 Chandana B. S1‡
Chandana B. S1‡ Rohit Kumar Mahto1,2‡
Rohit Kumar Mahto1,2‡ Rajesh Kumar Singh1‡
Rajesh Kumar Singh1‡ Ramachandra V3K. K. Singh4
Ramachandra V3K. K. Singh4 Sunita Kushwah5Gera Roopa Lavanya6
Sunita Kushwah5Gera Roopa Lavanya6 Himabindu Kudapa7Vinod Kumar Valluri7
Himabindu Kudapa7Vinod Kumar Valluri7 Anil Kumar Vemula7
Anil Kumar Vemula7 Raju Ratan Yadav8,9Lal Bahadur Yadav8,9H. D. Upadhyaya7,10
Raju Ratan Yadav8,9Lal Bahadur Yadav8,9H. D. Upadhyaya7,10 Aladdin Hamwieh11*†
Aladdin Hamwieh11*† Rajendra Kumar1*
Rajendra Kumar1*- 1Division of Genetics, ICAR-Indian Agricultural Research Institute, New Delhi, India
- 2School of Biotechnology, Institute of Science, BHU, Varanasi, India
- 3Department of Genetics and Plant Breeding, UAS, GKVK, Bengaluru, India
- 4ICAR-Indian Agricultural Research Institute (IARI) Regional Station, Pusa, Bihar, India
- 5Krishi Vigyan Kendra (KVK), Vaishali (Dr. Rajendra Prasad Central Agriculture University- Pusa), Bihar, Samastipur, India
- 6Department of Genetics and Plant Breeding, SHUATS, Naini, Prayagraj, India
- 7International Crops Research Institute for the Semi-Arid Tropics (ICRISAT), Patancheru, Telangana, India
- 8Department of Molecular Biology and Genetic Engineering, Govind Ballabh Pant University of Agriculture and Technology, Pantnagar, Uttarakhand, India
- 9Department of Plant Pathology, Govind Ballabh Pant University of Agriculture and Technology, Pantnagar, Uttarakhand, India
- 10Plant Genome Mapping Laboratory, University of Georgia, Athens, GA, United States
- 11International Center for Agricultural Research in the Dry Areas (ICARDA), Giza, Egypt
Chickpea (Cicer arietinum L.) is the second most important food legume crop, capable of converting atmospheric nitrogen (N2) into ammonia (NH3) in symbiotic association with Mesorhizobium cicero through a process called biological nitrogen fixation (BNF). BNF shows promise in effectively diminishing reliance on exogenous nitrogen applications, enhancing soil sustainability and productivity in pulse crops. Notably, there are limited studies on the molecular basis of root nodulation in chickpea. In order to identify new sources of highly nodulating genotypes and gain deep insights into genomic regions governing BNF, a diverse chickpea global germplasm collection (284) was evaluated for nodulation and yield traits in four different environments in an augmented randomized block design. The genotypes exhibited significant trait variation, encompassing all traits under study. Correlation analysis revealed a significant positive correlation of nodulation traits on yield within the chickpea population. The genotypes ICC 7390, ICC 15, ICC 8348, and ICC 2474 were identified as high nodulating across the locations. Genome-wide association studies (GWAS) identified noteworthy and stable marker–trait associations (MTAs) linked to the traits of interest. For the traits number of nodules (NON) and nodule fresh weight (NFW), 65 and 109 significant MTAs were identified, respectively. In addition, two SNPs, Ca1pos289.52482.1 and 6_33340878, identified in our earlier studies were validated by independent population studies, which are crucial in evaluating the accuracy and reliability of the projections. Subsequent analysis revealed that a substantial proportion of these MTAs were situated within intergenic regions, with the potential to modulate genes associated with the focal traits. The candidate genes identified could be converted to Kompetitive allele-specific PCR (KASP) markers and exploited in marker-assisted breeding, accentuating their impact on future chickpea breeding efforts.
Introduction
The globally grown third most important pulse chickpea (Cicer arietinum L.), a self-pollinated diploid crop having a 2n=2x=16 chromosome number, is mainly grown during the winter season as an annual crop. The crop is cultivated over an area of 14.84 million ha with an average yield of 1.0 t/ha and a total production of 15.08 million tons (FAOSTAT, 2023). India is the leading producer of chickpeas, with an annual production of 11.5 million tons (Grasso et al., 2022). The chickpea seed matrix is composed of carbohydrates (50%–58%), protein (15%–22%), moisture (7%–8%), fat (3.8%–10.20%), and micronutrients (<1%) (Milán-Carrillo et al., 2007; Misra et al., 2016; Katoch et al., 2016; Kumar et al., 2021; Mittal et al., 2023). Moreover, chickpea is a rich source of minerals including iron (Fe), zinc (Zn), and selenium (Se) (Sharma et al., 2013).
Chickpea, like other fellow legumes, has the ability to fix nitrogen through symbiotic association. Symbiotic nitrogen fixation (SNF), a form of biological nitrogen fixation (BNF), is an important biological event that provides 97% of a plant’s total N requirement and agronomical and environmental benefits, allowing legumes to grow efficiently under nitrogen-limiting conditions (Peoples and Craswell, 1992). Because of the unique ability of the host plant to form a symbiotic relationship with a group of nitrogen-fixing bacteria called rhizobia, legumes represent an important and diverse group of plants, harboring 50%–70% of BNF, leading to a terrestrial input of 40–50 million tons of nitrogen per year (Vitousek et al., 1997). Chickpea carries out SNF by forming a symbiotic interaction with Mesorhizobium ciceri (Greenlon et al., 2019). This process of symbiosis and nodulation leading to N2 fixation is quite complex and tightly regulated, but very scantily understood at the molecular level. Some significant microbiological work on chickpea root nodule symbiosis with a focus on phenotypic and genotypic diversity among symbiotic vs. non-symbiotic bacteria revealed that 55% of isolated bacteria belong to the Mesorhizobium genus (Benjelloun et al., 2019). Recently, experiments were conducted on root nodulation-specific chickpea genotypes in varied soils (Chandana et al., 2023; Plett et al., 2021). The Mesorhizobium strains such as LMG15046, CC1192, XAP4, XAP10, and LMG14989 were highly effective strains for nodulation and growth promotion in chickpea (Wanjofu et al., 2022). Frailey et al. (2022) reported two mutations of rn1 and rn4 in the genotypes of PM233 and PM 405 that result in the absence of nodulation. The BNF is necessary for an environmentally friendly and sustainable agricultural production.
Understanding genes and genomic regions that support BNF is essential to increase efficiency and utilize the benefits. There have been few large-scale studies comparing chickpea nodulation and nitrogen production across multiple genotypes. Recent advancements in next-generation sequencing technologies as well as the availability of applicable tools such as genome-wide association studies (GWAS) have aided in the identification of single-nucleotide polymorphisms (SNPs) for nutritional and agronomic traits (Roorkiwal et al., 2022; Istanbuli et al., 2024). The release of whole-genome sequencing (WGS) and whole-genome resequencing (WGRS) data of 3,366 chickpeas (Varshney et al., 2013, 2021) can facilitate the identification of genomic regions for traits associated with nodulation through genome-wide association mapping. In this regard, a newly constructed association panel for root nodulation traits is required by characterizing them through extensive phenotyping and genotyping. Understanding nodulation and N-fixation in chickpea is important to maximize the benefit of N-fixation and reduce reliance on nitrogenous fertilizers. With this background, we have carried out the current research on GWAS for root nodulation traits in chickpea as presented further.
Materials and methods
Plant material and experimental procedure
An association panel consisting of a collection of 284 diverse germplasms obtained and extracted from ICRISAT (Upadhyaya, 2003) along with four checks, BG 372, BG 3022, BG 547, and BG 1105, were used for GWAS analysis on nodulation traits. The experimental trials for the association panel were conducted at four environmental locations, namely, IARI, New Delhi (location 1; 28°38'24.0252” N latitude, 77°10'26.328” E longitude and 228.6 m AMSL) having sandy clay loam soils; Sam Higginbottom University of Agriculture, Technology and Sciences (SHUATS), Naini, Prayagraj (location 2; 25°24'41.27” N latitude, 81°51'3.42” E longitude and 98 m AMSL) with clay loam to sandy loam soil; KVK, Vaishali, Dr. Rajendra Prasad Central Agricultural University (RPCAU), Samastipur (location 3; 25°86'29.679”N latitude, “85°78'10.263”E longitude to accurately reflect the geographical location, and 52 m AMSL) with sandy loam soil; and IARI Regional station, Pusa, Bihar (location 4; 25°54'56.16” latitude, 85°40'24.9564” longitude, and 52 m AMSL) with alluvial soils during the year 2020–2021. Experimental trials consisted of 284 germplasm lines. The field in all the locations was divided into four blocks with four checks in each block. Each check was replicated three times in all the blocks. The experimental design was an augmented random block design with a row length of 5 m, a row spacing of 60 cm, and a plant-to-plant distance of 10 cm (Supplementary Table 1). Observations were taken for the following six traits associated with nodulation: number of nodules per plant (NON/plant), nodule fresh weight per plant [NFW (mg)/plant], number of pods per plant (NOP/plant), number of seeds per plant (NOS/plant), seed yield [SY (g)/plant], and 100 seed weight [100 SW (g)/plant]. Because NON is the ability of the nodulated plant to initiate symbiotic associations with rhizobia, a higher nodule count generally indicates enhanced opportunities for nitrogen fixation. Furthermore, efficiency also depends on nodule activity; therefore, observations on NFW were recorded. This trait is directly associated with metabolic activity and nitrogen fixation capacity. Larger and healthier nodules indicate effective rhizobial colonization and improved nitrogen assimilation.
Trait phenotyping for chickpea genotypes
The phenotyping for nodulation traits was done at 60 days after sowing (DAS), where plants were at 50% flowering stage. No external inoculation of Rhizobium was done to capture the natural variation of nodulation and associated traits. Five randomly selected plants from each genotype were uprooted with the adhered soil mass using a hand hoe by digging 20 cm or even deeper into the soil based on the plant growth and root length observed. Particular care was taken not to disturb the root nodule system during the sampling and removal of adhered soil. Root and shoot systems were separated. Roots with intact nodules were washed and counted for NON and stored in butter paper bags to further take NFW. NOP, NOS, SY, and 100 SW were taken by randomly selecting five plants for each genotype. Yield and 100 SW were measured in grams. The “corrplot” (Wei and Simko, 2021). R package was used to estimate Pearson’s correlation among the measured traits, while the “Factoextra” R package was used to undertake the principal component analysis (PCA) for the filtered data. The frequency distribution plots were generated using the “ggplot2” package in the R environment. Best linear unbiased predictors (BLUPs) were generated using Plant Breeding Tools Version 1.4 (PB Tools, 2014). Bartolome et al. (2014).
Genotyping
The genotypic/SNP marker data for the 284 accessions of the association panel were obtained in the HapMap format from the database of the Centre of Excellence in Genomics and Systems Biology (CEGSB), ICRISAT, Patencheru, Telangana (https://cegresources.icrisat.org/cicerseq/; Varshney et al., 2021). Out of a total of 2,470,880 raw SNPs, 355,546 SNPs were obtained for the association panel (284) and further analyzed. The following parameters were used for SNP filtering: missing data with less than 20%, minor allele frequency (MAF) cutoff at 0.05, and an additional filter for the rate of heterozygosity of less than 10% and after calculating the threshold value of the working set of SNPs (355,546) from the Bonferroni correction as 6.85. We utilized the vcf2gwas tool (Vogt et al., 2021) for filtering SNPs.
Marker–trait association study
Identification of marker–trait associations (MTAs) from genome-wide markers was conducted following GWAS analysis. The generated genotypic data were integrated with multi-locational phenotypic data recorded for the traits NON, NFW, NOP, NOS, SY, and 100 SW. During the phenotypic analysis of multi-location augmented trial design, the checks were considered as fixed effects while the remaining factors (location, block, and new entries/genotypes) were treated as random effects. The phenotypic data were first converted into BLUPs using statistical methods [analysis of variance (ANOVA)] performed using SAS v9.4 mixed procedure Cody, R., (2018). The population structure was assessed using neighbor-joining phylogenetic tree constructed through TASSEL software and visualized through ITOL software and PCA. For PCA, we considered the first three principal components as covariates in the Genome Association and Prediction Integrated Tool (GAPIT) in R software. The extent of linkage disequilibrium (LD) between the SNP markers was analyzed by calculating the LD (r2) values in TASSEL. The only r2 values with p < 0.05 within each chromosome were considered for LD decay analysis. LD decay plots were drawn by using r2 and physical distances (bp) measured by using the script following R version 4.1.1. Chromosome-wise SNP distribution with the “SNP density plot” was generated using the web tool SR-Plots (https://www.bioinformatics.com.cn/en). To identify significant MTAs, GWAS was carried out utilizing the GAPIT3 package Wang and Zhang, 2021) by employing Bayesian-information and Linkage-disequilibrium Iteratively Nested Keyway (BLINK) and fixed and random model circulating probability unification (FarmCPU) models (Liu et al., 2016). The Manhattan and quantile–quantile (Q–Q) plots were generated from qqman version 0.1.8 (Turner, 2018) using the rMVP package (0.99.17; https://github.com/xiaolei-lab/rMVP). The BLASTn search for the chickpea genome of GCA 000331145.1 on the NCBI database (https://blast.ncbi.nlm.nih.gov) was performed to investigate the genomic locations of the significant SNPs in order to find their corresponding genes.
Results
Dispersal and associations among nodulation and yield traits
ANOVA, frequency distribution, and correlation for the phenotypic data of the association panel collected under all the environments were statistically analyzed and the results are presented below (Table 1).
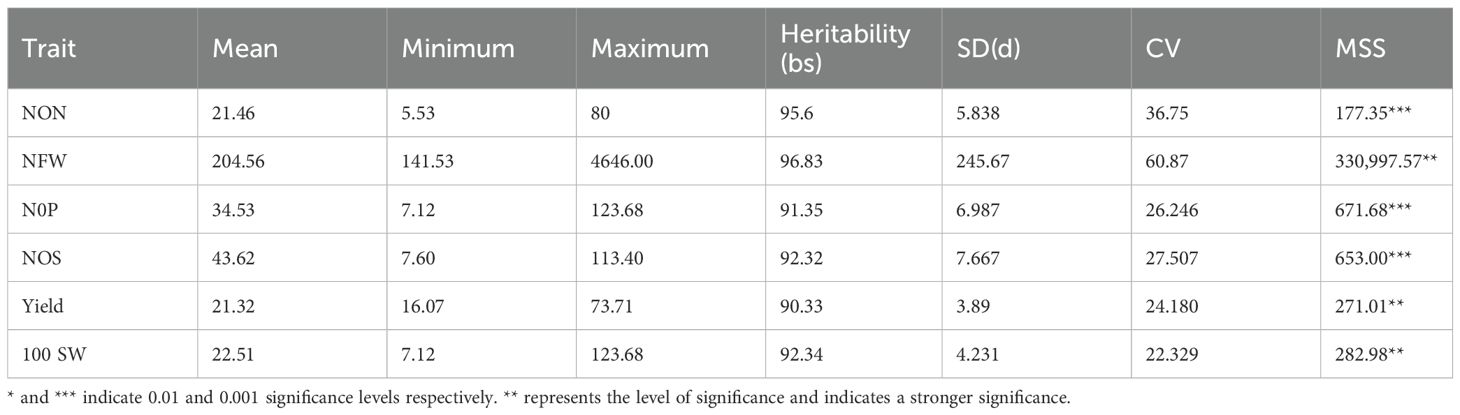
Table 1. Descriptive statistics and combined analysis of variance for the association panel.
Thus, notable statistically significant variations were observed for all the studied traits (p < 0.01). Frequency distribution depicted that all the studied traits were distributed normally in the population (Figure 1).
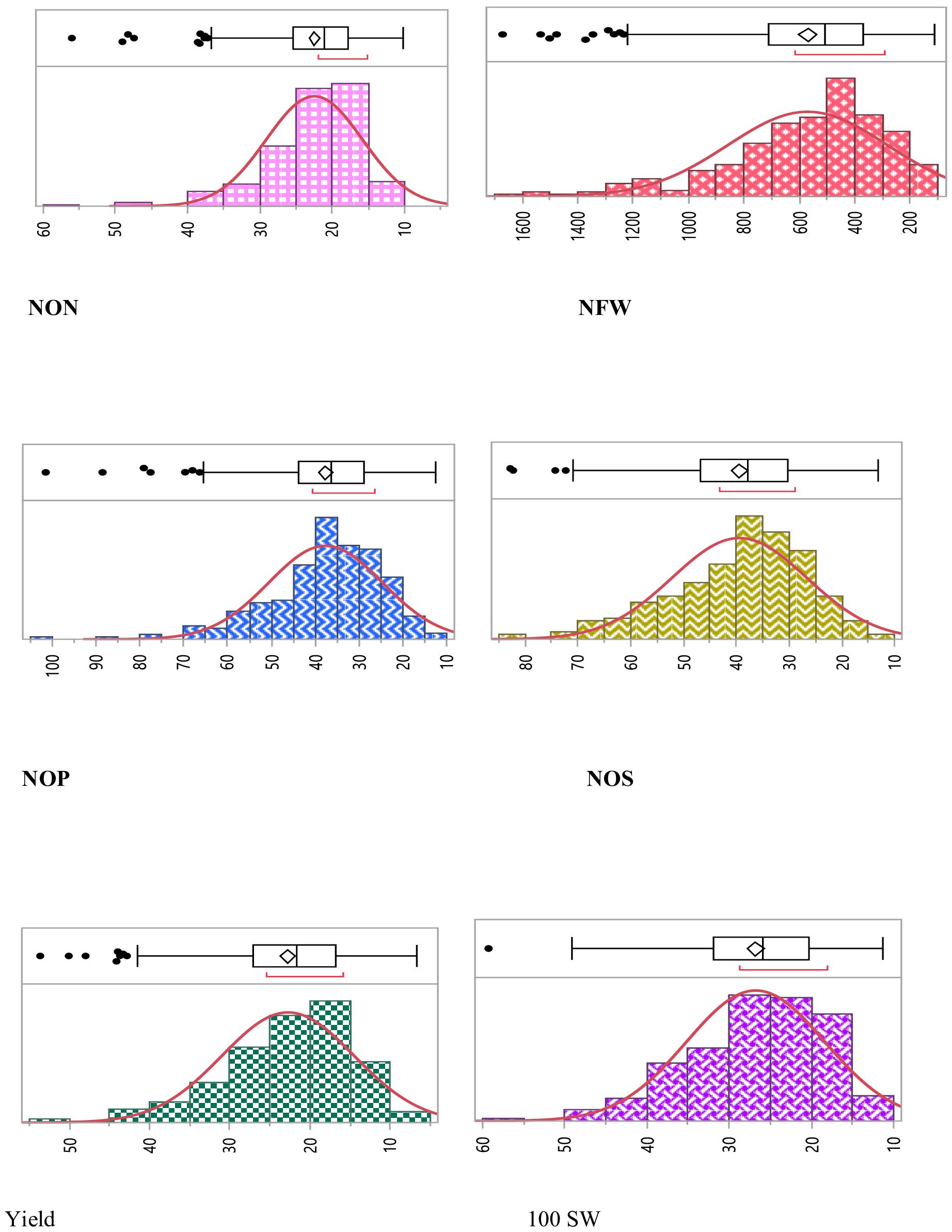
Figure 1. Phenotypic variation for studied traits assayed within the chickpea association panel. Within each histogram plot, the bold dashed line represents the median. The range and median for each trait are specified in the respective grid.
Combined descriptive statistics like minimum, maximum, mean, standard deviation, and coefficient of variation for all the studied traits for all environments were calculated. Based on the descriptive statistics, NON ranged from 5.53 to 80, NFW ranged from 141.53 to 4,646.0, NOP ranged from 7.12 to 123.68, NOS ranged from 7.60 to 113.40, SY ranged from 16.0 to 73.71, and 100 SW ranged from 7.12 to 123.68. Pearson correlation coefficient among different traits indicated that there was a significant positive correlation between NON and NFW with the yield of the studied genotypes along with positive correlation among all the studied traits (Figure 2).
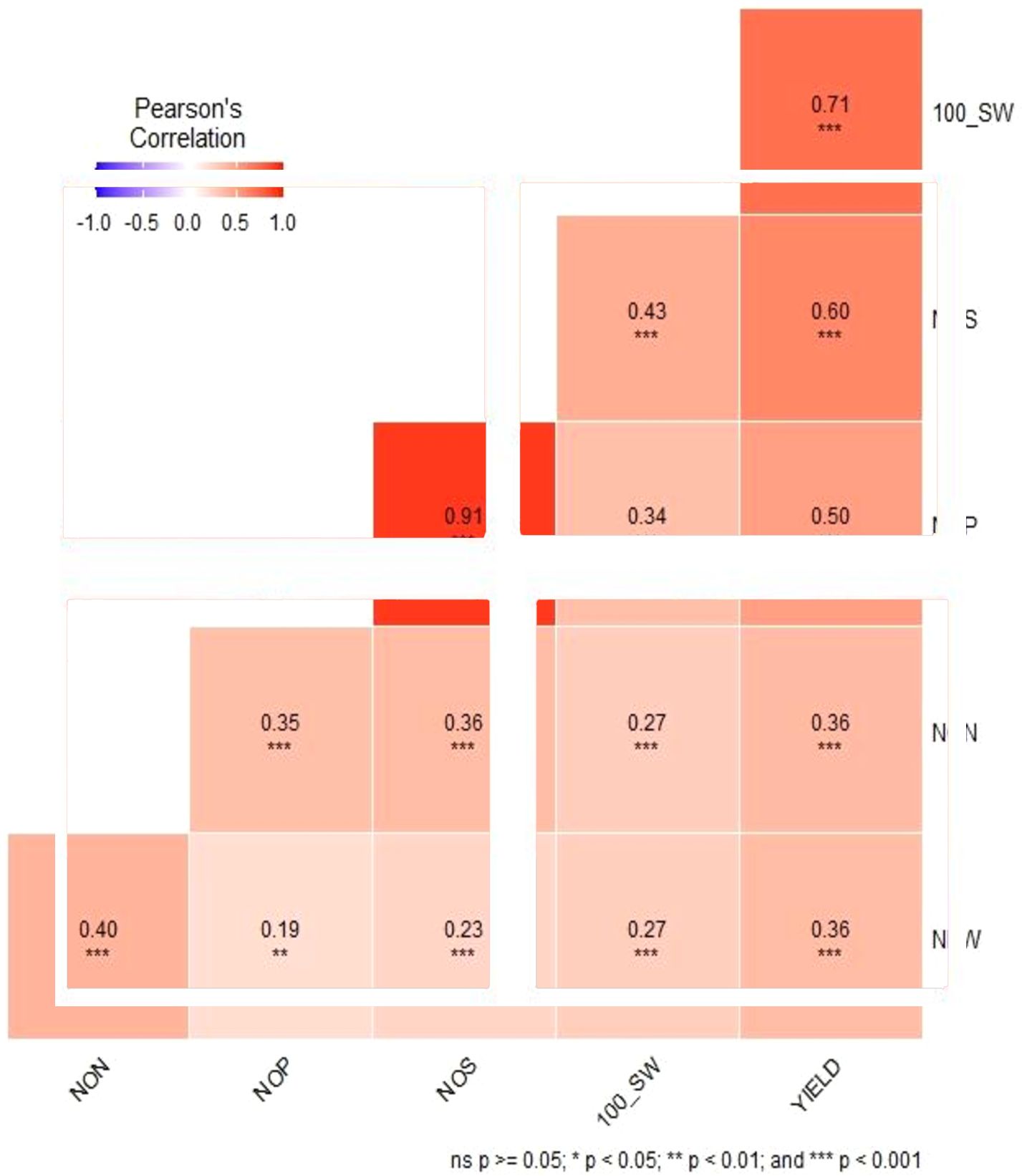
Figure 2. Pearson correlation analysis of six traits evaluated using the chickpea reference set. *Significant at the <0.05 level. Red indicates positive correlations, and blue indicates negative correlations among traits.
Based on mean of genotypes across the locations, the highly nodulating genotypes found in the association panel are presented (Figure 3). The premise was to identify a set of accessions (ICC 7390, ICC 15, ICC 8348, and ICC 2474) that can be incorporated in breeding programs for enhancing nodulation traits without adversely affecting other agronomic traits screened in different environments.

Figure 3. Genotypes identified as high nodulation and high yielding across the location within the chickpea association panel.
Genotypic characteristics and population diversity analysis
Population structure was assessed by admixture (Figure 4A), PCA (Figure 4B), and phylogenetic tree (Figure 4C). The admixture plot provides valuable insights into the genetic diversity and structure of the study population. The ancestral populations (K) on the X axis vs. cross-validation on the Y axis were used to determine the optimal number of genetic clusters or k in a data set when performing admixture analysis, and we found good CV at k = 16. PCA revealed that the population could be divided into three groups with significant genetic variations among the genotypes studied (Figures 4A, B). The presence of the three sub-populations was further confirmed from the neighbor-joining diversity tree (Figure 4C).
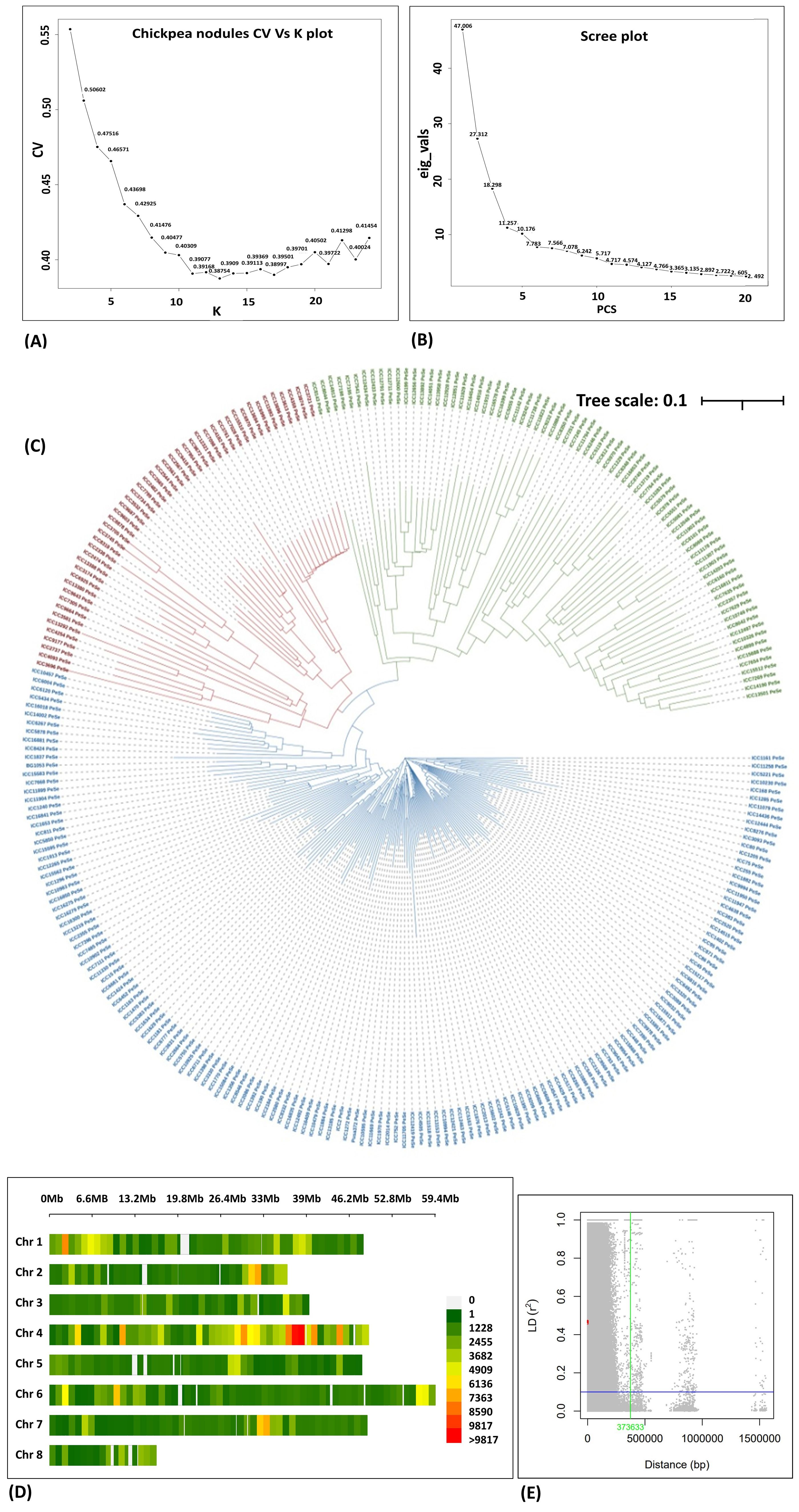
Figure 4. (A) Ancestry proportions from ADMIXTURE analysis (k = 3), optimal with the lowest cross-validation error. Each colored vertical line indicates the proportion of ancestral population k. for each accession. The numbers on the X axis represent the association panel accessions, and CV values are shown on the Y axis. (B) 2D plot of the principal component-based grouping is shown on the X axis, and eigen values are shown on the Y axis. (C) Diversity using the unweighted neighbor-joining tree method. The total number of genotypes is divided into three main clusters and represented in different colors. (D) SNP density plot indicating the distribution of filtered SNPs across the chromosomes in the association panel. (E) Linkage disequilibrium decay curve of the association panel plotting the measured r2 (Y axis) vs. the physical distance between pairs of SNP markers (X axis) (Plink 1.9).
Linkage disequilibrium and LD decay
The SNP density per chromosome ranged from 1,102 to 8,802 SNPs. The LD across the genome was estimated from the HapMap file containing 355,546 SNPs, and the average LD estimated across the genome was 635.9 kb (Figures 4D, E, respectively). Chromosome 4 contained a greater number of SNPs and chromosome 8 had the lowest number of SNPs. In our study, a huge number of high-quality SNPs were found in GWAS that increased the probability of detecting all the possible causal variants of the traits under study.
Marker–trait association for nodulation and yield traits
A genome-wide association analysis for traits under study was conducted using BLINK and FarmCPU models. The Bonferroni correction threshold value of –log10 > 7.0 (p-value) was used as the cutoff, which is highly significant to identify the significant SNPs associated with the studied traits. The markers considered to be significantly associated with tested traits were represented by illustrating the Manhattan plots (Figures 5, 6).
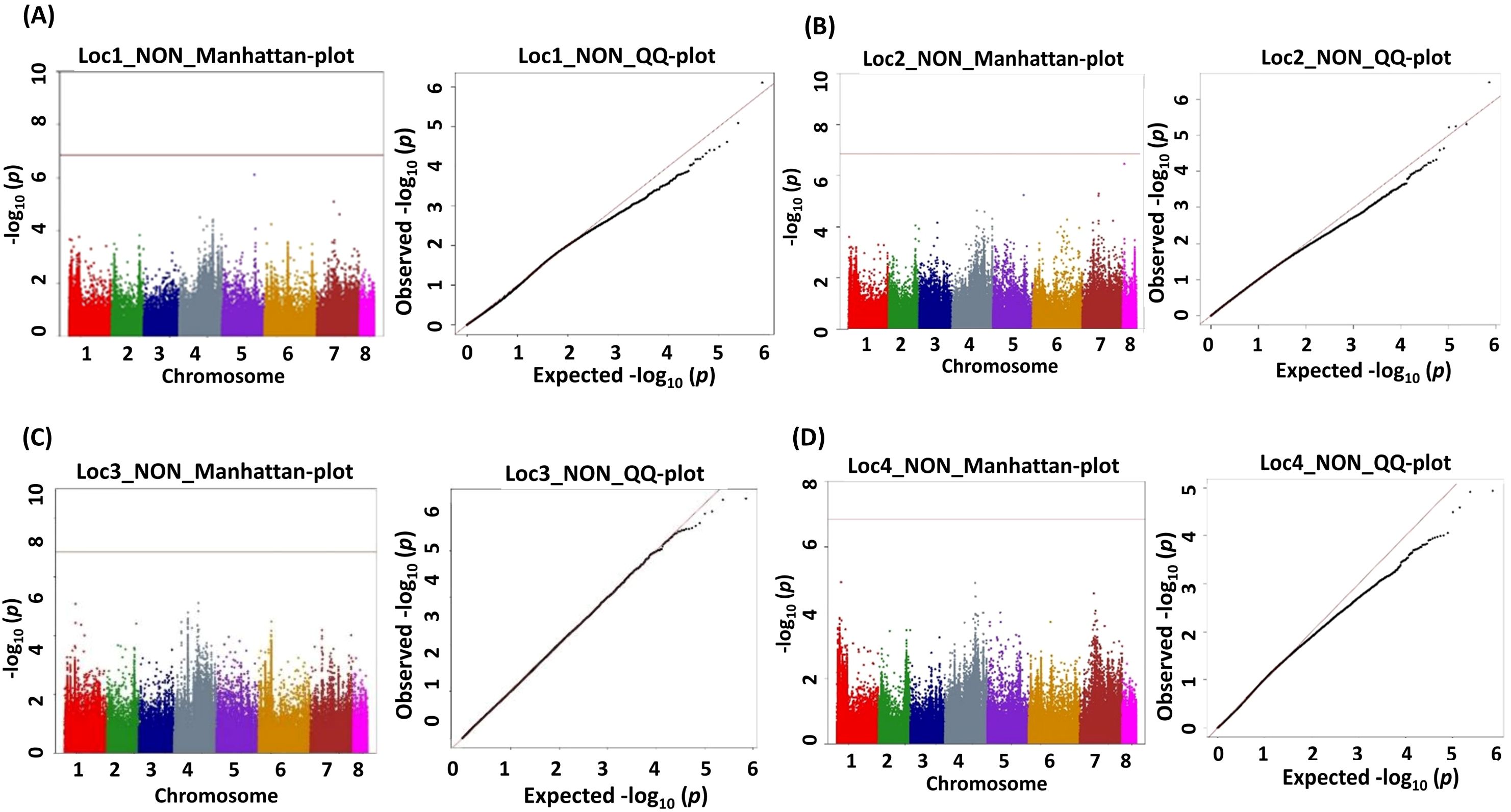
Figure 5. Manhattan (chromosome on the X axis and −log p-values on the Y axis) and quantile–quantile (Q–Q) plots of genome-wide association study (GWAS) signals illustrating SNPs linked to number of nodules (NON). (A) Location 1, (B) location 2, (C) location 3, and (D) location 4. Statistical significance threshold is shown with the green horizontal line along with their corresponding statistical significance represented by the Q–Q plot for the BLINK model.
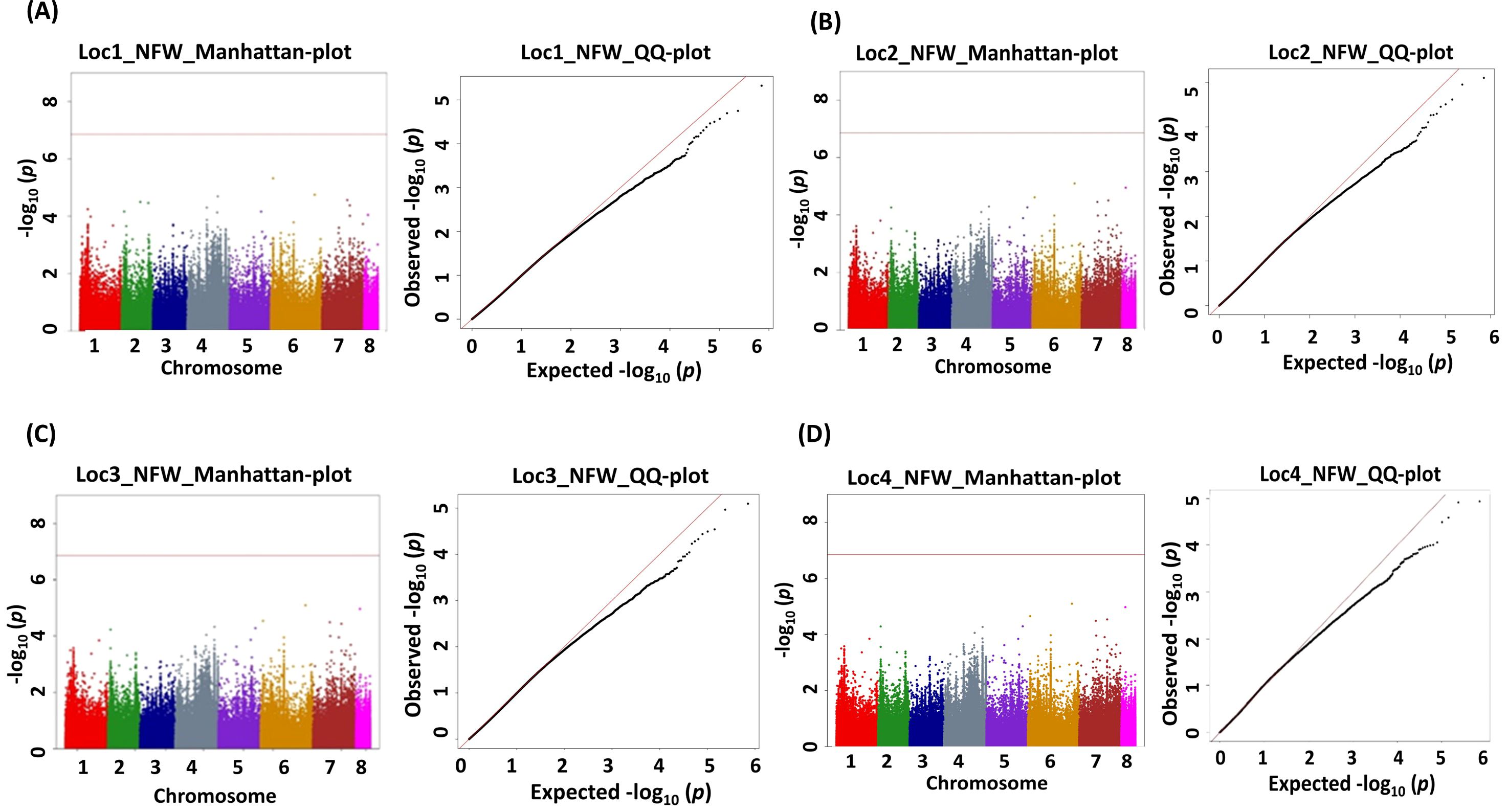
Figure 6. Manhattan (chromosome on the X axis and −log p-values on the Y axis) and quantile–quantile (Q–Q) plots of genome-wide association study (GWAS) signals illustrating SNPs linked to nodule fresh weight (NFW). (A) Location 1, (B) location 2, (C) location 3, and (D) location 4. Statistical significance threshold is shown with the red horizontal line along with their corresponding statistical significance represented by the Q–Q plot for the BLINK model.
This study identified a total of 632 significantly associated SNPs by both BLINK and FarmCPU models (Supplementary Tables 2–7) and Manhattan plots (Supplementary Figures 1–3). MTA for the trait NON resulted in the identification of 27 SNPs from BLINK and 38 SNPs from FarmCPU models having 15 markers common between these two models (Table 2).
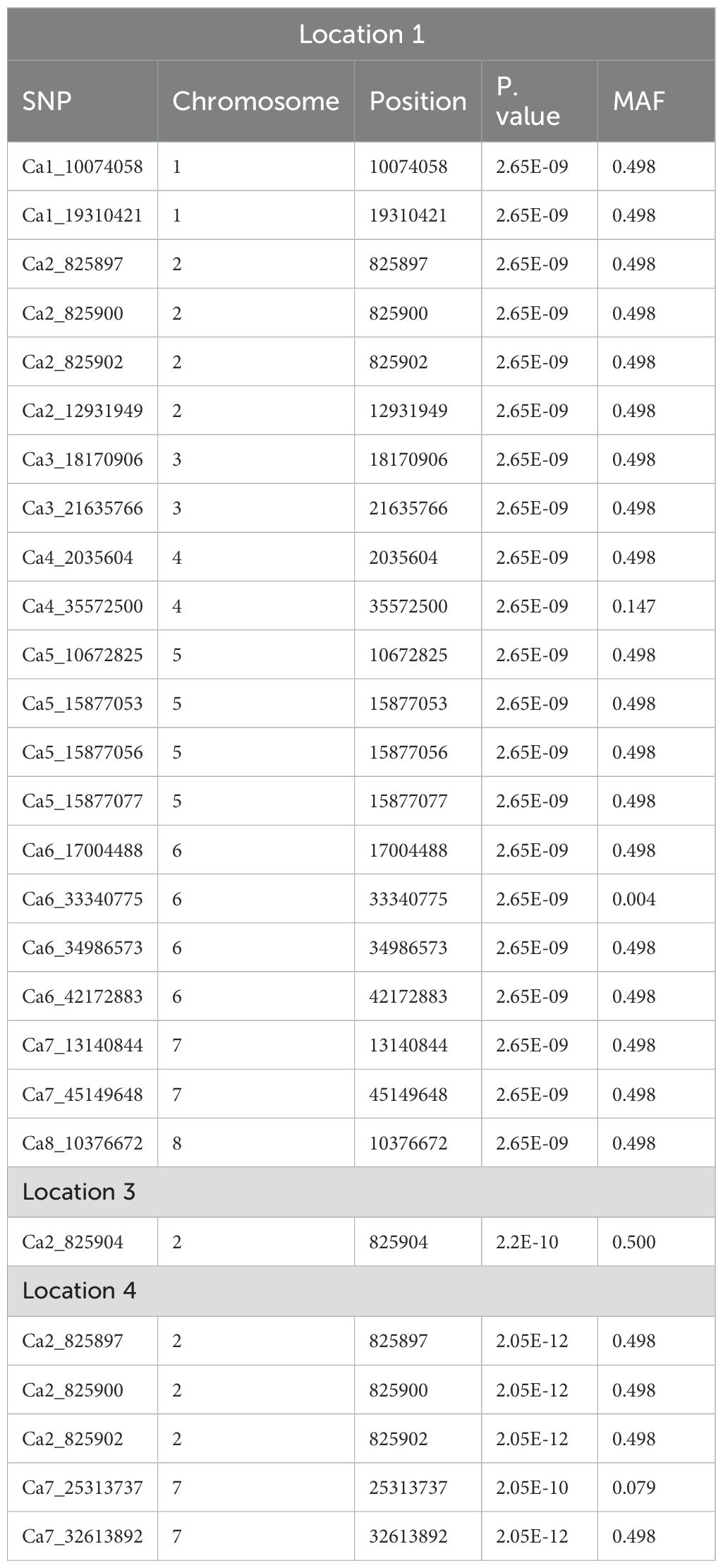
Table 2. Significant MTAs at Bonferroni-corrected p-values for NON in the association panel.
The MTAs Ca7_32613892, Ca2_825900, and Ca2_825902 explained phenotypic variance (PVE) with 48.80%, >25%, and >25%, respectively (Table 3). The MTAs of NFW demonstrated 96 SNPs from the BLINK model and 3 SNPs from the FarmCPU model (Supplementary Table 3), and the results from the BLINK model are shown in Table 4, as well as Manhattan plots along with Q–Q plots (Figures 5, 6).
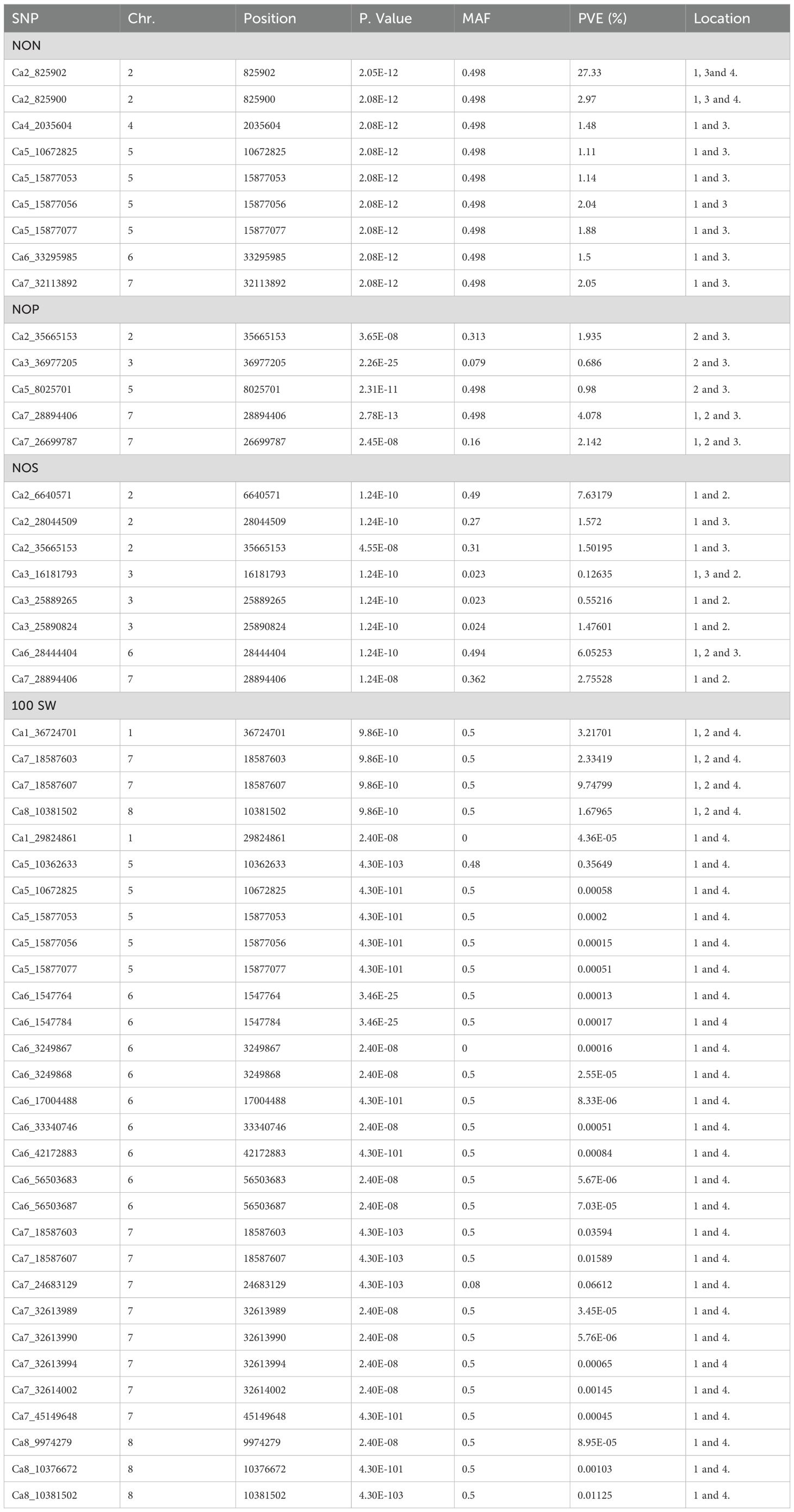
Table 3. List of stable SNPs expressed at more than one environment in the association panel.
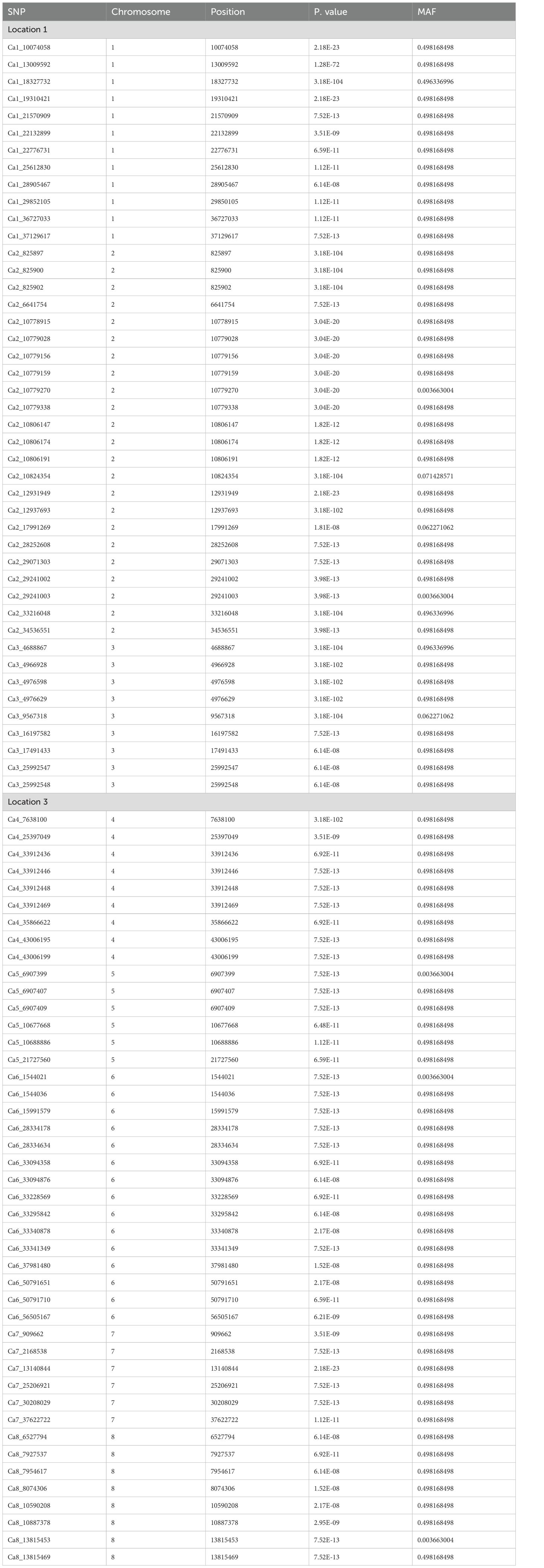
Table 4. Significant MTAs at Bonferroni-corrected p-values for NFW in the association panel.
The MTAs Ca1_28905467 and Ca1_29852105 explained 3.2% and 9.2% of PVE for respective SNPs of NFW. The MTAs of NOP recognized 43 SNPs from BLINK and 60 SNPs from FarmCPU, respectively, having 17 SNPs common with the BLINK model. MTAs of NOS identified 16 and 59 SNPs from the BLINK and FarmCPU models, respectively, with 12 SNPs being common. Furthermore, for the trait 100 SW, we have found 4 and 286 MTAs from the BLINK and farm CPU models, respectively. Thus, the maximum number of SNPs was identified for 100 SW followed by NFW, NON, and NOP.
Marker–trait associations expressed consistently across the environments
The SNPs found for more than two locations were considered as stable SNPs in our study (Table 3). The two SNPs Ca1_36724701 and Ca7_18587603 present on chromosomes 1 and 7, respectively, belonging to the trait NON were found to be stable at locations 1, 3, and 4. Seven SNPs were found to be stable at locations 1 and 3 for NON. The stable SNP 2_825902, which is found on chromosome 2, explained 27.33% of the PVE. Furthermore, 7 stable SNPs for NFW, 5 for NOP, 6 for NOS, and 32 for 100 SW were identified. The PVE explained by the stable MTAs ranged from 1.11% to 27.33% for NON, 5.7% to 34.46% for NFW, 1.12% to 8.95% for NOP, 1.23% to 11.71% for NOS, 1.23% to 31.45% for seed yield, and 0.92% to 28.65% for 100 SW. Variation in PVE % was observed across different locations. This may be attributed to the genotype-by-environment (G×E) interactions, which influence the relative performance of the genotypes under different environmental conditions.
Associated SNPs with two or more traits
The SNPs found common for two or more traits were considered as pleotropic (Table 5). The three SNPs (Ca1_10074058, Ca2_825900, and Ca2_825902) were found to be common for the traits NON and NFW. The SNP Ca7_18587607 located on chromosome number 7 was found to be common for the traits NOP, NOS, and 100 SW. We also found 11 significant and common SNPs for the traits NOP and 100 SW. Significant SNPs contribute substantially to the variability of the trait in the population being studied. Researchers prioritize these SNPs for further investigation and consider them as potential candidates for explaining the genetic basis of the trait.
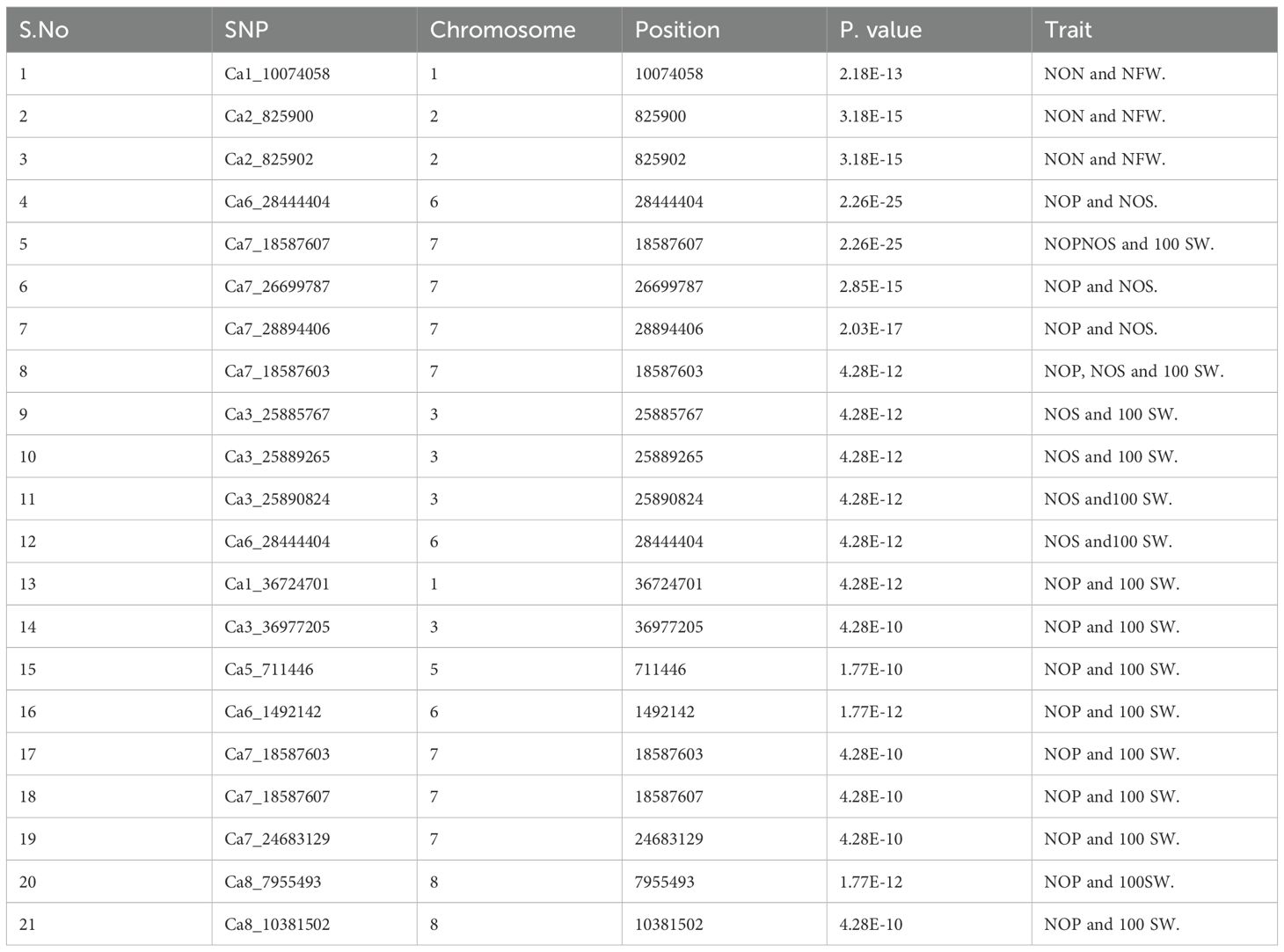
Table 5. List of the common MTAs identified for more than one trait.
Candidate genes and associated SNPs
The significantly associated SNPs with different traits were further used for the identification of putative candidate genes based on the position of SNPs and flanking regions (Kawahara et al., 2013). The 10-kb upstream and 10-kb downstream sequences from the SNP positions were retrieved from the NCBI database and functionally annotated based on CDC Frontier v1 functional annotations (Varshney et al., 2013). The SnpEff-4.3T open-source program was also used for variant annotation and prediction of significant SNP effects, and we found that majority of the SNPs in our study were present as intron variants. The Ca7_32113892 identified for NON was present within the genomic regions of the protein encoding a calumenin-like isoform X2 and a calumenin B-like isoform X1. These have a function in NOD factor export and, in turn, are shown to be involved in nodulation. The SNP located on Ca7_45149648 localized with the protein transcription factor GTE4-like protein that functions as an activator of gene expression upon infection with Pseudomonas syringae and helps in the upregulation of salicylic acid (SA)-mediated immune defense genes was associated with NON. The GTE4-like protein is closely associated with bacterial infection and may be involved in the regulation of the nodulation process. Some of the important proteins along with their molecular functions are listed in Table 6.
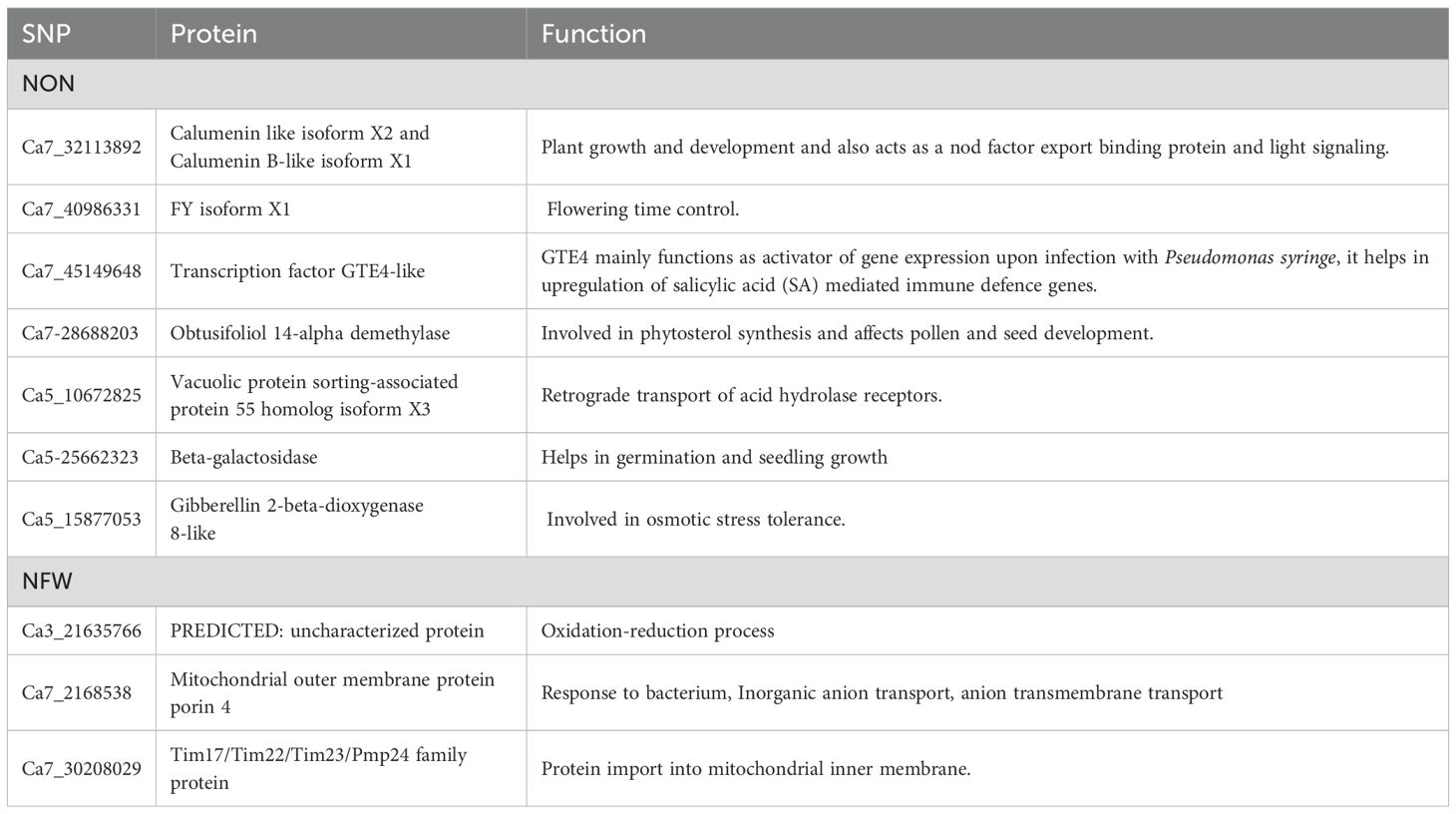
Table 6. Candidate genes identified at the 10-kb region of linked SNPs along with their molecular functions for NON and NFW in the association panel.
Discussion
The identification of chickpea germplasm with good nodulation parameters is key for developing cultivars for different end users and identifying high nodulation donors to help improve the BNF. The chickpea core collection also shows a considerable high amount of variation for nodulation traits among the studied accessions. It is believed that such genotypes exhibiting optimal nodulation play an important role in improving BNF. Phenotypic data analysis has shown that nodulation traits such as NON and NFW were positively correlated with yield per plant. Similar results were also reported by Istanbuli et al. (2024). The traits NOP and NOS were positively correlated with chickpea yield as reported earlier in several studies (Aarif et al., 2014; Dawane et al., 2020). In this study, we have identified chickpea accessions (ICC 7390, ICC 15, ICC8348, and ICC2474) across multiple locations that could be exploited as a base in breeding, especially for nodulation-associated traits. Genomics-assisted crop improvement of chickpea is usually hindered by its narrow genetic base and low intra-specific polymorphism among cultivated desi and kabuli accessions. To overcome this, large-scale discovery and genotyping of informative sequence-based markers such as SNPs differentiating the maximum number of desi and kabuli accessions and exhibiting high intra-specific potential among cultivated accessions using a user-friendly, rapid, and cost-effective genotyping assay at the genome-wide scale is essential (Bajaj et al., 2015).
Population structure analysis provides insight into genetic variation in chickpeas that has evolved through evolutionary processes such as genetic drift, demographic history, and natural selection (Andam et al., 2017). In our study population, stratification has been with three clusters/sub-populations independent of biological status and seed type. In accordance with the results obtained in this study, a study by Varshney et al. (2019) also reported the presence of three sub-populations using genome-wide SNP markers. In another study, four sub-populations were revealed in a diverse set consisting of 186 chickpea genotypes, using DArT-seq markers (Farahani et al., 2019). The presence of three sub-population aligns with a previous study on desi and kabuli chickpea genotypes, which emphasizes the importance of geographic origin and adaptive environments in genotype clustering (Basu et al., 2018). Further investigation revealed that most sub-populations were admixed, which could be useful in breeding programs to produce hybrids with desirable traits. These findings are supported by the pure and minimally admixed accessions studied by Bajaj et al. (2015). GWAS overcomes two common limitations of the traditional linkage mapping, viz., restricted allelic diversity and limited genetic resolution (Brachi et al., 2011). Owing to its high resolution, cost-effectiveness, and non-essential pedigrees, association mapping has been able to dissect many important traits in chickpea, such as concentration of mineral nutrients (Diapari et al., 2014; Upadhyaya et al., 2016; Fayaz et al., 2022; Samineni et al., 2022); seed yield (Basu et al., 2018); drought tolerance (Li et al., 2018); root morphology, phosphorous acquisition, and use efficiency (Thudi et al., 2021); and salinity tolerance (Ahmed et al., 2021).
In recent years, GWAS has become crucial for pinpointing SNPs in chickpeas and helping us identify genomic variations linked to specific traits (Roorkiwal et al., 2022). GWAS have been performed for location and trait-wise individual genotypes for nodulation and yield contributing traits. The present study identified SNPs that are common across two or more traits, which may indicate pleiotropy. However, majority of them controlled the related traits such 3 SNPs common for NON and NFW, 4 SNPs common for NOS and NOP, and 14 SNPs common for NOP and 100 SW, and some SNPs were found common for NOP, NOS, and 100 SW. Also, GWAS analysis of the association panel led to the identification of stable SNPs with good PVE values for all the traits under study as presented in the Results section. For the trait NON, SNP Ca2_825900 was found to be stable with 27.32% of PVE and the SNP Ca7_32613892 was found to explain 48.11% of PVE. We also found that most of the SNPs common across the models indicate the true association of markers with the trait of interest. SNP Ca1_19310421 associated with the NON trait is situated in a genomic region where the gene codes for an SNF1-related protein kinase regulatory subunit beta-3. This subunit is implicated in the regulation of protein kinase activity, cellular responses to nitrogen levels, and the intricate response to sucrose signaling (Jamsheer et al., 2022). These findings collectively contribute to our understanding of the genetic underpinnings of the observed traits and the multifaceted processes governing plant nutrient interactions and developmental response. MTAs identified for the NFW also had prominent candidate genes: Ca1_29852482 is located on chromosome number 1 that involves genes for the regulation of plant immunity (Ashraf et al., 2018), the SNP Ca1_28905467 located on chromosome 1 codes for environmental stress (Che et al., 2020), and the SNP Ca8_12988095 is located on the nearest gene that codes for plant–microbe interactions (Liu et al., 2023). The presence of nodulation-specific and other related candidate genes supports earlier findings on the discovery of SNPs Ca1pos289.52482.1 and 6_33340878 (Chandana et al., 2024).
Thus, this study may be considered as a complementary work of our previous accomplishments with a new set of association panel that also validates already identified markers for nodulation. The validated SNPs Ca1pos289.52482.1 and 6_33340878 can be converted to Kompetitive allele-specific PCR (KASP) markers and utilized in the marker-assisted breeding for genes related to nodulation and dissecting the BNF along with yield and productivity. Based on our study, we found that the genomic regions controlling NON are expected to be present on chromosome number 7 with a size of 286 to 451 mb. The MTAs have revealed that several genomic regions on chromosome number 5, with lengths ranging from 158 to 217 Mb, contain SNPs that contribute to the NON trait. The genomic regions controlling the nodules’ fresh weight are present on chromosome 6. The BNF is a sustainable and globally applicable means to supply nitrogen to agricultural systems. An effective strategy to augment BNF involves the breeding and utilization of legume genotypes possessing enhanced BNF capacity. The germplasm lines exhibited significant trait variations encompassing all traits under study. Correlation analysis revealed compelling insights, highlighting a significant positive correlation and the direct effect of nodulation traits on yield within the chickpea population. Based on nodulation and yield-related traits, the promising accessions identified in this study can serve as potential donors for designing nodulation-rich chickpea varieties for the future. Leveraging association studies, we successfully identified noteworthy and stable MTAs linked to the nodulation traits. Phylogenetic tree and genotypic PCA confirmed three sub-populations in the association panel. LD decay estimation revealed an average LD block size of 636.8 kb, which helped us find good MTAs in our study. Highly significant markers were reported for nodulation and yield traits in chickpea. Subsequent in silico analysis revealed that a substantial proportion of these MTAs were situated within intergenic regions with the potential to modulate genes associated with the focal traits.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Any further inquiries can be directed to corresponding authors.
Author contributions
CS: Investigation, Writing – original draft. RM: Investigation, Software, Writing – original draft. RS: Data curation, Investigation, Software, Writing – original draft. RV: Software, Writing – original draft. KS: Data curation, Investigation, Resources, Writing – review & editing. SK: Investigation, Resources, Writing – review & editing. GL: Investigation, Resources, Writing – review & editing. HK: Data curation, Formal Analysis, Software, Writing – review & editing. VK: Data curation, Formal Analysis, Software, Writing – review & editing. AV: Formal Analysis, Software, Writing – review & editing. RY: Investigation, Resources, Writing – review & editing. LY: Writing – review & editing, Data curation, Software. HU: Resources, Writing – review & editing. AH: Conceptualization, Funding acquisition, Resources, Writing – review & editing. RK: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Acknowledgments
The corresponding author sincerely acknowledges the DST-SERB for financial support through project CRG/2019/006273 entitled utilization of germplasm for identification of genes/QTLs for nodulation through association mapping in chickpea. All authors also sincerely acknowledge the ICAR-IARI, New Delhi; ICAR-IARI Regional Station, Pusa, Bihar; KVK, Vaishali (Dr RPCAU, Samastipur), Bihar; and the Department of Genetics and Plant Breeding, SHUATS, Naini, Prayagraj, Uttar Pradesh for providing facilities to carry out this research work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1652315/full#supplementary-material
References
Aarif, M., Rastogi, N. K., Johnson, P. L., and Chandrakar, P. K. (2014). Genetic analysis of seed yield and its attributing traits in kabuli chickpea (Cicer arietinum L.). J. Food Legumes. 27, 24–27.
Ahmed, S. M., Alsamman, A. M., Jighly, A., Mubarak, M. H., Al-Shamaa, K., Istanbuli, T., et al. (2021). Genome-wide association analysis of chickpea germplasms differing for salinity tolerance based on DArTseq markers. PloS One 16, 2. doi: 10.1371/journal.pone.0260709
Andam, C., Challagundla, L., Azarian, T., Hanage, W., and Robinson, D. (2017). Population structure of pathogenic bacteria in Genetics and evolution of infectious diseases. 2nd edi (Amsterdam: The Netherlands Elsevier), 51–70.
Ashraf, N., Basu, S., Narula, K., Ghosh, S., Tayal, R., Gangisetty, N., et al. (2018). Integrative network analyses of wilt transcriptome in chickpea reveal genotype dependent regulatory hubs in immunity and susceptibility. Sci. Rep. 8, 6528. doi: 10.1038/s41598-018-19919-5
Bajaj, D., Saxena, M. S., Kujur, A., Das, S., Badoni, S., Tripathi, S., et al. (2015). Genome-wide conserved non-coding microsatellite (CNMS) marker based integrative genetical genomics for quantitative dissection of seed weight in chickpea. J. Exp. Bot. 66, 1271–1129. doi: 10.1093/jxb/eru478
Bartolome, V. I., Sales, N. L., Morantte, R. I. Z. A., Raquel, A. M. V., and Ye, G. (2014). PB Tools (2014) Version 1.4. In: PB Tools User Manual. Biometrics and Breeding Informatics, PBGB Division, International Rice Research Institute (IRRI), Los Baños, Laguna, Manila, Philippines.
Basu, U., Srivastava, R., Bajaj, D., Thakro, V., Daware, A., Malik, N., et al. (2018). Genome-wide generation and genotyping of informative SNPs to scan molecular signatures for seed yield in chickpea. Sci. Rep. 8, 13240. doi: 10.1038/s41598-018-29926-1
Benjelloun, I., Thami Alami, I., Douira, A., and Udupa, S. M. (2019). Phenotypic and genotypic diversity among symbiotic and non-symbiotic bacteria present in chickpea nodules in Morocco. Front. Micro. 10, 1885. doi: 10.3389/fmicb.2019.01885
Brachi, B., Morris, G. P., and Borevitz, J. O. (2011). Genome-wide association studies in plants: the missing heritability is in the field. Genome bio. 12, 232. doi: 10.1186/gb-2011-12-10-232
Chandana, B. S., Mahto, R. K., Singh, R. K., Bhandari, A., Tandon, G., Singh, K. K., et al. (2024). Genome wide association mapping identifies novel SNPs for root nodulation and agronomic traits in chickpea. Front. Plant Sci. Sec 20, 900324. doi: 10.3389/fpls.2024.1395938
Chandana, B. S., Mahto, R. K., Singh, R. K., Singh, K. K., Kushwah, S., Lavanya, G. R., et al. (2023). Unclenching the potentials of global core germplasm for root nodulation traits for increased biological nitrogen fixation and productivity in chickpea (Cicer arietinum L.). Indian J. Genet. Plant Breed. 83, 526–534. doi: 10.31742/ISGPB.83.4.9
Che, Y., Kusama, S., Matsui, S., Suorsa, M., Nakano, T., Aro, E. M., et al. (2020). ArabidopsisPsbP-Like Protein 1 Facilitates the Assembly of the Photosystem II Super complexes and Optimizes Plant Fitness under Fluctuating Light. Plant Cell Physiol. 61, 1168–1180. doi: 10.1093/pcp/pcaa045
Dawane, J. E., Jahagirdr, J. E., and Shedge, P. (2020). Correlation studies and path coefficient analysis in chickpea (Cicer arietinum L.). Int. J. Curr. Microbiol. Appl. Sci. 9, 1266–1272. doi: 10.20546/ijcmas.2020.910.152
Diapari, M., Sindhu, A., Bett, K., Deokar, A., Warkentin, T. D., and Tar’an, B. (2014). Genetic diversity and association mapping of iron and zinc concentrations in chickpea (Cicer arietinum L.). Genome. 57, 459–468. doi: 10.1139/gen-2014-0108
FAOSTAT (Rome). Available online at: https://www.fao.org/faostat/es/data/QCL (Accessed December 17, 2023).
Farahani, S., Maleki, M., Mehrabi, R., Kanouni, H., Scheben, A., Batley, J., et al. (2019). Whole genome diversity population structure and linkage disequilibrium analysis of chickpea (Cicer arietinum L.) genotypes using genome-wide DArTseq-based SNP markers. Genes. 10, 676. doi: 10.3390/genes10090676
Fayaz, H., Tyagi, S., Wani, A. A., Pandey, R., Akhtar, S., Bhat, M. A., et al. (2022). Genome-wide association analysis to delineate high-quality SNPs for seed micronutrient density in chickpea (Cicer arietinum L.). Sci. Rep. 12, 11357. doi: 10.1038/s41598-022-14487-1
Frailey, D. C., Zhang, Q., Wood, D. J., and Davis, T. M. (2022). Defining the mutation sites in chickpea nodulation mutants PM233 and PM405. BMC Plant Biol. 22, 66. doi: 10.1186/s12870-022-03446-7
Grasso, N., Lynch, N. L., Arendt, E. K., and O'Mahony, J. A. (2022). Chickpea protein ingredients: A review of composition, functionality, and applications. Compr. Rev. Food Sci. Food Saf. 21, 435–452.
Greenlon, A., Chang, P. L., Damtew, Z. M., Muleta, A., Carrasquilla-Garcia, N., Kim, D., et al. (2019). Global-level population genomics reveals differential effects of geography and phylogeny on horizontal gene transfer in soil bacteria.) Chickpea protein ingredients: A review of composition, functionality, and applications. Compr. Rev. Food Sci. Food Saf. 21, 435–452. doi: 10.1073/pnas.190005611610.1073/pnas.1900056116
Istanbuli, T., Nassar, A. E., Abd El-Maksoud, M. M., Tawkaz, S., Alsamman, A. M., and Hamwieh, A. (2024). Genome-wide association study reveals SNP markers controlling drought tolerance and related agronomic traits in chickpea across multiple environments. Front. Plant Sci. 15, 1260690. doi: 10.3389/fpls.2024.1260690
Jamsheer, K. M., Jindal, S., Sharma, M., Awasthi, P., Sreejath, S., Sharma, M., et al. (2022). A negative feedback loop of TOR signaling balances growth and stress-response trade-offs in plants. Cell Rep. 39, 110631. doi: 10.1016/j.celrep.2022.110631
Kawahara, Y., de la Bastide, M., Hamilton, J. P., Kanamori, H., McCombie, W. R., Ouyang, S., et al. (2013). Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 6, 4. doi: 10.1186/1939-8433-6-4
Kumar, R., Singh, R. K., Misra, J. P., Yadav, A., Kumar, A., Yadav, R., et al. (2021). Dissecting proteomic estimates for enhanced bioavailable nutrition during varied stages of germination and identification of potential genotypes in chickpea. Legume Res. 45, 1082–1087. doi: 10.18805/LR-4531
Li, Y., Ruperao, P., Batley, J., Edwards, D., Khan, T., Colmer, T. D., et al. (2018). Investigating drought tolerance in chickpea using genome-wide association mapping and genomic selection based on whole-genome resequencing data. Front. Plant Sci. 9, 190. doi: 10.3389/fpls.2018.00190
Liu, Q., Cheng, L., Nian, H., Jin, J., and Lian, T. (2023). Linking plant functional genes to rhizosphere microbes: a review. Plant Biotechnol. J. 21, 902–917. doi: 10.1111/pbi.13950b
Liu, X., Huang, M., Fan, B., Buckler, E. S., and Zhang, Z. (2016). Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PloS Genet. 12, e1005767. doi: 10.1371/journal.pgen.1005767
Milán-Carrillo, J., Valdéz-Alarcón, C., Gutiérrez-Dorado, R., Cárdenas-Valenzuela, O. G., Mora-Escobedo, R., Garzón-Tiznado, J. A., et al. (2007). Nutritional properties of quality protein maize and chickpea extruded based weaning food. Plant Foods Hum Nutr. 62, 31–37. doi: 10.1007/s11130-006-0039-z
Misra, J. P., Yadav, A., Kumar, A., Yadav, R., and Kumar, R. (2016). Bio-chemical characterization of chickpea genotypes with special reference to protein. Res. J. Chem. Environ. 8), 38–43.
Mittal, N., Bhardwaj, J., Verma, S., Singh, R. K., Yadav, R., Kaur, D., et al. (2023). Disentangling potential polymorphic markers and genotypes for macro-micronutrients in Chickpea. Sci. Rep. 13, 10731. doi: 10.1038/s41598-023-37602-2
Katoch, O., Chauhan, U. S., Renu, Y., Yadav, S. S., Ashwani, K., and Ashwani, Y. (2016). Nitrate Reductase based phylogenetic analysis in chickpea. Res.J.Chem. Environ. 7), 1–8.
Peoples, M. B. and Craswell, E. T. (1992). Biological nitrogen fixation: investments expectations and actual contributions to agriculture. Plant Soil 141, 13–39. doi: 10.1007/BF00011308
Plett, K. L., Bithell, S. L., and Dando, A. (2021). Chickpea shows genotype-specific nodulation responses across soil nitrogen environment and root disease resistance categories. BMC. Plant Biol. 21, 310. doi: 10.1186/s12870-021-03102-6
Roorkiwal, M., Bhandari, A., Barmukh, R., Bajaj, P., Valluri, V. K., Chitikineni, A., et al. (2022). Genome-wide association mapping of nutritional traits for designing superior chickpea varieties. Front. Plant Sci. 13, 843911. doi: 10.3389/fpls.2022.843911
Samineni, S., Mahendrakar, M. D., Hotti, A., Chand, U., Rathore, A., and Gaur, P. M. (2022). Impact of heat and drought stresses on grain nutrient content in chickpea: genome-wide marker-trait associations for protein Fe and Zn. Environ. Exp. Bot. 194, 104688. doi: 10.1016/j.envexpbot.2021.104688
Sharma, S., Yadav, N., Singh, A., and Kumar, R. (2013). Nutritional and antinutritional profile of newly developed chickpea (Cicer arietinum L.) varieties. Inter. Food Res. J. 20, 805–810. doi: 10.5958/2229-4473.2016.00040.9
Thudi, M., Chen, Y., Pang, J., Kalavikatte, D., Bajaj, P., Roorkiwal, M., et al. (2021). Novel genes and genetic loci associated with root morphological traits phosphorus-acquisition efficiency and phosphorus-use efficiency in chickpea. Front. Plant Sci. 12, 636973. doi: 10.3389/fpls.2021.636973
Turner (2018). qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open-Source Software 3, 1731. doi: 10.21105/joss.00731
Upadhyaya, H. D. (2003). Geographical patterns of variation for morphological and agronomic characteristics in the chickpea germplasm collection. Euphytica 132, 343–352. doi: 10.1023/A:1025078703640
Upadhyaya, H. D., Bajaj, D., Narnoliya, L., Das, S., Kumar, V., Gowda, C. L. L., et al. (2016). Genome-wide scans for delineation of candidate genes regulating seed-protein content in chickpea. Front. Plant Sci. 7, 13. doi: 10.3389/fpls.2016.00302
Varshney, R. K., Roorkiwal, M., Sun, S., Bajaj, P., Chitikineni, A., Thudi, M., et al. (2021). A chickpea genetic variation map based on the sequencing of 3,366 genomes. Nature 599, 622–627. doi: 10.1038/s41586-021-04066-1
Varshney, R. K., Song, C., Saxena, R. K., Azam, S., Yu, S., Sharpe, A. G., et al. (2013). Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biot. 31, 240–246. doi: 10.1038/nbt.2491
Varshney, R. K., Thudi, M., Roorkiwal, M., He, W., Upadhyaya, H. D., Yang, W., et al. (2019). Resequencing of 429 chickpea accessions from 45 countries provides insights into genome diversity domestication and agronomic traits. Nat. Genet. 51, 857–864. doi: 10.1038/s41588-019-0401-3
Vitousek, P. M., Mooney, H. A., Lubchenco, J., and Melillo, J. M. (1997). Human domination of earth's ecosystems. Science 277, 494–499. doi: 10.1126/science.277.5325.494
Vogt, F., Shirsekar, G., and Weigel, D. (2021). vcf2gwas-python API for comprehensive GWAS analysis using GEMMA. Bioinform. 38, 839–840. doi: 10.1093/bioinformatics/btab710
Wang, J. and Zhang, Z. (2021). GAPIT version 3: boosting power and accuracy for genomic association and prediction. Genomics Proteomics Bioinf. 19, 629–640. doi: 10.1016/j.gpb.2021.08.005
Wanjofu, E. I., Venter, S. N., Beukes, C. W., Steenkamp, E. T., Gwata, E. T., and Muema, E. K. (2022). Nodulation and growth promotion of chickpea by mesorhizobium isolates from diverse sources. Microorganisms Cham, Switzerland: Springer 10, 2467. doi: 10.3390/microorganisms10122467
Keywords: chickpea, GWAS, KASP, MTAs, root nodulation
Citation: S CB, Mahto RK, Singh RK, V R, Singh KK, Kushwah S, Lavanya GR, Kudapa H, Kumar Valluri V, Vemula AK, Yadav RR, Yadav LB, Upadhyaya HD, Hamwieh A and Kumar R (2025) Genome-wide association studies identified novel SNPs associated with efficient biological nitrogen fixation in chickpea (Cicer arietinum L.). Front. Plant Sci. 16:1652315. doi: 10.3389/fpls.2025.1652315
Received: 26 June 2025; Accepted: 16 October 2025;
Published: 12 November 2025.
Edited by:
Muhammad Awais Farooq, University of Aberdeen, United KingdomReviewed by:
Kibrom B. Abreha, Swedish University of Agricultural Sciences, SwedenHaifeng Jia, Chinese Academy of Agricultural Sciences, China
Copyright © 2025 S, Mahto, Singh, V, Singh, Kushwah, Lavanya, Kudapa, Kumar Valluri, Vemula, Yadav, Yadav, Upadhyaya, Hamwieh and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajendra Kumar, cmFqZW5kcmFrNjRAeWFob28uY28uaW4=; Aladdin Hamwieh, YS5oYW13aWVoQGNnaWFyLm9yZw==; YWxhZGRpbi5oYW13aWVoQGZhby5vcmc=
†Present address: Aladdin Hamwieh, Plant Production and Protection Division, FAO, Rome, Italy
‡These authors have contributed equally to this work and share first authorship