 Fikru Tamiru Kenea
Fikru Tamiru Kenea Nan He
Nan He Xuqiang Lu
Xuqiang Lu Xiaowen Luo1
Xiaowen Luo1 Hongju Zhu
Hongju Zhu Wenge Liu
Wenge Liu- 1Henan Joint International Research Laboratory of South Asian Fruits and Cucurbits, Zhengzhou Fruit Research Institute, Chinese Academy of Agricultural Sciences, Zhengzhou, China
- 2Department of Horticulture, College of Agricultural and Natural Resources, Dilla University, Dilla, Ethiopia
- 3Zhongyuan Research Center, Chinese Academy of Agricultural Sciences, Xinxiang, China
Watermelon [Citrullus lanatus (Thunb.) Matsum. & Nakai] is a globally important vegetable crop valued for its taste, hydration, and nutritional benefits. Recent advances in multi-omics technologies have accelerated the identification of genes controlling key fruit metabolites that impact fruit quality traits such as sweetness, bitterness, sourness, aroma, texture, and color. This review synthesizes the current knowledge on watermelon genes regulating and transporting fruit metabolites, including sugars, cucurbitacin, organic acids, carotenoids, amino acids, flavonoids, and volatile organic compounds that impact fruit quality. Both forward and reverse genetics approaches, coupled with high-throughput phenotyping, have been instrumental in these gene discoveries. Breeding applications, including marker-assisted selection (MAS) and genomic selection (GS), are highlighted, emphasizing their potential to enhance fruit metabolites that improve fruit quality and nutritional value. Emerging technologies, such as CRISPR/Cas9-mediated gene editing, have been employed to uncover and validate CIVST1, PSY1, and PEPCK genes, enabling precision breeding for improved fruit metabolite profile. However, challenges persist due to the environmental sensitivity and polygenic nature of fruit metabolites, the narrow genetic base, and the limited adoption of molecular breeding methods like CRISPR/Cas9. Future directions emphasize leveraging wild germplasm, integrating AI-driven phenotyping, and applying precision breeding strategies. These approaches will enable the development of next-generation watermelon cultivars with improved multi-trait quality and nutritional profiles to meet evolving market demands.
1 Introduction
Watermelon (Citrullus lanatus) is a globally significant fruit crop known for its refreshing taste, high water content, and nutritional value (Sorokina et al., 2021; Dubey et al., 2023). As consumer demands evolve, there is increasing interest in developing watermelon cultivars with superior fruit quality traits, including sweetness, color, aroma, and nutritional content (Assefa et al., 2020; Ramirez et al., 2020). A key focus for maintaining this feature is examining the fruit metabolome, which encompasses all small molecules influencing the flavor and nutritional characteristics of the fruit. Though it is a time-consuming, expensive, and distractive approach, recent liquid chromatography–mass spectrometry (LC-MS/MS), ultra-high-performance liquid chromatography, gas chromatography-mass spectrometry (GC-MS), and nuclear magnetic resonance (NMR) spectroscopy have revolutionized watermelon fruit metabolome investigation. Furthermore, the cutting-edge and powerful non-destructive technique, Raman spectroscopy, possibly enhanced with artificial intelligence (AI), has been used to confirm the presence and intensity of carotenoids, particularly lutein and β-carotene, in watermelon fruit (Dhanani et al., 2022). Surprisingly, gradual adoption of generative AI, particularly by ChatGPT, compared with human perception for internal quality based on outer characteristics of watermelon fruit, has revealed significant acceptance of internal fruit quality expected by consumers (Ozdemir, 2025). Though it has drawbacks, exploring this breakthrough technology for other traits and genetic makeup will enable scientists to investigate thousands of fruits for desirable metabolomes and associated molecular mechanisms within a short period, at low cost, and without distracting the fruit.
The watermelon fruit metabolome consists of a diverse array of primary and secondary metabolites that influence taste, texture, appearance, and nutritional quality (Zhang and Hao, 2020; Gong et al., 2021b; Guo et al., 2015; Chu et al., 2022; Ning et al., 2024; Peng et al., 2025). Primary metabolites such as sugars and organic acids directly impact sweetness and sourness (Guo et al., 2019, Guo et al., 2015; Umer et al., 2020a, Umer et al, 2020b), while secondary metabolites like carotenoids (Guo et al., 2019; Ilahy et al., 2019; Sulaiman et al., 2020; Breniere et al., 2022; Lv et al., 2022; Peng et al., 2025) and volatiles affect color and aroma (Kyriacou et al., 2018; Bianchi et al., 2019; Gong et al., 2023, Gong et al., 2021a; Liu et al., 2018; Tripodi et al., 2019). Genetic and environmental factors tightly regulate the dynamic composition of these metabolites during fruit development (Soteriou and Kyriacou, 2015; Liu et al., 2018; Aslam et al., 2020; Trandel et al., 2021). Yuan et al. (2021a) has identified more than 431 metabolites, while Sorokina et al. (2021) reported 1,679 with different contents. This reveals that metabolite contents demand further investigation to exploit their potential among different germplasm and their causal genes.
Improving watermelon cultivars through conventional approaches has not only been time consuming but has also resulted in the inheritance of undesirable traits along with desired metabolites. Since the conventional breeding approaches have not been able to meet the demand arising from different dimensions, uncovering the genes of different metabolites has emerged to advance molecular breeding in watermelon fruit. Advances in high-throughput like multi-omics technologies have facilitated the identification of genes involved in the biosynthesis, degradation, and transportation of these metabolites (Kim and Buell, 2015; Gong et al., 2021b; Guo et al., 2015; Ren et al., 2021). This knowledge has been integrated into modern breeding programs through marker-assisted selection (MAS), genomic selection (GS), gene transformation, and gene editing, ultimately enabling the development of high-quality watermelon cultivars tailored to specific market and health demands (Fall et al., 2019; Guo et al., 2019; Mashilo et al., 2022; McGregor et al., 2023; Verma et al., 2024). The whole genome sequencing of watermelon, the “97103” elite cultivar (Guo et al., 2012) and its improved sequence (Guo et al., 2019), the “Charleston Gray” cultivar (Wu et al., 2019), the closest relative of the cultivated watermelon, the “Kordofan melon” (Renner et al., 2021), and the telomer-to-telomer gap-free genome of the elite watermelon inbred line “G42” (Deng et al., 2022) have all been used as a reference for watermelon gene discovery and genetic improvement. Furthermore, comparative analysis of 20 (Guo et al., 2012) and 414 resequenced germplasm (Guo et al., 2019b) and genomes of 1,365 accessions (Wu et al., 2019), as well as the super-pangenome from 547 different accessions, has been used to uncover candidate genes of various metabolites (Wu et al., 2023). For instance, the integration of metabolomics and genomics has identified the sugar transporter gene ClTST2 (Cla97C02G036390) and the synthase gene Cla97C10G194010 that have been harbored on chromosome 10 and associated with fruit sweetness (Guo et al., 2019). Similarly, the lycopene β-cyclase gene (LCYB, Cla97C04G070940) that manipulates the flesh color has been identified on Chromosome 4 (Liu et al., 2016; Guo et al., 2019).
By integrating metabolomics data with genomic tools such as quantitative trait locus (QTL) mapping and genome-wide association studies (GWAS), researchers have pinpointed specific genes responsible for desirable metabolic traits (Wang et al., 2021a; Liang et al., 2022; Zhan et al., 2023). Furthermore, these approaches have been used to reveal regulatory networks (Ren et al., 2014; Katuuramu et al., 2023; Vallarino et al., 2023). This systems-level understanding enables breeders to target metabolic pathways more effectively in the selection process. Similarly, the integration of other omics, such as transcriptomics and proteomics, with metabolomics has revolutionized gene discovery (Abdullah-Zawawi et al., 2022; Depuydt et al., 2022; Habibi et al., 2024; Shiratake and Suzuki, 2016). Transcriptomics provides insights into gene expression patterns during fruit development and ripening, while metabolomics quantifies the levels of various metabolites (Umer et al., 2020b; Gong et al., 2021a). The correlation between gene expression and quantified metabolite level has identified candidate genes that have been either upregulated or downregulated to alter the metabolite accumulation. For instance, upregulation of Cla97C03G064990 (sucrose synthase) and Cla97C03G068240 (citrate synthase) has been highly correlated with the accumulation of sucrose and citric acid, respectively, in watermelon fruit flesh (Umer et al., 2020b; Peng et al., 2025). Cla018406 (a chaperone protein, DNAJ-like protein) and Cla007686 (a zinc finger CCCH domain-containing protein) transcripts have been highly expressed in yellow- and red-fleshed watermelon, revealing high β-carotene and lycopene accumulation (Wang et al., 2021a; Yuan et al., 2021b). Systems biology approaches further enhance the understanding of the complex interactions between genes and metabolites, paving the way for more targeted breeding strategies. The use of multi-omics data thus represents a powerful approach for unraveling the molecular basis of the fruit metabolome.
The current review attempts to summarize and provide critical insights into recent watermelon metabolome gene discoveries that regulate key metabolic pathways. It aims to highlight recent advances in forward and reverse metabolomics-assisted uncovered genes and explore how cutting-edge tools, including MAS, GS, and gene editing, can be utilized to develop watermelon cultivars with a superior fruit metabolome. By addressing challenges such as limited resources, a shortage of skilled human power, insufficient investigation of multi-omics and AI integration, environmental sensitivity, and limited genetic diversity, this review also discloses the transformative potential of modern breeding strategies in enhancing the watermelon fruit metabolome.
2 Gene discovery approaches
To characterize the inherent and enormously diversified resources of plants for better management and efficient utilization in crop improvement, proper phenotyping and genotyping of a large set of plant genetic resources is crucial. Rapid and precise phenotypic assessment of thousands of breeding lines, clones, or populations over time under diverse environments is the only critical component for accelerating the development of new and improved cultivars through discovering elite genes (Fu, 2015; Sun et al., 2024). The plant phenotype refers to all morphological, physiological, and biochemical characteristics, which are controlled by genotype and environment, reflecting the structure, composition, and growth of a plant to identify key genes (Fiorani and Schurr, 2013; Johannsen, 2014; Pontarotti et al., 2022). This is satisfied through acquisition of high-dimensional phenotypic data [high-throughput phenotyping (HTP)] (Chawade et al., 2019; Kim, 2020) or “phenomics” (Houle et al., 2010; Zhao et al., 2019), technologies that help for the discovery of important genes. Complex characters that are relevant for plant selection (forward phenomics) and explanations of why the given genotypes stand out in a specific environment (reverse phenomics) are predicted from this method. Identifying genetic architectures through linking phenotypes and genotypes is used to regulate important traits that are used for plant breeding and development of plant genomics (Xiao et al., 2021).
Gene discovery can be done through forward genetics (phenotype to gene) and reverse genetics (gene to phenotype) approaches (Figure 1). Forward genetics (traditional), the phenotype gene approach, is an approach used to find the genetic basis of a particular phenotype of an organism (Brown et al., 2005). It identifies and characterizes genes (or sets of genes) responsible for mutant phenotypes or traits. In this approach, the phenotypic character of the plant is evaluated, and a novel plant genotype is selected (Jankowicz-Cieslak and Till, 2015). A plant with desired character is incorporated into breeding programs and/or used for further understanding of its genetic mechanisms and function validation. After the reference genome of watermelon was deciphered, several forward gene discovery methods, including genome-wide association analysis (GWAS), bulk segregant analysis, QTL mapping, and transcriptomics, were employed to uncover desirable genes for intended metabolites.
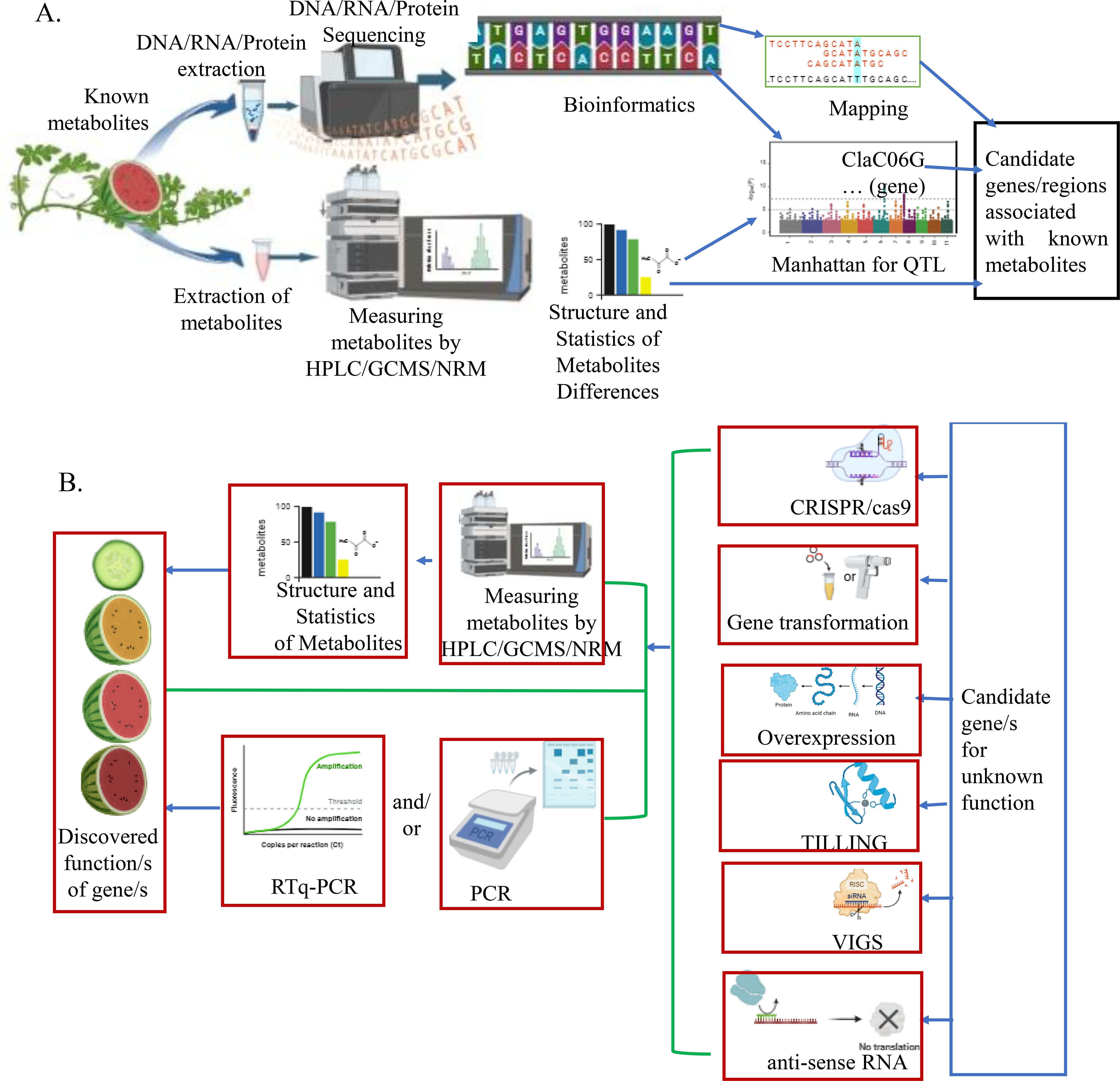
Figure 1. Examples of a short summary of forward and reverse genetics: (A) Forward gene discovery. (B) Reverse gene discovery (these figures were illustrated by biorender, an online tool).
Reverse genetics, which is known as gene to phenotype, is based on known gene sequences to find particular phenotypes (functions of genes) (Bouché and Bouchez, 2001). It involves finding the result of particular genetic sequences induced by a mutagen. This strategy works on the single gene which its function or the phenotype it creates is not known (Brown et al., 2005).
The first step for both forward and reverse genetics is obtaining a suitable mutagenized population through natural or induced mutagenesis (Serrat et al., 2014). The steps of forward genetics are getting a mutant which is a naturally varied or induced mutation, followed by selecting a phenotype to identify the gene and its function (Zakhrabekova et al., 2013). These authors revealed that the efficiency is increased through emerging technology like genetic mapping, quantitative trait loci mapping, and the exome sequencing approach. In the case of reverse genetics, the known sequence of a gene, whose function is unknown, is induced or altered in expression and followed by discovering the function of the gene by analyzing its phenotypic effects. Different tools and techniques used for reverse genetic approaches are genetic engineering, gene silencing, zinc finger nuclease, Targeting Induced Local Lesions in Genomes (TILLING), CRISPR/Cas9, T-DNA, transposons, mutagenesis, overexpression, and homologous chromosomes (Jankowicz-Cieslak and Till, 2015).
To manage diversity and prove effective in increasing long-term genetic gain, methodologies like genetic marker-based information have been developed (Jannink et al., 2010). It is important for developments in the field of plant breeding (Kebriyaee et al., 2012). It is a gene or DNA sequence with a known chromosome location controlling a particular gene or trait. Genetic markers act as signs or flags and closely related to the targeted genes (Collard et al., 2005). Genetic markers are broadly grouped into classical markers (morphological, cytological, and biochemical) and DNA/molecular markers. But some literature classifies it as a morphological and molecular (biochemical and DNA) marker. Restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), simple sequence repeats (SSRs), single nucleotide polymorphism (SNP), and diversity arrays technology (DArT) markers are some examples of molecular markers (Wenzl et al., 2006). Pyramiding all levels of information (different categories of traits measured at different times with environmental information) improves crop germplasm.
3 Watermelon fruit metabolome and their genes
3.1 Sugar-related genes
Sweetness is one of the most desirable traits in watermelon and is primarily determined by the accumulation of soluble sugars, such as sucrose, glucose, and fructose (Guo et al., 2019, Guo et al., 2015; Umer et al., 2020a, Umer et al., 2020b). Key genes involved in sugar metabolism include sucrose phosphate synthase (SPS), sucrose synthase (SUS), and various invertases (INVs), which regulate sucrose synthesis and degradation (Guo et al., 2019; Umer et al., 2020a, Umer et al., 2020b) (Table 1). Sweet watermelon varieties have exhibited higher unloading of raffinose and stachyose compared to non-sweet varieties, which has been correlated with increased sugar content (Liu et al., 2013). Gao et al. (2018b) reported that the cultivar “203Z” has produced higher sugar content than “SW,” while the higher acid content has been produced by “SW.” Different varieties of sweet watermelon have produced different levels of soluble sugar and enzymes that have been involved during their metabolism (Yativ et al., 2010). The sugar content of sweet watermelon is increased due to hydrolysis of raffinose and stachyose by galactosidases and is transported by ClVST1, ClSWEET3, and ClTST2 genes, which have been selected during evolution and domestication (Ren et al., 2021). Specific genes, such as ClAGA2, essential for the hydrolysis of oligosaccharides and sugar transport, influence the partitioning of carbohydrates in sweet watermelon fruits (Ren et al., 2021). For instance, nine α-galactosidase genes have been identified in the watermelon genome, and the insoluble acid invertase (IAI) gene (Cla020872) has been highly expressed in the mesocarp of elite 97103 than in PI296341-FR fruit flesh, resulting in high sugar accumulation in 97103 (Guo et al., 2015). Similarly, raffinose and stachyose, the major translocated sugars, have been less unloaded into non-sweet line PI296341-FR (C. lanatus subsp. lanatus) than elite sweet watermelon line 97103 (Citrullus lanatus subsp. vulgaris) fruit (Liu et al., 2013). On the other hand, insoluble acid invertase, the Cla020872 gene, which has been strongly correlated with enzyme activity in the flesh and the mesocarp of fruit, has been upregulated in 97103 but is at much lower levels in the flesh of PI296341-FR (Guo et al., 2015; Liu et al., 2013). This has resulted in fructose and glucose translocation and the intercellular sugar accumulation in 97103 watermelon fruit through extracellular sucrose degeneration (Guo et al., 2015). Some of the genes involved in the simple sugar formation pathway are illustrated in the following Figure 2A.
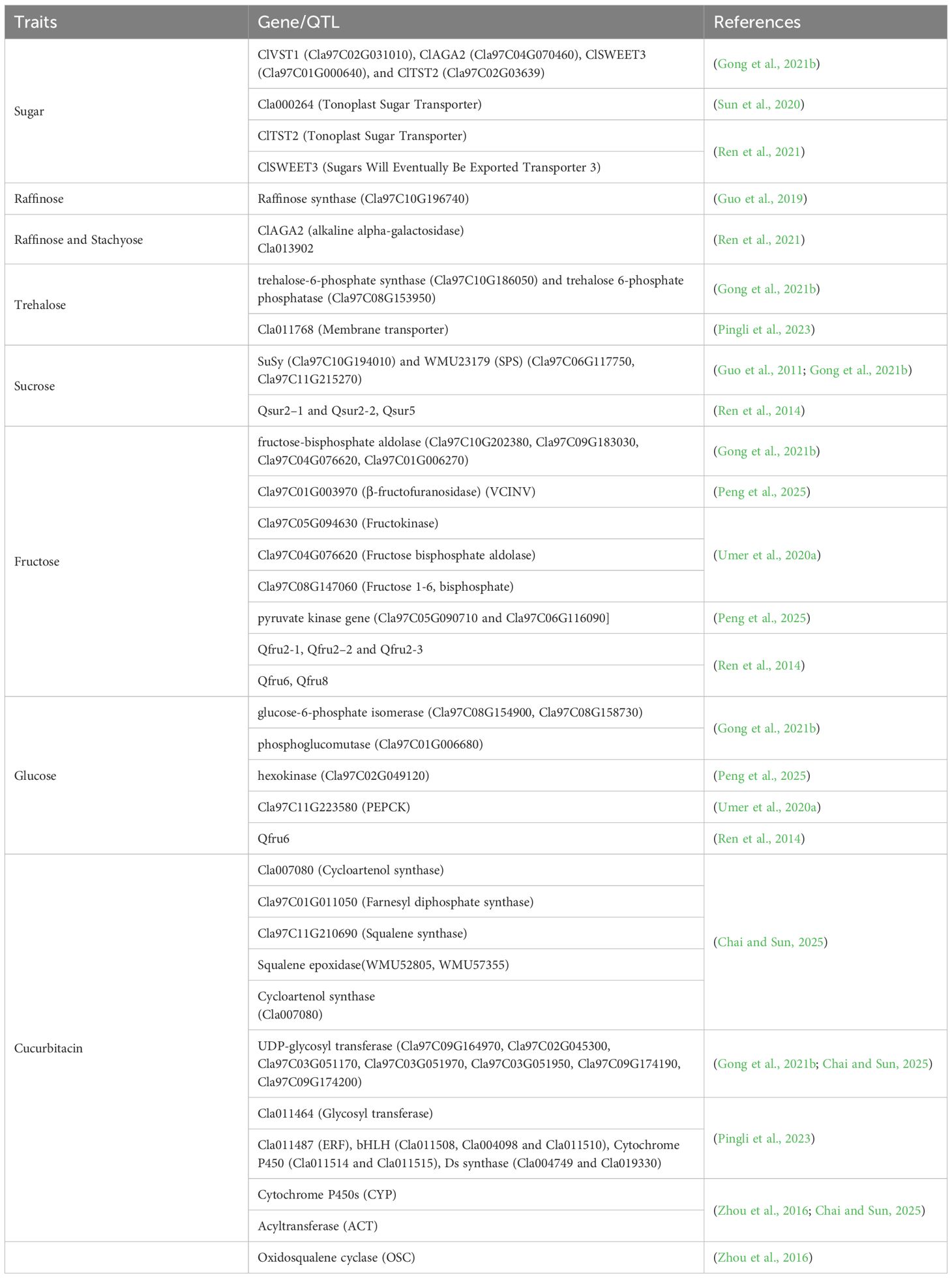
Table 1. Uncovered genes of sugars and cucurbitacin of watermelon fruits.
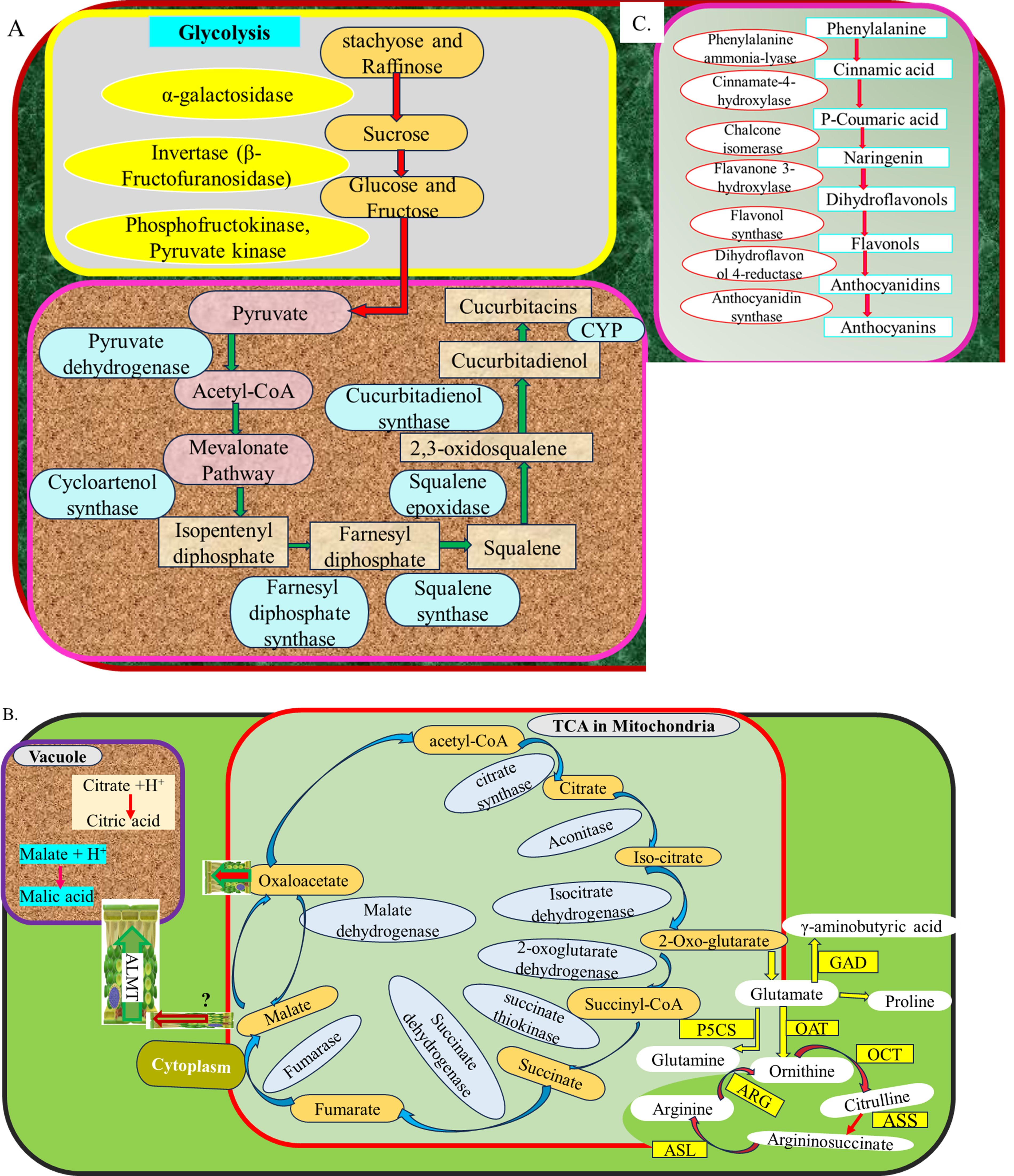
Figure 2. Genes involve in different metabolic networks in watermelon fruits (A) sugar and cucurbitacin biosynthesis (B) Organic acid and amino acid biosyntheis (C) Flavonoids biosynthesis.
The gene expression studies of specific genes, such as Cla97C01G000640 and Cla97C05G087120, which have been identified as sugar transporters, have been strongly correlated with sugar content (Umer et al., 2020a). The NAC transcription factor ClNAC68 has repressed the ClINV and ClGH3.6 genes and positively regulated sugar, thereby enhancing fruit sweetness (Wang et al., 2021b).
Genetic mapping studies have also identified major quantitative trait loci (QTLs) linked to sugar content on chromosome 10 (Guo et al., 2019). These findings will enable the development of molecular markers, allowing breeders to select high-sugar genotypes early in the breeding process, thereby significantly improving breeding efficiency. Moreover, unlocking these genes is the backbone of gene editing and mutant development, which requires more emphasis.
3.2 Cucurbitacin-related genes
Cucurbitacins, bitter-tasting compounds prevalent in wild watermelons, are mostly absent in cultivated varieties (Zhou et al., 2016). Deciphering the genes responsible for cucurbitacin biosynthesis and regulation is crucial for breeding efforts and enhancing fruit quality. Recent studies have uncovered critical genes and regulatory mechanisms that govern cucurbitacin production in watermelon fruit. Cucurbitacin biosynthesis in watermelon has been regulated by a coordinated network of structural genes, modifying enzymes, and transcriptional regulators. Key genes, such as the CGC1 cluster, cytochrome P450s, cucurbitadienol synthase, and various transferases, play central roles in the production and diversification of cucurbitacins (Zhou et al., 2016; Chai and Sun, 2025) as illustrated in Figure 2C. It was indicated from pyruvate to cucurbitacin in this figure. Fine mapping of F2 population of watermelon that has been developed from two inbred lines, “9904” (bitter flesh) and “Handel” (non-bitter flesh), with the parents, has enabled the uncovering of the locus of four candidate genes (Cla011507, Cla011508, Cla011509, and Cla011510) that synthesize watermelon fruit cucurbitacin (Gong et al., 2022). Pingli et al. (2023) have carried out genome-wide analysis and reported 42 watermelon fruit cucurbitacins with 8 major loci. Especially cytochrome P450s genes create diversity of cucurbitacins (Zhou et al., 2016). Through a biosynthesis of cucurbitacins, cucurbitadienol, which is the triterpenoid, has been utilized as a substrate for the production of cucurbitacins. It has been synthesized from 2,3-oxidosqualene by an oxidosqualene cyclase gene (Davidovich-Rikanati et al., 2015). Transcription factors of oxidosqualene cyclase (OSC), ACT, and CYP genes have also demonstrated high production of cucurbitacin in wild watermelon, while it has not been revealed in domesticated watermelon (Zhou et al., 2016). Similarly, SNP change in the transcription factor binding site (GCC-box) in promoter regions of cultivated watermelon has banned the production of cucurbitacins (Pingli et al., 2023). Furthermore, these authors reported the insertion of 244 amino acids in the CDS region of Cla019330 (ClOSC) in cultivated watermelon, while that insertion has been absent in wild watermelon and resulted in the absence of cucurbitacin production. This is deciphered as domestication has led to reduced expression and accumulation of these bitter compounds. The other reported genes have been revealed in the following Table 1. These genes need further validation and implementation for molecular breeding.
3.3 Carotenoid-related genes
Watermelon carotenoids are known for their antioxidant properties and potential health benefits (Edwards et al., 2003). Moreover, the flesh color of watermelon, a key quality attribute, has been determined by the presence of carotenoid pigments such as lycopene, β-carotene, violaxanthin, and lutein (Breniere et al., 2022; Ilahy et al., 2019; Lv et al., 2022; Peng et al., 2025; Sulaiman et al., 2020). Yuan et al (2021a) reported candidate genes of β-carotene, lycopene, and xanthophyll (such as violaxanthin and lutein) accumulation, which have been regulated by Cla018406 (a chaperone protein DNAJ-like protein), Cla007686 (a zinc finger CCCH domain-containing protein), and Cla021635 (photosystem I reaction center subunit II), have controlled the formation of orange, red, and yellow flesh-colored watermelon fruit, respectively. These color variations have been genetically regulated by key enzymes in the carotenoid pathway, including phytoene synthase (PSY1), lycopene β-cyclase (LCYB), and β-carotene hydroxylase (CHYB) (Nie et al., 2023) (Table 2).
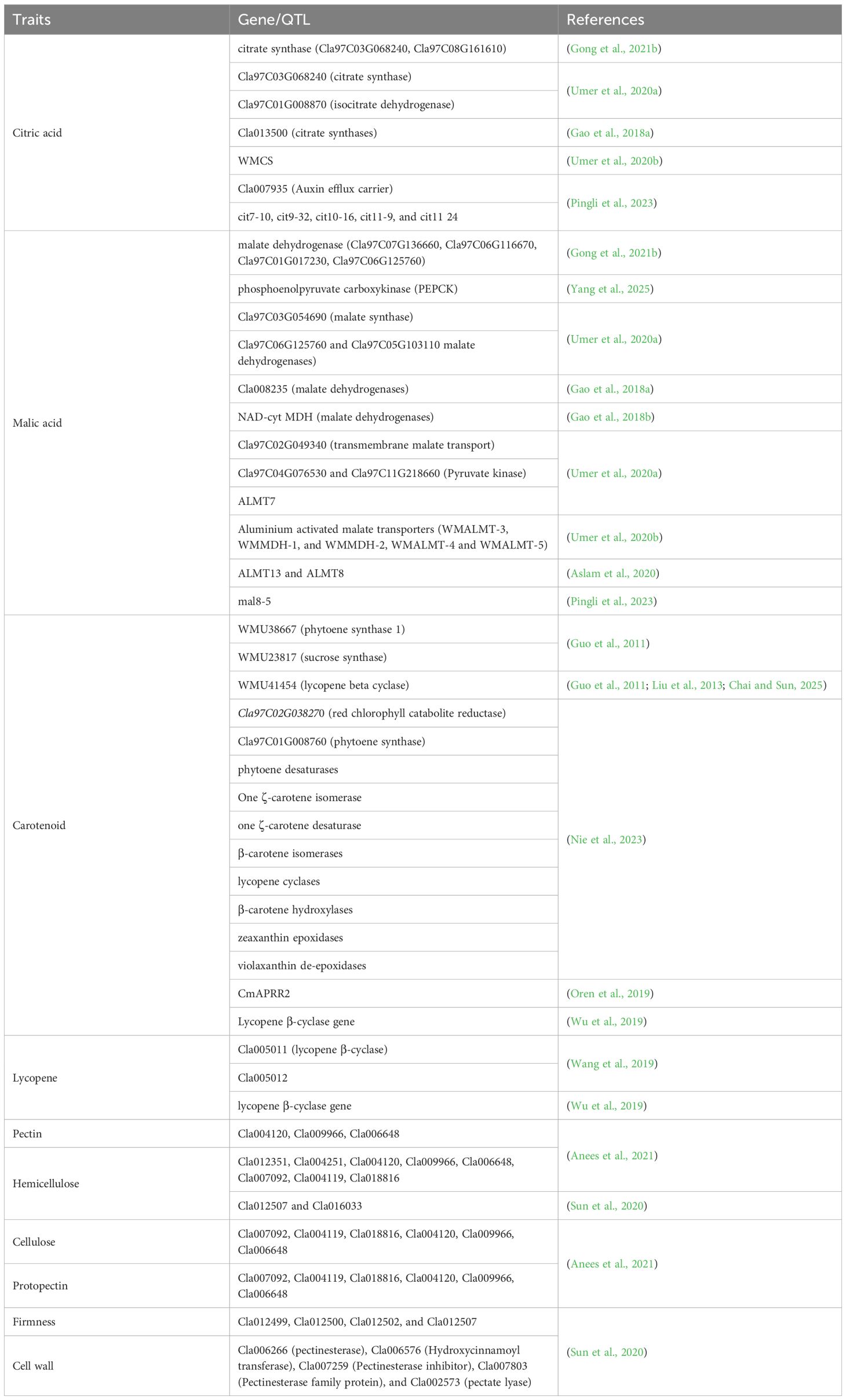
Table 2. Uncovered major genes of carotenoids, organic acids, and texture-related traits of watermelon fruits.
Natural allelic variations have also been reported for the observed diversity in watermelon pigmentation. In the coding region of lycopene β-cyclase (LCYB; Cla005011), two nonsense mutations, SNP1 (C/G) and SNP2 (A/C), revealed different colors of watermelon fruits. The varieties with the C allele have revealed increased β-carotene, and the ones with the G allele have shown increased β-apocarotenal production that leads to yellow- or orange-fleshed watermelon fruit (Pingli et al., 2023). On the other hand, in the SNP2 (A/C), the variety with the C allele has revealed more lycopene and has deciphered the red flesh color (Bang et al., 2007). Interestingly, these authors reported C1A2, G1A2, and C1C2 allele combinations of the above two SNPs that have been exhibited as pale, yellow, or orange and red or pink in wild, domesticated, and improved watermelon fruit, respectively. The development of molecular markers for PSY1 and lycopene β-cyclase (LCYB) has significantly advanced breeding efforts, enabling precise selection of flesh color traits to meet consumer preferences and enhance the marketability of new cultivars (Nie et al., 2023). As carotenoid is a polygenic trait, different genes have also been indicated as in the following Table 2.
3.4 Organic acid-related genes
Organic acids, such as malic acid, citric acid, and oxalic acid, contribute to the sourness and flavor complexity of watermelon fruit (Gao et al., 2018b; Umer et al., 2020b, Umer et al., 2020a). Their levels have been regulated by key genes, including phosphoenolpyruvate carboxylase (PEPC), malate dehydrogenase (MDH), citrate synthase (CS), and malate and citrate transporters, which have played essential roles in central carbon metabolism and organic acid transportation (Table 2). The genetic basis of organic acid content has been investigated through forward genetics, including transcriptomic, QTL, and GWAS approaches, and reverse genetics, including overexpression (Umer et al., 2020b) and CRISPR/Cas9 (Yang et al., 2025). During organic acid formation, several genes are involved in tricarboxylic acid (TCA) (Figure 2B). For example, comparative analysis of metabolomics and transcriptomics of watermelon cultivar “203Z” and its near-isogenic line “SW” has revealed the highly significantly expressed ALMT gene and its ortholog, Cla006064, the NAD-dependent malate dehydrogenase (NAD-cyt MDH) gene, and its ortholog, including Cla008235 and Cla011268, that have been positively correlated with malic acid accumulation (Gao et al., 2018b). These genes have synthesized acetyl CoA and malate or oxaloacetate. Similarly, these authors have reported upregulated citrate synthase (CS) that has manipulated citric acid contents of fruits. Transcriptomics and reverse transcription quantitative polymerase chain reaction (RT-qPCR) have also indicated high expression patterns of Cla97C07G128420 (ALMT), Cla97C03G068240 (CS), and Cla97C01G008870 (ICDH) genes that have been significantly correlated with malic acid and citric acid accumulations during key fruit developmental stages (Umer et al., 2020a). Further structural variation among different genotypes, validation, and utilization for molecular breeding are still the critical gaps that demand the engagement of researchers.
Gong et al. (2023) reported allelic variants that have been associated with reduced acidity. These discoveries are particularly valuable for breeding programs targeting markets that prefer milder-tasting fruit (Yang et al., 2025). By leveraging molecular markers linked to acid metabolism genes, breeders can more precisely fine-tune the flavor profiles of watermelon cultivars.
3.5 Amino acid-related genes
Watermelon is notable for its enormous amino acid content, including citrulline, arginine, glutamine, argininosuccinate, γ-aminobutyric acid, proline, valine, ornithine, phenylalanine, histidine, isoleucine, leucine, lysine, and alanine (Collins et al., 2007). Citrulline is a distinctive amino acid found in high concentrations in watermelon and has been recognized for its potential health benefits, including cardiovascular support and antioxidant activity (Burton-Freeman et al., 2021). Crimson Sweet watermelon has produced higher citrulline and arginine than “Dixielee” watermelon (Hartman et al., 2019). Citrulline content in watermelon shows moderate heritability, with estimates ranging from 38% to 43% in different populations, indicating a genetic basis for citrulline variation that can be exploited in breeding programs (Hartman, 2017). The genes ClCG05G018820 [ornithine carbamoyltransferase (OTC)] ClCG09G003180 [N-acetylornithine aminotransferase (N-AOA)], which have produced N-acetylornithine and N-acetylornithine deacetylase (AOD-3), have been involved in ornithine synthesis that has enhanced the accumulation of citrulline in watermelon fruit, while some genes involved in citrulline degradation include argininosuccinate synthases (ASS-1, ASS-2, and ASS-3), argininosuccinate lyases (ASL-1), ornithine decarboxylase (ODC), and arginine decarboxylase (ADC) to produce other amino acids (Joshi et al., 2019; Aslam et al., 2021). Some ornithine carbamoyltransferase genes, including Cla020511, Cla020512, and Cla020781 (ornithine carbamoyltransferase) have been candidate genes involved in the arginine accumulation (Fall et al., 2019). Moreover, the genes that have synthesized glutamate from 2-oxoglutarate of TCA to produce different amino acids are shown in Figure 2B. The genes that have been involved in different steps in the amino acid production pathway require further investigation to manipulate complete packages of amino acids. These discoveries facilitate the development of functional markers, enabling the breeding of nutrient-enriched watermelon varieties targeted at health-conscious consumers.
3.6 Volatile aromatic compound-related genes
The aroma of watermelon fruit is a crucial determinant of fruit quality, primarily governed by volatile organic compounds (VOCs) such as aldehydes, alcohols, esters, terpenes (Lu et al., 2024), apo-carotenoids, ketones, olefins, and furans (Bianchi et al., 2019). These compounds have been biosynthesized through fatty acid, amino acid, terpenoid, alcohol/aldehydes (ADH), ethyl acetate, acetaldehyde, tetradecanoic acid, and methyl acetate pathways regulated by key genes including lipoxygenases (LOX), alcohol dehydrogenases (ADH), and alcohol acyltransferases (AAT) (Gong et al., 2023). Watermelon genotypes have exhibited substantial diversity in their VOC profiles, resulting in distinct aromatic characteristics. Genes like lipoxygenases (LOX) (Liu et al., 2020), alcohol dehydrogenase genes (Cla97C01G013600, Cla97C05G089700, Cla97C01G001290, Cla97C05G095170, and Cla97C06G118330), and alcohol transferase genes (Gong et al., 2021a) have been involved in the metabolism of aromatic substances of watermelon fruit. There have been limited comparative transcriptome analyses involving wild and cultivated watermelon fruit to insight extensive variations in aroma development at various fruit developmental stages. While the genetic regulation of VOC biosynthesis remains complex, emerging research has identified some candidate genes linked to desirable aroma traits. These advances offer significant potential for targeted breeding strategies to improve flavor quality and sensory appeal in new cultivars.
3.7 Texture-related genes
Fruit texture is a critical quality trait that influences consumer acceptance and marketability of watermelon (Liu et al., 2024). Watermelon fruit texture is manipulated by cell wall components, including pectin, cellulose, and hemicellulose content, and identifying key genes involved in fruit texture regulation (Gao et al., 2020). Anees et al. (2021) identified key gene networks involving cell wall biosynthesis and the ethylene pathway (Table 2), providing insights for watermelon texture improvement in the future. Sun et al. (2020) also figured out multiple genes that have influenced watermelon fruit flesh firmness and consequently have affected cell wall components and hardness of ripened fruit (Table 2). Yang et al. (2023) reported that the Aux/IAA gene controls watermelon flesh firmness and influences fruit texture, with overexpression leading to increased flesh firmness and reduced ABA content. The Aux/IAA gene, Cla004102, has reduced flesh firmness of watermelon fruit when exposed to ethylene, and its expression has been decreased with fruit development stages (Anees et al., 2023). Sun et al. (2020) also reported fruit firmness controlling plant growth regulator genes, including Cla013991 (IAA-amido synthetase), Cla023158 (ethylene-responsive transcription factor), Cla016785 (ethylene-responsive transcription factor), Cla022055 (gibberellin receptor), Cla015407 (gibberellin 3-beta-hydroxylase), Cla005404 (9-cis-epoxycarotenoid dioxygenase), Cla016195 (ABA receptor), and Cla015981 (1-aminocyclopropane-1-carboxylate oxidase). These genes all need further validation and employment for fruit quality improvements through molecular breeding.
3.8 Flavonoids-related genes
Flavonoids in watermelon fruit contribute to its antioxidant properties, helping to protect cells from oxidative stress (Neglo et al., 2021). Gong et al. (2021b) reported more than 113 flavonoids that have been increased during key fruit developmental stages in wild watermelon “PI 632,751” and vice versa in cultivated watermelon “Cheng Lan” fruits. The flavonoid biosynthesis and accumulation have been regulated by genetic bases. Pingli et al. (2023) uncover the flavanone3-hydroxylase (Cla006682) gene, and Oren et al. (2019) figure out the CmAPRR2 gene that has been involved in flavonoid metabolism in watermelon fruit. Similarly, four genes, which are flavonoid 3 ′-monooxygenase (CYP75B1), phenolic glucoside malonyltransferase (PMAT1), isoflavone 2 ′-hydroxylase (CYP81E), and vestitone reductase (VR), have been deciphered (Peng et al., 2025). Flavonoids like luteolin and quercetin have been synthesized by the cytochrome P450 monooxygenase family gene, particularly CYP75B1 (Cla97C06G125240), in the flavonoid and flavonol pathways. Some of the unlocked genes have been involved in the phenylpropanoid pathway to regulate flavonoid accumulation in watermelon, as shown in Figure 2C. As it is one of the polygenic traits, further gene discovery needs to be employed and followed by validation and modern breeding approaches.
4 Applications in watermelon breeding
4.1 Marker-assisted selection
Marker-assisted selection (MAS) utilizes DNA markers linked to target traits to enhance breeding efficiency (Wang et al., 2019). The integrated genetic maps using SNPs, SSRs, insertion–deletion (InDels), and other markers have facilitated the identification and selection of desirable alleles for watermelon fruit metabolome improvements that have contributed to quality traits (Ren et al., 2014; Li et al., 2018). In watermelon fruit metabolite breeding programs, MAS has proven particularly effective for several key traits like sugar (using markers for INV and SPS genes), flesh color (via LCYB markers) (Wang et al., 2019), and citrulline content (through ASS1 markers) (Mashilo et al., 2022). This approach has offered significant advantages over conventional methods by enabling early selection of superior genotypes, thereby reducing both the time and costs associated with traditional phenotypic screening while simultaneously improving breeding accuracy. This method should also be employed for other genes to select genotype and improve the fruit metabolite profile.
4.2 Genomic selection
Genomic analysis deciphered the evolutionary history of watermelon fruit metabolites through identifying the gene/s that manipulate sweetness, bitterness, sourness, color, and texture during domestication and improvement, providing targets for genome selection (Guo et al., 2019; Katuuramu et al., 2023). Genomic selection (GS) utilizes genome-wide molecular markers and statistical prediction models to calculate breeding values (Wu et al., 2019). Genome-wide association analysis (GWAS) studies have identified QTL and SNP markers associated with soluble fruit metabolites that impact flesh color, flavor, and aroma to reveal phenotypic variation (Wu et al., 2019). When incorporated with metabolomic data, GS achieves greater predictive accuracy, particularly for complex polygenic traits such as flavor compounds and aroma profiles (Guo et al., 2012, Guo et al., 2019). This integrated approach empowers researchers to make data-driven selection decisions by combining genomic and metabolic insights, which significantly accelerates the development of superior cultivars, though limited validation studies exist for genomic prediction accuracy. The narrow genetic basis of watermelon also limits the application of gene discovery for genome selection (Wu et al., 2019).
4.3 Gene editing and precision breeding
Recent advances in gene editing technologies, particularly clustered regularly interspaced short palindromic repeats-associated protein 9 (CRISPR/Cas9) systems, have commenced in the transformation of precision breeding in watermelon by enabling targeted modifications of key metabolic genes (Wang et al., 2024). Unlike conventional transgenic methods, gene editing provides a non-GMO strategy for trait improvement, making it particularly valuable for markets with strict biotechnology regulations (Lemmon et al., 2018). The technique’s precision in editing specific nucleotide sequences without introducing foreign DNA addresses major consumer concerns about genetically modified organisms (Tsakirpaloglou et al., 2023). As regulatory frameworks worldwide increasingly recognize the distinction between gene-edited and transgenic crops, these technologies are emerging as powerful tools for developing improved watermelon metabolites. Knock-in, knockdown, and knockout of genes are three distinct CRISPR/Cas9 mechanisms to alter the function of the genes. The model of general steps is indicated in Figure 3. Even though knockout has been emerging, it has been prominently featured in watermelon fruit metabolome research when compared with knockdown and knockin, which has not yet been employed to manipulate these traits. For instance, knockout of phosphoenolpyruvate carboxykinase (PEPCK), an organic acid-synthesizing gene, has reduced the acidity of watermelon fruits by hindering the function of this gene (Yang et al., 2025). The ClIAA16 mutant, which has been developed through knockout, has exhibited premature termination of protein translation due to one base pair insertion, which has shown delayed watermelon fruit ripening. This mutation may contribute to extended shelf life, though it has reduced the fruit sugar content (Hu et al., 2023). Similarly, two ClVST1 mutants of the ZZJM watermelon cultivar, with 5 and 1 nucleotide deletions, have been developed through the knockout approach and have revealed reduced sugar contents (Ren et al., 2021). These examples indicate promising potential of this breakthrough approach, which allows precise regulation of gene function to reduce undesirable traits through knockout and to enhance desirable characteristics such as sweetness and flesh color through knock-in of sugar metabolism genes (e.g., SPS, SuSy, fructokinase) and modulation of pigmentation via lycopene biosynthesis genes such as ClPSY1 in watermelon fruit. By combining gene editing with traditional breeding methods, researchers can accelerate the creation of varieties with optimized metabolomes that can improve fruit metabolite profiles and enhance nutritional contents, positioning this technology as a cornerstone of future watermelon improvement programs. Still, this approach is very infantile and requires more attention for the improvement of watermelon fruit metabolites that pertain to fruit quality.
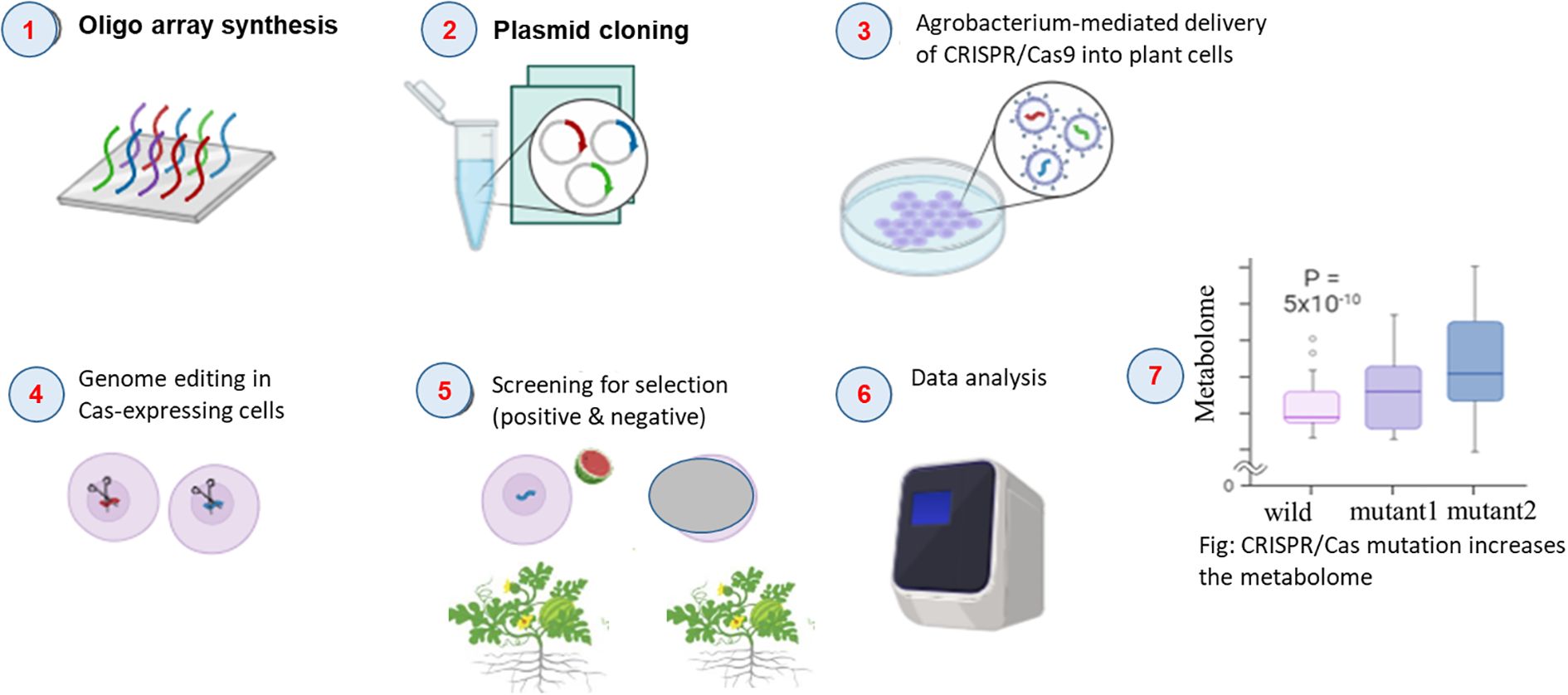
Figure 3. Model steps of CRISPR/Cas9 gene editing approach.
5 Challenges and future perspectives
5.1 Challenges and limitations
While substantial advances have been made, several key challenges persist in watermelon metabolome gene discovery and its application in breeding programs. Capturing a diverse and dynamic range of thousands of compounds requires multiple analytical platforms such as GC-MS, LC-MS, NMR, refractometers, and penetrometers, which are labor intensive, destructive, costly, and complex. Each metabolite or group of metabolites requires different sample preparation and measurement. The reproducibility and quantitative accuracy have still been hard to guarantee for comparative studies, as inconsistent annotation has been revealed. Differences in metabolite composition and their accumulation between tissues at different developmental stages have been reported in watermelon (Neglo et al., 2021), which demands layers of complex experiments to figure out causal genes. Furthermore, although thousands of metabolites have been characterized, only a few genes have been reported for some metabolites. Hence, the discovery of their pathways and genes underlying the enormous number of metabolites requires greater emphasis to exploit and utilize for breeding.
Uncovering genes of polygenic metabolites and utilizing them for breeding necessitates large breeding populations, sophisticated analytical models, and a number of validations for effective gene discovery and selection. Additionally, pinpointing the exact candidate gene of polygenic traits within QTL has been like finding a needle in a haystack. The combinations of these difficulties with the limited genetic diversity of elite watermelon cultivars have hindered the opportunities for novel gene and allele discovery (Levi et al., 2001; Kantor and Levi, 2018). However, candidate genes for some metabolites have been identified; their functional validation and utilization in molecular breeding, including MAS, genome selection, and gene editing, has remained limited due to inadequate infrastructure and skilled human resources.
The environmental sensitivity of metabolite expression, which often obscures genetic signals and complicates data interpretation, has been the other obstacle (He et al., 2022; Zhu et al., 2021; Chen et al., 2024). For instance, the ClPSY (Phytoene Synthase) gene, which is crucial for carotenoid biosynthesis, has been regulated by light and hormones to influence β-carotene accumulation (Wu et al., 2020). This makes it challenging to figure out and utilize such genes across multiple environments. The limited investigation of different genotypes with diverse metabolite background, due to limited germplasm, has also constrained efforts to robustly find enormous metabolite pathways and causal genes.
The lack of integration of genomics, transcriptomics, proteomics, and metabolomics data streams for comprehensive molecular mechanism insights of the watermelon fruit metabolome remains mysterious. On the other hand, the absence of AI-powered multi-omics investigation slowed progress in unlocking, validating, and utilizing target genes. Investigations into predicting gene expression and metabolite accumulation using remote sensing, based on climate and growth performance, are also lacking. Such approaches, combined with the early manipulation of gene expression to achieve desired metabolite levels, remain underexplored.
5.2 Future perspectives to accelerate gene discovery and breeding impact
Systematic integration of multi-omics is crucial to reveal gene-to-metabolite networks to provide a holistic view of how genetic information translates into the metabolic profile of fruits. To achieve this, several strategies are being pursued, including the implementation of high-throughput phenotyping platforms to capture precise trait measurements, collection and expansion of genetic resources through the characterization of wild relatives and cultivars (Guo et al., 2019; Wu et al., 2019), and the application of artificial intelligence (AI) to decipher complex genotype–phenotype relationships (Frey, 2018; Wu and Xie, 2024).
The development of AI-driven approaches to predict metabolomes and their molecular mechanisms, as well as the use of gene editing to enhance metabolite production without relying on labor-intensive, expensive, and complex wet-laboratory activities, will help overcome current limitations in gene discovery and advance molecular breeding strategies. The emerged x-ray imaging driven by AI that has been employed to classify seeds into viable seeds, nonviable seeds, and abnormal viable seeds demonstrates the potential of such technologies for innovation in fruit metabolome gene discovery. Moreover, establishing AI agents and integrating them into the multi-omics research pipelines, along with promoting partnerships and sharing data in digital approaches, can maximize the benefits of collaborative efforts. These integrated approaches hold the potential to accelerate the development of next-generation watermelon varieties with multiple enhanced metabolites. By digging out candidate genes and utilizing them for molecular breeding, researchers can generate cultivars with improved metabolome profiles and superior quality characteristics to fulfill evolving market demands.
6 Conclusions
The exploration of the watermelon fruit metabolome has entered a transformative era driven by multi-omics technologies, which have been instrumental in uncovering the genetic underpinnings of key metabolites such as sugar, cucurbitacin, carotenoids, volatile compounds, amino acids, organic acids, and flavonoids. The identified genes regulating sugars (SPS, INVs, SUS, ClTST2, ClSWEET3), cucurbitacin (OSC, ACT, CYP), carotenoids (LCYB, PSY1, CHYB), amino acids (OTC, N-AOA, AOD-3), organic acids (PEPCK, ALMT, ICDH, CS), and flavonoids (CmAPRR2, PMAT1, CYP81E, VR) provide a critical genetic background for modern breeding programs. The forward and reverse gene harvesting approach has revolutionized the sector. The application of these discovered genes through MAS, GS, and CRISPR/Cas9-mediated gene editing enables breeding to enhance desirable metabolites and suppress undesirable ones, offering an efficient pathway to develop superior cultivars. However, several significant challenges persist, including the polygenic and environmentally sensitive nature of metabolic traits, the limited genetic diversity of elite germplasms, the high cost and complexity of metabolomic phenotyping, and the limited application of molecular breeding. Future advancements hinge on systematically integrating multi-omics data, leveraging wild genetic resources to broaden diversity, and employing AI to decipher complex gene-to-metabolite networks and predict genetic bases and phenotypic outcomes, thereby accelerating the development of next-generation watermelon varieties with optimized fruit metabolites to meet evolving consumer and market demands.
Author contributions
FK: Writing – original draft, Conceptualization, Writing – review & editing. WL: Writing – review & editing, Project administration, Supervision, Conceptualization, Funding acquisition, Validation, Resources. HZ: Validation, Writing – review & editing, Conceptualization, Supervision, Resources. LQ: Resources, Validation, Writing – review & editing, Supervision. NH: Supervision, Validation, Resources, Writing – review & editing. LX: Resources, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was granted by Henan Province Key Research and Development Program (241111521500), the National Key Research and Development Program of China (2023YFE0206900), Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2021-ZFRI), China Agriculture Research System of MOF and MARA (CARS-25-03).
Acknowledgments
We thank the Chinese Government for providing full scholarship, and Dilla University as well as Ethiopian Ministry of Education to provid study leave and salary for the first author. Moreover, we thank all the members of the Polyploid Watermelon Research Group, Zhengzhou Fruit Research Institute, Chinese Academy of Agricultural Sciences (Zhengzhou, Henan, China). Furthermore, we acknowledge Henan Province Key Research and Development Program; the National Key Research and Development Program of China; Agricultural Science and Technology Innovation Program; and China Agriculture Research System of MOF and MARA, those provided us funds.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
This article has been corrected with minor changes. These changes do not impact the scientific content of the article.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdullah-Zawawi, M.-R., Govender, N., Harun, S., Muhammad, N., Zainal, Z., and Mohamed-Hussein, Z. (2022). Multi-omics approaches and resources for systems-level gene function prediction in the plant kingdom. Plants 11. doi: 10.3390/plants11192614
Anees, Q., Deepmala Sehgal, U., Gao, L., Umer, M. J., Pingli, Y., et al. (2021). Identification of key gene networks associated with cell wall components leading to flesh firmness in watermelon. Front. Plant Sci 12. doi: 10.3389/fpls.2021.630243
Anees, M., Gao, L., Gong, C., Umer, M. J., Yuan, P., Zhu, H., et al. (2023). Aux/IAA gene Cla004102, is involved in synergistic regulation of various endogenous hormones, regulating flesh firmness in watermelon. Scientia Hortic. 310. doi: 10.1016/j.scienta.2022.111719
Aslam, A., Shengjie, Z., Xuqiang, L., Nan, H., and Wenge, L. (2021). Rootstock mediates transcriptional regulation of citrulline metabolism in grafted watermelon; [Mediação do porta-enxerto na regulação transcricional do metabolismo da citrulina na melancia enxertada. Braz. J. Biol. 81, 125–136. doi: 10.1590/1519-6984.223633
Aslam, A., Zhao, S., Azam, M., Lu, X., He, N., Li, B., et al. (2020). Comparative analysis of primary metabolites and transcriptome changes between ungrafted and pumpkin-grafted watermelon during fruit development. PeerJ 8. doi: 10.7717/peerj.8259
Assefa, A. D., Hur, O.-S., Ro, N.-Y., Lee, J.-E., Hwang, A.-J., Kim, B.-S., et al. (2020). Fruit morphology, citrulline, and arginine levels in diverse watermelon (Citrullus lanatus) germplasm collections. Plants (Basel) 9, 1054. doi: 10.3390/plants9091054
Bang, H., Kim, S., Leskovar, D., and King, S. (2007). Development of a codominant CAPS marker for allelic selection between canary yellow and red watermelon based on SNP in lycopene β-cyclase (LCYB) gene. Mol. Breed. 20, 63–72. doi: 10.1007/s11032-006-9076-4
Bianchi, G., Provenzi, L., and Rizzolo, A. (2019). Evolution of volatile compounds in “Cuoredolce®” and “Rugby” mini- watermelons (Citrullus lanatus (Thunb.) Matsumura & Nakai) in relation to the ripening degree at harvest. J. Sci Food agriculture. 100, 945–952. doi: 10.1002/jsfa.10023
Bouché, N. and Bouchez, D. (2001). Arabidopsis gene knockout: phenotypes wanted. Curr. Opin. Plant Biol. 4 2, 111–117. doi: 10.1016/S1369-5266(00)00145-X
Breniere, T., Fanciullino, A. L., Brunel, B., Laugier, P., Landrier, J.-F., Riva, C., et al. (2022). Carotenoid content in mature tomato fruit under soil water deficit: accounting the role of pre-maturing processes through chlorophyll kinetics and cell microscopy. Acta Hortic. 1353, 1–8. doi: 10.17660/ActaHortic.2022.1353.1
Brown, D. M., Zeef, L., Ellis, J., Goodacre, R., and Turner, S. (2005). Identification of novel genes in arabidopsis involved in secondary cell wall formation using expression profiling and reverse genetics. Plant Cell Online 17, 2281–2295. doi: 10.1105/tpc.105.031542
Burton-Freeman, B., Freeman, M., Zhang, X., Sandhu, A. K., and Edirisinghe, I. (2021). Watermelon and l-citrulline in cardio-metabolic health: review of the evidence 2000–2020. Curr. Atheroscl. Rep. 23. doi: 10.1007/s11883-021-00978-5
Chai, Y. and Sun, Y. (2025). Advances in the biosynthesis, gene mining, and molecular mechanisms of cucurbitacin in Cucurbitaceae crops. V 5, 0–0. doi: 10.48130/vegres-0024-0039
Chawade, A., Ham, J., Blomquist, H., Bagge, O., Alexandersson, E., and Ortiz, R. (2019). High-throughput field-phenotyping tools for plant breeding and precision agriculture. Agronomy. 9(5). doi: 10.3390/AGRONOMY9050258
Chen, S., Zhong, K., Li, Y., Bai, C., Xue, Z., and Wu, Y. (2024). Joint transcriptomic and metabolomic analysis provides new insights into drought resistance in watermelon (Citrullus lanatus). Front. Plant Sci 15. doi: 10.3389/fpls.2024.1364631
Chu, S., Wang, S., Zhang, R., Yin, M., Yang, X., and Shi, Q. (2022). Integrative analysis of transcriptomic and metabolomic profiles reveals new insights into the molecular foundation of fruit quality formation in Citrullus lanatus (Thunb.) Matsum. & Nakai. Food Qual. Saf. 6. doi: 10.1093/fqsafe/fyac015
Collard, B., Jahufer, M. Z. Z., Brouwer, J., and Pang, E. (2005). An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 142, 169–196. doi: 10.1007/s10681-005-1681-5
Collins, J. K., Wu, G., Perkins-Veazie, P., Spears, K., Claypool, P. L., Baker, R. A., et al. (2007). Watermelon consumption increases plasma arginine concentrations in adults. Nutrition 23, 261–266. doi: 10.1016/j.nut.2007.01.005
Davidovich-Rikanati, R., Shalev, L., Baranes, N., Meir, A., Itkin, M., Cohen, S., et al. (2015). Recombinant yeast as a functional tool for understanding bitterness and cucurbitacin biosynthesis in watermelon (Citrullus spp.). Yeast 32, 103–114. doi: 10.1002/yea.3049
Deng, Y., Liu, S., Zhang, Y., Tan, J., Li, X., Chu, X., et al. (2022). A telomere-to-telomere gap-free reference genome of watermelon and its mutation library provide important resources for gene discovery and breeding. Mol. Plant. 15, 1268–1284. doi: 10.1016/j.molp.2022.06.010
Depuydt, T., Rybel, B. D., and Vandepoele, K. (2022). Charting plant gene functions in the multi-omics and single-cell era. Trends Plant Sci. 28, 283–296. doi: 10.1016/j.tplants.2022.09.008
Dhanani, T., Dou, T., Biradar, K., Jifon, J., Kurouski, D., and Patil, B. S. (2022). Raman spectroscopy detects changes in carotenoids on the surface of watermelon fruits during maturation. Front. Plant Sci 13. doi: 10.3389/fpls.2022.832522
Dubey, S., Rajput, H., and Batta, K. (2023). Utilization of watermelon rind (Citrullus lanatus) in various food preparations: A review. J. Agric. Sci Food Res. 14, 1–3. doi: 10.35248/2161-1025.23.14.141
Edwards, A. J., Wiley, E. R., Brown, E. D., Clevidence, B. A., Vinyard, B. T., Collins, J. K., et al. (2003). Consumption of watermelon juice increases plasma concentrations of lycopene and β-carotene in humans. J. Nutr. 133, 1043–1050. doi: 10.1093/jn/133.4.1043
Fall, L. A., Perkins-Veazie, P., Ma, G., and McGregor, C. (2019). QTLs associated with flesh quality traits in an elite × elite watermelon population. Euphytica 215. doi: 10.1007/s10681-019-2356-y
Fiorani, F. and Schurr, U. (2013). Future scenarios for plant phenotyping. Annu. Rev. Plant Biol. 64, 267–291. doi: 10.1146/annurev-arplant-050312-120137
Frey, L. (2018). Artificial intelligence and integrated genotype–phenotype identification. Genes 10. doi: 10.3390/genes10010018
Fu, Y.-B. (2015). Understanding crop genetic diversity under modern plant breeding. Theor. Appl. Genet. 128, 2131–2142. doi: 10.1007/s00122-015-2585-y
Gao, Y., Guo, Y., Su, Z., Yu, Y., Zhu, Z., Gao, P., et al. (2020). Transcriptome analysis of genes related to fruit texture in watermelon. SCIENTIA Hortic. 262. doi: 10.1016/j.scienta.2019.109075
Gao, L., Zhao, S., Lu, X., He, N., and Liu, W. (2018a). ‘SW’, a new watermelon cultivar with a sweet and sour flavor. HortScience 53, 895–896. doi: 10.21273/HORTSCI12857-18
Gao, L., Zhao, S., Lu, X., He, N., Zhu, H., Dou, J., et al. (2018b). Comparative transcriptome analysis reveals key genes potentially related to soluble sugar and organic acid accumulation in watermelon. PloS One 13, e0190096. doi: 10.1371/journal.pone.0190096
Gong, C., Diao, W., Zhu, H., Umer, M. J., Zhao, S., He, N., et al. (2021a). Metabolome and transcriptome integration reveals insights into flavor formation of ‘Crimson’ Watermelon flesh during fruit development. Front. Plant Sci 12. doi: 10.3389/fpls.2021.629361
Gong, C., He, N., Zhu, H., Anees, M., Lu, X., and Liu, W. (2023). Multi-omics integration to explore the molecular insight into the volatile organic compounds in watermelon. Food Res. Int. 166. doi: 10.1016/j.foodres.2023.112603
Gong, C., Li, B., Anees, M., Zhu, H., Zhao, S., He, N., et al. (2022). Fine-mapping reveals that the bHLH gene Cla011508 regulates the bitterness of watermelon fruit. Scientia Hortic. 292. doi: 10.1016/j.scienta.2021.110626
Gong, C., Zhu, H., Lu, X., Yang, D., Zhao, S., Umer, M. J., et al. (2021b). An integrated transcriptome and metabolome approach reveals the accumulation of taste-related metabolites and gene regulatory networks during watermelon fruit development. Planta 254, 35. doi: 10.1007/s00425-021-03680-7
Guo, S., Liu, J., Zheng, Y., Huang, M., Zhang, H., Gong, G., et al. (2011). Characterization of transcriptome dynamics during watermelon fruit development: sequencing, assembly, annotation and gene expression profiles. BMC Genomics 12. doi: 10.1186/1471-2164-12-454
Guo, S., Sun, H., Zhang, H., Liu, J., Ren, Y., Gong, G., et al. (2015). Comparative transcriptome analysis of cultivated and wild watermelon during fruit development. PloS One 10. doi: 10.1371/journal.pone.0130267
Guo, S., Zhang, J., Sun, H., Salse, J., Lucas, W. J., Zhang, H., et al. (2012). The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 45, 51–58. doi: 10.1038/ng.2470
Guo, S., Zhao, S., Sun, H., Wang, X., Wu, S., Lin, T., et al. (2019). Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat. Genet. 51, 1616–1623. doi: 10.1038/s41588-019-0518-4
Habibi, F., Boakye, D. A., Chang, Y., Casorzo, G., Hallman, L. M., Madison, M., et al. (2024). Molecular mechanisms underlying postharvest physiology and metabolism of fruit and vegeta bles through multi-omics technologies. Scientia Horticulturae. 324. doi: 10.1016/j.scienta.2023.112562
Hartman, J. (2017). Investigating citrulline in cucurbits: A survey of cucurbitaceae and heritability estimations in watermelon. Available online at: https://www.academia.edu/68133752/Investigating_Citrulline_in_Cucurbits_A_Survey_of_Cucurbitaceae_and_Heritability_Estimations_in_Watermelon (Accessed April 26, 2025).
Hartman, J. L., Wehner, T. C., Ma, G., and Perkins-Veazie, P. (2019). Citrulline and arginine content of taxa of cucurbitaceae. Horticulturae 5. doi: 10.3390/horticulturae5010022
He, N., Umer, M., Yuan, P., Wang, W., Zhu, H., Zhao, S., et al. (2022). Expression dynamics of metabolites in diploid and triploid watermelon in response to flooding. PeerJ. 10. doi: 10.7717/peerj.13814
Houle, D., Govindaraju, D., and Omholt, S. (2010). Phenomics: the next challenge. Nat. Rev. Genet. 11, 855–866. doi: 10.1038/nrg2897
Hu, Q., Yang, J., Meng, L., Liu, J., and Tian, S. (2023). Genome-wide identification of AUX/IAA genes in watermelon reveals a crucial role for clIAA16 during fruit ripening. Horticulturae 9. doi: 10.3390/horticulturae9111167
Ilahy, R., Tlili, I., Siddiqui, M. W., Hdider, C., and Lenucci, M. S. (2019). Inside and beyond color: comparative overview of functional quality of tomato and watermelon fruits. Front. In Plant Sci 10. doi: 10.3389/fpls.2019.00769
Jankowicz-Cieslak, J. and Till, B. (2015). Forward and reverse genetics in crop breeding. Cham, Switzerland: Springer Nature. 215–240. doi: 10.1007/978-3-319-22521-0_8
Jannink, J., Lorenz, A., and Iwata, H. (2010). Genomic selection in plant breeding: from theory to practice. Briefings Funct. Genomics 9 2, 166–177. doi: 10.1093/bfgp/elq001
Johannsen, W. (2014). The genotype conception of heredity. 1911. Int. J. Epidemiol. 43 4, 989–1000. doi: 10.1093/ije/dyu063
Joshi, V., Joshi, M., Silwal, D., Noonan, K., Rodriguez, S., and Penalosa, A. (2019). Systematized biosynthesis and catabolism regulate citrulline accumulation in watermelon. Phytochemistry 162, 129–140. doi: 10.1016/j.phytochem.2019.03.003
Kantor, M. and Levi, A. (2018). Utilizing genetic resources and precision agriculture to enhance resistance to biotic and abiotic stress in watermelon. Notulae Botanicae Horti Agrobotanici Cluj-napoca 10, 1–7. doi: 10.15835/NSB10110242
Katuuramu, D. N., Levi, A., and Wechter, W. (2023). Genome-wide association study of soluble solids content, flesh color, and fruit shape in citron watermelon. Plant Genome 16. doi: 10.1002/tpg2.20391
Kebriyaee, D., Kordrostami, M., Rezadoost, M. H., and Lahiji, H. (2012). QTL analysis of agronomic traits in rice using SSR and AFLP markers. Notulae Botanicae Horti Agrobotanici Cluj-napoca 4, 116–123. doi: 10.15835/NSB427501
Kim, J. Y. (2020). Roadmap to high throughput phenotyping for plant breeding. J. Biosyst. Eng. 45, 43–55. doi: 10.1007/s42853-020-00043-0
Kim, J. and Buell, C. R. (2015). A revolution in plant metabolism: genome-enabled pathway discovery. Plant Physiol. 169, 1532–1539. doi: 10.1104/pp.15.00976
Kyriacou, M. C., Leskovar, D. I., Colla, G., and Rouphael, Y. (2018). Watermelon and melon fruit quality: The genotypic and agro-environmental factors implicated. Scientia Hortic. 234, 393–408. doi: 10.1016/j.scienta.2018.01.032
Lemmon, Z. H., Reem, N. T., Dalrymple, J., Soyk, S., Swartwood, K., Rodríguez-Leal, D., et al. (2018). Rapid improvement of domestication traits in an orphan crop by genome editing. Nat. Plants 4, 766–770. doi: 10.1038/s41477-018-0259-x
Levi, A., Thomas, C., Wehner, T., and Zhang, X. (2001). Low genetic diversity indicates the need to broaden the genetic base of cultivated watermelon. Hortscience 36, 1096–1101. doi: 10.21273/HORTSCI.36.6.1096
Li, B., Lu, X., Dou, J., Aslam, A., Gao, L., Zhao, S., et al. (2018). Construction of A high-density genetic map and mapping of fruit traits in watermelon (Citrullus lanatus L.) based on whole-genome resequencing. Int. J. Mol. Sci. 19, 3268. doi: 10.3390/ijms19103268
Liang, X., Gao, M., Amanullah, S., Guo, Y., Liu, X., Xu, H., et al. (2022). Identification of QTLs linked with watermelon fruit and seed traits using GBS-based high-resolution genetic mapping. Scientia Hortic. 303. doi: 10.1016/j.scienta.2022.111237
Liu, S., Gao, P., Zhu, Q., Luan, F., Davis, A. R., and Wang, X. (2016). Development of cleaved amplified polymorphic sequence markers and a CAPS-based genetic linkage map in watermelon (Citrullus lanatus [Thunb.] Matsum. and Nakai) constructed using whole-genome re-sequencing data. Breed. Sci 66, 244–259. doi: 10.1270/jsbbs.66.244
Liu, J., Guo, S., He, H., Zhang, H., Gong, G., Ren, Y., et al. (2013). Dynamic characteristics of sugar accumulation and related enzyme activities in sweet and non-sweet watermelon fruits. Acta Physiol. Plant 35, 3213–3222. doi: 10.1007/s11738-013-1356-0
Liu, Y., He, C., and Song, H. (2018). Comparison of fresh watermelon juice aroma characteristics of five varieties based on gas chromatography-olfactometry-mass spectrometry. Food Res. Int. 107, 119–129. doi: 10.1016/j.foodres.2018.02.022
Liu, Y., Keefer, H., Watson, M., and Drake, M. (2024). Consumer perception of whole watermelons. J. Food Sci 89, 625–639. doi: 10.1111/1750-3841.16843
Liu, J., Zhou, Y., Li, J., Wang, F., and Yang, Y. (2020). Comprehensive genomic characterization and expression analysis of the lipoxygenase gene family in watermelon under hormonal treatments. Agriculture 10, 429. doi: 10.3390/agriculture10100429
Lu, H., Zhao, H., Zhong, T., Chen, D., Wu, Y., and Xie, Z. (2024). Molecular regulatory mechanisms affecting fruit aroma. Foods 13, 1870. doi: 10.3390/foods13121870
Lv, T., Zhao, L., Zhang, S., Guan, J., Liu, W., and Qi, H. (2022). ClPIF3-clHY5 module regulates clPSY1 to promote watermelon fruit lycopene accumulation earlier under supplementary red lighting. Int. J. Of Mol. Sci. 23. doi: 10.3390/ijms23084145
Mashilo, J., Shimelis, H., Ngwepe, R. M., and Thungo, Z. (2022). Genetic analysis of fruit quality traits in sweet watermelon (citrullus lanatus var. lanatus): a review. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.834696
McGregor, C., Rijal, S., Josiah, S., and Adams, L. (2023). “Genetics and genomics of fruit quality traits of watermelon,” in The watermelon genome. Eds. Dutta, S., Nimmakayala, P., and Reddy, U. K. (Springer International Publishing, Cham), 69–83. doi: 10.1007/978-3-031-34716-0_5
Neglo, D., Tettey, C. O., Essuman, E. K., Kortei, N. K., Boakye, A. A., Hunkpe, G., et al. (2021). Comparative antioxidant and antimicrobial activities of the peels, rind, pulp and seeds of watermelon (Citrullus lanatus) fruit. Sci. Afr. 11. doi: 10.1016/j.sciaf.2020.e00582
Nie, H., Kim, M., Lee, S., Lim, S., Lee, M. S., Kim, J. H., et al. (2023). High-quality genome assembly and genetic mapping reveal a gene regulating flesh color in watermelon (Citrullus lanatus). Front. In Plant Sci 14. doi: 10.3389/fpls.2023.1142856
Ning, K., Cai, X., Yan, L., Zhou, W., Xie, A., Wang, Y., et al. (2024). Transcriptomic and metabolomic analysis reveals improved fruit quality in grafted watermelon. Horticulturae 10. doi: 10.3390/horticulturae10121269
Oren, E., Tzuri, G., Vexler, L., Dafna, A., Meir, A., Faigenboim, A., et al. (2019). The multi-allelic APRR2 gene is associated with fruit pigment accumulation in melon and watermelon. J. Exp. Bot. 70, 3781–3794. doi: 10.1093/jxb/erz182
Ozdemir, S. (2025). Effectiveness of generative AI tool to determine fruit quality: watermelon case study. Horticulturae 11, 308. doi: 10.3390/horticulturae11030308
Peng, Z., Song, S., Fu, D., Zhou, J., Chang, H., Wang, B., et al. (2025). Combined transcriptome and metabolome analysis reveals the mechanism of fruit quality formation in different watermelon (Citrullus lanatus) cultivars. Scientia Hortic. 339, 113797. doi: 10.1016/j.scienta.2024.113797
Pingli, Y., Xu, C., He, N., Lu, X., Zhang, X., Shang, J., et al. (2023). Watermelon domestication was shaped by stepwise selection and regulation of the metabolome. Sci. China Life Sci. 66, 579–594. doi: 10.1007/s11427-022-2198-5
Pontarotti, G., Mossio, M., and Pocheville, A. (2022). The genotype–phenotype distinction: from Mendelian genetics to 21st century biology. Genetica 150, 223–234. doi: 10.1007/s10709-022-00159-5
Ramirez, J. L., Du, X., and Wallace, R. W. (2020). Investigating sensory properties of seven watermelon varieties and factors impacting refreshing perception using quantitative descriptive analysis. Food Res. Int. 138, 109681. doi: 10.1016/j.foodres.2020.109681
Ren, Y., Li, M., Guo, S., Sun, H., Zhao, J., Zhang, J., et al. (2021). Evolutionary gain of oligosaccharide hydrolysis and sugar transport enhanced carbohydrate partitioning in sweet watermelon fruits. Plant Cell 33, 1554–1573. doi: 10.1093/plcell/koab055
Ren, Y., McGregor, C., Zhang, Y., Gong, G., Zhang, H., Guo, S., et al. (2014). An integrated genetic map based on four mapping populations and quantitative trait loci associated with economically important traits in watermelon (Citrullus lanatus). BMC Plant Biol. 14. doi: 10.1186/1471-2229-14-33
Renner, S. S., Wu, S., Pérez-Escobar, O. A., Silber, M. V., Fei, Z., and Chomicki, G. (2021). A chromosome-level genome of a Kordofan melon illuminates the origin of domesticated watermelons. Proc. Natl. Acad. Sci. United States America 118. doi: 10.1073/pnas.2101486118
Serrat, X., Esteban, R., Guibourt, N., Moysset, L., Nogués, S., and Lalanne, E. (2014). EMS mutagenesis in mature seed-derived rice calli as a new method for rapidly obtaining TILLING mutant populations. Plant Methods 10, 5–5. doi: 10.1186/1746-4811-10-5
Shiratake, K. and Suzuki, M. (2016). Omics studies of citrus, grape and rosaceae fruit trees. Breed. Sci 66, 122–138. doi: 10.1270/jsbbs.66.122
Sorokina, M., McCaffrey, K. S., Deaton, E. E., Ma, G., Ordovás, J. M., Perkins-Veazie, P., et al. (2021). A catalog of natural products occurring in watermelon—Citrullus lanatus. Front. Nutr. 8. doi: 10.3389/fnut.2021.729822
Soteriou, G. A. and Kyriacou, M. C. (2015). Rootstock-mediated effects on watermelon field performance and fruit quality characteristics. Int. J. Vegetab le Sci 21, 344–362. doi: 10.1080/19315260.2014.881454
Sulaiman, F., Ahmad Azam, A., Ahamad Bustamam, M. S., Fakurazi, S., Abas, F., Lee, Y. X., et al. (2020). Metabolite profiles of red and yellow watermelon (Citrullus lanatus) cultivars using a 1H-NMR metabolomics approach. Molecules 25, 3235. doi: 10.3390/molecules25143235
Sun, L., Lai, M., Ghouri, F., Nawaz, M. A., Ali, F., Baloch, F., et al. (2024). Modern plant breeding techniques in crop improvement and genetic diversity: from molecular markers and gene editing to artificial intelligence—A critical review. Plants 13. doi: 10.3390/plants13192676
Sun, L., Zhang, Y., Cui, H., Zhang, L., Sha, T., Wang, C., et al. (2020). Linkage Mapping and Comparative Transcriptome Analysis of Firmness in Watermelon (Citrullus lanatus). Front. Plant Sci 11. Available online at: https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2020.00831 (Accessed February 16, 2024).
Trandel, M. A., Johanningsmeier, S., Schultheis, J., Gunter, C., and Perkins-Veazie, P. (2021). Cell wall polysaccharide composition of grafted ‘Liberty’ Watermelon with reduced incidence of hollow heart defect. Front. Plant Sci 12. doi: 10.3389/fpls.2021.623723
Tripodi, G., Condurso, C., Cincotta, F., Merlino, M., and Verzera, A. (2019). Aroma compounds in mini-watermelon fruits from different grafting combinations. J. Sci Food agriculture. 100, 1328–1335. doi: 10.1002/jsfa.10149
Tsakirpaloglou, N., Septiningsih, E., and Thomson, M. J. (2023). Guidelines for performing CRISPR/cas9 genome editing for gene validation and trait improvement in crops. Plants 12. doi: 10.3390/plants12203564
Umer, M. J., Bin Safdar, L., Gebremeskel, H., Zhao, S., Yuan, P., Zhu, H., et al. (2020a). Identification of key gene networks controlling organic acid and sugar metabolism during watermelon fruit development by integrating metabolic phenotypes and gene expression profiles. Hortic. Res. 7, 1–13. doi: 10.1038/s41438-020-00416-8
Umer, M. J., Gao, L., Kidanemariam, H., Safdar, L., Pingli, Y., Zhao, S., et al. (2020b). Expression pattern of sugars and organic acids regulatory genes during watermelon fruit development. Scientia Hortic. 265, 109102. doi: 10.1016/j.scienta.2019.109102
Vallarino, J. G., Hong, J., Wang, S., Wang, X., Sade, N., Orf, I., et al. (2023). Limitations and advantages of using metabolite-based genome-wide association studies: Focus on fruit quality traits. Plant Sci 333. doi: 10.1016/j.plantsci.2023.111748
Verma, S., Gupta, A. R. S. S. H., Yalla, S., Patel, P. J., Sharma, R., et al. (2024). “Integrating marker-assisted (MAS) and genomic selection (GS) for plant functional trait improvement,” in Plant functional traits for improving productivity. Eds. Kumar, N. and Singh, H. (Springer Nature, Singapore), 203–215. doi: 10.1007/978-981-97-1510-7_11
Wang, C., Luan, F., Liu, H., Davis, A., Zhang, Q., Dai, Z., et al. (2021a). Mapping and predicting a candidate gene for flesh color in watermelon. J. Integr. Agric. 20, 2100–2111. doi: 10.1016/s2095-3119(20)63487-6
Wang, C., Qiao, A., Fang, X., Sun, L., Gao, P., Davis, A. R., et al. (2019). Fine mapping of lycopene content and flesh color related gene and development of molecular marker–assisted selection for flesh color in watermelon (Citrullus lanatus). Front. Plant Sci 10. doi: 10.3389/fpls.2019.01240
Wang, Z., Wan, L., Ren, J., Zhang, N., Zeng, H., Wei, J., et al. (2024). Improving the genome editing efficiency of CRISPR/cas9 in melon and watermelon. Cells 13. doi: 10.3390/cells13211782
Wang, J., Wang, Y., Zhang, J., Ren, Y., Li, M., Tian, S., et al. (2021b). The NAC transcription factor ClNAC68 positively regulates sugar content and seed development in watermelon by repressing ClINV and ClGH3.6. Horticulture Res. 8, 214. doi: 10.1038/s41438-021-00649-1
Wenzl, P., Li, H., Carling, J., Zhou, M., Raman, H., Paul, E., et al. (2006). A high-density consensus map of barley linking DArT markers to RFLP and STS loci and agricultural traits. BMC Genomics 7, 206–206. doi: 10.1186/1471-2164-7-206
Wu, S., Sun, H., Gao, L., Branham, S. E., McGregor, C., Xu, Y., et al. (2023). A Citrullus genus super-pangenome reveals extensive variations in wild and cultivated watermelons and sheds light on watermelon evolution and domestication. Plant Biotechnol. J. 21, 1926–1928. doi: 10.1101/2023.06.08.544282
Wu, C., Sun, L., Lv, Y., Cui, H., Wang, X., Gao, P., et al. (2020). Functional characterization and in silico analysis of phytoene synthase family genes responsible for carotenoid biosynthesis in watermelon (Citrullus lanatus L.). Agronomy 10. doi: 10.3390/agronomy10081077
Wu, S., Wang, X., Reddy, U., Sun, H., Bao, K., Gao, L., et al. (2019). Genome of ‘Charleston Gray’, the principal American watermelon cultivar, and genetic characterization of 1,365 accessions in the U.S. National Plant Germplasm System watermelon collection. Plant Biotechnol. J. 17, 2246–2258. doi: 10.1111/pbi.13136
Wu, Y. and Xie, L. (2024). AI-driven multi-omics integration for multi-scale predictive modeling of genotype-environment-phenotype relationships. Comput. Struct. Biotechnol. J. 27, 265–277. doi: 10.1016/j.csbj.2024.12.030
Xiao, Q., Bai, X., Zhang, C., and He, Y. (2021). Advanced high-throughput plant phenotyping techniques for genome-wide association studies: A review. J. Advanced Res. 35, 215–230. doi: 10.1016/j.jare.2021.05.002
Yang, C., Shi, J., Qin, Y., Hua, S., Bao, J., Liu, X., et al. (2025). ClaPEPCK4: target gene for breeding innovative watermelon germplasm with low Malic acid and high sweetness. GM Crops Food 16, 156–170. doi: 10.1080/21645698.2025.2452702
Yang, D., Zhu, H., Anees, M., Zhao, Y., Yuan, P., Liu, W., et al. (2023). dentification of an aux/IAA regulator for flesh firmness using combined GWAS and bulked segregant RNA-seq analysis in watermelon. Hortic. Plant J. 10, 1198–1213. doi: 10.1016/j.hpj.2023.05.018
Yativ, M., Harary, I., and Wolf, S. (2010). Sucrose accumulation in watermelon fruits: Genetic variation and biochemical analysis. J. Plant Physiol. 167, 589–596. doi: 10.1016/j.jplph.2009.11.009
Yuan, P., He, N., Umer, M. J., Zhao, S., Diao, W., Zhu, H., et al. (2021a). Comparative Metabolomic Profiling of Citrullus spp. Fruits Provides Evidence for Metabolomic Divergence during Domestication. Metabolites 11, 78. doi: 10.3390/metabo11020078
Yuan, P., Umer, M. J., He, N., Zhao, S., Lu, X., Zhu, H., et al. (2021b). Transcriptome regulation of carotenoids in five flesh-colored watermelons (Citrullus lanatus). BMC Plant Biol. 21. doi: 10.1186/s12870-021-02965-z
Zakhrabekova, S., Gough, S., Lundh, L., and Hansson, M. (2013). Functional genomics and forward and reverse genetics approaches for identification of important QTLs in plants. Proceedings (News) of the Azerbaijan National Acad. Sci. — Biol. Med. Sci. 68, 23–28.
Zhan, Y., Hu, W., He, H., Dang, X., Chen, S., and Bie, Z. (2023). A major QTL identification and candidate gene analysis of watermelon fruit cracking using QTL-seq and RNA-seq. Front. Plant Sci 14. doi: 10.3389/fpls.2023.1166008
Zhang, C. and Hao, Y.-J. (2020). Advances in genomic, transcriptomic, and metabolomic analyses of fruit quality in fruit crops. Hortic. Plant J. 6, 361–371. doi: 10.1016/j.hpj.2020.11.001
Zhao, C., Zhang, Y., Du, J., Guo, X., Wen, W., Gu, S., et al. (2019). Crop phenomics: current status and perspectives. Front. Plant Sci 10. doi: 10.3389/fpls.2019.00714
Zhou, Y., Ma, Y., Zeng, J., Duan, L., Xue, X., Wang, H., et al. (2016). Convergence and divergence of cucurbitacin biosynthesis and regulation in Cucurbitaceae. Nat. Plants 2, 16183. doi: 10.1038/nplants.2016.183
Keywords: citrullus lanatus, metabolites, gene discovery, genomics, marker-assisted selection, fruit quality
Citation: Kenea FT, He N, Lu X, Luo X, Zhu H and Liu W (2025) Watermelon fruit metabolome gene discovery and its application in breeding: a review. Front. Plant Sci. 16:1687406. doi: 10.3389/fpls.2025.1687406
Received: 17 August 2025; Accepted: 28 September 2025;
Published: 17 October 2025; Corrected: 21 October 2025.
Edited by:
Vijay Sheri, Texas Tech University, United StatesReviewed by:
Sagar Datir, Naoroji Godrej Centre for Plant Research (NGCPR), IndiaPhanikanth Jogam, Kakatiya Medical College, India
Copyright © 2025 Kenea, He, Lu, Luo, Zhu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenge Liu, bGl1d2VuZ2VAY2Fhcy5jbg==; Hongju Zhu, emh1aG9uZ2p1QGNhYXMuY24=