 Cheng Fang1†
Cheng Fang1† Wei Ma2†Xiaotong Zhu1,3†Lingling Peng1,3†Liangzhi Tao1Zhiqiang Zhao4Fengbin Wang4
Wei Ma2†Xiaotong Zhu1,3†Lingling Peng1,3†Liangzhi Tao1Zhiqiang Zhao4Fengbin Wang4 Yafeng Ye1
Yafeng Ye1 Bojun Ma3
Bojun Ma3 Yuejin Wu1Jie Yuan4*
Yuejin Wu1Jie Yuan4* Binmei Liu1*
Binmei Liu1* Xifeng Chen3*
Xifeng Chen3* Weimin Cheng1*
Weimin Cheng1*- 1Key Laboratory of High Magnetic Field and Ion Beam Physical Biology, Hefei Institutes of Physical Science, Chinese Academy of Sciences, Hefei, China
- 2Huaihe River Basin Eco-Environmental Monitoring and Scientific Research Center of Huaihe River Basin Ecological Environmental Supervision and Administration Bureau, Bengbu, China
- 3College of and Life Sciences, Zhejiang Normal University, Jinhua, China
- 4Institute of Crop Sciences, Xinjiang Academy of Agricultural Sciences, Urumqi, China
Lesion mimic mutants (LMMs) are invaluable for uncovering the molecular mechanisms of programmed cell death (PCD) and plant immunity and identifying more LMMs expands our understanding of these complex processes. In this study, we characterized a novel rice LMM, spl39, identified through heavy-ion beam irradiation. The spl39 mutant exhibits reddish-brown lesions from the tillering stage, reduced plant height, grain size, and fertility. Chloroplast ultrastructure analysis revealed thylakoid disruption and membrane damage, contributing to reduced photosynthetic capacity. Excessive ROS accumulation and reduced antioxidative enzyme activities triggered PCD. Genetic mapping identified a 306-kb deletion in spl39, encompassing 33 genes, including 12 from the diterpenoid biosynthesis gene cluster. Transcriptomic analysis revealed upregulation of hormone signaling and defense-related pathways, consistent with elevated levels of SA, JA, auxins, and cytokinins. The spl39 mutant exhibited enhanced resistance to bacterial blight with reduced lesion lengths and increased expression of defense-related genes. These findings highlight the role of large genomic deletions in reprogramming plant metabolism and immunity, providing new insights into the mechanisms underlying lesion mimic phenotypes and disease resistance in rice.
1 Introduction
Plants have evolved intricate mechanisms to defend against biotic and abiotic stresses, among which the hypersensitive response (HR) is a central component of the immune system (Zurbriggen et al., 2010). HR is characterized by programmed cell death (PCD) at the site of pathogen infection, accompanied by reactive oxygen species (ROS) production, accumulation of antimicrobial compounds, and the expression of pathogenesis-related (PR) genes (Dickman and Fluhr, 2013). This localized response not only restricts pathogen spread but also triggers systemic acquired resistance (SAR), enhancing plant immunity (Lam et al., 2001; Wang et al., 2015).
Lesion mimic mutants (LMMs) are invaluable tools for studying HR-like PCD and plant immunity (Zeng et al., 2004; Qiao et al., 2010; Wang et al., 2017; Ma et al., 2019; Yuchun et al., 2021; Li et al., 2024). These mutants develop necrotic spots resembling pathogen-induced lesions without pathogen presence, providing insights into defense signaling pathways. To date, over 60 LMM-related genes have been identified across various plant species, including rice (Oryza sativa). These genes are implicated in diverse biological processes such as ROS homeostasis (Cui et al., 2021), calcium signaling (Zhang et al., 2023), protein ubiquitination (Zeng et al., 2004), and secondary metabolite biosynthesis (Li et al., 2024). Notably, LMMs often exhibit enhanced disease resistance, as demonstrated in rice mutants like spl28 and spl35, which show upregulated PR gene expression and increased resistance to bacterial blight (Xanthomonas oryzae pv. oryzae, Xoo) and blast (Magnaporthe oryzae) (Qiao et al., 2010; Ma et al., 2019).
The biosynthesis of diterpenoid phytoalexins is a critical aspect of rice immunity, mediated by specialized biosynthetic gene clusters (BGCs) (Kitaoka et al., 2021). One of the initial discoveries from the rice genome sequencing was the identification of two BGCs linked to the synthesis of antimicrobial phytoalexins (Wang et al., 2012). These phytoalexins were among the earliest defense compounds to be documented. For instance, the biosynthetic gene cluster on chromosome 2 (c2BGC) encodes essential enzymes, including cytochrome P450 monooxygenases (CYP76M7 and CYP76M8), which play a crucial role in the biosynthesis of phytocassanes antimicrobial diterpenoids that enhance resistance to fungal pathogens. The deletion or disruption of c2BGC genes not only compromises phytoalexin production but also leads to spontaneous lesion mimic phenotypes and immune reprogramming, highlighting its dual function in metabolism and defense signaling (Jiang et al., 2022; Li et al., 2022). Another major diterpenoid biosynthetic gene cluster, located on chromosome 4 (c4BGC), is primarily responsible for momilactone biosynthesis. This cluster comprises key genes such as OsCPS4, OsKSL4, CYP99A2, CYP99A3, and the dehydrogenase gene OsMAS, which collectively regulate the production of these potent defense compounds (Shimura et al., 2007).
In this study, we report a novel lesion mimic mutant derived from heavy-ion beam mutagenesis, characterized by a genomic alteration in the c2BGC region spanning approximately 306 kb. The mutant exhibits spontaneous HR-like lesions, increased resistance to Xoo, and significant upregulation of defense-related genes. Hormonal profiling revealed elevated levels of salicylic acid (SA), jasmonic acid (JA), and abscisic acid (ABA), aligning with enhanced immune responses. Transcriptome analysis further demonstrated the activation of multiple immune pathways, including PR gene networks and hormonal signaling cascades. These findings provide new insights into the complex interplay between secondary metabolism and immune signaling in rice. By elucidating the molecular mechanisms underlying lesion mimic phenotypes, this study advances our understanding of plant immunity and offers potential strategies for engineering disease-resistant crops.
2 Materials and methods
2.1 Plant materials and growth conditions
The spotted leaf 39 (spl39) mutant was derived from the japonica cultivar Wuyunjing7 (WYJ7) through heavy ion beam mutagenesis. To facilitate genetic mapping, the spl39 mutant was crossed with the indica cultivar 93-11, generating an F2 mapping population. All rice plants were cultivated under natural field conditions at the Hefei Institute of Physical Science, Chinese Academy of Sciences (Hefei, China), as well as in Sanya, Hainan Province, China.
2.2 Determination of chlorophyll contents
Leaves from both wild-type and spl39 mutant plants were collected at the tillering stage to assess chlorophyll content. Total chlorophyll was extracted using 80% acetone and quantified with a spectrophotometer. Absorbance readings at 470, 645, and 663 nm were used to estimate chlorophyll levels, following the method as previously described (Wellburn, 1994).
2.3 Investigation of various antioxidant indexes
Leaves were collected from wild-type and spl39 plants at the tillering stages for investigation of various antioxidant indexes. The kits used for investigating H2O2, MDA contents and SOD, CAT, APX, POD enzyme activities were provided by Solarbio company. All experiments were following the protocols of the kits.
2.4 Histochemical marker staining assay
Leaves from wild-type and spl39 plants with the lesion phenotype were used for DAB, NBT and trypan blue staining as described previously (Wu et al., 2016). DAB, NBT and trypan blue staining were detected for H2O2 accumulation, superoxide anion accumulation and dead cells respectively. Leaves were placed in DAB, NBT or trypan blue staining solution kept in the dark for more than 40 h, then cleared in 75% ethanol at 80°C to remove Chl for photographing.
2.5 Transmission electron microscopy analysis
Leaf samples from both the wild-type and spl39 mutant were collected from the sword leaves at the tillering stage in field-grown plants. The samples were fixed in a 2.5% glutaraldehyde-phosphate buffer solution and subsequently processed following the method as previously described (Wu et al., 2016).
2.6 TUNEL assays
Leaves from both wild-type and spl39 plants were collected at the tillering stage for TUNEL analysis. The TUNEL assay was performed according to the manufacturer’s protocol for the Fluorescein in Situ Cell Death Detection Kit (Roche). Sectioning and fluorescence labeling were carried out following the method as previously described (Wu et al., 2016).
2.7 Map-based cloning
The spl39 locus was mapped using 1,460 mutant plants from the F2 population as described above. Simple sequence repeat (SSR) markers distributed across the entire rice genome were utilized for the spl39 locus rough mapping. For fine mapping, both SSR markers and custom-designed Insertion/Deletion (Indel) markers were employed within the initially localized region. The precise size of the deleted fragment was determined using chromosome walking, a method that progressively extends known DNA sequences from flanking regions to accurately define the deletion breakpoints.
2.8 qRT-PCR and RNA-seq analysis
Total RNA was extracted from leaf samples using TRIGene Reagent (GeneStar). Complementary DNA (cDNA) was synthesized from total RNA using a cDNA synthesis kit (GeneStar). Quantitative RT-PCR was conducted using a qRT-PCR kit (GeneStar) on the LightCycler 96 system (Roche). For RNA-seq analysis, total RNA was extracted from leaves at the tillering stage, and sequencing was performed using the BGISEQ-500 platform (BGI) following standard RNA-seq protocols.
2.9 Disease evaluation
Eight Xanthomonas oryzae pv. oryzae (Xoo) strains were used to assess bacterial blight resistance. The WT and spl39 plants were cultivated in the greenhouse at the Hefei Institute of Physical Science, Chinese Academy of Sciences (Hefei, China). Inoculation was performed on flag leaves using the clipping method at the heading stage. Lesion lengths were measured 15 days post-inoculation, with each strain tested on more than six biological replicates per sample. Statistical significance of lesion length differences was analyzed using Welch’s t-test in GraphPad Prism 7.0.0.
2.10 Quantification of plant hormones
To analyze hormone levels, fresh leaf samples were collected, immediately frozen in liquid nitrogen, and stored at -80°C until processing. Total hormones, including IAA, CKs, ABA, ACC (ethylene precursor), SA, and JA, were extracted using 80% methanol (v/v) containing 1% formic acid, with stable isotope-labeled internal standards added for precise quantification. Samples were vortexed, incubated at 4°C for 12–24 hours, and centrifuged at 12,000 × g for 15 min at 4°C. The supernatant was collected, filtered through a 0.22 µm membrane, and purified using solid-phase extraction (SPE) columns when necessary. Hormone quantification was performed using UHPLC-MS/MS (Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry) with a reverse-phase C18 column. The mobile phase consisted of water (0.1% formic acid) and acetonitrile (0.1% formic acid), and detection was carried out in multiple reaction monitoring (MRM) mode. Absolute hormone concentrations were determined using external standard calibration curves and normalized to internal standard recovery rates. Data were expressed as ng/g fresh weight (FW), and statistical analysis was performed using one-way ANOVA in GraphPad Prism 7.0.0.
2.11 Growth conditions and salt treatment
For short-term hydroponic culture, rice seedlings were grown in 96-well plates, following the method as described previously (Cheng et al., 2024). For salt treatment, 14-day-old seedlings were exposed to Yoshida’s solution supplemented with 150 mM NaCl, while control seedlings were maintained in Yoshida’s solution without NaCl. For survival rate analysis, approximately 40–48 rice seedlings underwent 5 days of salt treatment, followed by a 7-day recovery period in Yoshida’s solution. The number of actively growing seedlings was counted, with experiments performed in triplicate. For growth and physiological parameter measurements, roots and shoots were collected from seedlings with or without salt treatment after 9 days. The samples were thoroughly rinsed with distilled water before further analysis.
3 Results
3.1 Phenotypic characteristics of the rice spl39 mutant
We isolated a lesion-mimic mutant (LMM) named spotted leaf 39 (spl39) from a rice mutant library (Oryza sativa L. japonica cv. Wuyunjing7) treated with heavy ion beam irradiation. No significant differences were observed between wild type (WT) and spl39 plants before the tillering stage. However, reddish-brown lesions began to appear on the older leaves of spl39 at the tillering stage (around 45 days) (Figures 1A, C). As the plants grew, the lesions became more numerous and larger, eventually spreading to all leaves (Figures 1B, D). The leaf shading experiment showed that the lesions formation depended on light exposure (Figure 1E).
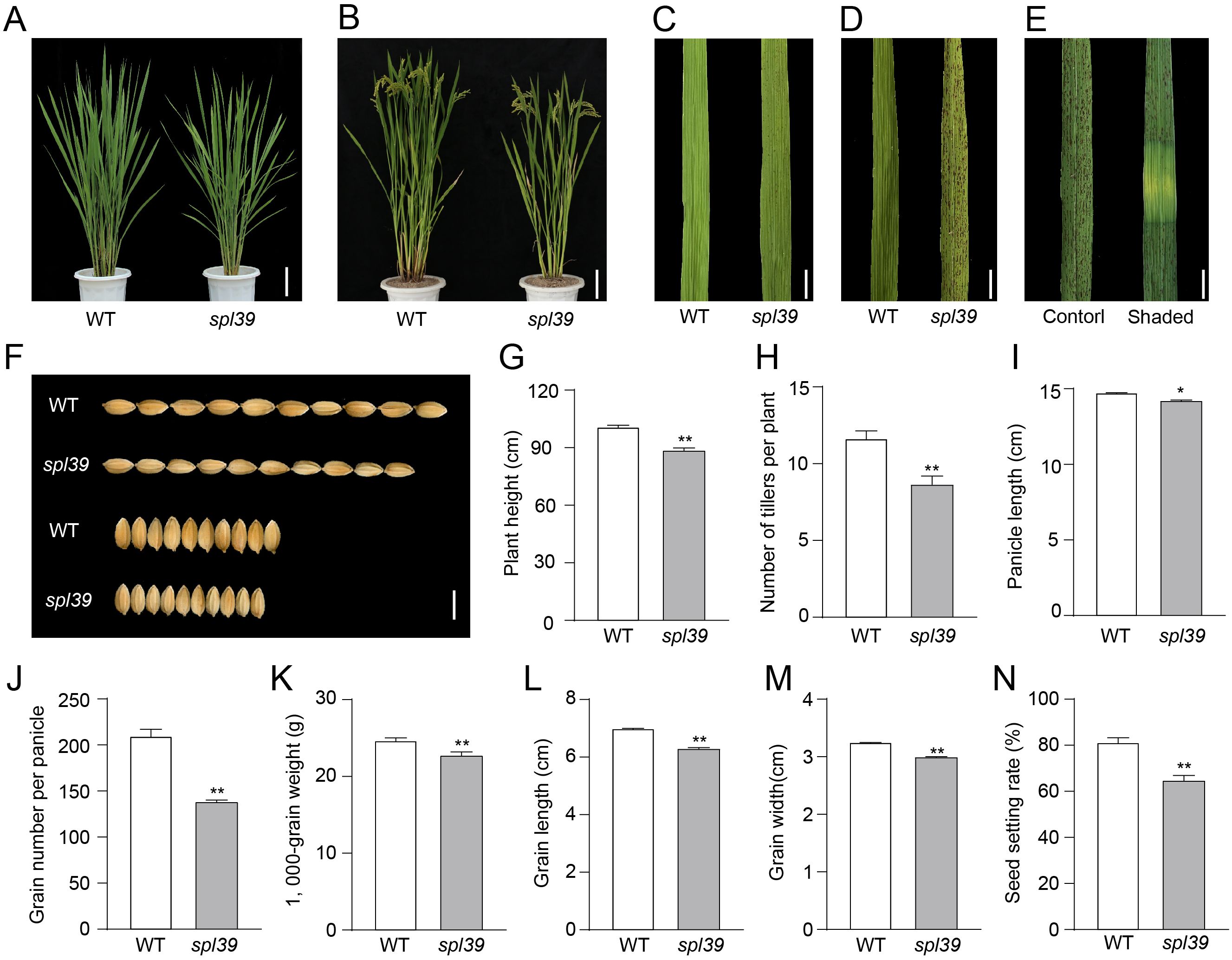
Figure 1. Phenotypic analysis of the WT and spl39 mutant. (A, B) Morphological comparison of WT and spl39 mutant plants at the tillering stage (A) and grain-filling stage (B). Bar=10 cm. (C, D) Leaf phenotype of WT and spl39 at the tillering stage (C) and heading stage (D), showing lesion-mimic characteristics in spl39. Bar=1 cm. (E) Lesion formation in spl39 leaves under control and shaded conditions. (F) Comparison of grains from WT and spl39, showing differences in grain size and shape. (G–N) Quantitative analysis of agronomic traits in WT and spl39: Plant height (G), Number of tillers per plant (H), Panicle length (I), Grain number per panicle (J), 1,000-grain weight (K), Grain length (L), Grain width (M), Seed setting rate (N). *P < 0.05, **P < 0.01 (Student’s t-test), error bars, ± SD (n=5).
In addition to the leaf lesion phenotype, the spl39 mutant exhibited significant differences in other agronomic traits compared to the WT, such as plant height, tiller number, grain number, grain size, grain weight, and setting rate (Figures 1F–N). At mature stage, spl39 plants were significantly shorter (Figures 1B, G), had fewer tillers, fewer grains, and lower spikelet fertility (Figures 1H, J, N). The grain size of spl39 was smaller than that of WT due to decreases in grain length (Figure 1L) and grain width (Figure 1M), resulting in a significantly lower 1,000-grain weight for spl39 (Figure 1K). These results suggest that the spl39 mutant has pleiotropic effects on plant growth and development.
To examine the physiological changes in spl39 plants, we measured chlorophyll (Chl a and Chl b) and carotenoid (Car) contents in flag leaves of spl39 and WT plants at heading stage. Both chlorophyll and carotenoid contents in spl39 plants were significantly decreased relative to the WT (Figure 2A). We also examined the ultrastructure of chloroplasts in the spl39 mutant using transmission electron microscopy. The results revealed a significant reduction in the number of thylakoids in the mutant, along with severe damage to the chloroplast membrane. These findings suggest that the disruption of chloroplast structure may be one of the factors contributing to the lesion mimic phenotype observed in spl39 mutant (Figures 2B–E).
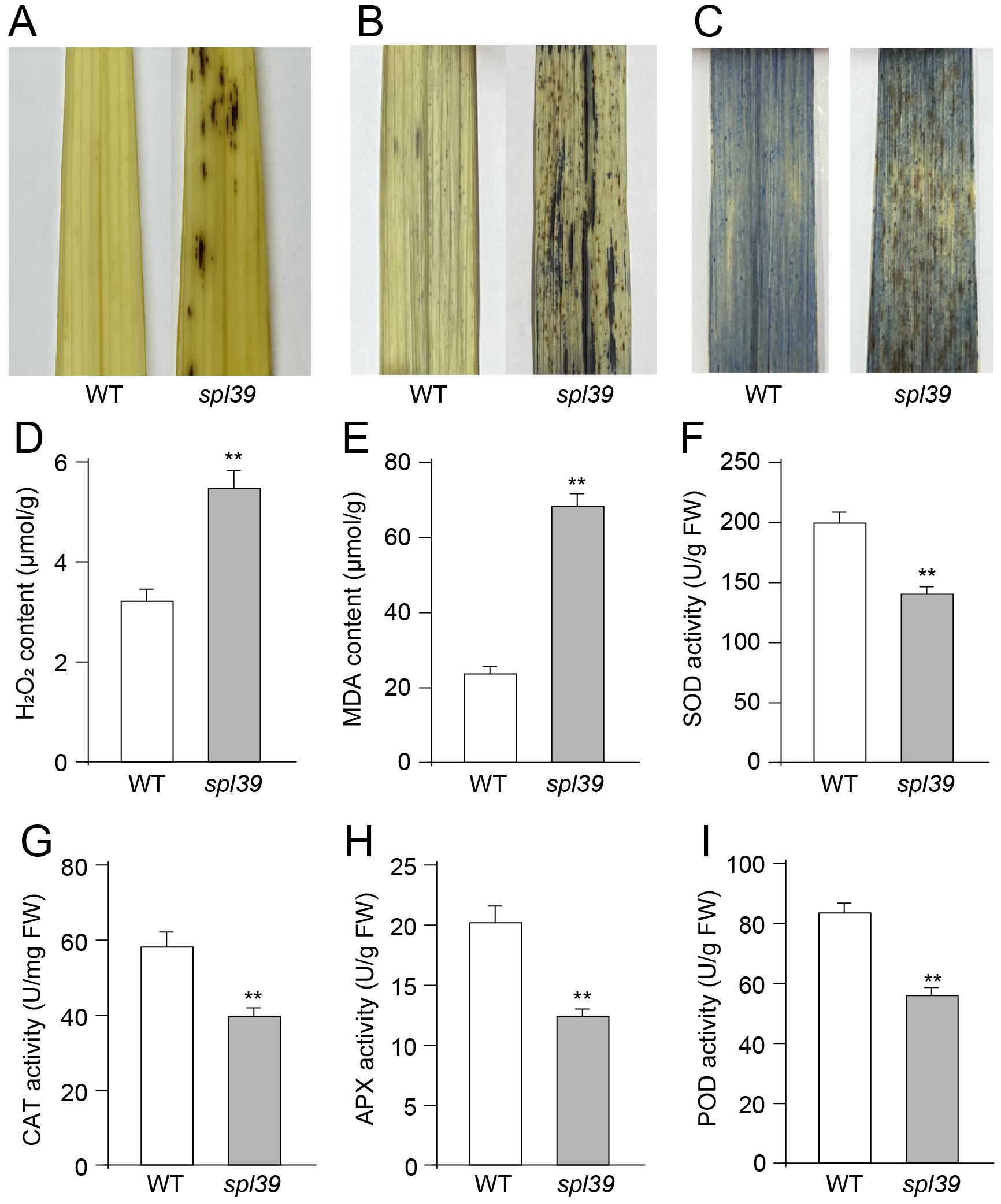
Figure 2. Increased oxidative damage and altered antioxidant enzyme activities in the spl39 mutant. (A) DAB staining in leaves. (B) NBT staining in leaves. (C) Trypan blue staining in leaves. (D) Statistical analysis of H2O2 content. (E) Statistical analysis of MDA content. (F) Statistical analysis of SOD activity. (G) Statistical analysis of CAT activity. (H) Statistical analysis of APX activity. (I) Statistical analysis of POD activity. **P < 0.01 (Student’s t-test), error bars, ± SD (n=5).
3.2 The spl39 mutant exhibits ROS accumulation and DNA damage
As previously reported, the overaccumulation of ROS, including superoxide and hydrogen peroxide, is associated with lesion formation and triggers PCD (Li et al., 2024). To assess the level of ROS in the spl39 mutant, several histochemical stains were employed to examine the content of H2O2 and O2-• in the leaves. For H2O2 accumulation, 3,3’-diaminobenzidine (DAB) staining was performed. The leaves with lesions in the spl39 mutant exhibited a deep brown-red coloration in the DAB staining, indicating the presence of H2O2. In contrast, no DAB staining was detected in the leaves of the WT (Figure 3A). Both nitro blue tetrazolium (NBT) staining for O2-• accumulation and trypan blue staining for cell death were conducted. The spl39 mutant displayed more intense staining around necrotic cells compared to the WT, indicating higher levels of O2-• accumulation (Figure 3B) and cell death (Figure 3C). We also conducted measurements of the concentrations of H2O2 and the lipid oxidation product malondialdehyde (MDA) during the tillering stage. H2O2 is a major ROS molecule, while MDA is a byproduct of lipid oxidation and serves as an indicator of cellular damage. The results show that the concentrations of H2O2 and MDA were obviously higher in spl39 mutant compared to the wild type. Collectively, these results suggest that the reddish-brown lesions observed in the spl39 mutant are a consequence of ROS accumulation. The excessive ROS production in the mutant likely contributes to the development of lesions and subsequent cell death.
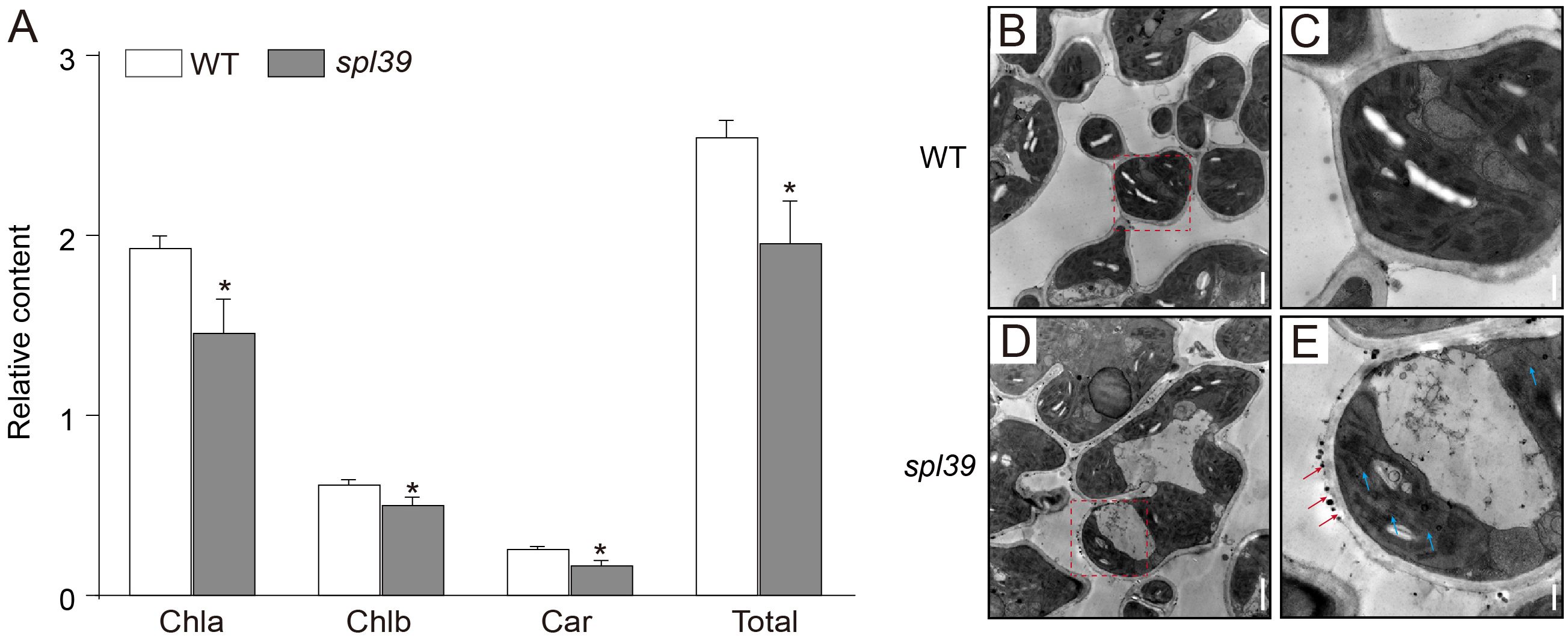
Figure 3. Reduced chlorophyll content and abnormal chloroplast ultrastructure in the spl39 mutant. (A) Relative chlorophyll a (Chla), chlorophyll b (Chlb), carotenoid (Car), and total chlorophyll content in WT and spl39 leaves. *P < 0.05 (Student’s t-test), error bars, ± SD (n=5). (B–E) Transmission electron microscopy (TEM) images of chloroplasts in WT (B, C) and spl39 (D, E) mesophyll cells. (B) WT chloroplasts show well-organized thylakoid membranes. (C) Higher magnification of a WT chloroplast with intact grana stacks. (D) spl39 chloroplasts displaying disrupted thylakoid structures and swollen stroma. (E) Higher magnification of a spl39 chloroplast, showing disorganized thylakoid membranes (red arrows) and disrupted grana stacks (blue arrows).
To gain further insight into the biochemical mechanism underlying ROS accumulation in the spl39 mutant, we analyzed the activities of several antioxidative enzymes, including superoxide dismutase (SOD), catalase (CAT), ascorbate peroxidase (APX), and peroxidase (POD), in both the spl39 mutant and the WT. Our findings revealed that the activities of these enzymes were significantly lower in the spl39 mutant leaves compared to the WT leaves (Figures 2F–I). This result suggested that the reduction in these enzymes activity may lead to the observed ROS accumulation in spl39 mutant.
Necrotic lesions observed in the leaves of the spl39 mutant indicate the occurrence of cell death, suggesting the presence of abnormal PCD. To investigate this further, we employed the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, which can sensitively detect DNA fragmentation associated with apoptosis signaling cascades. TUNEL analysis was performed on leaves of both the WT and spl39 mutant plants. As anticipated, the TUNEL signals in the nuclei of the spl39 mutant were robust and randomly distributed, indicating significant DNA fragmentation and apoptosis-like processes. In contrast, no evident TUNEL signals were detected in the WT leaves (Figures 4A–F). Additionally, we extracted leaf DNA from the spl39 mutant and WT plants and conducted gel electrophoresis, which revealed substantial DNA degradation in the spl39 mutant (Figure 4G). These findings suggest the presence of DNA damage and PCD in the spl39 mutant, ultimately contributing to the development of the lesion-mimic phenotype observed in spl39 plants.
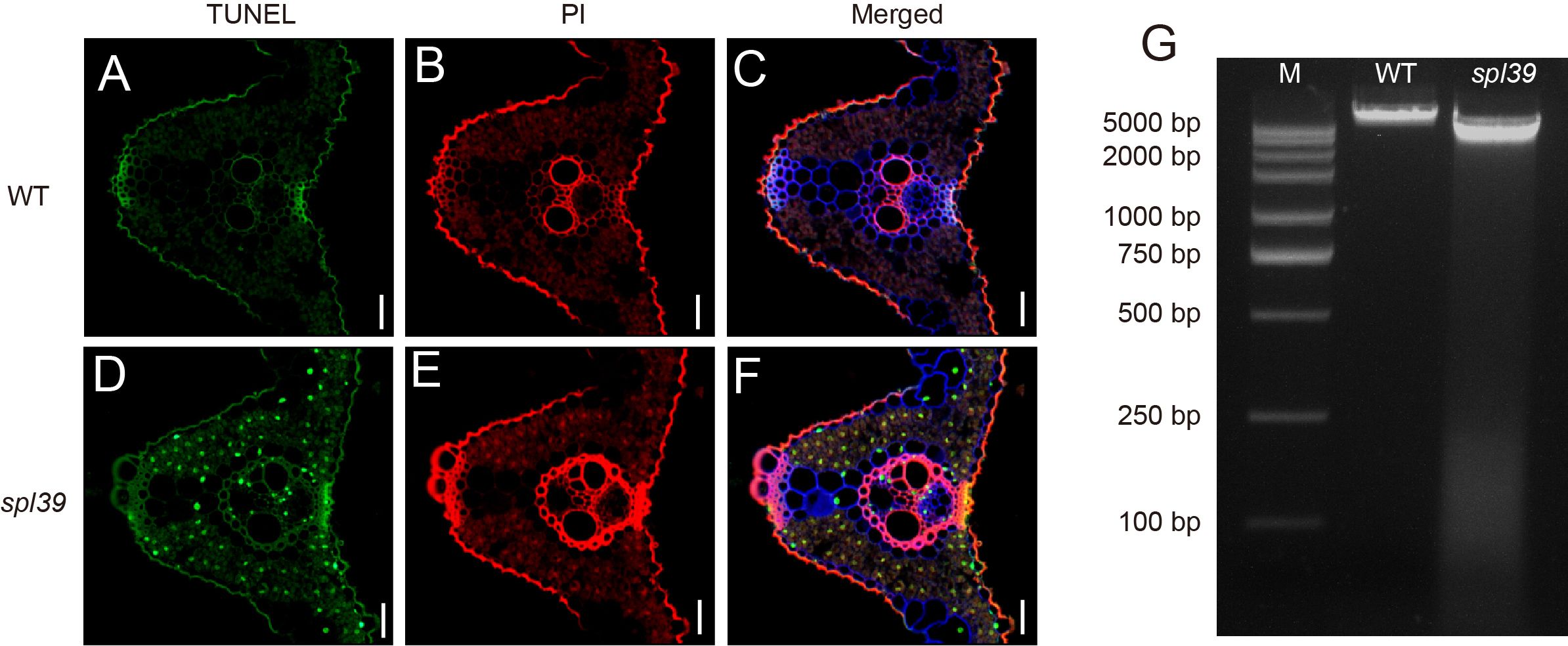
Figure 4. Increased DNA fragmentation and cell death in the spl39 mutant. (A–C) TUNEL assay in WT leaves. (D–F) TUNEL assay in spl39 leaves. Green represents TUNEL-positive signals, red signal indicates PI staining. Bar = 50 μm. (G) DNA fragmentation analysis by agarose gel electrophoresis. Lane M: DNA marker; Lane WT: Wild-type DNA showing intact genomic bands; Lane spl39: Mutant DNA exhibiting a characteristic laddering pattern, indicative of apoptotic-like DNA fragmentation.
3.3 Map-based cloning of SPL39
To investigate the molecular basis of the lesion phenotypes observed in the spl39 mutant, we conducted several genetic and molecular analyses. Firstly, we performed a cross between the spl39 mutant and its WT, and all F1 individuals exhibited the WT phenotype. In the F2 population, we observed 262 normal plants and 82 plants with leaf lesions. The segregation ratio (x2 [3:1] = 0.256 < x20.05 = 3.84; P > 0.05) suggested that the lesion phenotype in the spl39 mutant is controlled by a single nuclear recessive gene. To map the SPL39 locus, we crossed the spl39 mutant with 93-11, a wild type polymorphic indica cultivar, to generate a mapping population. Using a map-based cloning approach, we narrowed down the spl39 locus between molecular markers BSR22 and BSR25 on chromosome 2. Subsequently, we further narrowed it down to an approximate 410 kb region between Indels FM6 and FM7 (Figure 5A). Further fine mapping revealed that a specific marker primer failed to amplify a product in the spl39 mutant. This observation suggested the presence of a genomic deletion in spl39 mutant. To confirm and characterize the size and nature of this deletion, a chromosome walking strategy was employed (Figure 5B). Amplified fragments were sequenced and aligned with reference genome data (Figures 5C, D). The results revealed that the spl39 mutant harbors a deletion of approximately 306 kb (Figure 5). Within the deleted region, 33 genes were identified (Supplementary Table S1), including: 12 genes belonging to the diterpenoid biosynthesis gene cluster on chromosome 2 (c2BGC), which is crucial for the biosynthesis of phytoalexins. These metabolites are directly involved in resistance to rice blast and sheath blight disease. 18 genes encoding transposon proteins, hypothetical proteins, or proteins of unknown function. 4 additional genes with distinct functions: LOC_Os02g35970, which encodes an NPH3 domain-containing protein, a key signal transduction component in higher plant phototropism. LOC_Os02g36000, which encodes a putative zinc finger protein. LOC_Os02g36300, which encodes a zinc finger protein containing a C3HC4-type domain. LOC_Os02g36320, which encodes a putative RING-H2 finger protein. This deletion likely disrupts both diterpenoid biosynthesis and other important signaling or regulatory pathways, offering insights into the molecular basis of the observed lesion mimic phenotype and enhanced disease resistance in the spl39 mutant.
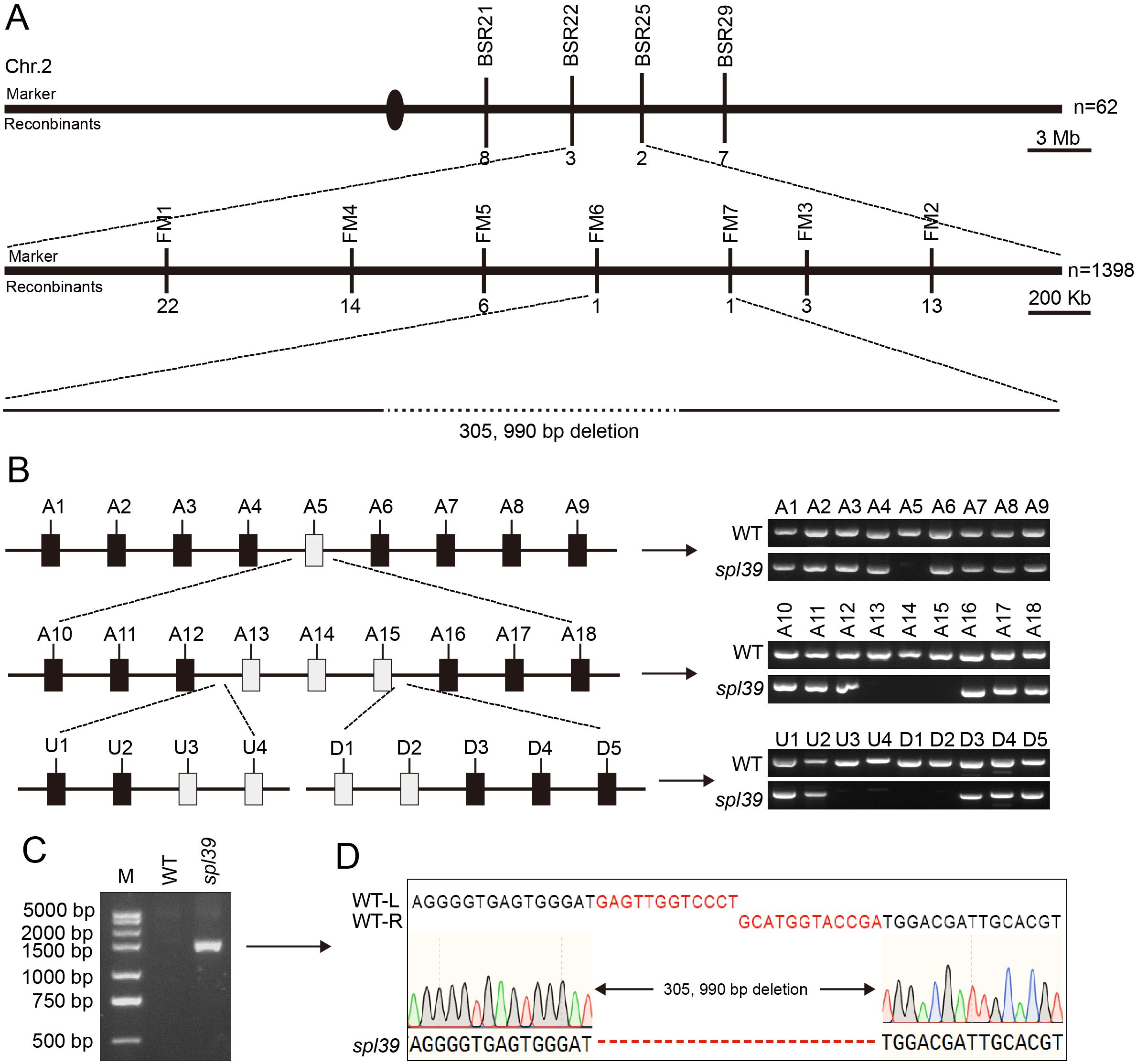
Figure 5. Mapping and identification of the deletion in spl39 mutant. (A) Fine mapping of the spl39 locus on chromosome 2. The rough mapping population (n = 62) identified a candidate region of ~3 Mb, which was further narrowed to a ~400 Kb region using a larger population (n = 1,398). A 305,990 bp deletion was identified in the spl39 mutant. (B) PCR verification of the deletion region. Schematic representation of the candidate region with primer sets (A1–A18, U1–U3, D1–D5) targeting various loci. PCR analysis confirmed the absence of amplicons in the spl39 mutant for specific loci within the deleted region. (C) Gel electrophoresis analysis of the deletion. A significantly shorter PCR fragment was detected in spl39, confirming the large deletion. (D) DNA sequencing of the deletion breakpoint. The sequences flanking the 305,990 bp deletion were aligned between WT and spl39, revealing the precise deletion junction.
3.4 Transcriptomic analysis of the spl39 mutant
To investigate the molecular basis of the lesion mimic phenotype in the spl39 mutant, we performed a comprehensive transcriptomic analysis comparing spl39 to WT plants. The results revealed significant alterations in gene expression, with distinct biological processes and pathways being either upregulated or downregulated in the mutant. A total of differentially expressed genes (DEGs) was identified in the spl39 mutant, as shown in the volcano plot (Figure 6A). A significant number of genes were either upregulated (2832) or downregulated (2128) in comparison to the WT, indicating profound transcriptomic reprogramming. These changes reflect disruptions in metabolic, developmental, and immune-related processes.
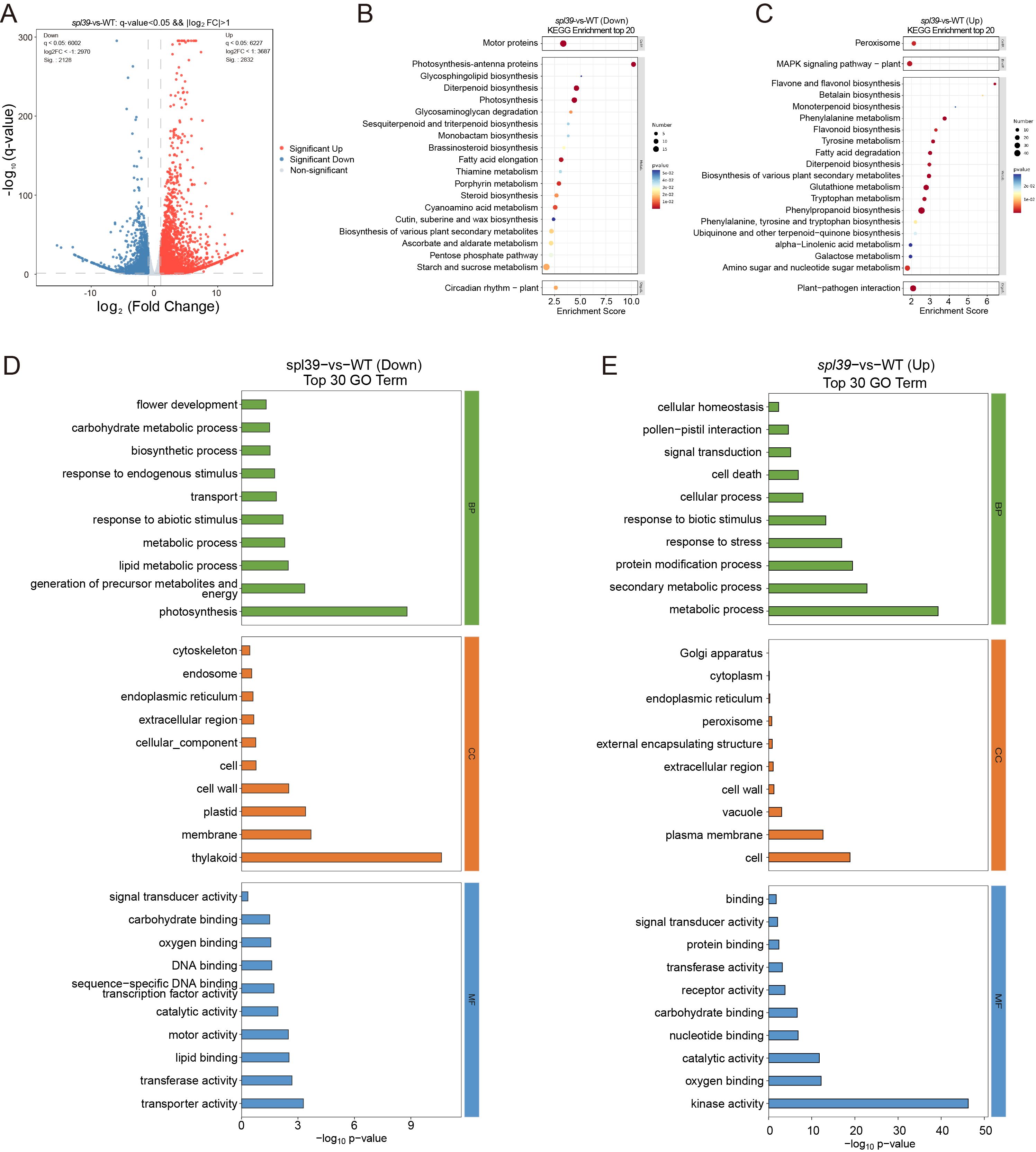
Figure 6. Differential gene expression and functional enrichment analysis in the spl39 mutant. (A) Volcano plot of differentially expressed genes (DEGs) in spl39 vs. WT. Genes significantly upregulated (red) and downregulated (blue) are highlighted. The horizontal axis represents the log2 (fold change), and the vertical axis represents the -log10 (p-value). (B, C) KEGG pathway enrichment analysis of downregulated (B) and upregulated (C) genes in spl39. Pathways with significant enrichment include photosynthesis, secondary metabolite biosynthesis, and hormone signaling pathways. The enrichment score represents the degree of pathway significance. (D, E) Gene Ontology (GO) analysis of downregulated (D) and upregulated (E) genes in spl39.
An enrichment analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways revealed distinct functional changes: 1. Downregulated Pathways (Figure 6B): The analysis highlighted a suppression of pathways associated with photosynthesis, thylakoid structure, fatty acid biosynthesis, and carbohydrate metabolism in the spl39 mutant. These findings align with the observed ultrastructural damage to chloroplasts and reduced photosynthetic capacity, as evident from the significant loss of thylakoids and membrane integrity in the mutant. 2. Upregulated Pathways (Figure 6C): Conversely, pathways related to plant-pathogen interactions, secondary metabolite biosynthesis, and stress-response signaling were significantly activated in the spl39 mutant. Notably, pathways involved in flavonoid and diterpenoid biosynthesis, MAPK signaling, and hormone signaling were enriched, indicating the mutant’s enhanced immune and stress responses.
To further understand the functional implications of DEGs, Gene Ontology (GO) enrichment analysis was performed, categorizing genes into biological processes, cellular components, and molecular functions. 1. Downregulated Genes (Figure 6D): Biological Processes: Significant suppression was observed in processes related to photosynthesis, carbohydrate metabolism, and lipid metabolism. These changes are consistent with the observed chloroplast damage and reduced photosynthetic activity. Cellular Components: Genes associated with plastids, thylakoids, and membranes were notably downregulated, further supporting the impaired chloroplast ultrastructure. Molecular Functions: A reduction in activities related to DNA binding, transferase activity, and transporter activity was detected, suggesting a decline in primary metabolic and regulatory processes. 2. Upregulated Genes (Figure 6E): Biological Processes: Genes involved in cell death, stress responses, and secondary metabolic processes were strongly enriched in the mutant. This highlights the activation of immune responses and metabolic reprogramming in the spl39 mutant. Cellular Components: Enrichment of genes associated with cellular structures like the Golgi apparatus, peroxisomes, and plasma membranes indicate the activation of stress-related processes and secondary metabolite transport. Molecular Functions: Enhanced activity of kinases, signal transduction proteins, and catalytic enzymes was observed, reflecting the heightened activation of immune signaling pathways.
Take together, the transcriptomic analysis revealed a complex interplay between metabolic suppression and immune activation in the spl39 mutant. The downregulation of photosynthesis-related pathways and damage to chloroplast structure likely contribute to the lesion mimic phenotype, while the upregulation of secondary metabolite biosynthesis, stress-response pathways, and immune signaling may underlie the mutant’s enhanced disease resistance.
3.5 Increased resistance to bacterial blight in spl39 mutant
Through the above RNAseq analysis, we observed that a large proportion of defense-related genes were significantly upregulated in the spl39 mutant compared to the wild type, suggesting that spl39 may play a role in disease resistance. To validate these findings, we inoculated both the spl39 mutant and WT plants with multiple virulent strains of Xanthomonas oryzae pv. oryzae (Xoo), including PXO61, PXO86, PXO79, PXO71, PXO112, PXO99, PXO145, and PXO280. The spl39 mutant exhibited significantly enhanced resistance, as evidenced by markedly smaller and fewer lesions compared to the WT (Figure 7A). Quantitative analysis further confirmed this observation, with lesion lengths in spl39 being significantly reduced across all tested strains (Figure 7B).
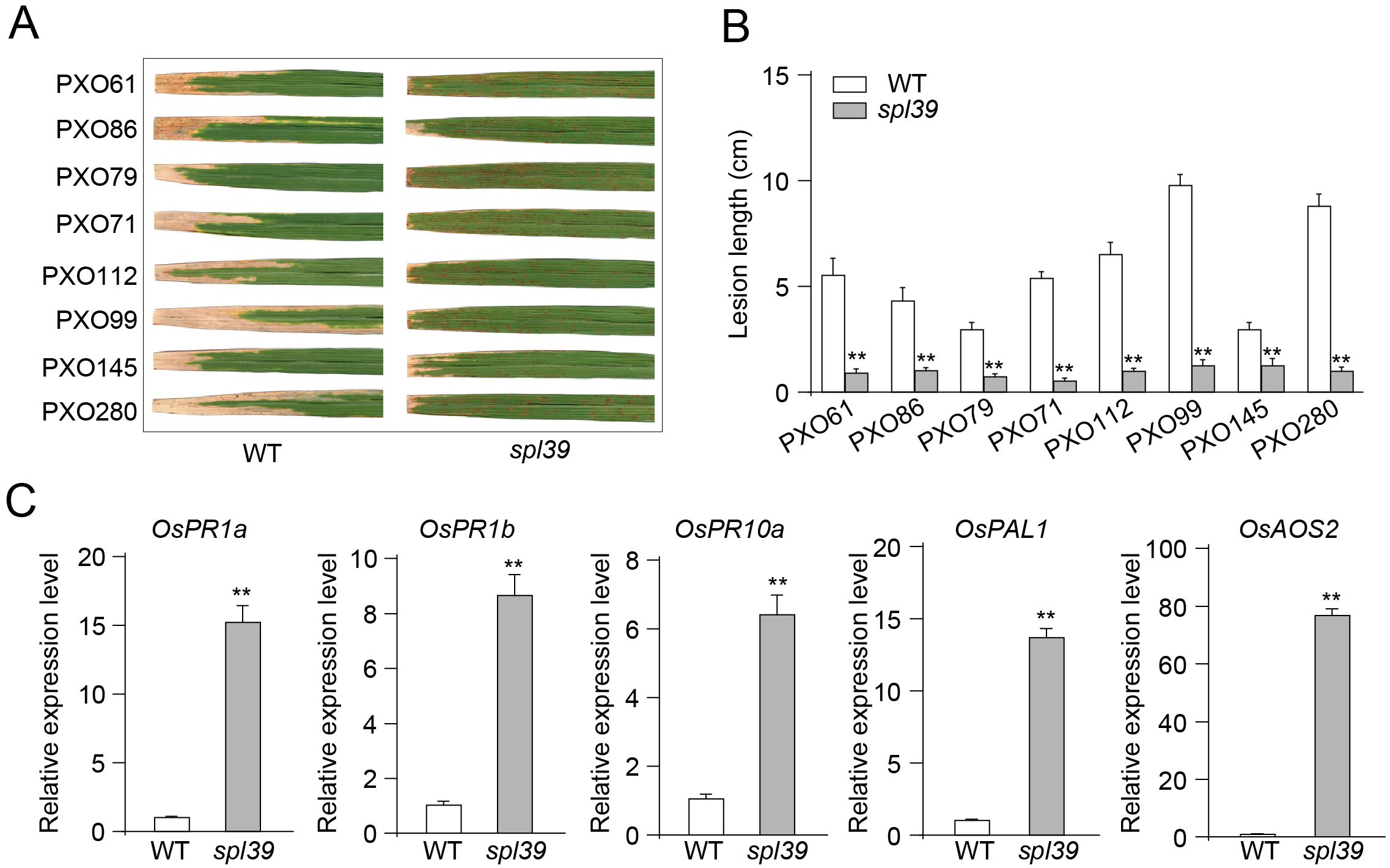
Figure 7. Enhanced resistance to Xanthomonas oryzae pv. oryzae (Xoo) and upregulation of defense-related genes in spl39 mutant. (A) Lesion phenotypes of WT and spl39 leaves after inoculation with eight different Xoo strains (PXO61, PXO86, PXO79, PXO71, PXO112, PXO99, PXO145, and PXO280). The spl39 mutant exhibits significantly reduced lesion formation compared to WT. (B) Quantification of lesion length (cm) in WT and spl39 after infection with Xoo strains. **P < 0.01 (Student’s t-test), error bars, ± SD (n=6). (C) Relative expression levels of defense-related genes in WT and spl39. The spl39 mutant shows significantly higher expression of these genes, indicating enhanced immune responses. Expression levels were normalized to reference genes. **P < 0.01 (Student’s t-test), error bars, ± SD (n=3).
To further investigate the molecular basis of this enhanced resistance, we analyzed the expression levels of key defense-related genes using quantitative PCR (qPCR). The results revealed that the expression of OsPR1a, OsPR1b, and OsPR10a, which are classic markers of defense responses, was significantly upregulated in spl39 mutant compared to the WT (Figure 7C). Additionally, OsPAL1, a gene involved in phenylpropanoid metabolism, and OsAOS2, a critical enzyme in the jasmonic acid biosynthesis pathway, also showed significantly higher expression levels in the mutant. These findings indicate that the spl39 mutant exhibits a pre-activated or heightened defense state, which likely contributes to its enhanced resistance to bacterial leaf blight.
3.6 Significant hormone accumulation in the spl39 mutant
Through the above RNAseq analysis, we observed that genes involved in hormone signaling pathways were significantly upregulated and enriched in spl39 mutant compared to the wild type. This suggests that the activation of hormone signaling may play a critical role in the enhanced disease resistance observed in the mutant. Since elevated hormone levels are known to be major contributors to disease resistance in rice, we further measured the hormone levels in the spl39 mutant to validate the transcriptomic findings and explore their potential roles in resistance mechanisms. To investigate the role of hormones in the enhanced disease resistance of the spl39 mutant, we quantified the levels of several phytohormones, including salicylic acid (SA), jasmonic acid (JA), abscisic acid (ABA), auxin (IAA), and cytokinins (e.g., trans-zeatin [tZ], cis-zeatin [cZ], isopentenyl adenine [iP], and isopentenyl riboside [iPR]). The results revealed significant alterations in hormone levels in the spl39 mutant compared to the WT, the hormone profiles demonstrated that most hormones were elevated in the spl39 mutant relative to the WT, except for ABA, which showed no significant change (Figure 8). This activation may contribute to the heightened immune response and enhanced disease resistance observed in the spl39 mutant. The accumulation of SA, JA, and their precursors likely plays a central role in the mutant’s enhanced resistance to bacterial pathogens, while the upregulation of auxins and cytokinins suggests a broader reprogramming of growth and defense trade-offs. These findings underscore the complex hormonal crosstalk that underlies the defense mechanisms in the spl39 mutant.
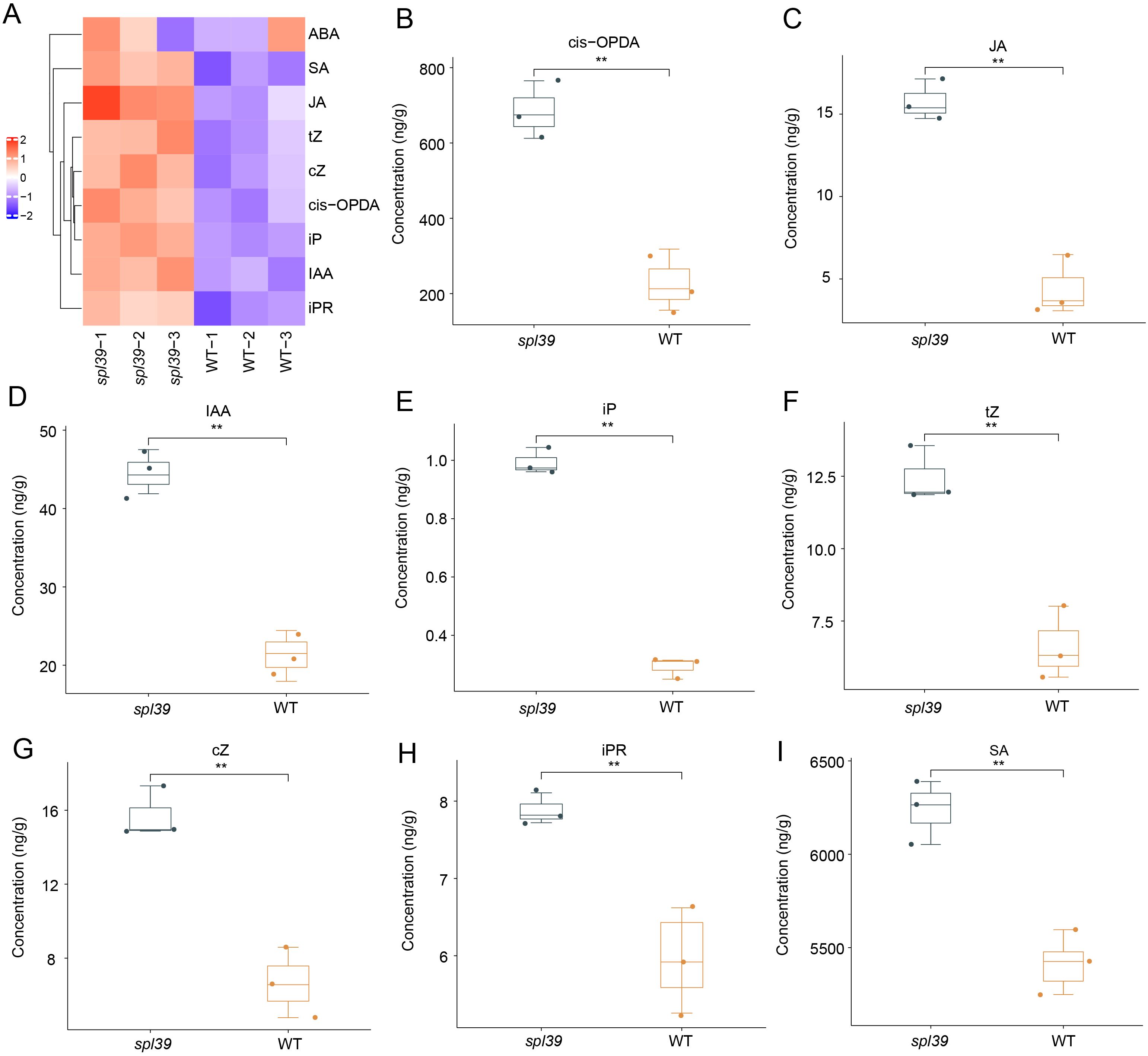
Figure 8. Altered phytohormone levels in the spl39 mutant. (A) Heatmap representation of hormone concentrations in WT and spl39. Hormones analyzed include abscisic acid (ABA), salicylic acid (SA), jasmonic acid (JA), cis-12-oxo-phytodienoic acid (cis-OPDA), trans-zeatin (tZ), cis-zeatin (cZ), isopentenyl adenine (iP), indole-3-acetic acid (IAA), and isopentenyl riboside (iPR). (B–I) Box plots showing significantly altered hormone concentrations in spl39 compared to WT. **P < 0.01 (Student’s t-test), error bars, ± SD (n=3).
3.7 Increased sensitivity to salt stress in spl39 mutant
The spl39 mutant is characterized by a large fragment deletion, raising the question of whether the deleted region includes genes involved in environmental stress responses. To address this, we subjected both WT and the spl39 mutant plants to NaCl treatment and evaluated their physiological and biochemical responses. Under normal conditions, no significant differences were observed between WT and spl39 plants. However, upon salt stress, the spl39 mutant exhibited severe growth inhibition and significantly lower survival rates compared to WT plants (Figures 9A, B). Biochemical analyses revealed that salt stress caused a marked increase in H2O2 (Figure 9C) and MDA (Figure D) levels in the spl39 mutant, indicating heightened oxidative damage. Further investigation of antioxidant enzyme activities showed significant alterations in the spl39 mutant under salt stress. The activities of SOD (Figure 9E) and CAT (Figure 9F) were notably reduced, while POD activity (Figure 9G) was significantly elevated. These results indicate that genes within the deleted spl39 region are likely critical for regulating oxidative stress responses and maintaining antioxidant enzyme activity under environmental stress.
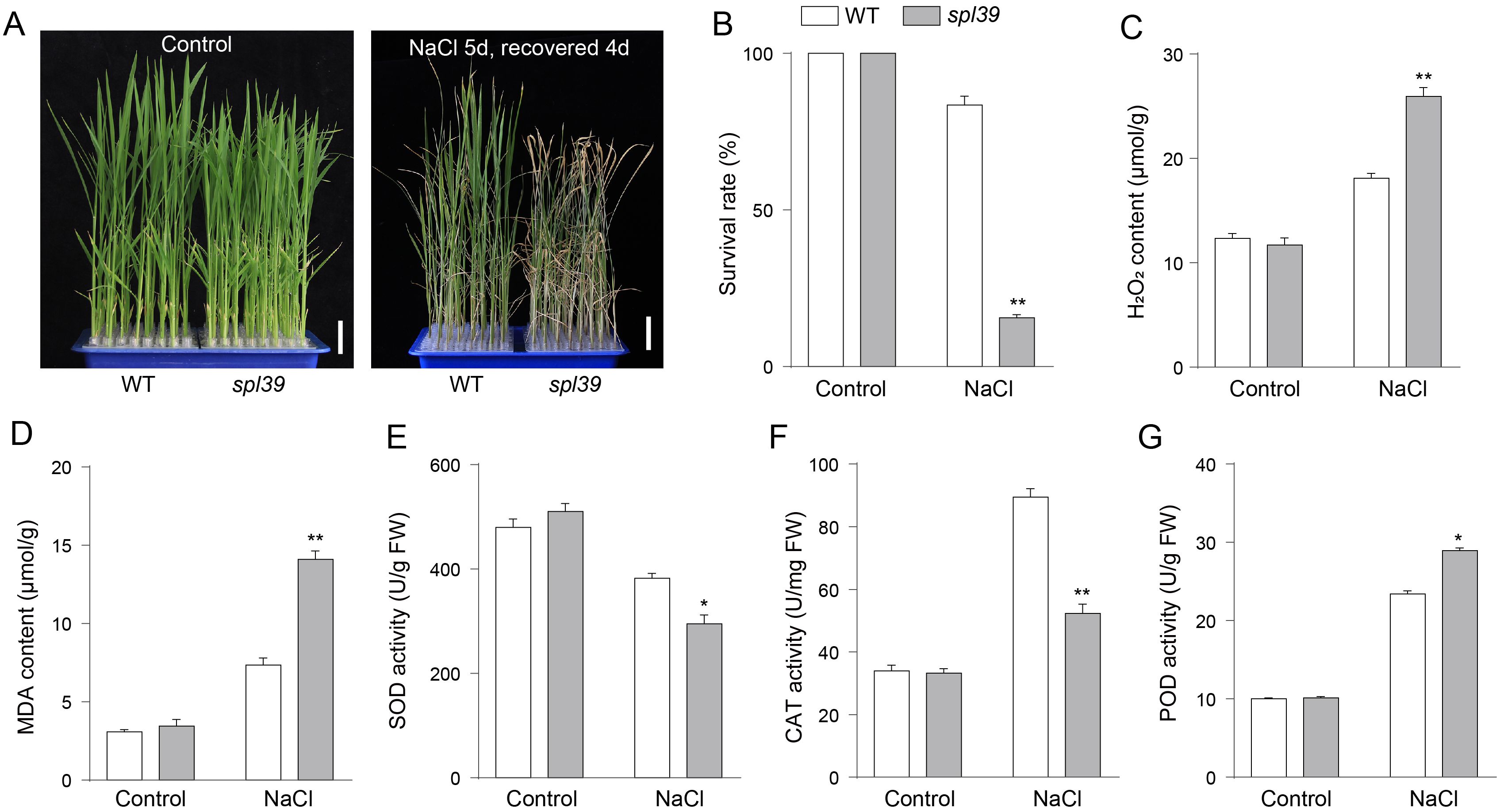
Figure 9. The spl39 mutant exhibits increased sensitivity to salt stress. (A) Phenotypic comparison of WT and spl39 seedlings under control conditions (left) and after 150 mM NaCl treatment for 5 days followed by a 4-day recovery (right). The spl39 plants showed severe wilting and chlorosis after salt stress. Scale bars = 2 cm. (B) Survival rate (%) of WT and spl39 under control and NaCl treatment conditions. (C, D) Quantification of H2O2 (C) and MDA (D) contents under both control and NaCl-treated conditions. (E–G) Antioxidant enzyme activities under control and NaCl conditions: (E) Superoxide dismutase (SOD), (F) Catalase (CAT), (G) Peroxidase (POD). *P < 0.05, **P < 0.01 (Student’s t-test), error bars, ± SD (n=3).
Taken together, our findings suggest that the large fragment deletion in spl39 includes genes essential for salt stress tolerance, as evidenced by the spl39 mutant’s compromised ability to manage oxidative stress. This study sheds light on the molecular functions associated with spl39 and its deleted region, providing valuable targets for improving stress resilience in rice.
4 Discussion
LMMs are commonly utilized in research to explore defense responses and PCD in plants as they can autonomously generate necrotic lesions without requiring pathogenic infection. In this study, we identified a novel lesion-mimic mutant, spl39, from a rice mutant library (Oryza sativa L. japonica cv. Wuyunjing7) treated with heavy ion beam irradiation. Map-based cloning revealed that the spl39 mutant harbors a 306-kb deletion encompassing 33 genes. Further analysis showed that this deletion led to the altered expression of numerous metabolism-related and hormone biosynthesis genes, conferring enhanced disease resistance in the spl39 mutant. However, the deletion also resulted in significantly reduced salt tolerance in the spl39 mutant, suggesting that the missing genes might be involved in regulating salt stress tolerance in rice. These findings provide a foundation for further functional studies to elucidate the roles of these genes in stress adaptation and plant growth regulation.
4.1 The spl39 mutant accumulates ROS and triggers cell death
Reactive oxygen species (ROS) play pivotal roles in plant development and stress responses, maintaining their functionality through a delicate balance between ROS production and scavenging mechanisms (Hu et al., 2011). Elevated ROS levels in rice can lead to oxidative damage of cell membranes, increasing permeability and ultimately causing cell death and spot-like lesions on leaves (Cui et al., 2021). Among these, H2O2, a key ROS molecule generated through endogenous metabolic pathways, is closely associated with programmed cell death (PCD) when excessively accumulated in plant cells. Our study demonstrated a marked increase in H2O2 levels in the spl39 mutant leaves, validated by histochemical staining and quantitative measurements (Figures 2A–D). Antioxidative enzymes, including SOD, CAT, APX and POD, are critical for maintaining ROS homeostasis by scavenging excessive ROS to support normal metabolic and signaling processes. In the spl39 mutant, the activities of these enzymes were significantly reduced, suggesting an impaired ROS scavenging capacity. This imbalance in ROS clearance likely contributed to ROS overaccumulation, leading to oxidative stress and eventual cell death. Therefore, we propose that the disruption of ROS homeostasis in the spl39 mutant is a key factor underlying its lesion mimic phenotype.
PCD in plants is a highly regulated, multi-step process orchestrated by genetic and biochemical factors (Locato and De Gara, 2018; Yan et al., 2022). LMMs, such as spl39, provide valuable tools for dissecting the molecular mechanisms of hypersensitive response (HR)-mediated cell death. Studies of LMMs have identified key factors associated with PCD, including excessive ROS accumulation, cellular structure damage, and DNA degradation (Wang et al., 2017). Our findings revealed that spl39 mutant exhibits hallmark features of PCD, such as increased ROS levels and pronounced DNA fragmentation. To further explore the involvement of spl39 in ROS-mediated cell death, we utilized multiple histochemical assays. Nitro blue tetrazolium (NBT) staining highlighted O₂⁻• accumulation, while diaminobenzidine (DAB) staining revealed high levels of H2O2 in the spl39 mutant leaves. Additionally, trypan blue staining demonstrated irreversible membrane damage (Figures 2A–C). Since DNA degradation is a defining feature of PCD, we employed the TUNEL assay to assess DNA fragmentation. The results revealed a significant increase in TUNEL-positive nuclei in spl39 mutant leaves, particularly near necrotic regions (Figure 4). This evidence strongly supports that the spl39 mutant plays an essential role in regulating ROS-mediated cell death in rice.
4.2 The spl39 mutant is a large fragment deletion mutant
The spl39 mutant was identified from a rice mutant library generated through heavy-ion beam mutagenesis. Observations of developmental phenotypes revealed that the spl39 mutant exhibits pleiotropic traits, including reduced plant height (Figures 1B, G), fewer tillers (Figure 1H), smaller grains (Figures 1F, K–M), lower seed setting rates (Figure 1N), and the appearance of distinct lesion mimic phenotypes after the tillering stage (Figures 1C, D). Additionally, the mutant displayed significant sensitivity to salt stress (Figure 9). Map-based cloning indicated that the spl39 mutation is characterized by a 306-kb deletion, resulting in the loss of 33 genes within this region (Figure 5). The absence of these genes likely contributes to the diverse phenotypic abnormalities observed in the mutant, including its heightened susceptibility to salt stress and the development of lesion mimic traits. The spl39 mutant provides a valuable resource for studying the functional roles of these deleted genes, particularly in processes related to plant growth, stress responses, and programmed cell death.
The spl39 mutant carries a 306-kb genomic deletion that primarily encompasses the chromosome 2 diterpenoid biosynthetic gene cluster (c2BGC). In rice, diterpenoid skeleton construction and modification are coordinately mediated by Terpene Synthase (TPS) genes and Cytochrome P450 (CYP) genes families. However, since diterpenoid biosynthesis genes are distributed not only on chromosome 2 but also on chromosomes 4, 6, and 11, the deletion of partial genes may not be lethal due to functional compensation by other homologous members. Therefore, functional redundancy among these genes likely explains why the large chromosomal deletion in spl39 mutant does not cause lethality.
The involvement of diterpenoid biosynthetic gene clusters (BGCs) within this deletion region underscores the critical role of secondary metabolites, such as phytoalexins, in balancing biotic and abiotic stress responses. Studies on related mutants, such as g380 and c2BGC deletion lines, highlight the significance of diterpenoid BGCs in regulating plant immunity (Jiang et al., 2022; Li et al., 2022). The absence of diterpenoid biosynthetic genes, particularly those coding for cytochrome P450 enzymes like CYP76M7 and CYP76M8, was shown to induce spontaneous lesion mimic phenotypes, elevate ROS levels, and activate defense-related pathways. Similarly, spl39 displayed increased bacterial blight resistance (Figure 7), which is likely linked to the upregulation of pathogenesis-related (PR) genes and enhanced salicylic acid (SA) and jasmonic acid (JA) signaling pathways (Figure 8). These observations emphasize how diterpenoid biosynthetic pathways contribute to immune reprogramming and resistance mechanisms in rice.
In contrast, the spl39 mutant exhibited heightened sensitivity to salt stress, potentially due to the loss of genes involved in ROS scavenging and oxidative stress regulation within the deleted region (Figure 9). This finding aligns with reports that large deletions affecting BGCs can disrupt ROS homeostasis, as seen in g380, where impaired ROS-scavenging enzyme activities led to oxidative stress and cell death. Such dual effects, enhanced immunity coupled with abiotic stress sensitivity, highlight the complexity of trade-offs mediated by secondary metabolites and stress-response pathways. The directional cross-cluster effects observed in c2BGC and c4BGC deletion mutants may also provide insights into the interactions between metabolic pathways in spl39. The deletion of multiple BGCs can alter the accumulation of intermediates, leading to phytotoxicity or modified stress responses. These findings suggest that the interplay between remaining metabolic pathways in spl39 could similarly influence its observed phenotypes.
Overall, the spl39 study, combined with evidence from related mutants, underscores the potential of heavy-ion beam mutagenesis for uncovering functional roles of large genomic regions, particularly in secondary metabolism and stress adaptation. Further research focusing on the specific functions of individual deleted genes within the spl39 locus will provide deeper insights into the molecular mechanisms driving these complex traits and could inform strategies for enhancing crop resilience and productivity. In the future, the spl39 mutant could be used as a platform to study plant responses under various environmental conditions, coupled with CRISPR-Cas9 technology to generate single-gene knockouts of the deleted genes. This approach would enable targeted functional analyses, uncover the precise roles of each gene and providing a comprehensive understanding of their contributions to stress responses, metabolism, and plant development.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
CF: Writing – original draft, Validation. WM: Writing – original draft, Investigation. XZ: Investigation, Writing – review & editing. LP: Data curation, Writing – original draft. LT: Validation, Writing – original draft. ZZ: Formal analysis, Writing – review & editing. FW: Methodology, Writing – review & editing. YY: Methodology, Writing – original draft. BM: Methodology, Writing – review & editing. YW: Methodology, Writing – review & editing. JY: Data curation, Writing – original draft. BL: Methodology, Writing – original draft. XC: Methodology, Writing – original draft. WC: Data curation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Key Scientific and Technological Innovation Cultivation Program of the Xinjiang Academy of Agricultural Sciences(xjkcpy-2022007), the Tianshan Innovation Team Program of the Department of Science and Technology of Xinjiang Uygur Autonomous Region (2023D14017), the Youth Innovation Promotion Association CAS (2022454), Joint research project of Yangtze River Delta science and technology innovation community (2024CSJZN1100), Anhui Provincial Key Research and Development Project (310180826033) and the Irradiation Terminal for Life Science and Shallow-Seated Tumor Therapy (TR4) of Heavy Ion Research Facility in Lanzhou (31111.02.HIRFL).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1639365/full#supplementary-material.
References
Cheng, W., Ren, Y., Wang, J., Chen, C., Fang, C., Peng, L., et al. (2024). Mutation of GLR2 confers enhanced glufosinate resistance and salt tolerance in rice. Plant Physiol. 197, 1–4. doi: 10.1093/plphys/kiae588
Cui, Y. J., Peng, Y. L., Zhang, Q., Xia, S. S., Ruan, B. P., Xu, Q. K., et al. (2021). Disruption of EARLY LESION LEAF 1, encoding a cytochrome P450 monooxygenase, induces ROS accumulation and cell death in rice. Plant J. 105, 942–956. doi: 10.1111/tpj.15079
Dickman, M. B. and Fluhr, R. (2013). Centrality of host cell death in plant-microbe interactions. Annu. Rev. Phytopathol. 51, 543–570. doi: 10.1146/annurev-phyto-081211-173027
Hu, L., Liang, W., Yin, C., Cui, X., Zong, J., Wang, X., et al. (2011). Rice MADS3 regulates ROS homeostasis during late anther development. Plant Cell 23, 515–533. doi: 10.1105/tpc.110.074369
Jiang, M., Yu, N., Zhang, Y., Liu, L., Li, Z., Wang, C., et al. (2022). Deletion of diterpenoid biosynthetic genes CYP76M7 and CYP76M8 induces cell death and enhances bacterial blight resistance in indica rice ‘9311’. Int. J. Mol. Sci. 23, 1–19. doi: 10.3390/ijms23137234
Kitaoka, N., Zhang, J., Oyagbenro, R. K., Brown, B., Wu, Y. S., Yang, B., et al. (2021). Interdependent evolution of biosynthetic gene clusters for momilactone production in rice. Plant Cell 33, 290–305. doi: 10.1093/plcell/koaa023
Lam, E., Kato, N., and Lawton, M. (2001). Programmed cell death, mitochondria and the plant hypersensitive response. Nature 411, 848–853. doi: 10.1038/35081184
Li, W. H., Cheng, W. M., Jiang, H. R., Fang, C., Peng, L. L., Tao, L. Z., et al. (2024). Mutation of rice EARLY LEAF LESION AND SENESCENCE 1 (ELS1), which encodes an anthranilate synthase α-subunit, induces ROS accumulation and cell death through activating the tryptophan synthesis pathway in rice. Plant J. 120, 2723–2737. doi: 10.1111/tpj.17141
Li, R., Zhang, J., Li, Z., Peters, R. J., and Yang, B. (2022). Dissecting the labdane-related diterpenoid biosynthetic gene clusters in rice reveals directional cross-cluster phytotoxicity. New Phytol. 233, 878–889. doi: 10.1111/nph.17806
Locato, V. and De Gara, L. (2018). Programmed cell death in plants: an overview. Methods Mol. Biol. 1743, 1–8. doi: 10.1007/978-1-4939-7668-3_1
Ma, J., Wang, Y. F., Ma, X. D., Meng, L. Z., Jing, R. N., Wang, F., et al. (2019). Disruption of gene SPL35, encoding a novel CUE domain-containing protein, leads to cell death and enhanced disease response in rice. Plant Biotechnol. J. 17, 1679–1693. doi: 10.1111/pbi.13093
Qiao, Y., Jiang, W., Lee, J., Park, B., Choi, M. S., Piao, R., et al. (2010). SPL28 encodes a clathrin-associated adaptor protein complex 1, medium subunit mu 1 (AP1M1) and is responsible for spotted leaf and early senescence in rice (Oryza sativa). New Phytol. 185, 258–274. doi: 10.1111/j.1469-8137.2009.03047.x
Shimura, K., Okada, A., Okada, K., Jikumaru, Y., Ko, K. W., Toyomasu, T., et al. (2007). Identification of a biosynthetic gene cluster in rice for momilactones. J. Biol. Chem. 282, 34013–34018. doi: 10.1074/jbc.M703344200
Wang, Q., Hillwig, M. L., Okada, K., Yamazaki, K., Wu, Y., Swaminathan, S., et al. (2012). Characterization of CYP76M5–8 indicates metabolic plasticity within a plant biosynthetic gene cluster. J. Biol. Chem. 287, 6159–6168. doi: 10.1074/jbc.M111.305599
Wang, S. A., Lei, C. L., Wang, J. L., Ma, J., Tang, S., Wang, C. L., et al. (2017). SPL33, encoding an eEF1A-like protein, negatively regulates cell death and defense responses in rice. J. Exp. Bot. 68, 899–913. doi: 10.1093/jxb/erx001
Wang, Z. H., Wang, Y., Hong, X., Hu, D. H., Liu, C. X., Yang, J., et al. (2015). Functional inactivation of UDP-N-acetylglucosamine pyrophosphorylase 1 (UAP1) induces early leaf senescence and defence responses in rice. J. Exp. Bot. 66, 973–987. doi: 10.1093/jxb/eru456
Wellburn, A. R. (1994). The spectral determination of chlorophyll-a and chlorophhyll-B, as well as total carotenoids, using various solvents with spectrophotometers of different resolution. J. Plant Physiol. 144, 307–313. doi: 10.1016/S0176-1617(11)81192-2
Wu, L. W., Ren, D. Y., Hu, S. K., Li, G. M., Dong, G. J., Jiang, L., et al. (2016). Down-regulation of a nicotinate phosphoribosyltransferase gene, osNaPRT1, leads to withered leaf tips. Plant Physiol. 171, 1085–1098. doi: 10.1104/pp.15.01898
Yan, J., Fang, Y., and Xue, D. (2022). Advances in the genetic basis and molecular mechanism of lesion mimic formation in rice. Plants (Basel) 11, 1–12. doi: 10.3390/plants11162169
Yuchun, R. A. O., Ran, J., Sheng, W., Xianmei, W. U., Hanfei, Y. E., Chenyang, P. A. N., et al. (2021). SPL36 encodes a receptor-like protein kinase that regulates programmed cell death and defense responses in rice. Rice (N Y) 14, 34. doi: 10.1186/s12284-021-00475-y
Zeng, L. R., Qu, S. H., Bordeos, A., Yang, C. W., Baraoidan, M., Yan, H. Y., et al. (2004). Spotted leaf11, a negative regulator of plant cell death and defense, encodes a U-box/armadillo repeat protein endowed with E3 ubiquitin ligase activity. Plant Cell 16, 2795–2808. doi: 10.1105/tpc.104.025171
Zhang, P., Ma, X. D., Liu, L. A., Mao, C. J., Hu, Y. K., Yan, B. X., et al. (2023). MEDIATOR SUBUNIT 16 negatively regulates rice immunity by modulating PATHOGENESIS RELATED 3 activity. Plant Physiol. 192, 1132–1150. doi: 10.1093/plphys/kiad120
Keywords: rice, spl39, lesion-mimic mutant, reactive oxygen species, genomic deletion, disease resistance
Citation: Fang C, Ma W, Zhu X, Peng L, Tao L, Zhao Z, Wang F, Ye Y, Ma B, Wu Y, Yuan J, Liu B, Chen X and Cheng W (2025) Large-scale genomic deletion in spl39 activates immune responses and confers resistance to rice bacterial blight. Front. Plant Sci. 16:1639365. doi: 10.3389/fpls.2025.1639365
Received: 02 June 2025; Accepted: 10 July 2025;
Published: 11 August 2025.
Edited by:
Dayun Tao, Yunnan Academy of Agricultural Sciences, ChinaReviewed by:
Yuxiang Zeng, Chinese Academy of Agricultural Sciences, ChinaWenhao Li, Zhejiang Normal University, China
Copyright © 2025 Fang, Ma, Zhu, Peng, Tao, Zhao, Wang, Ye, Ma, Wu, Yuan, Liu, Chen and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Yuan, eXVhbmppZTgwMTAyM0AxNjMuY29t; Binmei Liu, bGl1Ym1AaXBwLmFjLmNu; Xifeng Chen, eGZjaGVuQHpqbnUuY24=; Weimin Cheng, d21jaGVuZ0BpaW0uYWMuY24=
†These authors have contributed equally to this work