 Ranjeet R. Kumar1*†
Ranjeet R. Kumar1*† Suneha Goswami1
Suneha Goswami1 Vinutha Thimmegowda1
Vinutha Thimmegowda1 Nitin Singhal2
Nitin Singhal2 Ulaganathan Mabalirajan3
Ulaganathan Mabalirajan3 Gyan P. Mishra4
Gyan P. Mishra4 Girish K. Jha5
Girish K. Jha5 Gyanendra K. Rai6
Gyanendra K. Rai6 Shelly Praveen1
Shelly Praveen1 Chellapilla Tara Satyavathi7
Chellapilla Tara Satyavathi7- 1Division of Biochemistry, Indian Council of Agricultural Research (ICAR)-Indian Agricultural Research Institute, New Delhi, India
- 2Department of Biotechnology (DBT)-National Agri-Biotechnology Institute, Mohali, Punjab, India
- 3Council of Scientific and Industrial Research (CSIR)-Indian Institute of Chemical Biology, Kolkata, India
- 4Division of Seed Science and Technology, Indian Council of Agricultural Research (ICAR)-Indian Agricultural Research Institute, New Delhi, India
- 5CABin, Indian Council of Agricultural Research (ICAR)-Indian Agricultural Statistical Research Institute, New Delhi, India
- 6Sher-E-Kashmir University of Agriculture Science and Technology, Jammu, India
- 7Indian Council of Agricultural Research (ICAR)-Indian Institute of Millet Research, Telangana/Andhra Pradesh, Hyderabad, India
Pearl millet flour is highly nutritious but prone to rancidity, leading to off-odor development and reduced shelf life. To understand the molecular basis of this phenomenon, we performed de novo transcriptome sequencing on diverse pearl millet genotypes (landraces, hybrids, and composites) and identified 219,965 genes and 386,184 transcripts with functional annotation revealing key pathways linked to lipid and starch degradation. Differential gene expression (DGE) analysis identified significant upregulation of rancidity-linked genes [lipases (LIPs), lipoxygenases (LOXs), peroxidases (POXs), and polyphenol oxidases (PPOs)] in high-rancid genotypes. Data mining for characterizing rancid pathway showed the presence of 2,038 LIP, 209 Lox, 26 hydroperoxide lyase (HPL), 1,023 POX, and 17 PPO genes. Tissue-specific expression analysis of variants of Lip, Lox, Pox, and PPO during the different sub-stages of endosperm development showed an abundance of transcripts of Lip-2, LOX-3, and POX-4 during the seed hardening stage. Enzymatic assays confirmed increased LIP (up to 200.5 µmol h-1 g-1), LOX (184 nM HPOD min-1 mg-1), POX, and PPO activities in stored flour, correlating with rancidity progression. Notably, landraces exhibited lower expression of rancidity-linked genes compared to hybrids and composites, suggesting genetic variability in flour shelf life stability. Our study provides the first comprehensive transcriptomic resource for pearl millet rancidity, identifying candidate genes and enzymatic markers for future breeding programs aimed at improving flour storage quality.
Introduction
Pearl millet is considered as “God’s grain” because it has all the beneficial nutrients required in our balanced diet (Kumar et al., 2022b). This pseudo-grain crop was very much popular among our ancestors, but with the domination of cereals, it was slowly depleted from our plate (Satyavathi et al., 2021). In the post-green revolution era, millet fields have been replaced with fields that grow other cereal crops like wheat, rice, and maize. Food consumption pattern and the lack of diversity in food items lead to malnutrition in most parts of the world (Srivastava et al., 2022). The nutrient content of grains is depleting day-by-day due to adverse environmental conditions (Ashoka et al., 2023).
Pearl millet is a hardy crop and has inherent potential to survive under adverse environmental conditions with limited consumption of natural resources. The pearl millet grains are rich in carbohydrate (~65%–70%), lipid (~6%–7%), protein (10%–12%), fiber (~3%–3.5%), and micronutrients like Fe [~70 parts per million (ppm)] and Zn (~40 ppm) (Goswami et al., 2020). The short shelf life of the flour is one of the major hurdles in the production and utilization of pearl millet. Among all the millets and cereals, the pearl millet grain has maximum lipid content (~6%–7%) localized in the outer cover of the grains (Kumar et al., 2022a). The grains have very well-defined compartments for catabolic enzymes like α/β-amylases (Amy), lipases (Lip), proteases, lipoxygenase (LOX), peroxidase (POX), and polyphenol oxidase (PPO). These enzymes are localized in the germ tissue and are separated from their respective substrates dispersed in the endospermic tissue (Kumar et al., 2021; Aher et al., 2022). The micronutrients, especially iron and zinc, are localized in the outer cover of the grains (Srivastava et al., 2022). Recently, it has been reported that the quality of the pearl millet grains also deteriorates after long storage. Off-odor development, bitterness, and rancidity are exaggerated with the milling of grains into flour (Goswami et al., 2020). Upon milling, the catalytic enzymes localized in the germ tissue are exposed to their substrate like starch (α/β-amylases), lipid (lipase), and protein (proteases). Because of the high lipid content (~6%–7%), the lipase acts on these lipids and form free fatty acids (FFAs) along with glycerol (Kumar et al., 2021). FFAs are essential in our diet for various metabolic processes. FFAs serve as a substrate for the LOX enzyme, which oxidizes it into hydroperoxides. These hydroperoxides are further acted upon by hydroperoxide lyase (HPL) and POX to form various carbonyl compounds like aldehyde and alcohol. These carbonyl compounds are mainly responsible for the off-odor and bitterness of the flour. Even the browning of the flour happens due to the sequential reaction of PPO on polyphenols present in the flour (Ali et al., 2023a). The pearl millet flour has a very short shelf life [7–12 days after milling (DAM)], which restricts its use in food processing and manufacturing industries.
The advancement in basic tools and techniques has facilitated the characterization of various biological pathways in agriculturally important crops. It led to the discovery of key genes/proteins/metabolites associated with different processes within the plant system. The genes/enzymes responsible for the off-odor development and bitterness in pearl millet flour have not yet been elucidated. Few variants of rancidity-causing genes have been partially characterized in crops like rice, oats, soybean, and rye (Alomari et al., 2018).
Next-generation sequencing (NGS) has revolutionized the world of research and has provided immense opportunity to identify novel genes/proteins (Satam et al., 2023). Different omics tools have been used recently in crops to identify novel genes, hypothetical proteins, metabolites, and their involvement in different pathways (Gondane and Itkonen, 2023, Sinha et al. 2011). RNA sequencing (RNA-seq) is a method of characterizing or identifying the whole set of mRNA or genes present in a sample (Fan et al., 2013). It provides ample opportunity to identify the potential genes and understand their function and regulation. It also helps in mapping the pathway and understanding the mechanism of any biological process operating inside the system (Wang et al., 2009).
Here, our primary objective is to identify candidate genes responsible for encoding proteins/enzymes that contribute to the off-odor development and rancidity in pearl millet flour. We aimed to dissect the molecular mechanism underlying off-odor development and rancidity by correlating genes with enzymes, lipid degradation, and associated oxidative processes.
Materials and methods
Samples and growth conditions
We have selected two landraces (Chadhi Bajri and Damodhar Bajri), one hybrid (Pusa-1201), and one composite (Pusa-701) of pearl millet for the present investigation. The seeds were collected from different agro-climatic zones in India with the help of ICAR-All India Coordinated Research Project (AICRP) on pearl millet, Jodhpur, India. The seeds were first pre-treated with Bavistin at 0.25% and plants were grown in rectangular plastic pots of dimensions 66 × 21 × 15cm (L × W × H) inside the Nanaji Deshmukh Plant Phenomics Centre at ICAR-Indian Agricultural Research Institute, New Delhi, India. Equal quantities of farmyard manure (FYM) and sand were used for filling the pots, and standard procedures were followed for the inter-cultural operations (Kumar et al., 2020b). The experiment was carried out with three biological replicates. The plants were grown under 28 ± 2°C during vegetative/pollination and 32 ± 2°C during spike development/grain-filling with a 12-h light/dark cycle and a relative humidity of ~75% during different stages of growth and development. The samples (leaf, stem, and spike) for the de novo transcriptome sequencing were collected during the grain-filling (sub-stage seed hardening). Samples for the expression analysis were collected at different developmental stages. The collected samples were immediately frozen in liquid N2 and stored in a −80°C deep freezer for further downstream application.
De novo transcriptome sequencing
Total RNA isolation and library construction
The total RNA was isolated from different tissues (leaf, stem, and spikes) of Chadhi Bajri, Damodhar Bajri (landraces), Pusa-1201 (hybrid), and PC-701 (composite) using the Trizol method (Chomczynski, 1993). The purpose of using different tissues was to understand the gene network and expression dynamics operating in developing grains. The quality and integrity of the isolated RNA were checked using NanoDrop One (Thermo Fisher Scientific) and by resolving the sample on 1.2% agarose gel. The total RNA with two distinct bands and an OD260/OD280 ratio of ~2.0 was further used for pooling and library construction. Pooled RNA sample (~400 ng) was used to prepare RNA-seq libraries using the Illumina Truseq Stranded mRNA sample preparation kit (Illumina Inc., San Diego, CA, USA). The pooled samples were subjected to paired-end sequencing (2× 150 bp) on the Illumina HiSeq™ 2500 system (Illumina Inc., San Diego, CA, USA) following the protocols as mentioned in our earlier publication (Kumar et al., 2015).
De novo assembly and gene annotation
The quality of raw reads generated was checked using FASTQC v 0.11.9 (https://bioweb.pasteur.fr/; Andrews, 2010). We have used AdapterRemoval v2.3.3 (https://github.com/MikkelSchubert/adapterremoval/releases/tag/v2.3.3) to remove the adapter sequences and low-quality bases. We kept the minimum read length to 30 with a Phred Score of 20 in the trimming step. The raw data were aligned with the Silva database using Bowtie2 v 2.3.5.1 for rRNA removal. All trimmed paired reads data were used to assemble the transcriptome using Trinity v 2.15.1 with default parameters (https://github.com/trinityrnaseq/trinityrnaseq/releases/tag/Trinity-v2.15.1). We have further used the TrinityStats pearl script to examine the statistics of the assembly. The expression of the differentially expressed genes (DEGs) was carried out using Kallisto v 0.46.1 (https://github.com/pachterlab/kallisto) with default parameters. All assembled transcripts were subjected to annotation using the Rice Database [ftp://ftp.ensemblgenomes.org/pub/plants/release-46/fasta/oryza_sativa/pep/Oryza_sativa.IRGSP-1.0.pep.all.fa.gz] and NCBI ANNOTATIONX v 2.6.0+, and the search was performed against UniProt databases (https://www.uniprot.org/) based on homology. The annotation was carried out based on best hit and in-house pipeline. The differential gene expression (DGE) analysis was carried out using edgeR software v 3.19 (https://bioconductor.org/) with default parameters, and the results were filtered based on a log2 fold change of +2/−2 and p-value ≤ 0.05.
Validation of RNA-seq data using quantitative real-time PCR
To validate RNA-seq data, pooled samples from hybrid (Pusa-1201) collected during the grain-filling stage were used for the total RNA isolation, cDNA synthesis, and quantitative real-time PCR (qRT-PCR) (Kumar et al., 2020a). We have randomly selected 10 differentially expressed transcripts (DETs) from the RNA-seq data and designed transcript-specific primers to be used for the qRT-PCR (Supplementary Table S1). The DETs selected for the expression analysis were Serine/threonine kinase (STK; transcript no. DN2506), Lipase class-III (Lip-III; transcript no. DN63949), Heat shock protein 70 (HSP70; transcript no. DN1582), Glucoside hydrolase (GH; transcript no. DN15277), UDP glucosyltransferase (UGT; transcript no. DN51146), Phenyalanine lyase (PAL; transcript no. DN8260), Sucrose transporter (SUT; transcript no. DN48102), Glutamate dehydrogenase (GDH; transcript no. DN254), Mn-superoxide dismutase (MnSOD; transcript no. DN131007), and Lipoxygenase (LOX; transcript no. DN7868). The oligos were synthesized commercially and used for the qRT-PCR expression analysis following the steps mentioned in our earlier publication (Kumar et al., 2020a). The relative quantification of mRNA was performed on the CFX96 platform (Bio-Rad, UK) using three biological replicates. Actin-7 gene (accession no. KM105957.1) was used as an endogenous control gene for normalizing the Ct value and the fold expression was calculated using the Pfaffl method (Pfaffl, 2001). The fold expression (2−ddCt value) was converted to the absolute value of log2 fold change for comparative analysis with the RNA-seq dataset and for establishing the correlation between the RNA-seq and qPCR datasets.
Identification of genes associated with pathway responsible for flour rancidity
We have retrieved the coding sequences of some of the genes linked to rancidity, such as Lip, LOX, POX, and PPO from RAP-DB (Rice Annotation Project Database) and The Arabidopsis Information Resource database (TAIR, www.arabidopsis.org). The sequences were used as queries to search their orthologs in our transcriptome assembled sequences.
Expression analysis of rancidity-causing genes in endosperm and harvested grains
The potential transcripts showing homology with enzymes responsible for off-odor development in flour were identified, and furthermore, a few of them were selected (based on digital fold expression) for their expression analysis at different sub-stages of endosperm development (S0, button stage; S1, milky-ripe; S2, mealy-ripe; S3, seed hardening; S4, harvested grains) in pearl millet cv. Pusa 1201. The genes selected for the expression analysis were lipase [Lip-1 (accession no. OQ184871), Lip-2 (accession no. MZ590565), Lip-3 (accession no. MZ590564), and Lip-4 (accession no. OQ304605)], lipoxygenase [LOX-1 (accession no. OQ184873), LOX-2 (accession no. OQ184874), LOX-3 (TRINITY_DN10043_c0_g1_i1), and LOX-4 (TRINITY_DN118950_c0_g1)], peroxidase [POX-1 (TRINITY_DN109983), POX-2 (TRINITY_DN177283), POX-3 (accession no. PP171489) & POX-4 (TRINITY_DN115862)], and polyphenol oxidase [PPO-1 (accession no. PP765147), PPO-2 (TRINITY_DN183701_c0_g1), PPO-3 (TRINITY_DN167165_c0_g1), and PPO-4 (TRINITY_DN112679_c0_g1)]. The transcript-specific primers were designed using the Genefisher2.0 primer designing software (https://bibiserv.cebitec.uni-bielefeld.de/genefisher2) and were synthesized commercially (Supplementary Table S1). The tissues (different sub-stages of endosperm development) along with flour were used for the total RNA isolation using the RaFlex Total RNA Isolation Kit (Geneilabs). Other steps were followed as mentioned in our earlier publication (Kumar et al., 2020a). Actin-7 gene (accession no. KM105957.1) was used for normalizing the Ct value. The relative fold expression was calculated using the Pfaffl method (Pfaffl et al., 2002).
Activity assay of rancidity-causing enzymes in developing endosperm and flour
Based on our findings in the present investigation and the information retrieved from other literature, we performed activities of enzymes linked to off-odor development/rancidity like LIP, LOX, POX, and PPO in flour 0 (fresh flour, M0), 10 (M1), 30 (M2), 60 (M3), and 90 (M4) DAM.
Preparation of enzyme extract
We have optimized the extraction conditions (pH, molarity, and buffer) through a pilot experiment, and furthermore, 1g of flour was used for the enzyme extract preparation by homogenizing it in 10 mL of 0.2 M K2HPO4 buffer (pH 7.5). The homogenate was centrifuged at 10,000 rpm for 20min in a refrigerated centrifuge. The supernatant collected was filtered through four layers of cheese cloth and was used as enzyme extract for the activity assay. We have used the Bradford method for the estimation of total soluble protein (Bradford, 1976).
Activity assay of lipase and lipoxygenase enzymes
The activity assay of LIP was performed following the method of Itaya and Ui with some modifications (Itaya and Ui, 1966). A reaction mixture was prepared by adding 1 mL of substrate [0.98% (w/v) NaCl, 200 µL of Tween 20, and a mixture of olive oil and water in a 1:3 ratio] and 100 µL of enzyme extract and was incubated at 37°C for 15min; other steps were followed, as mentioned in the protocol. The LIP activity was calculated using the standard curve of oleic acid and expressed as nanomolar FFAs min−1 mg−1 soluble protein. The activity assay of the LOX enzyme was carried out as mentioned in our earlier publication (Goswami et al., 2020). The molar extinction coefficient of linoleic acid (25 mM−1 cm−1) was used for calculating the activity of LOX, and the result was presented as nanomolar hydroperoxide min−1 mg−1 soluble protein (nM HPOD min−1 mg−1 soluble protein).
Activity assay of polyphenol oxidase and peroxidase enzymes
We have assayed the PPO activity following the protocol of Almeida et al. (2019). In brief, a reaction mixture was prepared by adding 50 μL of enzyme extract along with 1,450 μL of phosphate buffer (0.2 M, pH 6.0) and 1,500 μL of catechol (0.2 M). The absorbance of RM was taken at 425 nm for 1, 2, and 3 min with 30-s intervals between the readings. The activity of PPO was calculated using the molar extinction coefficient of catechol (3,400 mM–1 cm–1) and was expressed as μmol min–1 g FW–1.
Similarly, the POX activity assay was performed following the method mentioned above (Almeida et al., 2019). In brief, a reaction mixture was prepared by adding 50 μL of enzyme extract along with 1,750 μL of phosphate buffer (0.2 M, pH 6.0), 100 μL of guaiacol (5 g L–1), and 100 μL of hydrogen peroxide (0.8g L–1). The absorbance was taken at 470 nm for every 1, 2, and 3 min. The activity of POD was calculated using the molar extinction coefficient of guaiacol (26.6 mM–1 cm–1) and expressed as μmol min–1 g FW–1.
Statistical analysis
We have used three biological replicates for the biochemical and molecular analysis. The data generated were characterized using one-way analysis of variance (ANOVA) conducted using SPSS (SPSS Inc., USA) (p ≤ 0.05).
Results and discussion
Pearl millet, often classified as an orphan crop, remains underexplored at the molecular level, particularly with respect to genes and proteins associated with key biological processes. One of the major challenges limiting its wider adoption is the rapid rancidity and off-odor development in its flour after milling (Ali et al., 2023b). However, there is a significant knowledge gap regarding the genetic, molecular, and proteomic basis of this phenomenon. The biochemical pathway responsible for the rancidity, along with its key molecular components, remains largely uncharacterized in pearl millet.
De novo transcriptome sequencing of pooled samples (leaf, stem, and spikes)
Here, we performed de novo transcriptome sequencing of pooled samples from diverse cultivars of pearl millet to identify candidate genes and their potential functional roles in flour rancidity. Total RNA was isolated from leaves, stem, and spike tissues during the seed hardening stage and used for library construction, following standard approaches reported for cereal transcriptomics (Varshney et al., 2017). The RNA-seq was performed using the Illumina Hiseq-4000 platform. We have generated ~8–10 Gb raw data per sample with GC% in the range of 46–49 and a Q30 value of 97 (Table 1). Base quality score distribution showed an average base quality Q30 of 97.32% (error probability ≥ 0.001). The findings were consistent with the high-quality sequencing benchmarks (Kulkarni et al., 2016). The high-quality reads from all the 12 samples were used to generate one pooled assembly using the Trinity assembler with default options, as successfully applied in several cereal de novo assemblies (Grabherr et al., 2011). The summary of transcriptome assembly results is presented in Supplementary Table S2. The length distribution for all assembled transcripts is presented in Supplementary Figure S1. Using the Trinity assembler, we have identified 219,965 genes and 386,184 transcripts in diverse genotypes of pearl millet with a ~GC of 47.98%. We have observed the average contig length of 1,208.13 with an N50 value of 2,614. The total number of assembled bases was 154,468,863 with a maximum average transcript length of 200–250 bp. RNA-seq has been used in the past to understand the regulation of hormones, seedling germination, and mechanism of abiotic and biotic stress tolerance in pearl millet (Wu, 1995; Shivhare et al., 2020).
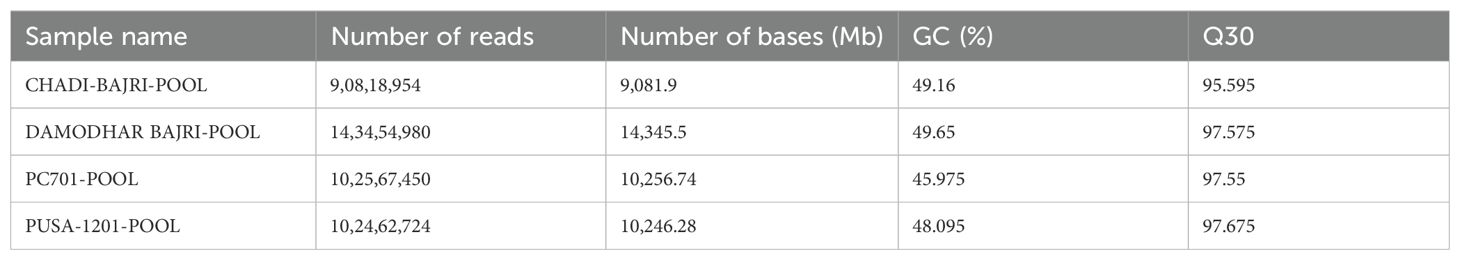
Table 1. Raw data summary generated from de novo transcriptome sequencing of pooled samples of landraces (Chadhi Bajri and Damodhar Bajri), Hybrid (Pusa-1201), and composite (PC-701) of pearl millet.
Annotation of transcripts generated using de novo transcriptome sequencing
All assembled transcripts were subjected to annotation using the Rice Database and NCBI ANNOTATIONX v 2.6.0+. We observed 42,355 coding sequences by mapping on the Rice Database. We observed 370,201 sequences (95.86%) based on homology using Top-hit (Supplementary Table S3). Furthermore, characterization for the identification of genes based on the Rice Database showed the presence of 370,201 genes (Supplementary Table S4). We have observed 34,890 rice coding sequences showing homology with Trinity sequences. The number of trinity sequences that remains unannotated was ~15,983.
Differential gene expression analysis
We have identified the DGEs by comparing the hybrid and composite with landraces (considered as the hidden treasure of genes). DGE was performed using the edgeR program; we used 386,184 transcripts for the analysis, and DEGs were filtered at log2 fold change with a cutoff of +2/−2 (p-value of ≤0.05) (Supplementary Table S5).
Comparative analysis among the landraces (Damodhar Bajri and Chadhi Bajri) showed the presence of 2,834 downregulated and 2,158 upregulated genes (Supplementary Table S6). We observed very significant differences in the expression of up- and downregulated genes (DEGs) among the landraces, with Chadhi Bajri having more DEGs, as compared to Damodhar Bajri. A similar pattern of expression was observed, when we compared the presence of DEGs in PC-701 (composite), as compared to Chadhi Bajri (landrace). The DEGs were observed to be maximum in landraces, as compared to composites. Comparative analysis of Pusa-1201 (hybrid) with Chadhi Bajri (landrace) showed maximum DEGs (downregulated—2,623) in hybrid, as compared to landrace. Similarly, comparative analysis of Pusa-1201 and Pusa-701 showed 2,186 upregulated and 2,077 downregulated genes. Stage-specific RNA-seq analysis showed the presence of 155 upregulated and 251 downregulated (during flowering stage) and 349 upregulated and 378 downregulated transcripts (during milking stage) linked to the regulation of Fe and Zn pathways (Satyavathi et al., 2021). We have also plotted a heatmap of the top 10 differentially expressed (DE) features within each pairwise comparison and observed maximum upregulated transcripts in PC-701 and downregulated transcripts in Pusa-1201 (Figure 1a). The expression of the top 10 DE showed significant variations among the landraces, hybrids, and composites. We also generated a heatmap of DEGs identified in the present investigation (Figure 1b). We observed the abundance of upregulated transcripts in Pusa-1201 and PC-701, whereas the downregulated transcripts were observed to be more abundant in landraces.
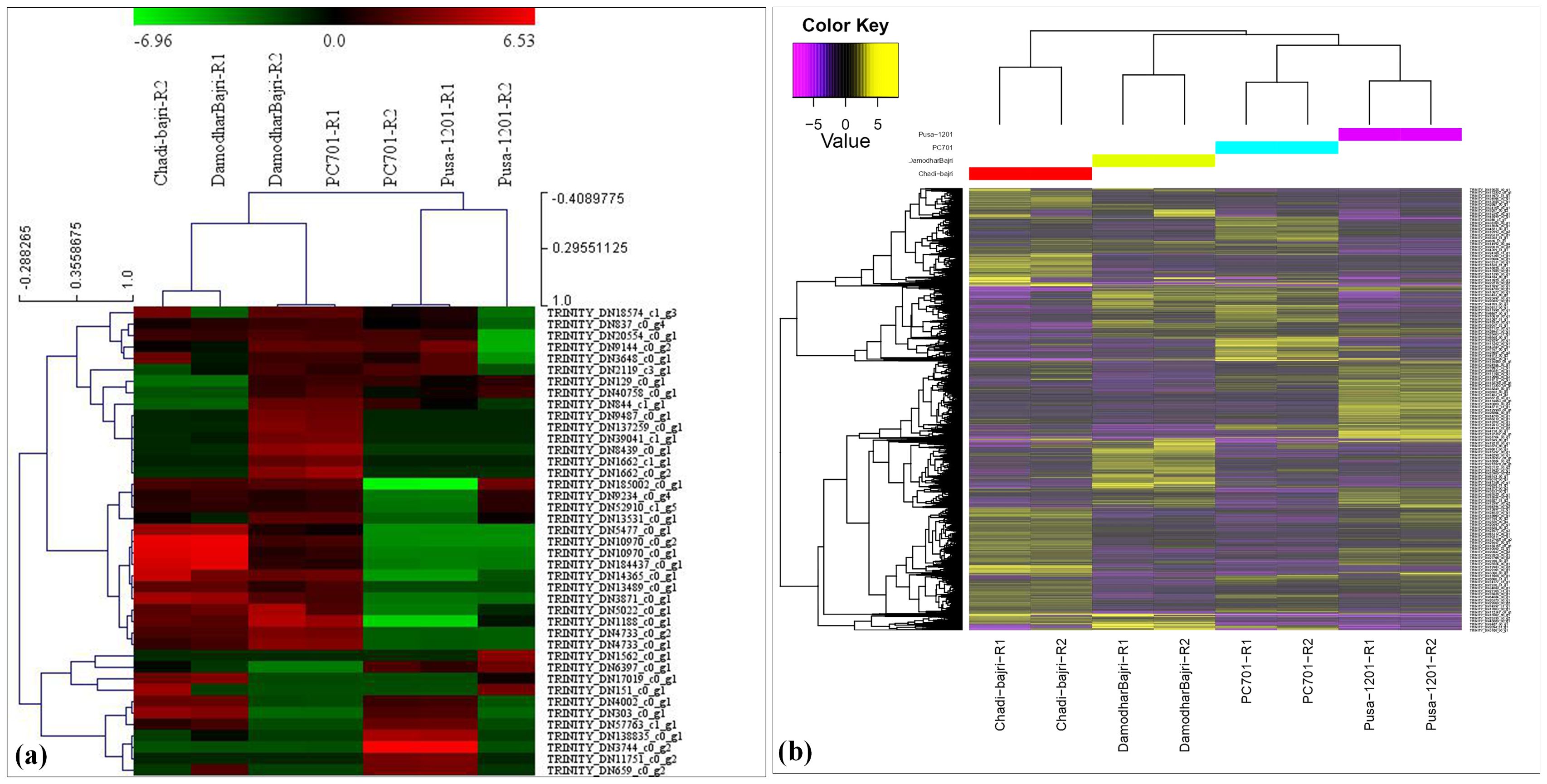
Figure 1. Differential gene expression (DGE) profiles in pearl millet genotypes. (a) Heatmap of the top 10 differentially expressed (DE) genes for each pairwise comparison (log2 FC ≥ 2, FDR ≤ 0.05). (b) Hierarchical clustering of all significant DEGs across landraces (Chadhi Bajri and Damodhar Bajri), hybrid (Pusa 1201), and composite (PC-701) genotypes. Columns indicate biological replicates; rows represent individual genes. Color scale: yellow/red = upregulation (log2 FC > 0), dark violet/green = downregulation (log2 FC < 0), dendrograms reflect Euclidean distance-based clustering of samples (columns) and genes (rows).
We have also generated a volcano plot in order to understand the pattern of DEGs among the landraces, hybrids, and composites. Volcano plot analysis of Damodhar Bajri with Chadhi Bajri showed a very discrete and concentrated appearance of DEGs (Supplementary Figure S2a). Similarly, volcano plot analysis of Pusa-1201 with Chadhi Bajri showed a very scattered pattern of up- and downregulated transcripts in hybrids, as compared to landraces (Supplementary Figure S2b). Comparative analysis of DEGs in PC-701 and Damodhar Bajri showed partially dispersed distribution of transcripts with the majority showing non-significant differences in expression (Supplementary Figure S2c). Volcano plot analysis of DEGs in Pusa-1201 with PC-701 showed a very wide distribution of downregulated transcripts, as compared to upregulated transcripts (Supplementary Figure S2d). Most of the DEGs (up- and downregulated) were observed to overlap in Pusa-1201 and PC-701 of pearl millet.
Comparison of the top 10 DE based on genes showed a much higher expression of genes in landraces compared with composites and hybrids, as evident from the color code. The findings are in conformity with the observation of Varshney et al. (2017). A de novo transcriptomic approach has been used to identify 1,396 up- and 936 downregulated and 1,000 up- and 1,591 downregulated transcripts in resistant inoculated/resistant control and susceptible inoculated/susceptible control of pearl millet (Kulkarni et al., 2016).
Gene Ontology analysis of annotated transcripts
The 370,201 transcripts identified were further subjected to functional annotation using the Gene Ontology (GO) analysis and were classified into three groups: GO (molecular function), GO (biological process), and GO (cellular components) (Figure 2; Supplementary Table S7). GO analysis of annotated transcripts based on molecular functions showed 3,862 transcripts to be involved in RNA binding [GO:0003723], followed by 1,744 transcripts in oxidoreductase activity [GO:0016491]. Very few transcripts were observed to be involved in fatty acid binding [GO:0005504]. GO analysis revealed the flavonoid pathway, lignin biosynthesis, and phenyl propanoid pathway to be more enriched in pearl millet during the flowering stage (Shivhare et al., 2020).
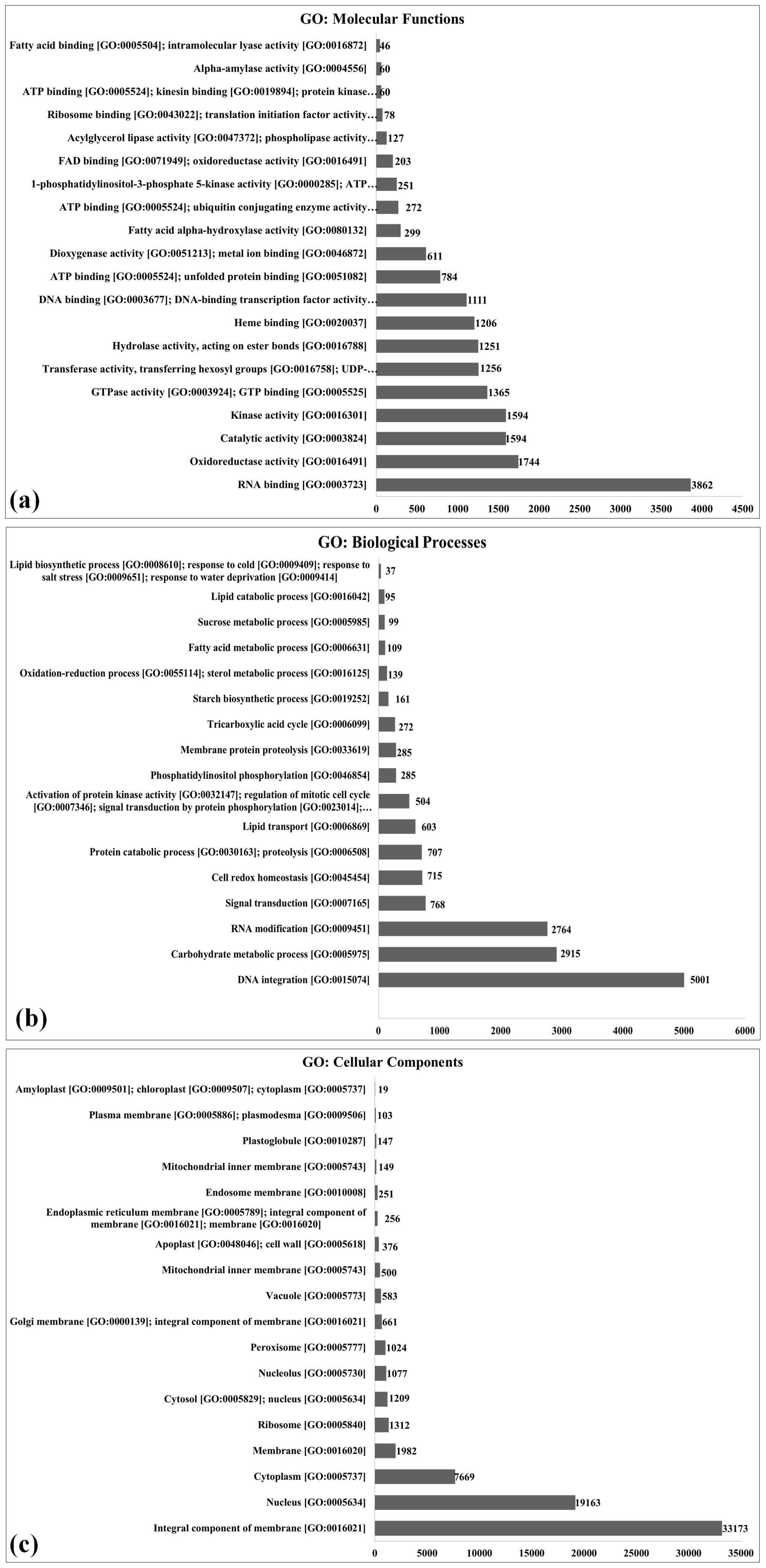
Figure 2. Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEGs) in pearl millet genotypes, Functional categorization of DEGs identified through de novo transcriptome sequencing is shown for (a) molecular function, (b) biological process, and (c) cellular component. The X-axis indicates the number of DEGs assigned to each GO term (count), while the Y-axis lists the significantly enriched GO terms (p ≤ 0.05); DEGs were derived from comparisons between landraces (Chadhi Bajri and Damodhar Bajri), hybrid (Pusa 1201), and composite (PC-701) genotypes.
Similarly, GO analysis of annotated transcripts based on biological processes showed a maximum of 5,001 transcripts to be involved in DNA integration [GO:0015074] followed by 2,915 transcripts involved in the carbohydrate metabolic process [GO:0005975]. A minimum number of transcripts were observed to be involved in the lipid biosynthetic process [GO:0008610]. The DE of a large number of genes linked to carbohydrate metabolism may be due to the fact that pearl millet leaves have very efficient photosynthetic and photosynthate transportation systems. It leads to the accumulation of a high content of carbohydrate (~70%–75%) in the grains. The genes linked to the lipid biosynthetic process were very low in spite of the established fact that the pearl millet grain has a very high content of TAGs (~6%–7%). This may be due to the fact that samples in the present investigation were collected during the grain-filling stage, a developmental phase in which the plant predominantly allocates resources to starch synthesis rather than lipid biosynthesis (Sreenivasulu et al., 2006). Other than the carbon assimilatory pathway, the secondary metabolic pathway, especially the phenylpropanoid pathway was observed to be most triggered in pearl millet under stress (Shivhare et al., 2020).
GO analysis of annotated transcripts based on cellular components showed a maximum of 33,173 transcripts to be associated with the integral component of the membrane [GO:0016021] followed by 19,163 transcripts with nucleus [GO:0005634] and 7,669 transcripts with cytoplasm [GO:0005737]. A minimum number of transcripts were observed to be associated with amyloplast [GO:0009501].
Transcripts expressing unique proteins in the grains of pearl millet
The identified transcripts generated through a de novo transcriptomic approach were further characterized for their unique appearance in landraces (Chadhi Bajri and Damodhar Bajri), hybrid (Pusa-1201), and composite (PC-701) of pearl millet. In the case of Chadhi Bajri, we observed the appearance of sugar/inositol transporter domain-containing protein (TRINITY_DN100083_c0_g1), similar to root hairless 1 (TRINITY_DN10016_c0_g1) and caleosin-related family protein (TRINITY_DN100212_c0_g1), among others (Supplementary Table S8). Similarly, Damodhar Bajri showed the presence of the BTB/POZ domain-containing protein (TRINITY_DN100005_c0_g1) and E3-ubiquitin ligase (TRINITY_DN100043_c0_g1), similar to GTP-binding protein (TRINITY_DN100058_c0_g1), among others, which were uniquely expressed in Damodhar Bajri and not observed in other landraces, hybrids, and composites of pearl millet. Similarly, 3,489 unique protein-coding transcripts were reported in the Indian barnyard millet (Jayakodi et al., 2019).
Hybrids of pearl millet are quite popular and demanding among the farmers because of their desirable traits and high yield, as compared to landraces. In the case of Pusa-1201, we observed the expression of unique proteins like wall-associated kinase-like protein (TRINITY_DN100329_c0_g1), transcriptional coactivator/pterin dehydratase family protein (TRINITY_DN100329_c0_g2), and F-box associated interaction domain-containing protein (TRINITY_DN100396_c0_g1) (Table S3). Similarly, PC-701 showed the expression of unique proteins like conserved hypothetical protein (TRINITY_DN100004_c0_g1), zinc finger POZ domain protein (fragment) (TRINITY_DN100388_c0_g1), and galactosyl transferase family protein (TRINITY_DN100575_c1_g1). These unique proteins, as annotated through their respective transcripts, provide a specific trait to the landraces (Chadhi Bajri and Damodhar Bajri), hybrid (Pusa-1201), and composite (PC-701). This is the reason behind the unique characteristics of these genetically diverse genotypes of pearl millet.
Identification of genes associated with rancidity/off-odor development
Rancidity/off-odor development in pearl millet flour has been considered as one of the major problems in its utilization, especially for the processing industries. Very limited work has been done in the past for the identification of genes/enzymes associated with rancidity/degradation of macromolecules in the flour.
Identification of putative lipase transcripts
LIP is considered the first enzyme of the rancidity pathway, catalyzing the hydrolysis of triacylglycerol into FFAs and glycerol. These FFAs are subsequently acted upon by downstream enzymes, leading to the formation of volatile compounds responsible for off-odors in flour (Kumar et al., 2021). In the present study, we identified 2,038 transcripts showing homology with LIP based on conserved domains (Supplementary Table S9). Representative putative LIP transcripts are presented in Table 2, with transcript sizes ranging from 275 to 937 bp. The majority of LIP transcripts were mapped to chromosome 8, followed by chromosomes 4 and 10, consistent with the distribution of LIP gene families reported in cereals (Lee and Park, 2019). Functional annotation revealed that these transcripts belong to diverse LIP classes, including GDSL-LIP, LIP class III, monoglyceride LIP, esterase/LIP, triacylglycerol LIP, and phospholipase. We have further cloned and characterized eight LIP genes from pearl millet (Kumar et al., 2025). The identified transcripts need validation, cloning, and biochemical characterization to confirm their roles in rancidity and to explore their potential as molecular markers in future pearl millet breeding programs. Based on the accumulation of FFAs in highly rancid lines, LIP was proclaimed as the real culprit for rancidity (Aher et al., 2022).
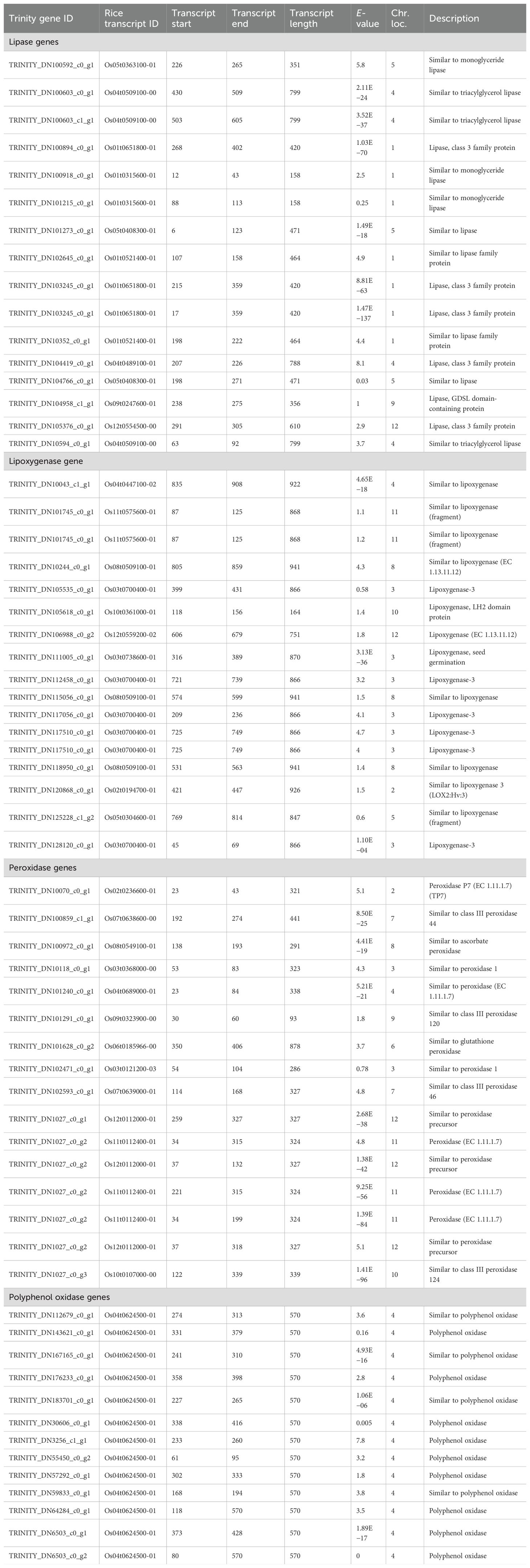
Table 2. List of different variants of putative lipase (LIP), lipoxygenase (LOX), peroxidase (POX), and polyphenol oxidase (PPO) genes identified in pearl millet landraces, hybrid, and composite genotypes through de novo transcriptome analysis.
Identification of putative lipoxygenase transcripts
Lipoxygenase uses FFAs as a substrate and form hydroperoxide and many other carbonyl compounds like aldehydes, alcohols, and ketonyl compounds. These products basically act as a substrate for the off-odor development in the flour (Sinha et al., 2020). LOX has a small N-terminal PLAT domain and a major C-terminal catalytic domain that were used in the present investigation to find out the transcripts showing homology with LOX. We have identified 209 putative transcripts showing homology with LOX reported from pearl millet (Supplementary Table S10). The size of the identified LOX transcripts was observed in the range of 125 to 941 bp. Most of the identified putative LOX transcripts were observed lying on Chr 3 followed by Chr 2 and Chr 4. The identified LOX was observed to be associated with different families like LOX2.3, LOX-3, and LOX LH2 domain-containing protein. Furthermore, we have cloned two LOX genes from pearl millet (LOX—accession no. OQ184873, LOX-6—accession no. OQ184874). In-depth kinetics study of these enzymes will help in regulating their activity and developing technology for enhancing the shelf life of pearl millet flour. Twelve LOX genes were identified from foxtail millet showing differential expression in response to different biological processes (Zhang et al., 2021).
Identification of putative hydroperoxide lyase transcripts
The hydroperoxide formed due to the activity of LOX enzyme on FFAs is used as a potential source for the production of carbonyl compounds like aldehyde, alcohol, and ketonyl by the HPL enzyme. Genes/proteins of this enzyme have not been characterized much in pearl millet. We have identified 26 transcripts showing homology with HPL (Supplementary Table S11). The size of the identified HPL genes was observed in the range of 480 to 510 bp. All the identified HPL was observed localized on Chr 2. Different variants of HAL have been identified and reported from different crops like soybean and rye. Many HPL genes have been identified and cloned from grape berries and characterized to play an important role in sugar metabolism and aroma volatiles (Šikuten et al., 2025).
Identification of putative peroxidase transcripts
POX is considered as a very versatile monomeric glycoprotein having two prominent domains—the heme group and the ferriprotoporphyrin prosthetic group. This enzyme is involved in different processes like detoxifying, neutralizing ROS, and producing various ketonyl compounds. Here, we identified 1,023 transcripts showing homology with POX from pearl millet (Supplementary Table S12). The size of the identified transcripts was in the range of 93–878 bp. The identified POX transcripts were observed localized on almost all the chromosomes with a maximum on Chr 7 followed by Chr 3 and Chr 4. Ma et al. (2022) identified 132 genes of class III POX in the whole genome of foxtail millet.
Identification of polyphenol oxidase transcripts
PPO catalyzes the oxidation of phenols to quinone, which in turn imparts a brown/dark color to the product. Here, we have identified 17 transcripts showing homology with the PPO gene based on domain search analysis and were observed to be lying on Chr 4 (Supplementary Table S13). Niranjan-Raj et al. (2019) identified and cloned a putative PPO gene of 395 bp from pearl millet. Four PPO genes were reported from virgin olive oil and one gene was characterized to play a role in catalyzing the hydroxylation of tyrosol to form hydroxytyrosol (Sánchez et al., 2023).
Identification of genes associated with starch degradation
Starches constitute a major share of nutrient composition of pearl millet grain and are stored in complex form inside the endospermic tissue. Various isoforms of amylases are involved in the degradation of starch forming maltose and simple sugars. It also invites various other pathogenic microorganisms compromising the quality of the flour. Here, we have identified 401 transcripts showing homology with different variants of amylases (Supplementary Table S14). The sizes of the transcripts were in the range of 244–1,121. Most of the genes were observed localized on Chr 8 followed by Chr 9 and Chr 7. Some of the variants of the amylases identified are glucoamylase, starch debranching enzyme, and α/β-amylases. Yadav et al. (2022) identified a single α/β-amylase gene from pearl millet through a genome-wide association study (GWAS).
Validation of RNA-seq data using qRT-PCR
To validate RNA-seq data, we have randomly selected 10 DETs and used their digital fold expression (log2 FC) from the RNA-seq dataset for comparative analysis with relative fold change (converted to log2 FC), as achieved through qPCR (Figure 3).
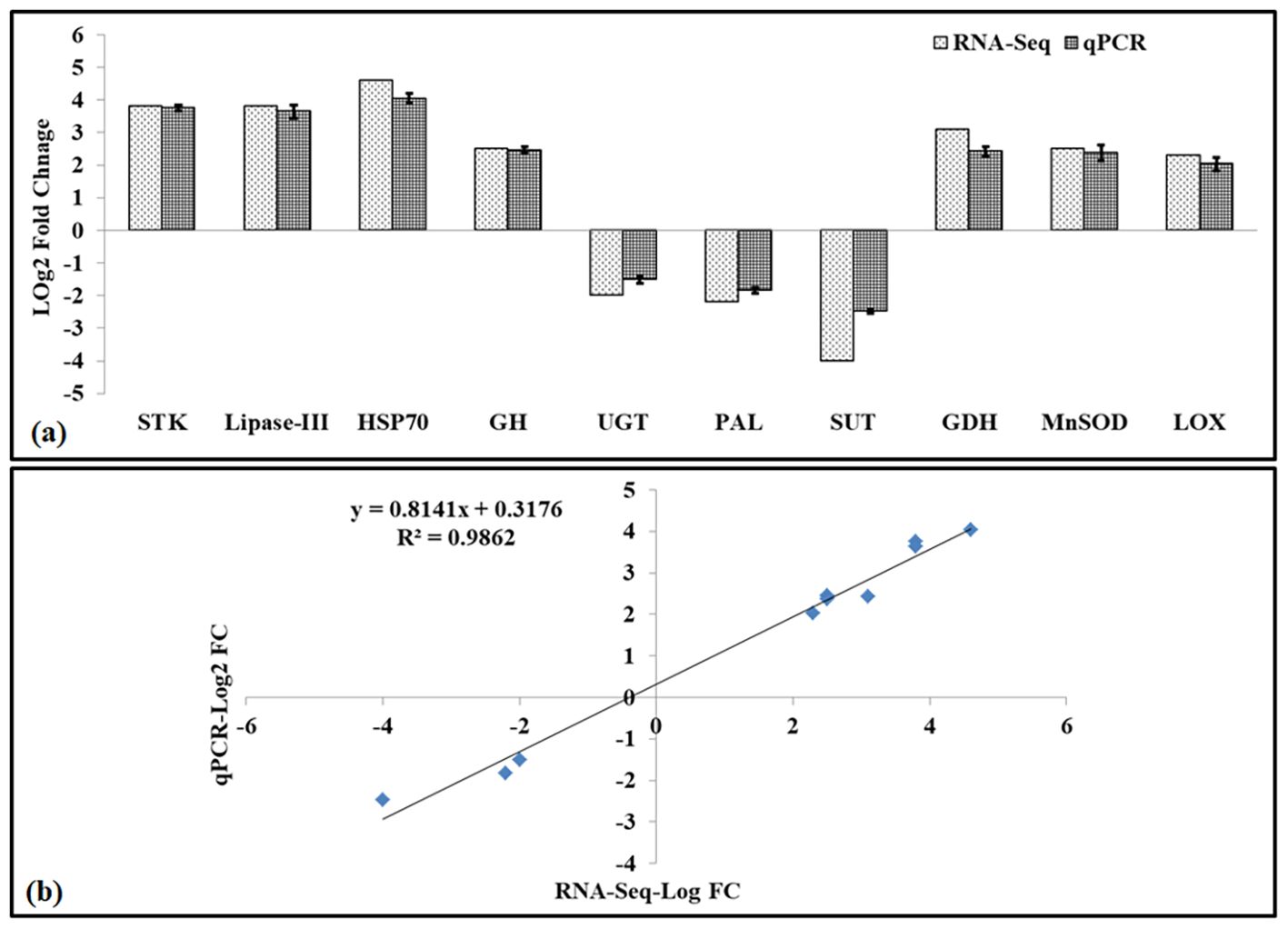
Figure 3. Validation of RNA-seq data using quantitative real-time PCR (qRT-PCR). (a) Log2 fold change comparison of RNA-seq (dotted bars) and quantitative real-time PCR (qRT-PCR, netted bars) of 10 differentially expressed genes (DEGs). (b) Correlation between RNA-seq and RT-PCR gene expression (R2 = 0.9862, p<0.05); lines above the qRT-PCR bar indicate a significant difference between treatments (p ≤ 0.05, one-way ANOVA).
The DETs selected for the comparative analysis were Serine/threonine kinase (STK, DN2506), Lipase class-III (Lip-III, DN63949), Heat shock protein 70 (HSP70, DN1582), Glucoside hydrolase (GH, DN15277), UDP glucosyltransferase (UGT, DN51146), Phenylalanine lyase (PAL, DN8260), Sucrose transporter (SUT, DN48102), Glutamate dehydrogenase (GDH, DN254), Mn-superoxide dismutase (MnSOD, DN131007), and Lipoxygenase (LOX, DN7868). The fold expression (2−ddCt value) was converted to log2 fold change for comparative analysis with RNA-seq dataset and for establishing the correlation between the RNA-seq and qPCR datasets. We observed significant variations (p ≤ 0.05) in the expression (upregulation and downregulation) of respective DEGs, as observed through RNA-seq data and qPCR data (Figure 3). A strong correlation (R2=0.98) was established between the RNA-seq dataset and qPCR dataset, which validates the quality of the data generated using the RNA-seq.
Expression analysis of genes linked to flour rancidity
Different variants of genes, as predicted through a domain-based homology search from de novo transcriptome sequencing data and few cloned in our lab, were used for the expression analysis: Lipase gene [Lip-1 (accession no. OQ184871), Lip-2 (accession no. MZ590565), Lip-3 (accession no. MZ590564), and Lip-4 (accession no. OQ304605)], Lipoxygenase [LOX-1 (accession no. OQ184873), LOX-2 (accession no. OQ184874), LOX-3 (TRINITY_DN10043_c0_g1_i1), and LOX-4 (TRINITY_DN118950_c0_g1)], Peroxidase [POX-1 (TRINITY_DN109983), POX-2 (TRINITY_DN177283), POX-3 (accession no. PP171489), and POX-4 (TRINITY_DN115862)], and Polyphenol oxidase [PPO-1 (accession no. PP765147), PPO-2 (TRINITY_DN183701_c0_g1), PPO-3 (TRINITY_DN167165_c0_g1), and PPO-4 (TRINITY_DN112679_c0_g1)].
We have identified four LIP (Lip-1, Lip-2, Lip-3, and Lip-4) genes to be used for the tissue-specific expression analysis. Lip-2 showed maximum relative fold expression (2.95-fold) during the S3 stage (seed hardening), as compared to the S1 stage (button stage) (Figure 4a). Lip-3 showed downregulation throughout the different sub-stages of endospermic tissue growth with minimum expression in harvested grains. All the Lip genes identified showed a significant (p ≤ 0.05) increase in the relative fold expression at different sub-stages of endospermic tissue growth with maximum expression during the seed hardening stage. The expression of Lip variants was observed to be very low in harvested grains, as compared to the button stage.
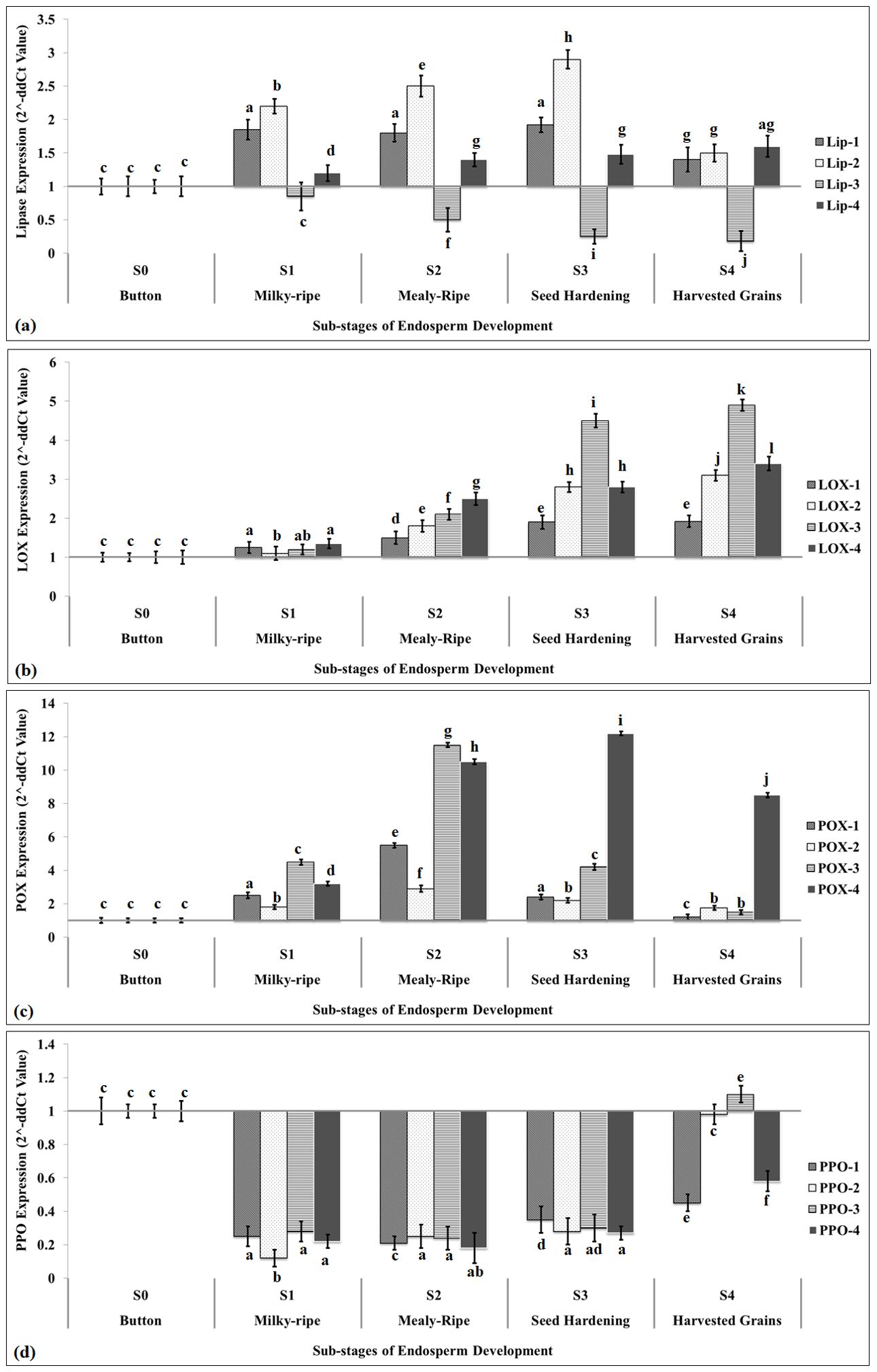
Figure 4. Tissue-specific expression analysis of identified rancidity-causing putative genes in endosperm of pearl millet using quantitative real-time PCR. (a) Expression of lipase gene, (b) expression of LOX gene, (c) expression of POX gene, and (d) expression of PPO gene. Sub-stages of endosperm used for the expression analysis: S0, button stage; S1, milky-ripe; S2, mealy-ripe; S3, seed hardening; S4, harvested grains; four different variants of LIP, LOX, POX, and PPO were used for the expression analysis; Actin-7 gene (acc. no. KM105957.1) was used as endogenous control; all data are presented as mean ± SE of three replicates; lines above each bar indicate a significant difference between treatments (p ≤ 0.05, one-way ANOVA).
We also identified four different variants of LOX genes (LOX-1, LOX-2, LOX-3, and LOX-4) and evaluated their tissue-specific expression pattern. LOX-3 showed maximum relative fold expression (5.1-fold) during the seed harvesting stage, as compared to the button stage (Figure 4b). We observed a significant (p ≤ 0.05) increase in the expression of all the four LOX variants at different stages of endospermic tissue growth and development. The abundance of transcript was observed minimum in the case of LOX-1 at different sub-stages of endospermic tissue growth compared with other variants of LOX.
We identified four different variants of POX (POX-1, POX-2, POX-3, and POX-4) genes and captured their expression pattern during the growth and development of endospermic tissue. Expression analysis showed maximum relative fold expression of POX-4 (12.1-fold) in harvested grains, as compared to the button stage (Figure 4c). We observed an abundance of transcripts of all the four variants of POX during different sub-stages of endosperm growth and development, except harvested grains, where the POX-4 transcript was observed at a maximum.
We selected four variants of PPO (PPO-1, PPO-2, PPO-3, and PPO-4) genes and captured their expression pattern in developing endosperm tissue of pearl millet. All the variants of PPO showed significant downregulation at different sub-stages of developing endosperm, as compared to the button stage (Figure 4d). PPO-3 showed upregulation (1.15-fold) in the harvested grain, as compared to the button stage. PPO-2 was observed to be highly downregulated during the milky-ripe stage (−0.1-fold), as compared to other variants of PPO. Although much information is not available on the expression pattern of these rancidity-causing genes in millets, some literature reported the DE of these genes in different crop species under other environmental factors. The tissue-specific expression pattern of 21 LOX genes was reported in Cannabis sativa (Fayaz et al., 2023).
Activity assay of rancidity-causing enzymes
We have observed four prominent enzymes (LIP, LOX, POX, and PPO) to be involved in off-odor development and lipid hydrolysis in pearl millet flour. The activities of all the four enzymes were analyzed in samples stored at different durations (0, 10, 30, 60, and 90 DAM).
LIP activity was observed to be at a minimum (88.5 µmol h−1 g−1 flour) in the flour of pearl millet cv. Chadhi Bajri stored for 90 DAM; a non-significant increase in the LIP activity was observed from 10 DAM onward (Figure 5a). The LIP activity was observed to be at a maximum (200.5 µmol h−1 g−1 flour) in cv. MPMH17 stored for 90 DAM. We observed a significant (p ≤ 0.05) increase in the activity of LIP enzyme, until M1 (10 DAM) in all the selected contrasting genotypes, and further non-significant (p ≤ 0.05) variations were observed with an increase in the number of DAM except cv. MPMH17. A similar pattern of LIP activity was observed in the flour of diverse germplasm of pearl millet stored at different durations (Goswami et al., 2020).
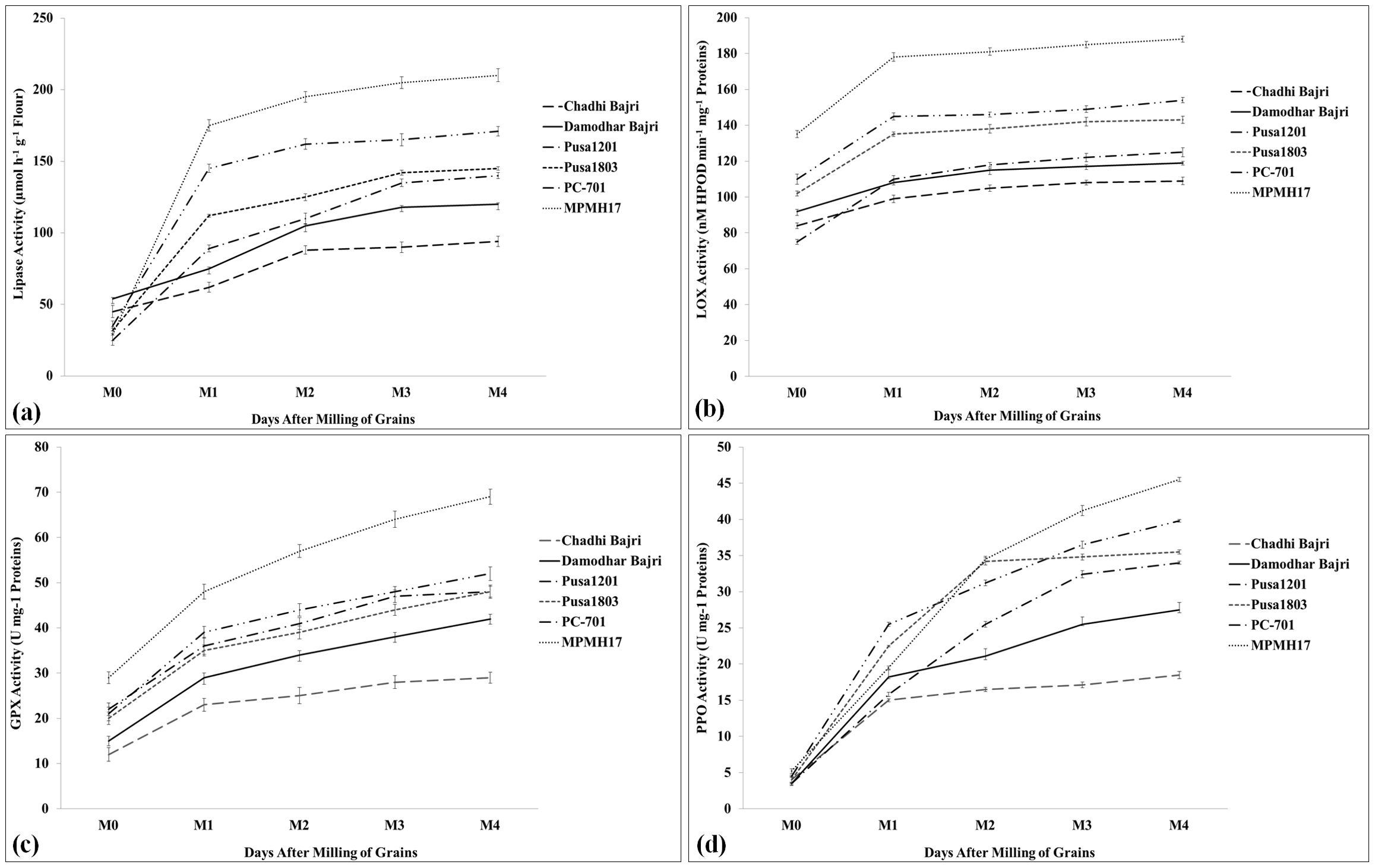
Figure 5. Activity assay of rancidity-causing enzymes in diverse genotypes of pearl millet. (a) Activity assay of lipase, (b) activity assay of lipoxygenase, (c) activity assay of guaiacol peroxidase, and (d) activity assay of polyphenol oxidase; six diverse genotypes were selected for the activity assay. Days after milling (DAM) of grains: M0, 0 DAM; M1, 10 DAM; M2, 30 DAM; M3, 60 DAM; M4, 90 DAM; all data are presented as mean ± SE of three replicates; lines above each bar indicate a significant difference between treatments (p ≤ 0.05, one-way ANOVA).
LOX assay showed minimum activity in the flour of pearl millet cv. Damodhar Bajri stored at different durations. The activity was observed to be at a minimum (103 nM HPOD min−1 mg−1 protein) during the M4 stage (90 DAM). The LOX activity was observed to be at a maximum (184 nM HPOD min−1 mg−1 protein) in pearl millet cv. Pusa-1803 during the M4 stage (90 DAM) (Figure 5b). Overall, the LOX activity was significantly (p ≤ 0.05) maximum in cv. Pusa-1803 analyzed in a sample stored at different durations, as compared to other contrasting cvs. We observed significant (p ≤ 0.05) variations in the LOX activity in samples stored up to 10 DAM, and further non-significant changes were observed in the stored flour up to 90 DAM. The findings are in conformity with the observation made by Goswami et al. (2020) and Kumar et al. (2025) in pearl millet.
A similar pattern of GPX activity was observed in the samples stored at different durations with maximum activity in the flour of cv. Pusa 1803 and minimum activity in cv. Damodhar Bajri. Pusa-1803 showed a gradual increase in the GPX activity until 90 DAM (Figure 5c). A very high activity of POX was observed in stored flour of pearl millet, and it was reported to be used as one of the biochemical markers for analyzing the shelf life of flour (Goyal et al., 2017).
PPO assay showed a very significant (p ≤ 0.05) increase in the activity during the first 10 DAM in all the selected cvs. The PPO activity was observed to be at a minimum in cv. Damodhar Bajri estimated at different time points (DAM), as compared to other cvs. The activity was observed to be at a maximum in cv. Pusa-1803 during M4 (90 DAM) and we observed an increased trend of PPO activity throughout the storage period (Figure 5d). A similar pattern of LIP, LOX, POX, and PPO activities was reported in the flour of contrasting pearl millet cvs (Kumar et al., 2024).
Remodeling the rancid pathway
Pearl millet has a very high content of lipid (~6%–7%) along with other beneficial nutrients in the grains. The pearl millet grains have numerous compartments packed with different macro- and micronutrients and catabolic enzymes responsible for rancidity (Aher et al., 2022; Kumar et al., 2025). The increase in the temperature upon milling triggered the activities of catabolic enzymes like α/β-amylases, proteases, and LIPs. This leads to the degradation of key nutrients accumulated inside the grains through oxidative/hydrolytic reactions (Ali et al., 2023a). We have identified almost 2,038 transcripts showing homology with LIP (Figure 6). Furthermore, we cloned eight LIP genes (accession nos. OQ184871, MZ590565, MZ590564, OQ304605, OQ304606, MW424397, PP107929, and PP114100) from contrasting pearl millet genotypes, and four were reported to be localized in the grains.
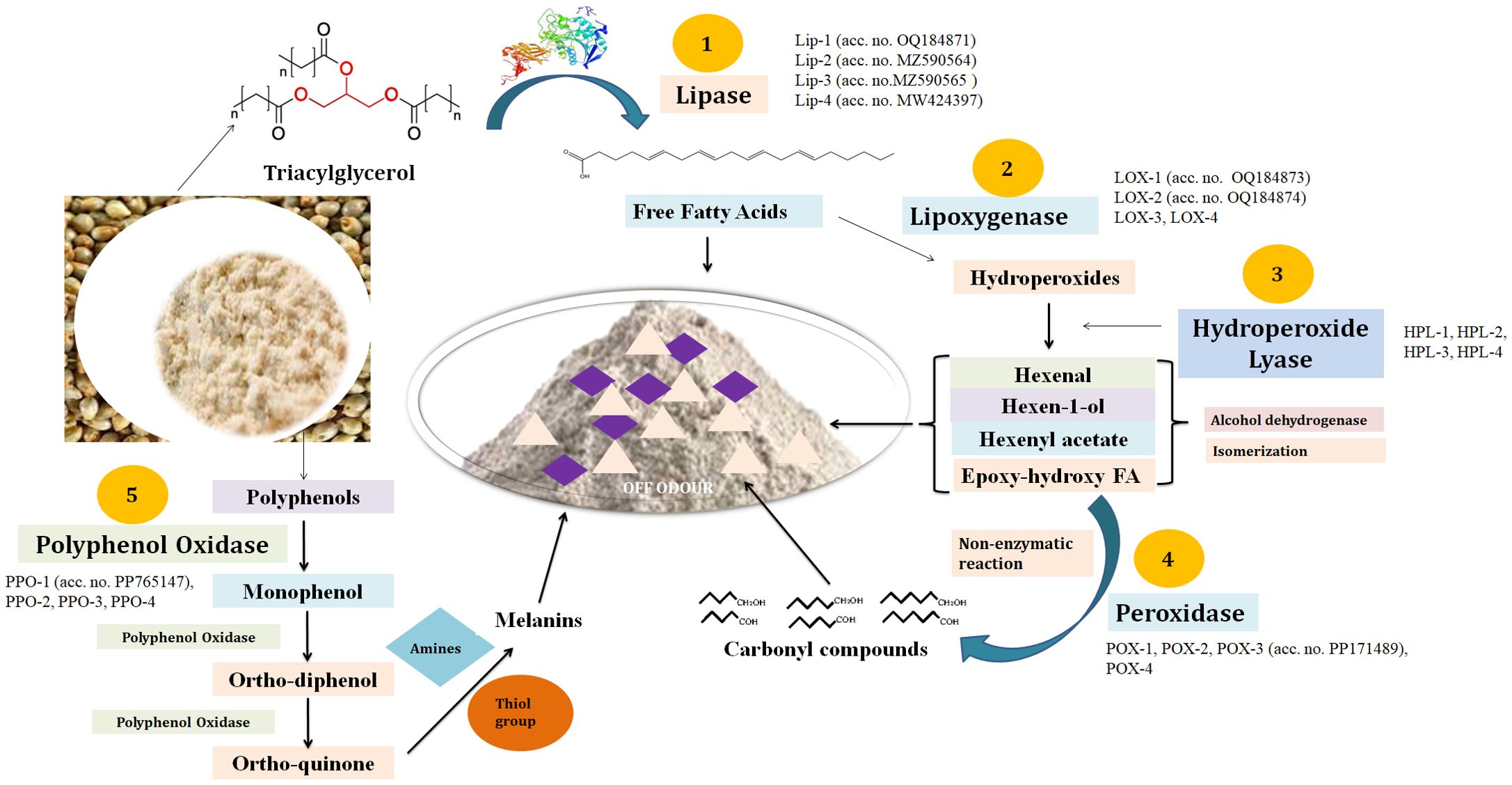
Figure 6. Molecular mechanism of rancidity development in pearl millet flour. The schematic integrates key genes, enzymes, and metabolites identified through transcriptomic and biochemical analyses: (1) Lipases (Lip 1–4) hydrolyze triacylglycerols (TAGs) into free fatty acids (FFAs); (2) lipoxygenases (LOX 1–4) oxidize polyunsaturated FFAs to hydroperoxides; (3) hydroperoxide lyases (HPLs) and peroxidases (POX 1–4) convert hydroperoxides into volatile aldehydes/ketones (hexanal, nonanal); and (4) polyphenol oxidases (PPO 1–4) catalyze phenolic oxidation, contributing to discoloration. Gray arrows indicate enzymatic reactions; colored boxes highlight differentially expressed genes (red: upregulated in high-rancid genotypes; blue: downregulated).
The product of triacylglycerol (TAG) degradation is FFAs and glycerol. The FFAs formed are further acted upon by LOX, which is a very stable, dynamic, and versatile enzyme (Figure 6). It acts on FFAs and forms hydroperoxides. We have identified 209 transcripts showing homology with the LOX gene and cloned two LOX genes (accession nos. OQ184873 and OQ184874) observed to be localized in the seeds. The hydroperoxides formed are further acted upon by the fatty acid HPL and forms short-chain aldehydes and oxo-acids like 3-hexenal, 11-formyl-9-undecenoic acid, and octanoic acid. These aldehydes, oxyacids, and ketonyl compounds are basically responsible for the off-odor development in pearl millet flour. We have identified 26 HPL genes in the present investigation and observed four to be localized in the endospermic tissue. We have not cloned any HPL gene from pearl millet. The POX enzyme acts on hydroperoxides and forms various carbonyl compounds mainly responsible for the off-odor and discoloration of the flour (Figure 6). We have identified 1,023 transcripts showing homology with the POX gene and cloned the putative POX-III gene (accession no. PP171489) from pearl millet cv. Pusa-1201 and was observed to be localized in the developing endosperm.
The pearl millet grain has an abundance of unique polyphenols that have numerous health benefits. Upon milling, these polyphenols are oxidized by the PPO enzyme and form a quinone compound that gives a brown/dark color to the flour upon storage. Here, we identified 17 putative transcripts showing homology with PPO and cloned a putative PPO gene (accession no. PP765147) from pearl millet cv. Pusa-1201. In silico characterization showed the respective protein to be localized in the grains. Combined metabolomics and transcriptomics showed the over-expression of SDR, FATA, FATB, and MFP responsible for the rancidity of FFAs in oil palm fruit (Zhang et al., 2023).
Overall, we predicted the genes responsible for the synthesis of rancidity-causing enzymes, their reactions, and metabolites, leading to off-odor development in flour. The findings are in conformity with the observation of Bhunia et al. (2023) in rice bran. They predicted enzymes and pathways responsible for the hydrolytic and oxidative rancidity of rice bran lipids. LIP initiates the TAG hydrolysis, and the product of the reaction is utilized by the LOX for oxidation and production of hydroperoxides—considered as the first definitive step in off-odor development. These hydroperoxides are further metabolized by HPL, POX, and alcohol dehydrogenases into aldehydes, acids, and peroxides, which are mainly responsible for the rancidity and bitterness of flour.
This degradation cascade occurs most rapidly within the first 10 DAM, after which it gradually slows down with storage. The identification and characterization of these key genes and enzymes laid the foundation for developing targeted strategies/technologies to regulate the activities of these enzymes in order to enhance the shelf life of pearl millet flour.
Conclusions
In conclusion, this study provides the first comprehensive de novo transcriptomic resource elucidating the molecular basis of rancidity in pearl millet flour. High-quality sequencing of diverse genotypes revealed over 219,000 genes and 386,000 transcripts, with differential expression analyses identifying key lipid- and starch-degrading enzymes—particularly LIPs, LOXs, POXs, and PPOs—as central drivers of post-milling deterioration. Landraces consistently exhibited lower expression of rancidity-associated genes than hybrids and composites, underscoring the potential of traditional germplasm for breeding shelf-stable varieties. Functional annotation and GO analysis linked the most enriched pathways to lipid catabolism, fatty acid oxidation, and oxidative browning, while tissue-specific expression profiling highlighted Lip-2, LOX-3, and POX-4 as major contributors during seed hardening and storage. Enzyme activity assays corroborated transcriptomic data, demonstrating early, sharp increases in hydrolytic and oxidative activities within 10 DAM, followed by sustained activity during storage. This integrative approach not only mapped the rancidity pathway—from triacylglycerol hydrolysis to aldehyde and ketone formation—but also pinpointed candidate genes and biochemical markers for targeted intervention. The results offer a genomic and biochemical foundation for pathway remodeling strategies, including breeding, genome editing, and post-harvest processing innovations, to mitigate rancidity without compromising nutritional quality. Ultimately, the findings pave the way for developing pearl millet cultivars and processing technologies with improved flour shelf life, thereby enhancing the grain’s marketability, nutritional impact, and role in food security.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/625418/, accession No. PRJNA625418.
Author contributions
RK: Conceptualization, Supervision, Validation, Writing – original draft, Writing – review & editing. SG: Data curation, Investigation, Methodology, Writing – review & editing. VT: Formal analysis, Validation, Data curation, Writing – review & editing. NS: Formal Analysis, Investigation, Validation, Writing – review & editing. UM: Methodology, Validation, Writing – review & editing. GM: Methodology, Supervision, Validation, Writing – review & editing. GJ: Data curation, Software, Supervision, Writing – review & editing. GR: Data curation, Investigation, Methodology, Writing – review & editing. SP: Formal Analysis, Validation, Visualization, Writing – review & editing. CT: Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Indian Council of Agricultural Research (ICAR) under the Global Centre of Excellence on Millets project (number 12–258 TG3361), NASF project grant (number NASF/MA-9014/2022-23), and grant from the Department of Science and Technology under the SHRI scheme.
Acknowledgments
The authors sincerely acknowledge the ICAR-All India Coordinated Research Project (AICRP) on Pearl Millet for providing the diverse pearl millet genotypes used in this study. We extend our gratitude to the Director and Project Coordinator (AICRP-Pearl Millet) for their continuous support and guidance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1677082/full#supplementary-material
Supplementary Figure 1 | Histogram showing the distribution of assembled transcripts length generated using de novo transcriptome sequencing of landraces, hybrid and composite of pearl millet.
Supplementary Figure 2 | Volcano plot analysis of DEGs identified in landraces, hybrid and composite of pearl millet using de novo transcriptome sequencing, a) Volcano plot analysis of DEGs in Damodhar Bajri vs Chadhi Bajri, b) Volcano plot analysis of DEGs in Pusa 1201 vs Chadhi Bajri, c) Volcano plot analysis of DEGs in PC701 vs Damodhar Bajri, and d) Volcano plot analysis of DEGs in Pusa 1201 vs PC701; Red is used to represent genes which were up-regulated, and blue represent genes which were down-regulated.
Abbreviations
NGS, next-generation sequencing; DEGs, differentially expressed genes; LIP, lipase; LOX, lipoxygenase; POX, peroxidase; PPO, polyphenol oxidase; PAL, phenylalanine lyase, DAM, days after milling; GO, Gene Ontology; TAG, triacylglycerol.
References
Aher, R. R., Reddy, P. S., Bhunia, R. K., Flyckt, K. S., Shankhapal, A. R., Ojha, R., et al. (2022). Loss-of-function of triacylglycerol lipases are associated with low flour rancidity in pearl millet [Pennisetum glaucum (L.) R. Br. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.962667
Ali, A., Kumar, R. R., Vinutha, T., Bansal, N., Bollinedi, H., Singh, S. P., et al. (2023a). Characterization of biochemical indicators and metabolites linked with rancidity and browning of pearl millet flour during storage. J. Plant Biochem. Biotechnol. 32, 121–131. doi: 10.1007/s13562-022-00787-0
Ali, A., Singh, T., Kumar, R. R., T, V., Kundu, A., Singh, S. P., et al. (2023b). Effect of thermal treatments on the matrix components, inherent glycemic potential, and bioaccessibility of phenolics and micronutrients in pearl millet rotis. Food Funct. 14, 1595–1607. doi: 10.1039/D2FO03143D
Almeida, S. L. D., Morais, M. A. D. S., Albuquerque, J. R. T. D., Barros Júnior, A. P., Simões, A. D. N., and Fonseca, K. S. (2019). Polyphenol oxidase and peroxidase enzyme assays in sweet potato cultivars harvested at different times. Rev. Caatinga 32, 537–542. doi: 10.1590/1983-21252019v32n226rc
Alomari, D. Z., Eggert, K., von Wirén, N., Alqudah, A. M., Polley, A., Plieske, J., et al. (2018). Identifying candidate genes for enhancing grain Zn concentration in wheat. Front. Plant Sci. 9. doi: 10.3389/fpls.2018
Andrews, S., Krueger, F., Segonds-Pichon, A., Biggins, L., Krueger, C., and Wingett, S. (2010). FastQC. A quality control tool for high throughput sequence data, 370.
Ashoka, P., Raut, D., Sudeepthi, B., Gawande, K. N., Reddy, Gurrala., S. V., Padhan, S. R., et al. (2023). Millet’s role as a climate resilient staple for future food security: A review. Int. J. Environ. Climate Change 13, 4542–4552. doi: 10.9734/ijecc/2023/v13i113634
Bhunia, R. K., Sinha, K., Kaur, R., Kaur, S., and Chawla, K. (2023). A holistic view of the genetic factors involved in triggering hydrolytic and oxidative rancidity of rice bran lipids. Food Rev. Int. 39, 441–466. doi: 10.1080/87559129.2021.1915328
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochem. 72, 248–254. doi: 10.1016/0003-2697(76)90527-3
Chomczynski, P. (1993). A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. BioTechniques 15, 532–534.
Fan, H., Xiao, Y., Yang, Y., Xia, W., Mason, A. S., Xia, Z., et al. (2013). RNA-Seq analysis of Cocos nucifera: transcriptome sequencing and de novo assembly for subsequent functional genomics approaches. PloS One 8, e59997. doi: 10.1371/journal.pone.0059997
Fayaz, M., Kundan, M., Gani, U., Sharma, P., Wajid, M. A., Katoch, K., et al. (2023). Identification of Lipoxygenase gene repertoire of Cannabis sativa and functional characterization of CsLOX13 gene. Plant Sci. 334, 111780. doi: 10.1016/j.plantsci.2023.111780
Gondane, A. and Itkonen, H. M. (2023). Revealing the history and mystery of RNA-seq. Curr. Issues Mol. Biol. 45, 1860–1874. doi: 10.3390/cimb45030120
Goswami, S., Asrani, P., Ansheef Ali, T. P., Kumar, R. D., Vinutha, T., Veda, K., et al. (2020). Rancidity matrix: development of biochemical indicators for analysing the keeping quality of pearl millet flour. Food Analytical Methods. doi: 10.1007/s12161-020-01831-2
Goyal, P., Chugh, L. K., and Berwal, M. K. (2017). Storage effects on flour quality of commonly consumed cereals. J. Appl. Natural Sci. 9, 551. doi: 10.31018/jans.v9i1.1228
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Itaya, K. and Ui, M. (1966). A new micromethod for the colorimetric determination of inorganic phosphate. Clinica Chimica Acta. doi: 10.1016/0009-8981(66)90114-8
Jayakodi, M., Lee, S. C., and Yang, T. J. (2019). Comparative transcriptome analysis of heat stress responsiveness between two contrasting ginseng cultivars. J. Ginseng Res. 43, 572–579. doi: 10.1016/j.jgr.2018.05.007
Kulkarni, K. S., Zala, H. N., Bosamia, T. C., Shukla, Y. M., Kumar, S., Fougat, R. S., et al. (2016). De novo transcriptome sequencing to dissect candidate genes associated with pearl millet-downy mildew (sclerospora graminicola sacc.) interaction. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00847
Kumar, R. R., Ahuja, S., Rai, G. K., Kumar, S., Mishra, D., Kumar, S. N., et al. (2022a). Silicon triggers the signalling molecules and stress-associated genes for alleviating the adverse effect of terminal heat stress in wheat with improved grain quality. Acta Physiologiae Plantarum 44, 30. doi: 10.1007/s11738-022-03365-y
Kumar, R. R., Babu H, P., Pandit, K., Kumar, A., Ranjan, A., T, V., et al. (2025). Characterizing the mono- and triacylglycerol lipase (MAGL and TAGL) genes from pearl millet (Pennisetum glaucum L.) and elucidating their dynamics with biochemical traits linked with rancidity. Planta 261, 57. doi: 10.1007/S00425-025-04621-4/FIGURES/8
Kumar, R. R., Bhargava, D. V., Pandit, K., Goswami, S., Mukesh Shankar, S., Singh, S. P., et al. (2021). Lipase – The fascinating dynamics of enzyme in seed storage and germination – A real challenge to pearl millet. Food Chem. 361, 130031. doi: 10.1016/j.foodchem.2021.130031
Kumar, R. R., Dubey, K., Goswami, S., Hasija, S., Pandey, R., Singh, P. K., et al. (2020a). Heterologous expression and characterization of novel manganese superoxide dismutase (Mn-SOD) – A potential biochemical marker for heat stress-tolerance in wheat (Triticum aestivum). Int. J. Biol. Macromolecules. doi: 10.1016/j.ijbiomac.2020.06.026
Kumar, R. R., Goswami, S., Sharma, S. K., Kala, Y. K., Rai, G. K., Mishra, D. C., et al. (2015). Harnessing next generation sequencing in climate change: RNA-seq analysis of heat stress-responsive genes in wheat (Triticum aestivum L.). OMICS: A J. Integr. Biol. 19, 632–647. doi: 10.1089/omi.2015.0097
Kumar, S., Hash, C. T., Singh, G., Basava, R. K., and Srivastava, R. K. (2020b). Identification of polymorphic SSR markers in elite genotypes of pearl millet and diversity analysis. Ecol. Genet. Genomics 14, 100051. doi: 10.1016/j.egg.2019.100051
Kumar, R. R., Singh, N., Goswami, S., Vinutha, T., Singh, S. P., Mishra, G. P., et al. (2024). Hydrothermal infra-red (HT-IR): the most effective technology for enhancing the shelf-life of pearl millet flour without compromising with the nutrient density and flour quality. J. Plant Biochem. Biotechnol. doi: 10.1007/s13562-024-00893-1
Kumar, R. R., Singh, S. P., Rai, G. K., Krishnan, V., Berwal, M. K., Goswami, S., et al. (2022b). Iron and Zinc at a cross-road: A trade-off between micronutrients and anti-nutritional factors in pearl millet flour for enhancing the bioavailability. J. Food Composition Anal. 111, 104591. doi: 10.1016/j.jfca.2022.104591
Lee, H.-J. and Park, O. K. (2019). Lipases associated with plant defense against pathogens. Plant Sci. 279, 51–58. doi: 10.1016/j.plantsci.2018.07.003
Ma, X. L., Xu, R. Q., Suo, X. M., Li, J. S., Gu, P. P., Yao, R., et al. (2022). Genome-wide identification of the Class III PRX gene family in foxtail millet (Setaria italica L.) and expression analysis under drought stress. Acta Agronomica Sinica(China) 48, 2517–2532. doi: 10.3724/SP.J.1006.2022.14185
Niranjan-Raj, S., Lavanya, S. N., and Nayaka, S. C. (2019). Molecular cloning and characterization of pearl millet polyphenol oxidase and its role in defense against downy mildew. J. Plant Prot. Res. 59, 423–427. doi: 10.24425/jppr.2019.129745
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. doi: 10.1093/nar/29.9.e45
Pfaffl, M. W., Horgan, G. W., and Dempfle, L. (2002). Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30, e36. doi: 10.1093/nar/30.9.e36
Sánchez, R., Arroyo, L., Luaces, P., Sanz, C., and Pérez, A. G. (2023). Olive polyphenol oxidase gene family. Int. J. Mol. Sci. 24, 3233. doi: 10.3390/ijms24043233
Satam, H., Joshi, K., Mangrolia, U., Waghoo, S., Zaidi, G., Rawool, S., et al. (2023). Next-generation sequencing technology: current trends and advancements. Biology 12, 997. doi: 10.3390/biology12070997
Satyavathi, C. T., Ambawat, S., Khandelwal, V., and Srivastava, R. K. (2021). Pearl millet: A climate-resilient nutricereal for mitigating hidden hunger and provide nutritional security. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.659938
Shivhare, R., Asif, M. H., and Lata, C. (2020). Comparative transcriptome analysis reveals the genes and pathways involved in terminal drought tolerance in pearl millet. Plant Mol. Biol. 103, 639–652. doi: 10.1007/s11103-020-01015-w
Šikuten, I., Štambuk, P., Marković, Z., Tomaz, I., and Preiner, D. (2025). Grape volatile organic compounds: analysis, biosynthesis, and profiling. J. Exp. Bot. 76, 3016–3037. doi: 10.1093/jxb/eraf082
Sinha, A. K., Jaggi, M., Raghuram, B., and Tuteja, N. (2011). Mitogen-activated protein kinase signaling in plants under abiotic stress. Plant Signaling Behavior. doi: 10.4161/psb.6.2.14701
Sinha, K., Kaur, R., Singh, N., Kaur, S., Rishi, V., and Bhunia, R. K. (2020). Mobilization of storage lipid reserve and expression analysis of lipase and lipoxygenase genes in rice (Oryza sativa var. Pusa Basmati 1) bran during germination. Phytochemistry 180, 112538. doi: 10.1016/j.phytochem.2020.112538
Sreenivasulu, N., Radchuk, V., Strickert, M., Miersch, O., Weschke, W., and Wobus, U. (2006). Gene expression patterns reveal tissue-specific signaling networks controlling programmed cell death and ABA- regulated maturation in developing barley seeds. Plant J. 47, 310–327. doi: 10.1111/j.1365-313X.2006.02789.x
Srivastava, R. K., Satyavathi, C. T., Singh, R. B., Mukesh Sankar, S., Singh, S. P., Soumya, S. L., et al. (2022). “Pearl millet: biofortification approaches in a micronutrient dense, climate-resilient nutri-cereal,” in Biofortification of Staple Crops. Singapore: Springer. doi: 10.1007/978-981-16-3280-8_7
Varshney, R. K., Shi, C., Thudi, M., Mariac, C., Wallace, J., Qi, P., et al. (2017). Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat. Biotechnol. doi: 10.1038/nbt.3943
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Wu, C. (1995). Heat shock transcription factor: Structure and regulation. Annu. Rev. Cell Dev. Biol. doi: 10.1146/annurev.cb.11.110195.002301
Yadav, C. B., Srivastava, R. K., Beynon, S., Englyst, K., Gangashetty, P. I., and Yadav, R. S. (2022). Genetic variability and genome-wide marker association studies for starch traits contributing to low glycaemic index in pearl millet. Food Energy Secur. 11, e341. doi: 10.1002/fes3.341
Zhang, S., Zhang, W., Martin, J. J. J., Qadri, R., Fu, X., Feng, M., et al. (2023). Differential analysis of transcriptomic and metabolomic of free fatty acid rancidity process in oil palm (Elaeis guineensis) fruits of different husk types. Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1132024
Keywords: rancidity, DEGs, differentially expressed transcripts, lipase, landrace, lipids
Citation: Kumar RR, Goswami S, Thimmegowda V, Singhal N, Mabalirajan U, Mishra GP, Jha GK, Rai GK, Praveen S and Tara Satyavathi C (2025) When genes turn traitor: de novo transcriptomics uncovers pearl millet’s rancidity machinery. Front. Plant Sci. 16:1677082. doi: 10.3389/fpls.2025.1677082
Received: 08 August 2025; Accepted: 27 October 2025;
Published: 17 November 2025.
Edited by:
María Serrano, Miguel Hernández University of Elche, SpainReviewed by:
Junpeng Niu, Northwest A&F University, ChinaNarayan Panda, Orissa University of Agriculture and Technology, India
Hao Zhang, Hebei Normal University, China
Copyright © 2025 Kumar, Goswami, Thimmegowda, Singhal, Mabalirajan, Mishra, Jha, Rai, Praveen and Tara Satyavathi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ranjeet R. Kumar, cmFuamVldF9iaW9jaGVtQGlhcmkucmVzLmlu
†ORCID: Ranjeet R. Kumar, orcid.org/0000-0002-2279-6340