 Long-Jiao Hu
Long-Jiao Hu Ji-Ping Xuan
Ji-Ping Xuan Yang Li1,2
Yang Li1,2 Li-Na Deng
Li-Na Deng Zheng-Hai Mo
Zheng-Hai Mo- 1Jiangsu Key Laboratory for the Research and Utilization of Plant Resources, Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing, Jiangsu, China
- 2Jiangsu Engineering Research Center for the Germplasm Innovation and Utilization of Pecan, Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing, Jiangsu, China
- 3School of Ocean and Biological Engineering, Yancheng Institute of Technology, Yancheng, Jiangsu, China
The anthracnose disease caused by Colletotrichum fructicola has widely occurred in pecan (Carya illinoinensis) in China, seriously affecting its fruit yield and quality. However, the details of the infection strategy of C. fructicola remain to be elucidated. In this study, unique molecular identifier-RNA sequencing (UMI RNA-seq) was used to analyze differentially expressed genes (DEGs) of C. fructicola and candidate effectors were predicted. Two candidate effectors were identified during the early infection stages of C. fructicola. There were 6,822 DEGs at three infection timepoints (6, 24, and 36 h post-inoculation), and these genes were involved in spore germination, nutrient uptake, detoxification, secretion of toxic substances (such as effectors and toxins), inhibition of the host’s immune response, and protein post-translational modification, which participated in the pathogenic process of C. fructicola. Moreover, 191 candidate effectors were predicted and their expression trends were divided into five clusters. Two candidate effectors Cf-ID1 and Cf-ID2 were selected for functional validation, and they were demonstrated to trigger cell death and immune response in Nicotiana benthamiana. Cf-ID1 and Cf-ID2 are located in both cytoplasm and nucleus and could suppress the infection of C. fructicola by eliciting defense responses in N. benthamiana. This study provided valuable information for in-depth research on the pathogenesis of C. fructicola.
Introduction
Pecan (Carya illinoinensis (Wangenh.) K. Koch), which belongs to the family Juglandaceae, genus Carya, is native to the United States and Mexico (Sparks, 2002). Due to the high economic value of its nuts and timber, pecan has been cultivated in many countries, including China, Australia, Argentina, and Peru (Bilharva et al., 2018). In China, this species is widely planted in Jiangsu and Anhui Provinces. With the continuous expansion of the planting area of pecan, disease severity has also increased, which has been a serious problem that has limited the development of the planting industry. Anthracnose caused by Colletotrichum spp. is one of the most serious diseases in pecan, which mainly infects the fruit and leaves (Chang et al., 2022; Meng et al., 2021). Among the numerous Colletotrichum spp. that infect pecan, the isolation rate of Colletotrichum fructicola is the highest, and it is a critical pathogen observed in pecan cultivation that impedes plant growth and overall fruit productivity (Meng, 2020).
C. fructicola has a broad host range and has been identified in over 50 plant species in the world, resulting in large economic losses (Liang et al., 2018). C. fructicola is a hemibiotroph with a multistage infection process (biotrophic and necrotrophic phases) (Liang et al., 2018). A previous study showed that factors contributing to Colletotrichum virulence, such as effectors and secondary metabolite (SM) biosynthetic enzymes, were generally expressed early in the infection process and functioned in host defense suppression or cellular activity manipulation (Baroncelli et al., 2016). At present, the widespread occurrence of apple anthracnose and Camellia anthracnose caused by C. fructicola has been further studied to explore the genes and pathways related to pathogenesis (Rockenbach et al., 2016; Liang et al., 2018). Chang et al. revealed the mechanisms of resistance to Colletotrichum fioriniae in two pecan varieties with different resistance levels through transcriptome sequencing (Chang et al., 2023). The potential pathways and genes involved in pecan resistance to Pestalotiopsis microspore were also explored (Chen et al., 2022). However, there are few studies on the interaction between C. fructicola and pecan thus far (Chang et al., 2022). With the increasingly serious occurrence of pecan anthracnose, exploring the pathogenesis of C. fructicola in pecan is greatly significant for the prevention and control of this disease.
In nature, plants are susceptible to various pathogens, including bacteria, fungi, oomycetes, viruses, and parasitic nematodes. To defend against pathogens, plants have developed an immune system that recognizes microbial invaders and activates defense mechanisms to prevent their proliferation. At the same time, pathogens secreted effectors to manipulate plant immunity pathways or create a favorable environment to facilitate pathogen colonization. The latest definition of an effector is: a pathogen-secreted molecule that functions to induce non-self-phenotypes in other organisms or the environment (Ai et al., 2024). Numerous studies have shown that the consequences of plant-pathogen interactions are largely determined by effectors (Jones and Dangl, 2006; Wang et al., 2018).
RNA-sequencing technology has been widely adopted to investigate host responses during pathogen infection (Foster et al., 2015; Li et al., 2019) or to identify the associated molecular cellular pathways and virulence factors (such as effectors) of pathogens secreted during the infection process (Zuluaga et al., 2016; Hu et al., 2021). Although RNA-seq is a powerful tool, sequence-dependent bias and the inaccuracy of PCR amplification limit its further applications (Chang et al., 2023). To solve this problem, in UMI RNA-seq, each cDNA molecule is labeled with a unique molecular identifier (UMI) before library construction (Smith et al., 2017).
In this study, UMI RNA-seq was applied to explore the infection strategy of C. fructicola in pecan during the early stages. Particular emphasis was placed on the identification of differentially expressed genes (DEGs), Gene Ontology (GO) enrichment, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of DEGs of C. fructicola. The candidate effectors of C. fructicola were predicted. To focus on the relationship between the candidate effectors and their corresponding enriched functions, the expression trends of the candidate effectors were divided. Then, two candidate effectors were selected for functional validation and were characterized via the transient expression in Nicotiana benthamiana. The abilities of the two effectors to trigger plant immunity and suppress C. fructicola infection, as well as their subcellular localization, were examined.
Materials and methods
Biological material
The C. fructicola isolate CZ102 was originally isolated from leaf spot lesions on pecan in Chuzhou City, Anhui Province, China. The isolate CZ102 was subcultured on potato dextrose agar (PDA) at 25°C and preserved using filter paper at -20°C in the Fungal Laboratory, Fruit Tree Research Center, Institute of Botany, Jiangsu Province and Chinese Academy of Sciences. Seedling progenies derived from an open-pollinated cultivar (‘Pawnee’) were used as plant materials. The seedlings were cultivated as previously described (Mo et al., 2023).
Sample preparation
In a preliminary study, the appressorium and primary hyphae (infection structures) of C. fructicola began to form on onion epidermis at 6 h and 24 h, respectively (Supplementary Figure S1). A previous study found that C. fructicola formed appressorium at 8 h after inoculation with cellophane, and formed a large number of primary hyphae at 36 h after inoculation (Liang et al., 2018). Thus, three infection timepoints [6, 24, and 36 h post-inoculation (hpi)] were finally determined for this study. Specifically, the C. fructicola isolate CZ102 was cultured in a complete medium (CM) to produce conidia as previously described. The CM composition per liter is: sucrose 20 g, yeast extract 6 g, and casamino acid 6 g (Huang et al., 2017). Conidial suspensions were adjusted to 1×106 spores/mL and sprayed on healthy leaves of 3-month-old pecan seedlings. The inoculated seedlings were kept in a moist chamber at 25°C (80% relative humidity and 12 h light and 12 h dark cycles). The leaves of pecan seedlings were collected after being inoculated with conidial suspensions for 6 h (CP_6 h, group B), 24 h (CP_24 h, group C), and 36 h (CP_36 h, group D). Purified conidia of C. fructicola were used as a control (group A). For each group, three independent samples were immediately frozen in liquid nitrogen and stored at -80°C for subsequent RNA extraction and sequencing.
RNA isolation, library preparation, and sequencing
The UMI RNA-seq experiment, the high throughput sequencing, and basic RNA-Seq data analysis were conducted by Seqhealth Technology Co., LTD (Wuhan, China).
The total RNA of all samples was extracted using TRIzol Reagent (Invitrogen, cat. No. 15596026)following the methods of Chomczynski et al (Chomczynski and Sacchi, 1987). DNA digestion was carried out using DNase I. RNA quality was determined by examining A260/A280 with a NanoDrop One C spectrophotometer (Thermo Fisher Scientifific, Inc.). The RNA integrity of 12 samples was confirmed using a 5300 Fragment Analyzer system (Agilent). Qualified RNAs of total samples were finally quantified by Qubit 3.0 with a Qubit RNA broad-range assay kit (Life Technologies, Q10210).
Two μg total RNAs were used for stranded RNA sequencing library preparation using a KC-DigitalTM Stranded mRNA Library Prep Kit (Catalog NO. DR08502, Wuhan Seqhealth Co., Ltd. China) following the manufacturer’s instructions. The kit eliminates duplication bias in PCR and sequencing steps by using a UMI of eight random bases to label the pre-amplified cDNA molecules. The library products corresponding to 200-500 bps were enriched, quantified, and finally sequenced on a DNBSEQ-T7 sequencer (MGI Tech Co., Ltd. China) with the PE150 model.
RNA-seq data analysis
Raw sequencing data of the four groups (A, B, C, and D) were first filtered using fastp (version 0.23.0). Then, the low-quality reads were discarded and the reads contaminated with adaptor sequences were trimmed. The clean reads were further treated with in-house scripts to eliminate duplication bias introduced in library preparation and sequencing. In brief, the clean reads were first clustered according to the UMI sequences, in which reads with the same UMI sequences were grouped into the same cluster. Reads in the same cluster were compared to each other by pairwise alignment, and then reads with sequence identity over 95% were extracted to a new sub-cluster. After all sub-clusters were generated, multiple sequence alignment was performed to get one consensus sequence for each sub-cluster. After these steps, any errors and biases introduced by PCR amplification or sequencing were eliminated.
The de-duplicated consensus sequences were used for standard RNA-seq analysis. They were mapped to the reference genome of C. fructicola from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/genome/57524?genome_assembly_id=750361) using STAR software (version 2.5.3a) with default parameters. The reads mapped to the exon regions of each gene were counted by featureCounts (Subread-1.5.1; Bioconductor) and then Fragments Per Kilobase of transcript per Million mapped reads (FPKM) was calculated. The DEGs between groups were identified using the edgeR package (version 3.12.1). An adjusted P-value < 0.05 and absolute log2 (fold change) ≥ 1 were used to judge the statistical significance of gene expression differences. Among the DEGs, upregulated genes of C. fructicola had a higher expression at least at one infection timepoint (B, C, or D) than at the conidia stage (A), and downregulated genes of C. fructicola had a lower expression at all the infection timepoints than at the conidia stage.
GO enrichment analysis of DEGs in C. fructicola
The GO enrichment analysis of C. fructicola upregulated genes from the above transcriptomic data (CP_6 h, CP_24 h, CP_36 h) was implemented by the GOseq R package, in which gene length bias was corrected (Young et al., 2014). Furthermore, DEGs were selected for biological process, cellular component, and molecular function analysis using the Rich factor as the screening indicator. GO terms with an adjusted P-value (i.e., false discovery rate, FDR) < 0.05 were considered significantly enriched.
KEGG enrichment analysis of DEGs in C. fructicola
The KEGG is a database resource for understanding the high-level functions and utilities of biological systems (http://www.genome.jp/kegg/) (Kanehisa et al., 2016). KOBAS software (version: 2.1.1) was used to determine the statistical enrichment (with adjusted P value < 0.05) of DEGs in KEGG pathways. Furthermore, to improve the annotation, a BLAST search was performed against the SWISS-Prot database.
Real-time quantitative PCR analysis
To validate the accuracy of UMI RNA-seq data, real-time quantitative polymerase chain reaction (RT-qPCR) assays were carried out in a CFX Opus 96 real-time PCR system (Bio-Rad, USA) with ChamQ SYBR qPCR Master Mix (Low ROX Premixed) (Vazyme, China) according to the manufacturer’s instructions. The relative expression levels of seven DEGs in C. fructicola were detected. PCR amplification was performed under the following conditions: 95°C for 30 s, followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. The alpha-tubulin (TUB) gene of C. fructicola was chosen as a constitutively expressed endogenous control gene (Liang et al., 2018; Shang et al., 2020). The relative quantifications were calculated based on the 2-ΔΔCt method. Primer sequences are provided in Supplementary Table S1.
Candidate effectors prediction and analysis
Potential effectors were screened according to several criteria (Stergiopoulos and De Wit, 2009). Briefly, they should be upregulated genes during the pathogen-host interaction stage. At the same time, there should be the presence of an N-terminal signal peptide and the absence of a transmembrane domain in the amino acid sequences of potential effectors. Moreover, the sequence length of potential effectors should be ≤ 300 amino acids (aa). Candidate effectors screened according to the above requirements were predicted to further distinguish between apoplastic and cytoplasmic effectors using EffectorP 3.0 (https://effectorp.csiro.au/) (Sperschneider and Dodds, 2022). The signal peptide and the transmembrane domain were predicted by SignalP5.0 (http:///services.healthtech.dtu.dk/services/SignalP-5.0/) (Almagro Armenteros et al., 2019) and TMHMM 2.0 (http://www.cbs.dtu.dk/services/TMHMM-2.0/) (Sonnhammer et al., 1998), respectively. A BLASTP search of all candidate effectors against the Non-Redundant (NR) database was conducted to predict their functions. The expression trends of candidate effectors were clustered using the R package Mfuzz and the membership degree represented the importance of the gene in the cluster to a certain extent.
Plasmid construction
Referring to the previously used method (Hu et al., 2019), three candidate effectors were cloned from the cDNA of C. fructicola using the specific primers listed in Supplementary Table S1. Subsequently, purified PCR products were ligated into the pBINGFP vector using the ApexHF HS DNA Polymerase FS (Accurate Biotechnology (Hunan) Co., LTD, Changsha, China) after confirmation by sequencing.
Transient expression assays in Nicotiana benthamiana
The constructed plasmids of the candidate effectors were inserted into Agrobacterium tumefaciens GV3101 by electroporation. The culture conditions of A. tumefaciens GV3101, carrying candidate effectors, components of washing buffer, and a final optical density (OD) of 600 nm of A. tumefaciens suspensions, were the same as those in our previous study (Hu et al., 2020). For agroinfiltration assays, the A. tumefaciens suspensions were infiltrated into the leaves of N. benthamiana using a needleless syringe. Symptoms of N. benthamiana were observed visually 7 days after infiltration. Both the empty vector pBINGFP and a candidate effector Cf-1 (Gene ID: CGMCC3_g6914), which has been previously validated to lack necrosis-inducing ability in N. benthamiana (unpublished data), were used as negative controls.
To test the suppression of C. fructicola infection by Cf-ID1 and Cf-ID2, pBINGFP-Cf-ID1, pBINGFP-Cf-ID2, and pBINGFP were infiltrated into the leaves of N. benthamiana. The leaves were detached at 36 h post-infiltration, and then inoculated on the infiltrated regions with 4 mm hyphae blocks of C. fructicola. Under dark conditions at 72 h post-infiltration, the leaves were illuminated with a handheld UV lamp (LUYOR-3410UV, Shanghai Luyor Biotechnology Co., Ltd, Shanghai, China) for clear observation of the lesion dimensions. At the same time, the disease lesions were photographed. For Cf-ID1 and Cf-ID2, the experiment was respectively performed three times, and three different plants with two inoculated leaves were used for each assay.
To determine whether candidate effectors Cf-ID1 and Cf-ID2 can trigger a PAMP-triggered immunity (PTI) response, the relative expressions of three PTI marker genes (NbAcre31, NbPTI5, and NbCyp71D20) (Nie et al., 2018) were detected at 3 h post-infiltration and 12 h post-infiltration with A. tumefaciens GV3101 carrying pBINGFP-Cf-ID1 and pBINGFP-Cf-ID2 in N. benthamiana, respectively. N. benthamiana infiltrated with pBINGFP was the control. NbEF1α of N. benthamiana was used as the constitutively expressed endogenous control gene (Ma et al., 2015). The amplification conditions of RT-qPCR were consistent with those mentioned above. The relative quantifications were calculated based on the 2-ΔΔCt method. The assay was performed three times. Primer sequences are provided in Supplementary Table S1.
Subcellular localization in N. benthamiana
The N. benthamiana leaves were agroinfiltrated with pBINGFP-Cf-ID1 and pBINGFP-Cf-ID2, and the P19 silencing suppressor in a 2:1 ratio at a final OD600 = 0.5 for each construct, respectively. The N. benthamiana leaves agroinfiltrated with empty vector pBINGFP were used as controls. Furthermore, 36 h after agroinfiltration, patches of N. benthamiana leaves were cut and mounted in water and analyzed using an LSM900 laser scanning microscope (Zeiss, Germany).
Results
Pathogenicity of Colletotrichum fructicola to pecan
Leaves of healthy 3-month-old pecan seedlings were sprayed with conidial suspensions of C. fructicola. Black spot symptoms appeared on the leaves of pecan 3 days after inoculation. We re-isolated, purified, and sequenced the pathogen from the spots, and the results showed that they were consistent with the initially inoculated C. fructicola. Therefore, the pathogenicity of C. fructicola to pecan was confirmed. The conidia morphology and susceptible pecan are shown in Supplementary Figure S2.
Summary data of the transcriptome for Colletotrichum fructicola
In total, we generated 1,064,785,506 raw reads and 886,242,942 clean reads from 12 samples by UMI RNA-seq. Sample A (C. fructicola conidia) produced no less than 7 Gb of data, and Samples B, C, and D (mixed samples of C. fructicola conidia and plant tissue) produced no less than 13 Gb of data. The percentage of Q30 of all samples reached more than 89.85%, and the GC content was 46.83%–54.58% (Supplementary Table S2). In the B, C, and D groups, a total of 678 million reads were obtained, of which approximately 1.36% were aligned to the C. fructicola genomes. For the A group, there were 112 million reads in three samples, and approximately 96.62% of the reads were aligned to the C. fructicola genomes. The complete list, annotations, and FPKM of all transcripts are shown in Supplementary Table S3.
Differentially expressed genes of Colletotrichum fructicola
In this study, gene expression levels were estimated via the FPKM method. The genes with a P-value < 0.05 and |log2 (fold change) | ≥1 were designated as DEGs between the early stages of infection (6, 12 and 24 h post-infection) and the conidia stage (0 h). Finally, there were 7,822 DEGs of C. fructicola at the three infection points, and the number of DEGs at 6 h, 24 h, and 36 h was 4,184 (1,867 upregulated genes and 2,317 downregulated genes), 4,827 (2,267 upregulated genes and 2,560 downregulated genes), and 5,019 (2,398 upregulated genes and 2,621 downregulated genes), respectively (Figure 1A). The list and annotation information and heatmap of all DEGs in C. fructicola are shown in Supplementary Table S4 and Supplementary Figure S3, respectively. According to the Venn diagram analysis, 3,648 genes were DEGs at two infection timepoints, and 2,015 genes were DEGs at all three infection periods (Figure 1B). The Venn diagram analysis of 4,284 upregulated genes in the C. fructicola transcriptome during the early stages of infection is shown in Supplementary Figure S4.
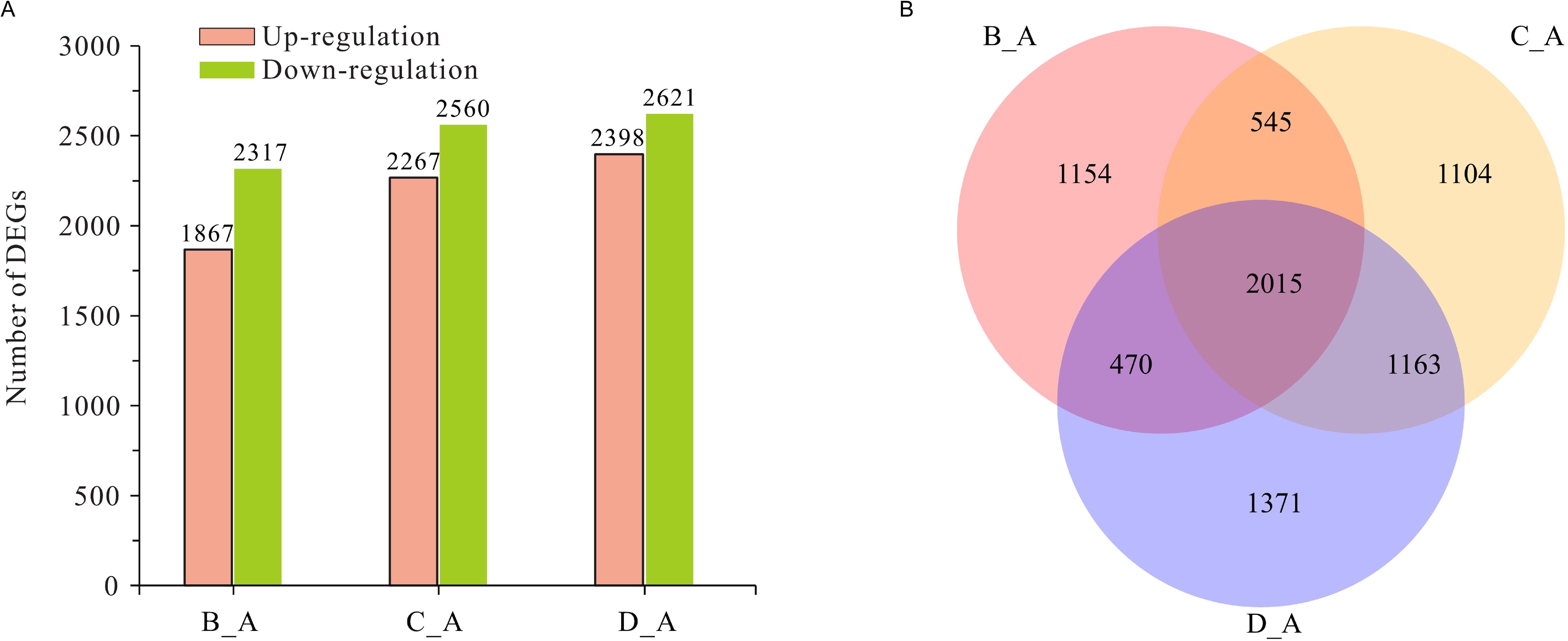
Figure 1. The analysis of differentially expressed genes (DEGs) in Colletotrichum fructicola during the early stages of pecan infection. (A) The numbers of DEGs of C. fructicola. (B) The Venn diagram analysis of DEGs of C. fructicola. A C. fructicola conidia. B 6 h post-inoculation (hpi). C 24 hpi. D 36 hpi.
GO functional annotation and KEGG functional enrichment analysis of DEGs of C. fructicola
In the C. fructicola transcriptomes at 6 hpi, the DEGs of C. fructicola showed significant enrichment in terms of post-transcriptional modifications, such as phosphorylation, tRNA methylation, and protein ubiquitination. Ribosome biogenesis, phosphorelay signal transduction system, signal transduction and response to oxidative stress, lipid storage, and phospholipid biosynthetic process were significantly enriched (Figure 2A). Moreover, the longevity regulating pathway, phosphonate and phosphinate metabolism, alanine aspartate and glutamate metabolism, phosphatidylinositol signaling system, cysteine and methionine metabolism, galactose metabolism, phenylalanine tyrosine and tryptophan biosynthesis, and MAPK signaling pathway were also significantly enriched (Figure 2B).
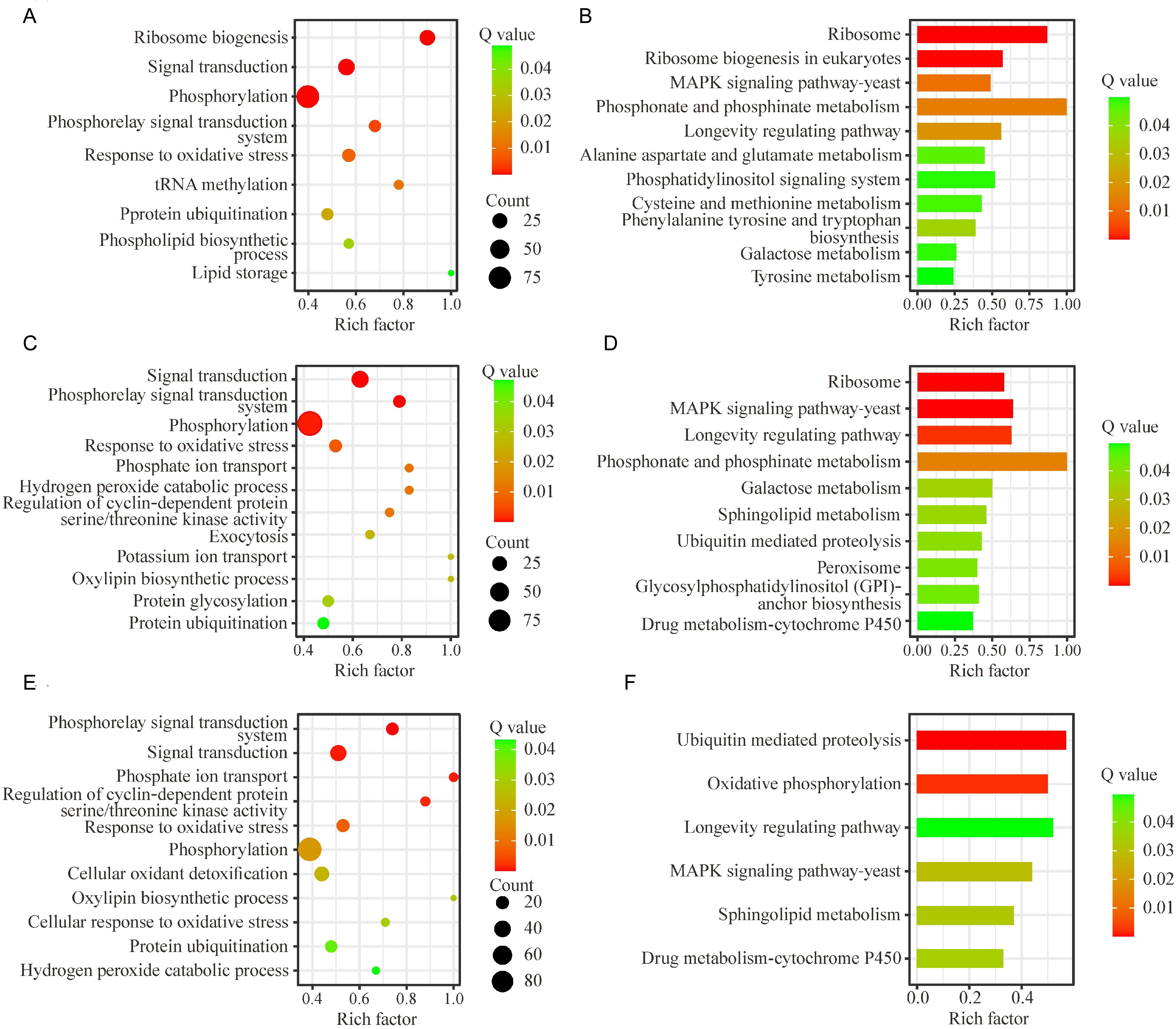
Figure 2. GO functional annotation and KEGG functional enrichment analysis of differentially expressed genes (DEGs) in Colletotrichum fructicola during the early stages of pecan infection. (A, C, E) Representative GO terms of DEGs in the C. fructicola transcriptomes at 6 h, 24 h, and 36 h post-infection, respectively. (B, D, F) Representative KEGG pathways of DEGs in the C. fructicola transcriptomes at 6 h, 24 h, and 36 h post-infection, respectively. A: C. fructicola conidia. B: 6 h post-inoculation (hpi). C: 24 hpi. D: 36 hpi.
In the C. fructicola transcriptomes at 24 hpi, the hydrogen peroxide catabolic process, oxylipin biosynthetic process, and response to oxidative stress were significantly enriched. Furthermore, exocytosis, potassium ion transport, phosphate ion transport, protein ubiquitination, and glycosylation were also significantly enriched (Figure 2C), which have been proven to be involved in nutrient intake, adaptation to new environment, and inhibition of the host’s immune response for successful infection and reproduction (Lamarche et al., 2008; Chen et al., 2014, 2017; Qiu and Luo, 2017). At the same time, pathways associated with sphingolipid metabolism, ubiquitin-mediated proteolysis, peroxisome, and glycosylphosphatidylinositol (GPI)-anchor biosynthesis, and cytochrome P450 were significantly enriched (Figure 2D).
In the C. fructicola transcriptomes at 36 hpi, the oxylipin biosynthetic process, response to oxidative stress, cellular oxidant detoxification, cellular response to oxidative stress, and hydrogen peroxide catabolic process were significantly enriched (Figure 2E). Metabolic pathways, such as ubiquitin-mediated proteolysis, oxidative phosphorylation, sphingolipid metabolism, and cytochrome P450, were significantly enriched (Figure 2F).
Notably, terms related to toxic response, survival and reproduction, and immune recognition, such as the phosphorelay signal transduction system, signal transduction and response to oxidative stress, phosphorylation, and protein ubiquitination, were significantly enriched at these three timepoints (Figures 2A, C, E). Moreover, the MAPK signaling pathway and longevity regulating pathway were significantly enriched at these three timepoints (Figures 2B, D, F). These results suggested that during the early stage of infection, C. fructicola might not only be forced to adapt to the host environment under oxidative stress but also actively initiates aggression (such as secreting toxic substances and evading host immune recognition).
Confirmation of the accuracy of the transcriptome data by qRT-PCR
To validate the accuracy of UMI RNA-seq, the transcript levels of seven DEGs from C. fructicola were determined by RT-qPCR. The trends obtained by RT-qPCR were highly consistent with the RNA-Seq results (Figure 3). This result indicated that the UMI RNA-seq data was reliable, and can be used for subsequent analysis.
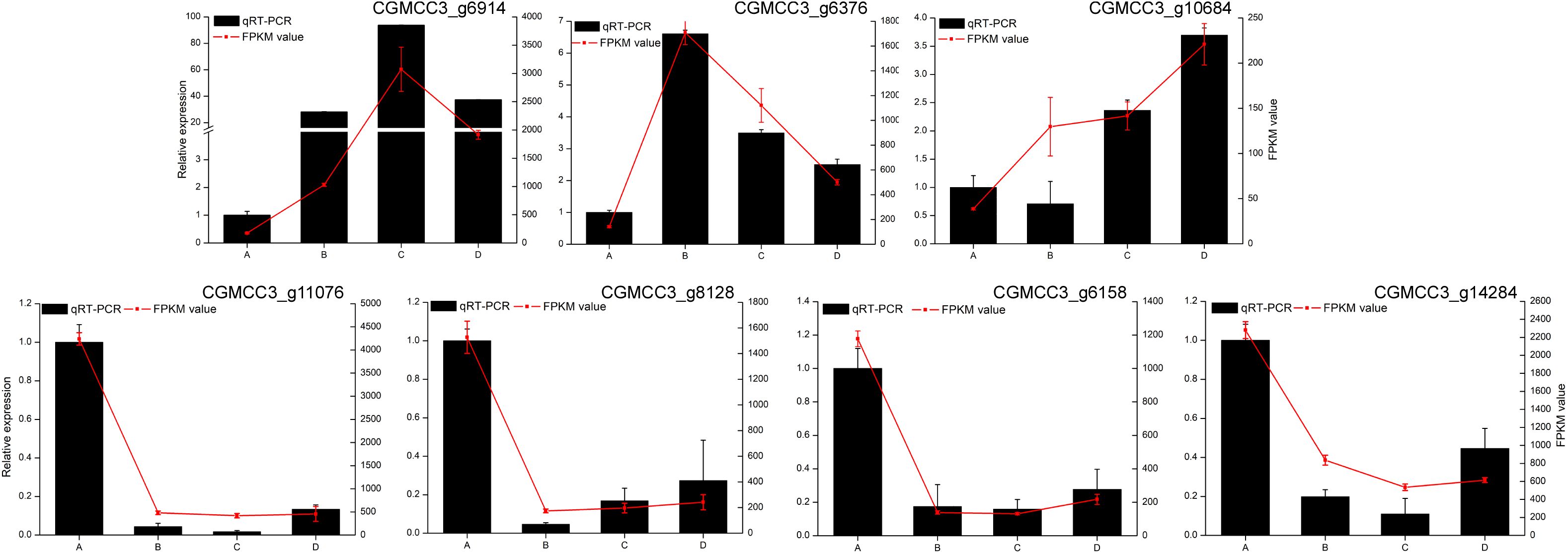
Figure 3. Validation of Colletotrichum fructicola transcriptome data. Bars: relative expression levels of differentially expressed genes (DEGs) in C. fructicola by real-time quantitative (RT-qPCR); red line: FPKM in the C. fructicola transcriptomes. Data are the means, and the error bars represent ± SD from three biological replicates. Different letters on top of the bars indicate statistically significant differences (p < 0.05, t-test) as measured by Duncan’s multiple range test.
Screen and functional annotation of candidate effectors of C. fructicola
Based on the screening criteria (Stergiopoulos and De Wit, 2009) for effectors, the amino acid sequences encoded by 666 upregulated genes contained signaling peptides, and 550 of them encoded amino acids without transmembrane domains. Thus, 550 genes were preliminarily screened from 4,284 upregulated genes of C. fructicola during the early stages of infection. Among them, there were 191 candidate effectors whose sequence length was less than 300 aa. The list of these candidate effectors is provided in Supplementary Table S5. Among them, 126 candidate effectors were predicted to be apoplastic or cytoplasmic effectors by EffectorP 3.0 (Supplementary Table S6). The 191 candidate effectors included several categories, such as pioneer genes without any annotation, putative toxins (including necrosis-inducing proteins, NLPs), small secreted proteins, glycohydrolase, cell wall degrading enzymes (pectate lyase), and surface-binding proteins (Supplementary Figure S5).
Moreover, the expression trends of 191 candidate effectors were further categorized to elucidate their potential functions. The result showed that they were divided into five clusters and the corresponding enriched biological processes of candidate effectors under these expression trends were also shown (Figure 4 and Supplementary Table S7). In the C1 and C3 clusters, the candidate effectors demonstrated maximal expression abundance at 36 hpi. In the C2 and C4 clusters, the candidate effectors demonstrated maximal expression abundance at 24 hpi. In the C5 cluster, these candidate effectors demonstrated maximal expression abundance at 6 hpi. GO enrichment analysis further indicated these candidate effectors were significantly enriched in the pathogenicity-related terms of cellular oxidant detoxification, RNA phosphodiester bond hydrolysis, translational elongation, xylan catabolic process, proteolysis, and ubiquitin-dependent ERAD pathway. It showed that C. fructicola secreted different effectors to participate in the pathogenic process at different timepoints and effectors were involved in various biological processes of C. fructicola in the process of infecting pecan.
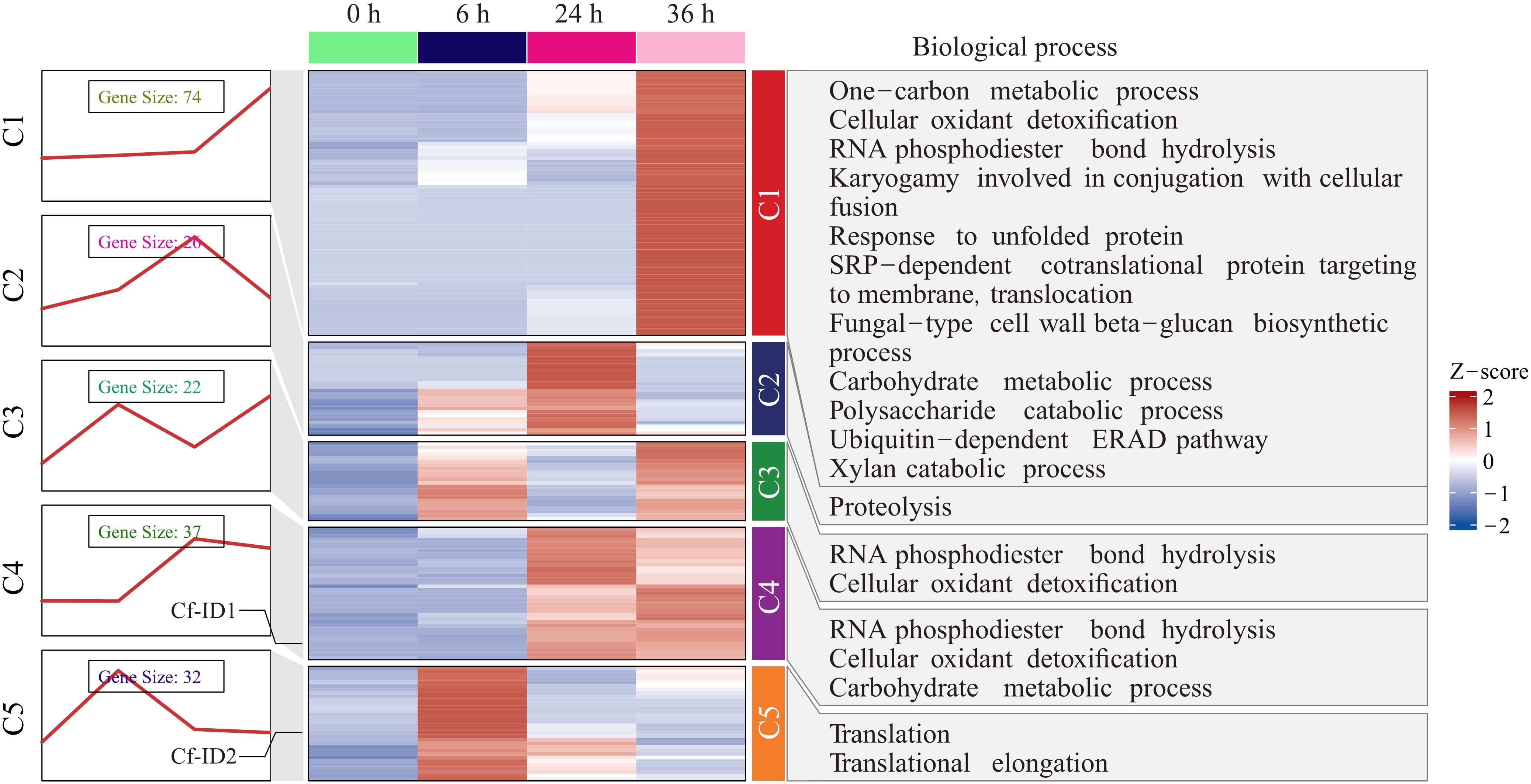
Figure 4. The expression trend analysis of 191 candidate effectors in Colletotrichum fructicola. Expression trends of 191 candidate effectors in C. fructicola were categorized into five clusters and biological processes with significant enrichment of candidate effectors were shown for each cluster.
Cf-ID1 and Cf-ID2 induce cell death and immune responses and suppress infection in Nicotiana benthamiana
Based on the expression trends and corresponding GO annotation information, the candidate effectors in the C4 and C5 clusters were prioritized for further analysis. Then, after a comprehensive analysis of FPKM and the membership in the corresponding clusters (Supplementary Table S7), two candidate effectors (Gene ID: CGMCC3_g883 and CGMCC3_g5994) and one candidate effector (Gene ID: CGMCC3_g12505) met the requirements (mean FPKM >100 and membership >0.75) in the C4 and C5 clusters, respectively. Finally, the one unannotated candidate effector, Cf-ID1 (Gene ID: CGMCC3_g883), and Cf-ID2 (Gene ID: CGMCC3_g12505) were selected for functional validation in N. benthamiana. Their annotation information is provided in Supplementary Table S8.
The transient expression assay result showed that Cf-ID1 and Cf-ID2 induce cell death of N. benthamiana (Figure 5A). The result of RT-qPCR analysis showed that the relative expressions of NbAcre31, NbPTI5, and NbCyp71D20 were significantly increased when infiltrated with pBINGFP-Cf-ID1 and pBINGFP-Cf-ID2 for 3 h and 12 h, respectively (Figures 5B, C).
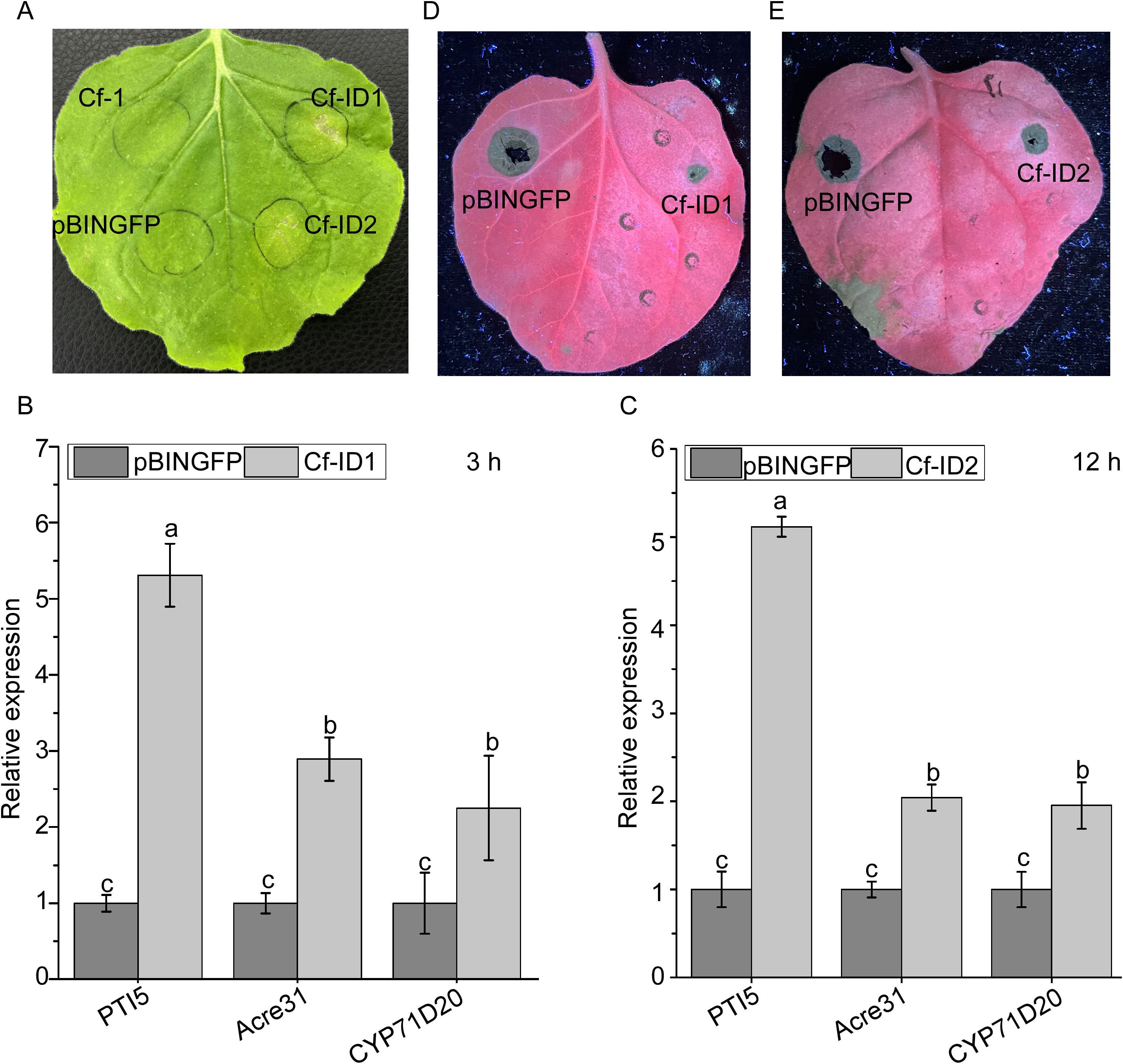
Figure 5. Cf-ID1 and Cf-ID2 could induce cell death and immunity response in Nicotiana benthamiana. (A) Representative N. benthamiana leaves at 7 days after agroinfiltration with Agrobacterium sp. strain GV3101 carrying pBINGFP-Cf-ID1 and pBINGFP-Cf-ID2. N. benthamiana infiltrated with GV3101 carrying the empty vector pBINGFP or the candidate effector Cf-1 (Gene ID: CGMCC3_g6914) were used as negative controls. Cf-1 has been previously validated to lack necrosis-inducing ability in N. benthamiana (unpublished data). These infiltration assays were separately performed three times, and three different plants with three inoculated leaves were used in each assay. (B) Transcriptional upregulation of three N. benthamiana PAMP-triggered immunity (PTI) marker genes infiltrated with pBINGFP-Cf-ID1 for 3 h (C) Transcriptional upregulation of N. benthamiana PTI marker genes infiltrated with pBINGFP-Cf-ID2 for 12 h N. benthamiana infiltrated with Agrobacterium sp. strain GV3101 carrying empty vector pBINGFP for 3 h and 12 h were used as controls. The experiment was repeated three times with similar results. Data are the means, and the error bars represent ± SD from three biological replicates. Different letters on top of the bars indicate statistically significant differences (p < 0.05, t-test) as measured by Duncan’s multiple range test. (D, E) Defense response induced by pretreatment with pBINGFP-Cf-ID1 and pBINGFP-Cf-ID2 and then inoculated 36 h later with C. fructicola in N. benthamiana. The left area of the leaves was separately infiltrated with GV3101 carrying pBINGFP-Cf-ID1 and pBINGFP-Cf-ID2, and the right area of the leaves was infiltrated with GV3101 carrying empty pBINGFP. The experiment was replicated three times with three plants per biological replicate and two leaves analyzed per plant. Disease lesions were assessed under UV light at 72 h post-infiltration.
In addition, to further assess whether Cf-ID1 and Cf-ID2 can suppress infection, N. benthamiana leaves were treated separately with pBINGFP-Cf-ID1 and pBINGFP-Cf-ID2 or with empty vector pBINGFP, and then 36 h later were inoculated with hyphae blocks of C. fructicola. At 72 h post-infiltration, the lesions that had been pretreated with the control were significantly larger than those of the pBINGFP-Cf-ID1 and pBINGFP-Cf-ID2-pretreated leaves in N. benthamiana (Figures 5D, E).
Cf-ID1 and Cf-ID2 are located in both the cytoplasm and nucleus in N. benthamiana
According to the EffectorP 3.0 software prediction, the possibilities of these two candidate effectors being cytoplasmic effectors were 70.1% and 88%, respectively. To determine the subcellular localization of Cf-ID1 and Cf-ID2, we expressed C-terminal GFP-tagged Cf-ID1 and Cf-ID2 (without a signal peptide). Upon the expression of pBINGFP-Cf-ID1 and pBINGFP-Cf-ID2 in N. benthamiana leaves, GFP-derived fluorescence was detected both in the nucleus and cytoplasm (Figure 6). The result showed that Cf-ID1 and Cf-ID2 were established in the nucleus and also in the cytoplasm.
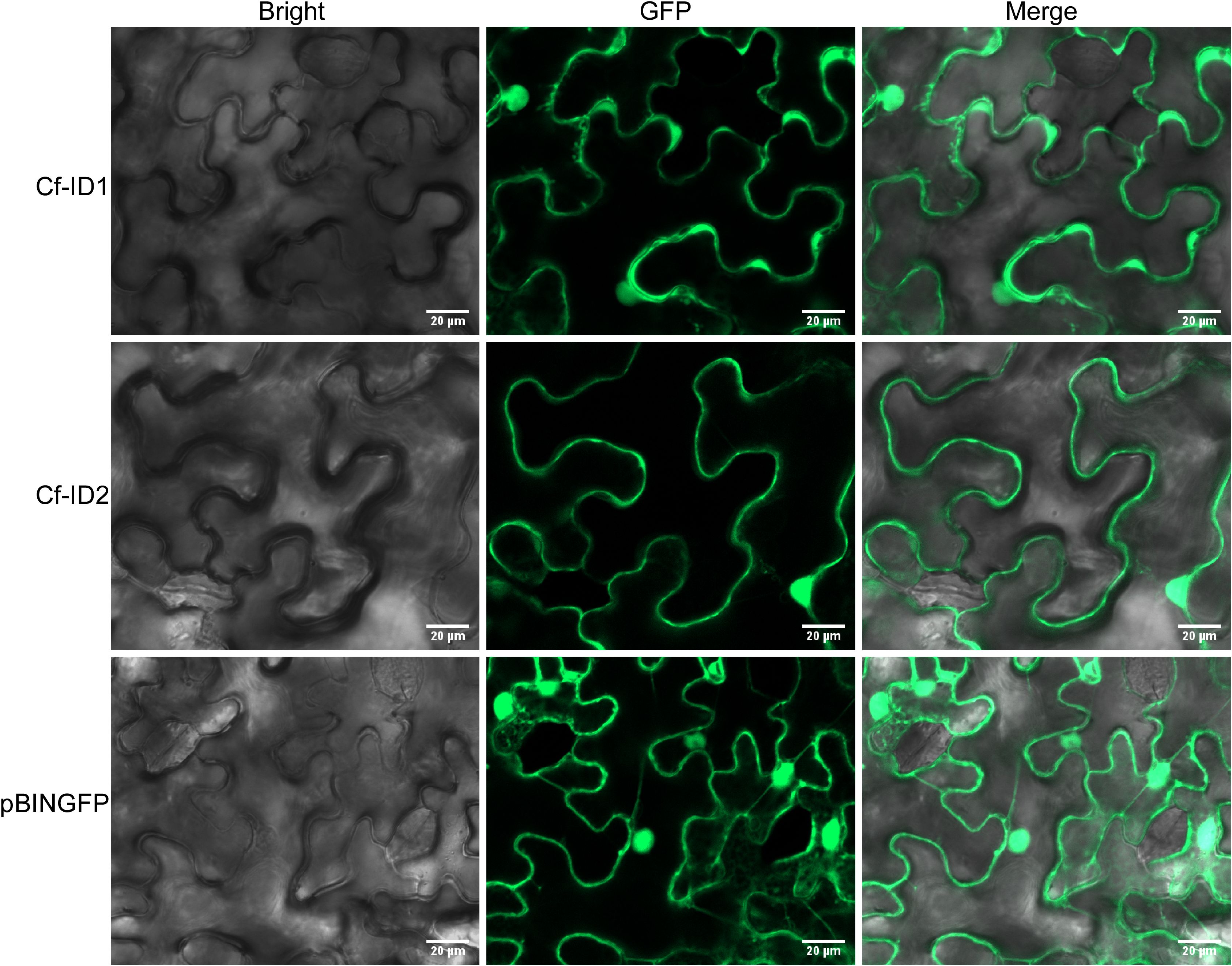
Figure 6. The subcellular localization of Cf-ID1 and Cf-ID2 in Nicotiana benthamiana. The subcellular localization of Cf-ID1 and Cf-ID2 were determined by transient expression of green fluorescent protein (GFP)-tagged proteins in N. benthamiana leaves. Confocal microscopy images were taken at 36 h post-infiltration.
Discussion
In this study, UMI-RNA-seq technology was used to explore the infection strategy of C. fructicola in pecan. Many terms and pathways associated with the pathogenic process and candidate effectors were found, which provided strong evidence for revealing the pathogenic mechanism of C. fructicola in pecan.
Ribosomes are the main site for protein synthesis in eukaryotes, providing various proteins necessary for cell growth and development (Li et al., 2019). Multiple amino acids play important roles in the growth and proliferation of pathogenic fungi, among which, tyrosine is involved in the synthesis of melanin (Ito et al., 2000). Based on the infection process of C. fructicola, the ribosomes and ribosome biogenesis in eukaryote pathways were significantly enriched, and various amino acid metabolisms, such as alanine aspartate and glutamate metabolism, cysteine and methionine metabolism, and tyrosine metabolism, were also enriched at 6 hpi. Therefore, we speculated that at this early stage of infection of pecan, ribosome-related pathways might be related to spore germination and mycelial growth of C. fructicola, and various amino acid metabolisms might promote the maturation and melanization of appressoria, which was one of the infection structures of C. fructicola.
In the transcriptome analysis of C. fructicola infecting apple leaves, the response to oxidative stress was significantly enriched during the appressorium formation stage and the apple leaf infection stage (Liang et al., 2018). In this study, the ROS-related terms (response to oxidative stress, hydrogen peroxide catabolic process, oxylipin biosynthetic process, cellular oxidant detoxification, and cellular response to oxidative stress) and metabolic pathways (peroxisome and oxidative phosphorylation) were all significantly enriched at 6, 24, and 36 hpi, which suggested that oxidative stress from pecan might be one of the major obstacles to the successful infection by C. fructicola during the early infection stages. Correspondingly, C. fructicola also hydrolyzes the ROS produced by pecan to survive.
Previous studies have shown that fungal cytochrome P450 not only mediated the detoxification of exogenous toxic compounds, enabling them to adapt to harsh environments, but also participated in the biosynthesis of fungal ergosterol and toxins, which was beneficial for fungal infection of plant hosts (Črešnar and Petrič, 2011; Jawallapersand et al., 2014). Moreover, in the transcriptome of the early stages of migratory phytoparasitic nematode Bursaphelenchus xylophilus infection in pine, the drug metabolism cytochrome P450 pathway was also significantly enriched during the infection stage (Hu et al., 2021). GPI anchoring was a common post-translational modification in eukaryotic cells, and reports have shown that GPI anchor biosynthesis-related proteins were essential for the nutritional growth and pathogenesis of Colletotrichum graminicola (Mei et al., 2022). In plant pathogenic fungi, sphingolipids, their derivatives, and the MAPK signaling pathway have been proven to regulate various cellular processes, in which pathogens adapted to their hosts and played a crucial role in the regulation of fungal pathogenicity (Gong et al., 2017; Zhu et al., 2023). In this study, at 24 and 36 hpi, not only was the drug metabolism cytochrome P450 metabolic pathway significantly enriched, but metabolic pathways such as the MAPK signaling pathway, GPI-anchor biosynthesis, and sphingolipid metabolism were also significantly enriched. This indicated that after infecting pecan, C. fructicola initiated its own detoxification strategy and nutrient uptake to adapt to the host environment. GPI anchor biosynthesis-related proteins and sphingolipids and their derivatives participated in the pathogenic process of C. fructicola.
A primary study showed that potassium transporter CgTrk1 of C. gloeosporioides was involved in invasive growth and full virulence (Wang et al., 2024). Two-component and phosphorelay signal transduction systems of pathogenic bacteria control the expression of genes encoding virulence factors and essential functions (Stephenson and Hoch, 2002). For example, the virulence of Pseudomonas syringae was inhibited via phosphorelay crosstalk with sense polyphenols (Xie et al., 2021). In our study, potassium ion transport, phosphorelay signal transduction system, and exocytosis were significantly enriched during the early infection stages. This suggested that C. fructicola might secrete toxic substances (such as effectors and toxins) to promote infection through the abovementioned pathways. Moreover, protein ubiquitination and glycosylation have been shown to be closely related to the survival, reproduction, and virulence of pathogens, and some effectors can evade recognition by the host immune system through ubiquitination and glycosylation (Chen et al., 2017; Qiu and Luo, 2017). In this study, protein ubiquitination and glycosylation were significantly enriched in the early stages of infection, which indicated that protein post-translational modification was also one of the important infection strategies of C. fructicola in pecan.
At present, several important effectors have been screened and validated using the method of combining transcriptome sequencing with transient expression technology in N. benthamiana (Hu et al., 2019; Shang et al., 2020; Wen et al., 2021). For example, the effector CfEC92 was first screened from the transcriptome data in C. fructicola and it inhibited BAX-triggered cell necrosis in N. benthamiana. Indeed, the contribution of CfEC92 to pathogenicity was subsequently confirmed by gene knockout. Similarly, in this study, 191 candidate effectors were predicted, and two effectors were successfully characterized using the same method. This indicated that this screening method was effective. It was noteworthy that most candidate effectors were pioneer genes without any annotation, which was consistent with a previous study (Espada et al., 2016). The types of candidate effectors in our study included those predicted by previous researchers, such as pioneer genes, NLPs, small secreted proteins, and surface-binding proteins (Liang et al., 2018). Moreover, other candidate effectors, including allergens, some cell wall-degrading enzymes (CWDEs, such as pectate lyase), hydrolases (such as SL-like lipase and glycosyl hydrolases), and ribosomal proteins have been also identified as candidate effectors in B. xylophilus (Tsai et al., 2016; Hu et al., 2021). In a previous study, allergens were proven to target plant signaling pathways and suppress host defenses (Haegeman et al., 2012). CWDEs were important for breaking down plant cell walls to establish infection (Sonah et al., 2016; Qu et al., 2021). Several lipases of pathogenic bacteria and fungi have been identified as virulence factors (Stehr et al., 2003). Some ribosomal proteins participated in the interaction between Meloidogyne incognita and Solanum lycopersicum (Shukla et al., 2018). Thus, it was speculated that C. fructicola secreted these candidate effectors to promote colonization in pecan in different ways, such as degradation of the host cell wall, suppression of host defenses, or being toxic to host cells. In addition, the candidate effectors we screened provided an important database for further identifying key effectors during the interaction progress between C. fructicola and pecan.
In primary studies, multiple effectors that can trigger plant cell necrosis and trigger plant immunity have been identified in plant pathogens, for example, Phytophthora sojae effectors PsXEG1 and Ps238 (Ma et al., 2015; Yang et al., 2017) and B. xylophilus effectors BxSapB1 and BxSapB2 (Hu et al., 2019; Zhao et al., 2020). At present, Han et al. have identified four effector genes, CfCE4, CfCE25, CfCE61, and CfCE66, secreted by C. fructicola during the infection of pear trees, which can trigger cell death in N. benthamiana (Han et al., 2024). This study identified two effectors, Cf-ID1 and Cf-ID2, of C. fructicola, which not only induced cell death, but also triggered the immune and defense responses to suppress infection of C. fructicola in N. benthamiana, thus adding two new members to such effectors. In the future, Cf-ID1 and Cf-ID2 are expected to be used in the development of immune inducers of pecan against anthracnose, which will provide new ideas for the prevention and control of anthracnose of pecan. Research on the mechanism of plant immune responses triggered by the effectors Cf-ID1 and Cf-ID2 of C. fructicola by gene knock-out and the target protein of pecan will be the focus of future research. Moreover, we will also identify and characterize effectors secreted by C. fructicola that inhibit plant immunity when interacting with pecan and research their function mechanism.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
L-JH: Conceptualization, Data curation, Formal Analysis, Investigation, Project administration, Software, Validation, Writing – original draft, Writing – review & editing. J-PX: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Resources, Writing – review & editing. YL: Formal Analysis, Investigation, Methodology, Software, Writing – original draft. MZ: Investigation, Methodology, Resources, Writing – review & editing. G-MW: Conceptualization, Investigation, Methodology, Writing – review & editing. L-ND: Conceptualization, Investigation, Methodology, Software, Supervision, Writing – review & editing. Z-HM: Conceptualization, Data curation, Formal Analysis, Resources, Software, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Doctoral Research Foundation of the Institute of Botany, Jiangsu Province and Chinese Academy of Sciences (JIBTF202203) and the Central Government Demonstration Project of Forestry Science and Technology (Su[2022]TG11).
Acknowledgments
We thank Prof. Yuanchao Wang (Departments of Plant Pathology, Nanjing Agricultural University, China) for providing the vector pBINGFP, the seeds of Nicotiana benthamiana, and technical assistance. We are also grateful to Prof. Yan Wang and Dr. Yeqiang Xia (Departments of Plant Pathology, Nanjing Agricultural University, China) for their helpful suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1551342/full#supplementary-material
References
Ai, G., Peng, H., Pan, W. Y., Li, Y. K., Wan, Z. R., Yin, Z. Y., et al. (2024). A catalogue of virulence strategies mediated by phytopathogenic effectors. Fundam. Res. 5, 663–673. doi: 10.1016/j.fmre.2023.10.026
Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/s41587-019-0036-z
Baroncelli, R., Amby, D. B., Zapparata, A., Sarrocco, S., Vannacci, G., Le Floch, G., et al. (2016). Gene family expansions and contractions are associated with host range in plant pathogens of the genus Colletotrichum. BMC Genomics 17, 555. doi: 10.1186/s12864-016-2917-6
Bilharva, M. G., Martins, C. R., Hamann, J. J., Fronza, D., De Marco, R., Malgarim, M. B. (2018). Pecan: from research to the Brazilian reality. J. Exp. Agric. Int. 23, 1–16. doi: 10.9734/JEAI/2018/41899
Chang, J., Wang, K., Zhang, C., Han, X., Zhang, X., Ren, H., et al. (2023). Transcriptome analysis of resistant and susceptible pecan (Carya illinoinensis) reveals the mechanism of resistance to black spot disease (Colletotrichum fioriniae). J. Agric. Food. Chem. 71, 5812–5822. doi: 10.1021/acs.jafc.2c08434
Chang, J., Zhai, F., Zhang, Y., Wang, D., Shu, J., Yao, X. (2022). Identification and characterization of Colletotrichum fioriniae and C. fructicola that cause anthracnose in pecan. Front. Plant. Sci. 13, 1043750. doi: 10.3389/fpls.2022.1043750
Chen, J., Lin, B., Huang, Q., Hu, L., Zhuo, K., Liao, J. (2017). A novel Meloidogyne graminicola effector, MgGPP, is secreted into host cells and undergoes glycosylation in concert with proteolysis to suppress plant defenses and promote parasitism. PloS Pathog. 13, e1006301. doi: 10.1371/journal.ppat.1006301
Chen, X. L., Shi, T., Yang, J., Shi, W., Gao, X., Chen, D., et al. (2014). N-glycosylation of effector proteins by an alpha-1,3-mannosyltransferase is required for the rice blast fungus to evade host innate immunity. Plant Cell 26, 1360–1376. doi: 10.1105/tpc.114.123588
Chen, Y., Zhang, S. J., Zhao, Y. Q., Mo, Z. H., Wang, W., Zhu, C. C. (2022). Transcriptomic analysis to unravel potential pathways and genes involved in pecan (Carya illinoinensis) resistance to Pestalotiopsis microspora. Int. J. Mol. Sci. 23, 11621. doi: 10.3390/ijms231911621
Chomczynski, P., Sacchi, N. (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, 156–159. doi: 10.1016/0003-2697(87)90021-2
Črešnar, B., Petrič, Š. (2011). Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta (BBA). Proteins Proteom. 1814, 29–35. doi: 10.1016/j.bbapap.2010.06.020
Espada, M., Silva, A. C., Eves Van Den Akker, S., Cock, P. J., Mota, M., Jones, J. T. (2016). Identification and characterization of parasitism genes from the pinewood nematode Bursaphelenchus xylophilus reveals a multilayered detoxification strategy. Mol. Plant Pathol. 17, 286–295. doi: 10.1111/mpp.2016.17.issue-2
Foster, A. J., Pelletier, G., Tanguay, P., Seguin, A. (2015). Transcriptome analysis of poplar during leaf spot infection with Sphaerulina spp. PloS One 10, e0138162. doi: 10.1371/journal.pone.0138162
Gong, X. D., Feng, S. Z., Zhao, J., Tang, C., Tian, L., Fan, Y. S., et al. (2017). StPBS2, a MAPK kinase gene, is involved in determining hyphal morphology, cell wall development, hypertonic stress reaction as well as the production of secondary metabolites in northern corn leaf blight pathogen Setosphaeria turcica. Microbiol. Res. 201, 30–38. doi: 10.1016/j.micres.2017.04.009
Haegeman, A., Mantelin, S., Jones, J. T., Gheysen, G. (2012). Functional roles of effectors of plant-parasitic nematodes. Gene 492, 19–31. doi: 10.1016/j.gene.2011.10.040
Han, M. Q., Wang, C. H., Zhu, W. H., Pan, Y. M., Huang, L. L., Nie, J. J. (2024). Extracellular perception of multiple novel core effectors from the broad host-range pear anthracnose pathogen Colletotrichum fructicola in the nonhost Nicotiana benthamiana. Horticult. Res. 11, uhae078. doi: 10.1093/hr/uhae078
Hu, L. J., Wu, X. Q., Ding, X. L., Ye, J. R. (2021). Comparative transcriptomic analysis of candidate effectors to explore the infection and survival strategy of Bursaphelenchus xylophilus during different interaction stages with pine trees. BMC Plant Biol. 21, 224. doi: 10.1186/s12870-021-02993-9
Hu, L. J., Wu, X. Q., Li, H. Y., Wang, Y. C., Huang, X., Wang, Y., et al. (2020). BxCDP1 from the pine wood nematode Bursaphelenchus xylophilus is recognized as a novel molecular pattern. Mol. Plant Pathol. 21, 923–935. doi: 10.1111/mpp.12939
Hu, L. J., Wu, X. Q., Li, H. Y., Zhao, Q., Wang, Y. C., Ye, J. R. (2019). An effector, BxSapB1, induces cell death and contributes to virulence in the pine wood nematode Bursaphelenchus xylophilus. Mol. Plant-Microbe Interact. 32, 452–463. doi: 10.1094/MPMI-10-18-0275-R
Huang, L., Zhang, S., Yin, Z., Liu, M., Li, B., Zhang, H., et al. (2017). MoVrp1, a putative verprolin protein, is required for asexual development and infection in the rice blast fungus Magnaporthe oryzae. Sci. Rep. 7, 41148. doi: 10.1038/srep41148
Ito, S., Wakamatsu, K., Ozeki, H. (2000). Chemical analysis of melanins and its application to the study of the regulation of melanogenesis. Pigment. Cell Res. 13, 103–109. doi: 10.1034/j.1600-0749.13.s8.19.x
Jawallapersand, P., Mashele, S. S., Kovačič, L., Stojan, J., Komel, R., Pakala, S. B., et al. (2014). Cytochrome P450 monooxygenase CYP53 family in fungi: comparative structural and evolutionary analysis and its role as a common alternative anti-fungal drug target. PloS One 9, e107209. doi: 10.1371/journal.pone.0107209
Jones, J. D., Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Lamarche, M. G., Wanner, B. L., Crepin, S., Harel, J. (2008). The phosphate regulon and bacterial virulence: a regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol. Rev. 32, 461–473. doi: 10.1111/j.1574-6976.2008.00101.x
Li, L., Zhu, X. M., Shi, H. B., Feng, X. X., Liu, X. H., Lin, F. C. (2019). MoFap7, a ribosome assembly factor, is required for fungal development and plant colonization of Magnaporthe oryzae. Virulence 10, 1047–1063. doi: 10.1080/21505594.2019.1697123
Liang, X., Shang, S., Dong, Q., Wang, B., Zhang, R., Gleason, M. L., et al. (2018). Transcriptomic analysis reveals candidate genes regulating development and host interactions of Colletotrichum fructicola. BMC Genomics 19, 557. doi: 10.1186/s12864-018-4934-0
Ma, Z., Song, T., Zhu, L., Ye, W., Wang, Y., Shao, Y., et al. (2015). A Phytophthora sojae glycoside hydrolase 12 protein is a major virulence factor during soybean infection and is recognized as a PAMP. Plant Cell 27, 2057–2072. doi: 10.1105/tpc.15.00390
Mei, J., Ning, N., Wu, H. X., Chen, X. L., Li, Z. Q., Liu, W. D. (2022). Glycosylphosphatidylinositol anchor biosynthesis pathway-related protein GPI7 is required for the vegetative growth and pathogenicity of Colletotrichum graminicola. Int. J. Mol. Sci. 23, 2985. doi: 10.3390/ijms23062985
Meng, K. (2020). Identification of pathogens and indoor drug screening for black spot disease of Pecan. Henan. Inst. Sci. Technol. 12, 28–32. doi: 10.27704/d.cnki.ghnkj.2020.000074
Meng, K., Zhang, Y. B., Chang, J., L, Z. H., Wang, D., Zhai, F. Y., et al. (2021). Toxicity test with 8 fungicides against 9 pathogens of pecan anthracnose (Colletotrichum spp.). For. Res. 34, 153–164. doi: 10.13275/j.cnki.lykxyj.2021.01.019
Mo, Z., Chen, Y., Zhai, M., Zhu, K., Xuan, J., Hu, L. (2023). Development and application of a virus-induced gene silencing system for functional genomics in pecan (Carya illinoinensis). Sci. Hortic. 310, 111759. doi: 10.1016/j.scienta.2022.111759
Nie, J., Yin, Z., Li, Z., Wu, Y., Huang, L. (2018). A small cysteine-rich protein from two kingdoms of microbes is recognized as a novel pathogen-associated molecular pattern. New Phytol. 222, 995–1011. doi: 10.1111/nph.15631
Qiu, J., Luo, Z. Q. (2017). Hijacking of the host ubiquitin network by Legionella pneumophila. Front. Cell Infect. Microbiol. 7, 487. doi: 10.3389/fcimb.2017.00487
Qu, Z., Zhang, H., Wang, Q., Zhao, H., Liu, X., Fu, Y., et al. (2021). Exploring the symbiotic mechanism of a virus-mediated endophytic fungus in its host by dual unique molecular identifier-RNA sequencing. mSystems 6, e0081421. doi: 10.1128/msystems.00814-21
Rockenbach, M. F., Velho, A. C., Gonçalves, A. E., Mondino, P. E., Alaniz, S. M., Stadnik, M. J. (2016). Genetic structure of Colletotrichum fructicola associated to apple bitter rot and Glomerella leaf spot in southern Brazil and Uruguay. Phytopathology 106, 774–781. doi: 10.1094/PHYTO-09-15-0222-R
Shang, S., Wang, B., Zhang, S., Liu, G., Liang, X., Zhang, R., et al. (2020). A novel effector CfEC92 of Colletotrichum fructicola contributes to glomerella leaf spot virulence by suppressing plant defences at the early infection phase. Mol. Plant Pathol. 21, 936–950. doi: 10.1111/mpp.12940
Shukla, N., Yadav, R., Kaur, P., Rasmussen, S., Goel, S., Agarwal, M., et al. (2018). Transcriptome analysis of root-knot nematode (Meloidogyne incognita)-infected tomato (Solanum lycopersicum) roots reveals complex gene expression profiles and metabolic networks of both host and nematode during susceptible and resistance responses. Mol. Plant Pathol. 19, 615–633. doi: 10.1111/mpp.2018.19.issue-3
Smith, T., Heger, A., Sudbery, I. (2017). UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 27, 491–499. doi: 10.1101/gr.209601.116
Sonah, H., Zhang, X., Deshmukh, R. K., Borhan, M. H., Fernando, W. G., Belanger, R. R. (2016). Comparative transcriptomic analysis of virulence factors in Leptosphaeria maculans during compatible and incompatible interactions with canola. Front. Plant. Sci. 7, 1784. doi: 10.3389/fpls.2016.01784
Sonnhammer, E. L., Von Heijne, G., Krogh, A. (1998). A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182.
Sparks, D. J. (2002). Rainfall governs pecan stand homogeneity in native, wild habitats. Am. Soc. Hortic. Sci. 127, 860–868. doi: 10.21273/JASHS.127.5.860
Sperschneider, J., Dodds, P. N. (2022). EffectorP 3.0: prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant-Microbe Interact. 35, 146–156. doi: 10.1094/MPMI-08-21-0201-R
Stehr, F., Kretschmar, M., Kröger, C., Hube, B., Schäfer, W. (2003). Microbial lipases as virulence factors. J. Mol. Catalysis. B.: Enzymatic. 22, 347–355. doi: 10.1016/S1381-1177(03)00049-3
Stephenson, K., Hoch, J. A. (2002). Two-component and phosphorelay signal-transduction systems. Curr. Opin. Pharmacol. 2, 507–512. doi: 10.1016/S1471-4892(02)00194-7
Stergiopoulos, I., De Wit, P. J. G. M. (2009). Fungal effector proteins. Annu. Rev. Phytopathol. 47, 233–263. doi: 10.1146/annurev.phyto.112408.132637
Tsai, I. J., Tanaka, R., Kanzaki, N., Akiba, M., Yokoi, T., Espada, M., et al. (2016). Transcriptional and morphological changes in the transition from mycetophagous to phytophagous phase in the plant-parasitic nematode Bursaphelenchus xylophilus. Mol. Plant Pathol. 17, 77–83. doi: 10.1111/mpp.2016.17.issue-1
Wang, Y., Xu, Y., Sun, Y., Wang, H., Qi, J., Wan, B., et al. (2018). Leucine-rich repeat receptor-like gene screen reveals that Nicotiana RXEG1 regulates glycoside hydrolase 12 MAMP detection. Nat. Commun. 9, 594. doi: 10.1038/s41467-018-03010-8
Wang, Z., Yang, J. Y., Sun, M. L., Pan, Y. T., Huang, L. (2024). Involvement of the transporter CgTrk1 in potassium uptake, invasive growth, and full virulence in Colletotrichum gloeosporioides. FORESTS 15, 1044. doi: 10.3390/f15061044
Wen, T. Y., Wu, X. Q., Hu, L. J., Qiu, Y. J., Rui, L., Zhang, Y., et al. (2021). A novel pine wood nematode effector, BxSCD1, suppresses plant immunity and interacts with an ethylene-forming enzyme in pine. Mol. Plant Pathol. 22, 1399–1412. doi: 10.1111/mpp.13121
Xie, Y., Ding, Y., Shao, X., Yao, C., Li, J., Liu, J., et al. (2021). Pseudomonas syringae senses polyphenols via phosphorelay crosstalk to inhibit virulence. EMBO Rep. 22, e52805. doi: 10.15252/embr.202152805
Yang, B., Wang, Q., Jing, M., Guo, B., Wu, J., Wang, H., et al. (2017). Distinct regions of the Phytophthora essential effector Avh238 determine its function in cell death activation and plant immunity suppression. New Phytol. 214, 361–375. doi: 10.1111/nph.2017.214.issue-1
Young, M. D., Wakefield, M. J., Smyth, G. K., Oshlack, A. (2014). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14. doi: 10.1186/gb-2010-11-2-r14
Zhao, Q., Hu, L. J., Wu, X. Q., Wang, Y. C. (2020). A key effector, BxSapB2, plays a role in the pathogenicity of the pine wood nematode Bursaphelenchus xylophilus. For. Pathol. 50, e12600. doi: 10.1111/efp.12600
Zhu, X. M., Li, L., Bao, J. D., Wang, J. Y., Daskalov, A., Liu, X. H., et al. (2023). The biological functions of sphingolipids in plant pathogenic fungi. PloS Pathog. 19, e1011733. doi: 10.1371/journal.ppat.1011733
Keywords: Colletotrichum fructicola, Carya illinoinensis, UMI RNA-seq, effectors, immune response, infection suppression
Citation: Hu L-J, Xuan J-P, Li Y, Zhai M, Wang G-M, Deng L-N and Mo Z-H (2025) Exploring the infection strategy of Colletotrichum fructicola in pecan and two effectors Cf-ID1 and Cf-ID2 were characterized using unique molecular identifier-RNA sequencing technology. Front. Plant Sci. 16:1551342. doi: 10.3389/fpls.2025.1551342
Received: 25 December 2024; Accepted: 21 March 2025;
Published: 17 April 2025.
Edited by:
Davide Giovanardi, University of Modena and Reggio Emilia, ItalyReviewed by:
Helena Gil Azinheira, University of Lisbon, PortugalPatrícia Fernandes, SUNY College of Environmental Science and Forestry, United States
Norliza Abu Bakar, Malaysian Agricultural Research and Development Institute (MARDI), Malaysia
Copyright © 2025 Hu, Xuan, Li, Zhai, Wang, Deng and Mo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zheng-Hai Mo, bW96aGVuZ2hhaUB5ZWFoLm5ldA==