 Huang Jin-Liang1†
Huang Jin-Liang1† Yu-Jie Shi2†
Yu-Jie Shi2† Fei-Fei Tian1
Fei-Fei Tian1 Jia-Xuan Mi3
Jia-Xuan Mi3 Fan Zhang4
Fan Zhang4 Shi-Xing Zhou1,5,6
Shi-Xing Zhou1,5,6 Xue-Qin Wan1*
Xue-Qin Wan1* Cong-De Huang1,5,6*
Cong-De Huang1,5,6*- 1College of Forestry, Sichuan Agricultural University, Chengdu, China
- 2College of Life Sciences, Zhejiang Provincial Key Laboratory of Plant Evolutionary Ecology and Conservation, Taizhou University, Taizhou, China
- 3College of Forestry, Southwest Forestry University, Kunming, China
- 4College of Landscape Architecture, Sichuan Agricultural University, Chengdu, China
- 5Sichuan Province Key Laboratory of Ecological Forestry Engineering on the Upper Reaches of the Yangtze River, Ya’an, Sichuan, China
- 6Sichuan Mt. Emei Forest Ecosystem National Observation and Research Station, Sichuan Agricultural University, Chengdu, China
Populus kangdingensis and P. ciliata are important tree species distributed across the Qinghai–Tibet Plateau, yet the structure and evolutionary characteristics of their mitochondrial genomes remain unclear. To elucidate these features and their phylogenetic relationships, we assembled the mitochondrial genomes of these two species using PacBio HiFi sequencing data with PMAT2, and annotated them with PMGA. The results showed that the mitochondrial genome of P. kangdingensis consists of three independent circular molecules with a total length of 785,824 bp, while that of P. ciliata exhibits a branched structure comprising two circular molecules and one linear molecule, totaling 798,807 bp. Both genomes contain 57 functional genes, including 34 highly conserved protein-coding genes. Comparative genomic analyses revealed differences in repeat sequences, RNA editing patterns, and chloroplast-derived sequences, suggesting species-specific evolution at the organellar genome level. Ka/Ks analysis identified genes such as atp4, ccmB, and mttB as potentially under positive selection, reflecting adaptation to high-altitude environments. Phylogenetic trees constructed using 30 mitochondrial and 58 chloroplast protein-coding genes confirmed the monophyly of Populus and Salix, and identified them as sister groups. However, topological inconsistencies observed within Populus suggest the influence of lineage sorting, hybridization, and differences in mutation rates. This study provides the first high-quality mitochondrial genomes of P. kangdingensis and P. ciliata, revealing the structural diversity of multi-circular and branched conformations in Populus mitochondrial genomes, and offering new insights into organellar genome evolution and high-altitude adaptation in this genus.
1 Introduction
Mitochondria are essential organelles in eukaryotic cells responsible for aerobic respiration and energy production. Beyond generating ATP through oxidative phosphorylation, they are also involved in critical physiological processes such as programmed cell death, metabolic regulation, cytoplasmic male sterility, and signal transduction (Chandel, 2014; Kim and Zhang, 2018). Compared with animal mitochondria, plant mitogenomes exhibit remarkable structural diversity and evolutionary complexity, characterized by large genome sizes, low coding density, frequent structural rearrangements, abundant repetitive sequences, and extensive incorporation of chloroplast and nuclear DNA fragments (Sloan et al., 2012; Petersen et al., 2015; Wynn and Christensen, 2019). Traditionally, plant mitochondrial genomes have been portrayed as single circular molecules. However, with the advent of high-throughput and long-read sequencing technologies, an increasing number of species have been found to possess multipartite, linear, or branched mitochondrial genome conformations (Alverson et al., 2011; Burger et al., 2012). These structural variations highlight the highly dynamic nature of plant mitochondrial genomes and provide important clues for exploring recombination mechanisms, replication regulation, and species evolution (Palmer et al., 2000; Rice et al., 2013; Sanchez-Puerta et al., 2017).
Populus is a genus of fast-growing deciduous trees with significant ecological, breeding, and molecular research value (Tuskan et al., 2006). In recent years, mitogenomes of several Populus species have been reported, including P. tremula, P. alba, P. davidiana, P. adenopoda, and P. simonii, with genome sizes ranging from 772 to 869 kb. These genomes typically contain over 30 protein-coding genes (PCGs), more than 20 tRNA genes, and 3 rRNA genes. Previous genome assemblies based on next-generation and long-read sequencing platforms, including Illumina, Ion Torrent, and PacBio—such as those for P. tremula (Kersten et al., 2016), P. alba (Brenner et al., 2019), and P. davidiana (Choi et al., 2017)—revealed a single linear mitochondrial genome structure, likely due to the relatively low coverage of long-read sequencing. In contrast, more recent assemblies using third-generation sequencing data, including those of P. trichocarpa, P. simonii (Bi et al., 2022), and P. deltoides (Qu et al., 2023), have revealed multipartite mitochondrial genomes, challenging the previously resolved structures. The species P. kangdingensis and P. ciliata are important montane Populus species native to the Qinghai–Tibet Plateau, exhibiting notable adaptations to extreme high-altitude environments such as low oxygen and high UV radiation (Shi et al., 2023, 2024b). However, to date, there has been no comprehensive study on the mitogenomes of these two species. Given the unique advantages of mitogenomes in maternal inheritance, species differentiation, and phylogenetic reconstruction, sequencing and comparative analysis of the mitogenomes of P. kangdingensis and P. ciliata will deepen our understanding of their adaptive evolution in plateau environments and fill a knowledge gap in Populus mitochondrial genomics.
In this study, we utilized PacBio HiFi long-read sequencing to assemble and annotate high-quality mitogenomes for these two species for the first time. We characterized their structural configurations, number of genes, RNA editing sites, codon usage bias, and repeat sequence distributions, compared them with mitogenomes of eight other Populus species, and conducted phylogenetic analyses to infer their evolutionary relationships. This study not only extends the known structural diversity of Populus mitogenomes to include linear conformations, but also provides foundational data and theoretical insights for understanding mitogenome evolution, multi-source DNA transfer, and lineage divergence in the genus Populus.
2 Materials and methods
2.1 Sample collection and sequencing data acquisition
Branches of P. kangdingensis and P. ciliata were collected from the Poplar Germplasm Repository at the College of Forestry, Sichuan Agricultural University (103°38′42″E, 30°33′97″N), and hydroponically acclimated in a controlled climate chamber for six weeks (He et al., 2025). Fresh young leaves were subsequently harvested, flash-frozen in liquid nitrogen, and stored at –80°C until use. Genomic DNA was extracted using a modified CTAB protocol (He et al., 2024), and DNA integrity and purity were assessed with a NanoDrop spectrophotometer and a Qubit v4 fluorometer (Invitrogen). High-quality samples (OD260/280 between 1.8 and 2.0, OD260/230 > 2.0, with distinct main bands and no observable degradation) were selected for library preparation. Single Molecule Real-Time (SMRT) sequencing was performed on the PacBio Sequel II platform. Raw reads were processed to remove adapter sequences and filtered for quality. Circular consensus sequences (HiFi reads) were then generated using the CCS software.
2.2 Mitochondrial genome assembly and annotation
The mitochondrial genomes were assembled using PMAT2 (Bi et al., 2024), generating initial assembly graphs. The assembly was visualized using Bandage v0.9.0 (Wick et al., 2015). Contigs suspected to be chloroplast- or nuclear-derived were first identified based on their distinctively higher or lower read coverage in the assembly graph, and then aligned against reference chloroplast sequences for confirmation. These non-mitochondrial contigs were subsequently removed from the assembly. The mitochondrial genome structures were resolved based on assembly graph topology and sequencing read coverage depth, with conformations parsed using Bandage visualization and guided by reference to the P. trichocarpa genome. Repeat regions were identified and analyzed, resulting in finalized mitochondrial genome assemblies. Annotation was performed using the Intelligent Plant Mitochondrial Genome Annotator (IPMGA; Li et al., 2025). The Plant Mitochondrial Genome map (PMGmap; Zhang et al., 2024) online tool was used to visualize the genome and generate cis- and trans-splicing gene maps. The mitochondrial genomes of P. kangdingensis and P. ciliata were then submitted to GenBank to obtain accession numbers.
2.3 Comparative analysis of mitochondrial genomes
The mitochondrial genomes of P. kangdingensis and P. ciliata were compared with those of eight other Populus species (P. trichocarpa, P. deltoides, P. alba, P. adenopoda, P. simonii, P. tremula, P. davidiana, and P. rotundifolia), as listed in Supplementary Table S1. All mitochondrial genomes were re-annotated using the same pipeline described in section 2.2 to ensure consistency across species. Genome metrics including total length, GC content, gene counts, and intron structure were calculated using the Genepioneer Cloud platform (http://cloud.genepioneer.com:9929/#/home). Protein-coding genes (PCGs) were extracted using custom Python scripts. The Ka/Ks ratios for 30 conserved mitochondrial PCGs across the 10 Populus species were calculated using KaKs_Calculator v2.0 (Wang et al., 2010).
2.4 Codon usage bias analysis
PhyloSuite v1.2.3 (Zhang et al., 2020) was used to calculate relative synonymous codon usage (RSCU) values and codon counts for each amino acid across the PCGs of the 10 species. Visualizations were produced via the Genepioneer Cloud. GC content at the first, second, and third codon positions (GC1, GC2, GC3) and the effective number of codons (ENC) were calculated for P. kangdingensis and P. ciliata. ENC-GC3 plots were used to assess the influence of selection and mutation bias on codon usage.
2.5 Repeat sequences and RNA editing site analysis
Simple sequence repeats (SSRs) were identified using the MISA online tool (Beier et al., 2017). Tandem repeats were identified using Tandem Repeat Finder (Benson, 1999), and four types of dispersed repeats were analyzed with REPuter (Kurtz and Schleiermacher, 1999), all using default parameters. C-to-U RNA editing sites in the 34 PCGs of both species were predicted using Deepred-mt (Edera et al., 2021) with default settings.
2.6 DNA transfer between mitochondria and chloroplasts
Previously assembled chloroplast genomes of P. kangdingensis and P. ciliata were used for homology searches against their mitochondrial genomes via BLAST, using an E-value threshold of 1e-5. Homologous segments with significant matches were identified as transferred mitochondrial plastid segments (MtPts). BEDTools v2.28.0 (Quinlan and Hall, 2010) was used to annotate MtPts based on GFF3 files, categorizing gene types and integrity (Complete/Partial). Circos v0.69-9 (Krzywinski et al., 2009) was used to visualize syntenic connections between mitochondrial and chloroplast genomes.
2.7 Synteny analysis
To investigate evolutionary relationships within Populus and among other genera from Malpighiales, synteny analysis was conducted between P. kangdingensis and representative species from six genera in Malpighiales (Supplementary Table S1), as well as among P. kangdingensis, P. ciliata, and previously published species from the sect. Tacamahaca, sect. Aigeiros, and sect. Populus (Zhao et al., 2024). Synteny and visualization were performed using NGenomeSyn v1.42 (He et al., 2023). Pairwise synteny was analyzed using the GetTwoGenomeSyn.pl script with minimap2, followed by manual configuration file preparation for visualization.
2.8 Phylogenetic analysis
All available mitochondrial and chloroplast genomes from 11 genera and 35 species in Malpighiales were downloaded from NCBI (Supplementary Table S1). Shared PCGs (19 mitochondrial, 58 chloroplast) were extracted using PhyloSuite v1.2.3, and gene trees for each were constructed using FastTree2 (Price et al., 2010). Species trees were then reconstructed using ASTRAL-IV (Zhang and Mirarab, 2022). Mitochondrial and chloroplast phylogenies were visualized using the tvBOT online tool (Xie et al., 2023).
3 Results and analysis
3.1 Mitochondrial genome features of P. kangdingensis and P. ciliata
HiFi sequencing using the PacBio Sequel II platform generated 93 GB and 39 GB of data for P. kangdingensis and P. ciliata, respectively. For P. kangdingensis, a total of 5,538,529 HiFi reads were obtained, with a maximum length of 59,758 bp and an average length of 16,803 bp. For P. ciliata, 2,337,475 HiFi reads were produced, with a maximum length of 51,421 bp and an average length of 17,003 bp. Mitogenomes were assembled using PMAT2 and manually curated. Bandage visualization showed multiple potential conformations in both P. kangdingensis and P. ciliata. We resolved one conformation by referring to the previously reported P. trichocarpa mitochondrial genome.The mitochondrial genome of P. kangdingensis consists of three circular molecules (Figure 1A), with a total length of 785,824 bp and a GC content of 44.74%. The sizes of the three subgenomes are 317,075 bp, 282,772 bp, and 185,977 bp, respectively (Supplementary Figure S1A). In contrast, the mitochondrial genome of P. ciliata exhibits a branched structure (Figure 1B), comprising two circular molecules and one linear molecule with a total length of 798,807 bp and a GC content of 44.73%. The two circular molecules are 327,191 bp and 206,251 bp in length, and the linear molecule is 265,365 bp (Supplementary Figure S1B). Annotation using PMGA identified 57 shared genes in both species, including 34 protein-coding genes (PCGs), 20 tRNA genes, and 3 rRNA genes (Supplementary Table S2). Among the PCGs, 24 are core genes and 10 are non-core. The core genes include five ATP synthase genes (atp1, atp4, atp6, atp8, atp9), four cytochrome c biogenesis genes (ccmB, ccmC, ccmFC, ccmFN), one cytochrome c reductase gene (cob), three cytochrome c oxidase genes (cox1, cox2, cox3), one maturase (matR), one membrane transporter (mttB), and nine NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, nad9). The non-core genes include three large subunit ribosomal proteins (rpl2, rpl10, rpl16), five small subunit ribosomal proteins (rps1, rps3, rps4, rps7, rps12), and two succinate dehydrogenase genes (sdh3, sdh4). Additionally, four cis-splicing genes (ccmFC, nad4, nad7, rps3) and three trans-spliced genes (nad1, nad2, nad5) were identified. Details of exons for each gene are provided in Supplementary Figures S2 and S3.
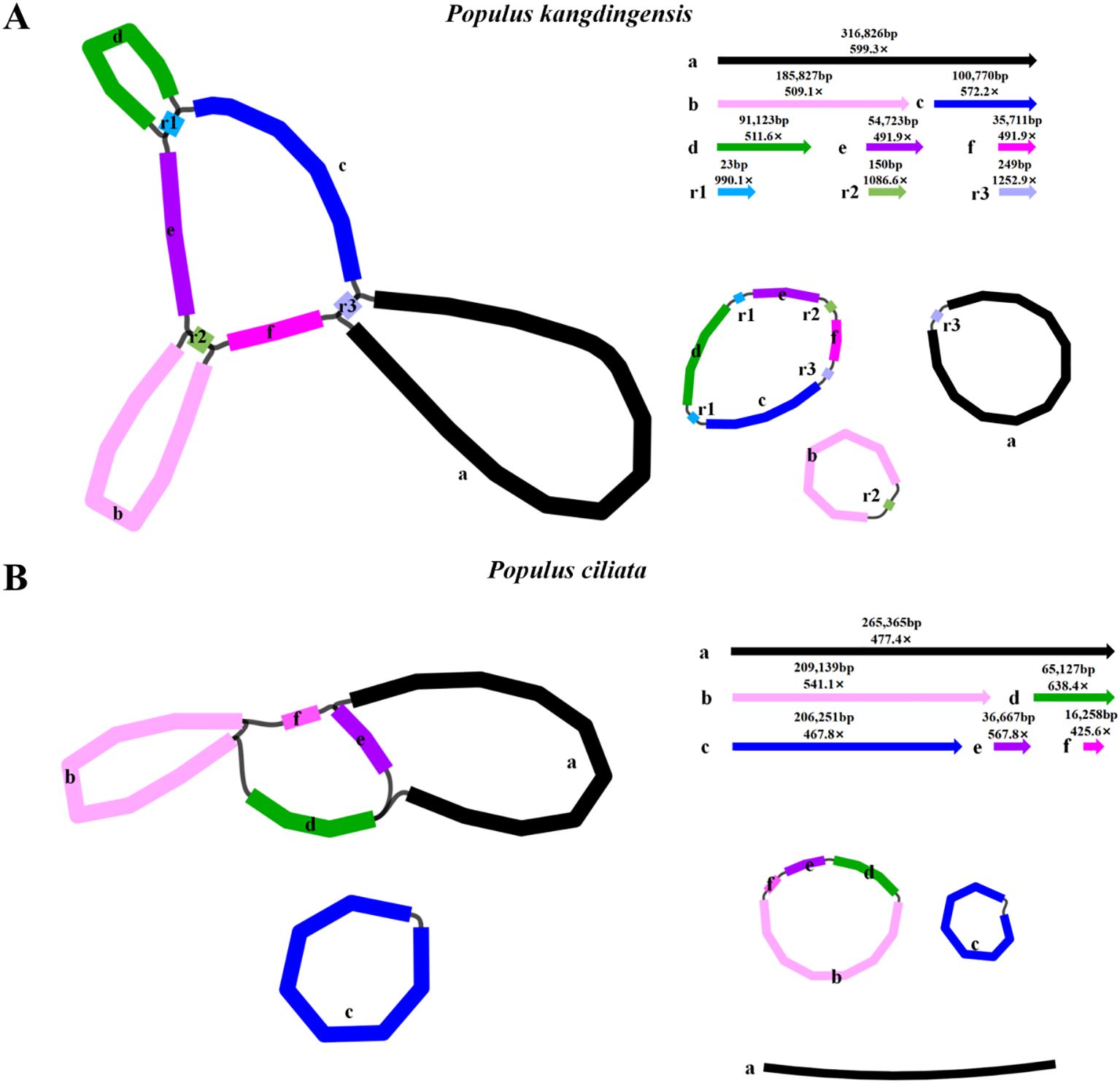
Figure 1. Structure of the mitochondrial genomes of P. kangdingensis and P. ciliata. (A) Circular configuration of the P. kangdingensis mitogenome, composed of three independent circular molecules. (B) Branched configuration of the P. ciliata mitogenome, consisting of two circular molecules and one linear molecule.
3.2 Comparative analysis of mitogenomes in Populus
To investigate the evolutionary features of Populus mitogenomes, we compared P. kangdingensis and P. ciliata with eight other species: P. simonii, P. trichocarpa, P. adenopoda, P. alba, P. davidiana, P. deltoides, P. rotundifolia, and P. tremula. The size of mitogenomes ranged from 772,549 bp (P. rotundifolia) to 869,108 bp (P. adenopoda), with GC content varying between 44.7% (P. adenopoda) and 44.85% (P. trichocarpa). Detailed values were listed in Supplementary Table S3. P. kangdingensis possessed the highest gene count (60), while P. rotundifolia had the fewest (54). The number of PCGs was relatively consistent among species, although intron numbers in NADH dehydrogenase genes varied. For instance, nad1, nad4, and nad7 in P. kangdingensis contained 1, 1, and 4 introns respectively, while in P. ciliata the counts were 2, 3, and 3. The genes rps14 were absent in P. kangdingensis, P. ciliata, and P. simonii, and sdh3 was only present in P. kangdingensis, P. ciliata, and P. adenopoda. All species had 3 rRNA genes, with most differences arising from the number of tRNA genes. To assess the effect of environmental pressure on mitogenome evolution, we calculated Ka/Ks ratios for 30 shared PCGs across all ten Populus species (Figure 2). Most genes had Ka/Ks < 1, indicating purifying selection. However, atp4, ccmB, ccmFN, and mttB had Ka/Ks > 1 in most species, suggesting positive selection, though in a few species these genes showed signs of purifying selection instead.
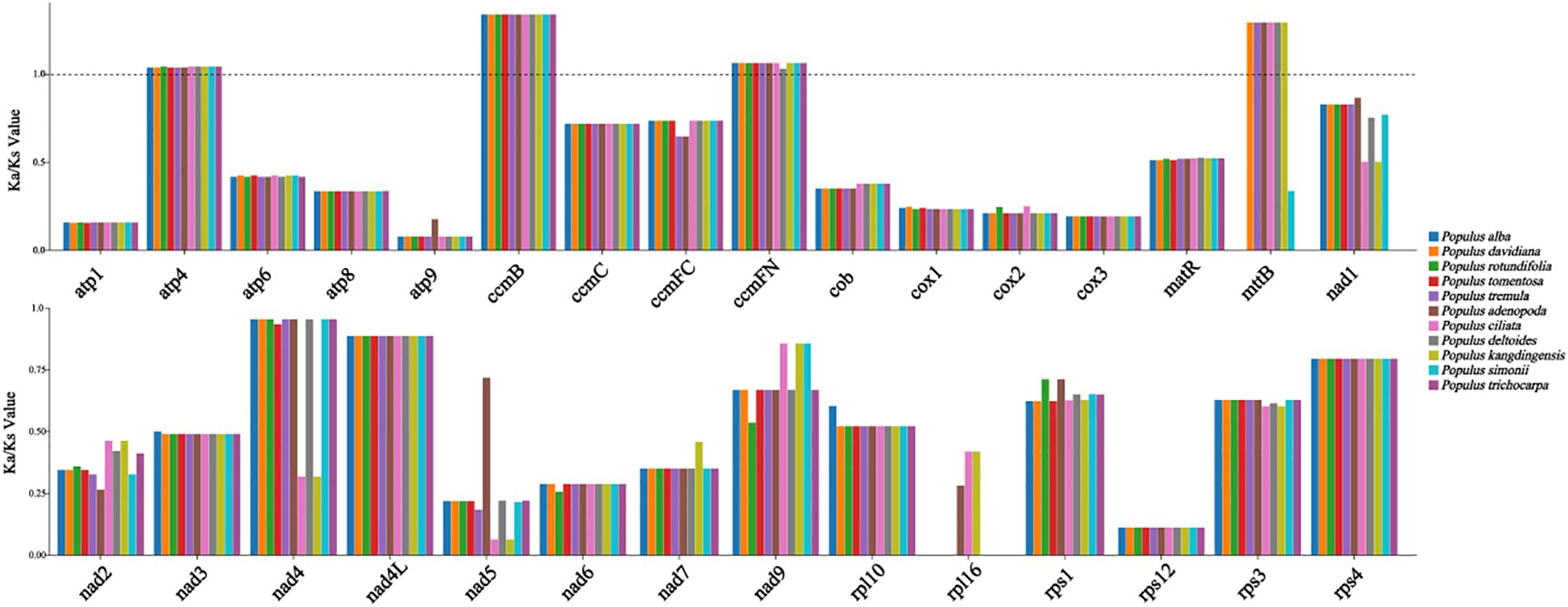
Figure 2. Ka/Ks ratios of 30 shared mitochondrial PCGs across 10 Populus taxa. Each bar represents the Ka/Ks ratio of a specific gene in a particular species. Colors denote different species, as indicated in the legend. The dashed line marks the reference value of Ka/Ks=1.
3.3 Codon usage bias in Populus mitochondrial genomes
In the ten Populus mitogenomes, codon usage analysis identified 64 codons encoding 21 amino acids (Figure 3D). The protein-coding genes (PCGs) comprised a total of 101,650 codons, with species-level counts ranging from 9,831 to 10,871. P. kangdingensis contained the fewest codons (9,831), followed by P. ciliata (9,851). Among all codons, UUU was the most frequently used across species. Leucine (Leu) and serine (Ser), each encoded by six synonymous codons, were the most abundant amino acids, accounting for 11,592 codons (11.4%) and 9,084 codons (8.94%), respectively. The mean relative synonymous codon usage (RSCU) values across species showed that 30 codons had RSCU > 1, while 32 codons had RSCU < 1. Methionine (AUG) and tryptophan (UGG) exhibited no codon usage bias RSCU = 1 (Figure 3A). In P. kangdingensis, RSCU patterns were consistent with the overall mean (Figure 3B). By contrast, in P. ciliata, 30 codons showed RSCU > 1 and 33 codons showed RSCU < 1, with a notable deviation in the CCA codon for proline (Pro) (Figure 3C). Collectively, except for AUG and UGG, most amino acids displayed codon usage bias.
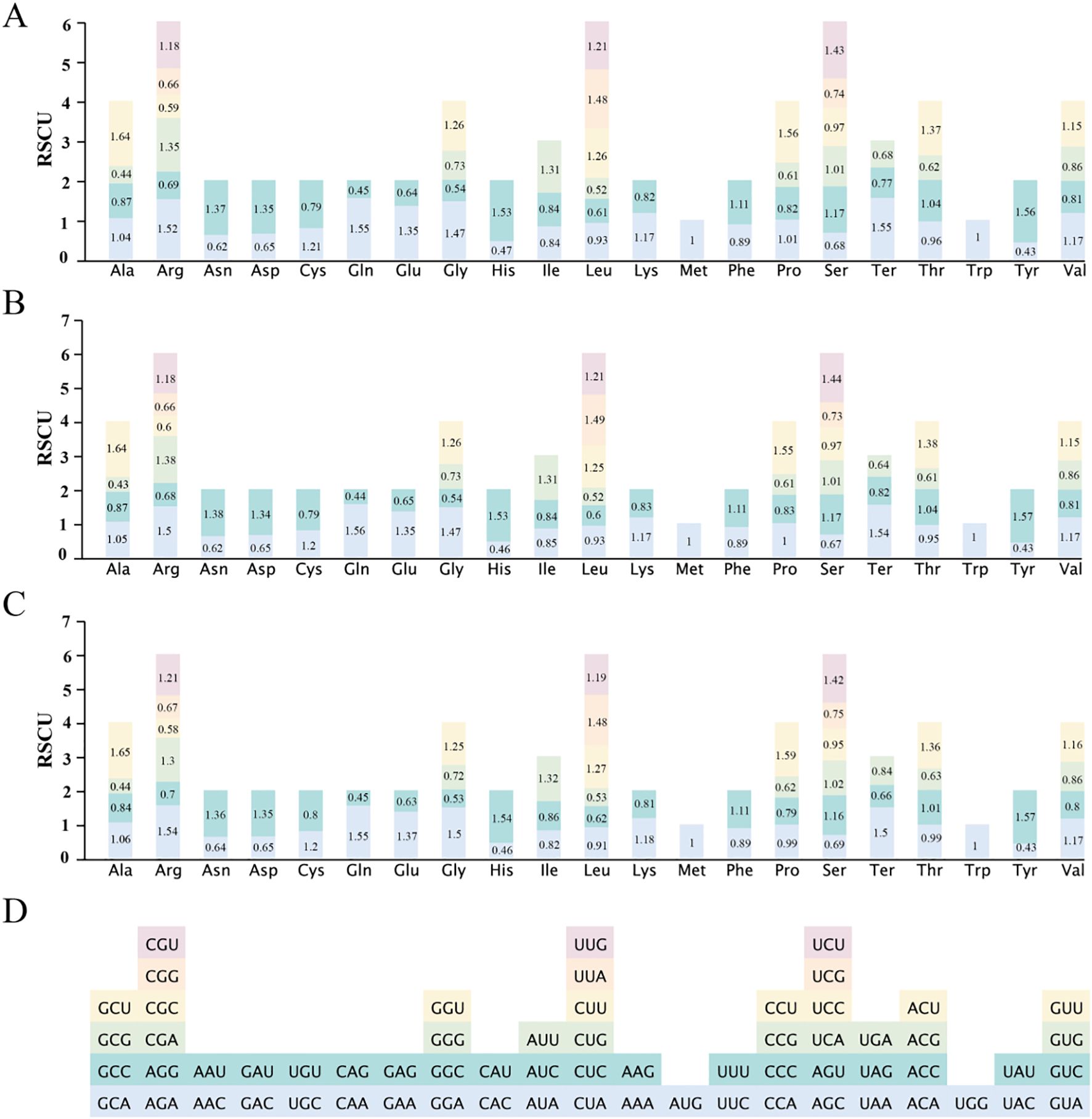
Figure 3. RSCU values of 64 codons in the mitogenomes of ten Populus species. (A) Mean RSCU values across the ten Populus species; (B) RSCU values in P. kangdingensis; (C) RSCU values in P. ciliata; (D) Codon color legend. Each stacked bar represents RSCU values for synonymous codons corresponding to a specific amino acid. RSCU > 1 indicates higher-than-expected usage; RSCU < 1 indicates lower-than-expected usage. Codons are color-coded and grouped under their respective amino acids.
To further examine codon usage patterns in P. kangdingensis and P. ciliata, we analyzed 34 PCGs, calculating GC content at the first (GC1), second (GC2), and third (GC3) codon positions, and the effective number of codons (ENC). GC1 ranged from 36.08% to 56.3%, GC2 from 30.3% to 55.47%, and GC3 from 25.55% to 50.5% (Supplementary Table S4), all averaging below 50%, suggesting a preference for A/T-ending codons. ENC values ranged from 28.47 to 57.36, with averages of 52.95 (P. kangdingensis) and 52.93 (P. ciliata), indicating weak codon usage bias (Supplementary Figure S4). ENC-GC3 plots showed that most PCGs deviated from the expected curve, suggesting codon bias is influenced by gene expression levels.
3.4 Repeat sequence analysis and RNA editing site prediction in P. kangdingensis and P. ciliata
Simple sequence repeats (SSRs) are widely distributed in the mitogenome. We identified 227 and 225 SSRs in the mitogenome of P. kangdingensis and P. ciliata, respectively (Figure 4A). Both species exhibited six types of SSR motifs, with similar distribution patterns across types. Notably, no Hexa-type SSRs were detected on the third chromosome in either species. Among these, tetranucleotide repeats were the most abundant, with 82 and 83 loci, accounting for 36.12% and 36.89% of the total SSRs, respectively, and were primarily located on the first chromosome. In contrast, hexanucleotide repeats were the least frequent, with 5 and 3 loci, representing 2.2% and 1.33% of the total SSRs, respectively, and were mainly found on the second chromosome. Tandem repeats were also identified, with 20 and 19 found in P. kangdingensis and P. ciliata (Figure 4A). Both species contained 150 dispersed repeats, including four types: forward (F), palindromic (P), reverse (R), and complement (C), with F and P being the most abundant. The length distribution of dispersed repeats was similar in both species, ranging from 17 to 262 bp and accounting for 84.64% and 84.11% of total repeat length. A total of 313 potential C-to-U RNA editing sites were predicted in each genome (Figure 4B). The nad4 gene exhibited the highest number of editing sites (39), followed by ccmB (31). Ribosomal protein genes generally had fewer editing sites.

Figure 4. Distribution of repeat sequences and RNA editing site predictions in the mitogenomes of P. kangdingensis and P. ciliata. (A) Integrated distribution of simple sequence repeats (SSRs), tandem repeats, and dispersed repeats across mitochondrial chromosomes. Dispersed repeats are classified into four types: forward (F), palindromic (P), reverse (R), and complement (C). (B) Predicted numbers of C-to-U RNA editing sites for each gene. In (A) the left side corresponds to P. kangdingensis and the right to P. ciliata.
3.5 Analysis of DNA fragment transfer between mitochondrial and chloroplast genomes
The transfer of genetic material from chloroplasts to mitochondria is a common phenomenon in the evolution of higher plants. However, these chloroplast-derived sequences often exhibit relatively low conservation. To explore such events in P. kangdingensis and P. ciliata, we identified homologous fragments between the chloroplast and mitochondrial genomes of both species. A total of 66 homologous fragments were identified in each species through sequence similarity analysis. In P. kangdingensis, the homologous fragments range from 29 bp to 2824 bp in length, with a total length of 46,481 bp, accounting for 5.91% of the mitochondrial genome (Figure 5A). These fragments display between 0 to 119 mismatches and 0 to 33 gaps (Supplementary Table S5). In P. ciliata, the fragment lengths ranged from 29 to 3845 bp, with a total length of 48,997 bp, constituting 6.13% of its mitogenome (Figure 5B). Mismatches ranged from 0 to 180, and gaps from 0 to 38 (Supplementary Table S6). These transferred fragments were termed mitochondrial plastid segments (MtPts). Annotation of MtPts in both species revealed 31 complete genes and 26 partial genes in each genome, suggesting high consistency. However, some differences were observed in gene composition and conservation. Twenty-six complete and twenty-five partial genes were shared between the two species. Unique complete genes in P. kangdingensis included rpl2, rpl23, trnI-CAU, trnL-CAA, and trnS-GCU, while P. ciliata had atpI, rpoC1, rps2, and fragments of trnL-CAA and trnS-GCU. Regarding partially overlapping genes, rpl2 was unique to P. kangdingensis, whereas rpoC1 was specific to P. ciliata. A total of 20 tRNA genes were identified: P. kangdingensis had 20 completes and 5 partial copies, whereas P. ciliata had 18 completes and 5 partial copies. These findings suggest that structural incompleteness or functional degradation of some tRNAs may have occurred during the transfer process. Despite similarities in the number of transferred genes, variations in gene type, integrity, and potential function reflect certain species-specific histories for the retention of exogenous gene fragments in mitochondrial genomes.
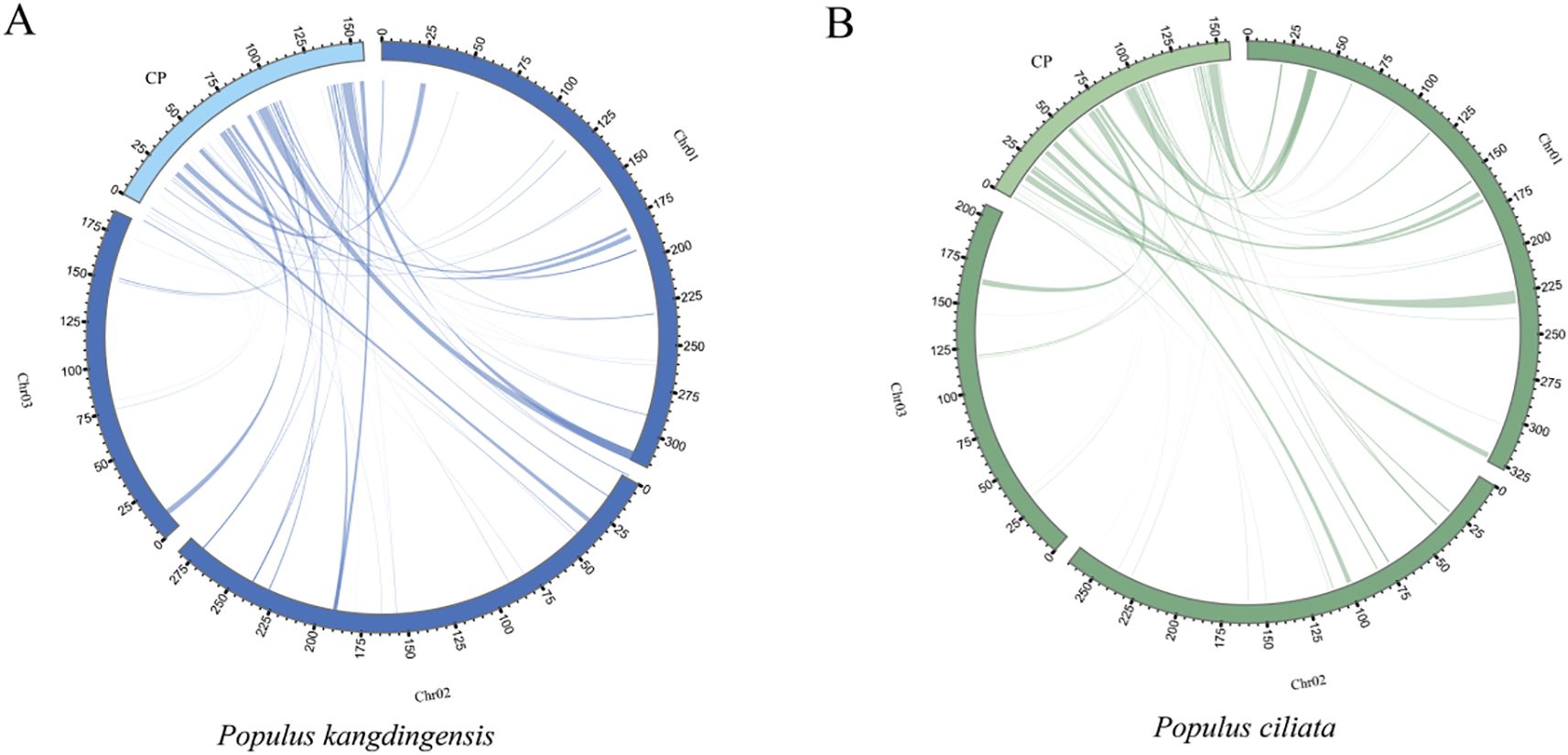
Figure 5. Homologous fragment analysis between the mitochondrial and chloroplast genomes of P. kangdingensis and P. ciliata. (A) Homologous segments between the mitochondrial and chloroplast genomes of P. kangdingensis. (B) Homologous segments between the mitochondrial and chloroplast genomes of P. ciliata. Lines represent plastid-to-mitochondrion transferred fragments (MtPts) identified via sequence similarity. In the circular plots, dark segments indicate mitochondrial genomes and light segments indicate chloroplast genomes.
3.6 Synteny analysis between P. kangdingensis, P. ciliata, and other species
We first examined syntenic relationships between P. kangdingensis and six representative species from different genera within Malpighiales (Figure 6A). The results revealed significant variability in synteny levels. P. kangdingensis and Salix purpurea exhibited the highest synteny, with 16 shared syntenic blocks spanning 735,077 bp, accounting for approximately 93.54% of the P. kangdingensis mitogenome. In contrast, Garcinia oblongifolia shared only 20 syntenic blocks spanning 62,665 bp, representing just 7.97% of the P. kangdingensis genome. These differences reflect frequent structural rearrangements in intergenic regions, which contribute to varying degrees of conservation in protein-coding regions across plant mitogenomes. Among these six analyzed species, overlaps between protein-coding regions and syntenic blocks varied significantly (Supplementary Table S7), with S. purpurea showing the highest overlap (99.76%) and G. oblongifolia the lowest (17.28%). These results underscore the structural instability of mitogenomes across species. We further analyzed synteny among seven representative species from three sections within Populus (Figure 6B). Marked differences in synteny levels were observed among these groups. The highest synteny was between the Sect. Tacamahaca and Sect. Aigeiros: P. ciliata and P. kangdingensis shared syntenic blocks with P. deltoides totaling 785,325 bp and 800,598 bp, accounting for 97.84% and 99.75% of the P. deltoides mitogenome, respectively. In contrast, synteny between the Sect. Tacamahaca and Sect. Populus was comparatively weaker, with average synteny lengths of 778,941 bp, covering 98.72% and 97.83% of the P. alba and P. tremula mitogenomes. Notably, synteny within each section of Populus was highly consistent, with coverage ranging from 97.83% to 99.98%, and most protein-coding regions overlapping with syntenic blocks.
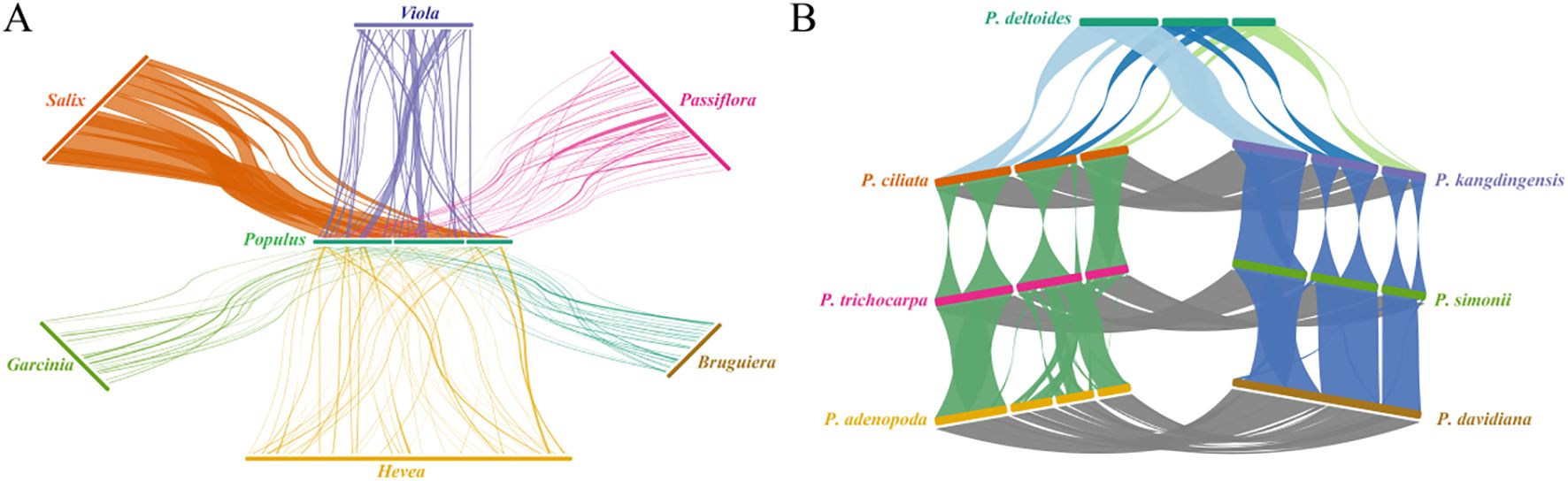
Figure 6. Mitogenome synteny analysis of P. kangdingensis, P. ciliata, and other species. (A) Syntenic relationships between P. kangdingensis and six representative species from different genera within Malpighiales. (B) Synteny among seven representative species from three Populus sections: Tacamahaca, Aigeiros, and Populus. Connecting lines represent syntenic blocks identified between species.
3.7 Phylogenetic analysis based on chloroplast and mitochondrial genomes in Malpighiales
We selected 35 representative species from 11 genera within Malpighiales (Populus, Salix, Passiflora, Viola, Garcinia, Calophyllum, Hevea, Manihot, Ricinus, Jatropha and Bruguiera) to construct phylogenetic trees using 33 mitochondrial and 58 chloroplast PCGs (Figures 7A, B). The two trees showed high topological consistency across most major clades, reflecting well-resolved intergeneric relationships. However, discrepancies in key lineages suggest organellar genomes may capture distinct evolutionary histories. Populus and Salix were strongly supported as monophyletic sister groups in both trees, indicating a close phylogenetic relationship. However, substantial differences emerged in intra-generic relationships between the two organelle-based trees. In the mitochondrial tree, P. adenopoda grouped with P. kangdingensis, P. ciliata, P. deltoides, and P. trichocarpa, forming a distinct clade. In contrast, the chloroplast tree placed P. adenopoda with P. alba and P. tomentosa in a clade, which then grouped with P. davidiana and P. tremula, highlighting discrepancies between mitochondrial and chloroplast phylogenetic signals. Among the complex clade composed of Hevea, Manihot, Ricinus, and Jatropha, branching patterns varied between trees. The chloroplast tree supported a stable monophyletic group, while the mitochondrial tree showed a more diffuse topology, with Hevea particularly deviating from monophyly. Additionally, Viola formed a sister group to the Hevea–Manihot–Ricinus–Jatropha clade in the chloroplast tree, but in the mitochondrial tree, it grouped with Garcinia and Calophyllum. Such conflicts may result from incomplete lineage sorting (ILS), ancient hybridization, or horizontal gene transfer between organelles. Some genera exhibited consistent phylogenetic placements in both trees. Bruguiera and Viola formed well-supported independent clades across both trees, suggesting minimal impact from conflicting phylogenetic signals. Garcinia and Calophyllum maintained a stable sister relationship in the chloroplast tree, while in the mitochondrial tree, their positions shifted slightly possibly reflecting differing evolutionary rates or lineage-specific biases in organelle genome history.
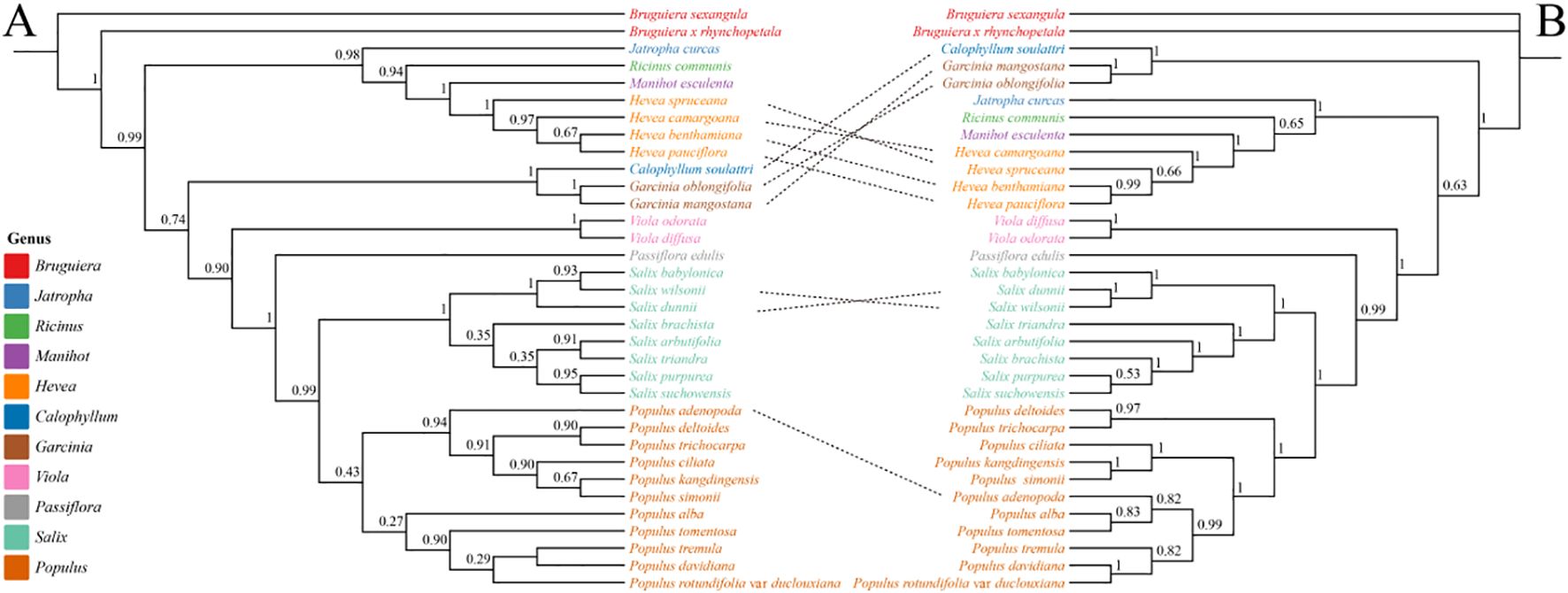
Figure 7. Phylogenetic relationships among different genera of Malpighiales based on chloroplast and mitochondrial genomes. (A) Phylogenetic tree constructed using 33 mitochondrial PCGs. (B) Phylogenetic tree constructed using 58 shared chloroplast PCGs. The trees include 35 representative species from 11 genera within Malpighiales. Genera are color-coded, and node values indicate bootstrap support levels.
4 Discussion
4.1 Structural diversity and functional conservation in Populus mitochondrial genomes
The mitogenomes of higher plants are structurally complex, frequently exhibiting multi-circular, branched, or linear configurations driven primarily by repetitive sequence-mediated recombination (Wu et al., 2022; Cai et al., 2024). Recent studies have also demonstrated that considerable structural differences can exist among mitochondrial genomes within the same genus (Kozik et al., 2019; Wang et al., 2024). In this study, we present the first sequencing, assembly, and characterization of mitogenomes for two representative Populus species from the Qinghai–Tibet Plateau: P. kangdingensis and P. ciliata. The P. kangdingensis mitogenome comprises three independent circular molecules, consistent with other species under the Tacamahaca section. In contrast, P. ciliata exhibits a previously unreported branched mitochondrial genome configuration, composed of two circular molecules and one linear segment distinct from the multi-circular structures described in other Populus. Notably, a similar configuration has recently been reported in Lindera aggregata, which contains a circular master circle and a linear segment (Shi et al., 2024a), further supporting the structural plausibility of this genome architecture.
Despite differences in genome architecture, both P. kangdingensis and P. ciliata contain 57 annotated genes, including 34 PCGs, 20 tRNAs, and 3 rRNAs. Notably, the composition of core genes (e.g., atp, cox, nad, ccm) is conserved between the two species. This functional conservation has been consistently observed across multiple Populus species. Interestingly, the sdh3 gene is present only in P. kangdingensis, P. ciliata, and P. rotundifolia, while the rps14 gene is absent in both P. kangdingensis and P. ciliata,. This phenomen may be attributed to their transfer to the nuclear genome, where they are subsequently expressed (Bonen, 2006; Sloan et al., 2010; Butenko et al., 2024).
4.2 Codon usage bias and selection pressure reveal clues to adaptive evolution
Ka/Ks ratio analysis is widely used to evaluate the selective pressures acting on genes during evolution and provides insights into species-specific adaptive changes and the direction of natural selection. Here, Ka/Ks analysis of mitochondrial PCGs in ten Populus species revealed that most genes are under strong purifying selection, reflecting the evolutionary conservation of protein function. This pattern is common among higher plants and suggests the structural and functional stability of key respiratory and energy metabolism enzyme complexes (Mower et al., 2012). Notably, a subset of genes (atp4, ccmB, ccmFN, and mttB) provide the evidence of positive selection across the ten species. As mitochondria play a central role in energy metabolism, these genes may be subject to directional selection under specific environmental conditions, potentially enhancing species’ adaptability and metabolic flexibility (Wang et al., 2021). (Lyu et al., 2024). conducted a selection analysis of mitochondrial genes across 58 species from different altitudes using the aBSREL model and identified three candidate genes potentially involved in high-altitude adaptation: atp4, atp9, and mttB. Among them, atp4 encodes a critical subunit of ATP synthase (Heazlewood et al., 2003), which may play a role in plant growth and development. In several high-altitude plant species, numerous transcriptional elements have been identified in the vicinity of mttB, which are likely induced by environmental pressures associated with high-altitude adaptation, suggesting a possible role of mttB in this process (Xiong et al., 2022). We analyzed the expression levels of 32 PCGs from the mitochondrial genomes of two Populus. The results showed that atp4 and mttB exhibited higher expression in P. kangdingensis, which is distributed at higher elevations (Supplementary Figure S5). Therefore, atp4 and mttB may be candidate genes for high-altitude adaptation in the mitochondrial genome of Populus and warrant further investigation.
Codons play a central role in the transmission of genetic information and protein synthesis. Due to factors such as gene mutation and natural selection, codon usage bias has emerged as an important evolutionary signature of genome evolution and selection pressure (Wang et al., 2018; Yengkhom et al., 2019). In this study, we systematically analyzed codon usage patterns for 34 PCGs in the mito genomes of P. kangdingensis and P. ciliata. Our results indicate a strong preference for A/T bases and A/T-ending codons in both species, consistent with the reported A/T bias in the P. deltoides mitogenome. This AT-rich tendency at the third codon position is a common feature in higher plants (Qu et al., 2023).
Further ENC-plot analysis revealed that most genes were located far from the theoretical curve, suggesting that codon usage is non-random and significantly associated with gene expression levels, implying strong natural selection. This trend aligns with findings in model plants such as Arabidopsis, rice, and wheat, supporting the view that natural selection is a key driver of codon usage bias (Sharp and Li, 1987; Wang and Hickey, 2007). Our findings suggest that codon usage bias in Populus mitochondrial genomes may reflect both functional constraints in coding regions and adaptive evolutionary patterns.
4.3 Species-specific differences in mitochondrial RNA editing and chloroplast-to-mitochondrion DNA transfer
RNA editing is a crucial post-transcriptional regulatory mechanism in plant mitochondrial genomes, primarily involving C-to-U conversions, which can alter amino acid codons and thus affect protein function (Hao et al., 2021). In both P. kangdingensis and P. ciliata, 313 RNA editing sites were detected, with genes such as nad4 and ccmB harboring the highest number of sites. These findings are consistent with results from species in the Populus section, such as P. tremula and P. alba, suggesting a central role for these genes in mitochondrial functional regulation (Sung et al., 2010). Conversely, rps exhibited fewer editing events, likely reflecting their functional conservation, which may negate the need for optimization through editing.
Transfer of genetic fragments from the chloroplast to the mitochondrion is a common feature of plant organelle genome evolution and has been observed across many plant taxa (Gualberto et al., 2014). In this study, we identified 66 plastid-derived transfer fragments (MtPts) in P. kangdingensis and P. ciliata, accounting for 5.91% and 6.13% of their mitochondrial genomes, respectively. These values are higher than those reported in the Aigeiros section species P. deltoides (4.11%) and in the Populus section species P. tomentosa (4.42%), and the fragments include both complete and partial tRNAs and PCGs. Since P. kangdingensis and P. ciliata belong to the more derived Sect. Tacamahaca compared to Sect. Populus, this observation may suggest an evolutionary trend toward increasing chloroplast-derived tRNA integration into mitochondrial genomes (Glover et al., 2001).
Although the two species share 31 identical transferred genes, most of these gene transfers are non-functional. For instance, the transferred gene rpl2 in P. kangdingensis is unique to this species and corresponds to its mitochondrial nad5 gene, while the species-specific transferred gene rpoC1 in P. ciliata is non-functional. Transcriptome data further reveal that only a subset of chloroplast-to-mitochondrion transferred genes (atp1, nad1, rps1, and nad5) are expressed in both species. This may reflect species-specific mechanisms of gene transfer and selective retention (Hertle et al., 2021; Kelly, 2021). These observations support the hypothesis that mitochondria exhibit a “selective acceptance” of foreign DNA fragments (Timmis et al., 2004; Smith and Keeling, 2015).
4.4 Phylogenetic topology conflicts reveal inconsistencies in lineage history
Using P. kangdingensis as a representative, we performed a comprehensive synteny analysis between its mitochondrial genome and those of six other genera within Malpighiales. Our results revealed substantial differences in synteny levels among genera. The highest synteny was observed between Populus and Salix, covering nearly the entire set of protein-coding regions; however, Garcinia exhibited the lowest synteny, suggesting high structural conservation in Salicaceae and a closer evolutionary relationship between its members. In contrast, the structural divergence observed between more distantly related genera may result from mechanisms such as recombination mediated by repeats and DNA loss (Gualberto and Newton, 2017).
Within Populus, differences in synteny levels were also observed between sections. The Sect. Tacamahaca and Aigeiros exhibited higher synteny compared to the Populus section. Nevertheless, all seven examined Populus species showed synteny levels exceeding 97%, indicating substantial structural conservation across the genus during mitochondrial genome evolution.
Both plastid phylogenetic trees strongly support the monophyly of Populus and Salix, identifying them as sister groups, a result consistent with the classification of Salicaceae proposed by the APG IV system (The Angiosperm Phylogeny Group et al., 2016). P. kangdingensis, P. ciliata, and P. simonii form a monophyletic clade, and the topologies of the two plastid phylogenies align with those of the nuclear gene tree for Populus on the Qinghai-Tibet Plateau reported by Mi (Mi et al., 2025), displaying high support values.
However, in the mitochondrial phylogeny, P. adenopoda (a member of the Sect. Populus) does not group with other Sect. Populus taxa as it does in the plastid phylogeny, but instead forms a clade with members of the Sect. Tacamahaca and Sect. Aigeiros. This result is inconsistent with the classification of Populus in the Flora of China. Previous studies (Wang et al., 2022) using nuclear gene trees showed that P. adenopoda clusters with other Sect. Populus species, in agreement with the plastid phylogeny in this study. Such conflict may be attributed to widespread introgression and incomplete lineage sorting (ILS) within Populus (Wang et al., 2020).
Overall, while these topological differences largely align with the APG IV framework at the intergeneric level, the species-level discrepancies within the genus underscore the complementary yet distinct evolutionary signals from organellar and nuclear phylogenies (Degnan and Rosenberg, 2009). Therefore, integrating mitochondrial, plastid, and nuclear genomic data is crucial for reconstructing a more accurate evolutionary history and phylogenetic relationships among species.
5 Conclusion
This study firstly reports the high-quality mitochondrial genome assemblies of P. kangdingensis and P. ciliata from the Qinghai-Tibet Plateau. The two species exhibit distinct genome structures multi-circular in P. kangdingensis and branched in P. ciliata—highlighting structural diversity within Populus. Both genomes contain 57 conserved functional genes, but show variation in intron distribution, repeat content, and codon usage bias. Widespread RNA editing and substantial chloroplast-to-mitochondrion DNA transfer were also observed. Phylogenetic analyses support the monophyly of Populus and Salix, but reveal discordance among other genera, suggesting organelle-specific evolutionary histories. These results provide valuable genomic resources and insights into the evolution and adaptation of Populus species in high-altitude environments.
Data availability statement
The mitochondrial genomes of P. kangdingensis have been deposited in GenBank under accession numbers PX116244, PX116245, and PX116246, while those of P. ciliata are available under accession numbers PX116247, PX116248, and PX116249. The raw sequence data have been deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA028946) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa/browse/CRA028946.
Author contributions
J-LH: Software, Funding acquisition, Writing – review & editing, Writing – original draft, Visualization, Supervision. Y-JS: Writing – review & editing. F-FT: Formal analysis, Writing – review & editing. J-XM: Writing – review & editing, Formal analysis. FZ: Software, Visualization, Writing – review & editing, Methodology. S-XZ: Software, Methodology, Visualization, Writing – review & editing. X-QW: Writing – review & editing, Funding acquisition. C-DH: Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Natural Science Foundation of Sichuan Province (2024NSFSC1190 to J-LH and 2025ZNSFSC0266 to S-XZ), National Natural Science Foundation of China (31870645 to X-QW).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1637726/full#supplementary-material
References
Alverson, A. J., Rice, D. W., Dickinson, S., Barry, K., and Palmer, J. D. (2011). Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 23, 2499–2513. doi: 10.1105/tpc.111.087189
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bi, C., Qu, Y., Hou, J., Wu, K., Ye, N., and Yin, T. (2022). Deciphering the multi-chromosomal mitochondrial genome of populus simonii. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.914635
Bi, C., Shen, F., Han, F., Qu, Y., Hou, J., Xu, K., et al. (2024). PMAT: an efficient plant mitogenome assembly toolkit using low-coverage HiFi sequencing data. Horticulture Res. 11, uhae023. doi: 10.1093/hr/uhae023
Bonen, L. (2006). Mitochondrial genes leave home. New Phytol. 172, 379–381. doi: 10.1111/j.1469-8137.2006.01904.x
Brenner, W. G., Mader, M., Müller, N. A., Hoenicka, H., Schroeder, H., Zorn, I., et al. (2019). High level of conservation of mitochondrial RNA editing sites among four populus species. G3 Genes|Genomes|Genetics 9, 709–717. doi: 10.1534/g3.118.200763
Burger, G., Jackson, C. J., and Waller, R. F. (2012). “Unusual Mitochondrial Genomes and Genes,” in Organelle Genetics: Evolution of Organelle Genomes and Gene Expression. Ed. Bullerwell, C. E. (Springer, Berlin, Heidelberg), 41–77. doi: 10.1007/978-3-642-22380-8_3
Butenko, A., Lukeš, J., Speijer, D., and Wideman, J. G. (2024). Mitochondrial genomes revisited: why do different lineages retain different genes? BMC Biol. 22, 15. doi: 10.1186/s12915-024-01824-1
Cai, Y., Chen, H., Ni, Y., Li, J., Zhang, J., and Liu, C. (2024). Repeat-mediated recombination results in Complex DNA structure of the mitochondrial genome of Trachelospermum jasminoides. BMC Plant Biol. 24, 966. doi: 10.1186/s12870-024-05568-6
Chandel, N. S. (2014). Mitochondria as signaling organelles. BMC Biol. 12, 34. doi: 10.1186/1741-7007-12-34
Choi, M. N., Han, M., Lee, H., Park, H.-S., Kim, M.-Y., Kim, J.-S., et al. (2017). The complete mitochondrial genome sequence of Populus davidiana Dode. Mitochondrial DNA Part B 2, 113–114. doi: 10.1080/23802359.2017.1289346
Degnan, J. H. and Rosenberg, N. A. (2009). Gene tree discordance, phylogenetic inference and the multispecies coalescen. Trends Ecol. Evol. 24, 332–340. doi: 10.1016/j.tree.2009.01.009
Edera, A. A., Small, I., Milone, D. H., and Sanchez-Puerta, M. V. (2021). Deepred-Mt: Deep representation learning for predicting C-to-U RNA editing in plant mitochondria. Comput. Biol. Med. 136, 104682. doi: 10.1016/j.compbiomed.2021.104682
Glover, K. E., Spencer, D. F., and Gray, M. W. (2001). Identification and structural characterization of nucleus-encoded transfer RNAs imported into wheat mitochondria. J. Biol. Chem. 276, 639–648. doi: 10.1074/jbc.M007708200
Gualberto, J. M., Mileshina, D., Wallet, C., Niazi, A. K., Weber-Lotfi, F., and Dietrich, A. (2014). The plant mitochondrial genome: Dynamics and maintenance. Biochimie. 100, 107–120. doi: 10.1016/j.biochi.2013.09.016
Gualberto, J. M. and Newton, K. J. (2017). Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 68, 225–252. doi: 10.1146/annurev-arplant-043015-112232
Hao, W., Liu, G., Wang, W., Shen, W., Zhao, Y., Sun, J., et al. (2021). RNA editing and its roles in plant organelles. Front. Genet. 12. doi: 10.3389/fgene.2021.757109
He, F., Niu, M.-X., Wang, T., Li, J.-L., Shi, Y.-J., Zhao, J.-J., et al. (2024). The ubiquitin E3 ligase RZFP1 affects drought tolerance in poplar by mediating the degradation of the protein phosphatase PP2C - 9. Plant Physiol. 196, 2936–2955. doi: 10.1093/plphys/kiae497
He, F., Wei, S.-Y., Wang, R.-Q., Tang, J. Y., Yang, P., Wang, T., et al. (2025). Females adapted better than males in Populus deltoides to simulated nitrogen deposition. Plant Soil. doi: 10.1007/s11104-025-07239-3
He, W., Yang, J., Jing, Y., Xu, L., Yu, K., and Fang, X. (2023). NGenomeSyn: an easy-to-use and flexible tool for publication-ready visualization of syntenic relationships across multiple genomes. Bioinformatics 39, btad121. doi: 10.1093/bioinformatics/btad121
Heazlewood, J. L., Whelan, J., and Millar, A. H. (2003). The products of the mitochondrial orf25 and orfB genes are FO components in the plant F1FO ATP synthase. FEBS Lett. 540, 201–205. doi: 10.1016/S0014-5793(03)00264-3
Hertle, A. P., Haberl, B., and Bock, R. (2021). Horizontal genome transfer by cell-to-cell travel of whole organelles. Sci. Adv. 7. doi: 10.1126/sciadv.abd8215
Kelly, S. (2021). The economics of organellar gene loss and endosymbiotic gene transfer. Genome Biol. 22, 345. doi: 10.1186/s13059-021-02567-w
Kersten, B., Rampant, P. F., Mader, M., Paslier, M.-C. L., Bounon, R., Berard, A., et al. (2016). Genome sequences of populus tremula chloroplast and mitochondrion: implications for holistic poplar breeding. PloS One 11, e0147209. doi: 10.1371/journal.pone.0147209
Kim, Y.-J. and Zhang, D. (2018). Molecular control of male fertility for crop hybrid breeding. Trends Plant Sci. 23, 53–65. doi: 10.1016/j.tplants.2017.10.001
Kozik, A., Rowan, B. A., Lavelle, D., Berke, L., Schranz, M. E., Michelmore, R. W., et al. (2019). The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PloS Genet. 15, e1008373. doi: 10.1371/journal.pgen.1008373
Krzywinski, M., Schein, J., Birol, İ., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kurtz, S. and Schleiermacher, C. (1999). REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics 15, 426–427. doi: 10.1093/bioinformatics/15.5.426
Li, J., Ni, Y., Lu, Q., Chen, H., and Liu, C. (2025). PMGA: A plant mitochondrial genome annotator. Plant Comm 6, 101191. doi: 10.1016/j.xplc.2024.101191
Lyu, Z. Y., Yang, G. M., Zhou, X. L., Wang, S. Q., Zhang, R., Shen, S. K., et al. (2024). Deciphering the complex organelle genomes of two Rhododendron species and insights into adaptive evolution patterns in high-altitude. BMC Plant Biol. 24, 1054. doi: 10.1186/s12870-024-05761-7
Mi, J.-X., Tian, F.-F., Huang, J.-L., Shi, Y.-J., Li, J., Meng, F.-Y., et al. (2025). A new pattern of species diversification revealed by populus on the qinghai-tibet plateau. Evolutionary Biol. 52, 71–83. doi: 10.1007/s11692-025-09647-w
Mower, J. P., Sloan, D. B., and Alverson, A. J. (2012). “Plant Mitochondrial Genome Diversity: The Genomics Revolution,” in Plant Genome Diversity Volume 1: Plant Genomes, their Residents, and their Evolutionary Dynamics. Eds. Wendel, J. F., Greilhuber, J., Dolezel, J., and Leitch, I. J. (Springer, Vienna), 123–144. doi: 10.1007/978-3-7091-1130-7_9
Palmer, J. D., Adams, K. L., Cho, Y., Parkinson, C. L., Qiu, Y.-L., and Song, K. (2000). Dynamic evolution of plant mitochondrial genomes: Mobile genes and introns and highly variable mutation rates. Proc. Natl. Acad. Sci. 97, 6960–6966. doi: 10.1073/pnas.97.13.6960
Petersen, G., Cuenca, A., Møller, I. M., and Seberg, O. (2015). Massive gene loss in mistletoe (Viscum, Viscaceae) mitochondria. Sci. Rep. 5, 17588. doi: 10.1038/srep17588
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PloS One 5, e9490. doi: 10.1371/journal.pone.0009490
Qu, Y., Zhou, P., Tong, C., Bi, C., and Xu, L. (2023). Assembly and analysis of the Populus deltoides mitochondrial genome: the first report of a multicircular mitochondrial conformation for the genus Populus. J. Forestry Res. 34, 717–733. doi: 10.1007/s11676-022-01511-3
Quinlan, A. R. and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rice, D. W., Alverson, A. J., Richardson, A. O., Young, G. J., Sanchez-Puerta, M. V., Munzinger, J., et al. (2013). Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm amborella. Science. 342, 1468–1473. doi: 10.1126/science.1246275
Sanchez-Puerta, M. V., García, L. E., Wohlfeiler, J., and Ceriotti, L. F. (2017). Unparalleled replacement of native mitochondrial genes by foreign homologs in a holoparasitic plant. New Phytol. 214, 376–387. doi: 10.1111/nph.14361
Sharp, P. M. and Li, W.-H. (1987). The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295. doi: 10.1093/nar/15.3.1281
Shi, Y., Chen, Z., Jiang, J., Wu, W., Yu, W., Zhang, S., et al. (2024a). The assembly and comparative analysis of the first complete mitogenome of Lindera aggregata. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1439245
Shi, Y.-J., Mi, J.-X., Huang, J.-L., He, F., Chen, L.-H., and Wan, X.-Q. (2023). Study on the origin and classification of two poplar species on the qinghai–tibet plateau. Forests 14, 1003. doi: 10.3390/f14051003
Shi, Y.-J., Mi, J.-X., Huang, J.-L., Tian, F.-F., He, F., Zhong, Y., et al. (2024b). A new species of Populus and the extensive hybrid speciation arising from it on the Qinghai-Tibet Plateau. Mol. Phylogenet. Evol. 196, 108072. doi: 10.1016/j.ympev.2024.108072
Sloan, D. B., Alverson, A. J., Chuckalovcak, J. P., Wu, M., McCauley, D. E., Palmer, J. D., et al. (2012). Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PloS Biol. 10, e1001241. doi: 10.1371/journal.pbio.1001241
Sloan, D. B., Alverson, A. J., Štorchová, H., Palmer, J. D., and Taylor, D. R. (2010). Extensive loss of translational genes in the structurally dynamic mitochondrial genome of the angiosperm Silene latifolia. BMC Evolutionary Biol. 10, 274. doi: 10.1186/1471-2148-10-274
Smith, D. R. and Keeling, P. J. (2015). Mitochondrial and plastid genome architecture: Reoccurring themes, but significant differences at the extremes. Proc. Natl. Acad. Sci. 112, 10177–10184. doi: 10.1073/pnas.1422049112
Sung, T.-Y., Tseng, C.-C., and Hsieh, M.-H. (2010). The SLO1 PPR protein is required for RNA editing at multiple sites with similar upstream sequences in Arabidopsis mitochondria. Plant J. 63, 499–511. doi: 10.1111/j.1365-313X.2010.04258.x
The Angiosperm Phylogeny Group, Chase, M. W., Christenhusz, M. J. M., Fay, M. F., Byng, J. W., Judd, W. S., et al. (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.12385
Timmis, J. N., Ayliffe, M. A., Huang, C. Y., and Martin, W. (2004). Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 5, 123–135. doi: 10.1038/nrg1271
Tuskan, G. A., DiFazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The genome of black cottonwood, populus trichocarpa (Torr. & Gray). Science 313, 1596–1604. doi: 10.1126/science.1128691
Wang, D., Zhang, Y., Zhang, Z., Zhu, J., and Yu, J. (2010). KaKs_Calculator 2.0: A Toolkit Incorporating Gamma-Series Methods and Sliding Window Strategies. Genom. Proteomics Bioinform. 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Wang, H.-C. and Hickey, D. A. (2007). Rapid divergence of codon usage patterns within the rice genome. BMC Evolutionary Biol. 7, S6. doi: 10.1186/1471-2148-7-S1-S6
Wang, Y., Huang, J., Li, E., Xu, S., Zhan, Z., Zhang, X., et al. (2022). Phylogenomics and biogeography of populus based on comprehensive sampling reveal deep-level relationships and multiple intercontinental dispersals. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.813177
Wang, L., Liu, X., Wang, Y., Ming, X., Qi, J., and Zhou, Y. (2024). Comparative analysis of the mitochondrial genomes of four Dendrobium species (Orchidaceae) reveals heterogeneity in structure, synteny, intercellular gene transfer, and RNA editing. Front. Plant Sci. 15. doi: 10.3389/fpls.2024.1429545
Wang, L., Xing, H., Yuan, Y., Wang, X., Saeed, M., Tao, J., et al. (2018). Genome-wide analysis of codon usage bias in four sequenced cotton species. PloS One 13, e0194372. doi: 10.1371/journal.pone.0194372
Wang, M., Zhang, L., Zhang, Z., Li, M., Wang, D., Zhang, X., et al. (2020). Phylogenomics of the genus Populus reveals extensive interspecific gene flow and balancing selection. New Phytol. 225, 1370–1382. doi: 10.1111/nph.16215
Wang, X., Zhou, S., Wu, X., Wei, Q., Shang, Y., Sun, G., et al. (2021). High-altitude adaptation in vertebrates as revealed by mitochondrial genome analyses. Ecol. Evol. 11, 15077–15084. doi: 10.1002/ece3.8189
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu, Z.-Q., Liao, X.-Z., Zhang, X.-N., Tembrock, L. R., and Broz, A. (2022). Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Systematics Evol. 60, 160–168. doi: 10.1111/jse.12655
Wynn, E. L. and Christensen, A. C. (2019). Repeats of unusual size in plant mitochondrial genomes: identification, incidence and evolution. G3 Genes|Genomes|Genetics 9, 549–559. doi: 10.1534/g3.118.200948
Xie, J., Chen, Y., Cai, G., Cai, R., Hu, Z., and Wang, H. (2023). Tree Visualization By One Table (tvBOT): a web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 51, W587–W592. doi: 10.1093/nar/gkad359
Xiong, Y. L., Yu, Q. Q., Xiong, Y., Zhao, J. M., Lei, X., Liu, L., et al. (2022). The complete mitogenome of Elymus sibiricus and insights into its evolutionary pattern based on simple repeat sequences of seed plant mitogenomes. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.802321
Yengkhom, S., Uddin, A., and Chakraborty, S. (2019). Deciphering codon usage patterns and evolutionary forces in chloroplast genes of Camellia sinensis var. assamica and Camellia sinensis var. sinensis in comparison to Camellia pubicosta. J. Integr. Agric. 18, 2771–2785. doi: 10.1016/S2095-3119(19)62716-4
Zhang, X., Chen, H., Ni, Y., Wu, B., Li, J., Burzyński, A., et al. (2024). Plant mitochondrial genome map (PMGmap): A software tool for the comprehensive visualization of coding, noncoding and genome features of plant mitochondrial genomes. Mol. Ecol. Resour. 24, e13952. doi: 10.1111/1755-0998.13952
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, C. and Mirarab, S. (2022). Weighting by gene tree uncertainty improves accuracy of quartet-based species trees. Mol. Biol. Evol. 39, msac215. doi: 10.1093/molbev/msac215
Keywords: Populus, mitochondrial genome, phylogenetic reconstruction, Qinghai –Tibet Plateau, adaptive evolution
Citation: Huang J-L, Shi Y-J, Tian F-F, Mi J-X, Zhang F, Zhou S-X, Wan X-Q and Huang C-D (2025) Structural diversity and phylogenomic insights from the mitochondrial genomes of two populus species from the Qinghai-Tibet Plateau. Front. Plant Sci. 16:1637726. doi: 10.3389/fpls.2025.1637726
Received: 29 May 2025; Accepted: 20 August 2025;
Published: 12 September 2025.
Edited by:
Luke R. Tembrock, Colorado State University, United StatesReviewed by:
SeonJoo Park, Yeungnam University, Republic of KoreaYanshu Qu, Jiangxi Academy of Forestry, China
Copyright © 2025 Huang, Shi, Tian, Mi, Zhang, Zhou, Wan and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cong-De Huang, aHVhbmdjb25nZGVAc2ljYXUuZWR1LmNu; Xue-Qin Wan, dy14dWVAMTYzLmNvbQ==
†These authors have contributed equally to this work