). For instance, species like Raphidocelis subcapitata and Desmodesmus subspicatus are recommended for ecotoxicological bioassays by the Organization for Economic Cooperation and Development (OECD) (TG201, http://www.oecd.org/) (Suzuki et al., 2018).
The rapid advancements in genome sequencing over the past two decades have made comparative genomics a key approach in biological research, which is instrumental in uncovering the origin and function of genes and gene families, as well as understanding the mechanisms that drive complexity and diversification during evolution (Goodwin et al., 2016). And comparative genomics has increasingly been applied to the study of eukaryotic algae, including Chlamydomonas (Chlorophyta) (Craig et al., 2021), Cladosiphon okamuranus (a brown alga) (Nishitsuji et al., 2020), diatoms (Jeong and Lee, 2024), red algae (Ho, 2020).
To date, 28 Sphaeropleales genomes have been sequenced and are available in public databases. Of these, 20 strains belong to four genera within the Scenedesmaceae, six strains are from three genera within the Selenastraceae, and additional genomes are available from Mychonastaceae and Scenedesmaceae. Most of these genomes have been assembled to the contig or scaffold level, with genome sizes ranging from 24 Mb to 208 Mb and contig N50 values ranging from 2.8 Kb to 6125 Kb. Among these 28 genomes, only eight have been annotated, including those of Monoraphidium neglectum, Monoraphidium minutum, Raphidocelis subcapitata, Scenedesmus sp. NREL 46B-D3, and two Tetradesmus obliquus strains (Bogen et al., 2013; Carreres et al., 2017; Dasgupta et al., 2018; Demirbas, 2011; Suzuki et al., 2018). The predicted gene counts for these species range from 7,092 to 17,867 genes. These sequenced Sphaeropleales genomes provide valuable insights into the biosynthesis of lipids and pigments, including triacylglycerol (TAG) and carotenoids, making these species suitable for biotechnological applications and production. Comparative genomics analyses have also revealed mechanisms underlying environmental adaptation, such as the capacity for both heterotrophic and mixotrophic lifestyles, as well as tolerance to salinity and low metal concentrations.
Nucleotide substitution rates are often used as the criterion to reflect selection pressure. While nonsynonymous substitution rates (dN) can cause amino acid change, synonymous substitution rates (dS) do not cause amino acid change. The dN/dS ratio is the measure of natural selection acting on the protein. According to Yang (Yang, 2007), dN/dS < 1 denotes negative purifying selection, dN/dS = 1 signifies neutral evolution, and dN/dS > 1 indicates positive selection (Xiong et al., 2021). As most of the plastid protein-coding genes undergo negative or purifying selection to maintain their function, they are conserved and have a low dN/dS ratio. However, some genes might undergo positive selection in response to environmental changes, consequently presenting relatively high dN/dS ratio (Henriquez et al., 2020; Iram et al., 2019; Smith, 2015).
In this study, we sequenced the genomes of seven Sphaeropleales strains, including the first two terrestrial Bracteacoccus species, and performed an in-depth analysis of these genomes together with the seven previously reported high-quality Sphaeropleales genomes. Functional annotation-based comparative genomic analysis revealed key insights into the environmental adaptations of this group. Phylogenetic and evolutionary analyses, based on gene families and low-copy orthologues, showed extensive gene family expansions and contractions across all species. Rapidly evolving and positively selected genes were identified in the terrestrial Bracteacoccus species, which contributed to the adaption to the terrestrial habitat. The findings of this study provide valuable information for understanding the environmental adaptations and evolutionary relationships within Sphaeropleales.
2 Materials and methods
2.1 Sampling, culture conditions, DNA extraction, library preparation, sequencing, genome assembly, and cleaning of the reads
We obtained seven Sphaeropleales strains from National Aquatic Biological Resource Center, Institute of Hydrobiology, Chinese Academy of Sciences. The lists were Bracteacoccus aerius (FACHB-895), Bracteacoccus engadinensis (FACHB-1300), four Tetradesmus obliquus strains (FACHB-14, FACHB-276, FACHB-417 and FACHB-1235), Monoraphidium contortum (FACHB-3677). Bracteacoccus aerius and Bracteacoccus engadinensis were terrestrial, and others were collected from freshwater. All these strains were grown at 25 °C in liquid BG11 medium under a 12/12-h light/dark cycle.
DNA was extracted using a Universal DNA Isolation Kit (Axygen, Suzhou, China). A NEB Next Ultra DNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, Massachusetts, USA) was used for preparing sequencing libraries which were sequenced on an DNBSEQ platform. The quality of the raw reads was initially assessed using FastQC v0.11.6 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ accessed on 24 June 2021). Data were trimmed using SOAPnuke software (Chen et al., 2017) and were then assembled by megahit v 1.0.3 (Li et al., 2015) with defaulted parameters. Anvio 7.0 (Eren et al., 2015) was used to remove prokaryotic sequences from the assembled genomes with defaulted parameters. Firstly, Bowtie2 (Langmead and Salzberg, 2012) was used to align the assembled genomes to the fq files, generating BAM files. Then, the ‘anvi-gen-contigs-database’ was employed to generate a contig database, followed by the use of ‘anvi-run-hmms’ to identify single-copy genes in the contig database. The ‘anvi-profile’ was used to import sample information into the database. Next, ‘anvi-interactive’ was used to visualize the results. Finally, ‘anvi-export-collection’ was used to export the eukaryotic bins, and ‘anvi-export-contigs’ was used to extract the sequences of the aimed bins, resulting in the final assembled genomes. The genomes of the seven Sphaeropleales trains were deposited in CNCB with accession numbers SAMC4339295 - SAMC4339301, respectively.
2.2 Gene prediction, genome annotation and evaluation
Ab initio gene prediction was performed by AUGUSTUS version 3.2.1 (Stanke et al., 2006) with four three-trained organism models: volvox, chlamy2011, and chlorella. GeMoMa-1.9 (Keilwagen et al., 2018) was employed for homolog-based gene prediction and integration of gene prediction results, with genomes and protein of Volvox carteri f. nagariensis (GCA_000143455.1), Gonium pectorale (GCA_001584585.1), Haematococcus lacustris (GCA_030144725.1), and Tetradesmus obliquus (GCA_030272155.1) from GenBank as the references. Functional annotation of predicted proteins was performed by InterProScan (Jones et al., 2014) and EggNOG-mapper (Huerta-Cepas et al., 2017) for GO term mapping and KEGG pathway analyses respectively. To assess the completeness of the genome assembly and annotation, BUSCO (Simao et al., 2015) (Benchmarking Universal Single-Copy Orthologs) was used, utilizing the chlorophyta_odb10 database for a quantitative evaluation.
2.3 Orthologous gene estimation, transporters identification and repeat composition
Orthologous gene groups for the Sphaeropleales samples (including those newly added in this study and those available in the public database with annotation completeness greater than 80%) were estimated using Orthofinder (Emms and Kelly, 2015) with default parameters to identify single-copy or low-copy orthologues. Additionally, V. carteri f. nagariensis (GCA_000143455.1) and D. salina (GCA_002284615.2) were included as outgroups to estimate shared orthologues for subsequent divergence time estimation and gene family evolution analysis. Transporters were identified using BLASTp, with the Transporter Classification Database (TCDB) as a query, with an e-value cut-off of <1E−5. Further, repeat content for each genome was determined by RepeatModeler and RepeatMasker with default parameters. A library of repeats was first created for each assembly using RepeatModeler (Version 2.0.1) and all repeats were masked using RepeatMasker (Version 4.1.0).
2.4 Phylogenomic analyses
The predicted protein sequences (PEP) sequences of each single-copy or low-copy orthologue were aligned using MAFFT v7.394 (Katoh and Standley, 2013) with the parameters -maxiterate 1000 and -globalpair. Regions with poor alignment were trimmed using TrimAl v1.2 (Capella-Gutierrez et al., 2009) with the -automated1 option. The resulting trimmed alignments of orthologous groups were then used for subsequent phylogenomic analysis. Coalescent-based analyses were employed to construct the phylogenetic tree. For these analyses, RAxML (Stamatakis, 2014) was used to perform maximum likelihood (ML) analysis of each single-copy orthologue, applying the PROTGAMMA GTR model. Additionally, ASTRAL (Zhang et al., 2018) was used to infer the coalescent-based species tree (ST) phylogeny.
The 18S rDNA sequences were aligned using MAFFT v7.0 (Katoh and Standley, 2013), and ambiguous regions were manually adjusted and refined using MEGA7 (Kumar et al., 2016). To locate the positions of 18S rDNA sequences within the sequenced genomes, nhmmer (Wheeler and Eddy, 2013) was used to align the final assembled genomes to the rDNA database, based on rDNA sequences downloaded from NCBI. Subsequently, SSU sequences were extracted using Perl scripts. To understand the phylogenetic relationships of the strains in this study, additional Sphaeropleales 18S rDNA sequences were downloaded from the public database. The 18S rDNA sequences were analyzed using jModelTest2 (Darriba et al., 2012) to select the best-fit model, which was found to be GTR + I + G. Bayesian inference (BI) method was applied to infer the phylogeny.
2.5 Gene family expansion and contraction estimation
PhyloSuite (Zhang et al., 2020) was used to concatenate all the shared single-copy orthologous groups from the Sphaeropleales and the outgroup, resulting in a concatenated sequence of 35,684 amino acids. This concatenated alignment, along with the species tree constructed by ASTRAL (Zhang et al., 2018), was used for Bayesian divergence time estimation using the approximate likelihood method described by dos Reis & Yang, implemented in MCMCtree v. 4.9 (Yang, 2007).
The number of gene copies per family, as determined by Orthofinder (Emms and Kelly, 2015), and the timetree estimated earlier by MCMCtree (Yang, 2007). were used to analyze gene family expansion and contraction using CAFE v. 5.1 (Mendes et al., 2020). Expanded and contracted genes were then extracted for Gene Ontology (GO) functional and KEGG pathway enrichment analyses. For GO enrichment, all PEP sequences were imported into InterProScan (Jones et al., 2014) for GO term mapping. The analysis was carried out using the clusterProfiler (Yu et al., 2012) package, with a significance cutoff of p < 0.05, and the false discovery rate (FDR) method was applied to adjust for multiple testing (Benjamini and Hochberg, 1995).
2.6 Evolutionary analysis
The CODEML program of PAML v4.9 (Yang, 2007) was used to estimate positive selection and rapidly evolving genes based on common orthologues were described as Xiong et al (Xiong et al., 2022, 2021). The branch model was employed in the calculation of dN/dS ratios for terrestrial Sphaeropleales species and aquatic ones with the two Bracteacoccus species labeled as foreground branches. A null model (model = 0), where one dN/dS ratio was fixed across all strains, was compared with an alternative model (model = 2), where Bracteacoccus species were allowed to have a different dN/dS ratio. Likelihood ratio tests were performed to examine model fit, a chi-squared test was used to analyze p values, and multiple testing was corrected using false discovery rate (FDR) in R. The genes were considered putative rapidly evolving genes if they had an FDR-adjusted p value < 0.05 and a higher dN/dS ratio in the foreground branch than in the background branches.
Branch-site model was utilized to find genes that possibly underwent positive selection. The improved branch-site model (model = 2, Nsites = 0) was used to detect signatures of positive selection on individual amino acids in a specific branch. The two Bracteacoccus species were set as the foreground branch. The null model assumed that no positive selection occurred on the foreground branch (fix_omega = 1, omega = 1), and the alternative model assumed that sites on the foreground branch were under positive selection (fix_omega = 0, omega = 1.5). LRT were used to test model fit and Chi-square test was applied for examining the P values in R. A correction was performed for multiple testing using an FDR criterion, and BEB method was employed to statistically identify sites under positive selection. Genes with an FDR-adjusted P < 0.05 were considered as putatively selected. For the genes belonging to both under positive selection and putative rapidly evolving were performed for gene ontology (GO) functional enrichment analyses as above.
3 Results and discussion
3.1 Phylogenetic analyses
The seven newly added Sphaeropleales strains were collected prior to 2000 and were deposited in National Aquatic Biological Resource Center, Institute of Hydrobiology, Chinese Academy of Sciences. Phylogenetic analysis based on 18S rDNA sequences extracted from the genomes was performed, and the results are shown in Supplementary Figure S1. The phylogenetic tree with high support values at the basal nodes confirmed species identification. We also performed a phylogenetic analysis using low-copy orthologues across the entire Chlorophyta, including core Chlorophyta and prasinophytes by Astral (Figure 1), which enables highly accurate phylogenomic estimation, even in the presence of high levels of gene tree conflict because of incomplete lineage sorting (Mirarab et al., 2014) or horizontal gene transfer (Davidson et al., 2015). The results were almost identical with the previous studies, supporting the phylogenetic relationships within core Chlorophyta and between prasinophytes. Notably, our analysis confirmed that P. coloniale (CCMP 1413) was distinct from both core Chlorophyta and prasinophytes, suggesting it belongs to a newly identified phylum, Prasinodermophyta (Li et al., 2020).
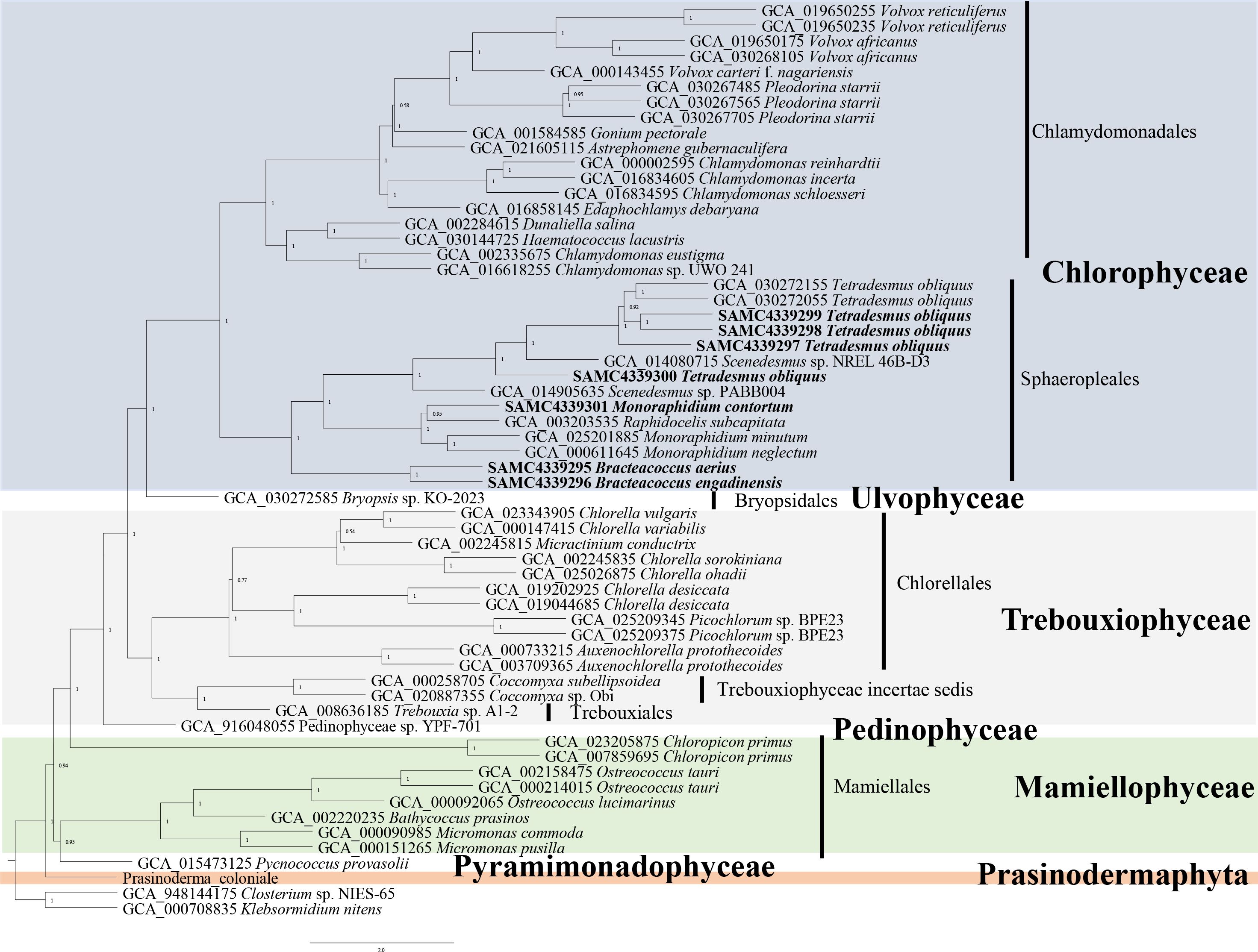
Figure 1. Phylogenetic tree of the Chlorophyta based on the low-copy orthologues to by ASTRAL. Numbers on branches represent support values of ASTRAL. Branch lengths are proportional to genetic distances, which are indicated by the scale bar. The species in bold indicates the newly added Sphaeropleales strains in this study.
3.2 Genome sequencing and characteristics
Using Illumina platforms, we sequenced and assembled draft genomes for B. aerius, B. engadinensis, M. contortum, S. obliquus, and three T. obliquus strains. These genomes, along with seven previously sequenced Sphaeropleales genomes, were summarized in Table 1. The contig N50 values of the seven newly sequenced genomes were nearly identical, ranging from 20.5 to 22.34 kb. The BUSCO completeness for all genomes exceeded 90%, indicating that the genome assemblies were sufficiently complete for comparative analyses of genomic and gene constituents. The genome sizes of the seven sequenced species ranged from 45.62 Mb to 109.8 Mb, with the GC content varied from 55.72% to 57.63%. Gene counts varied significantly among species, ranging from 6,874 to 13,568. The repeat composition covered 3.06% to 18,88% of the genome.
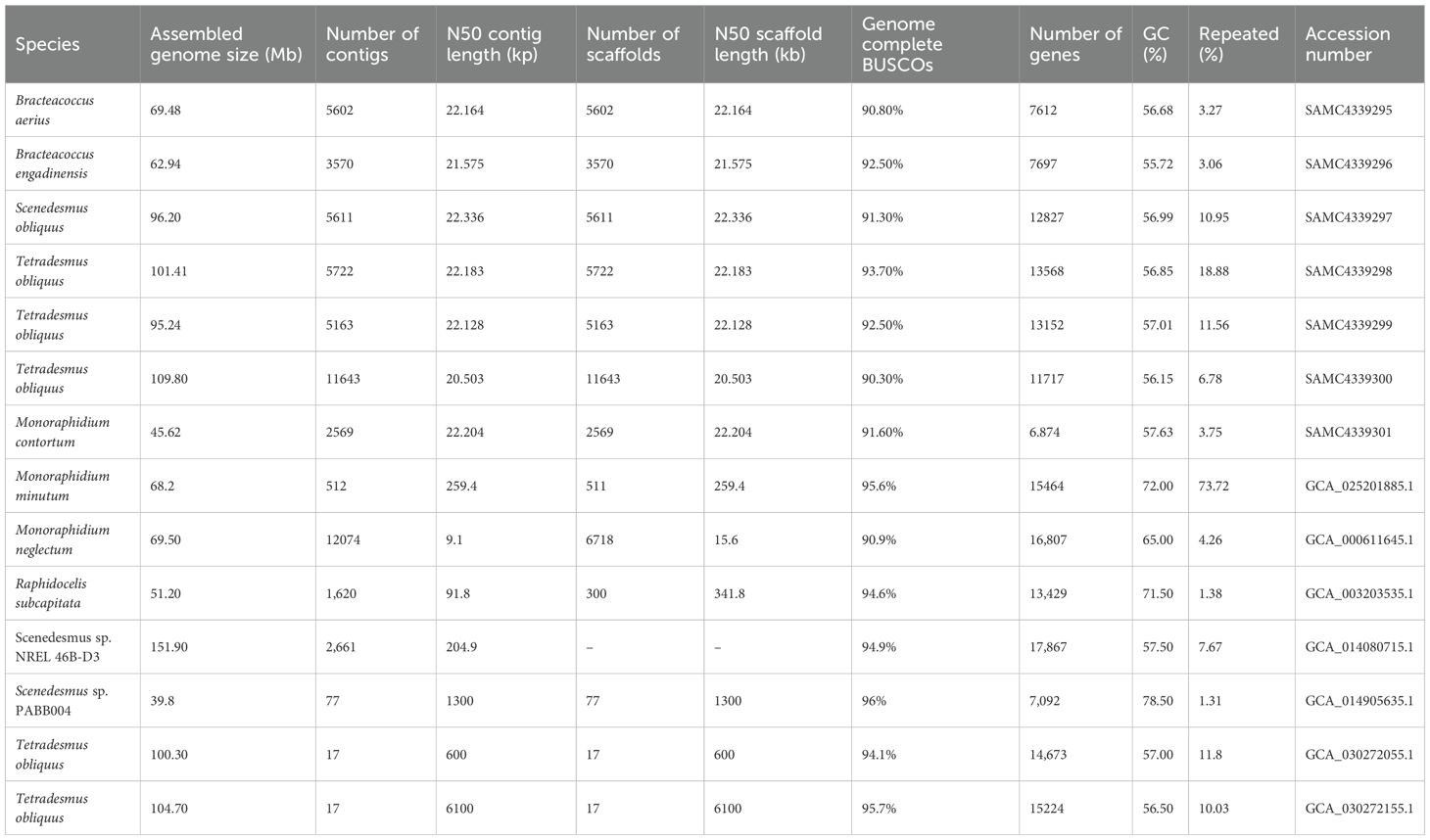
Table 1. Comparison of draft genome assemblies of seven species of Sphaeropleales algae.
Among all the Sphaeropleales species, the genomes size of Scenedesmus species exhibited significant variation, ranging from 39.8 Mb to 151.90 Mb, representing both the smallest and largest genomes within this group. In contrast, Monoraphidium, Raphidocelis, and Bracteacoccus displayed moderate genome sizes, ranging from 46.6 Mb to 69.5 Mb. And Tetradesmus species possessed relatively larger genomes that exceed 100 Mb. Most species have a GC content around 56%, exception for Monoraphidium and Scenedesmus sp. PABB004 greater than 70%. The repeat content varied considerably among all genomes examined, Scenedesmus sp. PABB004 and R. subcapitata showed the lowest repeat content about 1.3%, and T. obliquus (SAMC4339298) displayed the highest at 18.8%. Bracteacoccus and Monoraphidium have moderate repeat content, ranging from 3% to 4%, whereas Tetradesmus and most Scenedesmus species exhibit higher ranging 6.78% to 18.88%. minutum,
It is generally accepted that there is some degree of correlation between the genome size, the proportion of repetitive sequence, and the number of genes (Feng et al., 2017; Hou and Lin, 2009; Nishitsuji et al., 2020; Wang et al., 2021). These observations support the idea that larger genomes generally having more gene count, fewer repeats and lower GC content. While, it seemed larger genomes exhibited more gene count at the genus level of Sphaeropleales, the relationship the genomes size among the GC and repeat content showed no obvious pattern, more high-quality genomes will contribute to explore the relationships.
3.3 Analysis of orthologous gene families and comparative analysis of predicted gene function
We conducted orthologous gene family analysis based on the newly sequenced seven Sphaeropleales strains along with seven high-quality previously sequenced genomes at different taxonomic levels (Figure 2). The orthologous analysis revealed that 3,761 gene families were shared or conserved across all fifteen genomes (Figure 2A). Additionally, each strain contained unique gene families, comprising 2-3.5% of the total gene count: 2,009 families were unique to T. obliquus, 554 to S. obliquus, 411 to B. aerius, 257 to Scenedesmus sp. NREL 46B-D3, 247 to B. engadinensis, 151 to M. minutum, 144 to R. subcapitata, and 138 to M. contortum. At the genus level (Figure 2B), 6,733 gene families were shared or conserved across the five genera. Furthermore, 5,587 gene families were unique to Tetradesmus, 2,391 to Bracteacoccus, 1,432 to Monoraphidium, and 720 to Scenedesmus. At the family level (Figure 2C), 7,304 gene families were shared or conserved among the three families. Additionally, 10,656 gene families were unique to Scenedesmaceae, 3,149 to Selenastraceae, and 2,667 to Bracteacoccaceae. In whole, the Tetradesmus species, in particular, had the most unique gene families (5,587), likely due to their larger genomes.
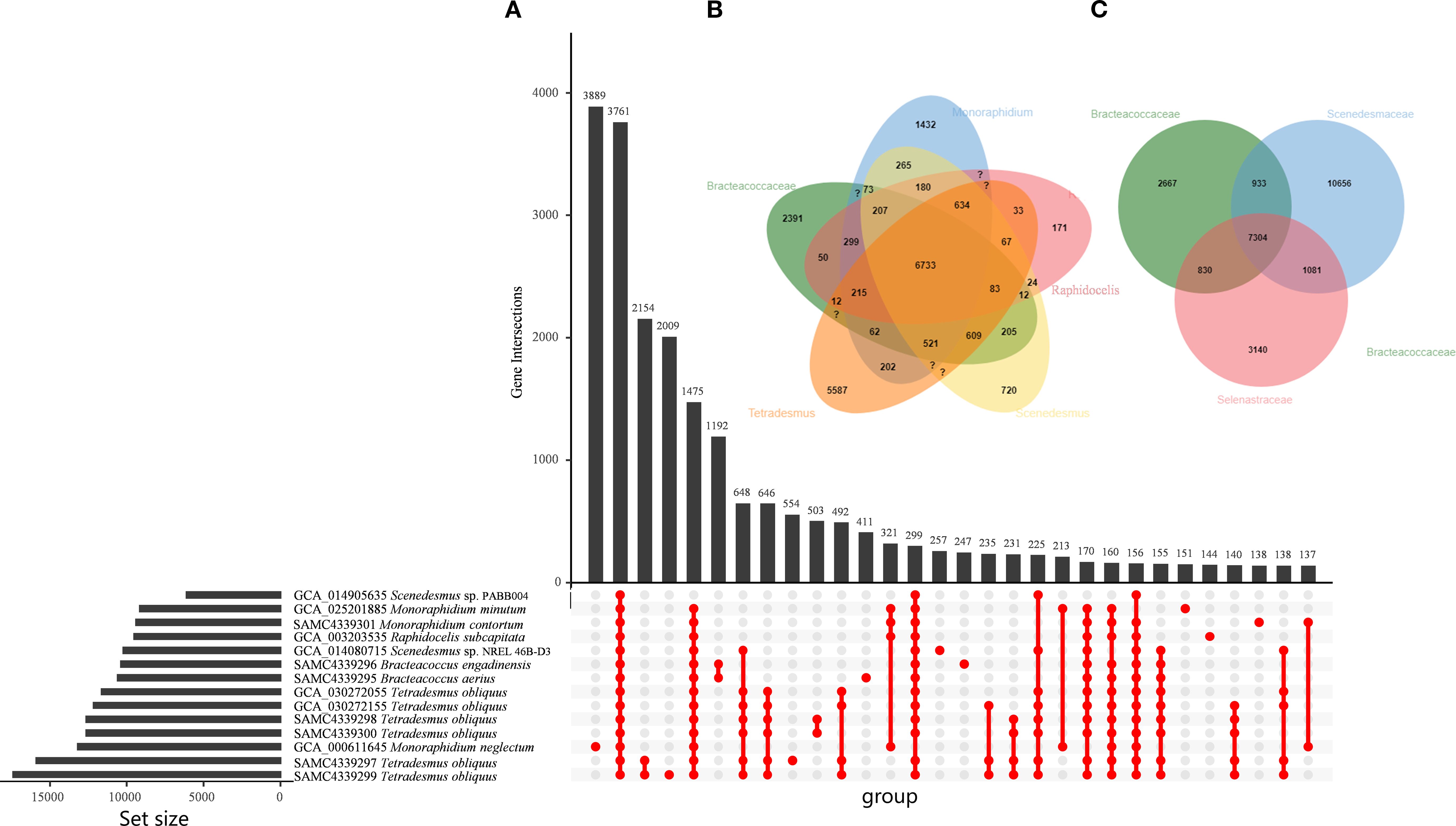
Figure 2. Orthologous gene analysis in genomes of the Sphaeropleales algae. (A) Shared families are shown on the horizontal axis and the number of patterns in the vertical axis. (B) Venn diagram showing the numbers of gene families shared among the Sphaeropleales algae at the level of genus. (C) Venn diagram showing the numbers of gene families shared among the Sphaeropleales algae at the level of family.
To further investigate gene functions within the Sphaeropleales, we classified all annotated proteins from 14 genomes into functional categories using the Gene Ontology (GO) database (Supplementary Figure S2). The predicted proteins were categorized into three primary GO domains: molecular function (MF), cellular component (CC), and biological process (BP). We compared the contents of each category and their corresponding percentages. Among the 11 strains, unique GO domains were identified, with T. obliquus FACHB-276 exhibiting the highest diversity, containing 278 distinct domains across the three GO categories. For the top ten functional categories at the CC and MF levels, most species shared nearly identical terms. We also calculated the PFAM domain categories and their percentages (Supplementary Figure S3) and KEGG pathway categories (Supplementary Figure S4) for all species, which showed that all the species shared similar top-ten categories and associated percentages.
3.4 Comparative analysis of predicted gene function
Based on the Transporter Classification Database, we analyzed the transporters in the Sphaeropleales genomes. A total of 320 transporters were identified, as shown in Supplementary Table S1. The transporters across the Sphaeropleales species exhibited similar patterns in terms of the top ten transporter categories and their respective numbers. As additional species were included in the analysis, the results were consistent with previous studies (Suzuki et al., 2018), which showed that Sphaeropleales species possess notably higher numbers of genes encoding for H+/hexose transporters (2.A.1.1, TCAD ID), amino acid permeases (2.A.18.2), peptide transporters (2.A.17.3), aquaporins (1.A.8.8), and metal-nicotianamine transporters (2.A.67.2) compared to Chlamydomonas reinhardtii (Table 2). The Sphaeropleales are a dominant group of freshwater algae, well-adapted to diverse environmental conditions (Baldisserotto et al., 2012; Fawley et al., 2004). These species also exhibit high sensitivity to exogenous substances (Nagai et al., 2016). Previous studies have suggested that Sphaeropleales have the ability to adapt to various environmental conditions for possessing a significantly greater number of genes related to H+/hexose transporters, amino acid permeases, peptide transporters, aquaporins, and metal-nicotianamine transporters comparing to C. reinhardtii (Suzuki et al., 2018). Aquaporins help algae adapt to high salt stress by facilitating the transport of small polar molecules, such as water, across cell membranes, thereby regulating intracellular osmotic pressure (Anderberg et al., 2011). Additionally, the presence of multiple metal-nicotianamine transporters, ABC transporters, and genes involved in heavy metal ion and xenobiotic transport (Supplementary Table S2) suggested that Sphaeropleales may have a high sensitivity to metals, positioning them as potential phytoremediation organisms for removing heavy metal pollution from aquatic environments (Benderliev and Ivanova, 1994, 1996; Murata et al., 2006; Schaaf et al., 2004). Genes related to H+/hexose cotransport, amino acid/peptide transporters, and nitrate/nitrite transporters are likely key to their rapid growth under varying nutrient conditions (Cho et al., 1981; Sauer et al., 1983).
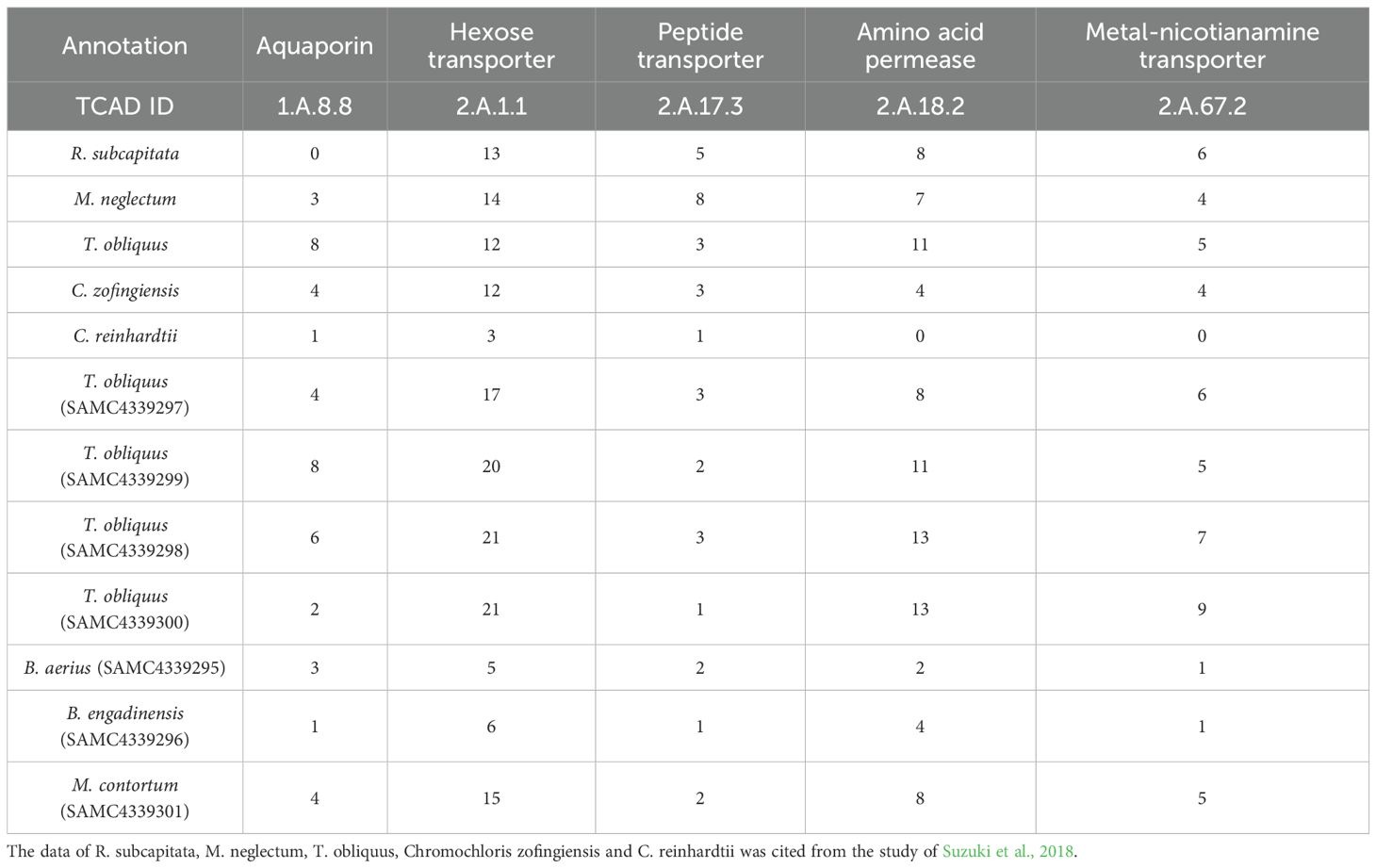
Table 2. The number of genes for several transporters.
Furthermore, Sphaeropleales species possessed a gene encoding pyrroline-5-carboxylate reductase, which synthesizes proline to alleviate osmotic stress under cold conditions (Liu et al., 2020). And pyrroline-5-carboxylate reductase is also associated with halotolerance (Arora et al., 2019; Kishor et al., 1995; Pérez-Arellano et al., 2010). Previous study indicated that copies of the pyrroline-5-carboxylate reductase gene of the halotolerant microalga Scenedesmus sp. NREL 46B-D3 were upregulated (Calhoun et al., 2021) in the cold stress, and pyrroline 5-carboxylate reductase was upregulated under the salt stress in the cold tolerant M. minutum 26B-AM and S. obliquus UTEX393 (Calhoun et al., 2022). They also exhibited genes encoding antioxidant enzymes (e.g., catalase, ascorbate peroxidase) and antioxidant biosynthesis pathways (e.g., glutamate, β-carotene) (Supplementary Table S3), which helped mitigate oxidative stress from excess reactive oxygen species (O2 and H2O2) under cold stress (Fryer et al., 1998; Liu et al., 2020; Prasad, 1997; Van Breusegem et al., 1999). The findings from this study, based on a larger set of sequenced Sphaeropleales genomes, supported with adaptation for different environmental condition.
All Sphaeropleales species showed the presence of genes involved in vitamin B biosynthetic pathways, including the vitamin B6 biosynthetic process, thiamine (vitamin B1) biosynthesis and metabolism, biotin (vitamin B7) synthase activity, and biotin biosynthesis (Supplementary Table S4), indicating the ability to synthesize these vitamins de novo, similar to C. reinhardtii, Cyanidioschyzon merolae, and Thalassiosira pseudonana (Croft et al., 2006). A previous study on the marine diatom Skeletonema costatum has shown that a mixture of vitamin B compounds plays a crucial role in mitigating the harmful effects of hypersalinity (Gede et al., 2017). Furthermore, the possession of a complete pathway for thiamine biosynthesis contributes to enhanced biotic and stress resistance (Almutairi et al., 2021). The biosynthesis of the essential amino acid methionine can occur via both B12-dependent and B12-independent isoforms of methionine synthase (MetH and MetE, respectively) (Croft et al., 2005, 2006; Helliwell et al., 2011). In this study, all the Sphaeropleales species showed no genes about cobalamin (vitamin B12) biosynthesis, while exhibited genes about cobalamin (vitamin B12) metabolic process, and cobalamin binding (Supplementary Table S4), indicating that methionine synthesis occurred solely via the VB12-independent pathway, namely MetE isoform in Sphaeropleales. A previous survey of 306 species aimed to determine whether these algae require vitamin B12 (Croft et al., 2006). The results indicated that none of the Sphaeropleales species showed a requirement for cobalamin, which acts as a cofactor for enzymes involved in rearrangement-reduction reactions and methyl transfer reactions. Vitamin B12 (VB12) can only be produced by bacteria (both eubacteria and archaea) in nature, and its concentration in the natural environment is typically lower than required in culture (Croft et al., 2006). Therefore, these species acquire VB12 or its precursors through a symbiotic relationship with bacteria. Such symbiotic interactions between bacteria and algae are widespread, as many algae species are capable of acquiring vitamin B12 from their bacterial partners (Croft et al., 2006; Daisley, 1969).
All species contained genes involved in the assembly, movement, and organization of cilia (Supplementary Table S5), as well as genes associated with meiosis (Supplementary Table S6). In the Sphaeropleales cell cycle, the stage with motility flagellates/) or meiosis are either not dominant or not well understood (Trainor and Burg, 1965; Yamagishi et al., 2017). Suzuki et al. proposed that immobility may force cells to adapt to different environmental conditions aided by their numerous transporters (Suzuki et al., 2018). Another possibility that a cryptic sexual cycle or previously unobserved motile life cycle stage with flagella may exist in these organisms, or these genes are nonfunctional in Sphaeropleales. This phenomenon has been reported in C. zofingiensi (Blanc et al., 2010; Roth et al., 2017).
3.5 Gene family expansion and contraction estimation and evolution analysis
To gain a comprehensive understanding of the orthogroups differences between species, particularly those with assignable functions, we performed gene family expansion and contraction analyses using CAFÉ, based on 29,073 gene families. As shown in Figure 3, all species and most ancestral nodes exhibited a substantial number of gene family expansions, with contractions being more prevalent. Specifically, B. engadinensis and B. aerius exhibited the highest number of contracted gene families, with 17,786 and 17,499 respectively, surpassing all other nodes, while T. obliquus (FACHB-276) displayed the most expanded gene families, with 2,142. In terms of ancestral nodes, the common ancestor of the Selenastraceae family had the largest number of expansions and contractions, with 10,832 contracted gene families and 471 expanded gene families. In contrast, the common ancestor of the Bracteacoccaceae family showed the smallest number, with only 11 expanded gene families and no contractions. The number of expanded or contracted gene families can be influenced by various factors, including gene duplication, de novo gene creation, gene loss, the functional roles of gene families, and environmental changes (Albalat and Canestro, 2016; Guo, 2013; Prachumwat and Li, 2008). Consequently, the observed differences in gene family expansions and contractions in Sphaeropleales are likely due to multiple causes. A previous study suggested that gene family contraction could contribute to genome reduction (Qiu et al., 2015). This may help explain why Scenedesmus sp. PABB004exhibited the largest number of gene family contractions, while simultaneously having the smallest genome size within the Scenedesmaceae, and the Bracteacoccus species showed relatively small genome size and almost the fewest gene count.
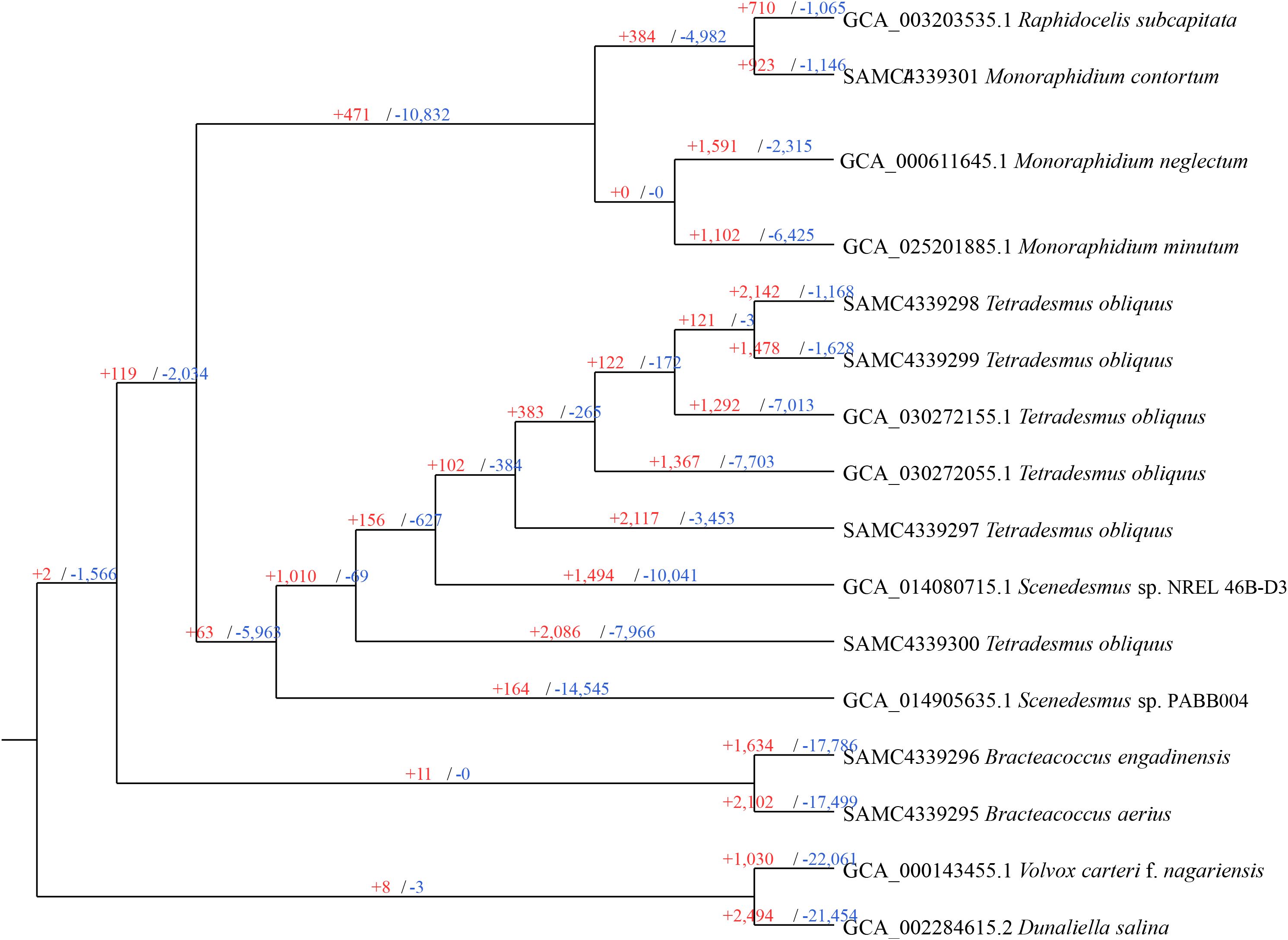
Figure 3. Gene family expansion or contraction in Sphaeropleales algae. Branch numbers indicate the number of gene families that have expanded (+) and contracted (−) since the split from the common ancestor.
To investigate whether gene family evolution correlates with habitat adaptation, we performed Gene Ontology (GO) enrichment analysis based on the expansions and contractions in the common ancestor of Bracteacoccaceae. The GO enrichment analysis revealed that the expanded families were enriched in six GO terms (Figure 4A), primarily related to methionine biosynthetic, cobalamin binding, tRNA Modification, pentosyltransferase activity, and metal ion binding.
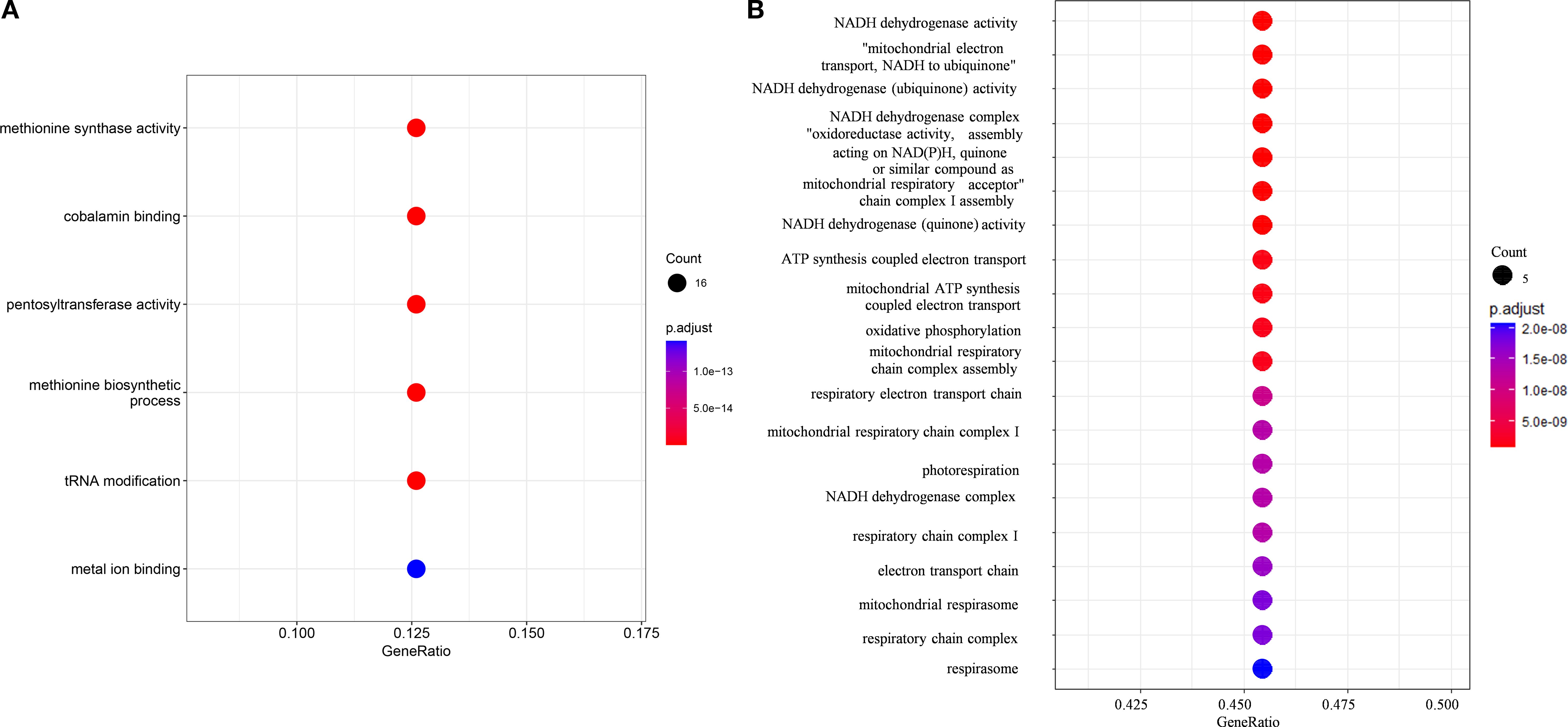
Figure 4. Dot plot showing the enrichment of the special orthologues about evolution. The dot sizes represent the numbers of genes, circle colors indicate different p values. (A) Dot plot showing the enrichment of the expanded orthologues in the common ancestor of Bracteacoccaceae. (B) Dot plot showing the enrichment of the orthologues experienced fast-evolving and positively selected.
The branch model of PAML was used to compare dN/dS ratios between terrestrial and aquatic Sphaeropleales species based on 3757 common orthologues. Among these, 3032 orthogroups showed significantly higher dN/dS ratios in the two terrestrial Bracteacoccaceae species, indicating the occurrence of rapid evolution (Supplementary Table S7). Positive selection analysis was performed based on branch-site model, and the null and alternative models were compared. The null model considered that the foreground branch only has dN/dS = 1, and the alternative model assumed that sites on the foreground branch have dN/dS > 1 (positive selection). When the two terrestrial Bracteacoccaceae species were labelled as the foreground branch, 13 orthogroups showed the FDR-adjusted P value less than 0.05, which indicated that these 13 orthogroups may possibly under positive selection (Supplementary Table S8), and they also under rapid evolution based on the result of Branch model. GO enrichment analysis showed these orthogroups were enriched in 87 GO terms (Figure 4B), among which 55 GO terms belonged to biological process category, 22 belonged to cellular component category and 10 belonged to molecular function category. Among all the three categories, there were some common functions such as oxidation-reduction process, the function of mitochondrial, the biosynthetic and metabolism of starch, polysaccharide and other organics (Figure 4B).
Bracteacoccaceae included only one genus Bracteacoccus, which are coccoid green algae that occurs in a wide range of soil types worldwide, spanning climates from polar to tropical and not avoiding even heavily polluted localities (Patova and Dorokhova, 2008; Ronquist et al., 2012; Stibal et al., 2006). Gene Ontology (GO) enrichment analysis, based on the expansions and contractions in the ancestral lineage of Bracteacoccaceae, revealed that the enriched GO terms were primarily associated with methionine biosynthesis, cobalamin binding, tRNA modification, pentosyltransferase activity, and metal ion binding. A previous study about C. reinhardtii indicated that methionine biosynthesis is an essential cellular mechanism for adaptation to thermal stress (Xie et al., 2013). Additionally, Zeng et al. identified key pathways in regulating natural variations in phenylpropanoid content, including flavone C-pentosyltransferase proteins, which are involved in UVB protection in Qingke (Tibetan hulless barley) (Zeng et al., 2020). It has also been reported that tRNA modifications is correlated with cold temperatures, drought, increased soil salinity, and developmental stages in vascular plants (Mmbando, 2024). Furthermore, the evolutionary analysis based on PAML showed the two species of Bracteacoccus exhibited genes experienced both rapidly evolution and positively selected. Among these, common functional categories included oxidation-reduction processes, mitochondrial functions, and the biosynthesis and metabolism of starch, polysaccharides, and other organic compounds. We considered these expanded gene families, rapidly evolution and positively selected were likely related to the adaption to the terrestrial habitat for Bracteacoccaceae.
4 Conclusions
The comprehensive genomic analysis of Sphaeropleales strains provides valuable insights into their evolutionary adaptations and genomic constituents. The phylogenetic analyses confirmed the distinctiveness of species and their relationships within the Chlorophyta. The genome sizes of Sphaeropleales species ranged from 39.8 to 151.9 Mb, with most having a GC content around 56%. Comparative analysis of orthologous gene families revealed conserved and unique gene families across species, with substantial expansions and contractions in all species and ancestral nodes. Functional annotation and analysis of transporter genes explained the importance of specific gene families in environmental adaptation. The gene family expansion and contraction analyses, along with positive selection studies, identified key functional categories associated with terrestrial adaptation in Bracteacoccaceae.
The work enriched the genomic data for Sphaeropleales, particularly the genus Bracteacoccus, enhancing our understanding of the ecological adaptations, evolutionary relationships, and biotechnological applications of this group of algae.
Data availability statement
The assembled data were deposited in the National Genomics Data Center (NGDC) databases BioProject PRJCA032465 (https://ngdc.cncb.ac.cn/bioproject/), and the accession numbers was SAMC4339295 - SAMC4339301, respectively.
Author contributions
QX: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. LQZ: Data curation, Writing – review & editing. QZ: Funding acquisition, Resources, Validation, Writing – review & editing. TL: Resources, Validation, Writing – review & editing. LLZ: Resources, Validation, Writing – review & editing. LS: Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Third Xinjiang Scientific Expedition Program (Grant No.2021xjkk0600) and the National Natural Science Foundation of China (Grant No. 32300177).
Acknowledgments
We thank the members of Protist 10,000 Genomes Project (P10K) consortium for their helpful suggestions. This research was supported by the Wuhan Branch, Supercomputing Center, Chinese Academy of Sciences, China.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1534646/full#supplementary-material
Supplementary Figure 1 | Phylogenetic tree of Sphaeropleales based on 18S rDNA sequences by Mrbayes. Numbers on branches represent support values of Bayesian posterior probabilities. Branch lengths are proportional to genetic distances, which are indicated by the scale bar. The species in bold indicates the newly added Sphaeropleales strains in this study.
Supplementary Figure 2 | The top ten functional categories of the 14 Sphaeropleales based on Gene Ontology (GO) database. The red, green and blue indicates biological process (BP), cellular component (CC), and molecular function (MF) respectively.
Supplementary Figure 3 | The top 20 PFAM domain categories of the 14 Sphaeropleales.
Supplementary Figure 4 | The top 10 KEGG pathway categories of the 14 Sphaeropleales.
Supplementary Table 1 | The analysis of the transporters in the Sphaeropleales genomes based on the Transporter Classification Database.
Supplementary Table 2 | The genes about heavy metal ions and xenobiotics in the Sphaeropleales genomes.
Supplementary Table 3 | The genes about encoding antioxidant enzymes (e.g., catalase, ascorbate peroxidase) and antioxidant biosynthesis pathways in the Sphaeropleales genomes.
Supplementary Table 4 | The genes about vitamins B in the Sphaeropleales genomes.
Supplementary Table 5 | The genes about cilium in the Sphaeropleales genomes.
Supplementary Table 6 | The genes about meiosis in the Sphaeropleales genomes.
Supplementary Table 7 | The fast-evolving genes based on the branch model of PMAL.
Supplementary Table 8 | The positively-selected genes based on branch-site model of PAML.
References
Albalat, R. and Canestro, C. (2016). Evolution by gene loss. Nat. Rev. Genet. 17, 379–391. doi: 10.1038/nrg.2016.39
Almutairi, A. W., El-Sayed, A.E.-K.B., and Reda, M. M. (2021). Evaluation of high salinity adaptation for lipid bio-accumulation in the green microalga Chlorella vulgaris. Saudi J. Biol. Sci. 28, 3981–3988. doi: 10.1016/j.sjbs.2021.04.007
Anderberg, H. I., Danielson, J. A. H., and Johanson, U. (2011). Algal MIPs, high diversity and conserved motifs. BMC Evol. Biol. 11. doi: 10.1186/1471-2148-11-110
Arora, N., Laurens, L. M., Sweeney, N., Pruthi, V., Poluri, K. M., and Pienkos, P. T. (2019). Elucidating the unique physiological responses of halotolerant Scenedesmus sp. cultivated in sea water for biofuel production. Algal Res. 37, 260–268. doi: 10.1016/j.algal.2018.12.003
Baldisserotto, C., Ferroni, L., Giovanardi, M., Boccaletti, L., Pantaleoni, L., and Pancaldi, S. (2012). Salinity promotes growth of freshwater Neochloris oleoabundans UTEX 1185 (Sphaeropleales, Chlorophyta): morphophysiological aspects. Phycologia 51, 700–710. doi: 10.2216/11-099.1
Benderliev, K. M. and Ivanova, N. I. (1994). High-affinity siderophore-mediated iron-transport system in the green-alga scenedesmus-incrassatulus. Planta 193 (2), 163–166.
Benderliev, K. M. and Ivanova, N. I. (1996). Determination of available iron in mixtures of organic chelators secreted by Scenedesmus incrassatulus. Biotechnology Techniques 10(7), 513–518.
Benjamini, Y. and Hochberg, Y. (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc B. 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Blanc, G., Duncan, G., Agarkova, I., Borodovsky, M., Gurnon, J., Kuo, A., et al. (2010). The Chlorella variabilis NC64A genome reveals adaptation to photosymbiosis, coevolution with viruses, and cryptic sex. Plant Cell 22, 2943–2955. doi: 10.1105/tpc.110.076406
Bogen, C., Al-Dilaimi, A., Albersmeier, A., Wichmann, J., Grundmann, M., Rupp, O., et al. (2013). Reconstruction of the lipid metabolism for the microalga Monoraphidium neglectum from its genome sequence reveals characteristics suitable for biofuel production. BMC Genomics 14. doi: 10.1186/1471-2164-14-926
Breuer, G., De Jaeger, L., Artus, V. P. G., Martens, D. E., Springer, J., Draaisma, R. B., et al. (2014). Superior triacylglycerol (TAG) accumulation in starchless mutants of Scenedesmus obliquus: (II) evaluation of TAG yield and productivity in controlled photobioreactors. Biotechnol. Biofuels 7. doi: 10.1186/1754-6834-7-70
Breuer, G., Lamers, P. P., Janssen, M., Wijffels, R. H., and Martens, D. E. (2015a). Opportunities to improve the areal oil productivity of microalgae. Bioresour. Technol. 186, 294–302. doi: 10.1016/j.biortech.2015.03.085
Breuer, G., Lamers, P. P., Martens, D. E., Draaisma, R. B., and Wijffels, R. H. (2012). The impact of nitrogen starvation on the dynamics of triacylglycerol accumulation in nine microalgae strains. Bioresour. Technol. 124, 217–226. doi: 10.1016/j.biortech.2012.08.003
Breuer, G., Martens, D. E., Draaisma, R. B., Wijffels, R. H., and Lamers, P. P. (2015b). Photosynthetic efficiency and carbon partitioning in nitrogen-starved Scenedesmus obliquus. Algal Res. 9, 254–262. doi: 10.1016/j.algal.2015.03.012
Calhoun, S., Bell, T. A. S., Dahlin, L. R., Kunde, Y., LaButti, K., Louie, K. B., et al. (2021). A multi-omic characterization of temperature stress in a halotolerant Scenedesmus strain for algal biotechnology. Commun. Biol. 4, 333. doi: 10.1038/s42003-021-01859-y
Calhoun, S., Kamel, B., Bell, T. A., Kruse, C. P., Riley, R., Singan, V., et al. (2022). Multi-omics profiling of the cold tolerant Monoraphidium minutum 26B-AM in response to abiotic stress. Algal Res. 66, 102794. doi: 10.1016/j.algal.2022.102794
Capella-Gutierrez, S., Silla-Martinez, J. M., and Gabaldon, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Carreres, B. M., De Jaeger, L., Springer, J., Barbosa, M. J., Breuer, G., Van Den End, E. J., et al. (2017). Draft genome sequence of the oleaginous green alga Tetradesmus obliquus UTEX 393. Genome Announc. 5, e01449-16. doi: 10.1128/genomeA.01449-16
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2017). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7. doi: 10.1093/gigascience/gix120
Chisti, Y. (2007). Biodiesel from microalgae. Biotechnol. Adv. 25, 294–306. doi: 10.1016/j.biotechadv.2007.02.001
Cho, B. H., Sauer, N., Komor, E., and Tanner, W. (1981). Glucose induces 2 amino-acid transport-systems in chlorella. PNAS 78, 3591–3594. doi: 10.1073/pnas.78.6.3591
Craig, R. J., Hasan, A. R., Ness, R. W., and Keightley, P. D. (2021). Comparative genomics of chlamydomonas. Plant Cell 33, 1016–1041. doi: 10.1093/plcell/koab026
Croft, M. T., Lawrence, A. D., Raux-Deery, E., Warren, M. J., and Smith, A. G. (2005). Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 438, 90–93. doi: 10.1038/nature04056
Croft, M. T., Warren, M. J., and Smith, A. G. (2006). Algae need their vitamins. Eukaryot. Cell 5, 1175–1183. doi: 10.1128/EC.00097-06
Daisley, K. W. (1969). Monthly survey of vitamin B12 concentrations in some waters of english lake district. Limnol. Oceanogr. 14, 224–&. doi: 10.4319/lo.1969.14.2.0224
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–772. doi: 10.1038/nmeth.2109
Dasgupta, C. N., Nayaka, S., Toppo, K., Singh, A. K., Deshpande, U., and Mohapatra, A. (2018). Draft genome sequence and detailed characterization of biofuel production by oleaginous microalga Scenedesmus quadricauda LWG002611. Biotechnol. Biofuels 11. doi: 10.1186/s13068-018-1308-4
Davidson, R., Vachaspati, P., Mirarab, S., and Warnow, T. (2015). Phylogenomic species tree estimation in the presence of incomplete lineage sorting and horizontal gene transfer. BMC Genomics 16. doi: 10.1186/1471-2164-16-S10-S1
De Jaeger, L., Verbeek, R. E. M., Draaisma, R. B., Martens, D. E., Springer, J., Eggink, G., et al. (2014). Superior triacylglycerol (TAG) accumulation in starchless mutants of Scenedesmus obliquus: (I) mutant generation and characterization. Biotechnol. Biofuels 7. doi: 10.1186/1754-6834-7-69
Demirbas, M. F. (2011). Biofuels from algae for sustainable development. Appl. Energy 88, 3473–3480. doi: 10.1016/j.apenergy.2011.01.059
Emms, D. M. and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16. doi: 10.1186/s13059-015-0721-2
Eren, A. M., Esen, O. C., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L., et al. (2015). Anvi’o: an advanced analysis and visualization platformfor ‘omics data. PeerJ 3. doi: 10.7717/peerj.1319
Fang, L., Leliaert, F., Zhang, Z.-H., Penny, D., and Zhong, B.-J. (2017). Evolution of the Chlorophyta: Insights from chloroplast phylogenomic analyses. J. Syst. Evol. 55, 322–332. doi: 10.1111/jse.12248
Fawley, M. W., Fawley, K. P., and Buchheim, M. A. (2004). Molecular diversity among communities of freshwater microchlorophytes. Microb. Ecol. 48, 489–499. doi: 10.1007/s00248-004-0214-4
Feng, R., Wang, X., Tao, M., Du, G., and Wang, Q. (2017). Genome size and identification of abundant repetitive sequences in Vallisneria spinulosa. PeerJ 5. doi: 10.7717/peerj.3982
Fryer, M. J., Andrews, J. R., Oxborough, K., Blowers, D. A., and Baker, N. R. (1998). Relationship between CO2 assimilation, photosynthetic electron transport, and active O2 metabolism in leaves of maize in the field during periods of low temperature (vol 116, pg 571, 1998). Plant Physiol. 117, 335–335. doi: 10.1104/pp.116.2.571
Fucikova, K., Leliaert, F., Cooper, E. D., Skaloud, P., D’hondt, S., De Clerck, O., et al. (2014). New phylogenetic hypotheses for the core Chlorophyta based on chloroplast sequence data. Front. Ecol. Evol. 2. doi: 10.3389/fevo.2014.00063
Gede, S., Alissa, D. P., Yovita, A. D., Fahma, F. N. A., Dea, I. A., Pingkan, A., et al (2017). Impact of Salinity and Light Intensity Stress on B Vitamins Content in Marine Diatom Skeletonema costatum. Journal of Fisheries and Aquatic Science, 12, 22–28.
Goodwin, S., Mcpherson, J. D., and Mccombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351. doi: 10.1038/nrg.2016.49
Guo, Y.-L. (2013). Gene family evolution in green plants with emphasis on the origination and evolution of Arabidopsis thaliana genes. Plant J. 73, 941–951. doi: 10.1111/tpj.12089
Helliwell, K. E., Wheeler, G. L., Leptos, K. C., Goldstein, R. E., and Smith, A. G. (2011). Insights into the evolution of vitamin B12 auxotrophy from sequenced algal genomes. Mol. Biol. Evol. 28, 2921–2933. doi: 10.1093/molbev/msr124
Henriquez, C. L., Abdullah, Ahmed, I., Carlsen, M. M., Zuluaga, A., Croat, T. B., et al. (2020). Molecular evolution of chloroplast genomes in Monsteroideae (Araceae). Planta 251. doi: 10.1007/s00425-020-03365-7
Ho, C.-L. (2020). Comparative genomics reveals differences in algal galactan biosynthesis and related pathways in early and late diverging red algae. Genomics 112, 1536–1544. doi: 10.1016/j.ygeno.2019.09.002
Hou, Y. and Lin, S. (2009). Distinct gene number-genome size relationships for eukaryotes and non-eukaryotes: gene content estimation for dinoflagellate genomes. PloS One 4. doi: 10.1371/journal.pone.0006978
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., Von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Iram, S., Hayat, M. Q., Tahir, M., Gul, A., Abdullah, and Ahmed, I. (2019). Chloroplast genome sequence of artemisia scoparia: comparative analyses and screening of mutational hotspots. Plants-Basel 8. doi: 10.3390/plants8110476
Jeong, Y. and Lee, J. (2024). Comparative analysis of organelle genomes provides conflicting evidence between morphological similarity and phylogenetic relationship in diatoms. Front. Mar. Sci. 10. doi: 10.3389/fmars.2023.1283893
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., Mcanulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Katoh, K. and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O., and Grau, J. (2018). Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinf. 19. doi: 10.1186/s12859-018-2203-5
Kishor, P. K., Hong, Z., Miao, G. H., Hu, C. A. A., and Verma, D. P. S. (1995). Overexpression of [delta]-pyrroline-5-carboxylate synthetase increases proline production and confers osmotolerance in transgenic plants. Plant Physiol. 108, 1387–1394. doi: 10.1104/pp.108.4.1387
Krienitz, L., Bock, C., Nozaki, H., and Wolf, M. (2011). SSU rRNA gene phylogeny of morphospecies affiliated to the bioassay alga “Selenastrum capricornutum” recovered the polyphyletic origin of crescent-shaped chlorophyta. J. Phycol. 47, 880–893. doi: 10.1111/j.1529-8817.2011.01010.x
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–U354. doi: 10.1038/nmeth.1923
Leliaert, F., Smith, D. R., Moreau, H., Herron, M. D., Verbruggen, H., Delwiche, C. F., et al. (2012). Phylogeny and molecular evolution of the green algae. Crit. Rev. Plant Sci. 31, 1–46. doi: 10.1080/07352689.2011.615705
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, L., Wang, S., Wang, H., Sahu, S. K., Marin, B., Li, H., et al. (2020). The genome of Prasinoderma coloniale unveils the existence of a third phylum within green plants. Nat. Ecol. Evol. 4, 1220–122+. doi: 10.1038/s41559-020-1221-7
Liu, C., Shi, X., Wu, F., Ren, M., Gao, G., and Wu, Q. (2020). Genome analyses provide insights into the evolution and adaptation of the eukaryotic Picophytoplankton Mychonastes homosphaera. BMC Genomics 21. doi: 10.1186/s12864-020-06891-6
Mendes, F. K., Vanderpool, D., Fulton, B., and Hahn, M. W. (2020). CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518. doi: 10.1093/bioinformatics/btaa1022
Mirarab, S., Reaz, R., Bayzid, M. S., Zimmermann, T., Swenson, M. S., and Warnow, T. (2014). ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30, I541–I548. doi: 10.1093/bioinformatics/btu462
Mmbando, G. S. (2024). The recent possible strategies for breeding ultraviolet-B-resistant crops. Heliyon 10. doi: 10.1016/j.heliyon.2024.e27806
Murata, Y., Ma, J. F., Yamaji, N., Ueno, D., Nomoto, K., and Iwashita, T. (2006). A specific transporter for iron(III)-phytosiderophore in barley roots. Plant J. 46, 563–572. doi: 10.1111/j.1365-313X.2006.02714.x
Nagai, T., Taya, K., and Yoda, I. (2016). Comparative toxicity of 20 herbicides to 5 periphytic algae and the relationship with mode of action. Environ. Toxicol. Chem. 35, 368–375. doi: 10.1002/etc.3150
Nishitsuji, K., Arimoto, A., Yonashiro, Y., Hisata, K., Fujie, M., Kawamitsu, M., et al. (2020). Comparative genomics of four strains of the edible brown alga, Cladosiphon okamuranus. BMC Genomics 21. doi: 10.1186/s12864-020-06792-8
Patova, E. N. and Dorokhova, M. F. (2008). Green algae in tundra soils affected by coal mine pollutions. Biologia 63, 831–835. doi: 10.2478/s11756-008-0107-y
Pérez-Arellano, I., Carmona-Álvarez, F., Martínez, A. I., Rodríguez-Díaz, J., and Cervera, J. (2010). Pyrroline-5-carboxylate synthase and proline biosynthesis: From osmotolerance to rare metabolic disease. Protein Sci. 19, 372–382. doi: 10.1002/pro.340
Prachumwat, A. and Li, W.-H. (2008). Gene number expansion and contraction in vertebrate genomes with respect to invertebrate genomes. Genome Res. 18, 221–232. doi: 10.1101/gr.7046608
Prasad, T. K. (1997). Role of catalase in inducing chilling tolerance in pre-emergent maize seedlings. Plant Physiol. 114, 1369–1376. doi: 10.1104/pp.114.4.1369
Qiu, H., Price, D. C., Yang, E. C., Yoon, H. S., and Bhattacharya, D. (2015). Evidence of ancient genome reduction in red algae (rhodophyta). J. Phycol. 51, 624–636. doi: 10.1111/jpy.12294
Rawat, I., Kumar, R. R., Mutanda, T., and Bux, F. (2013). Biodiesel from microalgae: A critical evaluation from laboratory to large scale production. Appl. Energy 103, 444–467. doi: 10.1016/j.apenergy.2012.10.004
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Roth, M. S., Cokus, S. J., Gallaher, S. D., Walter, A., Lopez, D., Erickson, E., et al. (2017). Chromosome-level genome assembly and transcriptome of the green alga Chromochloris zofingiensis illuminates astaxanthin production. PNAS 114, E4296–E4305. doi: 10.1073/pnas.1619928114
Sauer, N., Komor, E., and Tanner, W. (1983). Regulation and characterization of 2 inducible amino-acid-transport systems in chlorella-vulgaris. Planta 159. doi: 10.1007/BF00392075
Schaaf, G., Ludewig, U., Erenoglu, B. E., Mori, S., Kitahara, T., and Von Wirén, N. (2004). ZmYS1 functions as a proton-coupled symporter for phytosiderophore- and nicotianamine-chelated metals. J. Biol. Chem. 279, 9091–9096. doi: 10.1074/jbc.M311799200
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Smith, D. R. (2015). Mutation rates in plastid genomes: they are lower than you might think. Genome Biol. Evol. 7, 1227–1234. doi: 10.1093/gbe/evv069
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS:: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi: 10.1093/nar/gkl200
Stibal, M., Sabacka, M., and Kastovska, K. (2006). Microbial communities on glacier surfaces in Svalbard:: Impact of physical and chemical properties on abundance and structure of cyanobacteria and algae. Microb. Ecol. 52, 644–654. doi: 10.1007/s00248-006-9083-3
Sun, L., Fang, L., Zhang, Z., Chang, X., Penny, D., and Zhong, B. (2016). Chloroplast phylogenomic inference of green algae relationships. Sci. Rep. 6. doi: 10.1038/srep20528
Suzuki, S., Yamaguchi, H., Nakajima, N., and Kawachi, M. (2018). Raphidocelis subcapitata (=Pseudokirchneriella subcapitata) provides an insight into genome evolution and environmental adaptations in the Sphaeropleales. Sci. Rep. 8. doi: 10.1038/s41598-018-26331-6
Trainor, F. R. and Burg, C. A. (1965). Scenedesmus obliquus sexuality. Science 148, 1094–109+. doi: 10.1126/science.148.3673.1094
Turmel, M., De Cambiaire, J.-C., Otis, C., and Lemieux, C. (2016). Distinctive Architecture of the Chloroplast Genome in the Chlorodendrophycean Green Algae Scherffelia dubia and Tetraselmis sp CCMP 881. PloS One 11. doi: 10.1371/journal.pone.0148934
Turmel, M., Otis, C., and Lemieux, C. (2017). Divergent copies of the large inverted repeat in the chloroplast genomes of ulvophycean green algae. Sci. Rep. 7. doi: 10.1038/s41598-017-01144-1
Van Breusegem, F., Slooten, L., Stassart, J. M., Botterman, J., Moens, T., Van Montagu, M., et al. (1999). Effects of overproduction of tobacco MnSOD in maize chloroplasts on foliar tolerance to cold and oxidative stress. J. Exp. Bot. 50, 71–78. doi: 10.1093/jxb/50.330.71
Wang, D., Zheng, Z., Li, Y., Hu, H., Wang, Z., Du, X., et al. (2021). Which factors contribute most to genome size variation within angiosperms? Ecol. Evol. 11, 2660–2668. doi: 10.1002/ece3.7222
Wheeler, T. J. and Eddy, S. R. (2013). nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489. doi: 10.1093/bioinformatics/btt403
Wolf, M., Buchheim, M., Hegewald, E., Krienitz, L., and Hepperle, D. (2002). Phylogenetic position of the sphaeropleaceae (Chlorophyta). Plant Syst. Evol. 230, 161–171. doi: 10.1007/s006060200002
Xie, B., Bishop, S., Stessman, D., Wright, D., Spalding, M. H., and Halverson, L. J. (2013). Chlamydomonas reinhardtii thermal tolerance enhancement mediated by a mutualistic interaction with vitamin B12-producing bacteria. ISME J. 7, 1544–1555. doi: 10.1038/ismej.2013.43
Xiong, Q., Hu, Y., Dong, X., Chen, Y., Liu, G., and Hu, Z. (2022). Phylotranscriptomic and evolutionary analyses of Oedogoniales (Chlorophyceae, chlorophyta). Diversity 14, 157. doi: 10.3390/d14030157
Xiong, Q., Hu, Y., Lv, W., Wang, Q., Liu, G., and Hu, Z. (2021). Chloroplast genomes of five Oedogonium species: genome structure, phylogenetic analysis and adaptive evolution. BMC Genomics 22. doi: 10.1186/s12864-021-08006-1
Yamagishi, T., Yamaguchi, H., Suzuki, S., Horie, Y., and Tatarazako, N. (2017). Cell reproductive patterns in the green alga Pseudokirchneriella subcapitata (=Selenastrum capricornutum) and their variations under exposure to the typical toxicants potassium dichromate and 3,5-DCP. PloS One 12. doi: 10.1371/journal.pone.0171259
Yang, Z. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yu, G., Wang, L.-G., Han, Y., and He, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. Omics-a J. Integr. Biol. 16, 284–287. doi: 10.1089/omi.2011.0118
Zeng, X., Yuan, H., Dong, X., Peng, M., Jing, X., Xu, Q., et al. (2020). Genome-wide dissection of co-selected UV-B responsive pathways in the UV-B adaptation of Qingke. Mol. Plant 13, 112–127. doi: 10.1016/j.molp.2019.10.009
Zhang, D., Gao, F., Jakovlic, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Keywords: Sphaeropleales, comparative functional analysis, environmental adaptation, evolutionary analyses, gene family expansions and contraction
Citation: Xiong Q, Zheng L, Zhang Q, Li T, Zheng L and Song L (2025) Comparative genomics and evolutionary analyses of Sphaeropleales. Front. Plant Sci. 16:1534646. doi: 10.3389/fpls.2025.1534646
Received: 26 November 2024; Accepted: 17 September 2025;
Published: 16 October 2025.
Edited by:
Qing-Bo Yu, Shanghai Normal University, ChinaCopyright © 2025 Xiong, Zheng, Zhang, Li, Zheng and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lirong Song, bHJzb25nQGloYi5hYy5jbg==