 Qiang Li1*
Qiang Li1* Ran Wei2
Ran Wei2- 1College of Life Sciences, Nanjing Forestry University, Nanjing, Jiangsu, China
- 2College of Manufacturing Engineering, Maanshan University, Maanshan, China
Introduction: The genus Sorbus sensu lato (Sorbus s.l.) comprises over 260 species widely distributed across temperate regions of Asia, Europe, and North America. However, hybridization and polyploidization have posed significant challenges to phylogenetic and taxonomic studies within this genus.
Methods: Here, we assemble the first complete chloroplast and mitochondrial genomes of Karpatiosorbus bristoliensis to characterize organellar genomic features and establish a maternally inherited phylogenetic framework for Sorbus s.l.
Results and discussion: The mitochondrial genome of K. bristoliensis is circular (386,757 bp), encoding 55 genes, including 34 protein-coding genes, 18 rRNAs, and 3 tRNAs. Its chloroplast genome has a typical quadripartite structure (160,322 bp), containing 75 protein-coding genes, 29 tRNA genes, 4 rRNA genes, and one pseudogene (ycf1Ψ). Homologous gene transfer analysis of Sorbus s.l. species revealed inter-organellar gene transfer ranging from 3,021 to 3,846 bp. RNA editing analysis revealed 274–352 editing sites in Sorbus s.l., with nad4 containing the greatest number of editing sites across all protein-coding genes except those in K. bristoliensis. Simple sequence repeat (SSR) analysis detected 48–52 SSRs per species, predominantly mononucleotide repeats. Phylogenetic reconstruction on the basis of organellar genomes revealed that Karpatiosorbus is a sister to Torminalis. Plastome-based phylogeny revealed the non-monophyletic status of Sorbus s.s., attributed to the nested placement of the hybrid-origin genera Hedlundia and Scandosorbus within the genus. Additionally, Hedlundia austriaca and H. persica should be transferred to the nothogenus Sorbomeles. Mitochondrial genome collinearity analysis revealed extensive genomic structural rearrangements. Our findings not only delineate the structural characteristics of mitochondrial genomes across Sorbus s.l. taxa but also establish a high-resolution maternal phylogenetic framework for this genus.
Introduction
The genus Sorbus L. were primarily established by Linnaeus and included only two species (S. aucuparia L. and S. domestica L.) (Linnaeus, 1753). Subsequently, more species were transferred to this genus. Sorbus L. broadly (Sorbus L. sensu lato) contains approximately 260 species characterized by both simple-leaved species and pinnately compound-leaved species (Yü and Lu, 1974; Phipps et al., 1990; Lu and Stephen, 2003). Sorbus s.l. comprises trees or shrubs typically restricted to high altitudes, occupying diverse habitats including mountain valleys, streamsides, rocky slopes, and scrublands (Lu and Stephen, 2003). These plants are widely distributed across temperate Asia, Europe, and North America (Phipps et al., 1990). Many species exhibit ornamental value due to their conspicuous colorful fruits (McAllister, 2005).
Previous molecular and morphological evidence suggests that Sorbus s.l. is paraphyletic and raises six subgenera to the generic rank (Aria (Pers.) Host, Cormus Spach, Chamaemespilus Medikus, Micromeles Decaisne, Sorbus sensu stricto (Sorbus s.s.), and Torminalis Medikus) (Campbell et al., 2007; Zheng and Zhang, 2007; Potter et al., 2007; Lo and Donoghue, 2012). Among the six genera, the genera Aria, Chamaemespilus, Micromeles, and Torminalis all have simple leaves, whereas Cormus and Sorbus s.s. are characterized by compound leaves. However, the generic boundaries of these taxa have been continuously revised. For instance, Mezhenska et al. (2018) proposed transferring all Asian simple-leaved species to the genus Micromeles, including the previously taxonomically disputed genera Aria and Micromeles. In the same year, Rushforth (2018) alternatively classified Asian simple-leaved species within Micromeles and five newly established genera (Griffitharia, Alniaria, Thomsonaria, Dunnaria, and Wilsonaria) on the basis of morphological characteristics.
The primary factors contributing to the taxonomic complexity within Sorbus s.l. are hybridization and polyploidization. Notably, previous works highlighted that polyploidization, frequent natural hybridization and apomixis play crucial roles in the ongoing genetic diversification of Sorbus s.l. species (Robertson et al., 2010). Biparental, triparental and multiple-hybrid origins (Németh et al., 2020; Nelson-Jones et al., 2002; Robertson et al., 2004) contribute to allodiploid, triploid and tetraploid species, rendering the taxonomy complex. In addition, interspecific hybridization also promotes the innovation of morphological characteristics, such as the number of pairs of leaflets and fruit color (Nelson-Jones et al., 2002; Wu et al., 2024).
The taxonomic treatment of numerous hybrid-origin species remains unresolved, particularly with respect to their delimitation and placement within phylogenetic frameworks. These hybrid-origin species have been taxonomically elevated to subgenera (Rich et al., 2014) or genera (Sennikov and Kurtto, 2017). For example, Sennikov and Kurtto (2017) elevated certain hybrid-origin taxa within the genus Sorbus to five hybridogenous genera (Borkhausenia = Aria × Sorbus × Torminalis, Hedlundia = Aria × Sorbus, Karpatiosorbus = Aria × Torminalis, Majovskya = Aria × Chamaemespilus, Normeyera = Aria × Chamaemespilus × Sorbus).
Karpatiosorbus has a hybrid origin from Aria (Pers.) Host × Torminalis Medik, including one sexual hybrid (Karpatiosorbus × hybrida (Borkh.) Sennikov & Kurtto) and 84 apomictic species (Sennikov and Kurtto, 2017). Among these 85 species, Karpatiosorbus bristoliensis (Wilmott) Sennikov & Kurtto is a shrub or small tree approximately 30 ft high and has restricted areas of distribution in Avon Gorge, England. It inhabits open limestone outcrops, slopes, scrub, open grassland, quarry edges, and ancient woodland plateau soils, primarily within the Leigh Woods ecosystem (Rich et al., 2022). Despite its ornamental value, this species has been assessed as ‘Endangered’ under criterion D (IUCN, 2001) and on the Global Red List owing to its small population size (Rich et al., 2022). Using six chloroplast fragments, Chester et al. (2007) revealed that K. bristoliensis and Sorbus torminalis (L.) Crantz shared an identical haplotype (haplotype B), while proposing that K. bristoliensis may represent a case of in situ speciation. Employing microsatellite analysis, Robertson et al. (2010) determined the triploid hybrid origin of K. bristoliensis with diploid S. torminalis as the maternal progenitor and tetraploid S. eminens E.F.Warb. as the paternal progenitor (Robertson et al., 2010). Previous studies utilizing organellar genome have provided novel insights into the genetic diversity and conservation of endangered species (Zhang et al., 2024). However, the limited genomic information available for this endangered species hinders our understanding of its genomic structure and genomic diversity. The organellar genomes assembled in this study will lay the foundation for conservation genomics in K. bristoliensis.
The mitochondrial genome has received increasing attention in the field of plant phylogenomics (Lin et al., 2025). This is because of the slow evolutionary rate of mitochondrial genes compared with nuclear genes. Unlike the chloroplast genome, plant mitochondrial genomes are large (66 kb to 18.99 Mb) (Skippington et al., 2015; Huang et al., 2024a), exhibit frequent recombination, and often incorporate foreign DNA via horizontal gene transfer (HGT) from chloroplasts and nuclear (Wang et al., 2025; Zhang et al., 2025). These features result in highly variable genome sizes and architectures across plant lineages (Gualberto and Newton, 2017; Wu et al., 2022; Lu et al., 2025). The plant mitochondrial genome harbors abundant repetitive sequences, including simple sequence repeats (SSRs), tandem repeats, and interspersed repeats. These repetitive elements serve as a foundation for the development of diverse molecular markers to investigate population genetic diversity (Morgante et al., 2002). Moreover, the high abundance of repeats contributes significantly to the complex structural rearrangements observed in mitochondrial genomes, such as recombination-mediated inversions, duplications, and the formation of multipartite architectures (Bi et al., 2022).
The availability of extensive chloroplast genome sequencing data for the genus Sorbus s.l. enables the utilization of these datasets to address unresolved phylogenetic questions within this taxonomic group. Furthermore, recent advances in long-read sequencing have resolved complex plant mitochondrial genome structures. Taking advantage on the previously released PacBio HiFi sequencing data of Karpatiosorbus bristoliensis in the NCBI database, we utilized these datasets to perform the first de novo assembly of both chloroplast and mitochondrial genomes for the hybrid genus Karpatiosorbus. Our main goals are to (1) assemble and characterize the first mitochondrial and plastid genomes of this species; (2) compare the mitochondrial genomes of Sorbus s.l. species; (3) explore repetitive sequences and RNA editing sites; and (4) reconstruct the maternal phylogenetic framework within Sorbus s.l. utilizing organellar genomes.
Materials and methods
Plant material, mitochondrial genome assembly and annotation
We obtained PacBio-HiFi data for Karpatiosorbus bristoliensis (ERR13245294) from the SRA (https://www.ncbi.nlm.nih.gov/sra/?term=ERR13245294). The whole-genome sequencing of K. bristoliensis revealed a total of 50.3G bases. For mitogenome assembly, we employed PMAT v2.1.2 (https://github.com/aiPGAB/PMAT2/releases) (Bi et al., 2024), an efficient plant mitogenome assembly toolkit using low-coverage HiFi sequencing data, to perform de novo assembly of the K. bristoliensis mitogenome with the command “PMAT autoMito -I -o -st -g -m”. For plastid genome assembly, the ptGAUL v1.0.5 pipeline (https://github.com/Bean061/ptgaul) (Zhou et al., 2023) was applied to extract and assemble the plastid genome from PacBio-HiFi long-read data with standard parameters. We employed Bandage (Wick et al., 2015) to visualize the graphical representation of the mitochondrial and plastid genomes produced by the PMAT and ptGAUL pipelines, respectively. Mitogenome annotation was performed via the online program Plant Mitochondrial Genome Annotator (PMGA) (http://47.96.249.172:16084/index.html), with the database set as 319 mitogenomes (Li et al., 2024). This database uses multiple sequence alignment to identify genes. The plastid genome annotation was performed via the online program CPGAVAS2 (http://47.96.249.172:16019/analyzer/home) (Shi et al., 2019), with Sorbus helenae (NC_068536.1) as the reference genome. Manual corrections were performed via Sequin software. We utilized OGDRAW (v.1.3.1) (https://chlorobox.mpimp-golm.mpg.de/ogdraw.html) to generate plastid genome and mitogenome maps.
Analysis of repeat sequences
The prediction of simple sequence repeats (SSRs) was performed using the online program MISA-web (https://www.web-blast.ipk-gatersleben.de/misa/). The parameters of mono-, di-, tri-, tetra-, penta-, and hexanucleotides were set to 10, 5, 4, 3, 3, and 3, respectively. Tandem repeats within the five species were detected using Tandem Repeats Finder v4.09 (Benson, 1999) with default parameters. Dispersed repeats were identified using the online REPuter program (https://bibiserv.cebitec.uni-bielefeld.de/reputer) (Kurtz, 2001) with a minimal repeat size of 30 bp and a Hamming distance of 3.
Homologous fragment analysis
we employed the online BLAST tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to search highly similar sequences between the mitogenome and plastid genomes of K. bristoliensis, M. alnifolia (Siebold & Zucc.) Koehne, T. glaberrima (Gand.) Sennikov & Kurtto, S. aucuparia L., and Sorbus aucuparia subsp. pohuashanensis (Hance) McAll., respectively. The identification parameters for homologous sequences were set to ≥70% minimum similarity and ≤1e-5 E-value threshold.
Detection of RNA editing events within Sorbus s.l.
We employed the online program PMGA (http://47.96.249.172:16084/deepredmt.html) to predict RNA editing events in the mitogenomes of K. bristoliensis, M. alnifolia, T. glaberrima, S. aucuparia, and S. aucuparia subsp. pohuashanensis. This online program uses Deepred-Mt (Edera et al., 2021), a novel deep convolutional neural network, to detect potential RNA editing events. The cut-off value was set to 0.5 for accurate prediction.
Nucleotide diversity analysis
For plastid genome analyses, plastid genome sequences from a total of 59 species of Sorbus s.l. were downloaded. We used the command “cpstools Pi -d work_dir” from the CPStools package (Huang et al., 2024b) to calculate Pi values for the analyzed species. This package automatically extracts shared gene regions and intergenic sequences from GenBank-format files in work_dir, performs multiple-sequence alignments, and computes Pi values. For mitogenome analyses, we utilized PhyloSuite v1.2.3 (Zhang et al., 2020) to extract protein-coding genes from the five Sorbus s.l. species using default parameters. A total of 35 shared protein-coding genes were aligned using MAFFT (https://mafft.cbrc.jp/alignment/server/) (Katoh et al., 2018) with the default parameters. The nucleotide diversity (Pi) of each gene was subsequently calculated using DnaSP v6 (Rozas et al., 2017) with the option “compute variance of Pi”.
Phylogenetic analysis
The mitochondrial phylogenomic tree was reconstructed via 14 mitochondrial genomes, including those of five Sorbus s.l. species (Sorbus aucuparia subsp. pohuashanensis ON478177, Sorbus aucuparia MT648825, Micromeles alnifolia PP506330, Torminalis glaberrima MT610102, and Karpatiosorbus bristoliensis), with Rosa rugosa (PQ474155) designated as the outgroup. We employed PhyloSuite v1.2.3 (Zhang et al., 2020) to extract protein-coding genes that were shared by the 14 species. Multiple sequence alignment was subsequently performed using MAFFT (https://mafft.cbrc.jp/alignment/server/) (Katoh et al., 2018) with default parameters. The aligned sequence trimming was performed using the trimAl program within the PhyloSuite v1.2.3 (Zhang et al., 2020) with the default (-automated1) parameter. We performed phylogenetic reconstruction using maximum likelihood method in IQ-TREE v2 (Minh et al., 2020), with 1,000 bootstrap replicates (-bb 1000) and 1,000 replicates for the SH-like approximate likelihood ratio test (SH-aLRT; -alrt 1000). The optimal nucleotide substitution model was determined using ModelFinder (Kalyaanamoorthy et al., 2017) in IQ-TREE v2, which identified K3Pu+F+R4 as the best-fit model based on the Bayesian Information Criterion (BIC).
A total of 65 plastomes, including those of 59 Sorbus s.l. species, were utilized for phylogenetic analyses (Supplementary Table S1). The whole plastid genomes were aligned with MAFFT (https://mafft.cbrc.jp/alignment/server/) (Katoh et al., 2018) with default parameter. Phylogenetic reconstruction employed identical software and parameters as above, with the TIM+F+R4 nucleotide substitution model specified according to the Bayesian Information Criterion (BIC)
Collinearity analysis
We performed pairwise sequence alignments among five species of the Sorbus s.l genus using MUMmer v4.0.1 (Marçais et al., 2018), with the parameters set as: nucmer –maxmatch -c 100 -b 500 -l 50. Filtering of aligned sequences was performed with the following parameters: a minimum alignment identity of 90% and a minimum alignment length of 50 bp. Identification of collinear sequences and structural rearrangement events among mitochondrial genomes was conducted using SyRI v1.7.0 (Goel et al., 2019). Genome structural visualization was generated using Plotsr v1.1.0 (Goel and Schneeberger, 2022).
Results
General features of Karpatiosorbus bristoliensis mitogenome and plastome
We employed the autoMito program in PMAT to assemble the mitochondrial genome of K. bristoliensis. The average depth of the assembled results was 1408.7x (Supplementary Figure S1). The mitochondrial genome of K. bristoliensis was 386,757 bp in size. It encoded 55 unique genes, including 34 protein-coding genes, 18 tRNA genes and 3 rRNA genes (Figure 1A). The GC content was 45.3%. Three genes (ccmFC, trnE-UUC, trnS-GCU) had one intron, one gene (nad4) had three introns, and four genes (nad1, nad2, nad5, nad7) had four introns (Supplementary Tables S2, S3).
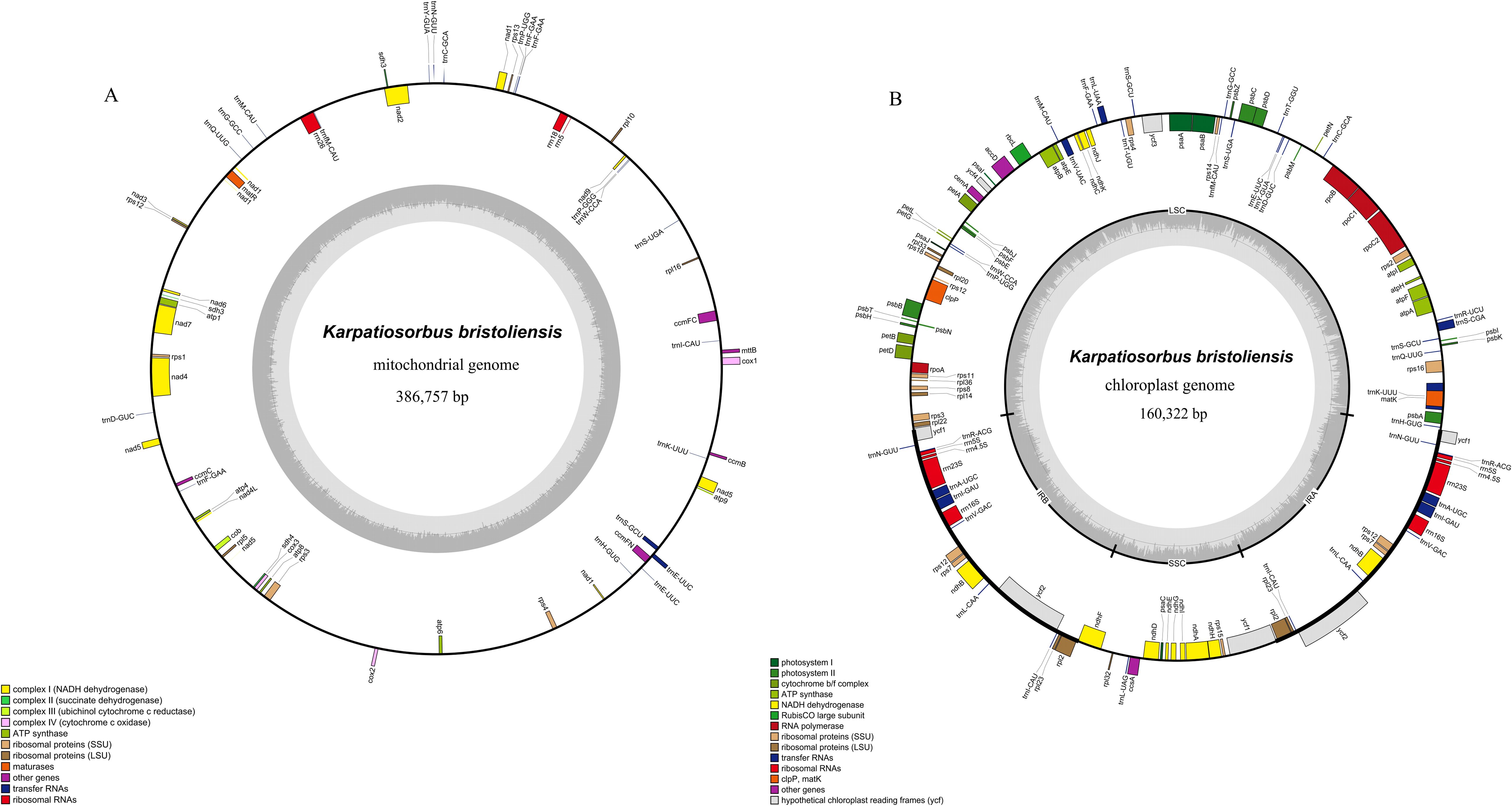
Figure 1. Mitochondrial and chloroplast genome diagram of K. bristoliensis. (A) mitogenome; (B) Plastome. Genes encoded on the forward strand are positioned on the outer circumference, while those on the reverse strand are located on the inner circumference. The gray inner ring represents the GC content.
The K. bristoliensis plastid genome has structures identical to those of most angiosperm plants, consisting of one large single copy (LSC), one small single copy (SSC), and two inverted repeats (IRs) (Figure 1B). The total length of the plastid genome was 160,322 bp, including 88,182 bp, 19,306 bp and 26,417 bp in the LSC, SSC and IR regions, respectively. The GC content was 36.50%. The plastid genome encodes 75 protein-coding genes, 29 tRNA genes, four rRNA genes, and one pseudogene (ycf1Ψ) for a total of 108 unique genes. Among these genes, eight genes (rps16, atpF, rpoC1, petB, petD, ndhB, rpl2, ndhA) have one intron, and two genes (ycf3, clpP) possess two introns (Supplementary Table S4).
Repeat sequence analyses
We detected 52, 53, 52, 48, and 51 simple sequence repeats (SSRs) in the mitochondrial genomes of K. bristoliensis, M. alnifolia, S. aucuparia subsp. pohuashanensis, S. aucuparia, and T. glaberrima, respectively (Figure 2A). All five species possess three types of SSRs: monomeric, dimeric, and trimeric repeats, with monomeric repeats being the most abundant. Notably, most SSRs were distributed in intergenic spacer regions, with fewer detected in protein-coding regions (Supplementary Table S5). Within protein-coding regions, SSRs primarily occurred in nad1, nad2, and nad3 genes (Figure 2B). We identified 50 dispersed repeats in each species, consisting of forward and palindromic repeats (Figure 2C). Repeat sequence lengths ranged from 91–428 bp (K. bristoliensis), 88–2745 bp (M. alnifolia), 76–26,130 bp (S. aucuparia subsp. pohuashanensis), 86–6452 bp (S. aucuparia), and 80–438 bp (T. glaberrima) (Figure 2C). Notably, S. aucuparia contained two large palindromic repeats (26,130 bp and 24,730 bp; Supplementary Table S6). A total of 24, 15, 22, 22, and 24 tandem repeats were identified in the mitochondrial genomes of K. bristoliensis, M. alnifolia, S. aucuparia subsp. pohuashanensis, S. aucuparia, and T. glaberrima, respectively (Figure 2D) (Supplementary Table S7).
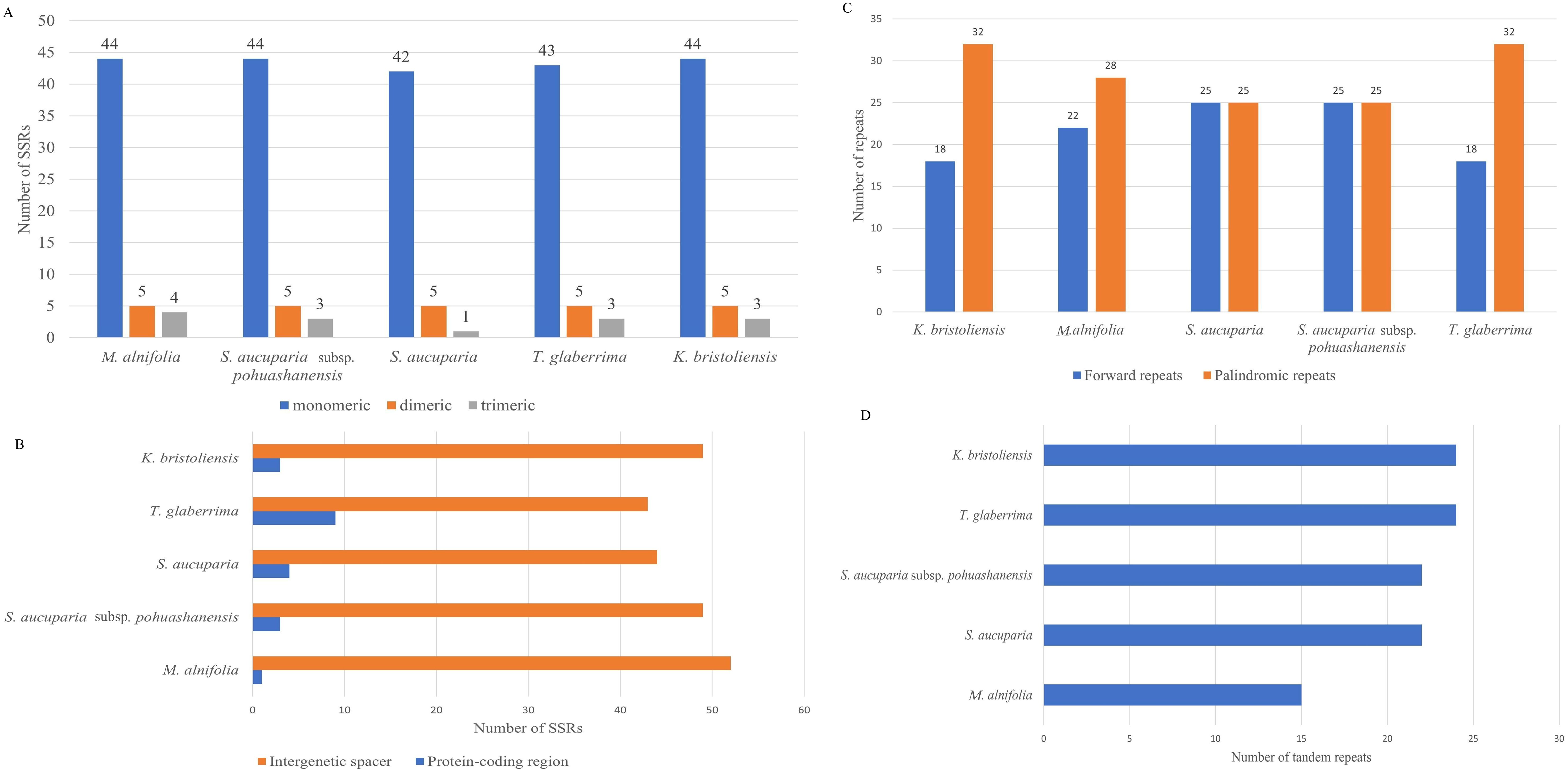
Figure 2. Distribution of repeat sequence in Sorbus s.l. mitogenome. (A) the number of SSRs in mitogenome; (B) distribution of SSRs in mitogenome; (C) the number of dispersed repeats; (D) the number of tandem repeats.
Analysis of inter-organellar gene transfer events
Mitochondrial genome analyses revealed 14, 13, 12, 12, and 14 chloroplast-derived DNA fragments in K. bristoliensis, M. alnifolia, T. glaberrima, S. aucuparia, and S. aucuparia subsp. pohuashanensis, respectively (Supplementary Table S8). The lengths of these transferred sequences exhibited limited variation, ranging from 3,846 bp (K. bristoliensis), 3,834 bp (M. alnifolia), 3,830 bp (T. glaberrima), 3,755 bp (S. aucuparia), and 3,021 bp (S. aucuparia subsp. pohuashanensis). Notably, the largest individual transferred fragment spanned 1,874 bp. Notably, K. bristoliensis, T. glaberrima, and S. aucuparia subsp. pohuashanensis each contained five fully transferred tRNA genes (trnW-CCA, trnD-GUC, trnH-GUG, trnN-GUU, and trnM-CAU). In contrast, M. alnifolia and S. aucuparia exhibited four completely transferred tRNA genes (trnW-CCA, trnH-GUG, trnD-GUC, and trnN-GTT). Furthermore, partial transfer events involving protein-coding genes were detected, including fragments of psbE, psbC, psaA, psbA, and atpA.
Prediction of RNA editing sites
A total of 442, 403, 474, 463, and 352 RNA editing sites were identified in the mitochondrial genomes of K. bristoliensis, M. alnifolia, S. aucuparia, S. aucuparia subsp. pohuashanensis, and T. glaberrima, respectively (Figure 3A). Comparative analysis revealed that three taxa (K. bristoliensis, S. aucuparia, and S. aucuparia subsp. pohuashanensis) each contained RNA editing modifications in 32 protein-coding genes, while M. alnifolia and T. glaberrima possessed 31 and 28 edited genes, respectively. Notably, nad4L exhibited the lowest frequency of RNA editing events among all protein-coding genes across the five species, each containing a single editing site. The nad4 gene demonstrated the highest RNA editing activity in four mitochondrial genomes (M. alnifolia, S. aucuparia, S. aucuparia subsp. pohuashanensis, and T. glaberrima), with 39 editing sites identified in M. alnifolia. In contrast, the K. bristoliensis mitochondrial genome showed unique editing patterns, where ccmB and ccmC collectively contained the highest number of RNA editing sites (31 total), representing a distinct regulatory feature in this species. All RNA editing sites belong to C to U. Notably, the start codons of cox1, nad1, rpl16, and rps4 in the mitochondrial genome of K. bristoliensis, through RNA editing, have changed from ACG to AUG. Additionally, RNA editing has contributed to stop codons for atp6 in the mitochondrial genomes of K. bristoliensis, S. aucuparia, and S. aucuparia subsp. pohuashanensis (Supplementary Table S9). The most frequent amino acid substitution was serine-to-leucine, except in M. alnifolia where proline-to-leucine predominated (Figure 3B).
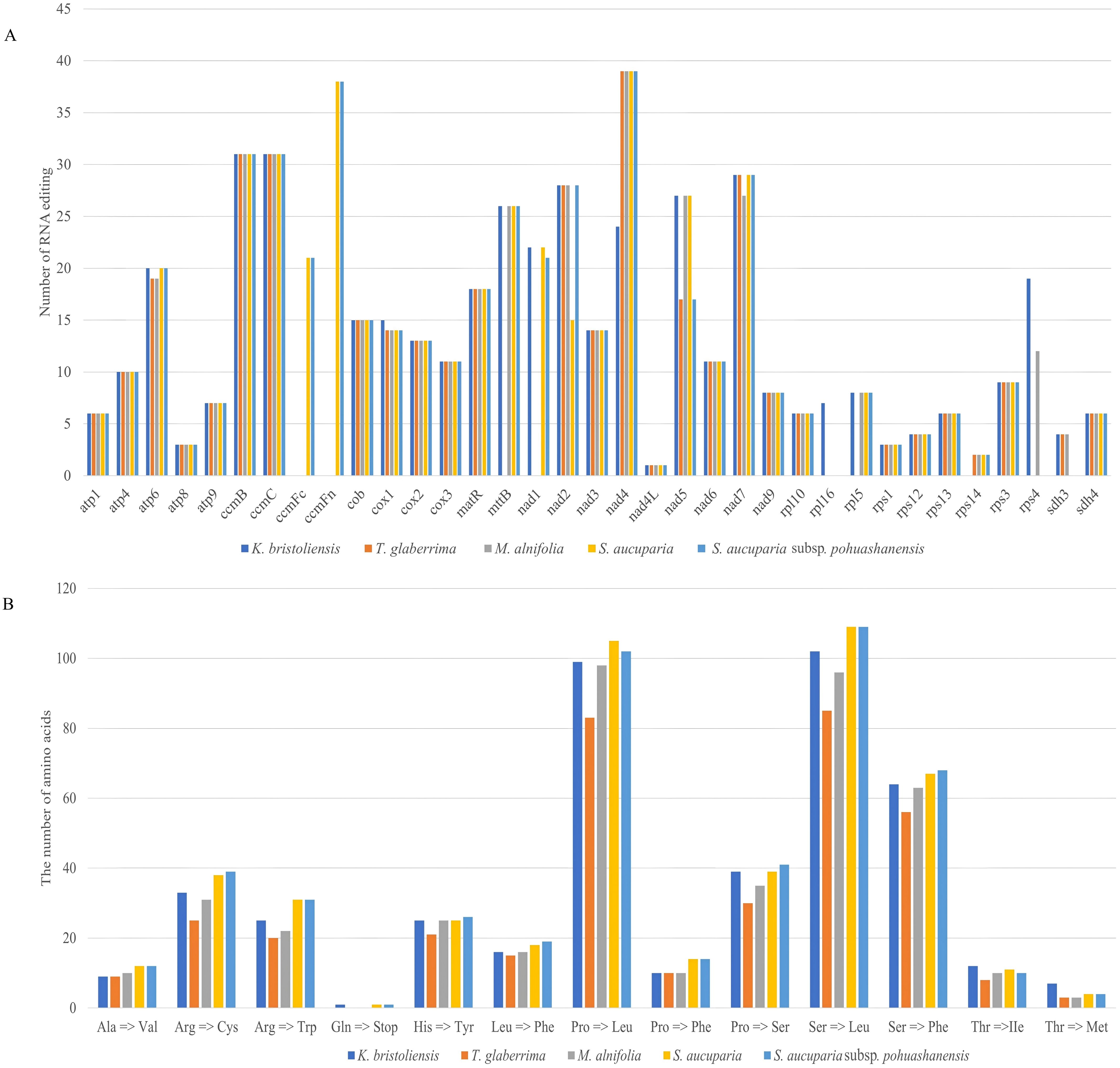
Figure 3. The number of RNA editing sites identified in the mitogenomes. (A) the number of RNA editing sites in Sorbus s.l. mitogenome; (B) number of types of RNA editing.
Nucleotide diversity analysis
Nucleotide diversity analysis was performed using DnaSP v6.0. For mitochondrial genomes, the Pi values ranged from 0 to 0.01808, with the nad4 gene exhibiting the highest diversity (0.01808) (Figure 4A). In plastid genomes, the Pi values showed greater conservation, spanning from 0 to 0.02672 (petG-trnW-CCA) (Figures 4B–D). Comparative analysis identified seven hypervariable regions as potential molecular markers. Seven regions with relatively high Pi values may be designed for DNA barcodes, including trnH-GUG-psbA, trnT-GUG-trnL-UAA, petG-trnW-CCA, ndhC-trnV-UAC-exon2, trnW-CCA-trnP-UGG, rpl33-rps18.
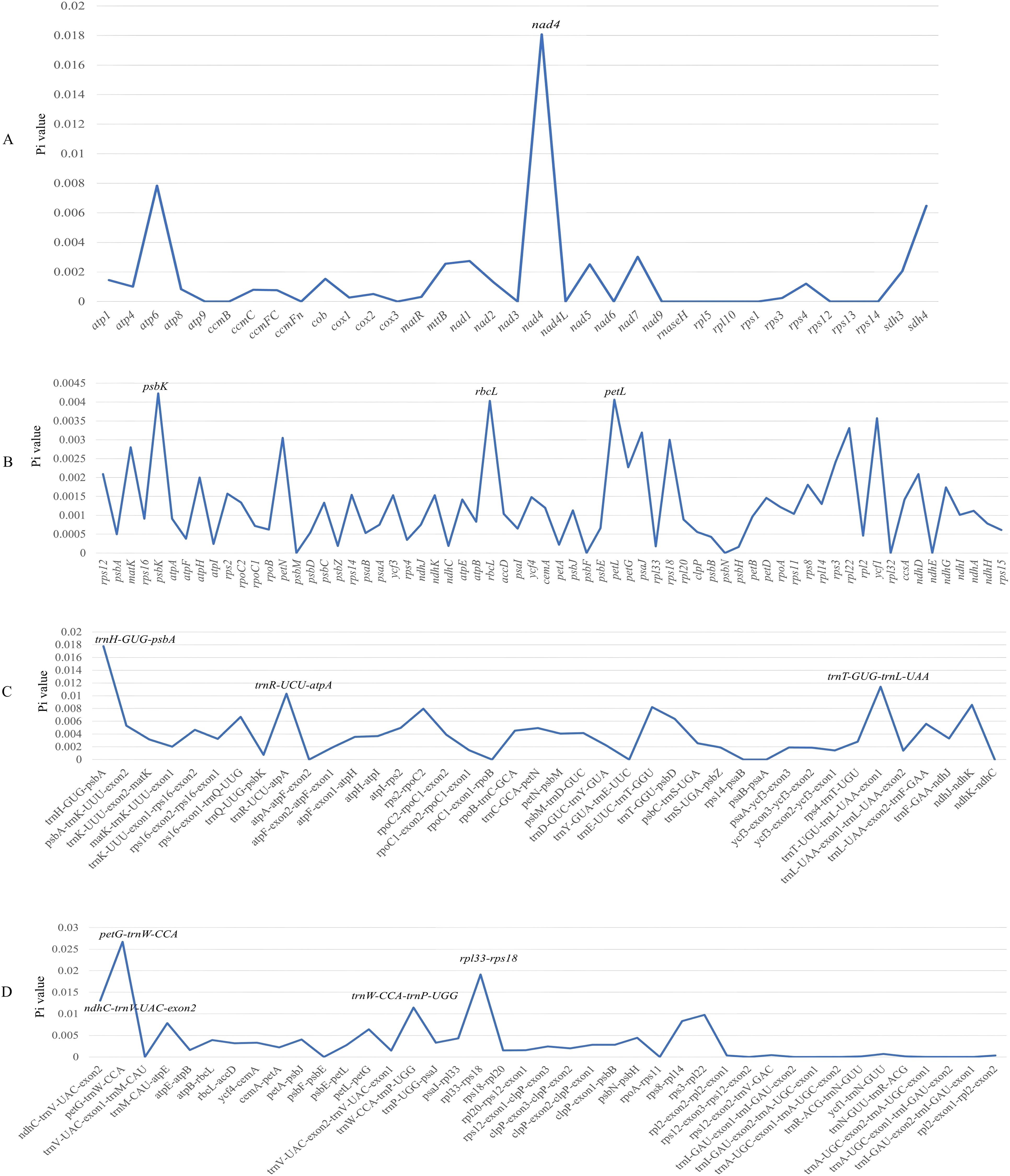
Figure 4. Nucleotide diversity analysis of Sorbus s.l. species. (A) mitogenome; (B–D) plastome.
Phylogenomic analyses
We recovered a plastome-based phylogeny of Sorbus s.l., which included an ingroup of 59 accessions (Figure 5). Our plastome-based phylogeny yielded strong bootstrap values (> 90%) for the vast majority of branches. With robust node support, we recovered eight major clades (A, B, C, D, E, F, G, H). Among these clades, Sorbus s.l. was dispersed in six clades. Clade A contains four genera (Chamaemespilus, Aria, Torminalis, and Karpatiosorbus) of Sorbus s.l. Karpatiosorbus, which are closely phylogenetically related to Torminalis. The genus Aria was found to be nonmonophyletic, with one species embedded in the genus Chamaemespilus. Clade C contains one species, Cormus domestica. Clade E corresponds to the genera Micromeles and Hedlundia. Micromeles are not monophyletic, with Hedlundia embedded in them. Clades F-H correspond to Sorbus s.s. and are expected to include the hybrid-origin genus Hedlundia and Scandosorbus.
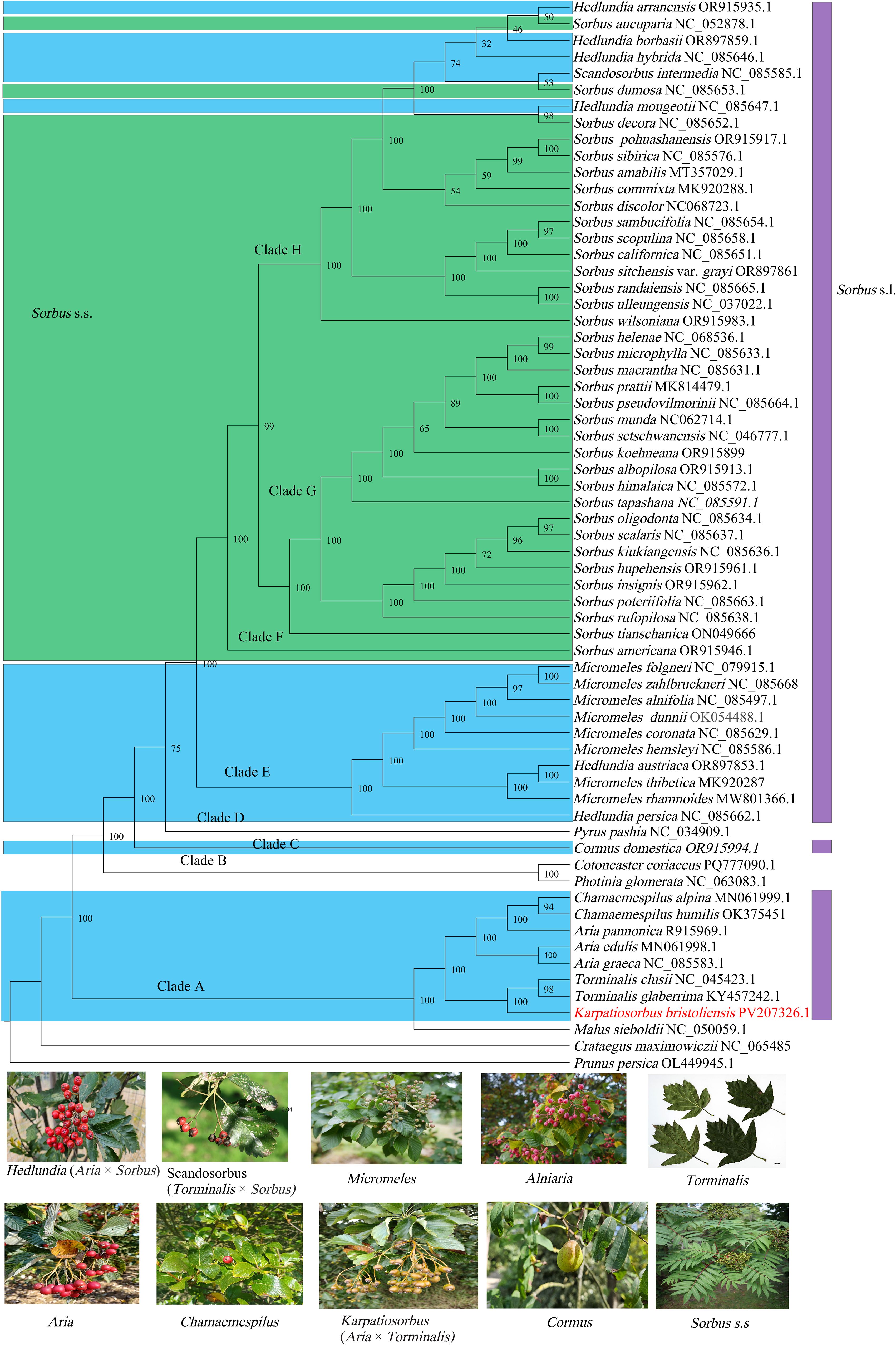
Figure 5. The maximum likelihood phylogeny reconstructed with IQ-TREE v2 displays bootstrap values on branches. Colors in the figure denote Sorbus s.l. lineages, with green designating Sorbus s.s. clades. Botanical images beneath the phylogeny represent constituent genera of Sorbus s.l. included in this study, with parenthetical notation indicating documented hybrid origins.
To better understand the mitochondrial genome evolution of Sorbus s.l., we also utilized the maximum likelihood method to construct mitochondrial genome-based phylogenetic trees based on 35 shared protein-coding genes (Figure 6). The Prunus genus was used as the outgroup. Most nodes of the phylogenetic tree presented relatively lower bootstrap values than those of the plastome-based phylogenetic tree did. Consistent with the plastome-based phylogeny, the genus Karpatiosorbus is a sister to Torminalis.
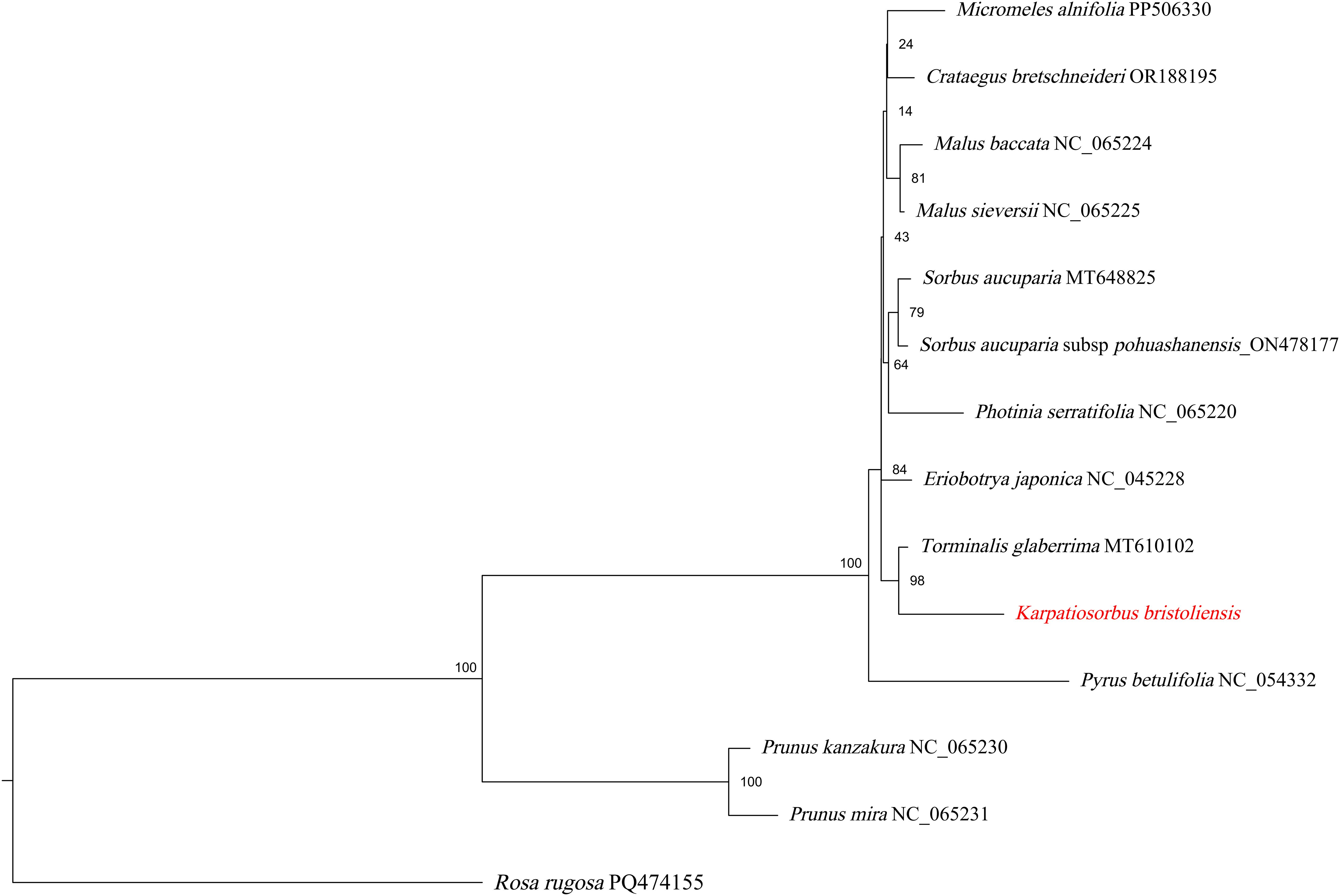
Figure 6. ML phylogeny (IQ-TREE v2; 34 shared protein-coding genes) showing bootstrap values on branches.
Analysis of mitogenome collinearity and structural rearrangement
Analysis of mitochondrial genome sequences and structures across five Sorbus species revealed extensive collinear regions, encompassing structural homology, translocations, and inversions (Figure 7). The longest sequence-similarity segments between species pairs were: 84,781 bp (M. alnifolia vs S. aucuparia), 97,089 bp (S. aucuparia vs S. aucuparia subsp. pohuashanensis), 229,635 bp (K. bristoliensis vs T. glaberrima), and 101,625 bp (T. glaberrima vs M. alnifolia). The highest degree of syntenic segment was observed between K. bristoliensis and T. glaberrima (310,482 bp), accounting for 80.28% of the K. bristoliensis mitochondrial genome length. Additionally, a 76,099 bp inversion structure was identified in the K. bristoliensis and T. glaberrima (Supplementary Table S10).

Figure 7. Collinearity Analysis of Mitochondrial Genomes in Five Sorbus s.l. Species. The gray arcs represent structurally homologous sequences, while other colors indicate structural rearrangements: inversions (green), transpositions (blue), and duplications (orange).
Discussion
Structure and size variation of chloroplast and mitochondrial genomes
This study reports the first complete mitochondrial and plastid genome assemblies for the endangered tree species K. bristoliensis. The circular mitochondrial genome spans 386,757 bp, demonstrating near-identical size conservation (1 bp difference) with its maternal progenitor Torminalis glaberrima (386,756 bp), consistent with its hybrid origin (Aria × Torminalis). The chloroplast genome of K. bristoliensis is 68 bp smaller than that of Torminalis glaberrima. Compared with those of other species within Sorbus s.l., the mitochondrial genome of K. bristoliensis was smaller than those of M. alnifolia (45,5361 bp) and S. aucuparia subsp. pohuashanensis (396,857 bp) but larger than that of Sorbus aucuparia (384,977 bp). Previous studies have reported that the most minor mitochondrial genome in Rosaceae is Taihangia rupestris var. ciliata T.T.Yu & C.L.Li, with a size of 265,633 bp (Li et al., 2025), and the largest one is Prunus mume (Siebold) Siebold & Zucc. (535,727 bp) (Sun et al., 2022). The mitochondrial genome size of Sorbus s.s. falls within the range of mitochondrial genome size variation observed in Rosaceae. The mitochondrial genome of K. bristoliensis exhibited a GC content of 45.3%, aligning closely with the conserved genomic composition observed across Rosaceae mitogenomes (range: 43.31%–45.62%); (Lu et al., 2023).
Characteristics of repetitive sequences in the mitochondrial genome of Sorbus s.l.
SSRs exhibit high polymorphism and codominant inheritance, thus frequently used in population genetics studies (Hu et al., 2019). This study identified a large number of SSRs within the mitochondrial genome of Sorbus s.l., which exhibit potential utility for population genetic investigations. The number of SSRs in the mitochondrial genome of Sorbus s.l. is comparable to that observed in other Rosaceae species such as Rubus idaeus L (Zhang et al., 2025), yet demonstrates a lower frequency compared to members of the Malus genus within the same family (Wang et al., 2024). Consistent with previous findings, monomeric repeats predominated among the three SSR motif categories, exhibiting the highest numerical abundance (Niu et al., 2024; Sun et al., 2024). Tandem repeats may contribute to mitochondrial genome size expansion, particularly in species with exceptionally large genomes (Wynn and Christensen, 2019). Some tandem repeats exhibit species-specific amplification, serving as molecular markers for phylogenetic studies. A total of 107 tandem repeats were detected among the five species, which will be helpful for future studies. Dispersed repeats are nonadjacent homologous sequences ranging from tens to thousands of base pairs and are often distributed throughout the mitochondrial genome. These repeats facilitate homologous recombination, a key driver of structural rearrangements such as inversions, duplications, and the generation of subgenomic circles (Wu and Sloan, 2019). Previous studies have demonstrated prevalent mitochondrial genome rearrangements in Rosaceae species, as exemplified by members of the genus Malus (Wang et al., 2024) and Fragaria (Fan et al., 2022). Notably, one large palindromic repeat (26,130 bp) and forward repeat (24,730 bp) were detected in the mitochondrial genome of S. aucuparia. In addition, the S. aucuparia subsp. pohuashanensis mitogenome also presented one forward repeat exceeding 6,000 bp (6452 bp). Whether these large repeats are involved in mitochondrial genome rearrangement and other structural evolution remains to be further studied.
Gene transfer between mitochondrial and chloroplast genomes
Horizontal gene transfer (HGT) from chloroplasts to mitochondrial genomes represents a notable phenomenon in plant organelle evolution, reflecting complex inter-organellar genomic interactions (Yang et al., 2022). Although chloroplast-to-mitochondrion gene transfers occur less frequently than chloroplast-to-nuclear transfers do, accumulating evidence reveals their evolutionary significance in mitogenome evolution (Hong et al., 2021). In this study, we identified some chloroplast fragments transferred to the mitochondrial genome, with sizes ranging from 3,021 bp to 3,856 bp. Compared with previous studies in other Rosaceae, such as Rubus idaeus L. (46,456 bp transferred from chloroplasts to mitochondria) (Zhang et al., 2025), we found that homologous fragment transfer between the plastome and mitogenome in Sorbus s.l. was relatively rare. These findings indicate that the mitochondrial and chloroplast genomes of Sorbus s.l. are relatively highly conserved. Consistent with previous studies (Wang et al., 2007), we also found that the most frequently transferred genes were tRNAs, with a total of five tRNA genes (trnW-CCA, trnD-GUC, trnH-GUG, trnN-GUU, and trnM-CAU) shared by the plastid genome and the mitochondrial genome.
RNA editing in Sorbus s.l. mitochondrial genome
RNA editing is frequently reported in plant mitochondrial genomes (Liu et al., 2024). It is a crucial posttranscriptional modification process that plays a crucial role in controlling gene expression and functionality (Liu et al., 2020). At present, the common type of RNA editing is C–U editing, and G-to-C and T-to-A editing can also occur (Liu et al., 2024; Ichinose and Sugita, 2016; Yang et al., 2022). In this study, all RNA editing sites in Sorbus s.l. were associated with C–U editing. The number of RNA editing sites differed greatly in Sorbus s.l., ranging from 352 (T. glaberrima) to 463 (S. aucuparia subsp. pohuashanensis). Compared with those in other Rosaceae species, the number of RNA editing sites in Sorbus s.l. species was greater than that in Prunus pedunculata (262) (Liu et al., 2023) but less than that in Rubus chingii var. suavissimus (492) (Shi et al., 2024) and Taihangia rupestris var. ciliata (470) (Li et al., 2025). Overall, RNA editing sites exhibit significant variations across different species and even within the same genus, such as S. aucuparia subsp. pohuashanensis and S. aucuparia. Notably, the RNA editing sites appear to exhibit a gene preference, with editing events occurring most frequently within the NADH dehydrogenase genes. Similar results have been reported in other species, such as Prunus pedunculata (Pall.) Maxim. (Liu et al., 2023) and Gelsemium elegans (Gardner & Champ.) Benth (You et al., 2023). We also observed that RNA editing influenced the start and termination codons of protein-coding genes. For example, the start codons of cox1, nad1, rpl16, and rps4 changed from ACG to AUG through RNA editing, whereas RNA editing led to the termination codons of atp6. RNA editing events that alter initiation and termination codons are frequently observed, as exemplified by modifications to the nad1 initiation codon and ccmFc termination codon in the mitochondrial genome of Garcinia mangostana L (Wee et al., 2022).
Capture of polymorphic loci
The mitochondrial and chloroplast genomes harbor a substantial number of polymorphic sequences that can be developed as DNA barcodes for phylogenetic and population genetic studies (Li and Wei, 2022). This study analyzed the mitochondrial genomes of five Sorbus s.l. species and revealed that the nad4 gene presented the greatest number of nucleotide polymorphisms. These findings suggest its potential utility as an effective DNA barcode for investigating phylogenetic relationships within Sorbus s.l. Through comparative analysis of chloroplast genomes, we identified six plastid gene regions exhibiting high genetic polymorphism, which could serve as chloroplast DNA barcodes for investigating phylogenetic relationships within Sorbus s.l. Furthermore, the ycf1 gene, conventionally employed as a universal barcode for angiosperms (Dong et al., 2015), demonstrated lower nucleotide polymorphism in this study.
Maternal lineage phylogenetic analysis of the Sorbus s.l.
In this study, we provide a high-resolution maternal genetic framework for Sorbus s.l. Aria pannonica (Kárpáti) Sennikov & Kurtto, which is a hybrid-origin species with a ploidy level of triploid (Somlyay and Sennikov, 2015). A. pannonica originated from intragenus hybridization because of its morphological similarity to A. nivea Host and A. graeca (Spach) M.Roem (Somlyay and Sennikov, 2015). However, A. pannonica is more closely related to the genus Chamaemespilus in our plastome-based phylogeny. Therefore, we inferred that the female parent of this hybrid species may have originated from Chamaemespilus. This fact indicated that Aria pannonica should treat as the member of Majovskya. The genus Karpatiosorbus is thought to originate from the hybridization of Aria and Torminalis (Pellicer et al., 2012). Both our mitogenome- and platome-based phylogenetic data supported that Karpatiosorbus and Torminalis clustered into one branch. These findings indicate that the female parent of this genus is Torminalis. The genus Hedlundia contains one sexual hybrid and 39 apomictic species and originates from crosses between various species of Aria and Sorbus s.l (Sennikov and Kurtto, 2017). The genus Hedlundia is separated into two clades: one clade is closely related to Micromeles, and the other clade is embedded in Sorbus s.s, indicating the complex origin of the hybrid genus. Sennikov and Kurtto (2017) proposed the establishment of the new nothogenus Sorbomeles to accommodate hybrids of Sorbus and Micromeles origin. In the present study, molecular evidence suggests Micromeles likely served as the maternal progenitor of Hedlundia austriaca (Beck) Sennikov & Kurtto and H. persica (Hedl.) Mezhenskyj. Consequently, these two taxa are hereby formally proposed for reclassification into the hybrid genus ×Sorbomeles. A previous study indicated that hybridization between Aria and Sorbus s.s. contributed to the genus Micromeles. Both our results and those of previous plastome-based studies support that Micromeles is a sister to Sorbus s.s., whereas this is inconsistent with nuclear phylogenetic analyses (Zhang et al., 2023). Therefore, hybridization occurs between Micromeles and Sorbus s.s. Scandosorbus intermedia (Ehrh.) Sennikov originates from hybridization among Sorbus s.s., Aria, and Torminalis (Pellicer et al., 2012). It involves a complex species formation process. In this study, S. intermedia was a sister to S. dumosa. These findings indicate that the female parent of this species is S. dumosa House or a related species.
Phylogenetic analyses have consistently recovered Sorbus s.s. as a monophyletic clade distinct from other simple-leaved genera and the compound-leaved genus Cormus in previous studies (Campbell et al., 2007; Zheng and Zhang, 2007; Potter et al., 2007; Lo and Donoghue, 2012). This limitation arises from inadequate sampling in prior studies, which excluded phylogenetically critical hybrid-origin taxa. However, our current phylogenomic reconstruction reveals that Hedlundia and Scandosorbus (simple-leaved hybrid-origin genera) nests within Sorbus s.s., thereby rendering the latter non-monophyletic. This phylogenetic incongruence results from the hybrid origin of Hedlundia and Scandosorbus, which inherited its maternal genome from ancestral Sorbus s.s. lineages. Sorbus s.s. is one of the most typical examples of taxonomic complexity arising from the combined effects of hybridization, polyploidy and apomixis in the Rosaceae (Robertson et al., 2010). Previous research has demonstrated that both insect-pollinated and bird-pollinated are present in the genus Sorbus s.s.; moreover, mammals also play an important role in the spread of Sorbus s.s. seeds (Raspé et al., 2000; Bednorz et al., 2006). Some species of this genus are self-incompatible (Raspé et al., 2000). These factors strongly increase the frequency of interspecific hybridization in Sorbus s.s. as well as hybrid with other genera. Therefore, elucidating taxonomic challenges in Sorbus s.s requires resolving hybridization patterns and clarifying phylogenetic placements of hybrid-origin taxa within the genus.
Frequent structural rearrangements of mitogenome
Mitochondrial genome structural complexity arises from extensive repetitive sequences and frequent structural rearrangements. Such mitochondrial genome rearrangements have also been frequently reported in other genera of Rosaceae, notably Prunus (Zhai et al., 2025) and Malus (Wang et al., 2024). This study revealed that K. bristoliensis shares substantial homologous segments with T. glaberrima (species from its maternal genus), while exhibiting limited homology with phylogenetically distant genera. This pattern aligns with their close phylogenetic origin and the hybrid origin of Karpatiosorbus. Despite the extensive shared homology, a 76,099 bp inversion structure was identified between their mitochondrial genomes, suggesting incipient mitogenomic divergence between the two genera.
Conclusions
In this study, we reported the first complete mitochondrial and plastid genomes of the endangered tree Karpatiosorbus bristoliensis utilizing PacBio-HiFi long reads. K. bristoliensis mitochondrial and plastid genomes consists of one circular chromosome structures with the length of 386,757 bp bp and 160,322 bp, respectively. We analyzed repeat sequences, RNA editing, inter-organellar gene transfer, and pi value in the mitochondrial genome of Sorbus s.l., enriching our knowledge of mitochondrial genome evolution of this genus. We also conducted phylogenetic analyses within Sorbus s.l. utilizing mitochondrial and plastid genome. We emphasize that resolving hybridization dynamics in Sorbus s.l. is critical for achieving a taxonomically coherent delineation of the genus. Overall, our study provided a high-resolution maternal framework for Sorbus s.l.
Data availability statement
The mitochondrial and plastid genome have been deposited in GenBank under the accession numbers: PV207325 and PV207326.
Author contributions
QL: Funding acquisition, Conceptualization, Investigation, Supervision, Writing – review & editing, Resources, Software, Writing – original draft, Project administration, Validation, Formal Analysis, Data curation, Methodology, Visualization. RW: Methodology, Writing – review & editing, Software, Visualization.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We are grateful to Dr Maarten J.M. Christenhusz for providing the raw data of Karpatiosorbus bristoliensis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1619267/full#supplementary-material
Supplementary Figure 1 | Bandage visual mitochondrial genome assembly map.
References
Bednorz, L., Myczko, L., and Kosiński, P. (2006). Genetic variability and structure of the wild service tree (Sorbus torminalis (L.) crantz) in Poland. Silvae Genet. 55, 197–202. doi: 10.1515/sg-2006-0027
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., and Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of populus simonii. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.914635
Bi, C., Shen, F., Han, F., Qu, Y., Hou, J., Xu, K., et al. (2024). PMAT: an efficient plant mitogenome assembly toolkit using low-coverage HiFi sequencing data. Hortic. Res. 11, uhae023. doi: 10.1093/hr/uhae023
Campbell, C. S., Evans, R. C., Morgan, D. R., Dickinson, T. A., and Arsenault, M. P. (2007). Phylogeny of subtribe Pyrinae (formerly the Maloideae, Rosaceae): Limited resolution of a complex evolutionary history. Plant Syst. Evol. 266, 119–145. doi: 10.1007/s00606-007-0545-y
Chester, M., Cowan, R. S., Fay, M. F., and Rich, T. C. G. (2007). Parentage of endemic Sorbus L. (Rosaceae) species in the British Isles: Evidence from plastid DNA. Bot. J. Linn. Soc 154, 291–304. doi: 10.1111/j.1095-8339.2007.00669.x
Dong, W., Xu, C., Li, C., Sun, J., Zuo, Y., Shi, S., et al. (2015). ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 5, 8348. doi: 10.1038/srep08348
Edera, A. A., Small, I., Milone, D. H., and Sanchez-Puerta, M. V. (2021). Deepred-Mt: Deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 136, 104682. doi: 10.1016/j.compbiomed.2021.104682
Fan, W., Liu, F., Jia, Q., Du, H., Chen, W., Ruan, J., et al. (2022). Fragaria mitogenomes evolve rapidly in structure but slowly in sequence and incur frequent multinucleotide mutations mediated by microinversions. New Phytol. 236, 745–759. doi: 10.1111/nph.18334
Goel, M. and Schneeberger, K. (2022). plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics 38, 2922–2926. doi: 10.1093/bioinformatics/btac196
Goel, M., Sun, H., Jiao, W.-B., and Schneeberger, K. (2019). SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20, 277. doi: 10.1186/s13059-019-1911-0
Gualberto, J. M. and Newton, K. J. (2017). Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 68, 225–252. doi: 10.1146/annurev-arplant-043015-112232
Hong, Z., Liao, X., Ye, Y., Zhang, N., Yang, Z., Zhu, W., et al. (2021). A complete mitochondrial genome for fragrant Chinese rosewood (Dalbergia odorifera, Fabaceae) with comparative analyses of genome structure and intergenomic sequence transfers. BMC Genomics 22, 672. doi: 10.1186/s12864-021-07967-7
Hu, Y., Wang, X., Zhang, X., Zhou, W., Chen, X., and Hu, X. (2019). Advancing phylogeography with chloroplast DNA markers. Biodivers. Sci. 27, 219–234. doi: 10.17520/biods.2018319
Huang, K., Xu, W., Hu, H., Jiang, X., Sun, L., Zhao, W., et al. (2024a). The Mitochondrial Genome of Cathaya argyrophylla Reaches 18. 99 Mb: Analysis of Super-Large Mitochondrial Genomes in Pinaceae. arXiv. Available online at: https://arxiv.org/pdf/2410.07006.
Huang, L., Yu, H., Wang, Z., and Xu, W. (2024b). CPStools: A package for analyzing chloroplast genome sequences. iMetaOmics 1, e25. doi: 10.1002/imo2.25
Ichinose, M. and Sugita, M. (2016). RNA editing and its molecular mechanism in plant organelles. Genes (Basel). 8, 5. doi: 10.3390/genes8010005
IUCN (2001). IUCN Red List Categories and Criteria: Version 3.1 (Gland, Switzerland: IUCN Species Survival Commission). Available online at: https://portals.iucn.org/library/sites/library/files/documents/RL-2001-001.pdf (Accessed February 9, 2000).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., Rozewicki, J., and Yamada, K. D. (2018). MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kurtz, S. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Li, J., Ni, Y., Lu, Q., Chen, H., and Liu, C. (2024). PMGA: A plant mitochondrial genome annotator. Plant Commun. 6, 101191. doi: 10.1016/j.xplc.2024.101191
Li, Z.-Z., Wang, Y., He, X.-Y., and Li, W.-G. (2025). The Taihangia mitogenome provides new insights into its adaptation and organelle genome evolution in Rosaceae. Planta 261, 59. doi: 10.1007/s00425-025-04629-w
Li, Q. and Wei, R. (2022). Comparison of boraginales plastomes: insights into codon usage bias, adaptive evolution, and phylogenetic relationships. Diversity 14, 1104. doi: 10.3390/d14121104
Lin, D., Shao, B., Gao, Z., Li, J., Li, Z., Li, T., et al. (2025). Phylogenomics of angiosperms based on mitochondrial genes: insights into deep node relationships. BMC Biol. 23, 45. doi: 10.1186/s12915-025-02135-9
Linnaeus, C. (1753). Species plantarum Vol. 2 (Stockholm: impensis Laurentii Salvii). Holmiae. doi: 10.5962/bhl.title.669
Liu, R., Cao, S.-K., Sayyed, A., Yang, H.-H., Zhao, J., Wang, X., et al. (2020). The DYW-subgroup pentatricopeptide repeat protein PPR27 interacts with ZmMORF1 to facilitate mitochondrial RNA editing and seed development in maize. J. Exp. Bot. 71, 5495–5505. doi: 10.1093/jxb/eraa273
Liu, Q., Wu, Z., Tian, C., Yang, Y., Liu, L., Feng, Y., et al. (2023). Complete mitochondrial genome of the endangered Prunus pedunculata (Prunoideae, Rosaceae) in China: characterization and phylogenetic analysis. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1266797
Liu, G. H., Zuo, Y. W., Shan, Y., Yu, J., Li, J. X., Chen, Y., et al. (2024). Structural analysis of the mitochondrial genome of Santalum album reveals a complex branched configuration. Genomics 116, 110935. doi: 10.1016/j.ygeno.2024.110935
Lo, E. Y. Y. and Donoghue, M. J. (2012). Expanded phylogenetic and dating analyses of the apples and their relatives (Pyreae, Rosaceae). Mol. Phylogenet. Evol. 63, 230–243. doi: 10.1016/j.ympev.2011.10.005
Lu, L. and Stephen, A. ,. S. (2003). Sorbus L. Eds. Wu, Z. Y., Raven, P. H., and Hong, D. Y.Flora of China (SaintLouis: Science Press, Beijing & Missouri Botanical Garden Press), 144–170.
Lu, G., Zhang, K., Que, Y., and Li, Y. (2023). Assembly and analysis of the first complete mitochondrial genome of Punica granatum and the gene transfer from chloroplast genome. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1132551
Lu, G., Zhang, C., Wu, Q., Sun, T., Yang, S., Wei, E., et al. (2025). Multiple chromosomal configurations and phylogenetic implications in Saccharum. J. Integr. Agric. doi: 10.1016/j.jia.2025.02.018
Marçais, G., Delcher, A. L., Phillippy, A. M., Coston, R., Salzberg, S. L., and Zimin, A. (2018). MUMmer4: A fast and versatile genome alignment system. PloS Comput. Biol. 14, e1005944. doi: 10.1371/journal.pcbi.1005944
McAllister, H. (2005). The genus Sorbus Mountain Ash and other Rowans (London: Royal Botanical Gardens).
Mezhenska, L., Mezhenskyj, V., and Yakubenko, B. (2018). NULESU Collections of Fruit and Ornamental Plants (Kiev: Lira-K). КОЛЕКЦІЯ НУБІП УКРАЇНИ ПЛОДОВИХ І ДЕКОРАТИВНИХ РОСЛИН.
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Morgante, M., Hanafey, M., and Powell, W. (2002). Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 30, 194–200. doi: 10.1038/ng822
Nelson-Jones, E. B., Briggs, D., and Smith, A. G. (2002). The origin of intermediate species of the genus Sorbus. Theor. Appl. Genet. 105, 953–963. doi: 10.1007/s00122-002-0957-6
Németh, C., Papp, N., Nosková, J., and Höhn, M. (2020). Speciation by triparental hybridization in genus Sorbus (Rosaceae). Biol. Futur. 71, 209–222. doi: 10.1007/s42977-020-00003-x
Niu, Y., Gao, C., and Liu, J. (2024). Mitochondrial genome variation and intergenomic sequence transfers in Hevea species. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1234643
Pellicer, J., Clermont, S., Houston, L., Rich, T. C. G., and Fay, M. F. (2012). Cytotype diversity in the Sorbus complex (Rosaceae) in Britain: Sorting out the puzzle. Ann. Bot. 110, 1185–1193. doi: 10.1093/aob/mcs185
Phipps, J. B., Robertson, K. R., Smith, P. G., and Rohrer, J. R. (1990). A checklist of the subfamily Maloideae (Rosaceae). Can. J. Bot. 68, 2209–2269. doi: 10.1139/b90-288
Potter, D., Eriksson, T., Evans, R. C., Oh, S., Smedmark, J. E. E., Morgan, D. R., et al. (2007). Phylogeny and classification of rosaceae. Plant Syst. Evol. 266, 5–43. doi: 10.1007/s00606-007-0539-9
Raspé, O., Saumitou-Laprade, P., Cuguen, J., and Jacquemart, A. L. (2000). Chloroplast DNA haplotype variation and population differentiation in Sorbus aucuparia L. (Rosaceae: Maloideae). Mol. Ecol. 9, 1113–1122. doi: 10.1046/j.1365-294X.2000.00977.x
Rich, T. C. G., Green, D., Houston, L., Lepší, M., Ludwig, S., and Pellicer, J. (2014). British Sorbus (Rosaceae): six new species, two hybrids and a new subgenus. New J. Bot. 4, 2–12. doi: 10.1179/2042349714Y.0000000036
Rich, T. C. G., Houston, L., Wray, N., King, C., Brown, A. P., and Fay, M. F. (2022). 1049. SORBUS BRISTOLIENSIS: rosaceae. Curtis’s Bot. Mag. 39, 737–751. doi: 10.1111/curt.12483
Robertson, A., Newton, A. C., and Ennos, R. A. (2004). Multiple hybrid origins, genetic diversity and population genetic structure of two endemic Sorbus taxa on the Isle of Arran, Scotland. Mol. Ecol. 13, 123–134. doi: 10.1046/j.1365-294X.2003.02025.x
Robertson, A., Rich, T. C. G., Allen, A. M., Houston, L., Roberts, C., Bridle, J. R., et al. (2010). Hybridization and polyploidy as drivers of continuing evolution and speciation in Sorbus. Mol. Ecol. 19, 1675–1690. doi: 10.1111/j.1365-294X.2010.04585.x
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sennikov, A. N. and Kurtto, A. (2017). A phylogenetic checklist of Sorbus s.l. (Rosaceae) in Europe. Memo. Soc pro Fauna Flora Fenn. 93, 1–78.
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi: 10.1093/nar/gkz345
Shi, Y., Chen, Z., Jiang, J., Wu, W., Xin, Y., and Zeng, W. (2024). Assembly and comparative analysis of the complete mitogenome of Rubus chingii var. suavissimus, an exceptional berry plant possessing sweet leaves. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1504687
Skippington, E., Barkman, T. J., Rice, D. W., and Palmer, J. D. (2015). Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. 112, E3515–E3524. doi: 10.1073/pnas.1504491112
Somlyay, L. and Sennikov, A. N. (2015). Taxonomic Interpretation and Typification of Sorbus pannonica (Rosaceae), a Presumed Intermediate between S. aria and S. graeca from Hungary. Ann. Bot. Fenn. 52, 274–287. doi: 10.5735/085.052.0322
Sun, M., Zhang, M., Chen, X., Liu, Y., Liu, B., Li, J., et al. (2022). Rearrangement and domestication as drivers of Rosaceae mitogenome plasticity. BMC Biol. 20, 1–19. doi: 10.1186/s12915-022-01383-3
Sun, N., Han, F., Wang, S., Shen, F., Liu, W., Fan, W., et al. (2024). Comprehensive analysis of the Lycopodium japonicum mitogenome reveals abundant tRNA genes and cis-spliced introns in Lycopodiaceae species. Front. Plant Sci. 15, 1446015. doi: 10.3389/fpls.2024.1446015
Wang, S., Qiu, J., Sun, N., Han, F., Wang, Z., Yang, Y., et al. (2025). Characterization and comparative analysis of the first mitochondrial genome of Michelia (Magnoliaceae). Genomics Commun. 2, e001. doi: 10.48130/gcomm-0025-0001
Wang, X., Wang, D., Zhang, R., Qin, X., Shen, X., and You, C. (2024). Morphological Structure Identification, Comparative Mitochondrial Genomics and Population Genetic Analysis toward Exploring Interspecific Variations and Phylogenetic Implications of Malus baccata ‘ZA’ and Other Species. Biomolecules 14, 912. doi: 10.3390/biom14080912
Wang, D., Wu, Y.-W., Shih, A. C.-C., Wu, C.-S., Wang, Y.-N., and Chaw, S.-M. (2007). Transfer of chloroplast genomic DNA to mitochondrial genome occurred at least 300 MYA. Mol. Biol. Evol. 24, 2040–2048. doi: 10.1093/molbev/msm133
Wee, C.-C., Nor Muhammad, N. A., Subbiah, V. K., Arita, M., Nakamura, Y., and Goh, H.-H. (2022). Plastomes of Garcinia mangostana L. and comparative analysis with other Garcinia species. Plants 12, 930. doi: 10.3390/plants12040930
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Z., Liao, X., Zhang, X., Tembrock, L. R., and Broz, A. (2022). Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Syst. Evol. 60, 160–168. doi: 10.1111/jse.12655
Wu, Z. and Sloan, D. B. (2019). Recombination and intraspecific polymorphism for the presence and absence of entire chromosomes in mitochondrial genomes. Heredity (Edinb). 122, 647–659. doi: 10.1038/s41437-018-0153-3
Wu, Y., Yu, X., Tang, W., Yang, W., Fu, Q., Zheng, Y., et al. (2024). Morphological and molecular evidence for natural hybridization between Sorbus pohuashanensis and S. discolor (Rosaceae). J. For. Res. 35, 1–13. doi: 10.1007/s11676-023-01659-6
Wynn, E. L. and Christensen, A. C. (2019). Repeats of unusual size in plant mitochondrial genomes: identification, incidence and evolution. G3 Genes|Genomes|Genetics 9, 549–559. doi: 10.1534/g3.118.200948
Yang, Z., Ni, Y., Lin, Z., Yang, L., Chen, G., Nijiati, N., et al. (2022). De novo assembly of the complete mitochondrial genome of sweet potato (Ipomoea batatas [L.] Lam) revealed the existence of homologous conformations generated by the repeat-mediated recombination. BMC Plant Biol. 22, 1–12. doi: 10.1186/s12870-022-03665-y
You, C., Cui, T., Zhang, C., Zang, S., Su, Y., and Que, Y. (2022). Assembly of the Complete Mitochondrial Genome of Gelsemium elegans Revealed the Existence of Homologous Conformations Generated by a Repeat Mediated Recombination. Int. J. Mol. Sci. 24, 527. doi: 10.3390/ijms24010527
Yü, T. and Lu, L. (1974). “Sorbus L,” in Flora Reipublicae Popularis Sinicae, vol. 36 . Ed. Yü, T. T. (Science Press, Beijing), 283–344.
Zhai, T., Zhao, Z., Fu, C., Huang, L., Jiang, C., Li, M., et al. (2025). De novo assembly and comparative analysis of cherry (Prunus subgenus Cerasus) mitogenomes. Front. Plant Sci. 16. doi: 10.3389/fpls.2025.1568698
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, L., Morales-Briones, D. F., Li, Y., Zhang, G., Zhang, T., Huang, C. H., et al. (2023). Phylogenomics insights into gene evolution, rapid species diversification, and morphological innovation of the apple tribe (Maleae, Rosaceae). New Phytol. 240, 2102–2120. doi: 10.1111/nph.19175
Zhang, Q.-J., Wu, X.-Y., Wang, X., Yang, A.-S., Zhang, X.-Y., Zhao, W.-M., et al. (2024). Evolutionary history and population dynamics of a rare and endangered medicinal plant Bergenia scopulosa (Saxifragaceae): Evidences from chloroplast genomes and ecological niche analysis. Glob. Ecol. Conserv. 54, e03097. doi: 10.1016/j.gecco.2024.e03097
Zhang, H., Yan, M., Li, L., Jiang, Z., Xiong, Y., Wang, Y., et al. (2025). Assembly and comparative analysis of the complete mitochondrial genome of red raspberry (Rubus idaeus L.) revealing repeat-mediated recombination and gene transfer. BMC Plant Biol. 25, 85. doi: 10.1186/s12870-024-05969-7
Zheng, D. and Zhang, M. (2007). A cladistic and phenetic analysis of the infrageneric relationships of Sorbus s.l. (Maloideae, Rosaceae) based on the morphological characters. Acta Hortic. Sinnica 34, 723–728.
Keywords: Karpatiosorbus bristoliensis, Sorbus s.l., mitochondrial genome, RNA editing events, gene transfer, phylogenetic analysis
Citation: Li Q and Wei R (2025) Complete plastome and mitogenome assembly of endangered tree Karpatiosorbus bristoliensis reveals phylogenetic architecture for Sorbus sensu lato (Rosaceae). Front. Plant Sci. 16:1619267. doi: 10.3389/fpls.2025.1619267
Received: 27 April 2025; Accepted: 09 June 2025;
Published: 08 July 2025.
Edited by:
Zhiqiang Wu, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Zhechen Qi, Zhejiang Sci-Tech University, ChinaGuilong Lu, Henan Institute of Science and Technology, China
Luan Rabelo, Vale Technological Institute (ITV), Brazil
Copyright © 2025 Li and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Li, bGlxaWFuZzk1MDNAbmpmdS5lZHUuY24=