 Xuan Du1†
Xuan Du1† Hongmei Zhang
Hongmei Zhang Na Liu
Na Liu- 1Shanghai Academy of Agricultural Sciences, Shanghai Key Laboratory of Protected Horticultural Technology, Shanghai, China
- 2Fengxian District Agricultural Technology Extension Center, Shanghai, China
Bottle gourd (Lagenaria siceraria)belongs to cucurbit crop and hasunique semi-autonomous organelle genome. Using Illumina short-read and Nanopore long-read sequencing data, we sequenced and annotated the complete mitochondrial genome of L. siceraria. And a comparative phylogenetic analysis was conducted with its close relatives. The mitochondrial genome of bottle gourd is a circular sequence of 357,496 bp with a GC content of 45.03%. It contains 63 genes, including 34 mRNAs, 24 tRNAs, 4 rRNAs, and 1 pseudogene. The rps19 gene is present, but rpl10 is absent. 22,294 bp (6.24%) are repetitive sequences. 497 RNA editing sites were identified. 45 homologous fragments (40,579 bp, 11.35%) were shared with the chloroplast genome. Phylogenetic analysis revealed that C. maxima, C. sativus, C. lanatus, and L. acutangula are closely related to bottle gourd. Gene arrangement analysis indicated that L. acutangula exhibits the highest collinearity with L. siceraria compared to other cucurbit crops. However, genome size and repetitive sequences are most similar to watermelon. Nearly all Ka/Ks ratios <1.0 suggest stabilizing selection in protein-coding genes. These findings provide a foundation for further understanding the evolutionary relationships within cucurbit crops.
Introduction
Plant mitochondria, like chloroplasts, are crucial organelles in plant cell activities, with genomes that are independent of nuclear genomes, exhibiting semi-autonomous genetic characteristics (Rodríguez-Moreno et al., 2011; Wang et al., 2025). Mitochondria play a vital role in plant growth and development plants (Srivastava et al., 2018; Wang et al., 2024a), primarily through their involvement in energy metabolism, providing ATP for cell growth, division, differentiation, metabolism, and apoptosis via oxidative phosphorylation (Møller et al., 2021; Xu et al., 2022). During evolution, plant mitochondrial (mt) genomes have undergone significant changes in gene sequence, genome structure, and sequence migration from other organelles (Greiner and Bock, 2013; Timmis et al., 2004; Chevigny et al., 2020; Kubo and Newton, 2008; Wang et al., 2024a). Consequently, plant mt genomes are 100 to 10,000 times larger than those of animals and exhibit greater structural complexity (Best et al., 2020; Christensen, 2013; Wu et al., 2025). Mitochondrial genomes vary not only among plant species, but also within the same species (O’Conner and Li, 2020; Kozik et al., 2019), in contrast to the highly conserved structure of plant chloroplast genomes (Niu et al., 2023). As a result, mt genomes have become a valuable source of genetic information and have been widely used in phylogenetic studies to understand basic cellular processes (Cao et al., 2023; Xu et al., 2013; Wang et al., 2024b).
Bottle gourd (Lagenaria siceraria) (2n = 2x =22), also known as long calabash, belongs to the Cucurbitaceae family, which comprises 95 genera and 942–978 species (Tanaka et al., 2013), including cucumber (Cucumis sativus), melon (Cucumis melo), watermelon (Citrullus lanatus), pumpkin (Cucurbita moschata) and zucchini (Cucurbita pepo). The economic importance of cucurbit crops is second only to that of the Solanaceae family (Rodríguez-Moreno et al., 2011).Cucurbit crops are known to possess unique semi-autonomous organelle genomes (mitochondria and chloroplast genomes), with significant differences observed among different species (Levi et al., 2006; Rodríguez-Moreno et al., 2011). Organelle genes in cucurbit crops are associated with critical metabolic pathways such as photosynthesis and respiration, as well as important traits like cold resistance (Olechowska et al., 2022)and sex differentiation (Levi et al., 2006). Mitochondrial genome data can enhance cucurbit breeding programs by identifying conserved genes linked to stress tolerance or yield. Additionally, comparative analyses aid biodiversity conservation by clarifying genetic relationships among species and detecting adaptive traits in wild relatives.
With the advancement of long-read sequencing technologies, organelle genome sequencing has become more accessible. In this study, we constructed and annotated the complete mitochondrial genome of bottle gourd using a combination second- and third-generation sequencing technologies, performed phylogenetic analyses, and compared the mitochondrial genomes of bottle gourd with other cucurbit crops. These results provide insights into the characteristics of the bottle gourd mitochondrial genome and offer a theoretical foundation for further studies on organelle genome differences, evolutionary relationships, and mitochondrial genetic patterns among cucurbit crops.
Materials and methods
Plant materials and DNA sequencing
The bottle gourd variety “BG-54” used in this study was obtained commercially from Zhongziku APP (http://www.zhongziku.cc/). The plants were cultivated under controlled conditions at the Zhuanghang Comprehensive Experimental Station(E 121°28′, N 30°57′) of the Shanghai Academy of Agricultural Sciences. The photon flux density ranged from 650 to 850 W.m-2 with temperatures between 10–25°C and relative humidity of 50–70%. Fresh leaves were frozen in liquid nitrogen and stored at -80°C. Total DNA was isolated following the protocol for the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) and the Oxford Nanopore PromethION (Oxford Nanopore Technologies, Oxford Science Park, UK).
Raw data from second-generation sequencing were filtered using fastp software (version 0.20.0, https://github.com/OpenGene/Fastp) (Chen et al., 2018). Third-generation sequencing data of mitochondrial reads were filtered using Filtlong (version 0.2.1, https://github.com/rrwick/Filtlong). The filtered third-generation data were aligned to the reference gene sequence using Minimap2 (version 2.1) (Li and Birol, 2018), and sequences with alignment lengths greater than 50 bp were selected. Sequences with overlaps greater than 1 kb and similarity greater than 70% were chosen as seed sequences. The original data were iteratively compared to the seed sequences to obtain all third-generation sequencing data of the mitochondrial genome. The third-generation assembly software Canu (Koren et al., 2017) was used to correct the obtained data. The corrected sequences were then aligned with the second-generation data using Bowtie2 (v2.3.5.1), and Unicycler (v0.4.8) was used to assemble the second- and third-generation data. Due to the complex physical structure of the mitochondrial genome, including subrings and non-circular forms, the corrected third-generation sequencing data were manually compared with the contigs obtained in the second step using Minimap2 to determine the branching direction and obtain the final assembly result (Figure 1).
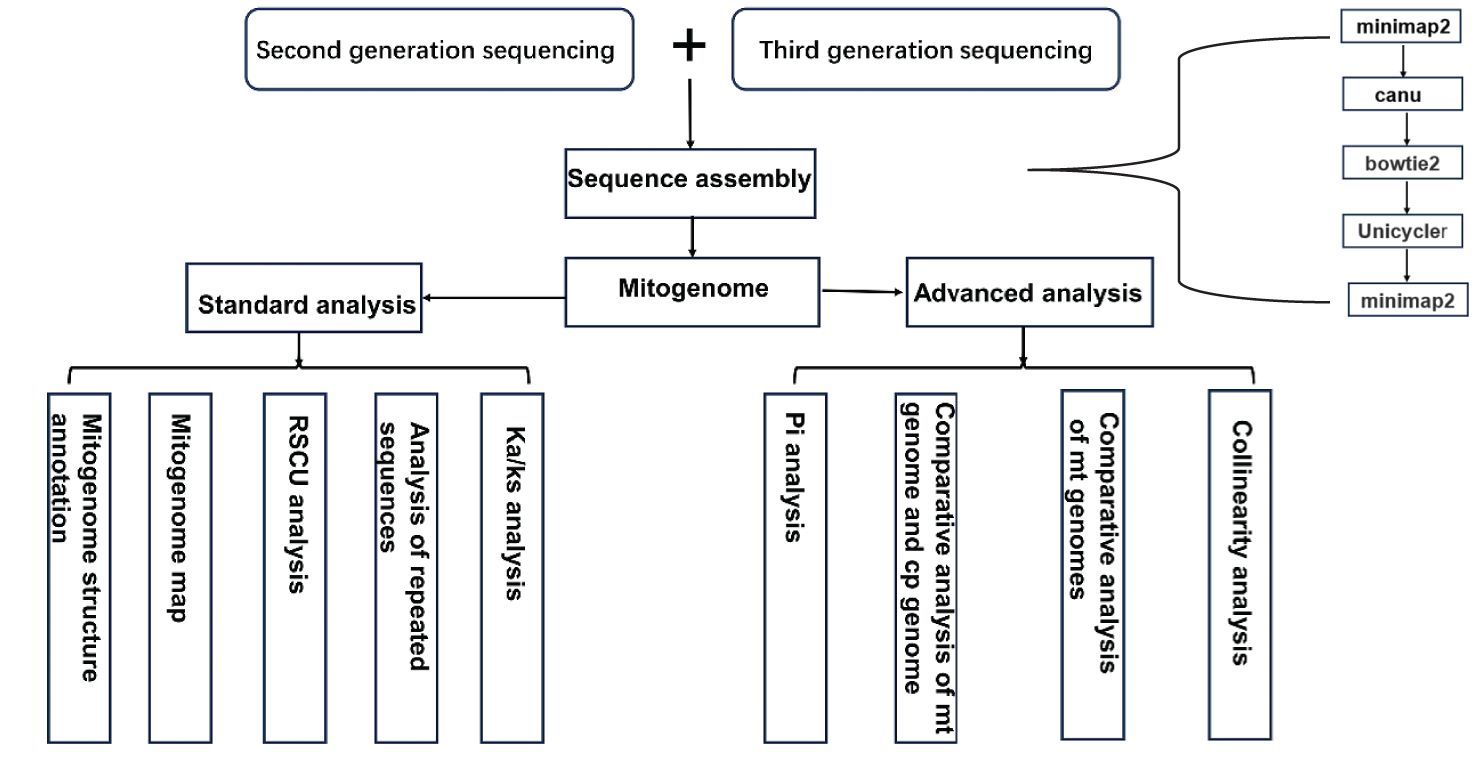
Figure 1. A flow diagram of experiment and analysis.
Mitogenome annotation
Protein-coding genes and rRNA sequences were annotated by comparing them with published plant mitochondrial sequences using BLAST, followed by manual adjustments based on related species. Transfer RNA (tRNA) genes were annotated using tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/) (Chan and Lowe, 2019). Open Reading Frames (ORFs) were identified using the Open Reading Frame Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html), with the minimum length set to 102 bp. Redundant sequences and those overlapping with known genes were excluded. Sequences with alignments longer than 300 bp were annotated against the nr library. Potential RNA editing sites in the protein-coding genes (PCGs) of bottle gourd were predicted using the online Predictive RNA Editor for Plant Mitochondrial Genes (PREP-Mt) suite (http://prep.unl.edu/) (Mower, 2005). The physical circular map of the mitochondrial genome was generated using the Organellar Genome DRAW (OGDraw) v1.2 program (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html). Relative synonymous codon usage (RSCU) was calculated using the CAI Python package developed by Lee (Lee, 2018),and codon frequencies were determined using the Codon Usage tool in the Sequence Manipulation Suite (bioinformatics.org/sms2/codon_usage.html) (Stothard, 2000).
Analysis of repeated sequences
Three types of repeats—simple sequence repeats (SSRs), tandem repeats, and dispersed repeats—were identified in the bottle gourd mitochondrial genome. SSRs were detected using the MIcroSAtellite identification tool (v1.0, parameters: 1-10 2-5 3-4 4-3 5-3 6-3) implemented in a Perl script (Thiel et al., 2003). Tandem repeats (>6 bp repeat units) were identified using Tandem Repeats Finder v4.09 (trf409.linux64, parameters: 27 7 80 10 50 2000 -f -d -m) (http://tandem.bu.edu/trf/trf.submit.options.html) (Benson, 1999). Dispersed repeats were detected using BLASTn (v2.10.1) with the following parameters: -word_size 7 and E-value 1e-5.
Ka/Ks analysis
Gene sequences were aligned using MAFFT V7.310 (https://mafft.cbrc.jp/alignment/software/), and the nonsynonymous-to-synonymous substitution ratio (Ka/Ks) was calculated using the Ka/Ks Calculator V2.0 (https://sourceforge.net/projects/kakscalculator2/). The MLWL method was employed for the calculations.
Pi analysis
Nucleotide diversity (Pi) was used to assess sequence variation among different species, with regions of high variation serving as potential molecular markers for population genetics. Homologous gene sequences from different species were globally aligned using MAFFT software (v7.427, –auto mode). The aligned sequences were concatenated, trimmed using trimAl (v1.4.rev15, parameter: -gt 0.7), and analyzed with DNAsp5 to calculate Pi values for each gene.
Homologous sequence analysis of chloroplast and mitochondria
Homologous sequence analysis between chloroplast and mitochondrial genomes were conducted using BLAST, with a similarity threshold of 70% and an E-value of 1e-5. The results were visualized using Circos v0.69-5.
Phylogenetic tree construction and sequence collinearity analysis
Phylogenetic analysis was conducted using the mitochondrial genomes of bottle gourd and 32 other species representing 24 families. Sequences from different species were aligned using MAFFT software (v7.427, –auto mode). The aligned sequences were concatenated, trimmed with trimAl (v1.4.rev15, parameter: -gt 0.7), and the best-fit evolutionary model (GTR) was determined using jModelTest-2.1.10. A maximum likelihood phylogenetic tree was constructed using RAxML V8.2.10 (https://cme.h-its.org/exelixis/software.html) under the GTRGAMMA model with 1,000 bootstrap replicates.
Collinearity analysis of the bottle gourd mitochondrial genome was performed using two methods. The first method involved comparing genomes using nucmer (4.0.0beta2) with the –maxmatch parameter to generate dot-plot diagrams. The second method utilized BLASTn (v2.10.1+) with parameters set to -word_size 7 and E-value 1e-5. Fragments with alignment lengths greater than 300 bp were screened, and collinearity maps were generated by comparing the assembled species with selected species.
Results
Features of the bottle gourd mitogenome
The Illumina MiSeq and Nanopore sequencing produced 29,675,595 and 1,406,000 reads, respectively, with a mean read length of 7,433 bp for Nanopore sequencing. The complete mitochondrial genome of bottle gourd is a circular sequence of 357,496 bp with a GC content of 45.03% (Figure 2). The sequence has been submitted to the GenBank database (accession number: PP727017). The mitochondrial genome contains 63 genes, including 34 mRNAs, 24 tRNAs, 4 rRNAs, and 1 pseudogene (Table 1). Notably, three copies of the nad1 and nad5 genes were identified. Additionally, three tRNA genes located in repeat regions were found in two or three copies (trnp-TGG, trnM-CAT, and trnW-CCA) (Figure 2).
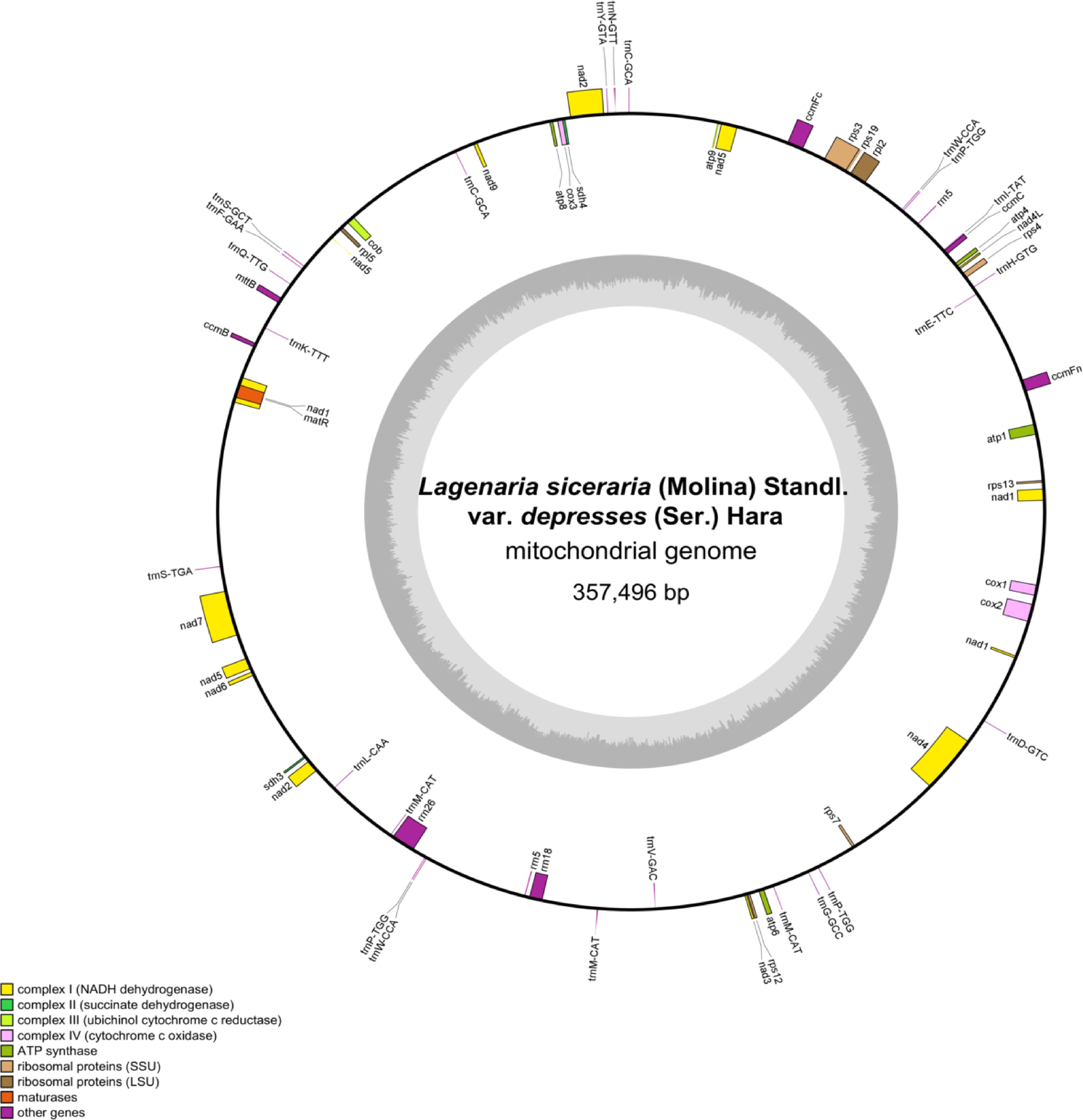
Figure 2. L. siceraria mitogenome gene map. Genes shown on the outside and inside of the circle are transcribed clockwise and counterclockwise, respectively. The dark gray region in the inner circle depicts GC content.
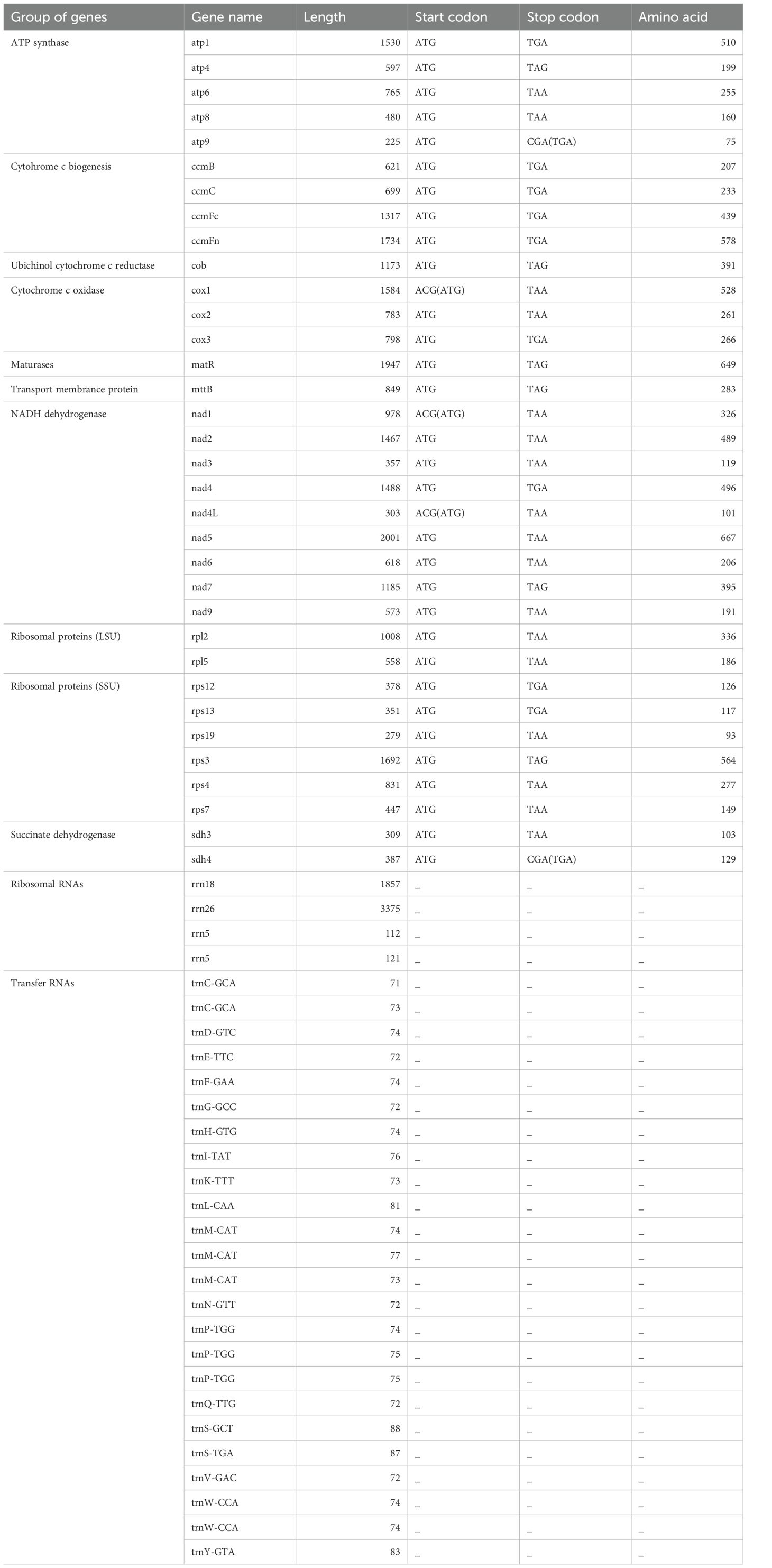
Table 1. Gene profile and organization of the bottle gourd mitogenome.
Codon usage analysis of PCGs
In the mitochondrial (mt) genome of bottle gourd, the protein-coding genes (PCGs) can be categorized into 10 functional groups. These include ATP synthases (5 genes), cytochrome C biogenesis accessory proteins (4 genes), ubiquinol cytochrome C reductases (1 gene), cytochrome C oxidases (3 genes), maturases (1 gene), transport membrane proteins (1 gene), NADH dehydrogenases (9 genes), ribosomal proteins (LSU) (2 genes), ribosomal proteins (SSU) (6 genes), and succinate dehydrogenases (2 genes). Most PCGs utilize the typical ATG start codon, while cox1, nad1, and nad4L begin with ACG, likely due to C-to-U RNA editing at the second codon position (Table 1). Four types of stop codons were identified: TGA, TAG, TAA, and CGA. RNA editing from C to U was observed in the stop codons of atp9 and sdh4 (Table 1). The usage frequencies of these stop codons were 26.47% (TGA), 17.65% (TAG), 50% (TAA), and 5.88% (CGA), with TAA being the most frequently used stop codon.
The coding sequence (CDS) length of the bottle gourd mitochondrial genome is 30,212 bp, encoding 10,104 codons. Among these, 31 codons exhibited a relative synonymous codon usage (RSCU) value greater than 1, indicating a higher usage frequency compared to other synonymous codons. Analysis of RSCU for 24 PCGs in the bottle gourd mitogenome revealed that all NNT and NNA codons had RSCU values exceeding 1.0, except for the termination codon TGA (0.97) and the alanine codon GCA (0.98) (Figure 3). Codon usage in the bottle gourd mitogenome showed a strong bias toward A or T(U) at the third codon position, a pattern commonly observed in the mitochondrial genomes of land plants.
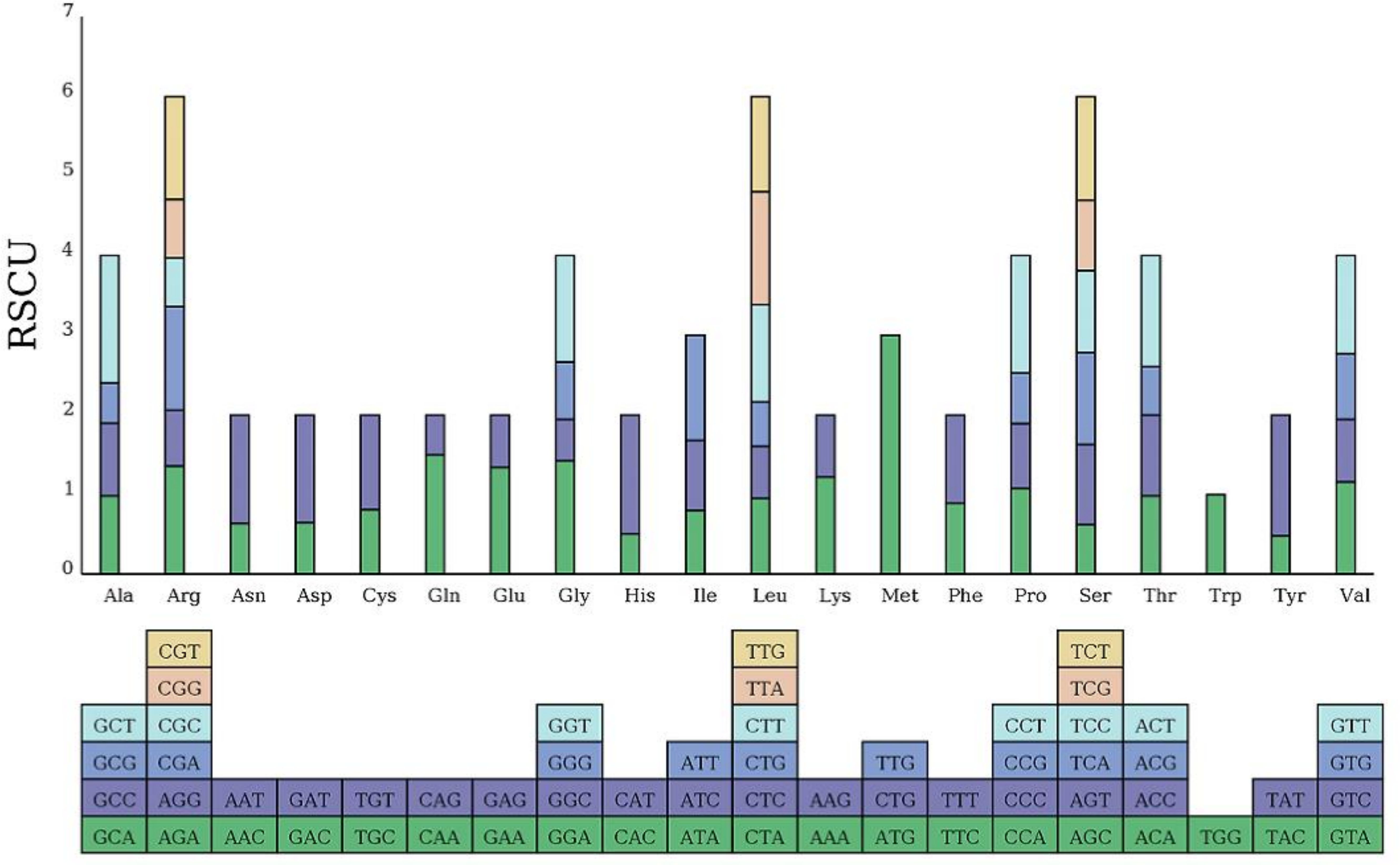
Figure 3. Relative synonymous codon usage (RSCU) in the L. siceraria mitogenome. Codon families are shown on the x-axis. RSCU values are the number of times a particular codon is observed relative to the number of times that codon would be expected for a uniform synonymous codon usage.
Prediction of RNA editing sites
In this study, a total of 497 RNA editing sites were predicted across 34 protein-coding genes (PCGs) in the mitochondrial (mt) genome of Lagenaria siceraria (Table 2; Figure 4). Among these, the genes rps19, rps7, and sdh3 had the fewest predicted editing sites, with only 2 each. In contrast, ccmFn and nad4 contained the highest number of predicted editing sites, with 38 each. Following RNA editing, the hydrophobicity of 60.76% of the amino acids remained unchanged. However, 7.85% of hydrophobic amino acids were converted to hydrophilic, while 30.99% of hydrophilic amino acids became hydrophobic. All RNA editing events in the bottle gourd mt genome involved C-to-U conversions, with editing occurring at both the first and second positions of the triplet codon. This resulted in the conversion of proline (CCC) to phenylalanine (TTC or TTT). Notably, RNA editing in the coding genes atp9 and sdh4 led to premature termination of the coding process.
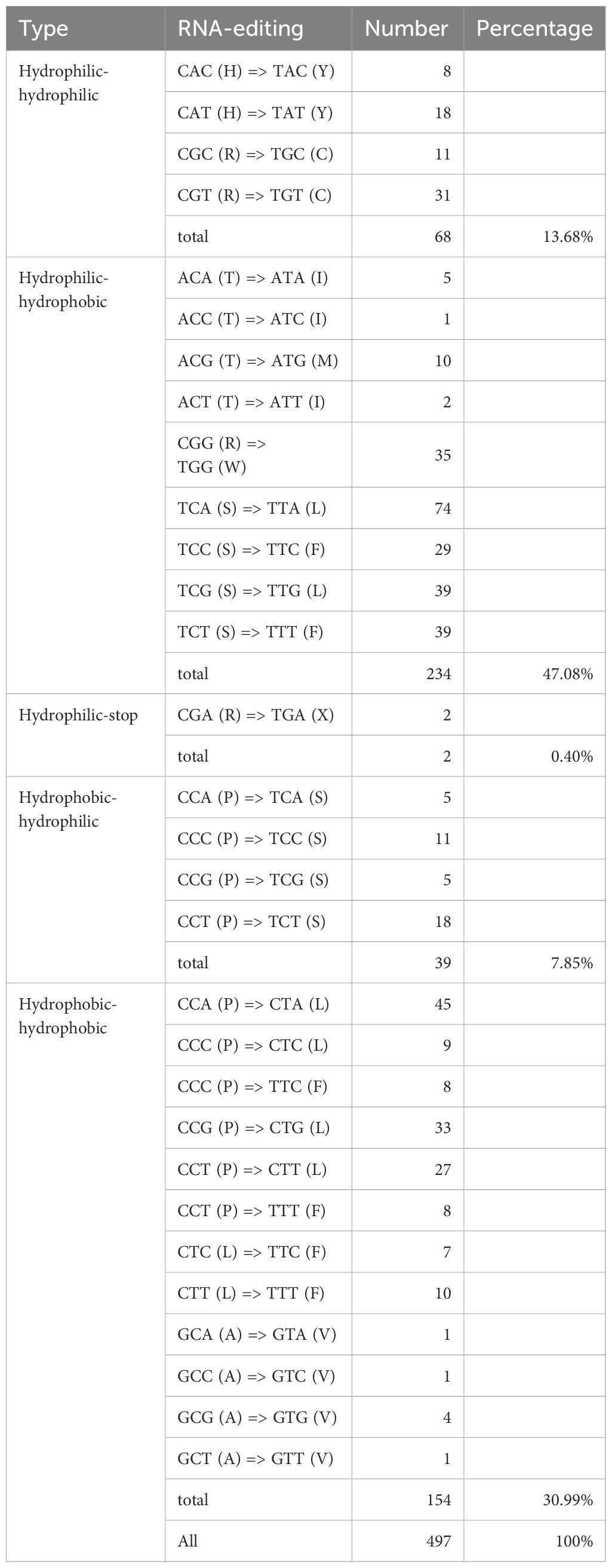
Table 2. Prediction of RNA editing sites.
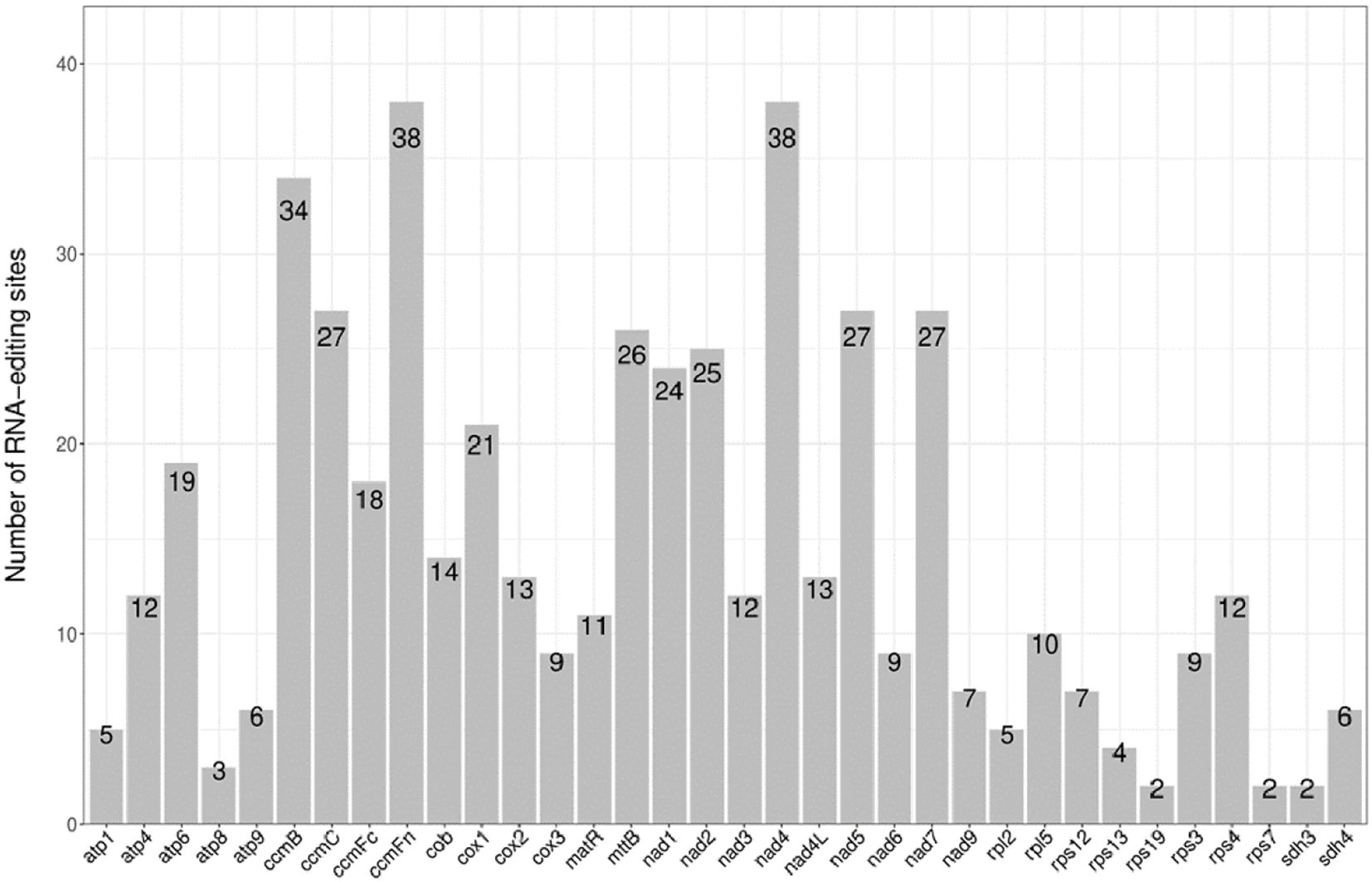
Figure 4. The distribution of RNA editing sites in mitogenome protein-coding genes of bottle gourd. The x-axis is the name of the gene. The y axis indicates the number of editing sites.
Analysis of repeats in the bottle gourd mitogenome
In the mitochondrial (mt) genome of bottle gourd, we identified a total of 260 interspersed repeats with lengths of 29 bp or greater. Among these, 123 were forward repeats, and 137 were palindrome repeats. The longest forward repeat sequence measured 2,349 bp, while the longest palindrome repeat sequence was 1,689 bp. As illustrated in Figure 5, forward repeats were most abundant in the 30–39 bp range, whereas palindrome repeats were most abundant in the 40–49 bp range.
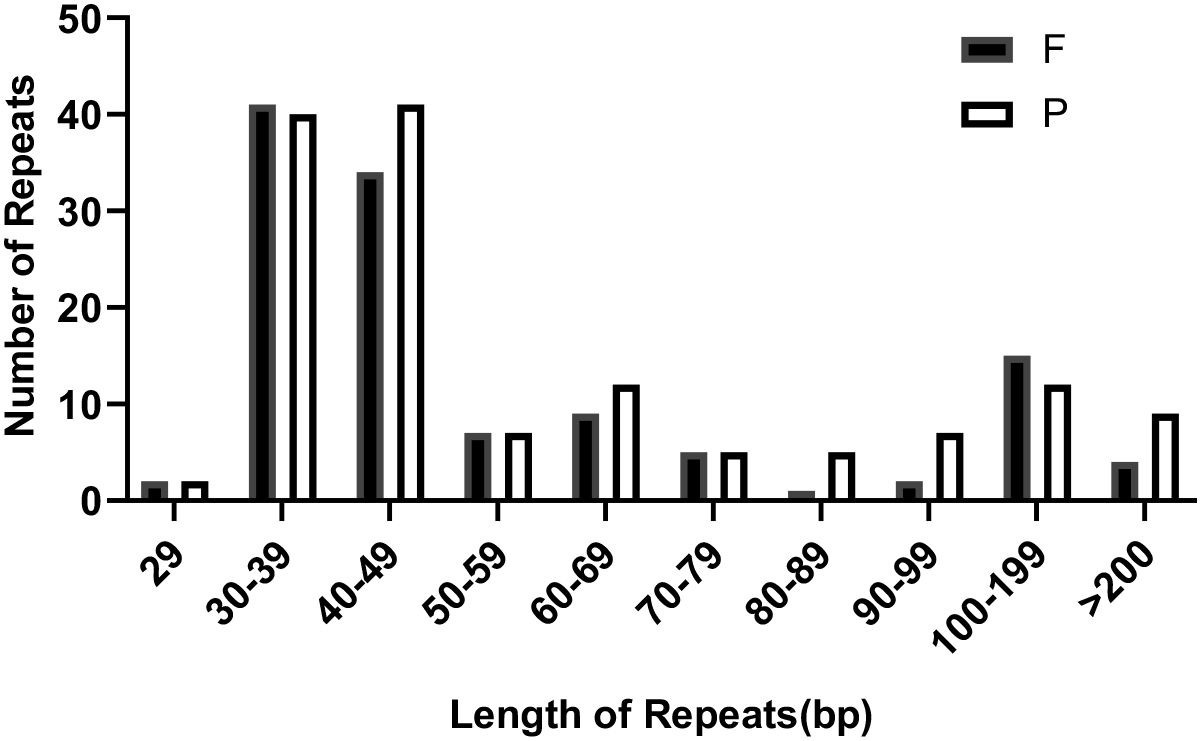
Figure 5. The length distribution of reverse and inverted repeats in the L. siceraria mt genome. F, Forward; P, Palindromic.
A total of 100 simple sequence repeats (SSRs) were detected in the bottle gourd mitogenome. These included 32 (32%) mononucleotide repeats, 25 (25%) dinucleotide repeats, 9 (9%) trinucleotide repeats, 30 (30%) tetranucleotide repeats, and 4 (4%) pentanucleotide repeats (Table 3). Mononucleotide, tetranucleotide, and dinucleotide repeats were the most abundant types. Further analysis of SSR repeat units revealed that 90.63% of mononucleotide repeats consisted of A/T bases, and 72% of dinucleotide repeats were AT/TA. The high AT content of these SSRs contributes to the overall AT richness (54.97%) of the bottle gourd mitogenome. Additionally, as shown in Table 4, a total of 9 tandem repeats, ranging in length from 12 to 39 bp and with a match degree greater than 80%, were identified in the bottle gourd mitogenome.
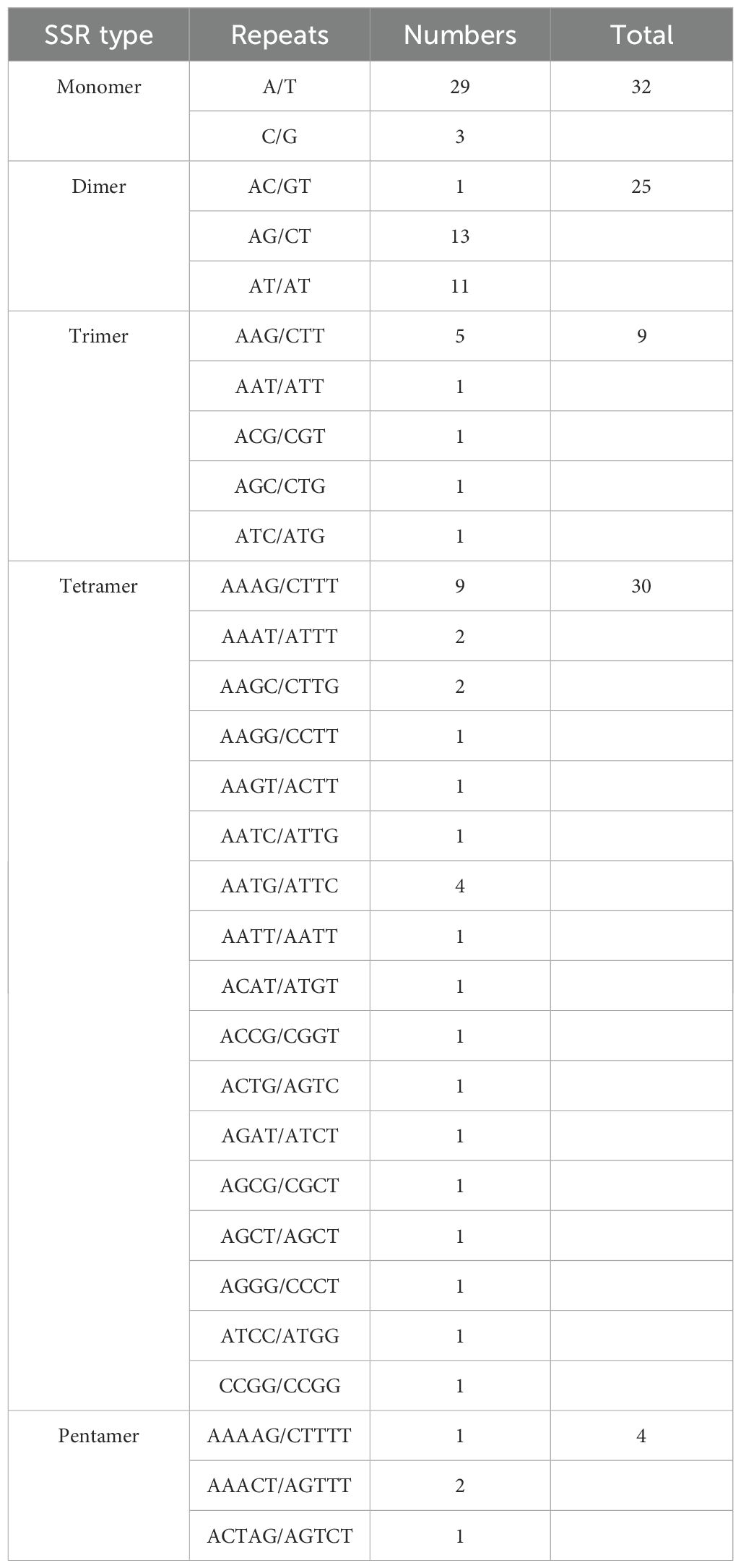
Table 3. Distribution of SSRs in L. siceraria mt genome.
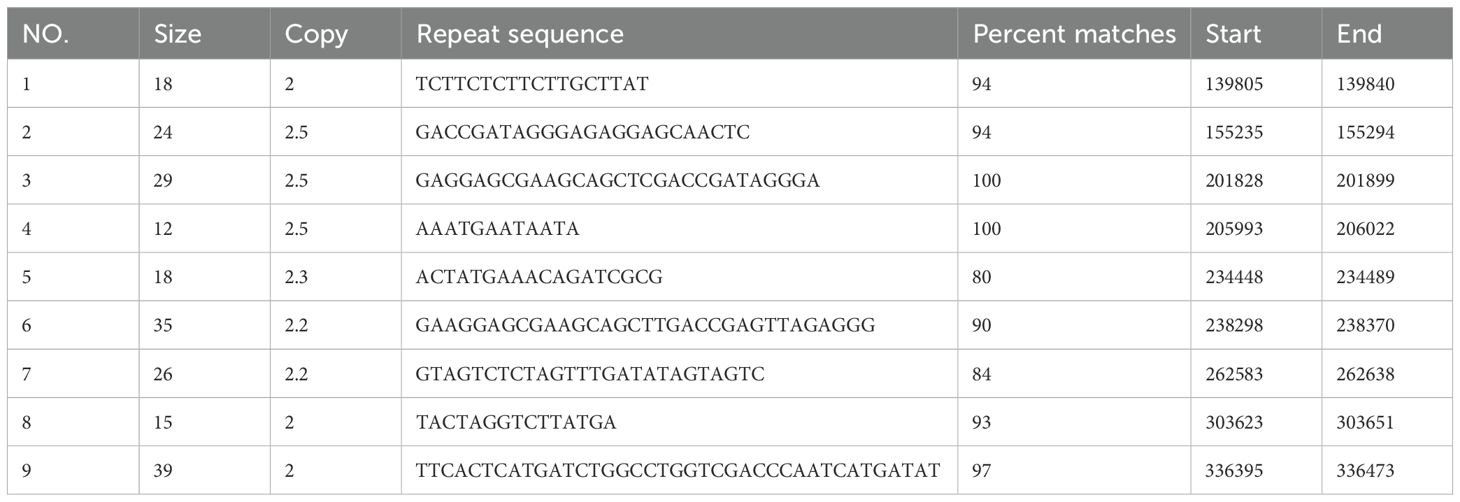
Table 4. Distribution of tandem repeats in L. siceraria mt genome.
Ka/Ks analysis
In genetics, the nonsynonymous-to-synonymous substitution ratio (Ka/Ks) is a key metric for understanding the evolutionary dynamics of genes. The Ka/Ks ratio helps determine whether a protein-coding gene (PCG) is under selective pressure during evolution. Under neutral selection, Ka = Ks, resulting in a Ka/Ks ratio of 1. If Ka > Ks (Ka/Ks > 1), it indicates positive selection, whereas if Ks > Ka (Ka/Ks < 1), it suggests negative (purifying) selection. In this study, the Ka/Ks ratio was calculated for 38 PCGs shared among L. siceraria, C. lanatus, C. sativus, L. acutangula, and C. maxima. As shown in Figure 6, when comparing the mitochondrial (mt) genome of bottle gourd with that of C. lanatus, 16 PCGs exhibited Ka/Ks values < 1. In comparison to C. sativus, 24 PCGs had Ka/Ks values < 1, while 7 PCGs had Ka/Ks values > 1. Relative to L. acutangula, 15 PCGs showed Ka/Ks values < 1. When compared to C. maxima, 29 PCGs had Ka/Ks values < 1, and 3 PCGs had Ka/Ks values > 1. Notably, nearly all Ka/Ks ratios were less than 1.0, indicating that most PCGs were under stabilizing (purifying) selection during evolution. In contrast, two genes (atp8 and rps10) had Ka/Ks ratios > 1.0, suggesting they underwent positive selection. Additionally, three genes (atp4, rpl10, rpl2, rps19, and rps4) had Ka/Ks ratios close to 1.
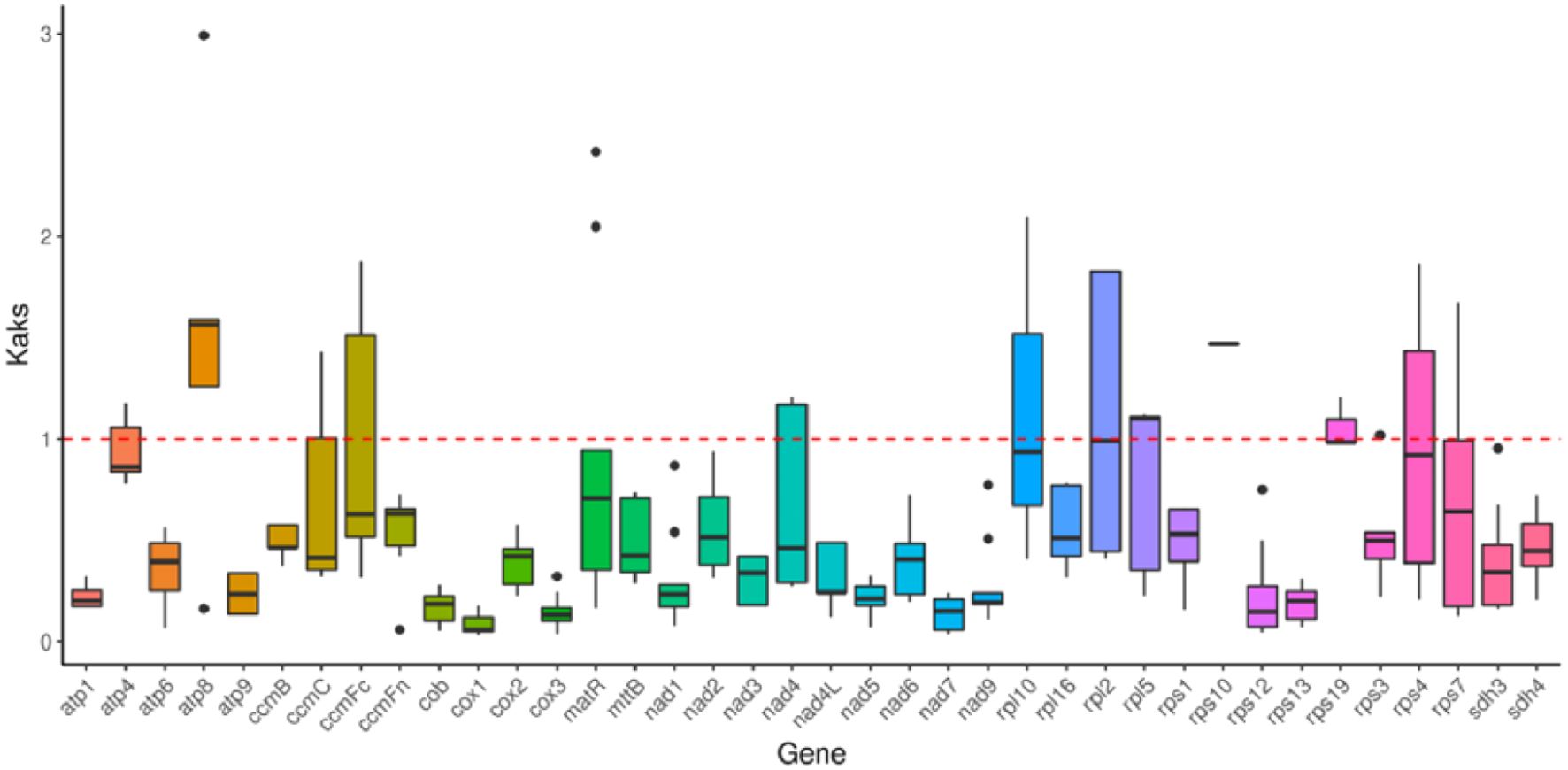
Figure 6. Ka/Ks ratios of 38 protein-coding genes in L. siceraria, C. lanatus, C. sativus, L. acutangular and C. maxima. Ka/Ks=1 means neutral selection. Ka/Ks > 1 indicates positive selection. Ka/Ks < suggests negative (purifying) selection.
Pi analysis
Nucleotide diversity (Pi) was calculated for 37 genes to assess sequence variation. A total of 1,338 polymorphic sites were identified (Supplementary Table S1). Among these, the maximum Pi value was 0.05028, corresponding to 65 polymorphic sites, while the minimum Pi value was 0.00594, associated with 4 polymorphic sites (Figure 7).
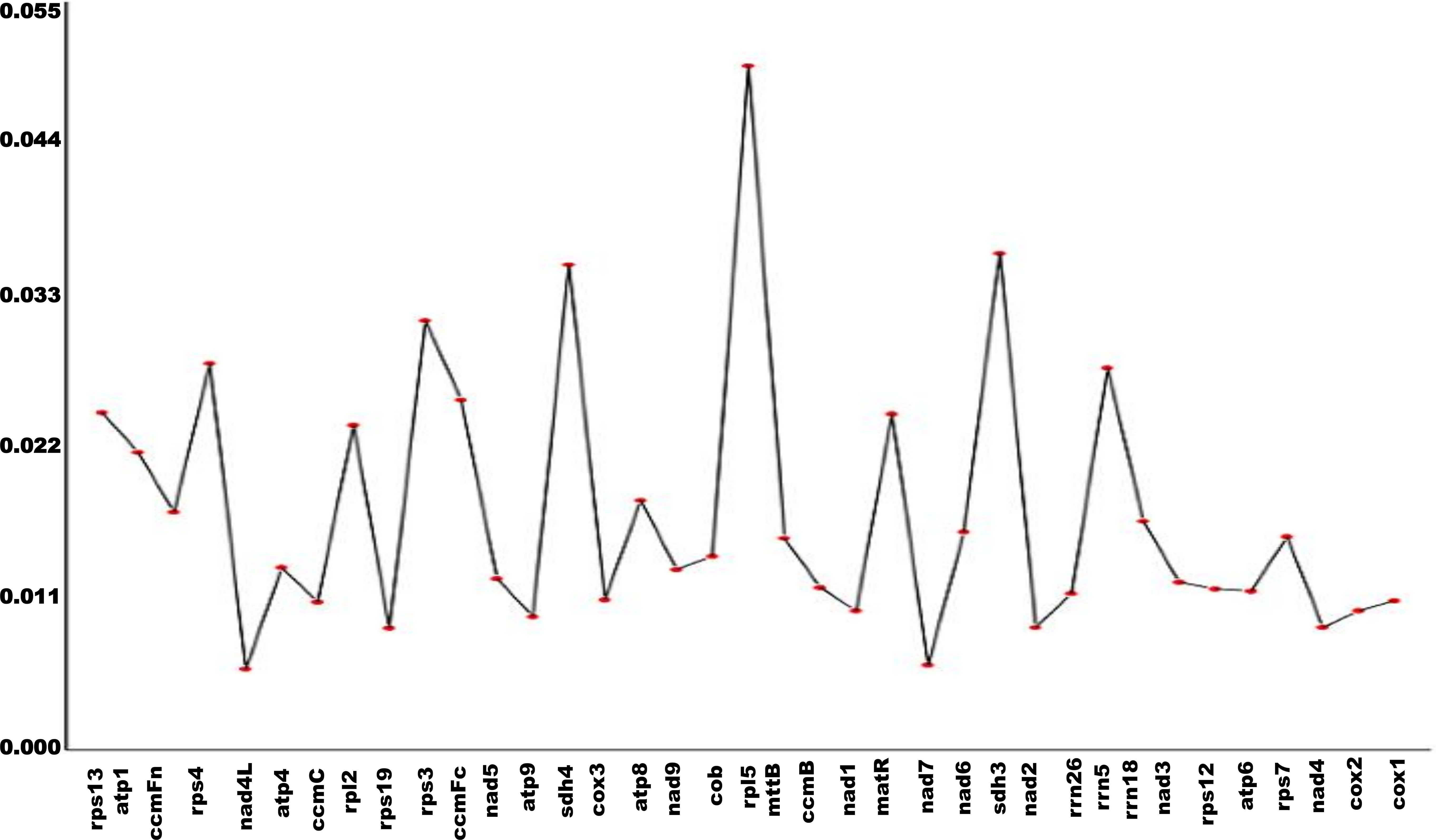
Figure 7. Nucleic acid diversity of genes in L. siceraria.The x-axis represents the gene name and the y-axis represents the pi value.
Analysis of homologous fragments between mitochondria and chloroplasts
We identified 45 homologous fragments between the mitochondrial (mt) and chloroplast (cp) genomes, with a total length of 40,579 bp, accounting for 11.35% of the mt genome (Figure 8, Table 5). These homologous fragments included 8 annotated genes, of which 6 were tRNA genes (trnL-CAA, trnM-CAT, trnN-GTT, trnD-GUC, trnP-TGG, and trnV-GAC) and 2 were ribosomal protein (SSU) genes (rps7 and rps12).
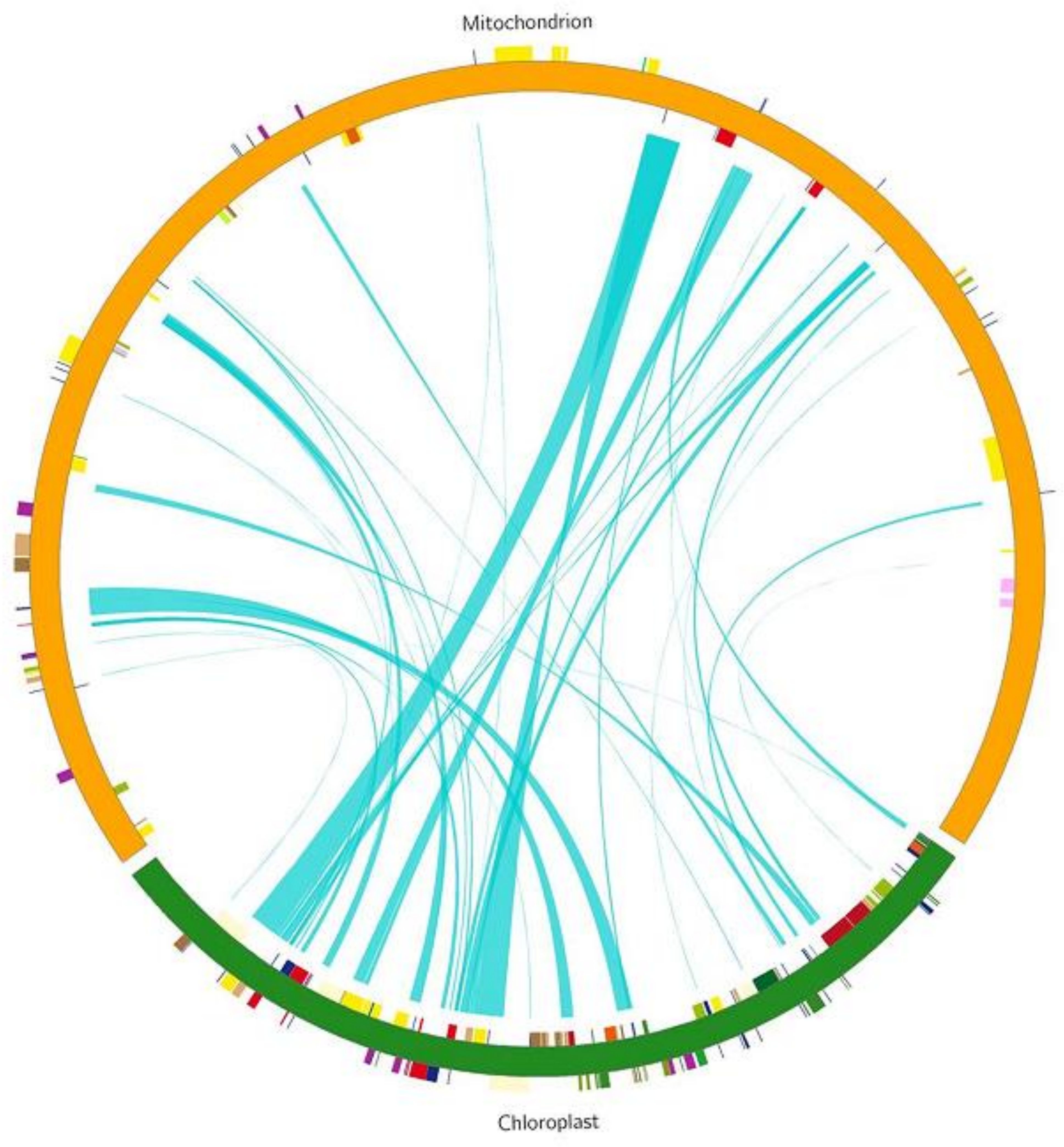
Figure 8. DNA and gene transfer between Chloroplast and Mitochondrial genomes in L. siceraria. The track shows complete genomes of cp and mt in green and orange respectively. The blue line segment in the circle connects the start and end points of the transferred gene fragments. The width of the blue line segment represents the size of the transferred fragment,.
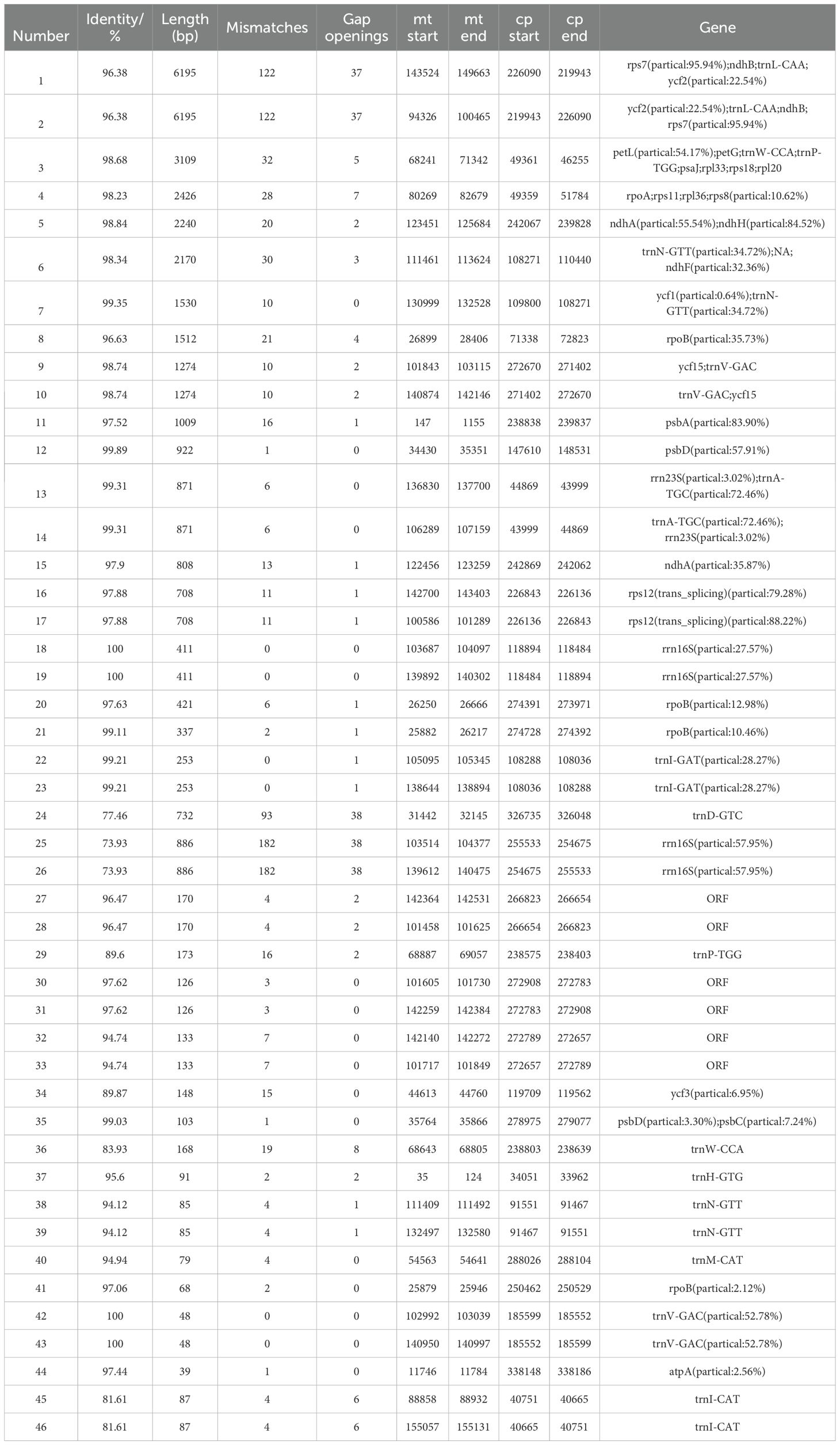
Table 5. Comparison of a homologous fragment in the L. siceraria chl genome to that in the mt genome.
Phylogenetic analysis and gene arrangement analysis
Phylogenetic trees were constructed using the maximum likelihood method to explore the evolutionary relationships between the bottle gourd mt genome and the published mt genomes of 32 plant species. The selected species and their details are listed in Table 6. The results revealed that C. maxima, C. sativus, C. lanatus, and L. acutangula were closely clustered with bottle gourd (Figure 9).
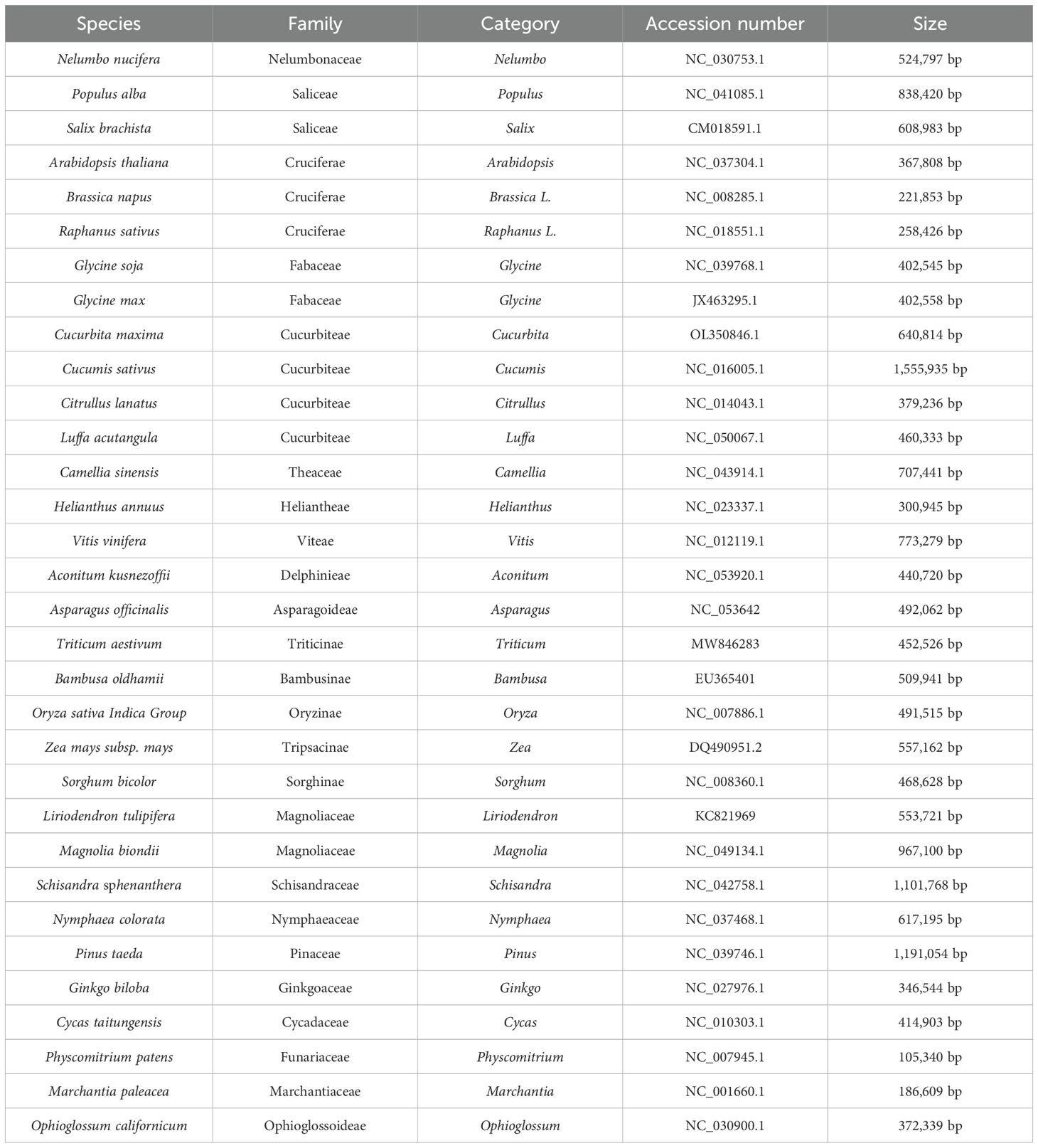
Table 6. NCBI accession numbers of mt genomes used in this study.
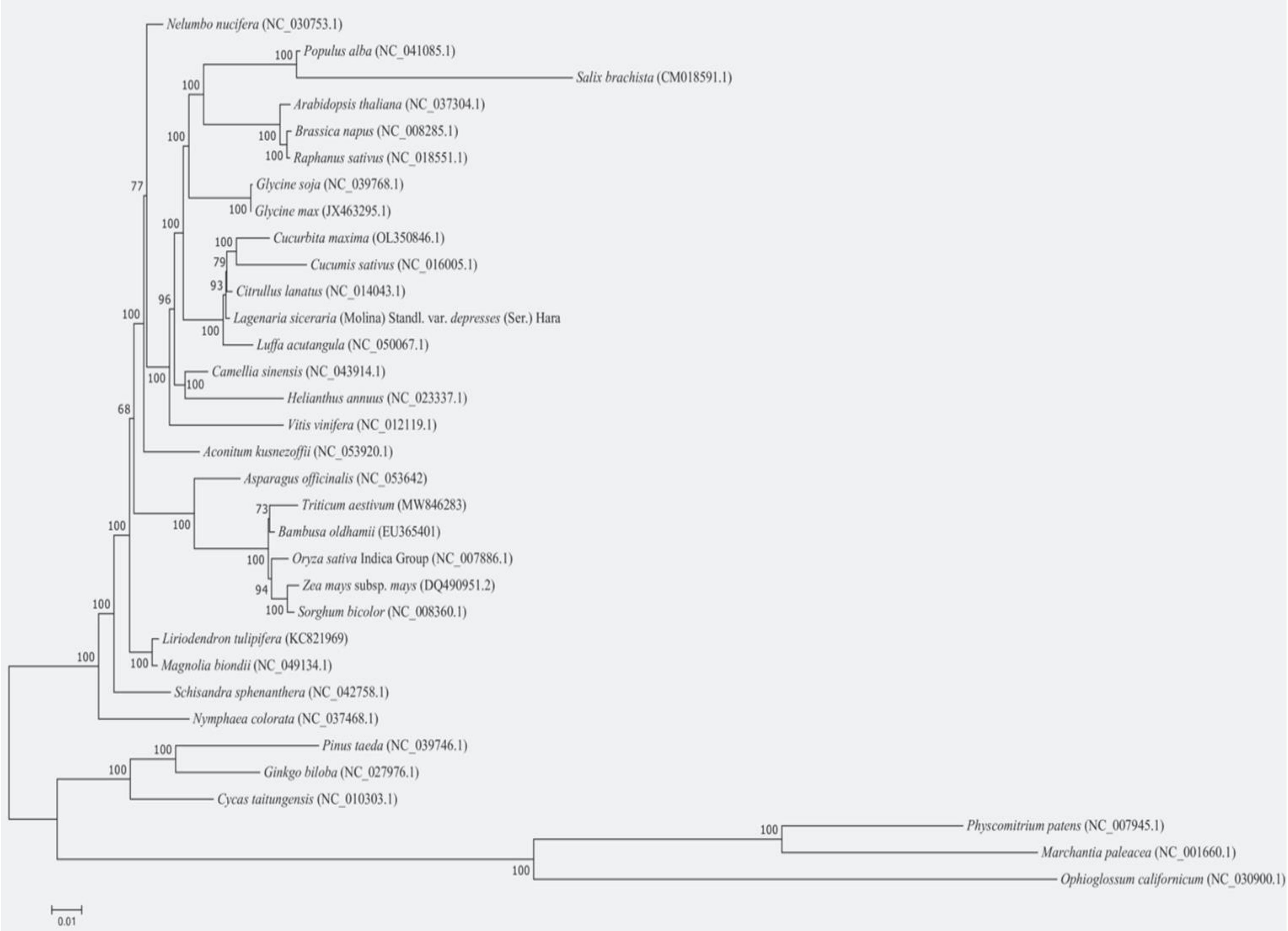
Figure 9. The phylogenetic relationships of L. siceraria with other 32 plant species. C. maxima, C. sativus, C. lanatus, and L. acutangula were closely clustered with bottle gourd.
Based on the phylogenetic tree, the 32 plant species were grouped into three major clusters: angiosperms, gymnosperms, and spore plants. The clustering pattern in the phylogenetic tree aligns with the traditional taxonomic relationships at the family and genus levels, demonstrating the reliability of mt genome-based phylogenetic analysis.
Dot plot analysis revealed only sporadic collinear regions between C. sativus and L. siceraria, indicating poor collinearity (Figure 10B). In contrast, C. maxima, C. lanatus, and L. acutangula exhibited better collinearity with L. siceraria (Figures 10A, C, D). These findings were further supported by BLASTn collinearity analysis (Figure 10E).
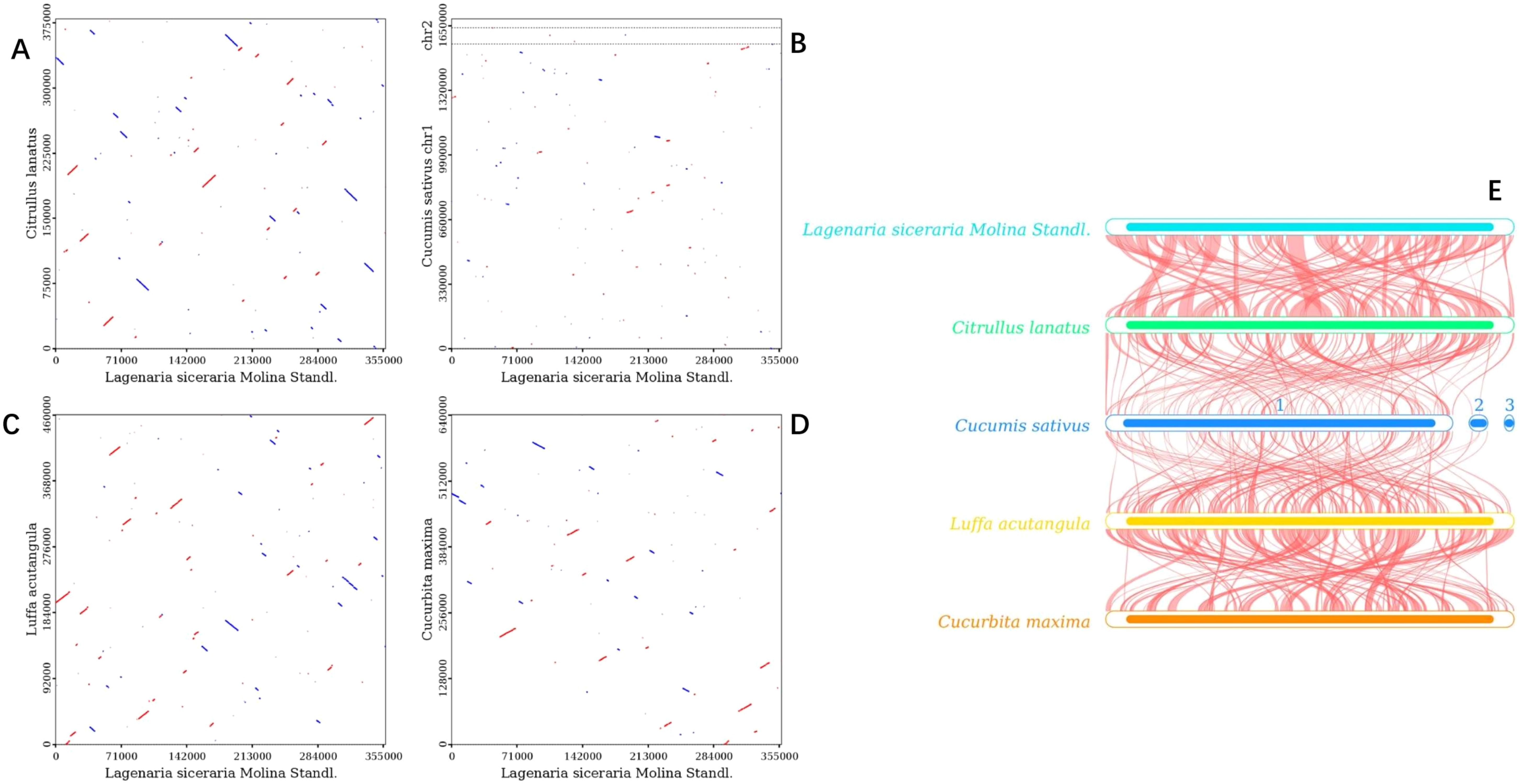
Figure 10. Collinearity analysis of the mitogenomes of L. siceraria, C. sativus, C. maxima, C. lanatus and L. acutangular. (A–D) are dot plots of C. lanatus, C. sativus, L. acutangular, and C. maxima with L. siceraria, respectively. (E) L. siceraria mitogenome synteny. The box in each row represents a genome, and the connecting line in the middle represents homology regions.
Discussion
The size of mitochondrial genomes varies significantly among different species. Previous studies have shown that angiosperms possess larger mitochondrial genomes than animals (Best et al., 2020; Christensen, 2013). To date, Silene conica (11.3 Mb) has the largest known mitochondrial genome in plants. Among cucurbit crops, the mitochondrial genome size ranges from 379 kb to 2,936 kb, with C. melo having the largest genome (Alverson et al., 2010). The mitochondrial genome of bottle gourd is 357,496 bp, smaller than that of watermelon (379,236 bp), making it the smallest mitochondrial genome among cucurbit crops. Although plant mitochondrial genomes are large, they typically contain only 50–60 coding genes, as the coding regions account for only 7–17% of the total genome, with the remainder consisting of intergenic regions. In bottle gourd, the coding region constitutes 8.48% of the mitochondrial genome, with 34 protein-coding genes. Watermelon, in contrast, has 37 protein-coding genes, similar to bottle gourd (Alverson et al., 2010). GC content is another important indicator for species evaluation (Liu et al., 2023). The GC content of cucurbit crops generally ranges from 44.1% to 44.6%, with cucumber mitochondrial genomes having a GC content of 44.2–44.6%, Cucumis hystrix at 44.5%, and Cucumis melo at 44.1%. However, bottle gourd has a higher GC content of 45.03%, the highest among known cucurbit crops.
Previous comparative analyses of mitochondrial genome sequences in cucurbit crops have revealed the presence of unique conserved sequences. A comparative analysis of mitochondrial genome composition between bottle gourd and other cucurbit species showed that bottle gourd possesses the rps19 gene, which is present in most species. However, the rpl10 gene, found in C. melo, C. hystrix, and C. sativus (Xia et al., 2022) is absent in the mitochondrial genome of bottle gourd. The mitochondrial rpl10 gene has become a pseudogene in some plants and has been entirely lost from the mitochondrial genome in others. The lost mitochondrial rpl10 gene has been replaced by an extra copy of the nuclear gene that normally encodes chloroplast rpl10 protein (Kubo and Arimura, 2009). The loss of rpl10 in the mitochondria of bottle gourd and its existence in the others indicate that the evolution of rpl10 within cucurbit crops has taken some unexpected and interesting turns. Additionally, the number of tRNA genes varies significantly among species, with 40 in C. melo, 13 in C. pepo, and 24 in bottle gourd. This suggests that tRNA genes have undergone substantial changes during the evolution of cucurbit crops. The presence of extra tRNA and rps genes in bottle gourd, which originated from chloroplast horizontal gene transfer, distinguishes it from other cucurbit species. This implies that sequence transfer between plastid genomes is a frequent occurrence during the evolution of flowering plants (Notsu et al., 2002; Xia et al., 2022). These transfer events contribute to the acquisition of functional tRNA genes and help explain the genetic variation observed in mitochondrial genomes across higher plants (Alverson et al., 2011; Xia et al., 2022).
Codon usage analysis indicates that, as in most other plants, Leu, Ser, and Arg are the most common amino acids in bottle gourd, while Met and Trp are much less frequent (Figure 2) (Ma et al., 2022). The preference for codons ending in A/T in the bottle gourd mitochondrial genome aligns with the codon usage patterns of most dicotyledons, in contrast to monocotyledons, which favor codons ending in G/C (Mazumdar et al., 2017). RNA editing, another critical factor influencing gene expression in plant mitochondrial genomes, plays a significant role in plant evolution (Edera et al., 2018). In cucurbit crops, RNA editing typically occurs at one of the first two positions of the codon, with the number of editing sites ranging from 444 to 501. In bottle gourd, 497 RNA editing sites were identified, a number similar to that found in C. hystrix (501) (Xia et al., 2022). RNA editing can take various forms, such as C-to-U, U-to-C, and A-to-I conversions (Small et al., 2019). However, in bottle gourd, all RNA editing events involve C-to-U conversions (Table 2), consistent with the pattern observed in C. hystrix. High-frequency RNA editing serves as a critical strategy for mitochondria to cope with genomic reduction, environmental stress, and complex regulatory demands, reflecting the profound evolutionary significance of post-transcriptional regulation in bottle gourd. This mechanism balances the stability and flexibility of genetic information, holding key value for understanding cellular metabolism and evolution.
Repetitive sequences in plant mitochondria play a crucial role in determining genome size, structure, and recombination (Cole et al., 2018). In bottle gourd, we identified multiple interspersed repeats, simple sequence repeats (SSRs), and tandem repeats. The total length of repetitive sequences in the bottle gourd mitochondrial genome is 22,294 bp, accounting for 6.24% of the genome. Compared to other cucurbit crops, bottle gourd has the fewest repetitive sequences, which may explain why its mitochondrial genome is the smallest among cucurbit species.
The results of Ka/Ks analysis of the mt genomes of L. siceraria, C. lanatus, C. sativus, L. acutangula, and C. maxima that most of the genes were negatively selected during the evolution process, indicating that the protein-coding genes of the bottle gourd mt genome are relatively well-conserved. However, the positive selection on atp8 and rps10 may enhance energy metabolism efficiency and translational capacity, thereby improving adaptability to growth or environmental stress in bottle gourd. This hypothesis requires further validation through combined experimental and evolutionary analyses, offering new insights into the domestication mechanisms of mitochondrial genes in crops.
DNA transfer between organelles, as well as between nuclear genomes and species, is a common phenomenon in plants. However, the extent of such transfers varies significantly among species (Timmis et al., 2004). Reported cases range from 50 kb in A. thaliana to 1.1 Mb in Oryza sativa subsp. Japonica. In this study, we identified 40,579 bp of DNA transferred from the chloroplast (cp) genome to the mitochondrial (mt) genome, accounting for 11.35% of the mt genome. This proportion is higher than that observed in other crops, such as Bupleurum chinense DC (2.56%), Acer truncatum (2.36%), and Suaeda glauca (5.18%) (Qiao et al., 2022).
The mitochondrial genome serves as a valuable source of genetic information for phylogenetic research (Xia et al., 2022). In this study, C. maxima, C. sativus, C. lanatus, L. acutangular, and L. siceraria were grouped together in the Cucurbiteae family. The topology of the mitochondrial DNA-based phylogenetic tree aligns with the Angiosperm Phylogeny Group classification. The clustering of these 32 species on the evolutionary tree is consistent with their traditional taxonomic relationships, demonstrating the congruence between traditional and molecular taxonomy. While cucumber and bottle gourd fruits are typically used as vegetables, and watermelon fruits are consumed as fruits, evolutionary analysis reveals that bottle gourd is more closely related to watermelon than to cucumber. This is further supported by similarities in genome size, composition, and the number of repetitive sequences between bottle gourd and watermelon.
Conclusion
We present the first complete mitochondrial genome assembly and annotation of a cucurbit crop, bottle gourd. The mitochondrial genome of gourd is also the smallest among cucurbitaceae crops so far.Comparative analysis of gene structure, codon usage, repeat regions, and RNA editing sites in the bottle gourd mitochondrial genome were analyzed, contributing to our understanding of bottle gourd. Repeat sequences, RNA editing edits, and the horizontal gene transfer events in the bottle gourd mitochondrial genome were analyzed, contributing to our understanding of bottle gourd.We found that bottle gourd is closely related to watermelon in size, but L. acutangula exhibits the highest collinearity with L. siceraria according to gene arrangement analysis. Further resolution of mitochondrial genomic information could contribute to our knowledge of the unique mitochondrial revolution of bottle gourd. The well-conserved protein-coding genes in mitochondrial genome of the bottle gourd could potentially serve as molecular markers in phylogenetic studies. This study provides extensive information about the mitochondrial genome for L. siceraria, facilitating the deciphering of evolutionary and genetic relationships within the cucurbit crops.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
XD: Formal analysis, Writing – original draft, Conceptualization. KW: Writing – original draft, Methodology, Data curation, Validation. YT: Writing – review & editing, Software. JW: Writing – review & editing, Software. XY: Resources, Writing – review & editing. HZ: Data curation, Writing – review & editing. ZZ: Funding acquisition, Methodology, Writing – review & editing. NL: Methodology, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was financially supported by Shanghai Agriculture Applied Technology Development Program, China (Grant No.X2022-02-08-00-12-F01105) and the SAAS Program for Excellent Research Team (2025-030).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1599596/full#supplementary-material
References
Alverson, A. J., Rice, D. W., Dickinson, S., Barry, K., and Palmer, J. D. (2011). Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell. 23, 2499–2513. doi: 10.1105/tpc.111.087189
Alverson, A. J., Wei, X., Rice, D. W., Stern, D. B., Barry, K., and Palmer, J. D. (2010). Insights into the Evolution of Mitochondrial Genome Size from Complete Sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evolution. 27, 1436–1448. doi: 10.1093/molbev/msq029
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Best, C., Mizrahi, R., and Ostersetzer-Biran, O. A.-O. (2020). Why so complex? The intricacy of genome structure and gene expression, associated with angiosperm mitochondria, may relate to the regulation of embryo quiescence or dormancy-intrinsic blocks to early plant life. LID - 10.3390/plants9050598 [doi] LID - 598. Plants (Basel). 9, 598. doi: 10.3390/plants9050598
Cao, Y., Yin, D., Pang, B., Li, H., Liu, Q., Zhai, Y., et al. (2023). Assembly and phylogenetic analysis of the mitochondrial genome of endangered medicinal plant Huperzia crispata. Funct. Integr. Genomics 23, 295. doi: 10.1007/s10142-023-01223-9
Chan, P. P. and Lowe, T. M. (2019). “tRNAscan-SE: searching for tRNA genes in genomic sequences,” in Gene prediction: methods and protocols. Ed. Kollmar, M. (Springer New York, New York, NY), 1–14. doi: 10.1007/978-1-4939-9173-0_1
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34, i884-i890. doi: 10.1101/274100
Chevigny, N., Schatz-Daas, D., Lotfi, F., and Gualberto, J. M. (2020). DNA repair and the stability of the plant mitochondrial genome. Int. J. Mol. Sci. 21, 328. doi: 10.3390/ijms21010328
Christensen, A. C. (2013). Plant mitochondrial genome evolution can be explained by DNA repair mechanisms. Genome Biol. Evolution. 5, 1079–1086. doi: 10.1093/gbe/evt069
Cole, L. W., Guo, W., Mower, J. P., and Palmer, J. D. (2018). High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol. Biol. Evolution. 35, 2773–2785. doi: 10.1093/molbev/msy176
Edera, A. A., Gandini, C. L., and Sanchez-Puerta, M. V. (2018). Towards a comprehensive picture of C-to-U RNA editing sites in angiosperm mitochondria. Plant Mol. Biol. 97, 215–231. doi: 10.1007/s11103-018-0734-9
Greiner, S. and Bock, R. (2013). Tuning a ménage à trois: Co-evolution and co-adaptation of nuclear and organellar genomes in plants. BioEssays. 35, 354–365. doi: 10.1002/bies.201200137
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PloS Genet. 15, e1008373. doi: 10.1371/journal.pgen.1008373
Kubo, N. and Arimura, S. (2009). Discovery of the rpl10 gene in diverse plant mitochondrial genomes and its probable replacement by the nuclear gene for chloroplast RPL10 in two lineages of angiosperms. DNA Res. 17, 1–9. doi: 10.1093/dnares/dsp024
Kubo, T. and Newton, K. J. (2008). Angiosperm mitochondrial genomes and mutations. Mitochondrion. 8, 5–14. doi: 10.1016/j.mito.2007.10.006
Lee, B. D. (2018). Python implementation of codon adaptation index. J. Open Source Software 3, 96. doi: 10.21105/joss.00905
Levi, A., Thomas, C. E., Thies, J. A., Simmons, A. M., Ling, K.-S., Harrison, H. F., et al. (2006). Novel Watermelon Breeding Lines Containing Chloroplast and Mitochondrial Genomes derived from the Desert Species Citrullus colocynthis. HortScience HortSci. 41, 463–464. doi: 10.21273/HORTSCI.41.2.463
Li, H. and Birol, I. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Liu, D., Qu, K., Yuan, Y., Zhao, Z., Chen, Y., Han, B., et al. (2023). Complete sequence and comparative analysis of the mitochondrial genome of the rare and endangered Clematis acerifolia, the first clematis mitogenome to provide new insights into the phylogenetic evolutionary status of the genus. Front. Genet. 13. doi: 10.3389/fgene.2022.1050040
Ma, Q., Wang, Y., Li, S., Wen, J., Zhu, L., Yan, K., et al. (2022). Assembly and comparative analysis of the first complete mitochondrial genome of Acer truncatum Bunge: a woody oil-tree species producing nervonic acid. BMC Plant Biol. 22, 29. doi: 10.1186/s12870-021-03416-5
Mazumdar, P., Binti Othman, R., Mebus, K., Ramakrishnan, N., and Ann Harikrishna, J. (2017). Codon usage and codon pair patterns in non-grass monocot genomes. Ann. Botany. 120, 893–909. doi: 10.1093/aob/mcx112
Møller, I. M., Rasmusson, A. G., and Van Aken, O. (2021). Plant mitochondria – past, present and future. Plant J. 108, 912–959. doi: 10.1111/tpj.15495
Mower, J. P. (2005). PREP-Mt: predictive RNA editor for plant mitochondrial genes. BMC Bioinf. 6, 96. doi: 10.1186/1471-2105-6-96
Niu, Y., Qin, Q., Dong, Y., Wang, X., Zhang, S., and Mu, Z. (2023). Chloroplast genome structure and phylogenetic analysis of 13 lamiaceae plants in tibet. Front. Biosci. (Landmark Ed). 28, 110. doi: 10.31083/j.fbl2806110
Notsu, Y., Masood, S., Nishikawa, T., Kubo, N., Akiduki, G., Nakazono, M., et al. (2002). The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genomics 268, 434–445. doi: 10.1007/s00438-002-0767-1
O’Conner, S. and Li, L. (2020). Mitochondrial fostering: the mitochondrial genome may play a role in plant orphan gene evolution. Front. Plant Science. 11. doi: 10.3389/fpls.2020.600117
Olechowska, E., Słomnicka, R., Kaźmińska, K., Olczak-Woltman, H., and Bartoszewski, G. A.-O. X. (2022). The genetic basis of cold tolerance in cucumber (Cucumis sativus L.)-the latest developments and perspectives. J. Appl. Genet. 63, 597–608. doi: 10.1007/s13353-022-00710-2
Qiao, Y., Zhang, X., Li, Z., Song, Y., and Sun, Z. (2022). Assembly and comparative analysis of the complete mitochondrial genome of Bupleurum chinense DC. BMC Genomics 23, 664. doi: 10.1186/s12864-022-08892-z
Rodríguez-Moreno, L., González Vm Fau - Benjak, A., Benjak A Fau - Martí, M. C., Martí Mc Fau - Puigdomènech, P., Puigdomènech P Fau - Aranda, M. A., Garcia-Mas, A.M., et al. (2011). Determination of the melon chloroplast and mitochondrial genome sequences reveals that the largest reported mitochondrial genome in plants contains a significant amount of DNA having a nuclear origin. BMC Genomics 20, 424. doi: 10.1186/1471-2164-12-424
Small, I. D., Schallenberg-Rüdinger, M., Takenaka, M., Mireau, H., and Ostersetzer-Biran, O. (2019). Plant organellar RNA editing: what 30 years of research has revealed. Plant J. 101, 1040–1056. doi: 10.1111/tpj.14578
Srivastava, S., Upadhyay, M., Srivastava, A., Abdelrahman, M., Suprasanna, P., and Tran, L.-S. (2018). Cellular and subcellular phosphate transport machinery in plants. Int. J. Mol. Sci. 19, 1914. doi: 10.3390/ijms19071914
Stothard, P. (2000). The sequence manipulation suite: javaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques. 28, 1102–1104. doi: 10.2144/00286ir01
Tanaka, K., Akashi, Y., Fukunaga, K., Yamamoto, T., Aierken, Y., Nishida, H., et al. (2013). Diversification and genetic differentiation of cultivated melon inferred from sequence polymorphism in the chloroplast genome. Breed. Science. 63, 183–196. doi: 10.1270/jsbbs.63.183
Thiel, T., Michalek, W., Varshney, R., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Timmis, J. N., Ayliffe Ma Fau - Huang, C. Y., Huang Cy Fau - Martin, W., and Martin, W. (2004). Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 5, 123–135. doi: 10.1038/nrg1271
Wang, J., Kan, S., Liao, X., Zhou, J., Tembrock, L. R., Daniell, H., et al. (2024a). Plant organellar genomes: much done, much more to do. Trends Plant Science. 29, 754–769. doi: 10.1016/j.tplants.2023.12.014
Wang, S., Qiu, J., Sun, N., Han, F., Wang, Z., Yang, Y., et al. (2025). Characterization and comparative analysis of the first mitochondrial genome of Michelia (Magnoliaceae). Genomics Commun. 2, 0–0. doi: 10.48130/gcomm-0025-0001
Wang, J., Zou, Y., Mower, J. P., Reeve, W., and Wu, Z. (2024b). Rethinking the mutation hypotheses of plant organellar DNA. Genomics Commun. 1, 0–0. doi: 10.48130/gcomm-0024-0003
Wu, Y., Sun, Z., Liu, Z., Qiu, T., Li, X., Leng, L., et al. (2025). Assembly and analysis of stephania japonica mitochondrial genome provides new insights into its identification and energy metabolism. BMC Genomics 26, 185. doi: 10.1186/s12864-025-11359-6
Xia, L., Cheng, C., Zhao, X., He, X., Yu, X., Li, J., et al. (2022). Characterization of the mitochondrial genome of Cucumis hystrix and comparison with other cucurbit crops. Gene. 823, 146342. doi: 10.1016/j.gene.2022.146342
Xu, Y., Dong, Y., Cheng, W., Wu, K., Gao, H., Liu, L., et al. (2022). Characterization and phylogenetic analysis of the complete mitochondrial genome sequence of Diospyros oleifera, the first representative from the family Ebenaceae. Heliyon. 8, e09870. doi: 10.1016/j.heliyon.2022.e09870
Keywords: Lagenaria siceraria, mitochondrial genome, phylogenetic analysis, cucurbit, evolutionary analysis
Citation: Du X, Wang K, Tang Y, Wu J, Yang X, Zhang H, Liu N and Zhang Z (2025) Characterization and phylogenetic analysis of the complete mitochondrial genome sequence of Lagenaria siceraria, a cucurbit crop. Front. Plant Sci. 16:1599596. doi: 10.3389/fpls.2025.1599596
Received: 27 March 2025; Accepted: 27 June 2025;
Published: 22 July 2025.
Edited by:
Zhiqiang Wu, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Carlos I. Arbizu, Universidad Nacional Toribio Rodríguez de Mendoza de Amazonas, PeruZhechen Qi, Zhejiang Sci-Tech University, China
Copyright © 2025 Du, Wang, Tang, Wu, Yang, Zhang, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaohui Zhang, emhhbmd6aGFvaHVpQHNhYXMuc2guY24=; Na Liu, bGl1bmEyMDIzMDNAMTI2LmNvbQ==
†These authors have contributed equally to this work