 Zongqing Qiu1
Zongqing Qiu1 Liangliang Hu
Liangliang Hu- 1College of Horticulture, Xinjiang Agricultural University, Urumqi, China
- 2Postdoctoral Station of Horticulture, Xinjiang Agricultural University, Urumqi, China
Introduction: The TALE gene family acts as key regulators of plant growth, development, and stress adaptation. However, systematic characterization of this family in watermelon (Citrullus lanatus L.), an economically important cucurbit crop susceptible to abiotic stresses like drought and cold, is lacking. This gap hinders understanding of watermelon’s stress-responsive mechanisms and the breeding of stress-resilient varieties.
Methods: ClTALE genes were comprehensively identified using the watermelon genome database. Bioinformatics analyses (phylogenetic classification, genomic structure annotation, conserved motif detection, cis-acting element prediction) were performed. Protein-protein interactions were inferred via STRING. qRT-PCR detected expression profiles under drought, low potassium (LK), and melatonin + cold (MT+CT) treatments. Subcellular localization of candidate genes was analyzed by transient expression, and yeast heterologous expression verified stress tolerance under PEG-simulated drought.
Results: A total of 22 ClTALE members were identified, clustering into seven subclades (KNOX-I/STM, KNOX-II, BELL-I to BELL-V). Their promoters contain abundant hormone-related (abscisic acid, jasmonic acid) and abiotic stress-related (drought, cold) cis-acting elements. ClTALE proteins may interact with core growth and development transcription factors. ClTALE2, 3, 8, 11, and 20 were significantly upregulated under drought; ClTALE2 and 3 showed cross-response to LK and MT+CT. ClTALE3 localizes to the nucleus, and its overexpression enhanced yeast tolerance to PEG stress.
Discussion: This study is the first systematic characterization of the watermelon ClTALE family, clarifying its genomic features, evolutionary relationships, and stress-responsive patterns. ClTALE2 and 3 (especially ClTALE3) exhibit potential as key stress adaptation regulators. These findings provide a theoretical basis and genetic resources for elucidating watermelon’s stress-resistance mechanisms and breeding stress-tolerant varieties.
1 Introduction
The three amino acid loop extension, known as TALE, is a category of transcription factors found in eukaryotes, affecting key regulatory processes (Bürglin, 1997). A highly conserved sequence, hereafter referred to as the homeobox (Gehring, 1987). The family gene encodes proteins with KNOX and BLH/BELL domains, which form physically and functionally analogous heterodimers (Jia et al., 2023b). TALE family genes encode homeobox domains consisting of 63 amino acids. This family is referred to as the homeobox protein superfamily because of the three-amino acid loop extension linking the first and second helices of the homeodomain (Ma et al., 2019). The transcription factors of Arabidopsis thaliana (AtTALE) are categorized into two subfamilies: BELL and KNOX. Plants have KNOX1, KNOX2, ELK, and Homeobox KN domains (Jia et al., 2023a).
The first homeobox found in plants was Kn-1 (Knotted-1) (Jia et al., 2023b). The mutation in STM (SHOOTMERISTEMLESS) severely decreased SAM synthesis (Scofield et al., 2013). The KNAT7 is involved in the production of the secondary cell wall. The synthesis of cotton fiber is regulated by GhKNL1, a member of the TALE family (Ahmad et al., 2024). The regulation of flowers, xylem differentiation, and hormonal treatment may be influenced by the Arabidopsis KNOX Class I family genes (STM, KNAT2, BREVIPEDICELLUS (BP)/KNAT1, and KNAT6) (Guo et al., 2022). Class I KNOX genes also exert inhibitory effects on secondary cell wall (SCW) production. Conversely, Class II KNOX genes appear to exert opposing effects on the regulation of stem elongation and SCW buildup compared to Class I. KNAT3 binds to SCW-forming transcription factor NST1/2 to control Ferulate 5-Hydroxylase (F5H) and enhance syringyl lignin formation (Qin et al., 2020). KNAT7 also activates the expression of IRXs in Arabidopsis, which positively regulates SCW biosynthesis (He et al., 2018). The KNOX genes regulate several target genes that are responsible for regulating hormone homeostasis. The expression of the Abscisic acid (ABA)-responsive gene ABI3 can be directly up-regulated by KNAT3. ABA treatment increased the expression and promoter activity of MdKNOX19 in apples (Jia et al., 2023b). MdKNOX19 directly binds to and upregulates ABI5 to transmit ABA perception. These findings provide more evidence for a regulatory feedback loop involving KNOX and ABA signaling (Jia et al., 2023b).
Drought stress is a detrimental abiotic stress that affects physiological and biochemical systems, reducing plant growth and yield (Sharif et al., 2022a, 2022b; Yan et al., 2024). The RNAi antisense lines of MtKNOX3-like in Medicago truncatula displayed compromised response to drought stress (Iannelli et al., 2023). In another study, the expression of GhBLH5-A05 in cotton was stimulated by drought stress. The overexpression of GhBLH5-A05 in both Arabidopsis and cotton enhanced drought tolerance, while its silencing led to greater sensitivity. The GhBLH5-A05 binds to increase the expression of GhRD20-A09 and GhDREB2C-D05. GhBLH5-A05 interacts with the KNOX transcription factor GhKNAT6-A03. The co-expression of GhBLH5-A05 and GhKNAT6-A03 enhanced the transcription of GhRD20-A09 and GhDREB2C-D05. Altogether, GhKNAT6-A03-GhBLH5-A05 functions as a regulatory element in cotton’s response to drought stress by triggering the expression of the drought-responsive genes GhRD20-A09 and GhDREB2C-D05 (Zhang et al., 2024). VIGS silencing of GhKNOX4-AGh and GhKNOX22-D genes affected cotton seedling growth and development under salt and drought (Sun et al., 2023). Despite their distinctive role in stress biology, TALE genes have yet to be explored in watermelon.
A lack of potassium (K+) severely limits the quantity and quality of crops, making it one of the most important nutrients for plants (Zhong et al., 2018). It is well known that low K in plant tissues exacerbates the impacts of drought stress by affecting the photosynthetic carbon metabolism and the osmoregulation process. Melatonin (MT), also known as N-acetyl-5-methoxytryptamine, is a biological substance that is non-toxic and is produced in the pineal gland of animals and in certain cells of plants (Sharif et al., 2018). Under moderate and severe drought stress, the development of soybean plants was greatly enhanced by the exogenous application of MT, which inhibited membrane damage and reduced ROS concentrations (Sharif et al., 2018).
Watermelon is an economically important fruit crop and is counted among the world’s top five most consumed fresh fruits. Nevertheless, it is highly susceptible to various unfavorable environmental factors, which lead to a decline in its quality and yield. While TALE genes play a central role in plant stress signaling and have been studied in many species, they remain uncharacterized in watermelon.
Our study comprehensively examined the TALE gene family in the watermelon genome. Bioinformatics analysis, including phylogeny, conserved motifs, protein interactions, and cis-elements, was performed. Moreover, the expression of ClTALE genes was analyzed. The expression response of ClTALE genes to drought stress was examined. Additionally, the expression under LK and MT was also examined. By offering a comprehensive analysis of watermelon ClTALE genes, these studies enable future functional investigations and the potential application of novel candidates for crop improvement in stress resistance, growth, and development.
2 Materials and methods
2.1 Plant material and experimental treatments
In northwest China, Xinjiang is situated in the midlatitude inland region (34°20′11–49°10′55 N, 73°29′54–96°23′03 E). The watermelon line ‘97103’ (obtained from the Xinjiang Academy of Agriculture Science) was grown in the greenhouse of Xinjiang Agriculture University. The plants were then moved to a solution that included 20% (W/V) PEG-6000 (Mahmoud et al., 2023). After being subjected to simulated drought stress, RNA was extracted from samples collected at 6, 12, 18, and 24 h from both the treated and untreated watermelon seedlings. The watermelon line ‘97103’ seeds were heated to 30°C and planted in a sponge. After a week, homogeneous seedlings were grown in a controlled environment chamber using hydroponics. Blue, opaque plastic boxes measuring 320 × 240 × 140 mm were used as hydroponic growing containers. The lids made of polystyrene foam had six holes punched into them, each 25 mm in diameter. Soaps were placed around each hole with the intention of supporting a single seedling. Nine liters of nutritional solution with a sufficient amount of K+ (CK) were then added to each box. For the K+ starvation treatment (LK), half of the seedlings were moved to a nutrient solution devoid of K+ (lacking KCl) after five days, while the other half were moved to the CK nutrient solution as a control (Fan et al., 2014). The samples were collected at different timepoint (120, 144, and 168 hours) for qRT-PCR expression analysis. Additionally, the watermelon seedlings were treated with MT (Melatonin) 150 μM and CT (Cold treatment) 8°C (Chang et al., 2021). The samples were collected 24 hours after treatment for qRT-PCR expression analysis. There were three biological and four technical replicates for each sample.
2.2 RNA extraction and qPCR analysis
The RNAprep Pure Plant Kit (Tiangen, Beijing, China) was used to extract total RNA from the roots and leaf of watermelon ‘97103’, respectively. HiScript II Q RT SuperMix (Vazyme, Nanjing, China) was used for subsequent reverse transcription. Using the SYBR Green PCR Master Mix kit (TransGen, Beijing, China), quantitative PCR (qPCR) was carried out. As previously described, the 2^-ΔΔCT approach was used to assess the relative expression levels of the ClTALE genes. The Actin (Cla97C01G014580) was used as the internal reference in all qPCR reactions (Zhang K, et al., 2023). Each sample was set with three biological and three technical replications. The specific primers employed in these investigations are detailed in Supplementary Table S1.
2.3 Identification and isolation of ClTALE genes from the watermelon genome
The BLAST algorithm in the Watermelon genome database was used to query against the TALE proteins of Arabidopsis thaliana to discover all sequences related to the TALE family. To ensure that the conserved domains were present, the non-redundant sequences of TALE were examined using the CDD search and the Pfam database following the method of (Ahmad et al., 2023b). The instability index (GRAVY), isoelectric points (pI), and molecular weight (MW) of ClTALE were predicted using the ProtParam tool. This study used Plant-mPLoc (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/#) as a tool to determine the subcellular localization of all ClTALE genes and proteins.
2.4 Physical location and synteny of ClTALE genes
To characterize the physical location and synteny of ClTALE genes, a standardized analytical workflow was implemented. Gff3-format files were first retrieved from the watermelon genome database, which contains comprehensive genomic annotations including gene coordinates, exon-intron boundaries, and associated feature attributes—critical data for precise gene mapping. The extracted gff3 files were subsequently mapped to watermelon chromosomes using TBtools (Toolbox for biologists) (v0.6655). This bioinformatics platform was selected for its robust capabilities in genomic data visualization and positional analysis, enabling high-precision localization of ClTALE genes to their respective chromosomal regions (Chen et al., 2020).
2.5 Phylogenetic analysis of ClTALE proteins
To reconstruct the evolutionary relationships among TALE proteins, we obtained amino acid sequences from watermelon, cucumber, wax gourd, and the model plant Arabidopsis thaliana. A multiple sequence alignment was performed using Clustal-Omega, which served as the input for phylogenetic tree construction (Ahmad et al., 2023a; Tan et al., 2023). Phylogenetic relationships were inferred from the Clustal-Omega alignment using the Maximum Likelihood (ML) method implemented on the IQ-TREE web server. The robustness of the tree topology was assessed with 1,000 bootstrap replicates. The final phylogenetic tree was visualized and rendered using the Interactive Tree of Life (iTOL v.5) platform (Ahmad et al., 2022).
2.6 Conserved motif analysis of ClTALE proteins
The conserved motifs in ClTALE proteins were identified using the MEME suite (http://meme-suite.org) with the following parameters: a maximum of 10 motifs, a motif width ranging from 6 to 100 amino acids, and a limit of one occurrence per sequence. The resulting motifs were visualized using TBtools (v0.6655) (Chen et al., 2020, 2023).
2.7 Interactive protein partners
A protein-protein interaction network for the ClTALE proteins was generated using the STRING database (v11.5), with the number of interactors limited to 5 for the first shell and 10 for the second shell. The network structure was subsequently visualized and rendered using Cytoscape v3.8.2.
2.8 Gene ontology analysis of ClTALE genes
Additionally, ClTALE protein sequences were analyzed using the GO tool Blast2GO (Version 2.7.2) (http://www.blast2go.com) (accessed on 3rd July 2024) (Ullah et al., 2022). By repeating the steps outlined earlier, we were able to reassemble the three categories into which the cellular component GO categorization, molecular functions, and biological processes fell.
2.9 Promoter analysis of ClTALE genes
The 2.0 kb promoter regions upstream of the ATG start site for each ClTALE gene were analyzed using PlantCARE to identify known cis-acting elements. The identified elements were then categorized based on their predicted functions in growth, hormone response, and stress the tool is referenced by (Chen et al., 2020, 2023).
2.10 Subcellular localization and yeast overexpression assay of ClTALE3 protein
To determine the subcellular localization of ClTALE proteins, the coding sequence (CDS) of ClTALE3 was PCR-amplified and cloned into the Nco I/Bgl II restriction sites of the overexpression vector pCAMBIA1302-EGFP, resulting in the recombinant construct 35S::ClTALE3-EGFP (Supplementary Table S1). The plasmid was transformed into Agrobacterium tumefaciens strain GV3101 via electroporation, and the transformed cells were cultured on solid LB medium supplemented with appropriate antibiotics at 30°C for 48 hours. A single colony of transformed A. tumefaciens was inoculated into liquid LB medium and grown overnight at 30°C with shaking at 200 rpm. The culture was centrifuged at 6,000 ×g for 5 min, and the pellet was resuspended in infiltration buffer (10 mM MgCl2, 10 mM MES-KOH, pH 5.6, 150 μM acetosyringone) to an OD600 of 0.2. The bacterial suspension was incubated at 28°C for 2–3 h with gentle agitation to induce virulence gene expression. Following activation, the Agrobacterium suspension was pressure-infiltrated into fully expanded leaves of 4-week-old Nicotiana benthamiana plants using a 1 mL syringe without a needle. Infiltrated plants were maintained in a growth chamber at 22°C under a 16 h light/8 h dark photoperiod for 48 h post-infiltration. The subcellular localization of the ClTALE3-EGFP fusion protein was visualized using a laser scanning confocal microscope (Olympus FV3000). The nuclear marker Mcherry was co-expressed to delineate the nuclear compartment. Images were acquired using a 60× oil immersion objective and processed with FV10-ASW software.
The cDNA of ClTALE3 was subcloned into the yeast expression vector pYES2. The resulting plasmid, pYES2-ClTALE3, was used to transform the W303a strain using the lithium acetate method, with transformants selected on uracil-deficient (SD -Ura) medium. Yeast cells expressing the ClTALE3 protein were incubated for 3 days at 30°C. Subsequently, the cells were harvested, diluted to OD600 values of 0.1 and 0.01 with distilled water, and 5 µL of each dilution was spotted onto SD-Gal (2% galactose) solid medium containing 20% PEG. The plates were incubated at 30°C for 2–3 days before being photographed.
2.11 Statistical analysis
The statistical analyses were performed using SPSS software. Data are expressed as mean ± standard deviation (SD). Differences between groups were assessed by one-way ANOVA with Tukey’s honest significant difference (HSD) test for multiple comparisons. A p-value of less than 0.05 was considered statistically significant, and different lowercase letters are used to denote these differences in the figures.
3 Results
3.1 Genome-wide characterization of ClTALE genes
To identify TALE family genes in watermelon, we performed a BLASTP search of the watermelon genome using known Arabidopsis thaliana TALE protein sequences as queries. The resulting candidate sequences were further validated using the HMMER software to confirm the presence of conserved TALE domains. After removing redundant and incomplete sequences, a non-redundant set of 22 genes was identified and designated as ClTALE1 to ClTALE22 based on their chromosomal locations (or: accession numbers), as detailed in Table 1.

Table 1. Physiochemical properties of ClTALE proteins.
3.2 Chromosomal localization of ClTALE genes
The 22 ClTALE genes were distributed across 10 of the 11 watermelon chromosomes (Figure 1). Chromosomes 1, 2, and 8 contained the highest number of genes, with four ClTALE genes each. This was followed by chromosome 10, which contained two genes (ClTALE20 and ClTALE21). The remaining genes were singly located on chromosomes 3, 4, 9, and 11.
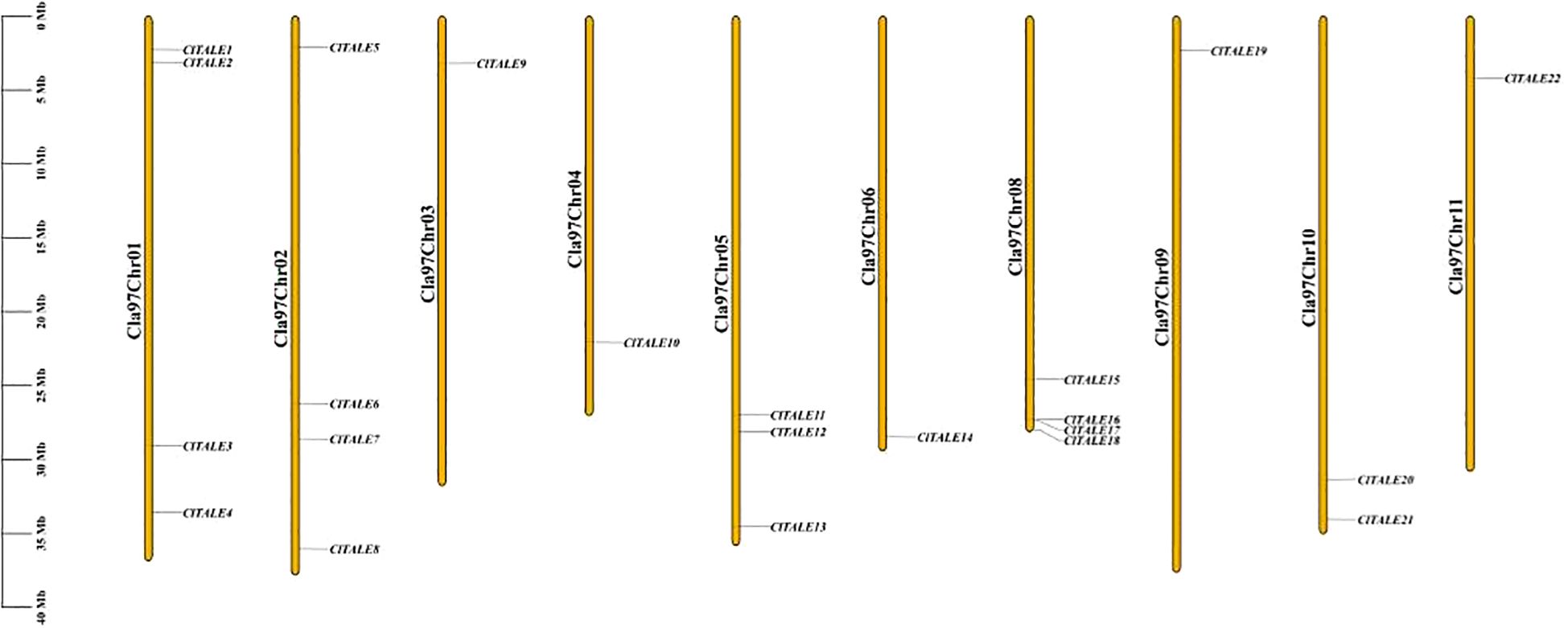
Figure 1. Chromosomal localization of ClTALE genes. The chromosomal number denoted at the tip of each chromosome. The schematic representation was created via TBtools-II (Version 1.098765).
3.3 Evolutionary and conserved domain analysis of TALE proteins
The TALE protein sequences from watermelon, Arabidopsis, wax gourd, and cucumber were identified and aligned using MEGA 6.0. The resulting phylogenetic analysis classified the proteins into distinct subfamilies (Figure 2A). In the KNOX group, the STM subfamily was the largest, followed by KNOX-II. The BELL group was subdivided into five subfamilies (BELL-I to BELL-V), with BELL-I and BELL-V containing the highest number of genes and the others (BELL-II, -III, and -IV) exhibiting a moderate representation. We also analyzed the conserved domains of these proteins (Figure 2B). KNAT family members were found to contain KNOX1, KNOX2, ELK, and HOX domains, whereas BEL family members featured POX and HOX domains (Figure 2B, C).
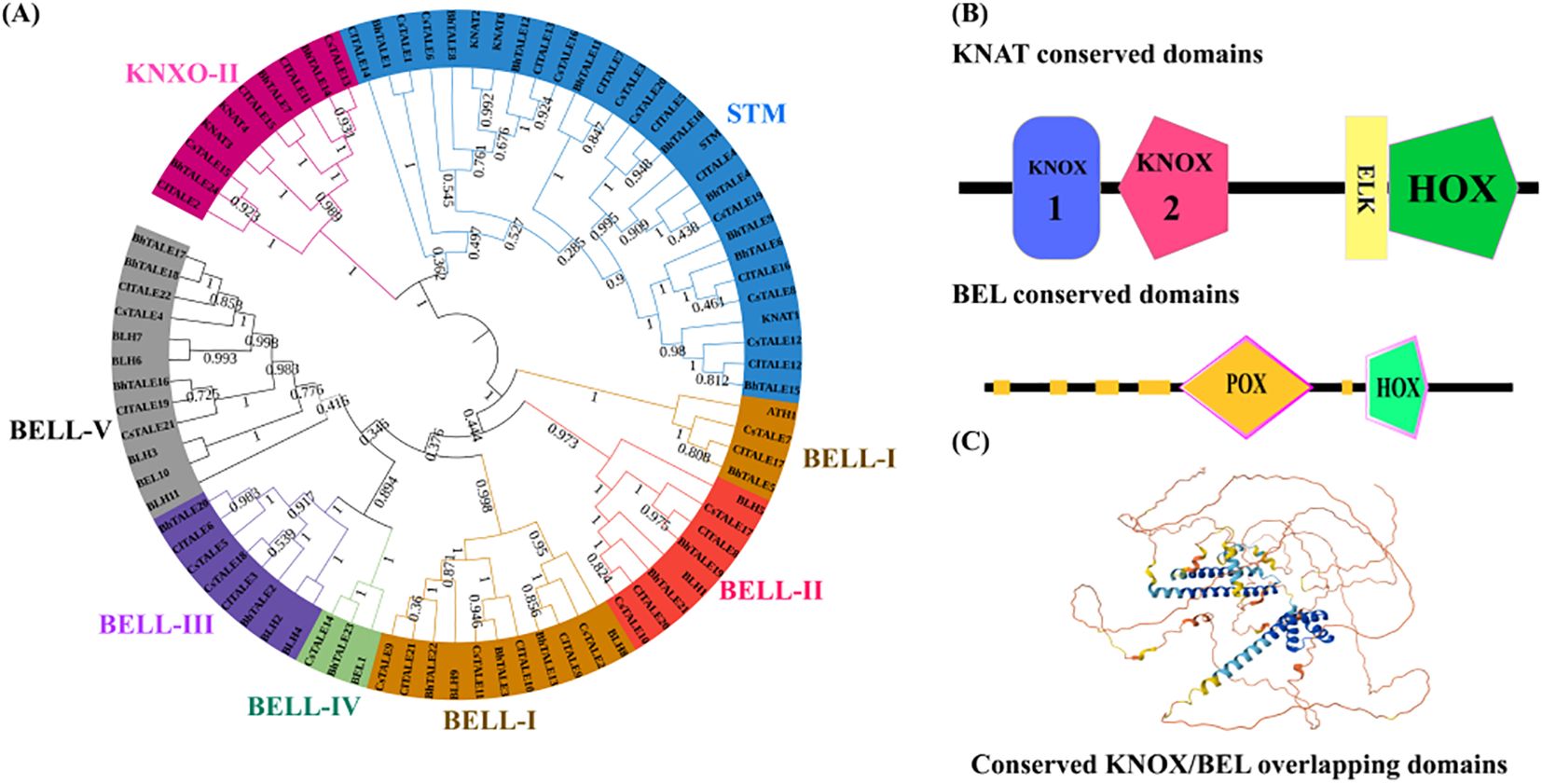
Figure 2. Phylogenetic and structural analysis of the ClTALE gene family. (A) Phylogenetic tree of ClTALE proteins constructed using the Maximum Likelihood method in MEGA 6.0 based on full-length amino acid sequences. Bootstrap values from 1,000 replicates are shown at key nodes. The TALE superfamily is divided into two major classes: the KNOX family (further subdivided into KNOX I and KNOX II) and the BELL family (subdivided into BELL I-V). The tree was visualized and annotated using the Interactive Tree of Life (iTOL) platform. (B) Conserved protein domain architecture of representative ClTALE members. Key domains including KNOX1, KNOX2, ELK, Homeobox (HOX), and POX are indicated by distinct colored boxes. (C) Representative three-dimensional (3D) protein structure model of a conserved region, highlighting the spatial overlap and arrangement of the KNOX and BEL domains. The model was generated using SWISS-MODEL.
3.4 Gene structure and conserved motif analysis of ClTALE
Gene structure analysis provides key insights into evolutionary relationships. Using genomic and coding sequence (CDS) data, we mapped the structure of the ClTALE genes. The analysis revealed diverse architectures, with most genes containing between 4 and 5 exons and 2 to 3 introns (Figure 3A). This variation in intron-exon organization reflects the structural divergence within the gene family.
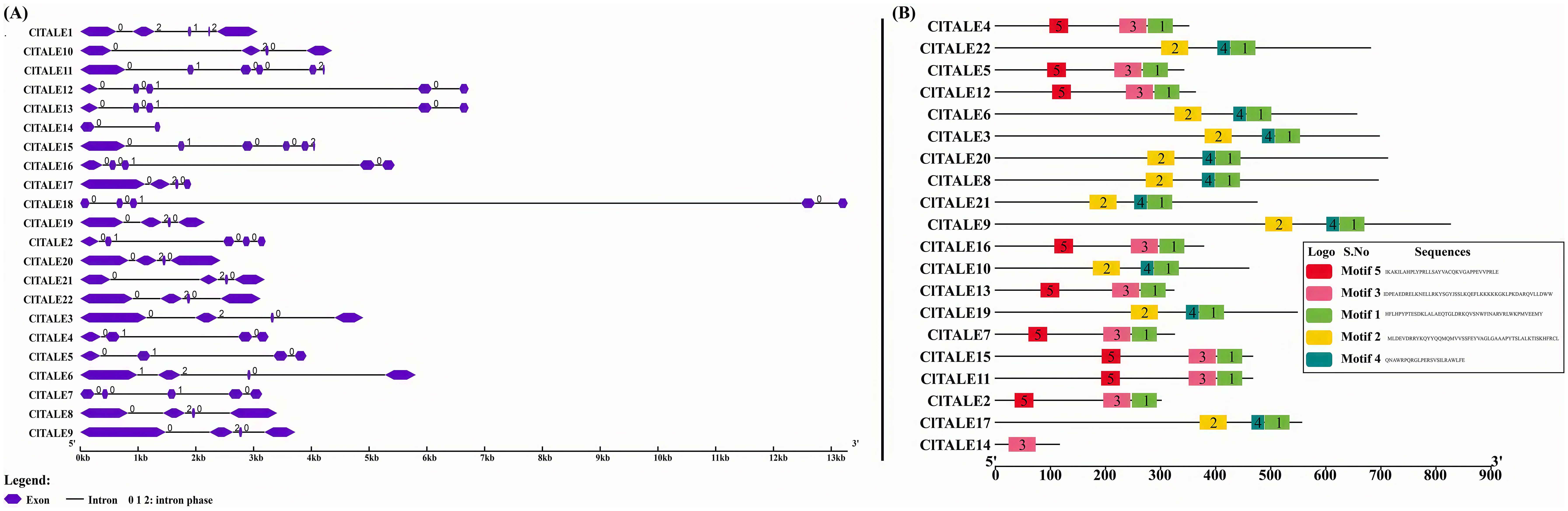
Figure 3. (A) Diagram depicting the structure of a gene. The gene display structure served as the basis for the gene structure analysis. The blue boxes representing exons whereas the grey lines represent introns. (B) Conserved motif analysis of ClTALE proteins in watermelon genome database.
To obtain a comprehensive understanding of the functional diversity of ClTALE genes, we utilized the MEME web server (http://meme-suite.org/tools/meme) to project conserved motifs in ClTALE proteins (Figure 3B). Five unique motifs were recognized in the ClTALE proteins. Six members of the ClTALE gene family (ClTALE1, ClTALE8, ClTALE12, ClTALE16, ClTALE19, ClTALE20) had the greatest abundance of KNOX1, KNOX2, ELK, Homeobox_KN, and POX Superfamily motifs. Furthermore, only ClTALE proteins, namely ClTALE3, ClTALE13, and ClTALE15, have KNOX1, KNOX2, and Homeobox_KN domains, while ClTALE6 features ELK and Homeobox_KN domains. The highest concentration of ClTALE proteins was detected inside the Homeobox_KN and POX Superfamily domains (Figure 3B).
3.5 Interactive protein analysis
Protein-protein interaction (PPI) is an ideal way to understand the function of our target protein in concert with another. Here, we used the String online server to predict the target interactive partners of our reference protein. We used the ClTALE2 as a reference and identified a cluster of important interactive partners such as C17SL2, ClGATA11, and ClTBL37. Other key interactive proteins, such as ClKNAT1/2/5 and ClKNATM, were also identified (Figure 4A). Further, we performed the enrichment analysis of reference protein ClTALE1 and its interactive partners (Figure 4B). The enrichment analysis suggested that these proteins are predominantly involved in transcriptional regulation of target genes.
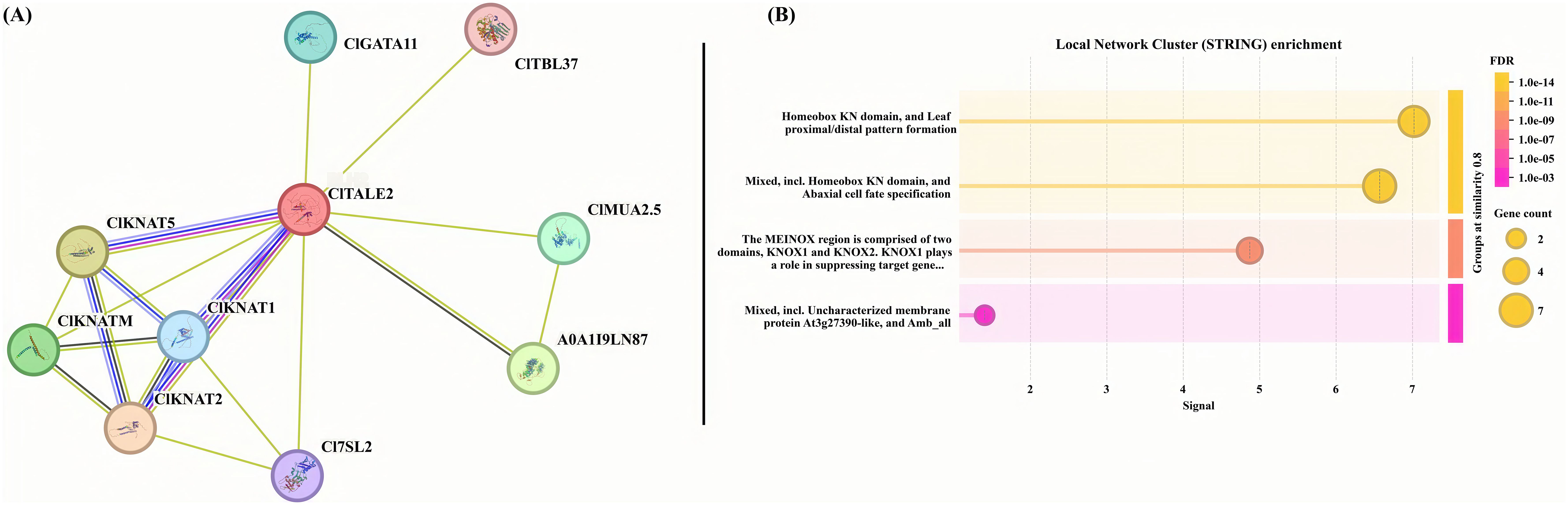
Figure 4. (A) The predictive functional associates of ClTALE proteins. The ClTALE2 served as a reference in the string online tool https://cn.string-db.org/ functional predictive analysis. (B) Local cluster analysis of ClTALE proteins together with their interactive partners.
3.6 Gene ontology analysis and Cis-acting elements of ClTALE genes in watermelon
Gene ontology (GO) analysis is an ideal way to predict the function of particular genes in biological, molecular, and cellular processes. Herein, we examined the protein sequences of ClTALE genes for possible GO terms to further understand their role in watermelon growth (Figure 5A). For instance, the dominant GO terms were related to floral induction, stimulus to hormones, and biotic stress regulation (Figure 5A). We also annotated GO terms associated with molecular function, including regulation of transcription.
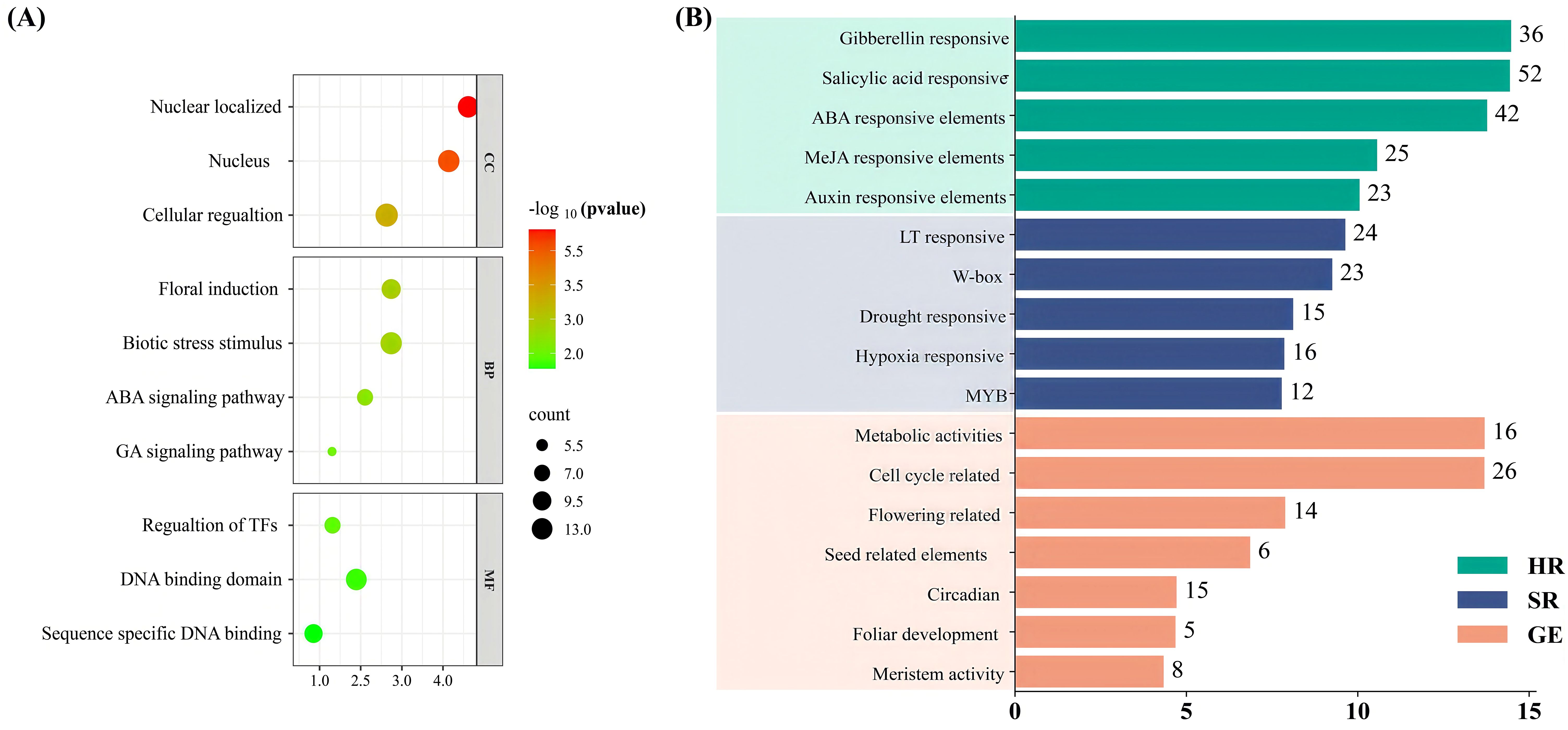
Figure 5. (A) ClTALE gene GO enrichment analysis in watermelon. The data depicted biological processes, molecular activities, cellular components, and their respective localization proportions. (B) ClTALE genes’ cis-acting components in watermelon. The data depicted hormone responsive (HR), stress response (stress responsive), and growth enriched (GE).
To investigate the probable regulatory mechanisms of expression for ClTALE family members, we found the cis-acting elements associated with each member (Figure 5B). The selected cis-acting elements pertain to plant hormones, plant development, stress responses, and light responses for investigation. Initially, the 2,000-bp promoter sequence was analyzed, revealing stress-related elements such as LTR, MBS, ARE, and other components, along with transcriptional regulatory elements including MYB, W-box, and MYB-also. Furthermore, we identified and confirmed other components associated with plant hormones, including P-box, TGA-element, ABRE, and additional elements. The predominant motif is the cis-acting element associated with ABA reactivity, comprising 30% of the analyzed hormone response motifs. The methyl jasmonate (MeJA) reactivity-related cis-acting components of the TGACG motif made up about 13%. Additionally, we discovered that the TCA element in response to Salicylic acid (SA) occurred 29 times and accounted for 10% of the 19 ClTALE gene promoters. Drought stress was associated with the MBS factor, accounting for 12% (Figure 5B). These results imply that these hormones and drought stress may influence ClTALE gene transcription.
3.7 Synteny analysis of ClTALE genes
To investigate the potential functions of ClTALE genes, syntenic relationships among watermelon, cucumber, and Arabidopsis genomes were analyzed. As shown in Figure 6A, approximately 70% of ClTALE genes exhibited synteny with Arabidopsis, while about 50% showed syntenic relationships with cucumber. Likewise, it was shown that watermelon (~65%) and cucumber (~75%) have strong syntenic connections (Figure 6B). These broad gene-level synteny associations can show the watermelon chromosomes’ substantial rearrangement events and close evolutionary linkages throughout genome evolution.
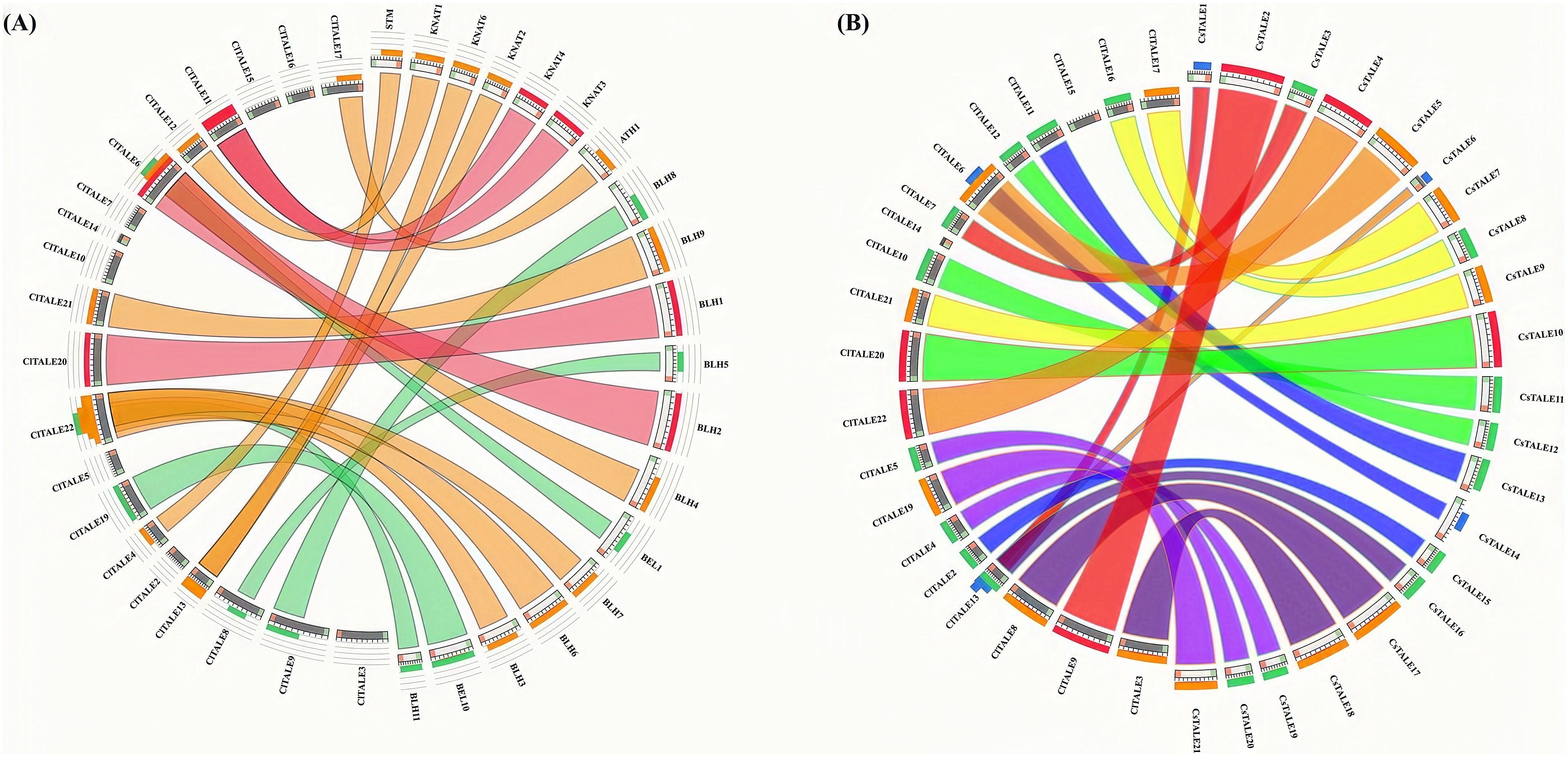
Figure 6. Synteny study of TALE genes between (A) watermelon and Arabidopsis and (B) watermelon and cucumber. The chromosomes of watermelon, Arabidopsis, and cucumber are organized in a circular formation. Colored lines denote syntenic occurrences of TALE genes.
3.8 Expression analysis of ClTALE genes under PEG treatment
The expression of ClTALE genes was investigated in watermelon seedlings subjected to drought (PEG) stress. We performed the expression of all 22 ClTALE genes, the expression of ClTALE2 reached its maximum of 4-fold at 12h compared to that of control (0h) (Figure 7). The ClTALE3 following drought stress increased sharply at 6 and 18h. Compared to control (0h), higher expression of ClTALE3 was also recorded at other timepoints. The ClTALE4/5/6 followed a similar expression trend and decreased significantly following drought stress treatment. The ClTALE8/11/20 all triggered after drought stress and reached a maximum of 4, 13, and 22 folds, respectively (Figure 7).
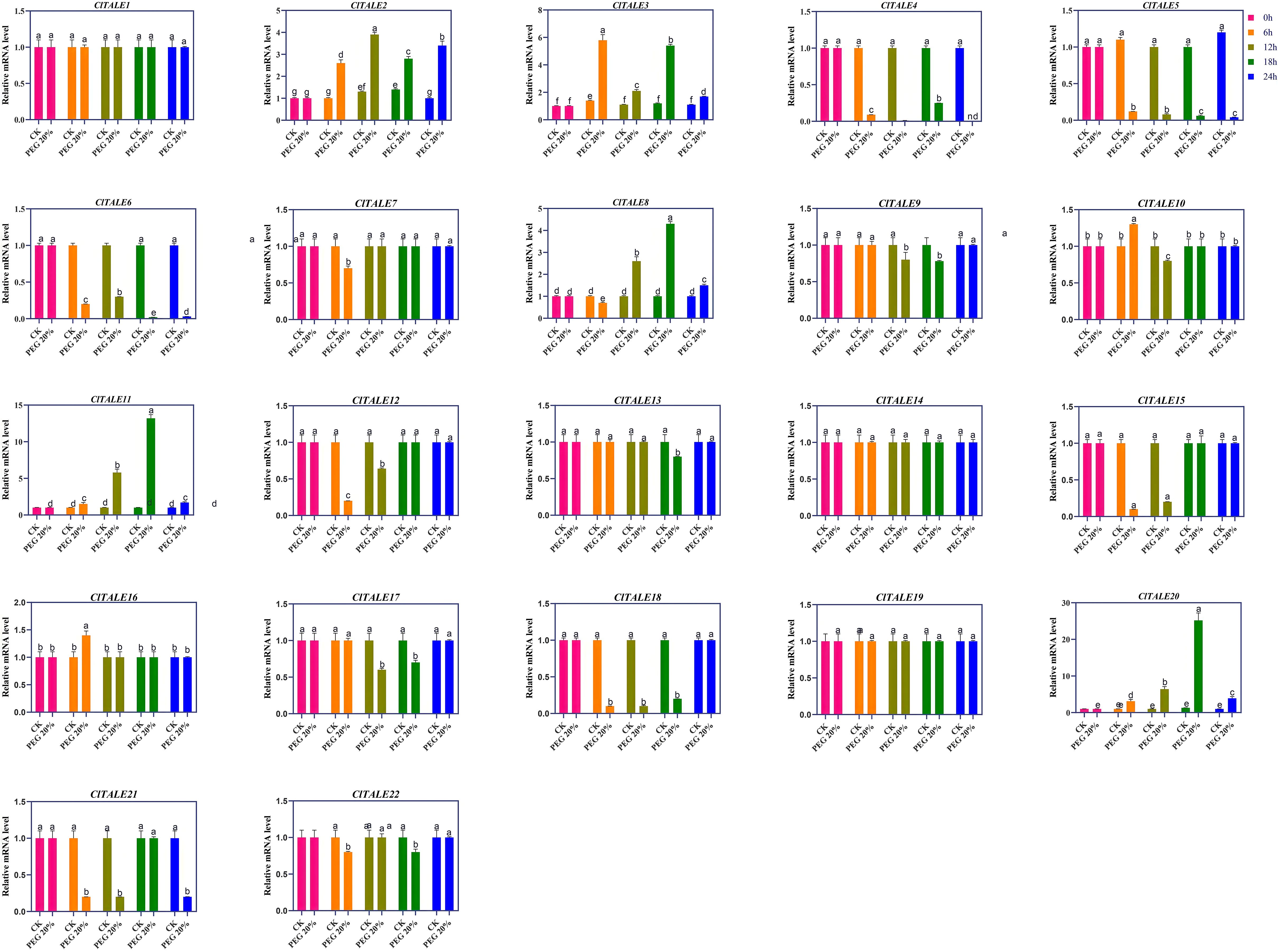
Figure 7. Expression profiles of selected ClTALE genes under drought stress (simulated by PEG treatment). Expression levels were analyzed at 0, 6, 12, 18, and 24 hours post-treatment.The CK represents the normal plants without PEG treatment. The Data are shown as mean ± SEM (n = 4). Different lowercase letters above the bars indicate statistically significant differences (p < 0.05) as determined by one-way ANOVA followed by Tukey’s HSD test.
3.9 Expression analysis under potassium, melatonin and cold stress
The expression analysis of ClTALE was further analyzed under low potassium (LK) and sufficient potassium conditions. Additionally, the melatonin (MT) hormone was sprayed on watermelon seedlings subjected to cold stress (CT). We examined the tendency of the ClTALE genes under MT and MT+CT.
Given that melatonin (MT) is a master regulator of plant stress responses, profiling the expression of ClTALE genes under MT treatment provides critical insight into their potential function in watermelon stress adaptation. Our analysis revealed that the expression of ClTALE1 was significantly induced by CT, MT, and their combined treatment (MT+CT). A similar upregulation pattern was observed for ClTALE15, ClTALE16, and ClTALE18. In contrast, ClTALE22 was constitutively upregulated across all conditions, including the control (CK), MT, CT, and MT+CT. Conversely, several other genes, including ClTALE7, ClTALE8, ClTALE9, ClTALE14, and ClTALE20, exhibited a marked downregulation under both CK and MT conditions (Figure 8A). These distinct expression patterns suggest specific and divergent roles for ClTALE family members in mediating melatonin- and cold-triggered stress signaling pathways.
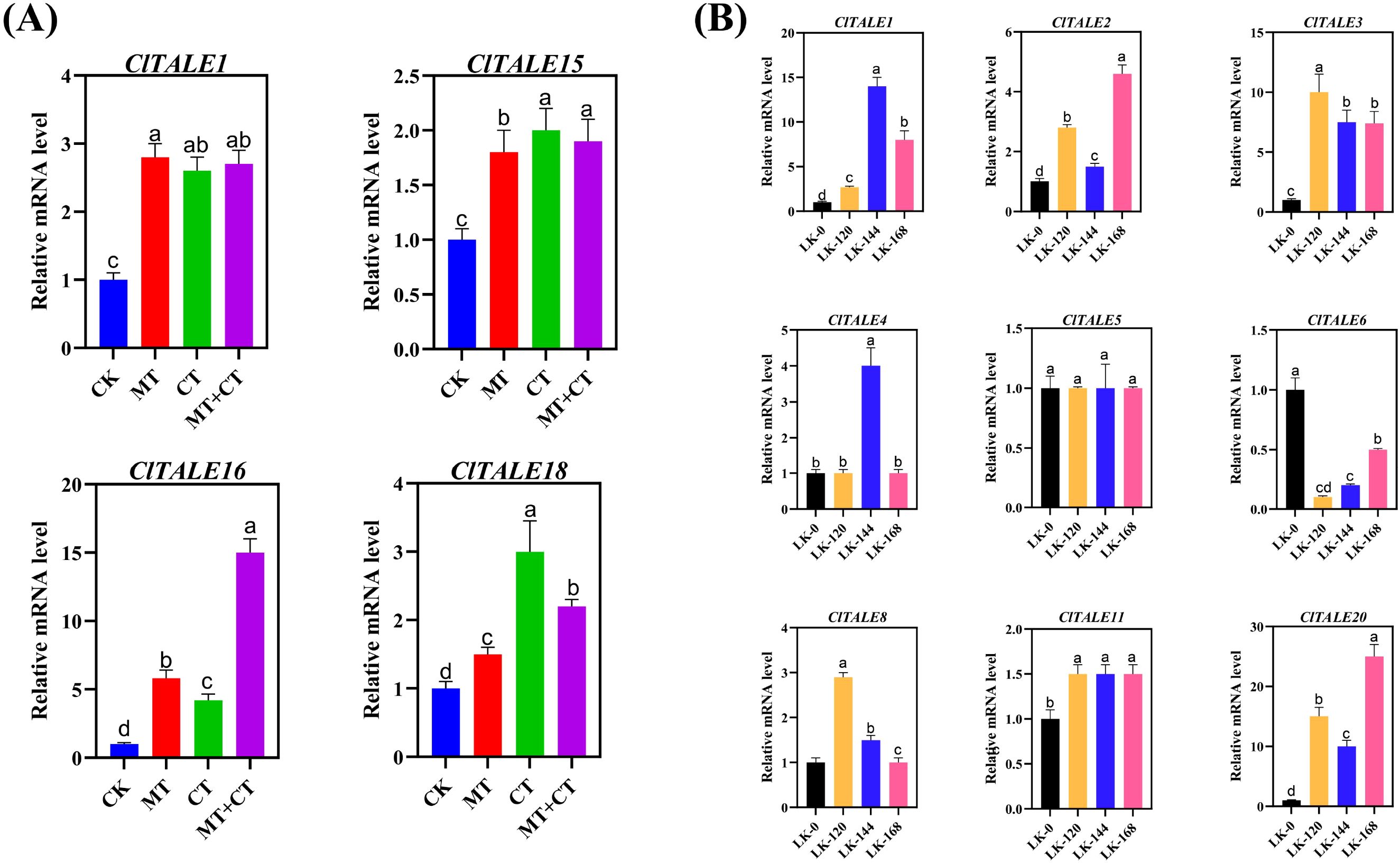
Figure 8. (A) Expression analysis of ClTALE genes under abiotic stress. (A) Heatmap of ClTALE gene expression profiles in response to co-treatment with melatonin (MT) and cold (CT). The heatmap was generated by hierarchical clustering of log2-transformed FPKM values using TBtools. red indicates high relative expression; blue indicates low relative expression. (B) Time-course qRT-PCR analysis of selected ClTALE genes under low-potassium (LK) stress. Samples were collected at 0, 120, 144, and 168 hours after treatment initiation. Gene expression levels were normalized to the internal reference gene and are presented relative to the 0-hour control using the 2^–ΔΔCT method. Data are shown as mean ± SD from four biological replicates. Different lowercase letters above the bars indicate statistically significant differences (p < 0.05) as determined by one-way ANOVA followed by Tukey’s HSD test.
To further investigate the role of the ClTALE gene family under low potassium (LK) stress, we conducted a time-course qRT-PCR analysis (Figure 8B). The qRT-PCR results validated the microarray data, confirming a consistent expression trend. Specifically, ClTALE1 expression was significantly induced at 120, 144, and 168 hours post-treatment. A similar upregulation was observed for ClTALE2, ClTALE3, ClTALE8, ClTALE11, and ClTALE20. In contrast, ClTALE4 exhibited a transient expression pattern, with a significant increase only at the 144-hour time point.
3.10 Subcellular localization of ClTALE3 protein
To experimentally validate the predicted nuclear function of ClTALE3, we investigated its subcellular localization through transient expression in Nicotiana benthamiana. We co-infiltrated leaves with Agrobacterium strain GV3101 carrying either pCAMBIA1302-EGFP-ClTALE3 or an mCherry-tagged nuclear marker. As shown in Figure 9, the EGFP fluorescence from ClTALE3 exclusively overlapped with the nuclear mCherry signal, confirming its nuclear localization.
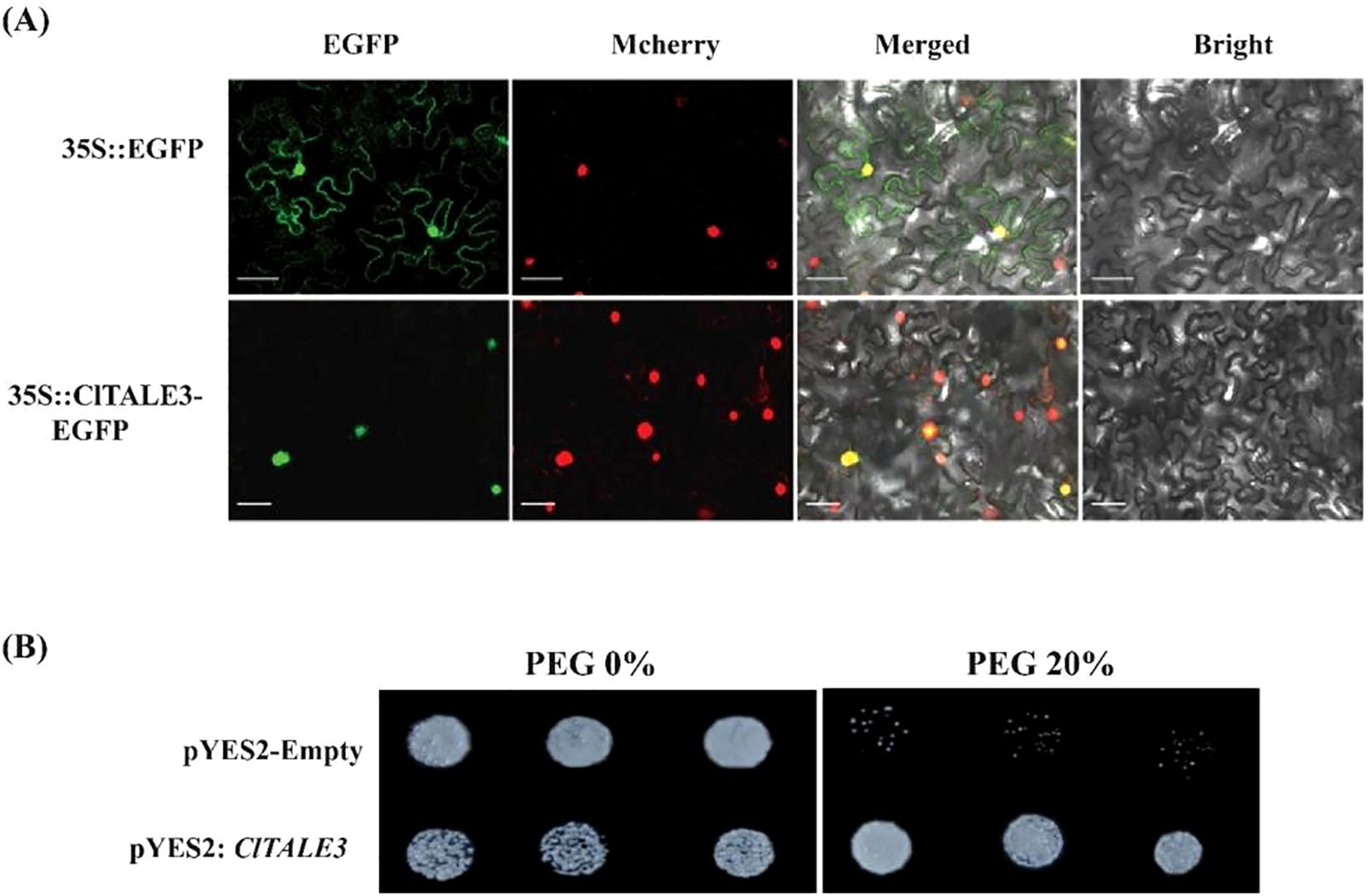
Figure 9. (A) Subcellular localization of ClTALE3 in Nicotiana tabacum leaf epidermal cells. EGFP, green fluorescent reporter protein; Mcherry, nuclear marker; BF, brightfield imaging; Merged, an integrated visualization incorporating EGFP, chloroplast auto-luminescence, and brightfield signals. Separate EGFP driven by the 35S promoter was used as a control. Scale bar= 50 μm. (B) Heterologous overexpression of ClTALE3 in yeast cell displayed resistance to PEG stress.
A heterologous yeast overexpression assay was used to assess the function of ClTALE3 in drought stress response. Compared to the empty vector control (pYES2-empty), which showed progressively inhibited growth on PEG-supplemented medium (20%), yeast expressing ClTALE3 (pYES2-ClTALE3) maintained normal growth (Figure 9B). This indicates that ClTALE3 enhances tolerance to drought-simulating osmotic stress.
4 Discussion
4.1 ClTALE genes are widely distributed in watermelon genome
The early 20th century saw a surge in research on TALE (Jia et al., 2023b; Hussain et al., 2024), a eukaryotic gene family that is widely distributed. This work proposes a thorough bioinformatic investigation of 22 ClTALE genes extracted from the watermelon genome database. They are all expected to be dispersed randomly among the seven chromosomes (Figure 1) and to be located in the nucleus (Table 1). To look into the evolutionary links of the proteins that the 22 genes encode, we constructed an unrooted phylogenetic tree. Unlike previous studies (Han et al., 2022; Wang et al., 2022), we classified these proteins into seven separate categories (Figure 2A). While the BELL genes are further divided into five categories, the KNOX genes have been separated into two groups (Figure 2A). Our classification results are also supported by the ML-phylogenetic tree built with the best model and the annotations of the SMART database (Ullah et al., 2022; Fei et al., 2024). These gene are widely documented for their role in stress response. For instance, the induction of PagKNAT2/6b, a gene from the STM clade, under drought conditions was linked to a marked improvement in drought resistance. Transgenic poplars overexpressing this gene exhibited a stunted growth architecture, characterized by shorter internodes, smaller leaves, and short or absent petioles, yet demonstrated greater survival under both acute and prolonged water deficit (Song et al., 2021). In Arabidopsis thaliana, ATH1 from BELL-I clade plays a key role in regulating light-induced gene expression and photomorphogenesis. Its expression is positively regulated by light and negatively regulated by COP1 and DET1, as evidenced by the increased ATH1 levels in dark-grown cop1 det1 double mutants. Genetic analysis further positions ATH1 as a critical downstream component of the COP1 and DET1 signaling pathway (Niu and Fu, 2022). Evidence from multiple species underscores the importance of BLH genes in abiotic stress regulation. In Arabidopsis, BLH1 negatively regulates salt tolerance during early development, as shown by the increased tolerance of a blh1 mutant and the hypersensitivity of an overexpressor (Niu and Fu, 2022). Another member, BLH8, is specifically required for leaf ion tolerance, with mutants showing chlorosis under Na+/K+ stress. The functional conservation of these genes is highlighted by rice OsBIHD1, which, when overexpressed in tobacco, confers increased sensitivity to salt and oxidative stress, the latter inducing cell membrane damage (Niu and Fu, 2022). Certain domains or combinations of domains stand in for each class. An intron is present in every ClTALE gene, according to gene structural research. All members of the same family share a high degree of genomic conservation. Phylogenetic tree members that are closely related also have fairly comparable exon lengths (Figure 3B). Protein motif analysis and annotation revealed that members of the same class have identical protein motifs, which is in line with previous studies on the poplar TALE family (Zhao et al., 2019).
Gene transcription is controlled by the cis-elements’ selective binding of transcription factors at the gene promoter region. Numerous cis-elements linked to hormonal responses and abiotic stress were identified in the ClTALE promoter sequence, including methyl jasmonate, abscisic acid, and gibberellin (Figure 5B), corroborating other studies (Jia et al., 2023a; Wang et al., 2020). The findings indicated a conservative component within the ClTALE gene promoter. ABRE has been connected in studies to high salt stress, drought, and ABA induction in plants (Zareen et al., 2024). Additionally, ARE, MBS, and LTR are all components that are associated with stress. Involvement of the ClTALE gene family in watermelon abiotic stress was found to be significant. Predictions of gene functions and analysis of protein-protein networks point to the ClTALE family as a critical regulator of ovule and inflorescence establishment. Consistent with the earlier research, ClTALE2 interacts with several proteins found in floral organs, as shown by protein-protein network analysis (Kim et al., 2022).
4.2 ClTALE genes are crucial abiotic stresses regulators
Several plant species have shown that members of the TALE gene family can react to environmental stresses (Jia et al., 2023b). The transcriptome analysis and cloning of eleven KNOX genes in Camellia japonica L. showed that these genes significantly affect drought and salinity tolerance (Dai et al., 2023). Hormonal treatments and stressful environmental factors, such as drought, ABA, MeJA, and SA, can affect the 19 KNOX genes in Dendrobium huoshanense, according to studies (Li et al., 2023). The GA pathway gene PagGA20ox1 may be influenced by Poplar’s PagKNat2/6b, which in turn mediates drought responses (Song et al., 2021). The promoters of the TALE gene family in soybean contain stress-responsive cis-elements, suggesting that salt and drought may cause alterations in the expression of GmTALE (Wang et al., 2021). There is evidence that certain TALE genes have a role in stress adaptation in cotton, as they are upregulated in response to abiotic stimuli (Sun et al., 2023). In Arabidopsis, the homologous gene KNAT7 is linked to the ZmTALE1, ZmTALE37, and ZmTALE38 genes in maize (Razzaq et al., 2020). Reportedly, this gene has the ability to control lignin synthesis in Arabidopsis, which in turn influences the development of vascular tissue (Qin et al., 2020). According to research, plants’ ability to withstand drought and cold is greatly influenced by the process of lignin production (Zhang Q, et al., 2023; Yadav and Chattopadhyay, 2023). In addition, the ZmTALE24 gene, which is thought to be similar to Arabidopsis’s KNAT1, controls gibberellin activity and, by extension, vascular tissue development through its interactions with DELLA, a negative component of the gibberellin signaling pathway (Qian et al., 2024). The leaves of Toona sinensis exhibited increased salt sensitivity and enhanced tolerance to osmotic stress following the transient expression of the TsBLH4 and TsKNOX6 genes (Chen et al., 2025). The repression of the GhKNOX4-AGh and GhKNOX22-D genes in cotton seedlings exposed to salt and drought had a notable impact on their growth and development (Sun et al., 2023). By controlling stomata opening and oxidative stressors, GhKNOX4-A and GhKNOX22-D might play a role in drought response (Sun et al., 2023). Concurrently with the reported studies, we observed varied expression trends of ClTALE genes following drought stress (Figure 7). For instance, the ClTALE2/3/8/11/20 sharply induced following drought stress. On the other hand, the ClTALE4/5/6 declined in drought-stressed watermelon seedlings comparing to control. Low temperature constitutes a significant abiotic stress that adversely affects plant development, growth, and eventually, agricultural production (Liu J, et al., 2025; Guan et al., 2025; Zhang et al., 2025). According to the study, RNAi-silenced lines of CmBLH2 showed a reduced ability to scavenge reactive oxygen species, whereas its overexpression increased the antioxidant system’s activity and decreased cellular damage under cold stress (Liu P, et al., 2025). While the molecular narrative of TALE genes is rich and growing, their role in the plant’s response to K stress is undermined. To the best of our knowledge, this narrative is unexplored by any study to date. In this context, our discovery of ClTALE3 is particularly intriguing. Heterologous overexpression of ClTALE3 in yeast cell displayed resistance to PEG stress (Figure 9B). This gene emerged as a dynamic respondent, its expression dramatically elevated under a gauntlet of abiotic stresses including drought, low K, and cold. Such versatile responsiveness marks ClTALE3 as a compelling candidate for a central character in the stress tolerance network, whose role begs for deeper investigation through transgenic techniques. When we look upstream, the promoter of ClTALE is enriched with a high number of ABA-responsive cis-elements. This is a finely tuned regulatory circuit, poised to direct the plant’s response to water scarcity. This molecular potential resonates deeply with a pressing real-world challenge. In the farmlands of Xinjiang, P.R. China, drought is not just a scientific concept but a major threat to agriculture. Understanding how ClTALE3 fine-tunes watermelon’s defense could therefore provide the script for building resilience where it is most urgently needed (Yang et al., 2024). Xinjiang is a quintessential arid and semiarid region, serving as the principal economic zone of the Silk Road in China. Between 1961 and 2000, it encountered 26 severe droughts. The rise in temperatures and decline in precipitation have exacerbated local evapotranspiration (ET), intensified drought conditions and affecting vegetation (Yang et al., 2024; Han et al., 2023). Compounding the challenge of drought, Xinjiang’s climate is also marked by unpredictable cold spells. Our findings on ClTALE gene expression provide fresh insights into the molecular mechanisms that may underpin watermelon adaptation to this combined abiotic stress. The pronounced upregulation of specific ClTALE members in response to both drought and cold positions them as key candidates for developing climate-resilient crops. Consequently, these genes serve as excellent molecular markers for precision breeding programs aimed at generating new watermelon lines capable of thriving in the arid and thermally volatile conditions of Xinjiang. Finally, the yeast heterologous overexpression assay is an excellent initial, reductionist tool for characterizing gene function and provide platform for in planta research.
5 Conclusion
Our analysis identifies 22 ClTALE genes in the watermelon genome, which we have phylogenetically divided into seven groups. The presence of numerous stress-responsive cis-elements suggests these genes are central to balancing growth and immunity, a hypothesis supported by their expression patterns under stress. Specifically, ClTALE genes are implicated in the response to low potassium (LK) and the acquisition of MT+CT. Among them, ClTALE3 demonstrates a striking upregulation across three major abiotic stresses: drought, LK, and MT+CT. The heterologous overexpression assay of ClTALE3 augmented yeast growth in PEG 20% media. This makes it an exceptionally promising target for in-depth functional characterization and a strategic molecular marker for developing watermelon lines with broad-spectrum abiotic stress resistance.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
ZQ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft. JD: Conceptualization, Data curation, Formal analysis, Methodology, Software, Validation, Visualization, Writing – original draft. LC: Data curation, Investigation, Software, Writing – review & editing. LZ: Methodology, Software, Writing – review & editing. LH: Formal analysis, Software, Writing – review & editing. HW: Data curation, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was supported by the Key research and development project of Xinjiang Uygur Autonomous Region (2023B02017-2); ‘Tianchi Talent’ Project of XinJiang province; Natural Science Foundation of Xinjiang Uygur Autonomous Region (2023D01B29); National Natural Science Foundation of China (32302560); China Postdoctoral Science Foundation (2023MD734232); the Earmarked Fund for XJARS (XJARS-06).
Acknowledgments
We are thankful to Rahat Sharif (South China Agriculture University, Guangzhou, P.R. China) for revising the language of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2025.1711607/full#supplementary-material
References
Ahmad, S., Ali, S., Shah, A. Z., Khan, A., and Faria, S. (2023a). Chalcone synthase (CHS) family genes regulate the growth and response of cucumber (Cucumis sativus L.) to Botrytis cinerea and abiotic stresses. Plant Stress. 8, 100159. doi: 10.1016/j.stress.2023.100159
Ahmad, S., Chen, Y., Shah, A. Z., Wang, H., Xi, C., Zhu, H., et al. (2022). The Homeodomain-Leucine Zipper Genes Family Regulates the Jinggangmycin Mediated Immune Response of Oryza sativa to Nilaparvata lugens, and Laodelphax striatellus. Bioengineering. 9, 398. doi: 10.3390/bioengineering9080398
Ahmad, S., Jeridi, M., Siddiqui, S., Ali, S., and Shah, A. Z. (2023b). Genome-wide identification, characterization, and expression analysis of the Chalcone Synthase gene family in Oryza sativa under Abiotic Stresses. Plant Stress. 9, 100201. doi: 10.1016/j.stress.2023.100201
Ahmad, S., Khan, K., Saleh, I. A., Okla, M. K., Alaraidh, I. A., AbdElgawad, H., et al. (2024). TALE gene family: identification, evolutionary and expression analysis under various exogenous hormones and waterlogging stress in Cucumis sativus L. BMC Plant Biol. 24, 564. doi: 10.1186/s12870-024-05274-3
Bürglin, T. R. (1997). Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res. 25, 4173–4180. doi: 10.1093/nar/25.21.4173
Chang, J., Guo, Y., Li, J., Su, Z., Wang, C., Zhang, R., et al. (2021). Positive interaction between H2O2 and Ca2+ mediates melatonin-induced CBFpathway and cold tolerance in watermelon (Citrullus lanatus L.). Antioxidants. (Basel). 10, 1457. doi: 10.3390/antiox10091457
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools, an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, S., Jia, Y., Yang, Y., Liu, H., Chen, H., Liu, J., et al. (2025). Genome-wide analysis of the TsBLH gene family reveals TsBLH4 involved the regulation of abiotic stresses by interacting with KNOX6 in Toona sinensis. Plant Stress. 15, 100721. doi: 10.1016/j.stress.2024.100721
Chen, C., Wu, Y., Li, J., Wang, X., Zeng, Z., Xu, J., et al. (2023). TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742. doi: 10.1016/j.molp.2023.09.010
Dai, H., Zheng, S., Zhang, C., Huang, R., Yuan, L., and Tong, H. (2023). Identification and expression analysis of the KNOX genes during organogenesis and stress responseness in Camellia sinensis (L.) O. Kuntze. Mol. Genet. Genomics 298, 1559–1578. doi: 10.1007/s00438-023-02075-5
Fan, M., Huang, Y., Zhong, Y., Kong, Q., Xie, J., Niu, M., et al. (2014). Comparative transcriptome profiling of potassium starvation responsiveness in two contrasting watermelon genotypes. Planta. 239, 397–410. doi: 10.1007/s00425-013-1976-z
Fei, L., Liu, J., Liao, Y., Sharif, R., Liu, F., Lei, J., et al. (2024). The CaABCG14 transporter gene regulates the capsaicin accumulation in Pepper septum. Int. J. Biol. Macromol. 280, 136122. doi: 10.1016/j.ijbiomac.2024.136122
Gehring, W. J. (1987). Homeo boxes in the study of development. Science. 236, 1245–1252. doi: 10.1126/science.2884726
Guan, Y., Wu, H., Manda, T., Li, R., Lu, Y., Gao, M., et al. (2025). Evolution of MYC-type bHLH transcription factors in green plants and functional role of inducer of CBF expression 1b from Liriodendron chinense in enhancing cold tolerance. Int. J. Biol. Macromol. 320, 145986. doi: 10.1016/j.ijbiomac.2025.145986
Guo, C., Quan, S., Zhang, Z., Kang, C., Liu, J., and Niu, J. (2022). Genome-wide Identification, Characterization and Expression profile of TALE gene family in (Juglans regia L.). Sci. Hortic-Amsterdam. 297, 110945. doi: 10.1016/j.scienta.2022.110945
Han, W., Guan, J., Zheng, J., Liu, Y., Ju, X., Liu, L., et al. (2023). Probabilistic assessment of drought stress vulnerability in grasslands of Xinjiang, China. Front. Plant Sci. 14, 1143863. doi: 10.3389/fpls.2023.1143863
Han, Y., Zhang, L., Yan, L., Xiong, X., Wang, W., Zhang, X., et al. (2022). Genome-wide analysis of TALE superfamily in Triticum aestivum reveals TaKNOX11-A is involved in abiotic stress response. BMC Genomics 23, 89. doi: 10.1186/s12864-022-08324-y
He, J. B., Zhao, X. H., Du, P. Z., Zeng, W., Beahan, C. T., Wang, Y. Q., et al. (2018). KNAT7 positively regulates xylan biosynthesis by directly activating IRX9 expression in Arabidopsis. J. Integr. Plant Biol. 60, 514–528. doi: 10.1111/jipb.12638
Hussain, S., Chang, J., Li, J., Chen, X., Xie, D., and Zhang, B. (2024). Transcriptome wide identification and expression analysis revealed bhTALE gene family regulates wax gourd (Benincasa hispida) response to low calcium and magnesium stress. Horticulturae. 10, 1083. doi: 10.3390/horticulturae10101083
Iannelli, M. A., Nicolodi, C., Coraggio, I., Fabriani, M., Baldoni, E., and Frugis, G. (2023). A novel role of medicago truncatula KNAT3/4/5-like class 2 KNOX transcription factors in drought stress tolerance. Int. J. Mol. Sci. 24, 12668. doi: 10.3390/ijms241612668
Jia, P., Sharif, R., Li, Y., Sun, T., Li, S., Zhang, X., et al. (2023a). The BELL1-like homeobox gene MdBLH14 from apple controls flowering and plant height via repression of MdGA20ox3. Int. J. Biol. Macromol. 242, 124790. doi: 10.1016/j.ijbiomac.2023.124790
Jia, P., Wang, Y., Sharif, R., Dong, Q., Liu, Y., Luan, H., et al. (2023b). KNOTTED1-like homeobox (KNOX) transcription factors - Hubs in a plethora of networks: A review. Int. J. Biol. Macromol. 253, 126878. doi: 10.1016/j.ijbiomac.2023.126878
Kim, K., Lee, J., Kim, B., Shin, J., Kang, T., and Kim, W. (2022). GATA25, a novel regulator, accelerates the flowering time of Arabidopsis thaliana. Appl. Biol. Chem. 65, 28. doi: 10.1186/s13765-022-00698-7
Li, G., Manzoor, M. A., Wang, G., Chen, C., and Song, C. (2023). Comparative analysis of KNOX genes and their expression patterns under various treatments in Dendrobium huoshanense. Front. Plant Sci. 14, 1258533. doi: 10.3389/fpls.2023.1258533
Liu, J., Chen, C., Chen, L., Sharif, R., Meng, J., Gulzar, S., et al. (2025). The banana MaFLA27 confers cold tolerance partially through modulating cell wall remodeling. Int. J. Biol. Macromol. 290, 138748. doi: 10.1016/j.ijbiomac.2024.138748
Liu, P., Tang, J., Lei, Y., Zhang, L., Ye, J., Wang, C., et al. (2025). Construction of the KNOX-BELL interaction network and functional analysis of CmBLH2 under cold stress in Chrysanthemum morifolium. Int. J. Biol. Macromol. 293, 139365. doi: 10.1016/j.ijbiomac.2024.139365
Ma, Q., Wang, N., Hao, P., Sun, H., Wang, C., Ma, L., et al. (2019). Genome-wide identification and characterization of TALE superfamily genes in cotton reveals their functions in regulating secondary cell wall biosynthesis. BMC Plant Biol. 19, 432. doi: 10.1186/s12870-019-2026-1
Mahmoud, A., Qi, R., Chi, X., Liao, N., Malangisha, G. K., Ali, A., et al. (2023). Integrated bulk segregant analysis, fine mapping, and transcriptome revealed QTLs and candidate genes associated with drought adaptation in wild watermelon. Int. J. Mol. Sci. 25, 65. doi: 10.3390/ijms25010065
Niu, X. and Fu, D. (2022). The roles of BLH transcription factors in plant development and environmental response. Int. J. Mol. Sci. 23, 3731. doi: 10.3390/ijms23073731
Qian, B., Wang, Q., Zhang, C., Guo, J., Yu, Z., Han, J., et al. (2024). Exploring the roles of TALE gene family in maize drought stress responses. Agronomy. 14, 1267. doi: 10.3390/agronomy14061267
Qin, W., Yin, Q., Chen, J., Zhao, X., Yue, F., He, J., et al. (2020). The class II KNOX transcription factors KNAT3 and KNAT7 synergistically regulate monolignol biosynthesis in Arabidopsis. J. Exp. Bot. 71, 5469–5483. doi: 10.1093/jxb/eraa266
Razzaq, A., Ashraf, J., Malik, W., Shaban, M., Zhang, R., Liang, C., et al. (2020). In silico analyses of TALE transcription factors revealed its potential role for organ development and abiotic stress tolerance in cotton. Int. J. Agric. Biol. 23, 1083–1094. doi: 10.17957/IJAB/15.1389
Scofield, S., Dewitte, W., Nieuwland, J., and Murray, J. A. H. (2013). The Arabidopsis homeobox gene has cellular and meristem-organisational roles with differential requirements for cytokinin and CYCD3 activity. Plant J. 75, 53–66. doi: 10.1111/tpj.12198
Sharif, R., Su, L., Chen, X., and Qi, X. (2022a). Involvement of auxin in growth and stress response of cucumber. Veg Res. 2, 1–9. doi: 10.48130/VR-2022-0013
Sharif, R., Su, L., Chen, X., and Qi, X. (2022b). Hormonal interactions underlying parthenocarpic fruit formation in horticultural crops. Hortic. Res. 9, uhab024. doi: 10.1093/hr/uhab024
Sharif, R., Xie, C., Zhang, H., Arnao, M. B., Ali, M., Ali, Q., et al. (2018). Melatonin and its effects on plant systems. Molecules. 23, 2352. doi: 10.3390/molecules23092352
Song, X., Zhao, Y., Wang, J., and Lu, M. (2021). The transcription factor KNAT2/6b mediates changes in plant architecture in response to drought via down-regulating GA20ox1 in Populus alba× P. glandulosa. J. Exp. Bot. 72, 5625–5637. doi: 10.1093/jxb/erab201
Sun, R., Qin, T., Wall, S. B., Wang, Y., Guo, X., Sun, J., et al. (2023). Genome-wide identification of KNOX transcription factors in cotton and the role of GhKNOX4-A and GhKNOX22-D in response to salt and drought stress. Int. J. Biol. Macromol. 226, 1248–1260. doi: 10.1016/j.ijbiomac.2022.11.238
Tan, Y., Xiao, L., Zhao, J., Zhang, J., Ahmad, S., Xu, D., et al. (2023). Adenosine monophosphate-activated protein kinase (AMPK) phosphorylation is required for 20-hydroxyecdysone regulates ecdysis in apolygus lucorum. Int. J. Mol. Sci. 24, 8587. doi: 10.3390/ijms24108587
Ullah, U., Shalmani, A., Ilyas, M., Raza, A., Ahmad, S., Shah, A. Z., et al. (2022). BZR proteins: identification, evolutionary and expression analysis under various exogenous growth regulators in plants. Mol. Biol. Rep. 49, 12039–12053. doi: 10.1007/s11033-022-07814-2
Wang, L., Yang, X., Gao, Y., and Yang, S. (2021). Genome-wide identification and characterization of TALE superfamily genes in soybean (Glycine max L.). Int. J. Mol. Sci. 22, 4117. doi: 10.3390/ijms22084117
Wang, J., Zhao, P., Cheng, B., Zhang, Y., Shen, Y., Wang, X., et al. (2022). Identification of TALE transcription factor family and expression patterns related to fruit chloroplast development in tomato (Solanum lycopersicum L.). Int. J. Mol. Sci. 23, 4507. doi: 10.3390/ijms23094507
Wang, Y., Zhao, Y., Yan, M., Zhao, H., Zhang, X., and Yuan, Z. (2020). Genome-wide identification and expression analysis of TALE gene family in pomegranate (Punica granatum L.). Agronomy. 10, 829. doi: 10.3390/agronomy10060829
Yadav, S. and Chattopadhyay, D. (2023). Lignin: the building block of defense responses to stress in plants. J. Plant Growth Regul. 42, 6652–6666. doi: 10.1007/s00344-023-10926-z
Yan, W., Sharif, R., Sohail, H., Zhu, Y., Chen, X., and Xu, X. (2024). Surviving a double-edged sword: response of horticultural crops to multiple abiotic stressors. Int. J. Mol. Sci. 25, 5199. doi: 10.3390/ijms25105199
Yang, J., Li, Y., Zhou, L., Zhang, Z., Zhou, H., and Wu, J. (2024). Effects of temperature and precipitation on drought trends in Xinjiang, China. J. Arid Land. 16, 1098–1117. doi: 10.1007/s40333-024-0105-0
Zareen, S., Ali, A., and Yun, D. (2024). Significance of ABA biosynthesis in plant adaptation to drought stress. J. Plant Biol. 67, 175–184. doi: 10.1007/s12374-024-09425-9
Zhang, Q., Ahmad, N., Li, Z., He, J., Wang, N., Naeem, M., et al. (2023). CtCYP71A1 promotes drought stress tolerance and lignin accumulation in safflower and Arabidopsis. Environ. Exp. Bot. 213, 105430. doi: 10.1016/j.envexpbot.2023.105430
Zhang, K., Gao, W., Zhou, Y., Zhao, H., Xia, Y., Zhang, M., et al. (2023). Allelic variations of ClACO gene improve nitrogen uptake via ethylene-mediated root architecture in watermelon. Theor. Appl. Genet. 136, 199. doi: 10.1007/s00122-023-04448-1
Zhang, Z., Li, S., Sun, S., Li, H., Zhang, Q., Li, Y., et al. (2025). The 14–3–3 gene AaGRF1 positively regulates cold tolerance in kiwifruit. Plant Sci. 353, 112403. doi: 10.1016/j.plantsci.2025.112403
Zhang, J., Wang, Y., Zhang, S., Cheng, F., Zheng, Y., Li, Y., et al. (2024). The BEL1-like transcription factor GhBLH5-A05 participates in cotton response to drought stress. Crop J. 12, 177–187. doi: 10.1016/j.cj.2023.10.011
Zhao, K., Zhang, X., Cheng, Z., Yao, W., Li, R., Jiang, T., et al. (2019). Comprehensive analysis of the three-amino-acid-loop-extension gene family and its tissue-differential expression in response to salt stress in poplar. Plant Physiol. Bioch. 136, 1–12. doi: 10.1016/j.plaphy.2019.01.003
Zhong, Y., Chen, C., Nawaz, M. A., Jiao, Y., Zheng, Z., Shi, X., et al. (2018). Using rootstock to increase watermelon fruit yield and quality at low potassium supply: A comprehensive analysis from agronomic, physiological and transcriptional perspective. Sci. Hortic-Amsterdam. 241, 144–151. doi: 10.1016/j.scienta.2018.06.091
Keywords: watermelon, ClTALE gene family, low potassium, cold, drought
Citation: Qiu Z, Dong J, Chen L, Zhao L, Hu L and Wang H (2025) Genome-wide characterisation of the three amino acid loop extension gene family of watermelon in response to abiotic stresses. Front. Plant Sci. 16:1711607. doi: 10.3389/fpls.2025.1711607
Received: 23 September 2025; Accepted: 30 October 2025;
Published: 18 November 2025.
Edited by:
Diaa Abd El Moneim, Arish University, EgyptReviewed by:
Klára Kosová, Crop Research Institute (CRI), CzechiaBen Zhang, Shanxi University, China
Copyright © 2025 Qiu, Dong, Chen, Zhao, Hu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huilin Wang, d2FuZ2h1aWxpbkAxMjYuY29t; Liangliang Hu, SHVsbDA1MDFAeGphdS5lZHUuY24=