 Muralikrishna Narra
Muralikrishna Narra Anamika Ray2
Anamika Ray2 Brittany Polley
Brittany Polley Hui Yang
Hui Yang Pankaj K. Bhowmik
Pankaj K. Bhowmik- 1Aquatic and Crop Resource Development, National Research Council of Canada (NRC), Saskatoon, SK, Canada
- 2Anandi Botanicals Inc., Lethbridge, AB, Canada
The advent of artificial intelligence (AI) holds great promise for revolutionizing the fields of plant tissue culture and genome editing. Plant tissue culture is recognized as a powerful tool for rapid multiplication and crop improvement. However, the complex interactions between genetic and environmental factors generate large volumes of data, posing challenges for traditional statistical analysis methods. To address this, researchers are now employing machine learning (ML)-based and artificial neural networks (ANN) approaches to predict and optimize in vitro culture protocols thereby improving precision, sustainability, and efficiency. Integrating AI technologies such as machine learning (ML), artificial neural networks (ANN), and deep learning (DL) can significantly advance the development of data-driven models for CRISPR/Cas9 genome editing. Today, AI-driven methods are routinely applied to enhance precision in predicting on- and off-target sequence locations and editing outcomes. Additionally, predicting protein structures can provide a directed evolution framework that facilitates the creation of improved gene editing tools. However, the application of AI-based CRISPR modeling in plants is not yet fully explored. In this context, we aim to examine representative ML/DL/ANN models of CRISPR/Cas based editing employed in various organisms. This review significantly compiles a diverse set of studies and provides a clear overview of how AI is transforming the fields of plant tissue culture and genome editing. It emphasizes AI’s potential to increase the efficiency and precision of biotechnological practices, making them more accessible and cost-effective. While outlining current findings, the paper sets the stage for future research, encouraging further exploration into the integration of AI with plant biotechnology.
1 Introduction
In recent years, plant biotechnology has made significant strides, notably by the integration of artificial intelligence (AI) with plant tissue culture and genome editing, that represents a transformative advancement in plant biotechnology, offering powerful tools to accelerate crop improvement. Plant tissue culture and CRISPR/Cas genome editing have each revolutionized plant breeding and genetic engineering (Ibrahim et al., 2023). Incorporating AI techniques further enhances these technologies by enabling faster, more accurate, and cost-effective optimization of culture protocols and precise genetic modifications. AI models excel at deciphering complex, non-linear patterns in large biological datasets, enhancing predictions of growth responses, culture conditions, and editing outcomes (Kaushik et al., 2025). The continued advancement and adoption of AI methodologies are essential to overcoming existing technical and biological limitations. Harnessing the full potential of AI, in conjunction with plant tissue culture and genome editing, will drive the development of sustainable, precision agriculture solutions, supporting global food security, promoting environmental sustainability, and ushering in a new era of innovation in plant sciences.
Plant tissue culture is the sterile cultivation of plant explants on defined media under controlled conditions, leveraging totipotency, the ability of a single cell to regenerate into a whole plant (Phillips and Garda, 2019). It is fundamental to micropropagation, crop improvement, metabolite production, and conservation. To achieve efficient in vitro regeneration, factors such as explant type, nutrient composition and growth regulators must be carefully optimized (Loyola-Vargas and Ochoa-Alejo, 2018). Traditional statistical methods are limited in handling the complexity and non-linearity of in vitro systems, making optimization time-consuming and labor-intensive (Niazian and Niedbała, 2020). Artificial intelligence, especially machine learning and artificial neural networks, enables reliable modeling of complex biological interactions using experimental data. It facilitates accurate prediction and optimization of plant tissue culture stages, while reducing time, cost, and experimental load (Hesami et al., 2020a). The CRISPR/Cas system has emerged as a leading genome engineering platform due to its programmability and targeting flexibility (Liu et al., 2022a; Liu et al., 2022b). Its broad applicability has enabled its use across a wide range of applications, from gene disruption and modification to gene regulation and RNA editing. Despite challenges like off-target effects and variable efficiency, the focus has shifted from feasibility to optimization (Ran et al., 2013, 2015). Machine learning (ML)/Deep learning (DL) offers powerful methods for modeling complex biological systems. By learning patterns from data, AI enables accurate prediction of CRISPR editing outcomes, surpassing the limits of traditional experimentation (Chuai et al., 2017). The integration of deep learning into genomics and genetic engineering presents new opportunities for precision and innovation in gene editing (Eraslan et al., 2019).
While previous reviews have acknowledged the promise of AI)in plant biotechnology, most focus narrowly either on ML applications in plant tissue culture or on CRISPR/Cas-based genome editing in non-plant systems. For instance, certain studies (Hesami and Jones, 2021; Ibrahim et al., 2023; Kaushik et al., 2025) offer valuable overviews of AI in tissue culture optimization but do not address the convergence of AI with gene editing platforms like CRISPR/Cas9. This review is uniquely positioned at the intersection of two critical domains and emphasizes the integrative role of AI across plant tissue culture and genome editing. In addition to summarizing current models and algorithms, this review outlines challenges in data availability, model interpretability, and experimental validation, and proposes concrete future research directions to bridge these gaps. This positions the review as both a resource and a roadmap for researchers aiming to operationalize AI in plant biotechnology.
2 Machine learning-based approaches in plant tissue culture
Plant tissue culture (micropropagation or in vitro cell and tissue culture), is a technique used to grow plants in a nutrient-rich medium under controlled, sterile conditions. It begins with culturing various explants such as leaves, stems, or roots, which due to their totipotency capacity, can regenerate into whole plants and produce multiple plantlets (Loberant and Altman, 2010). This technique has diverse applications, including mass propagation of elite plants, genetic modification, germplasm conservation, and production of disease-free plants (Brown and Thorpe, 1995). In vitro cultures are influenced by several factors, including nutrient media composition, plant genotype, explant type and age, plant growth regulators (PGRs), and phytohormone concentrations (Sudheer et al., 2022). Traditional regression methods and analysis of variance (ANOVA) have been employed for analyzing the data. However, the complexity and non-linearity of biological systems often limit the effectiveness of these techniques (Compton, 2024). In contrast, AI and computer-assisted tools can process continuous, binomial, discrete, and incomplete data, even those generated through unstructured, trial-and-error experiments. This ultimately reduces the need for extensive laboratory experimentation, saving time and resources in optimizing conditions for in vitro culture (Sarker, 2022).
ML has emerged as a powerful tool for addressing this challenge, enabling computers to learn from data and make accurate predictions and classifications (Ray, 2019). ML techniques include supervised learning, where models are trained on labeled datasets for precise prediction and classification (Alloghani et al., 2020); unsupervised learning, which identifies patterns in unlabeled data for clustering and analysis (Alloghani et al., 2020); reinforcement learning, which improves performance through trial-and-error interactions with the environment (Szepesvári, 2010); and semi-supervised learning, that integrates limited labeled data with abundant unlabeled data to enhance model accuracy, particularly when labeling is costly or time-consuming (Zhu and Goldberg, 2009) (Figure 1). In plant tissue culture research, recent advances have leveraged ML to interpret complex, nonlinear data. Hybrid approaches combining ML with optimization algorithms have been used to analyze the relationships between variables. As a result, data-driven approaches are increasingly used to model and optimize tissue culture conditions, including media composition and other critical parameters (Sarker, 2022). A comprehensive literature review was conducted to collect the data specific to plant tissue culture. Using online databases, google scholar, pubmed and scopus were searched for relevant studies with keywords such as “MACHINE LEARNING IN PLANT TISSUE CULTURE”, “ANN IN PLANT TISSUE CULTURE”. Selected peer-reviewed publications with machine learning application in plant tissue culture techniques were screened and considered for writing this review article (Table 1).
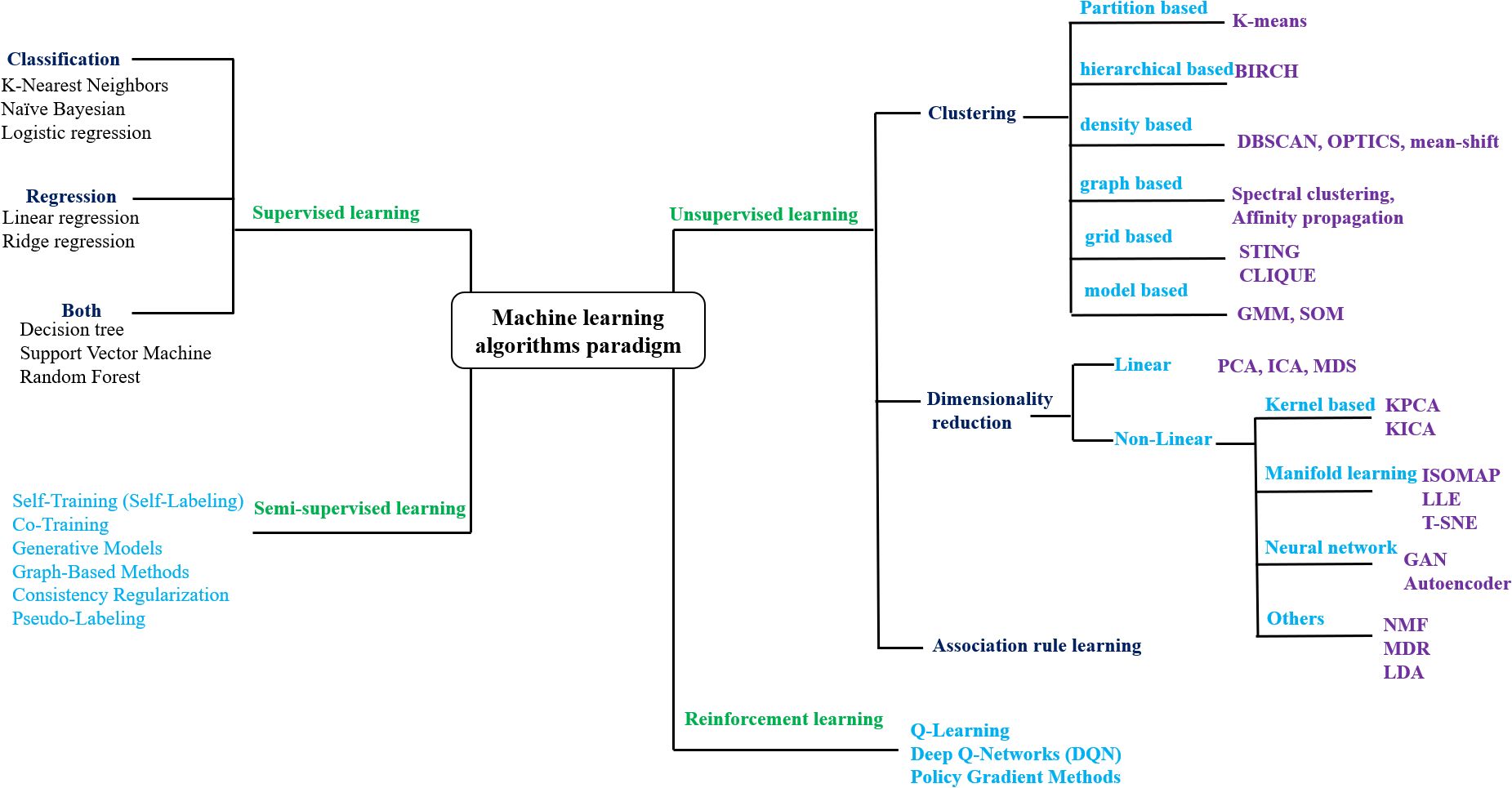
Figure 1. Taxonomy of machine learning algorithm paradigms. BIRCH, Balanced Iterative Reducing and Clustering Using Hierarchies; DBSCAN, Density-Based Spatial Clustering of Applications with Noise; OPTICS, Ordering Points To Identify the Clustering Structure; STING, STatistical INformation Grid; CLIQUE), CLustering In QUEst; GMM, Gaussian Mixture Models; SOM, Self-Organizing Map; PCA, Principal Component Analysis; ICA, Independent Component Analysis; MDS, MultiDimensional Scaling; KPCA, Kernel PCA; KICA, Kernel ICA; ISOMAP, ISOmetric feature MAPping; LLE, Locally Linear Embedding; t-SNE, t-distributed Stochastic Neighbor Embedding; NMF, N on-negative Matrix Factorization; MDR, Multifactor Dimensionality Reduction; LDA, Latent Dirichlet Allocation; GAN, Generative Adversarial Network.
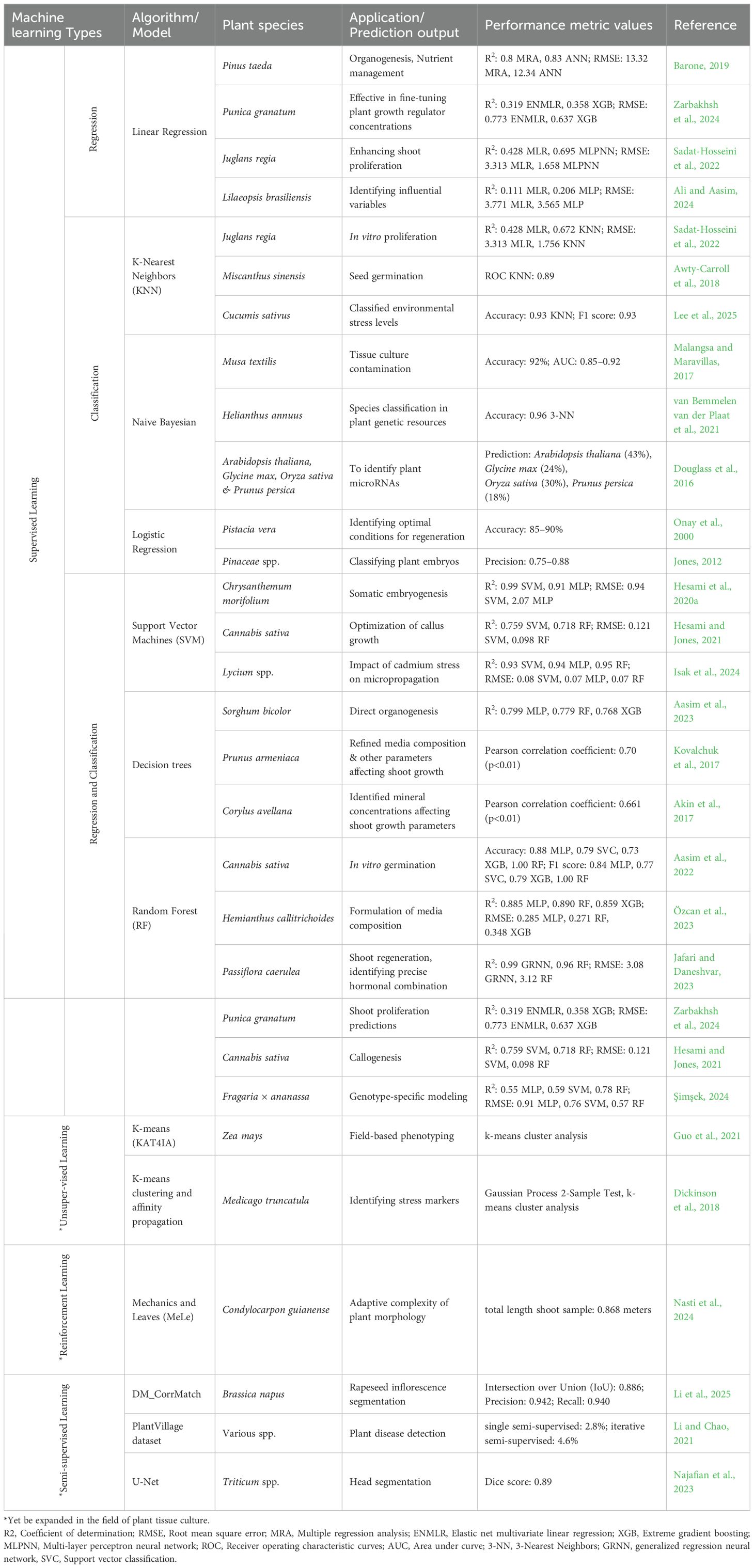
Table 1. Applications of machine learning-based approaches/models used in plant tissue culture and other prediction studies.
2.1 Supervised learning
Supervised learning is a subset of machine learning that employs labeled datasets to train algorithm models for analysis and prediction in plant tissue culture research. The success of tissue culture is influenced by variety of factors, including sterilization techniques, plant genotype, growth conditions and media composition. Supervised learning algorithms help to model and predict these outcomes. The process begins with model training by using a dataset that includes input data (features) and corresponding output data (labels or target variables). Once the training is complete, the model is evaluated on a test dataset to assess its accuracy and performance. During the learning process, the algorithm analyzes the relationship between the input features and the output labels (Hastie et al., 2009a). The model’s performance is then fine-tuned by adjusting parameters and cross-validation to balance bias and variance, this sequentially helps to ensure that the model generalizes effectively to new, unseen data. Based on the nature of the output variables, supervised learning can be broadly categorized into (i) Regression, that deals with continuous, numerical outputs without predefined labels, for example, predicting growth rate, biomass accumulation, or shoot multiplication based on factors like temperature, light, or media compositions. (ii) Classification on the other hand, involves categorical outputs with defined labels, such as classifying tissue culture outcomes into categories like successful regeneration, no regeneration, or diseased (Hastie et al., 2009a) (Figure 2A).
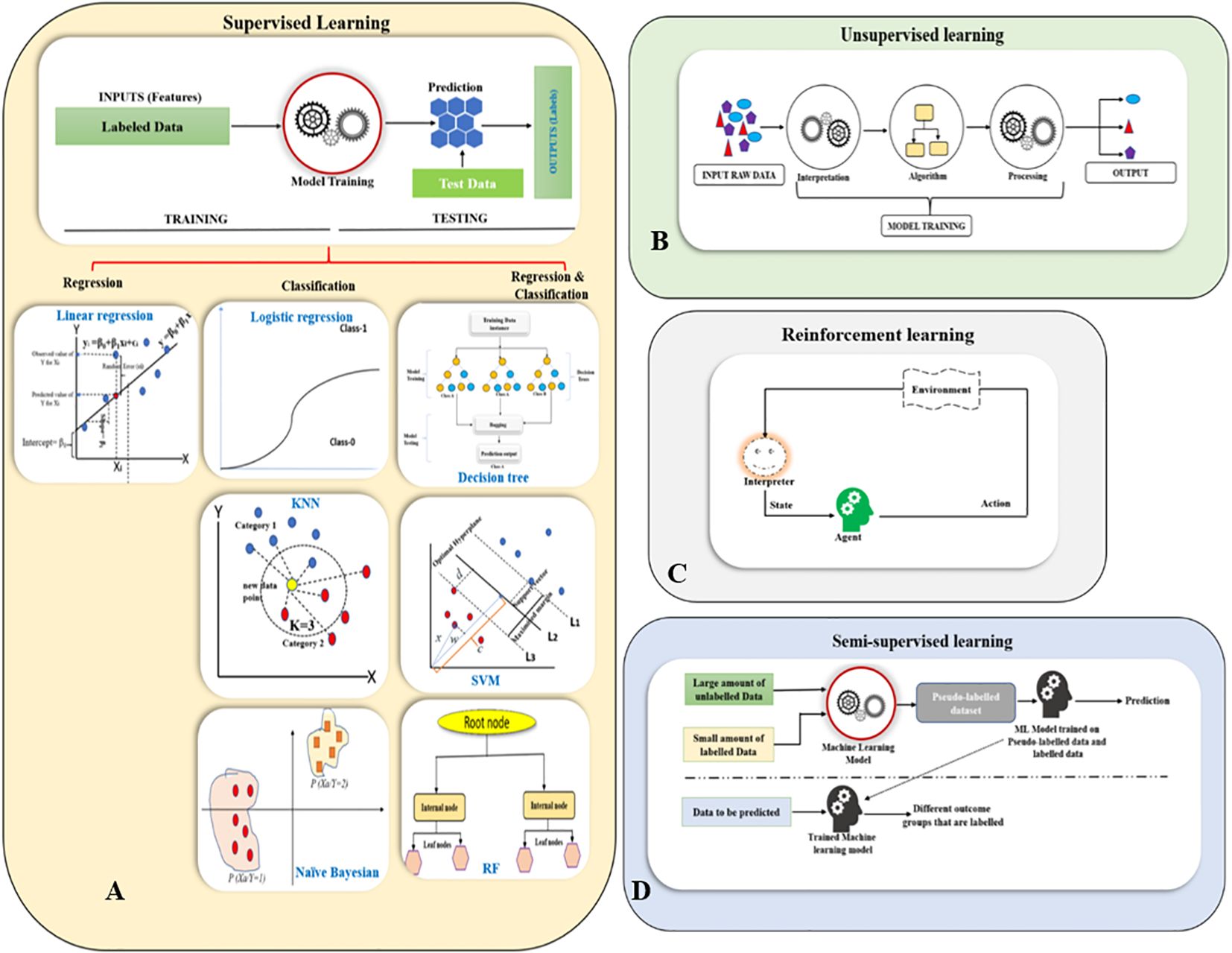
Figure 2. Overview of machine learning paradigms. (A) Supervised Learning (Linear Regression, Logistic Regression, Decision Trees, K-Nearest Neighbors (KNN), Support Vector Machines (SVM), Naïve Bayes, and Random Forests (RF); (B) Unsupervised Learning; (C) Reinforcement Learning; (D) Semi-supervised learning.
2.1.1 Linear regression
This is used to predict continuous variables based on one or more independent variables. It computes the linear relationship between the dependent variable and one or more independent features by fitting a linear equation to the observed data. The primary objective of linear regression is to find the best-fit line, which minimizes the error between predicted and actual values. Here the dependent variables (Y), the output you want to predict (e.g., plant growth rate, biomass) and independent variables (X), the factors affecting the output (e.g., temperature, humidity, light intensity) (Equation 1).
Where:
β0 is the intercept (value of y when x=0).
β1 is the coefficient (effect of x on y).
ϵi is the error term (difference between predicted and actual values).
ϵi = y(predicted) – yi
where y(predicted) = β0+β1xi
The equation of this best-fit line represents the relationship between the dependent and independent variables, with the slope indicating how much the dependent variable changes for a unit change in the independent variable(s). Linear regression can be classified further into (i) simple linear regression, that models the relationship between one independent variable and one dependent variable by fitting a straight line. The line is defined by an intercept (β0) and a slope (β1), where the slope indicates the change in the dependent variable for each unit increase in the independent variable, and the intercept represents the expected value of the dependent variable when the independent variable is zero. The model aims to minimize the difference between actual and predicted values using a cost function, typically mean squared error (MSE) (Equation 2).
Model performance is also evaluated using metrics like the R-squared (which shows the proportion of explained variance), residual standard error (RSE) and root mean squared error (RMSE). Valid regression requires assumptions such as linearity, independence, normality, and constant variance of residuals. (ii) multiple linear regression, extends this concept to include multiple independent variables (Equation 3):
It follows the same assumptions as simple linear regression, with added concerns like multicollinearity, overfitting, and the need for feature selection. The bias-variance tradeoff is crucial where underfitting occurs when the model is too simple, while overfitting happens when the model captures noise instead of the true pattern, especially in high-dimensional or collinear datasets. Techniques like cross-validation, regularization, and careful feature engineering help strike the right balance. In summary, linear regression is a foundational tool in data science and machine learning, valued for its interpretability and effectiveness. It not only helps make accurate predictions but also lays the groundwork for more complex modeling approaches (Hastie et al., 2009b).
Research application
The reviewed studies collectively highlight the exponential use of integrated regression-based machine learning techniques in optimizing plant tissue culture protocols. In pomegranate, a combination of Bayesian-tuned ensemble stacking regression and non-dominated sorting genetic algorithm-II (NSGA-II) proved effective in fine-tuning plant growth regulators (PGR) concentrations, enhancing shoot proliferation while minimizing somaclonal variation (Zarbakhsh et al., 2024). Similarly, in Pinus taeda, multiple regression and neural networks revealed nitrogen concentration as a key factor in organogenesis, emphasizing the need for precise nutrient management (Barone, 2019). The comparative modeling in persian walnut (Juglans regia) revealed that although linear regression offers baseline predictions, advanced models like genetic programming (GEP) and multilayer perceptron neural network (MLPNN) deliver superior accuracy in forecasting in vitro proliferation. The optimization of macronutrient compositions for pear rootstocks leveraged both stepwise regression and AI methods to pinpoint critical factors influencing explant growth, showcasing the synergy between statistical and algorithmic tools (Sadat-Hosseini et al., 2022). Lastly, the application of response surface methodology and regression analysis in brazilian micro sword (Lilaeopsis brasiliensis) regeneration highlighted the practical utility of these models in identifying influential variables. Overall, these studies validate the robustness of regression and hybrid modeling approaches in refining tissue culture conditions across diverse plant species (Ali and Aasim, 2024).
2.1.2 K-nearest neighbors
K-Nearest neighbors (KNN) machine learning algorithm was generally used for classification but can also be applied to regression tasks. It works by finding the “K” closest data points (neighbors) to a given input and makes predictions based on the majority class for classification or the average value for regression (Mucherino et al., 2009). Since KNN makes no assumptions about the underlying data distribution, it is considered as a non-parametric and instance-based learning method. K-nearest neighbors is also called a lazy learner algorithm because it does not learn from the training set immediately; instead, it stores the dataset and performs computations on it only at the time of classification. Cross-validation is a reliable method for choosing the optimal “K” in KNN by dividing the dataset into k parts, training on some, and testing on others, then selecting the k with the highest average accuracy. The k-nearest neighbors (KNN) algorithm functions with selecting an optimal value for K, which denotes the number of neighbors to consider when making predictions. Next, the algorithm calculates the distance between the target data point and all points in the training set, commonly using Euclidean distance as the measure of similarity, which is a straight-line distance between two points in a plane or space. It then identifies the “K” data points with the smallest distances to the target, designating these as the nearest neighbors (Equation 4) (James et al., 2021).
Research application
Optimizing plant tissue culture media is a complex process influenced by multiple factors including genotype, mineral nutrients, plant growth regulators (PGRs), and vitamins, often resulting in inefficiencies and physiological disorders like browning of callus, shoot tip necrosis and vitrification. In this context, predictive modeling using ML offers a promising solution. In walnut (Juglans regia L.) proliferation, three ML models, multi-layer perceptron neural network (MLPNN), KNN, and gene expression programming (GEP) were evaluated against multiple linear regression (MLR) for their predictive accuracy. All ML models outperformed MLR, with GEP showing the highest R² (0.802 in Chandler and 0.428 in Rayen varieties) and subsequently optimized using particle swarm optimization (PSO). This highlights GEP-PSO as a powerful hybrid tool, while MLPNN and KNN also demonstrated strong estimation abilities (Sadat-Hosseini et al., 2022). Similarly, in miscanthus seed germination, KNN improved phenotype scoring accuracy when compared to human assessments, especially under challenging conditions such as low germination rates and presence of mold. The model achieved a ROC-AUC of 0.69–0.7, improving to 0.89 on optimized image sets, confirming its utility for consistent, automated germination analysis (Awty-Carroll et al., 2018). In cucumber seedlings, ML models combined with image-based feature extraction (color, texture, morphology) effectively classified environmental stress levels. Among the tested models, KNN achieved the highest accuracy (94%), emphasizing its effectiveness in early stress detection and its potential application in precision agriculture for real-time crop monitoring (Lee et al., 2025).
2.1.3 Naive Bayesian
Naive Bayesian is a classification algorithm that uses probability to predict which category a data point belongs to, assuming that all features are unrelated. The naive Bayesian classifier is based on bayes theorem, which describes the probability of an event, based on prior knowledge of conditions that might be related to the event (Webb, 2011) (Equation 5).
The general formula for Bayes’ theorem is:
Where:
P(y/x) is the posterior probability of class y given the features X.
P(x/y) is the likelihood, i.e., the probability of observing the features X given class y.
P(y)is the prior probability of class y.
P(x) is the evidence or the total probability of observing the features X under all classes.
The term “naive” comes from the assumption that all features are independent of each other, given the class label. This assumption simplifies the computation of the likelihood P(x/y), as we can decompose it into the product of individual feature probabilities. Naïve Bayesian classifiers can be broadly categorized based on the type of data they handle. (i) The gaussian naive Bayesian is used when the features are continuous and assumes that these features follow a gaussian (normal) distribution. In this case, the probability P(y∣ X) is estimated using the probability density function (PDF) of the normal distribution. (ii) The multinomial naïve Bayesian, is commonly used for text classification where the features are discrete counts, such as word frequencies in documents. It models the probability of each feature’s occurrence given a particular class using a multinomial distribution. (iii) Lastly, bernoulli naïve Bayesian is employed when the features are binary, representing the presence or absence of a feature (e.g., 0 or 1). It assumes that each feature follows a bernoulli distribution, where the outcomes are binary. Preprocessing steps like color normalization and data augmentation enhance naïve bayes performance by reducing variability and improving generalizability in image-based tissue culture analysis.
Research application
The performances of naïve bayes and KNN classifiers were compared for grading contamination in abaca tissue culture specimens. The methodology involved capturing images with a masking technique, extracting features from mean RGB values and binary images. Specimens were classified as healthy or contaminated, and classifier performance was evaluated using accuracy, precision, and recall. Naïve Bayes outperformed KNN, achieving 76% accuracy, compared to 68% for KNN at K = 3 and 58% at K = 7 (Malangsa and Maravillas, 2017). Different machine learning classifiers like random forest (RF), neighbor-joining (NJ), KNN, and naïve bayes (NB) were evaluated for species classification in plant genetic resources collections in sunflower. The authors found the KNN classifier to be the most reliable, especially for datasets with variability and uncertainty. The study also highlighted the importance of marker selection for improving classifier accuracy and introduced a method to enhance suboptimal datasets, particularly valuable for genebanks with limited high-quality references (van Bemmelen van der Plaat et al., 2021). Douglass et al. (2016) developed a naïve bayes classifier to identify plant microRNAs (miRNAs), which are key regulatory molecules in eukaryotes. Traditional methods, relying on stem-loop structures, often miss low-count miRNAs. Their probabilistic approach uses sequence length, observation counts, miRNA sequence presence, and other features, and was tested on small RNA data from soybean, peach, Arabidopsis, and rice.
2.1.4 Logistic regression
Logistic regression is primarily used for classification tasks (Sperandei, 2014). Unlike linear regression, which predicts continuous values, logistic regression estimates the probability that an input belongs to a specific class. It is mainly employed for binary classification problems, where the output consists of two possible outcomes, such as Yes/No; True/False; or 0/1. The algorithm utilizes a sigmoid function to convert inputs into a probability between 0 and 1. In contrast, multinomial logistic regression is applied when the dependent variable has three or more unordered categories. It extends the binary logistic regression approach to accommodate multiple classes. Lastly, ordinal logistic regression is used when the dependent variable consists of three or more categories with a natural order or ranking, like ratings of “low,” “medium,” and “high.” This model accounts for the inherent order in the categories when making predictions. The logistic regression model is represented by the following equation (Equation 6):
Where:
p(y=1/X) is the probability that the instance belongs to class 1 given the feature vector X.
wTX is the linear combination of the features (i.e., weights and features).
b is the bias term (also called the intercept).
Σ(sigma) is the sigmoid function that maps the linear combination to a probability.
To minimize the loss function, gradient descent is commonly used, iteratively adjusting the model parameters (weights and bias) to reduce the error. In addition to the primary cost function, regularization techniques like L1 (lasso) and L2 (ridge) regularization are often applied to prevent overfitting, especially in cases with high-dimensional data.
Research application
Linear logistic models were used to assess the significance of the treatments and identify optimal conditions for both processes, highlighting BAP’s superior role in regeneration. This study has explored the effects of 6-benzylaminopurine (BAP), abscisic acid (ABA), and sucrose on the germination and plantlet regeneration of pistachio somatic embryos. Germination rates increased with longer culture durations, with BAP and ABA concentrations influencing outcomes, while sucrose had little effect. Similarly, plantlet regeneration improved over time but was inhibited at higher BAP or ABA levels, with ABA reducing the likelihood of regeneration, particularly during extended maturation periods (Onay et al., 2000). A method for classifying plant embryos (Pseudotsuga and Pinus) based on quality using a penalized logistic regression (PLR) model is disclosed. First, image or spectral data are collected from plant embryos with known quality. Next, each data set is assigned a class label corresponding to the embryo’s quality. Then, metrics are calculated from these image or spectral data sets (Jones, 2012).
2.1.5 Support vector machines
A support vector machine is a powerful ML-algorithm used for both classification and regression tasks. It works by finding the optimal line (or hyperplane) that separates data into distinct groups, while maximizing the distance between the closest data points (support vectors) of each group (Vapnik, 2000). A larger margin typically results in better generalization, allowing the model to perform well on new, unseen data. The optimal hyperplane, also known as the “hard margin,” is the one that maximizes this distance, ensuring a clear separation between the classes. A soft margin, on the other hand, allows for some misclassifications or violations of the margin, which helps improve generalization. This optimizes the following equation to balance margin maximization and penalty minimization (Equation 7).
The penalty for violations is typically the hinge loss, which behaves as follows, if a data point is correctly classified and lies within the margin, there is no penalty (loss = 0) and if a data point is misclassified or violates the margin, the hinge loss increases in proportion to the distance of the violation.
During the training phase, the algorithm is fed with labeled data points from both classes. This is for to determine the optimal hyperplane that maximizes the margin between the two classes, ensuring that no data points fall between the hyperplane and the support vectors. Once the model is trained, it enters the testing phase, where it is presented with new, unseen data points. The model then assesses the position of these points relative to the hyperplane and classifies them based on which side of the hyperplane they are located. A kernel is a function that allows support vector machines to handle non-linear data by implicitly mapping inputs to a higher-dimensional space. Common types include the linear kernel, suitable for linearly separable data; the polynomial kernel, which captures more complex relationships using polynomial functions; and the RBF (Radial basis function) kernel, which measures similarity based on the distance between data points.
Research application
Recent studies have demonstrated the effective application of SVM in plant tissue culture, particularly for modeling and optimizing processes such as somatic embryogenesis and callus development. Support vector regression (SVR) was employed to model somatic embryogenesis in chrysanthemum. The SVR model outperformed multilayer perceptron (MLP) models, achieving an R² value greater than 0.92. Furthermore, integrating SVR with the NSGA-II led to optimization of the culture medium, resulting in a 99.09% embryogenesis rate and an average of 56.24 somatic embryos per explant (Hesami et al., 2020a). SVM in combination with random forest (RF) and genetic algorithm (GA) to model and optimize callus growth in Cannabis sativa. This hybrid approach improved prediction accuracy and provided valuable insights into the influence of various factors on callus development (Hesami and Jones, 2021). More recently, a study by Isak et al. (2024) utilized multiple machine learning algorithms, including SVM, to evaluate the impact of cadmium stress on goji berry micropropagation. The SVM model effectively predicted plant growth parameters under different stress conditions, contributing to the development of strategies for mitigating abiotic stress in plant tissue culture.
2.1.6 Decision tree
A decision tree is used for both classification and regression. It models data using a tree-like structure where each internal node represents a decision based on a feature, branches represent outcomes of those decisions, and leaf nodes provide the final prediction (Thomas et al., 2020). The process starts from a root node that represents the entire dataset, and splits it into subsets based on selected features using specific splitting criteria: (i) Entropy (Information theory) measures the impurity or uncertainty in a dataset (Equation 8);
For a dataset D with k classes
Where:
pi is the proportion of samples in class i in dataset D.
Entropy is 0 when all samples belong to one class (pure), and maximum when classes are equally distributed.
(ii) Information Gain (IG) that is used in ID3 algorithm, measures the reduction in entropy after a dataset is split on an attribute A (Equation 9);
Where:
D = dataset before split. = subset of D where attribute A has value . = weight of the subset.
(iii) Gini impurity in the classification and regression trees (CART) algorithm, is another alternative to entropy to measure impurity of a dataset. Like entropy, Gini is 0 for pure datasets and it’s computationally simpler than entropy. These criteria assess how well a feature separates the data. Once the optimal feature is chosen, the dataset is partitioned based on that feature, and the process is repeated recursively on each subset until a stopping condition is met, such as reaching a maximum tree depth, or having a minimum number of samples per node. Since decision trees can overfit the training data, pruning techniques are employed to simplify the model. Common algorithms include iterative dichotomiser 3 (ID3) (using information gain), C4.5 (using gain ratio), CART (using gini impurity or MSE), and chi-squared automatic interaction detector (CHAID) (using Chi-square tests).
Research application
Aasim et al. (2023) successfully established an efficient in vitro regeneration protocol for Sorghum bicolor using direct organogenesis from mature zygotic embryo explants. The use of MS-medium supplemented with varying concentrations of BAP alone or in combination with IBA or NAA significantly influenced shoot count and shoot length, with optimal results observed for 2 mg/L BAP + 0.25 mg/L NAA and 2.0 mg/L BAP, respectively. Statistical analyses using factorial regression, pareto charts, and surface modeling confirmed the substantial effects of cytokinin–auxin interactions. Moreover, the integration of artificial intelligence-based models, including multilayer perceptron (MLP), random forest (RF), and XGBoost, demonstrated the superior predictive accuracy of MLP, highlighting the potential of AI in optimizing tissue culture protocols. In parallel, studies on wild apricot (Prunus armeniaca) (Kovalchuk et al., 2017) and hazelnut (Corylus avellana) employed decision tree algorithms such as CART, CHAID, and exhaustive CHAID to refine media composition and identify key mineral concentrations affecting shoot growth parameters. Particularly, CART proved effective in modeling complex nonlinear relationships and establishing specific cutoff points for components such as KH2PO4, MgSO4, and CuSO4. These findings underscore the relevance of decision tree-based modeling in deciphering multifactorial effects in plant tissue culture, offering a robust framework for media optimization across diverse species (Akin et al., 2017, 2018). The synergy between traditional statistical methods, AI models, and decision tree algorithms enhanced the ability to develop genotype-independent, efficient micropropagation systems, vital for crop improvement and biotechnological applications.
2.1.7 Random forest
Random forest algorithm combines multiple decision trees to make more accurate predictions. Each decision tree in the random forest examines different random subsets of the data. The results from these trees are then combined via majority voting for classification (to determine the final class) or by averaging the predictions for regression (Breiman, 2001). This approach helps improve accuracy and reduce errors. Another key aspect of RF is the depth of the trees. In most cases, the individual decision trees are grown to their full depth without any pruning, making them very complex and potentially prone to overfitting if considered independently. However, this risk is mitigated in RF because the trees are aggregated, and their combined predictions help prevent overfitting. The two common ensemble techniques used in random forests to improve the performance of models are (i) bootstrap aggregating (bagging) is a core principle behind random forests, and it helps improve model accuracy and robustness. Samples are trained based on different decision trees. After training, the predictions from all the trees are aggregated: for classification, the final class is determined by majority voting (i.e., the class chosen by most trees), and for regression, the predictions are averaged.
Formula (Majority voting in random forest) (Equation 10):
Where:
● I (yb = c)is an indicator function that returns 1 if yb=c and 0 otherwise,
● B is the number of trees in the forest.
(ii) boosting, on the other hand, focuses on reducing bias by sequentially training models. Each new tree in a boosting algorithm is trained to correct the mistakes made by the previous trees, with more attention given to misclassified data points. The final prediction is a weighted combination of the trees’ outputs, where trees with better performance have more influence. The other boosting techniques like gradient boosting machine (GBM), extreme gradient boosting machine (XGBM), LightGBM, and CatBoost become essential to confront the machine learning more deliverable with accuracy and precision.
Research application
In vitro propagation is an essential technique for conserving and mass-producing economically valuable plant species, yet it faces persistent challenges such as low germination rates, contamination, and genotype-specific responses. For instance, the in vitro germination of Cannabis sativa, historically difficult due to low germination and high contamination, was optimized using five ML models. Random Forest (RF) outperformed others with F1 scores between 0.98–1.00, identifying an optimal hydrogen peroxide concentration of ~2.2% (Aasim et al., 2022). Similarly, in aquatic plants like Hemianthus callitrichoides, RF and MLP models demonstrated high accuracy in predicting growth based on media composition (Özcan et al., 2023). In Passiflora caerulea, RF and GRNN, coupled with genetic algorithms (GA), successfully modeled shoot regeneration, identifying a precise hormonal combination for optimal results (Jafari and Daneshvar, 2023). Furthermore, in Punica granatum, RF and XGBoost, supported by novel tools like the global performance indicator (GPI) and NSGA-II, showed high fidelity in shoot proliferation predictions (Zarbakhsh et al., 2024). Finally, ML applications in drought-stressed Fragaria × ananassa highlighted genotype-specific modeling, with RF achieving the highest overall accuracy (Şimşek, 2024). Collectively, these examples underscore the versatility and predictive power of ML, particularly RF and other machine learning algorithms, in optimizing tissue culture protocols across diverse species.
2.2 Unsupervised learning
Unsupervised learning is a type of machine learning that works with unlabeled data. These algorithms are designed to identify patterns and relationships within the data on their own, without any prior knowledge of what the data represents (Nasrabadi, 2007). There are three primary types of unsupervised learning algorithms. (i) Clustering algorithms group data points based on similarity to uncover patterns without predefined labels. Common methods include K-means, which partitions data into K-clusters based on distance; hierarchical clustering, which creates a tree-like structure of nested clusters; and density-based clustering (DBSCAN) which identifies dense regions while treating sparse points as noise. Mean-shift clustering shifts points toward high-density areas to form clusters, while spectral clustering uses graph theory to group data based on relationships between points. (ii) Association rule learning identifies relationships between variables in large datasets, commonly used in market basket analysis to find items frequently purchased together. Key algorithms include Apriori, which iteratively finds frequent item sets, which improves efficiency by avoiding candidate generation; and Eclat, which uses set intersections. (iii) Dimensionality reduction techniques reduce the number of features in a dataset while preserving meaningful information, improving model simplicity, computational efficiency, and visualization. Principal component analysis (PCA) transforms data into uncorrelated components that capture maximum variance, while linear discriminant analysis (LDA) finds projections that enhance class separability (Tipping and Bishop, 1999; Hastie et al., 2009b) (Figure 2B).
Research application
A high-throughput phenotyping method was developed to efficiently collect trait data using imaging systems during key crop growth stages. To reduce dependence on human-labeled data for image-based trait extraction, KAT4IA is introduced, a self-supervised learning pipeline that applies K-means clustering to greenhouse images to automatically generate training data for field-based phenotyping (Guo et al., 2021). Metabolomics was integrated with transcriptomic analysis, to detect metabolic responses to combined stress in Medicago truncatula. LC-HRMS data from roots and leaves were analyzed using the gaussian process 2-sample test, K-means clustering, and affinity propagation for temporal clustering. Results revealed known stress markers, including altered sucrose and citric acid levels, with combined stress amplifying drought effects. While leaf responses were more pronounced, fusarium-related changes were also observed in roots (Dickinson et al., 2018).
2.3 Reinforcement learning
Reinforcement learning is a branch of machine learning where an agent learns to make decisions by interacting with an environment to maximize cumulative rewards over time. The agent observes the current state, takes actions, and receives feedback in the form of rewards, adjusting its strategy, or policy, to improve future outcomes (Szepesvári, 2010). A central challenge in RL is balancing exploration (trying new actions) and exploitation (leveraging known rewarding actions). This process is often modeled using markov decision processes (MDPs), which define states, actions, transition probabilities, and rewards. There are several categories of RL algorithms. Value-based methods, such as Q-learning and state-action-reward-state-action (SARSA), focus on estimating value functions that predict the expected future rewards of actions taken in given states (Watkins and Dayan, 1992). More recently, deep reinforcement learning leverages neural networks to handle complex, high-dimensional environments, with algorithms such as deep Q-networks (DQN) and proximal policy optimization (PPO) achieving state-of-the-art results. Overall, reinforcement learning provides a powerful framework for training agents to make sequential decisions in uncertain and dynamic environments (Hastie et al., 2009b) (Figure 2C).
Research application
The use of RL in plant biology and agriculture presents a promising avenue for addressing both fundamental biological questions and applied agricultural challenges. Across three distinct domains, plant organ development, crop breeding, and field management, RL has demonstrated its versatility and effectiveness as a decision-making and optimization framework and yet be expanded in the field of plant tissue culture. The reinforcement learning was used to model the biomechanics of the “searcher shoot”, a plant organ specialized for spatial exploration. By framing mass distribution and structural constraints as a markov decision process (MDP), the authors created the Searcher-Shoot environment to simulate adaptive growth strategies. Results showed consistent shoot tapering, suggesting that plants may naturally adopt efficient mass allocation to optimize elongation without surpassing stress limits. The close match between simulated and empirical data highlights the potential of RL to model the adaptive complexity of plant morphology (Nasti et al., 2024). RL was applied to crop breeding, a domain challenged by slow generation turnover, high-dimensional decision spaces, and increasing environmental pressures due to climate change. By introducing a suite of Gym environments tailored for breeding simulations, the authors trained RL agents to make selection and crossing decisions based on real-world genomic data (Younis et al., 2024). In a parallel application, Balderas et al. (2025) examined RL’s potential in optimizing crop production management. Using the gym-DSSAT environment, a well-established crop simulation framework, they evaluated two widely used RL algorithms, proximal policy optimization (PPO) and deep Q-networks (DQN), across key agricultural tasks: fertilization, irrigation, and integrated management. Their findings revealed that PPO generally performed better in single-task settings (fertilization and irrigation), whereas DQN excelled in the mixed management task.
2.4 Semi-supervised learning
Semi-supervised learning is a hybrid approach that lies between supervised and unsupervised learning, combining both labeled and unlabeled data. This method is particularly useful when labeled data is scarce or expensive to obtain, but a large amount of unlabeled data is available, helping to improve model performance through various strategies (Zhu and Goldberg, 2009). Self-training iteratively labels unlabeled data using an initial model trained on labeled data. Co-training leverages multiple classifiers, each trained on different data views, to label unlabeled examples. Generative models, like gaussian mixture models (GMMs) and hidden markov models (HMMs) predict labels based on learned data distributions (Hastie et al., 2009b) (Figure 2D).
Research application
Pure self-supervised learning (SSL) methods, like FixMatch and others, haven’t been widely adopted in plant tissue culture research. This is largely because of the small dataset sizes and the challenges in reliably generating pseudo-labels for in-vitro outcomes. Instead, most recent studies have focused on label-efficient models, approaches that combine regression or classification techniques with limited experimental data or utilize self- and semi-controlled setups. A semi-supervised framework, DM_CorrMatch, was proposed for rapeseed inflorescence segmentation. It integrates data augmentation with a denoising diffusion probabilistic model (DDPM) to address limited annotated data. The Mamba-Deeplabv3+ network used in the study captures both local and global features, enhancing segmentation accuracy despite complex backgrounds and variable inflorescence poses. Validated on the rapeseed flower segmentation dataset (RFSD), the model achieved an intersection over union (IoU) of 0.886, precision of 0.942, and recall of 0.940 (Li et al., 2025). A study focused on plant leaf disease recognition using semi-supervised few-shot learning approach was carried out to address the challenge of limited labeled data in plant pathology. By combining source and target domains from the PlantVillage dataset, the iterative semi-supervised method achieved an accuracy improvement of up to 4.6% (Li and Chao, 2021). Najafian et al. (2023) explored the use of deep learning for semantic segmentation in agriculture with minimal annotation. By applying domain adaptation techniques, the authors achieved impressive segmentation results for wheat heads with just two annotated images, demonstrating the power of synthesized datasets and the effectiveness of using limited labeled data.
3 Artificial neural networks in plant tissue culture
Artificial neural networks have been extensively applied in plant tissue culture to predict, analyze, and optimize processes including plant growth, tissue regeneration, callus formation, and disease detection (Prasad and Gupta, 2008) (Table 2). Several types of ANNs can be employed for different purposes in plant tissue culture research (Figure 3). Some of the most common types include.
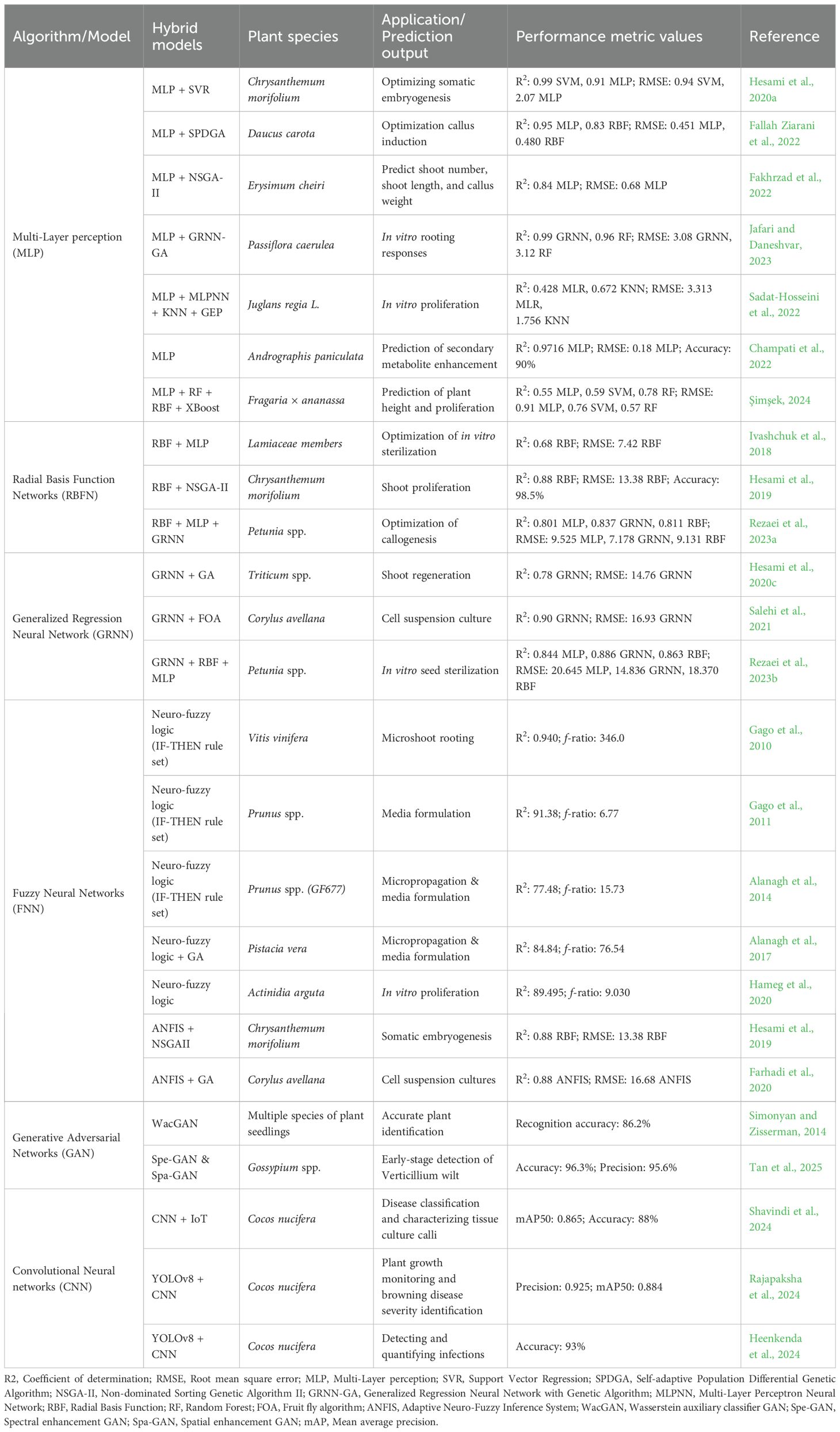
Table 2. Applications of artificial neural networks (ANNs) approaches/models used in plant tissue culture and other prediction studies.
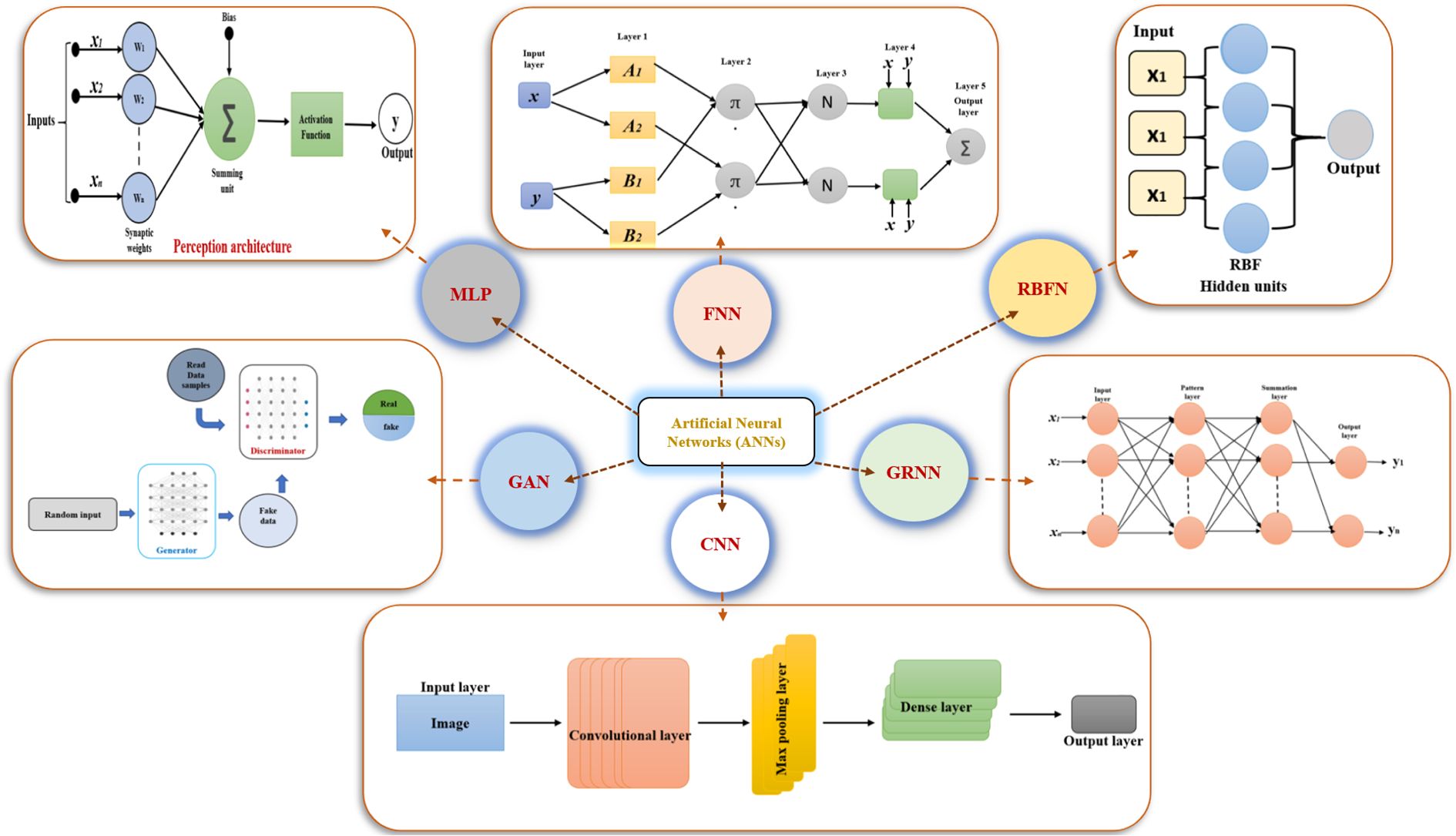
Figure 3. Architectural variants of artificial neural networks (ANNs). MLP, Multilayer Perceptron; FNN, Fuzzy Neural Network; RBFN, Radial Basis Function Network; GAN, Generative Adversarial Network; CNN, Convolutional Neural Network; GRNN, General Regression Neural Network.
3.1 Multi-layer perception
A Multi-layer perceptron is consisting of fully connected layers that transform input data through various dimensions. It is called “multi-layer” because it includes an input layer, one or more hidden layers, and an output layer. The input layer contains neurons representing each feature in the data, while the hidden layers process the information passed from the input. The number of hidden layers and neurons in each can vary. Finally, the output layer generates the prediction or result, with a number of neurons corresponding to the number of outputs. The main purpose of an MLP is to model complex relationships between inputs and outputs (Christopher, 1995). Each neuron in the hidden layers processes the input by first calculating a weighted sum of the inputs, given by the formula (Equation 11).
Where xi is the input feature, wi is the corresponding weight and b is the bias term. This weighted sum z, is then passed through an activation function to introduce non-linearity. Once the network generates an output, the next step is to compute the loss using a loss function, which compares the predicted output to the actual label. For a classification task, the binary cross-entropy loss function is typically used (Equation 12):
where yi is the actual label, is the predicted label, and N is the number of samples.
For regression problems, mean squared error (MSE) is often used (Equation 13):
Where is the average label value at node t, ∣t∣ is the number of samples at node t.
The goal of training an MLP is to minimize the loss function by adjusting the network’s weights and biases through backpropagation. MLPs use optimization algorithms to iteratively update weights and biases during training. Stochastic Gradient Descent (SGD) updates weights using individual samples or small batches, while the Adam optimizer enhances SGD by incorporating momentum and adaptive learning rates, enabling more efficient and effective training through dynamic adjustment of learning rates (Osama et al., 2015).
Research application
The critical role of optimizing somatic embryogenesis for successful gene transformation in chrysanthemum was analyzed by comparing two ML models, multilayer perceptron (MLP) and support vector regression (SVR). The study found that SVR outperformed MLP, achieving a higher predictive accuracy (R² > 0.92 vs. R² > 0.82). When combined with the NSGA-II optimization algorithm, the SVR model led to exceptional results including 99.09% embryogenesis efficiency and an average of 56.24 embryos per explant. This study highlighted the potential of integrating machine learning with evolutionary algorithms to enhance plant tissue culture outcomes (Hesami and Jones, 2020b). Similarly, Fallah Ziarani et al. (2022) employed an MLP model integrated with a single point discrete genetic algorithm (SPDGA) to optimize callus induction in carrots. The MLP model outperformed the radial basis function (RBF) network, achieving R² values around 0.95. Sensitivity analysis revealed MS-salt concentration as the most influential factor, underscoring the value of targeted ML optimization in improving regeneration protocols. In Erysimum cheiri, a lesser-studied medicinal plant, Fakhrzad et al. (2022) developed MLP-based models to predict shoot number, shoot length, and callus weight, with strong predictive performance (R²=0.84–0.99). Optimization using NSGA-II helped fine-tune hormone concentrations, further validating the reliability of the model. A focused study on rooting in Passiflora caerulea, was carried out by applying a hybrid general regression neural network–genetic algorithm (GRNN-GA) model (Jafari and Daneshvar, 2023). This study demonstrated the GRNN-GA model’s effectiveness in capturing complex in vitro rooting responses. GRNN achieved excellent accuracy (R² > 0.92), and the optimization process produced favorable rooting outcomes based on auxin types and explant sources. The application of MLP-based artificial neural network was successful to correlate soil nutrient levels with andrographolide content in Andrographis paniculata across 150 accessions in eastern India (Champati et al., 2022). Their best-performing model (14-12–1 architecture) achieved a high accuracy (R² = 0.9716), and the optimization process raised andrographolide content from 3.38% to 4.90%. This study showcased the broader utility of ML in site selection and secondary metabolite enhancement, extending its value beyond traditional tissue culture. Sadat-Hosseini et al. (2022) compared multiple ML models, including MLPNN, K-nearest neighbors (KNN), and gene expression programming (GEP), against traditional multiple linear regression (MLR) for predicting in vitro proliferation in persian walnut (Juglans regia). GEP, particularly when optimized with particle swarm optimization (PSO), provided the most accurate predictions (R² = 0.802 for the ‘chandler’ cultivar, significantly outperforming MLR (R² = 0.412). The study also validated KNN as a simple yet effective tool, highlighting the range of viable ML approaches depending on data complexity and application needs. Şimşek (2024) extended ML applications to genotype-specific optimization in lavender and strawberry cultivars. In lavender, random forest (RF) outperformed other models (MLP, RBF, XGBoost, and Gaussian process) in predicting root traits, particularly for the ‘Festival’ and ‘Fortuna’ genotypes. Meanwhile, MLP and GP models were more effective in predicting traits for ‘Rubygem’. Similarly, in strawberries under PEG-induced drought stress, RF achieved the highest accuracy (91.16%) for root trait prediction, while MLP and GP better predicted plant height and proliferation. These findings emphasize the need to tailor model selection to genotype and trait specificity.
3.2 Radial basis function networks
Radial basis function network is designed primarily for tasks such as function approximation, classification, and regression. This is particularly effective for problems where the relationship between input and output is nonlinear. At their core, RBFN is a form of feedforward neural network consisting of three layers: the input layer, which receives the input features; the hidden layer, which contains radial basis function neurons that transform the input into a higher-dimensional space; and the output layer, which generates the final output after the transformation (Lin et al., 2020a). A radial basis function is a real-valued function whose output depends only on the distance between the input and a specific point (often referred to as a center). The output of the function is maximum at the center and decreases as the input moves farther away from it.
The output of the network can be expressed as (Equation 14):
Where:
y(x) the output of the network, wi are the weights between the hidden neurons and the output layer, ϕ(∥x−ci∥, σi) is the output of the radial basis function at neuron i, ci is the center of the ith radial basis function, σi is the spread (or width) of the ith radial basis function.
Commonly, optimization algorithms like gradient descent, least squares, or LMS (Least mean squares) are used to minimize the error between the predicted and actual outputs.
Research application
Ivashchuk et al. (2018) applied neural networks to model and optimize the sterilization stage for plant seeds, specifically targeting rare and medicinal plants. The study used both MLP and RBF networks to predict the optimal sterilization parameters (sterilant type, concentration, and exposure time). The combination of radial basis function (RBF) networks and the non-dominated sorting genetic algorithm-II (NSGA-II) was used to model and optimize the medium compositions for shoot proliferation in chrysanthemum (Hesami et al., 2019). The study is focused on four outputs, proliferation rate (PR), shoot number (SN), shoot length (SL), and basal callus weight (BCW), based on the concentrations of four variables: BAP, IBA, phloroglucinol (PG), and sucrose. The results showed high prediction accuracy with R² values of 0.88, 0.91, 0.97, and 0.76 for PR, SN, SL, and BCW, respectively. Importantly, the predicted and actual outcomes showed negligible differences, validating RBF-NSGAII as a reliable and efficient tool for optimizing in vitro organogenesis. Rezaei et al. (2023a) focused on the optimization of callogenesis in petunia by developing a predictive model using three machine learning algorithms: multilayer perceptron (MLP), radial basis function (RBF), and generalized regression neural network (GRNN). The goal was to optimize the concentrations of phytohormones to enhance callus formation rate (CFR) and callus fresh weight (CFW). Among the models, GRNN outperformed MLP and RBF with R² values of ≥83%. The study also employed sensitivity analysis, which revealed that IBA was the most influential phytohormone for callogenesis. By integrating GRNN with a genetic algorithm (GA), the study identified an optimized set of phytohormone concentrations that maximized CFR to 95.83%. Experimental validation confirmed the accuracy of the predicted optimal conditions, showing no significant difference between the experimental and GA-predicted results. This approach demonstrates the successful integration of machine learning, sensitivity analysis, and genetic algorithms to optimize tissue culture conditions in a controlled and efficient manner.
3.3 Generalized regression neural network
The generalized regression neural network is a special form of radial basis function (RBF) networks and functions as a non-parametric, memory-based model that provides a smooth approximation of the underlying relationship between inputs and outputs. One of its main advantages is its ability to approximate any arbitrary function directly from training data without requiring an assumed functional form. GRNN consists of four layers: the input layer, which contains neurons representing the features of the input vector and simply passes the data forward without processing; the pattern layer (or radial basis layer), where each neuron corresponds to a training sample and calculates the euclidean distance between the input and that sample, then applies a gaussian kernel to convert this distance into a similarity measure (Equation 15).
Where x is input vector, xi is training sample vector and σ is smoothing parameter (spread or bandwidth).
This structure allows GRNN to provide smooth and effective regression estimates directly from the data, making it a powerful tool for nonlinear function approximation (Specht, 1991).
Research application
Hesami et al. (2020c) used GRNN-GA to model shoot regeneration in wheat, addressing genotype-dependent variability. Based on 10 input variables, GRNN-GA achieved good predictive performance (R² = 0.78) and identified 2,4-D, explant type, and genotype as key factors, supporting more efficient, genotype-independent regeneration protocols. A cost-effective method for paclitaxel production in Corylus avellana was demonstrated using cell suspension culture (CSC) enhanced by fungal elicitors (Salehi et al., 2021). In this study, a general regression neural network optimized by the fruit fly algorithm (GRNN-FOA) was applied to predict and optimize paclitaxel biosynthesis and biomass production, using four input variables: cell extract (CE), culture filtrate (CF), elicitor adding day, and harvesting time. GRNN-FOA showed higher accuracy (R² = 0.88–0.97) than traditional regression (R² = 0.57–0.86), and performed comparably to MLP-GA, with slight advantages for MLP-GA in total and extracellular paclitaxel prediction. GRNN-FOA optimization predicted a maximum paclitaxel yield of 372.89 µg L-¹ under specific conditions, closely matching observed values, validating its potential in optimizing secondary metabolite production in vitro. Rezaei et al. (2023b) evaluated six disinfectants and immersion times on Petunia seed sterilization and germination. Among MLP, RBF, and GRNN models, GRNN performed best. They applied NSGA-II for multi-objective optimization, demonstrating that GRNN-NSGA-II effectively balances contamination control and germination success, offering a robust tool for plant tissue culture optimization.
3.4 Fuzzy neural networks
Neuro-fuzzy logic is a hybrid system that integrates neural networks with fuzzy logic principles to solve complex problems involving uncertainty and imprecision. By combining the learning capabilities of neural networks with the reasoning power of fuzzy logic, it proves especially effective in fields such as control systems, pattern recognition, decision-making, and adaptive systems (Ishibuchi, 1996). Unlike classical logic, where variables are strictly true or false, fuzzy logic allows for a continuum of truth values between 0 and 1. This approach enables systems to model fuzzy rules while simultaneously learning from data. The system operates through four main steps (i) fuzzyfication, where input data is transformed into fuzzy values using membership functions (ii) rule formation, where fuzzy rules describe input-output relationships and can adapt automatically during learning (iii) adaptive learning, where the neural network adjusts the fuzzy rule parameters, like membership functions and rule weights, using algorithms such as backpropagation to minimize errors; and finally, (iv) defuzzification, where fuzzy outputs are converted into crisp values through methods like the centroid technique. Various neuro-fuzzy system implementations exist, notably the Adaptive Neuro-Fuzzy Inference System (ANFIS), known for its efficiency in function approximation. Another approach, the Fuzzy Neural Network (FNN), combines fuzzy logic with neural networks and often uses Mamdani-type rules, which enhance interpretability but increase computational cost. Additionally, Fuzzy C-Means, to partition the input space into clusters, enabling more effective decision-making by training neural networks on these fuzzy clusters (Talpur et al., 2023).
Research application
Recent advances in plant tissue culture optimization increasingly utilize AI, particularly neuro-fuzzy systems and hybrid models combining with evolutionary algorithms, to model complex relationships and enhance culture protocols. Initially, Gago et al. (2010) applied neuro-fuzzy logic to grapevine microshoot rooting, uncovering interactions, such as auxin-sucrose effects, not previously identified by statistical methods. This deepened the understanding of the micropropagation process and demonstrated the rapid applicability of AI modeling. The effective application of neuro-fuzzy logic to mine apricot micropropagation databases has revealed meaningful IF-THEN rules linking cultivars, mineral nutrients, and plant growth regulators with growth parameters (Gago et al., 2011). Their approach validated and extended traditional statistical findings by generating interpretable, reusable knowledge that supports future media optimization. In line with these applications, Alanagh et al. (2014) employed neuro-fuzzy logic to model macronutrient effects on GF677 peach × almond rootstock micropropagation. Their model pinpointed key ion interactions, such as NO3− × Ca2+, that significantly affect shoot quality and development, providing a powerful tool to infer optimal nutrient combinations and mitigate physiological disorders like hyperhydricity. Neuro-fuzzy logic and hybrid AI techniques to dissect nutrient and growth regulator influences on pistachio (Pistacia vera) micropropagation was given by Alanagh et al. (2017). By reducing complex media component combinations via design of experiments (DOE) and applying AI modeling, they uncovered critical ion interactions affecting shoot proliferation, quality, and physiological disorders, enabling more rational design of culture media. Hameg et al. (2020) applied neurofuzzy logic to kiwi (Actinidia arguta) micropropagation, revealing that BAP concentration predominantly influences shoot number, while a combination of BAP and GA3 affects shoot length. Their model also underscored the importance of the number of subcultures and media composition, highlighting AI’s role in interpreting multifactorial effects and guiding protocol refinement. Similarly, Hesami et al. (2019) introduced a hybrid adaptive neuro-fuzzy inference system combined with the non-dominated sorting genetic algorithm-II (ANFIS-NSGAII) for modeling somatic embryogenesis in chrysanthemum. Their results showed high predictive accuracy (R² > 92%) and identified optimal media compositions and light conditions to maximize embryogenesis frequency and somatic embryo number. Sensitivity analysis further highlighted 2,4-D as a critical factor, illustrating how hybrid AI models can guide precise optimization in complex biological systems. Farhadi et al. (2020) modeled paclitaxel production in Corylus avellana cell suspension cultures using ANFIS combined with genetic algorithms. Their model outperformed traditional regression approaches and successfully predicted optimal elicitor and methyl-β-cyclodextrin concentrations and timing to maximize paclitaxel yield, showcasing AI’s utility in bioproduct optimization.
3.5 Generative adversarial networks
Generative adversarial networks are a class of machine learning frameworks designed to generate new data instances that resemble a given dataset. They consist of two neural networks, the generator and the discriminator, that are trained simultaneously in a competitive setting. The unique aspect of GANs lies in their adversarial nature, where the generator creates data, and the discriminator tries to distinguish between real and fake data. Over time, as these networks “battle,” the generator learns to produce increasingly convincing data, while the discriminator improves its ability to differentiate between real and fake samples. As this adversarial process continues, both networks improve iteratively, with the generator producing more realistic data and the discriminator becoming more adept at distinguishing the two (Goodfellow et al., 2020).
Research application
Madsen et al. (2019) addressed the challenge of high intra-class variance and low inter-class variance in plant seedlings, an issue that hinders accurate plant identification using deep learning. To mitigate limited training data, they employed generative adversarial networks (GANs) to generate synthetic images across nine plant species. While the GAN-augmented model reached high recognition accuracy, misclassifications arose mainly during the dicotyledonous growth stage, where visual similarities between species and subtle shape differences confused the model. Notably, the synthetic data proved effective for pretraining a classification model, which performed well even before fine-tuning. Further fine-tuning with real data yielded only marginal improvement, indicating the robustness of the pretrained model. The early detection challenges of verticillium wilt (VW) in cotton, was addressed as a major concern for cotton yields globally (Tan et al., 2025). The study proposed an innovative method integrating GANs with hyperspectral imaging to enhance early-stage detection. Two models, Spe-GAN (spectral enhancement) and Spa-GAN (spatial enhancement), were developed to capture subtle symptoms of VW, a task complicated by limited data. The results showed that these GAN-based models significantly outperformed traditional machine learning (RF, SVM) and deep learning methods (LSTM, ResNet18), with Spe-GAN achieving 94.52% accuracy and Spa-GAN 91.78%. The approach’s success was largely attributed to its ability to augment limited data and enhance model interpretability, offering a new approach for early disease detection in cotton and potentially other plants.
3.6 Convolutional neural networks
A convolutional neural network is designed specifically for processing grid-like data, such as 2D images. Unlike fully connected neural networks (FCNs), CNNs use local receptive fields and shared weights to extract spatial hierarchies of features through convolutions. The input layer, is typically a 3D tensor (height × width × channels). Convolutional Layer, applies a number of filters (kernels) that scan across the input image to extract feature maps (Simonyan and Zisserman, 2014). A filter is a small matrix that slides over the input, performing element-wise multiplication and summing the results to form a single output value per position. Parameters include stride (the number of pixels the filter moves across the image) and Padding (Adding zero borders to control the spatial size of the output). In CNNs, pooling layers perform down sampling to reduce the spatial dimensions of feature maps, lowering computational complexity and helping to prevent overfitting. Max pooling is the most common method, selecting the maximum value within a local window to preserve dominant features. After several convolution and pooling stages, the network transitions to fully connected layers, where flattened feature maps are used for high-level reasoning. The final output layer, also fully connected, uses activation functions suited to the task, softmax for multi-class classification and sigmoid for binary or multilabel classification.
Research application
The integration of deep learning, image processing, and IoT technologies in coconut tissue culture monitoring, as demonstrated by Shavindi et al. (2024), presents a transformative shift in agricultural biotechnology. By automating the classification of callus tissues and enabling continuous culture monitoring, the study addresses critical inefficiencies in traditional tissue culture methods. Complementing this work, Rajapaksha et al. (2024) focused on automating the measurement of plant growth parameters using deep learning models, particularly YOLOv8 and CNN architectures. Their research achieved high accuracy in tasks such as flask detection (precision: 0.990, mAP@50: 0.995), leaf and stem segmentation, and root classification. Moreover, InceptionV3 achieved 96% accuracy in browning disease classification, demonstrating the potential of CNNs in plant health diagnostics. Heenkenda et al. (2024) further highlighted the limitations of conventional tissue culture techniques, especially in early disease detection. Their proposed machine learning-based approach for identifying and predicting bacterial and fungal infections through image analysis offers a critical advancement. The integration of technological tools with agronomical expertise bridges the gap between traditional farming practices and modern precision agriculture, fostering sustainable cultivation and timely disease management.
4 Advancements in genome editing technologies
Genome editing is a revolutionary technology that enables precise modifications to an organism’s DNA, including insertions, deletions, and replacements, to modify biological traits. Several genome editing tools have been developed, each with distinct mechanisms and limitations. Meganucleases are natural endodeoxyribonucleases that recognize and cleave DNA sequences typically 20–30 base pairs long. Although their recognition sites can be re-engineered for site-specific editing, the structural complexity and time-consuming modification process have limited their widespread use (Ashworth et al., 2006; Grizot et al., 2009). Zinc Finger Nucleases (ZFNs) combine engineered zinc finger proteins with the FokI endonuclease to generate site-specific DNA breaks (Kim et al., 1996), while Transcription Activator-Like Effector Nucleases (TALENs) use TALE-derived DNA-binding domains and FokI to induce targeted double-stranded breaks (DSBs) (Christian et al., 2010). Despite their proven efficacy, the intricate and labor-intensive design and assembly of meganucleases, ZFNs, and TALENs have restricted their adoption.
In contrast, CRISPR/Cas systems (Makarova et al., 2011), particularly CRISPR/Cas9 and CRISPR/Cas12a, have gained prominence due to their simplicity, flexibility, and efficiency. Originating as an adaptive immune system in bacteria and archaea, CRISPR/Cas uses guide RNAs to direct Cas proteins to specific DNA sequences adjacent to a PAM (protospacer adjacent motif), enabling targeted cleavage (Garneau et al., 2010). CRISPR/Cas systems introduce DSBs, which are repaired by either non-homologous end joining (NHEJ), an error-prone process leading to insertions or deletions or homology-directed repair (HDR), which allows precise gene correction using a template. Early CRISPR studies in plants demonstrated successful gene knockouts and modifications, highlighting its powerful potential in plant biology (Feng et al., 2013; Li et al., 2013; Nekrasov et al., 2013). Recent advancements include base editing and prime editing. Base editors combine a Cas9 nickase or dead Cas9 (dCas9) with a deaminase enzyme to convert specific nucleotides without inducing DSBs (Komor et al., 2016). Prime editors fuse dCas9 with reverse transcriptase and use a prime editing guide RNA (pegRNA) to direct edits at a target site through a templated reverse transcription mechanism (Anzalone et al., 2019). These next-generation tools have been successfully applied across various plant species, significantly advancing precision breeding and crop improvement (Lin et al., 2020b) (Figure 4).
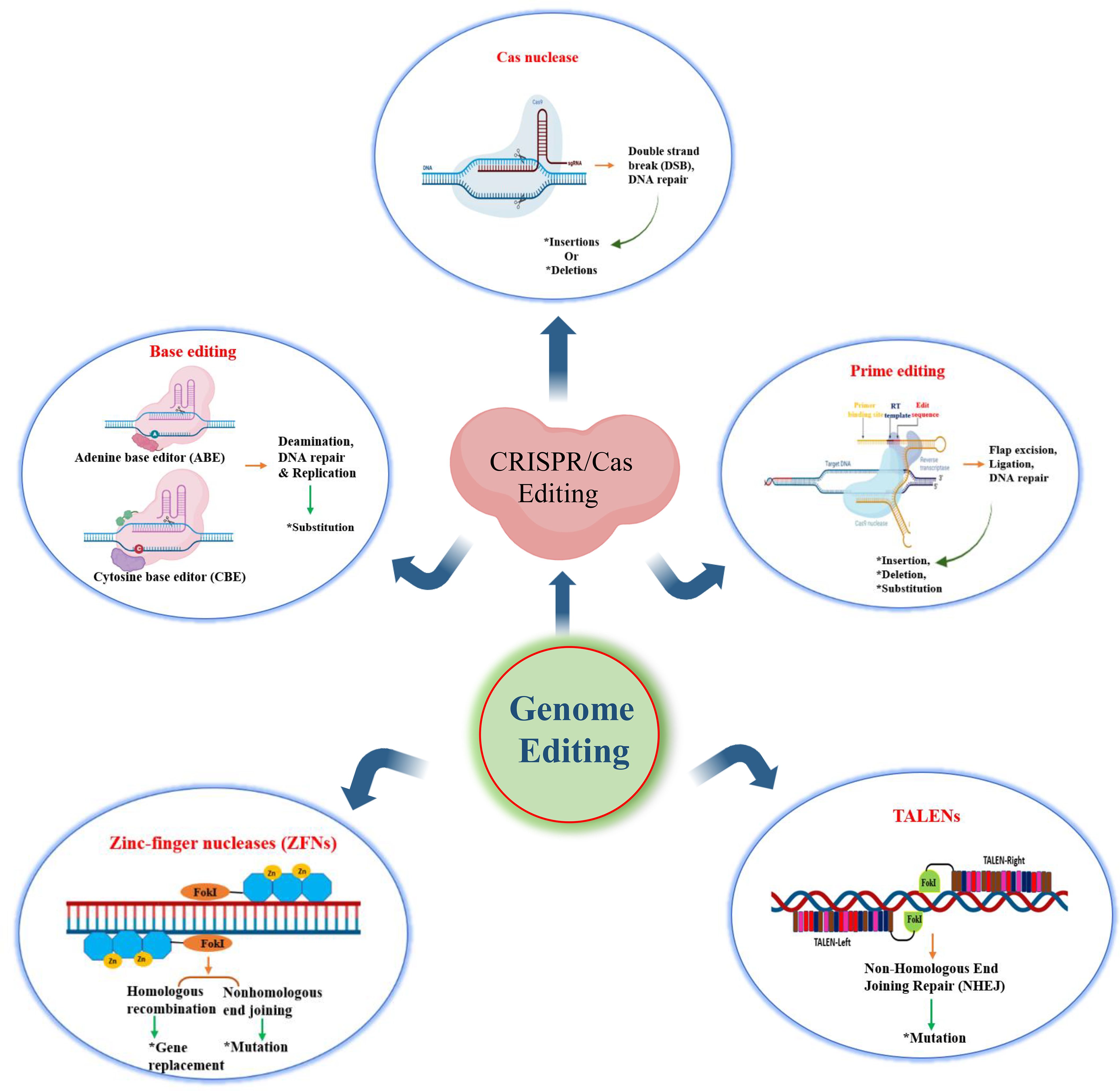
Figure 4. Overview of genome editing techniques: CRISPR/Cas and other nuclease-based approaches (This figure was drawn using free version of BioRender; https://www.biorender.com).
4.1 Searching and screening strategy
The identification phase involves constructing search queries using combinations of keywords, that include: “CRISPR EDITING USING MACHINE LEARNING”, “DEEP LEARNING AND CRISPR EDITING”, “MACHINE AND DEEP LEARNING AND CRISPR EDITING”. This study draws from multiple online databases like pubmed central, scopus and google scholar, compiling research on the application of machine learning (ML) and deep learning (DL) in genome editing technologies. The articles were selected based on their relevance and uniqueness. Peer-reviewed research papers and review articles were considered for analysis. Initial screening was conducted at the abstract level, followed by data extraction and full-text analysis.
4.2 Role of machine learning/deep learning in CRISPR-Cas9 genome editing
The accuracy and accessibility of genome-editing tools particularly CRISPR-associated Cas9 protein has revolutionized the biological research and therapeutic development. A key objective is improving guide RNA (gRNA) design to enhance on-target efficiency (OTE) and minimize off-target effects (OFTE). This review explores recent advances in computational methods, particularly ML and DL, for predicting gRNA performance. It also highlights the role of AI in advancing base editing, prime editing and epigenome editing as well as discusses about tools for predicting gene editing outcome’s and optimizing editing proteins.
4.2.1 sgRNA-DNA sequence encoding
To serve as input for AI models, sgRNA–DNA sequence data must first undergo preprocessing, commonly referred to as “sequence encoding”. This step transforms nucleotide sequences, composed of letters (A, C, G, T), into numerical formats that ML and DL algorithms can process and interpret (Figure 5). Proper encoding is critical for enhancing predictive accuracy. The two most widely adopted encoding methods in CRISPR-Cas9 research are (i) In one-hot encoding, each nucleotide is represented as a binary vector; e.g., A = [1, 0, 0, 0], C = [0, 1, 0, 0]), forming a 4×L matrix for a sequence of length L. Chuai et al. (2018) introduced DeepCRISPR, augmenting the standard one-hot encoding to a (4+n)×23 matrix by appending n epigenetic features per position, optimized for convolutional denoising networks. Lin and Wong (2018) proposed a unified 4×23 matrix encoding both sgRNA and target DNA for use in feedforward and convolutional networks. Charlier et al. (2021) developed a bijective mapping combining separate 4×23 matrices into an 8×23 input for FNNs, CNNs, and RNNs. Zhang et al. (2020a) further expanded the encoding to a 20×L multi-channel matrix incorporating bulges and mismatches, improving off-target prediction with CNNs and data augmentation. (ii) Word embedding, by contrast, assigns each substring (or k-mer) a dense vector in a continuous space, capturing semantic or contextual relationships between sequences. A widely used embedding method is Word2Vec (Mikolov et al., 2013), a neural network-based technique originally developed for natural language processing. Liu et al. (2020a) combined word embedding with transformers, CNNs, and FNNs, achieving performance comparable to advanced one-hot methods. They later used GloVe to generate dense sgRNA vectors fed into bidirectional LSTM and CNN models, reaching state-of-the-art off-target prediction.
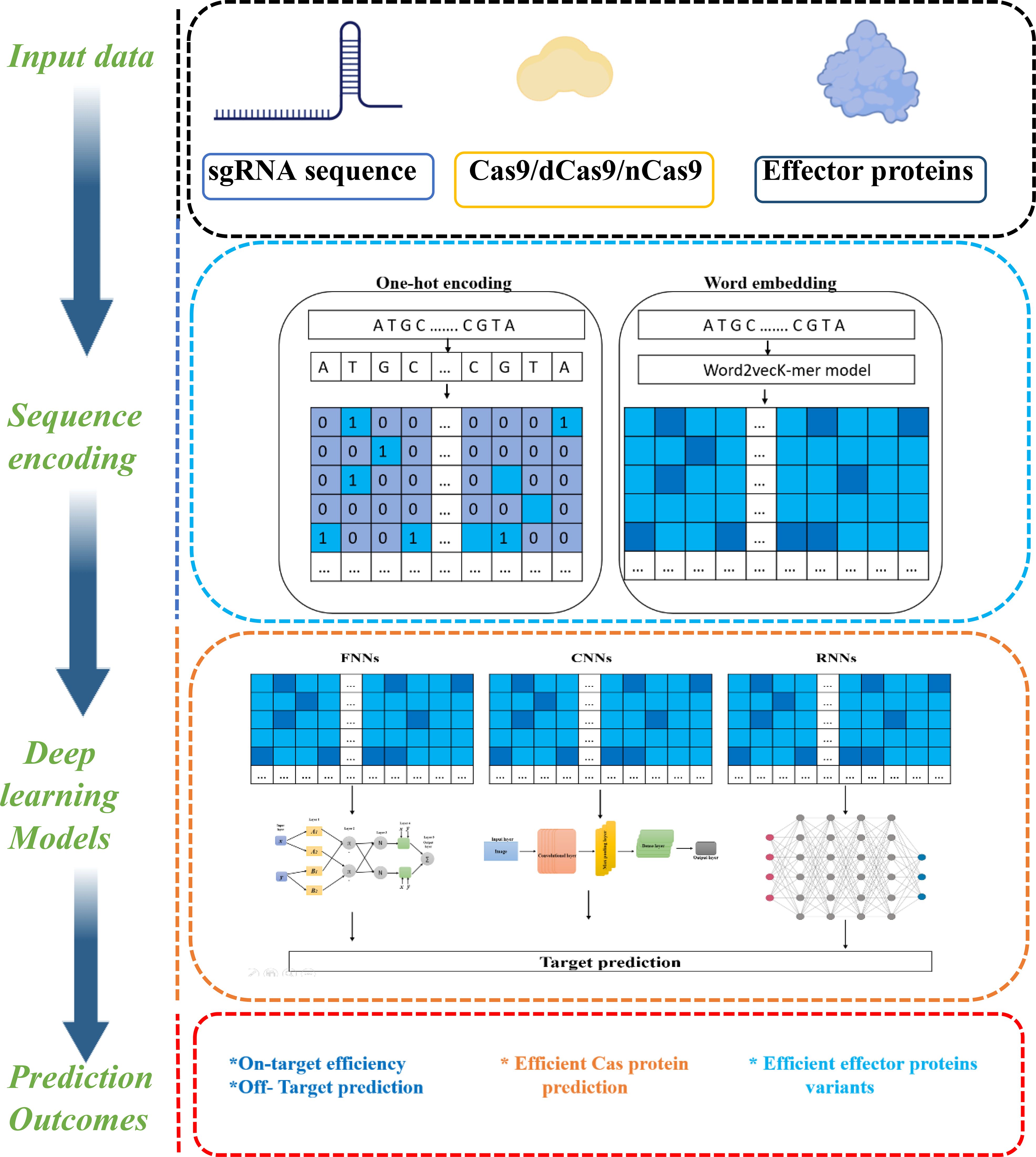
Figure 5. Schematics of deep learning framework for CRISPR-Cas system prediction using sequence encoding techniques (This figure was drawn using free version of BioRender; https://www.biorender.com).
Beyond encoding, traditional ML models rely on engineered features such as nucleotide frequencies, GC content, thermodynamic properties (Doench et al., 2014; Xu et al., 2015), RNA secondary structure and n-gapped dinucleotides (Rahman and Rahman, 2017). Biological annotations like exon positions, amino acid traits, and domain occupancy also enhance predictions (Schoonenberg et al., 2018). Studies have highlighted the importance of positional and thermodynamic features, structural elements, accessibility, mismatch counts, and allele information (Muhammad Rafid et al., 2020). Statistical methods identify key predictors (Hiranniramol et al., 2020), and factors like frameshift probability and amino acid sensitivity improve sgRNA efficiency forecasts (He et al., 2021). Although deep learning models can automatically learn feature representations, manually engineered features remain essential for the performance of conventional machine learning models (Figure 6).
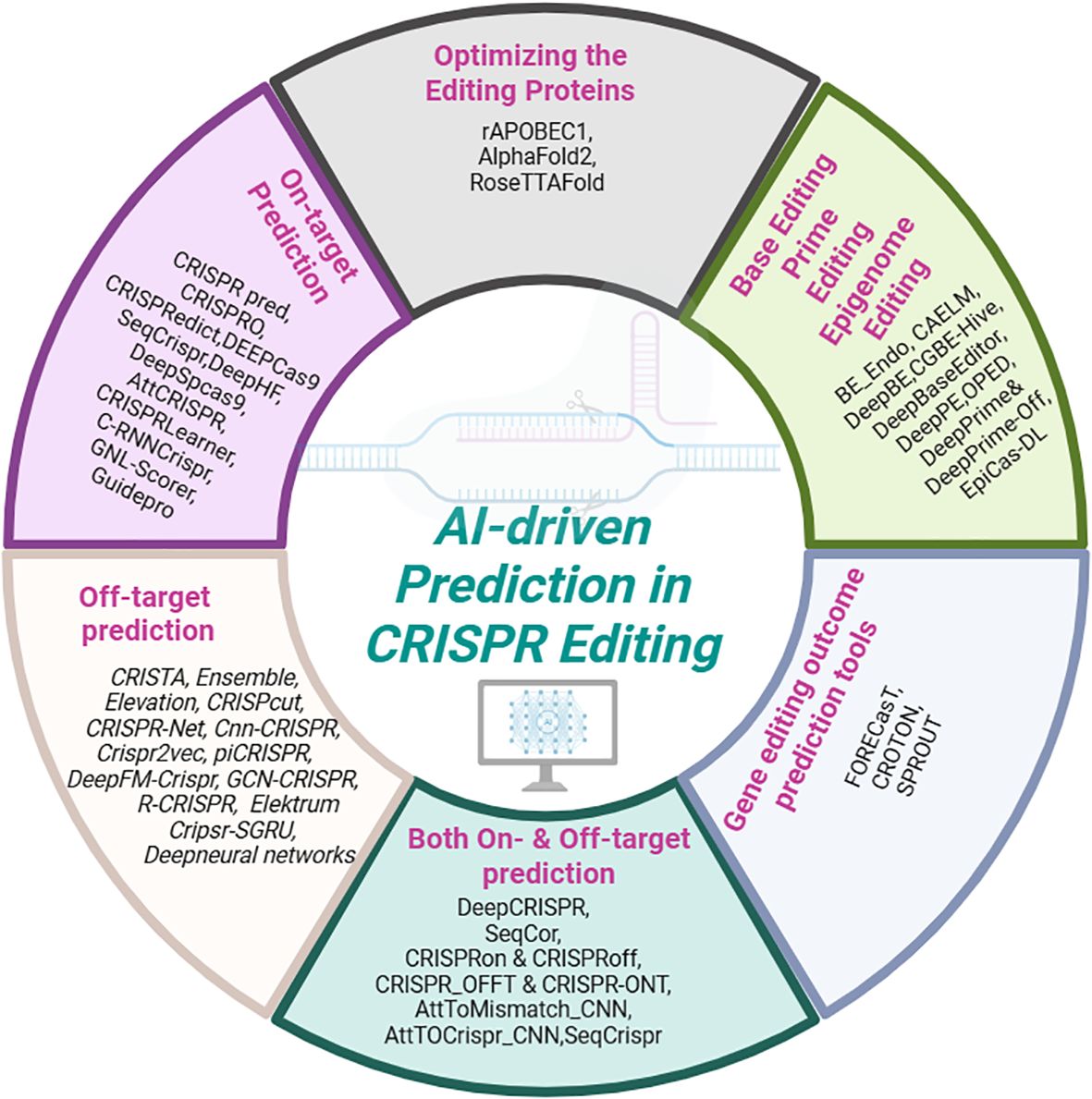
Figure 6. AI-driven prediction tools and applications in CRISPR genome editing (This figure was drawn using free version of BioRender; https://www.biorender.com). .
4.2.2 Off-target prediction in CRISPR/Cas9 editing
In CRISPR gene editing, the single guide RNA (sgRNA) directs the Cas9 protein to a specific genomic site for targeted modification. However, Cas9 can sometimes cleave unintended sites, leading to off-target effects that may disrupt gene sequences and interfere with normal gene function. These unintended effects are influenced by factors such as the structure and length of the sgRNA (Chao and Fei, 2023). Off-target effects are typically categorized into mismatches between the sgRNA and DNA, RNA bulges (insertions), and DNA bulges (deletions). To mitigate off-target activity, AI-based prediction models are employed, primarily using two approaches. In the classification approach, genomic sites are labeled as “1” for off-target and “0” for on-target or non-off-target sites. In the regression approach, a continuous score is assigned to each site, representing the likelihood or severity of off-target activity. Reliable benchmark datasets are essential for training and evaluating these prediction models (Table 3).

Table 3. A summary of studies applying traditional machine learning and deep learning models/methods for off-target prediction in CRISPR/Cas9 editing.
Benchmark dataset and prediction algorithms
GUIDE-seq, developed by Tsai et al. (2015), was one of the first genome-wide methods for off-target detection and remains a benchmark. Using sgRNAs targeting vascular endothelial growth factor (VEGFA), fanconi anemia -associated gene (FANCF), and HEK293 loci, it identified 28 off-target sites (≥0.1% modification) from 403 candidates. Later, Tsai et al. (2017) introduced CIRCLE-seq, a high-throughput in vitro method that profiled 10 gRNAs and detected 7,371 active off-targets, including mismatches, insertions, and deletions. CRISTA (Abadi et al., 2017) is a random forest-based method that uses features such as genomic rigidity and PAM proximity to predict Cas9 cleavage sites. The model was trained on datasets from three unbiased genome-wide profiling methods: GUIDE-seq (Tsai et al., 2015), HTGTS (Frock et al., 2015), and BLESS (Ran et al., 2015; Slaymaker et al., 2016), covering 33 sgRNAs and 872 confirmed off-targets. Peng et al. (2018) utilized datasets comprising 215 and 527 sgRNA–DNA pair, derived from data sources such as GUIDE-seq (Tsai et al., 2015), HTGTS (Frock et al., 2015), CIRCLE-seq (Tsai et al., 2017), and Digenome-seq (Kim et al., 2015, 2016). To handle class imbalance, they applied under-sampling and trained an ensemble SVM classifier, achieving superior off-target prediction performance. Listgarten et al. (2018) developed Elevation, a two-layer regression model for gRNA–target scoring. The first layer predicts off-target activity for single mismatches; the second uses penalized linear regression to combine multiple mismatches. Guide-level scores are generated by aggregating target scores via boosted regression trees, incorporating features like gene context to improve accuracy.
CRISPEY (Chen et al., 2019) dataset consisting of 23,936 samples, distinguishing 306 effect and 23,630 no-effect entries. They evaluated logistic regression, SVM, random forest, and DNN models, with SVM achieving the highest recall (64%) and logistic regression showing the highest accuracy (94%). Zhang et al. (2019) used an ensemble learning approach that combined scores from tools like CCTop (Stemmer et al., 2015), MIT (Haeussler et al., 2016), CFD (Doench et al., 2014), and Cropit (Singh et al., 2015), along with chromatin and conservation data. Their AdaBoost based model demonstrated superior performance on a dataset of 25,332 candidates, including 152 confirmed off-targets. Lazzarotto et al., 2020 trained a gradient boosting model using encoded vectors derived from their CHANGE-seq data. Their model emphasized PAM and protospacer features and outperformed GUIDE-seq and CIRCLE-seq in terms of specificity. Lin and Wong (2018) applied transfer learning to improve predictions on GUIDE-seq data using a model trained on CRISPOR data. While their initial encoding method (a 4×23 matrix) resulted in information loss, a subsequent improvement with a 7×23 lossless encoding significantly boosted model performance. In a follow-up study by Lin et al., 2020c, the authors introduced CRISPR-Net, an LRCN model with inception modules and bidirectional LSTM layers, which further improved off-target prediction accuracy.
Ji et al. (2021) developed DNA-BERT by adapting the original BERT model for DNA sequence analysis. To overcome limited training data, they pretrained DNA-BERT on extensive genomic datasets. The resulting embeddings were combined with handcrafted features, including mismatches and secondary structure, and used with LightGBM to build classification and regression models. DL-CRISPR (Zhang et al., 2020b), a deep learning framework which tackled data imbalance using a novel augmentation method and an ensemble strategy. Charlier et al. (2021) proposed a novel 8×23 matrix encoding for sgRNA–DNA pairs, testing various FNN, CNN, RNN, and traditional classifiers (e.g.,Random Forest, naive Bayes, logistic regression). Using transfer learning on GUIDE-seq and CRISPOR datasets, their framework improved prediction accuracy, raising AUCROC scores by up to 35% over previous methods. Störtz and Minary (2021) introduced piCRISPR, incorporating physically informed features through four encoding schemes (target-guide, target-mismatch, mismatch-type, and target-OR-guide, and used SHAP to evaluate feature importance. Their results on the crisprSQL dataset highlighted the roles of sequence context and chromatin accessibility in cleavage prediction. Niu et al. (2021) proposed R-CRISPR, encoding sgRNA targets into binary matrices fed into a CNN with Rep-VGG layers, followed by bidirectional LSTM for precise off-target prediction.
CRISPR-IP (Zhang and Jiang, 2022) is a deep learning model designed to enhance off-target activity prediction by encoding detailed sequence pair information. It employs a novel encoding scheme that separates functional regions through a function channel and distinguishes bases from base pairs using type channels. The model integrates CNN, BiLSTM, and attention mechanisms to effectively extract features from the encoded sequences. Elektrum (Zhang et al., 2023) is a deep learning framework for modeling biochemical reaction kinetics with high accuracy and interpretability. It first trains kinetically interpretable neural networks (KINNs) on in vitro assay data to predict reaction rates. These KINNs are then embedded into deeper convolutional networks and fine-tuned via transfer learning to predict in vivo outcomes. Crispr-SGRU (Zhang et al., 2024) is a deep learning model for predicting off-target activity with mismatches and indels. It combines inception and stacked BiGRU architectures and uses a dice loss function to handle class imbalance. Interpretability analyses via Deep-SHAP and knowledge distillation reveal that the model effectively captures sequence patterns linked to off-target effects. Bao and Liu (2024) developed DeepFM-Crispr, as a versatile and robust deep learning model, originally developed for the Cas13d system but adaptable to other CRISPR-Cas platforms. By leveraging large language model techniques, it captures complex genetic interactions and sequence features critical for accurate genome editing.
4.2.3 On-target prediction in CRISPR/Cas9 editing
When the guide RNA (gRNA) is designed to target a specific DNA sequence, it forms a complex with the Cas9 protein, guiding the CRISPR system precisely to that genomic locus. Upon binding, Cas9 induces a double-stranded break (DSB) at the targeted site. This break initiates the cell’s intrinsic DNA repair mechanisms, predominantly through two pathways: non-homologous end joining (NHEJ), a quick but error-prone repair process that directly ligates the broken DNA ends, often resulting in small insertions or deletions (indels) at the cleavage. In contrast, homology-directed repair (HDR) uses a homologous DNA template to accurately repair the break, allowing precise editing but generally occurring at lower efficiency (Lackner et al., 2023). While these repair mechanisms enable genome editing, they also pose significant challenges due to their variability and unpredictability, which can lead to unintended genetic alterations at the target site. To mitigate these issues and enhance the precision of genome editing, AI approaches have been developed to predict the efficiency and specificity of gRNAs, thereby forecasting their on-target activity and minimizing off-target risks (Table 4).
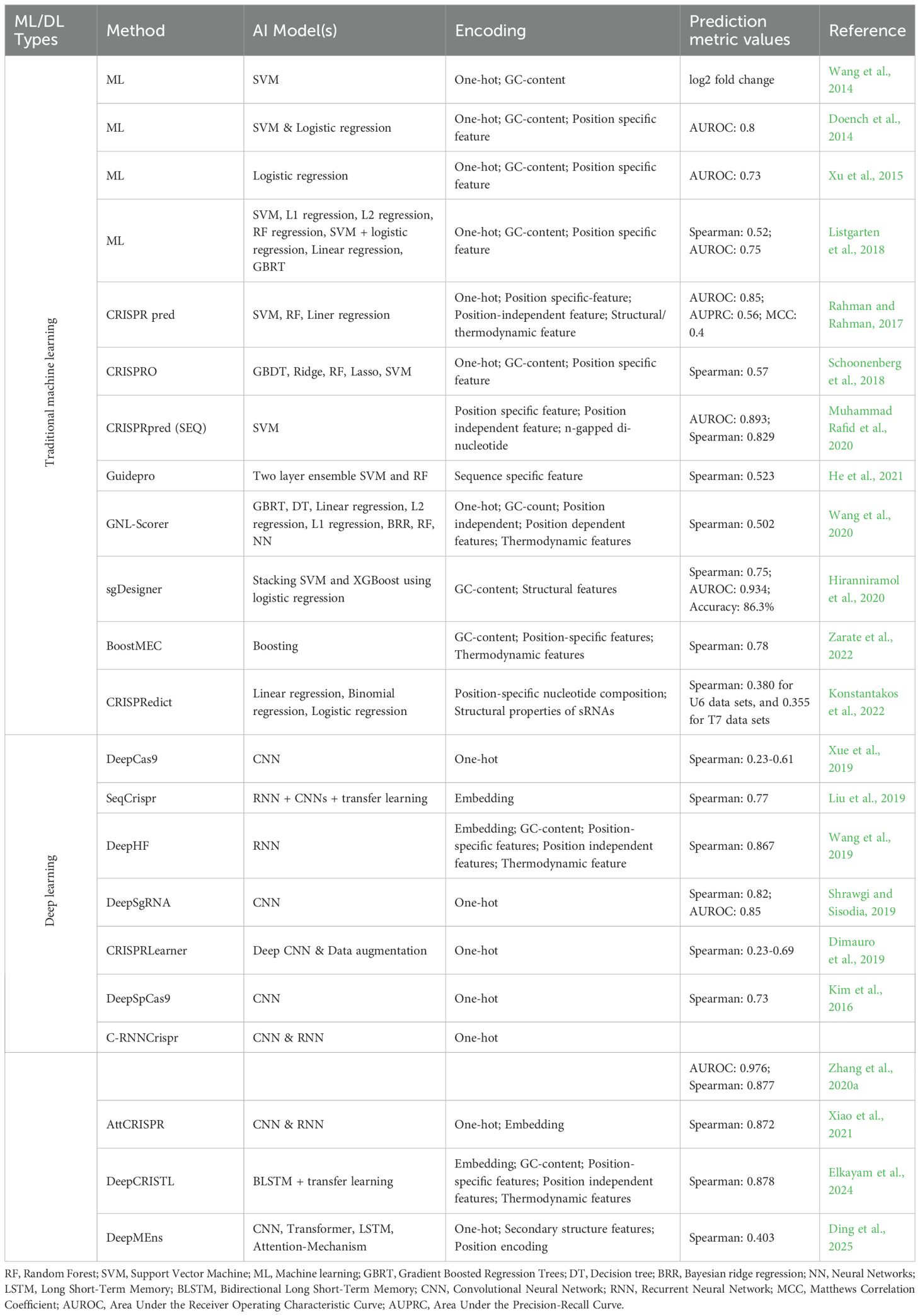
Table 4. A summary of studies applying traditional machine learning and deep learning models/ methods for on-target prediction in CRISPR/Cas9 editing.
Benchmark dataset and prediction algorithms
Early machine learning approaches for on-target prediction include Wang et al. (2014), employed SVMs with fold-change data to classify sgRNA efficacy from a library of 73,000 sgRNAs targeting ribosomal and non-ribosomal genes in HL-60 and KBM7 cells; Doench et al. (2014), used logistic regression to distinguish top and bottom activity quintiles and Xu et al. (2015), applied elastic-net regression, outperforming previous models. The studies from Rahman and Rahman, 2017 and Muhammad Rafid et al., 2020, emphasized feature engineering’s importance, developing SVM-based tools (CRISPRpred and CRISPRpred(SEQ)) that rival deep learning models like DeepCRISPR. CRISPRO, a computational pipeline developed by Schoonenberg et al. (2018), links functional scores of guide RNAs to genomic, transcript, and protein-level coordinates and structures. Xue et al. (2019) introduced DeepCas9, a CNN-based framework outperforming traditional methods. DeepHF method was used to generate the largest dataset, measuring indel rates of over 50,000 gRNAs across ~20,000 genes for three SpCas9 variants (Wang et al., 2019). Deep learning advancements include the DeepSgRNA CNN model, which eliminates manual feature engineering and achieves state-of-the-art accuracy on GenomeCRISPR data (Shrawgi and Sisodia, 2019).
CRISPRLearner (Dimauro et al., 2019) utilizes a deep convolutional neural network to automatically extract sequence features and predict sgRNA efficiency. It can operate using either pre-trained models or be trained on custom datasets. The CNN employs linear regression to estimate efficiency from the training data, with ten different models developed across ten unique gene datasets. GuidePro, a two-layer ensemble model was introduced by He et al. (2021) to prioritize sgRNAs for protein knockout by integrating diverse predictive features. When tested on independent datasets, GuidePro consistently outperformed existing tools in predicting protein loss-of-function phenotypes, highlighting its reliability and effectiveness across various CRISPR/Cas9 knockout applications. GNL-Scorer is used for cross-species sgRNA activity prediction by combining multiple datasets and models (Wang et al., 2020). Hiranniramol et al. (2020) developed the sgDesigner plasmid library, consisting of 12,472 oligonucleotides, to assess sgRNA efficiency using machine learning. Zarate et al. (2022) developed BoostMEC, a LightGBM-based gradient boosting model that outperformed many deep learning methods while providing greater interpretability.
CRISPRedict an interpretable web tool employing regression models and nucleotide composition features, achieving competitive accuracy (Konstantakos et al., 2022). C-RNNCrispr, a hybrid model introduced by Zhang et al. (2020a), integrates convolutional neural networks (CNNs) and bidirectional gated recurrent units (BGRUs) to predict CRISPR/Cas9 sgRNA on-target activity. The model has two branches, an sgRNA branch and an epigenetic branch—that process a binary-encoded sgRNA sequence and four epigenetic features, respectively, ultimately producing a regression score. AttCRISPR, a deep learning approach designed for intrinsically interpretable on-target activity prediction. By combining encoding-based and embedding-based methods through an ensemble learning strategy, it improves both interpretability and prediction accuracy (Xiao et al., 2021). DeepMEns, is a deep learning ensemble model for predicting sgRNA on-target activity (Ding et al., 2025). It combines five sub-regression models, each with three components utilizing different encoding methods and neural network architectures. By integrating multi-feature representation, ensemble learning, and attention mechanisms, DeepMEns achieved superior performance on three independent test sets, WT-SpCas9, eSpCas9(1.1), and SpCas9-HF1.
4.2.4 Both on- and off-target prediction in CRISPR/Cas9 editing
The benchmark datasets included in this context contains both on-target and off-target information (Table 5). Doench et al. (2016) developed optimized genome-wide sgRNA libraries for human and mouse using machine learning models to predict on- and off-target activity. Evaluated models included linear regression, L1/L2-regularized regression, L1 logistic regression, linear-kernel SVM, hybrid SVM-logistic regression, random forest, and gradient-boosted regression trees. DeepCRISPR dataset introduced by Chuai et al. (2018), has approximately 680 million sgRNA sequences from 13 human cell lines (e.g., HEK293, MCF-7, K562, HCT116), enriched with cell type–specific epigenetic information. They also developed DeepCRISPR, a deep learning framework capable of jointly predicting on-target knockout efficiency and off-target cleavage. The model utilizes a deep convolutional denoising neural network (DCDNN) for feature extraction, followed by a hybrid neural network that performs both classification (via softmax) and regression (via an identity function). Liu et al. (2019) introduced two transformer-based models, AttnToMismatch_CNN for off-target specificity and AttnToCrispr_CNN for on-target efficiency. Key innovations include attention-based architectures for sequential genomic data, integration of cell-specific network-based gene properties, a novel matrix encoding for sgRNA–DNA pairs, and a universal feature ranking algorithm for model interpretability. SeqCor, an open-source tool based on random forest, designed to extract sequence features affecting both sgRNA efficiency and off-target activity, while minimizing bias from library design (Liu et al., 2020b). Two attention-based deep learning models: CRISPR-ONT for predicting sgRNA efficiency and CRISPR-OFFT for off-target specificity was developed by Zhang et al. (2021). CRISPR-ONT combines a CNN with attention to emphasize PAM-proximal regions crucial for Cas9 cleavage. CRISPR-OFFT integrates CNN and attention to extract multi-level features from sgRNA-DNA pairs for off-target prediction. Xiang et al. (2021) developed CRISPRon and CRISPRoff, two deep learning models designed for predicting gRNA activity. CRISPRon estimates on-target efficiency for gRNAs with NGG PAM sequences, while CRISPRoff evaluates specificity and the relative likelihood of off-target cleavage. Both models combine convolutional neural networks (CNNs) with gradient boosting trees and incorporate position-specific, position-independent, and thermodynamic features.
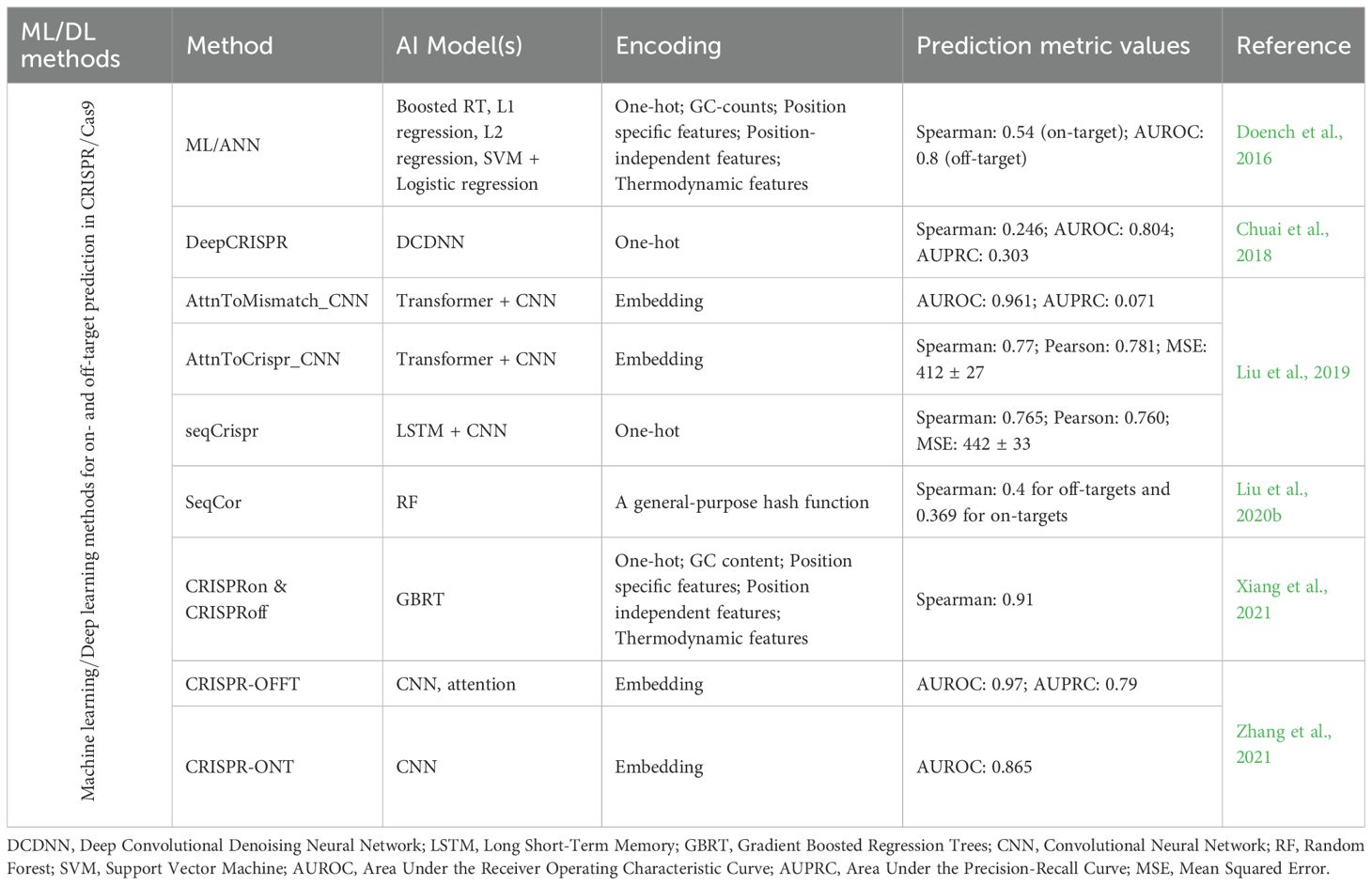
Table 5. A summary of studies applying traditional machine learning and deep learning models/methods for on- and off-target prediction in CRISPR/Cas9 editing.
4.2.5 Performance metrics for on-target and off-target detection
Accurate evaluation of on-target and off-target effects in CRISPR/Cas9 systems relies on robust performance metrics. Commonly used measures include AUPRC (Area under precision recall curve), PR-AUC (Precision recall area under curve), MCC (Matthews correlation coefficient), Spearman and Pearson correlations, and the CFD (Cutting frequency determination) score, each with specific strengths and limitations. AUC-ROC (Area under the receiver operating characteristic curve metrics) provides an overall measure of classification performance but may be less informative in highly imbalanced datasets. PR-AUC, by contrast, is more effective in highlighting rare off-target events due to its focus on precision and recall. MCC considers all elements of the confusion matrix and is well-suited for imbalanced data, though it can be sensitive to small sample sizes. Spearman and Pearson correlations assess the agreement between predicted and observed outcomes, with Spearman capturing monotonic trends and Pearson evaluating linear relationships; both, however, can be affected by outliers and skewed distributions. The CFD score, specific to CRISPR, estimates the likelihood of off-target cleavage but may not fully account for sequence context in rare events. A combined use of these metrics offers a more comprehensive assessment of CRISPR editing accuracy, particularly for detecting low-frequency off-target effects (Doench et al., 2014).
4.2.6 Gene editing outcome prediction tools
About five computational tools have been developed for predicting CRISPR-Cas9 editing outcomes, each introducing novel benchmark datasets. inDelphi (Shen et al., 2018) was the first, predicting 90 microhomology (MH) deletions, 59 non-MH deletions, and 4 one-base insertions using data from 1,095 target sites across human and mouse cell lines (HEK293, K562, HCT116, mESCs, U2OS). FORECasT, predicts gRNA-induced mutations using synthetic constructs, with over 31,000 samples and ~440 mutation types (Allen et al., 2018). SPROUT, provides detailed editing outcome statistics, including indel rates, lengths, edit diversity, inserted bases, and efficiency (Leenay et al., 2019). CROTON, employs CNNs and neural architecture search to predict 1 bp indels, deletion frequencies, and frameshifts from raw sequences, trained on FORECasT and evaluated on SPROUT’s endogenous T-cell data (Li et al., 2021a). Apindel (Liu et al., 2022a) integrates FORECasT and Lindel data to predict 557 mutation types, covering insertions from 1 bp to ≥29 bp across multiple classes.
4.2.7 Tools for optimizing the editing proteins
In addition to designing highly specific guide RNAs, off-target effects in genome editing can be reduced by modifying or discovering novel editing proteins. New bioinformatics approaches have led to the identification of new proteins like LrCas9 (Zhong et al., 2023), Cas13X, and Cas13Y (Xu et al., 2021). Protein engineering methods have further optimized existing editors such as VQR-Cas9 (Kleinstiver et al., 2015), SpRY (Walton et al., 2020), xCas9 (Hu et al., 2018), SpCas9-NG (Nishimasu et al., 2018), eSpCas9 (Slaymaker et al., 2016), LZ3 Cas9 (Schmid-Burgk et al., 2020) offering improvements over SpCas9 in terms of size, PAM compatibility, specificity, and efficiency. Many of these enhanced systems have already been applied successfully in plant genome editing (Hua et al., 2019; Li et al., 2021b). Traditional protein discovery and optimization methods are time-consuming and costly, often requiring detailed structural knowledge. However, advances in machine learning, particularly structure prediction tools like AlphaFold (Senior et al., 2020), AlphaFold2 (Jumper et al., 2021), and RoseTTAFold (Baek et al., 2021), have significantly improved the prediction of protein structures and functions. For example, AlphaFold2 was used to compare wild-type Cas12a with its active variants, revealing structural changes that explain functional differences and guiding the design of more specific Cas variants (Ma et al., 2022). Huang et al. (2023) applied AlphaFold2 to predict the structures of proteins containing deaminase domains, grouped them by structural similarity, and experimentally validated members of the SCP1.201 clade.
4.2.8 Role of AI in enhancing base editing, prime editing and epigenome editing
In recent years, the development of ML and DL models has significantly advanced the ability to predict the outcomes and efficiency of base editing in CRISPR-Cas9 systems (Table 6). BE-DICT, an attention-based DL model that takes protospacer sequences as input and outputs editing probabilities per nucleotide, achieving AUC scores of 0.86 for ABEmax, 0.94 for CBE4max, 0.66 for ABE8e, and 0.97 for TargetAID (Marquart et al., 2021). Pallaseni et al. (2022) introduced an ML model that used DNA sequence and positional information to predict editing outcomes for various base editors. The model achieved precision scores between 0.49 and 0.72, and highlighted the challenge of predicting editing efficiency and bystander mutations due to strong sequence dependence around the target base. CAELM, proposed by Li et al. (2022), uses chromatin accessibility and sequence features to predict CBE efficiency. Because the available dataset was relatively small (1,134 targets), they opted for the XGBoost algorithm, which often outperforms deep networks on limited data. Their model achieved a pearson correlation of 0.64. Chen et al. (2022) introduced a new class of C•G-to-G•C base editors (CGBEs), with editing profiles evaluated across 10,638 genomic sites, enabling over 90% precision and up to 70% efficiency in correcting 546 disease-related transversion SNPs. Park and Kim (2023) presented DeepABE and DeepCBE, DL-based web tools for ABE and CBE outcome prediction.
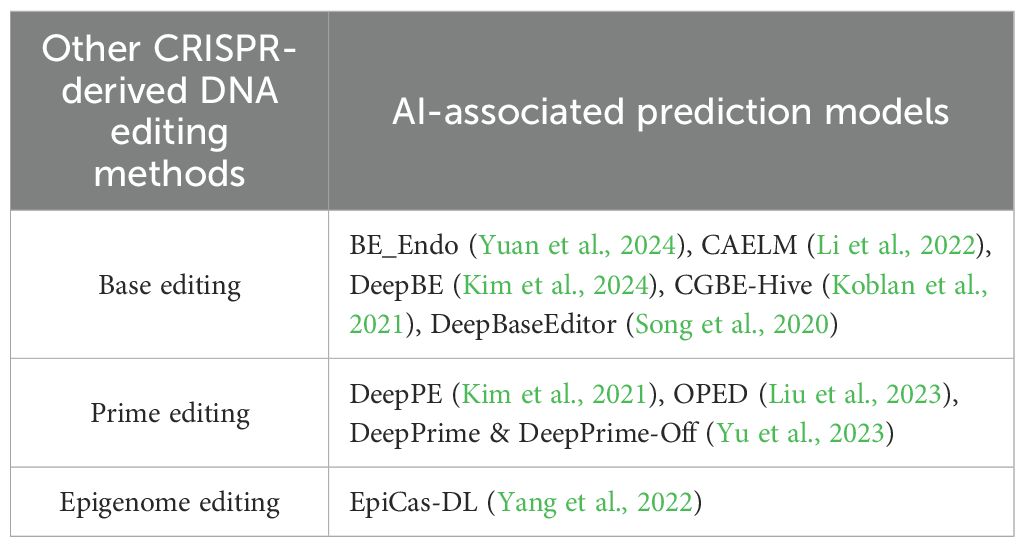
Table 6. List of other CRISPR-derived DNA editing methods and their AI-associated prediction models.
Several ML and DL models have been developed to predict the outcomes of prime editing (PED). Koeppel et al. (2023) analyzed factors influencing PED insertion efficiency and developed an ML model that incorporates features such as nucleic acid structure, insert length, secondary structure, and the expression levels of TREX1 and TREX2, enzymes known to degrade the 3′ DNA flap essential for successful insertions. Their findings showed that insertion efficiency is strongly influenced by the insert sequence’s length, composition, and structural properties. In addition, Mathis et al. (2023) introduced PREDICT, a deep learning model based on a recurrent neural network (RNN), trained on over 90,000 PED experiments. PREDICT accurately forecasts editing outcomes and rates for small genomic alterations, achieving Spearman correlation values of 0.85 for intended edits and 0.78 for unintended ones.
CRISPR/Cas-based epigenome editing is a powerful method for regulating gene expression without altering the underlying DNA sequence. This is achieved by targeting specific genomic regions with CRISPR/Cas systems fused to epigenetic modifiers (Goell and Hilton, 2021). Studies by Rauschert et al. (2020) and Machnicka and Wilczynski (2020) demonstrate how AI techniques can aid in interpreting epigenetic mechanisms and reconstructing the epigenetic code. To support CRISPR-mediated epigenome editing (epi-GED) specifically, dedicated AI tools are beginning to emerge. One such tool is EpiCas-DL (Yang et al., 2022), a deep learning model designed to predict the activity of sgRNAs used in epigenetic editing. EpiCas-DL incorporates four key epigenetic features, gene expression, DNA methylation, chromatin accessibility, and the distance to the transcription start site, to enhance prediction accuracy.
4.2.9 Current research gaps and prospective advances in AI-driven genome editing
Current CRISPR/Cas9 prediction models primarily focus on SpCas9, limiting their applicability across other CRISPR systems. Generalization to proteins like SaCas9 or Cpf1 remains underexplored. Advanced architectures such as LSTM, GRU, transformers, and embedding-based methods (e.g; DNA2vec k-mer representations) (Ng, 2017), offer potential for learning universal patterns applicable across systems and broader genomic contexts, including non-coding regions and whole-genome analysis. Performance is constrained by limited, imbalanced, and poorly labeled datasets. Addressing off-target effect (OFTE) prediction remains challenging. Integration of multi-omics data (genomic, epigenomic, transcriptomic, proteomic) can enhance predictive accuracy, especially by capturing features like DNA methylation and chromatin accessibility (Chuai et al., 2018; Sun et al., 2024). Deep learning models, particularly deep neural networks (DNNs), are composed of multiple layers with numerous neurons, making the design of efficient architectures and the tuning of hyperparameters both critical and challenging tasks. To optimize model performance in CRISPR/Cas9 applications, techniques such as evolutionary strategies, random search, exhaustive grid search, and Bayesian optimization (Goodfellow et al., 2016; Cho et al., 2020) should be systematically explored. Alongside architectural improvements, enhancing the explainability and interpretability of these models has become increasingly important (Goodfellow et al., 2016). Recent methods (Chou et al., 2021; Vilone and Longo, 2021) aim to address this by providing better insights into model behavior, which is particularly valuable for understanding on- and off-target effects in genome editing, a key factor for clinical translation. In terms of input representation, feature engineering has shown strong potential in improving prediction accuracy.
Emerging approaches include the use of hybrid deep learning models (e.g., CNN–RNN combinations), epigenetic information, and advanced DL architectures such as transformers. Transfer learning and pretraining on large-scale datasets like encyclopedia of DNA elements (ENCODE) or genotype-tissue expression (GTEx) (The ENCODE Project Consortium, 2012) could reduce reliance on extensive CRISPR-specific annotations. Engineered features like epigenetic markers, microhomology properties, and RNA fold scores have proven useful (Wang et al., 2019; Shrawgi and Sisodia, 2019), while CNNs and LRCNs offer automated feature extraction directly from sequence data, reducing biases associated with manual selection (Lin et al., 2020c; Shrawgi and Sisodia, 2019; Niu et al., 2021). Tools like SHapley Additive exPlanations (SHAP) (Lundberg and Lee, 2017), Tree-SHAP (Lundberg et al., 2018), and Deep-SHAP (Lundberg and Lee, 2017) further support model interpretability by quantifying each feature’s impact using Shapley value-based approaches. Model reliability can also be improved through uncertainty quantification. This involves assessing aleatoric uncertainty (stemming from data noise) and epistemic uncertainty (from model limitations) (Mazoure et al., 2022).
5 Key notes for applying AI and machine learning in plant biotechnology
Effective deployment of AI and ML in plant biotechnology hinges upon rigorous data management and understanding the intrinsic characteristics of the datasets involved.
5.1 Data considerations for AI/ML in plant biotechnology
The quality and quantity of data critically determine the reliability and applicability of machine learning models in biological research. Large, diverse datasets are essential for training complex models, particularly deep learning architectures, to prevent overfitting and enhance generalization (Kaushik et al., 2025). Standardized data collection, aided by automated sensors and uniform protocols, ensures consistency and reduces variability from experimental or operator differences (Ibrahim et al., 2023). Proper sampling strategies, such as stratified sampling, capture sufficient diversity across genotypes and environments, minimizing bias and supporting robust model training. Accurate labeling is vital for supervised learning, and expert annotation with iterative validation reduces error propagation (Hesami and Jones, 2021).
Data preprocessing steps such as cleaning or noisy observations and handling outliers ensure dataset integrity, while normalization and feature scaling prevent dominance of large-magnitude features and accelerate model convergence. Partitioning data into training, validation, and test sets through cross-validation enables fair evaluation and tuning (Hesami et al., 2020a; Niazian and Niedbała, 2020). Effective feature engineering, including biologically meaningful variable extraction and appropriate categorical encoding (e.g., one-hot encoding of nucleotide sequences), enhances model interpretability and performance. Finally, interpreting model outputs using comprehensive metrics (Liu et al., 2020a). Addressing data imbalances and avoiding overfitting or confounding biases require continuous vigilance throughout model development and deployment.
5.2 Choosing the most appropriate AI/ML approach
Selecting an optimal AI/ML approach requires assessing data volume, type, complexity, and the desired outcome (Hesami et al., 2020a). In tissue culture optimization, such as media formulation or growth prediction, regression and ensemble methods like random forests offer robustness to nonlinear and multivariate relationships (Aasim et al., 2022). Simpler models (e.g., linear regression) suit smaller, well-curated datasets, providing interpretability and lower computational cost (Zarbakhsh et al., 2024). Conversely, complex or high-dimensional problems benefit from neural networks or hybrid models integrating evolutionary algorithms to address biological complexity and data heterogeneity. For classification and detection tasks, particularly those involving images, convolutional neural networks (CNNs) excel by capturing hierarchical spatial features (Shavindi et al., 2024). When data are limited, tree-based or probabilistic classifiers (e.g., decision trees, naïve Bayes) serve as interpretable, data-efficient alternatives (Aasim et al., 2023). Reinforcement learning holds promise for dynamic or sequential decision-making, though its application in plant tissue culture remains in early exploration (Nasti et al., 2024). Across all scenarios, model selection should account for computational scalability, interpretability, and the practicality of experimental validation. Hybrid workflows that couple AI models with domain expertise and iterative experimentation typically produce more reliable and impactful results.
Applying machine learning (ML) to CRISPR genome editing begins with clearly defining the problem type. Predicting on-target efficiency constitutes a regression task (Wang et al., 2014), while off-target prediction involves classification or ranking (Chao and Fei, 2023). sgRNA design is addressed through generative modeling or optimization, base-editing outcome prediction through multiclass classification, and Cas variant selection through classification or recommendation models (Hiranniramol et al., 2020). The choice of ML method depends on data size and nature. Sequence-based tasks typically use CNNs or transformers, feature-based analyses favor gradient boosting or random forests, structure-based studies employ graph neural Networks (GNNs), and optimization problems leverage reinforcement learning or genetic algorithms. Data representation is equally critical (Liu et al., 2020a). sgRNA sequences are encoded as one-hot vectors, k-mers, or learned embeddings for deep models; structural information is modeled as graphs for GNNs; and epigenetic or chromatin features are expressed as numerical vectors for tree-based or neural models (Chuai et al., 2018). PAM sequences and experimental outcomes, such as indel frequencies, are incorporated into regression or classification models as relevant predictors (Zhang et al., 2020a).
6 Applications of artificial intelligence in bioinformatics
Artificial intelligence (AI) has become an essential tool in bioinformatics and computational molecular biology. With a broad array of AI algorithms now available, researchers are able to develop systems to classify, analyze, and mine biological data, resulting breakthroughs in genomics, drug discovery, and personalized medicine. Deep learning models such as AlphaFold have revolutionized protein structure prediction (Palvadi and Kadiravan, 2025). AI also assists in analyzing genomic, transcriptomic, and metabolomic data to identify disease biomarkers and predict outcomes (Venkadesh et al., 2025). However, challenges such as data quality and interpretability remain, highlighting the need for continued development (Bukhari et al., 2025). Furthermore, the wide variety of available methods makes it difficult to select the most appropriate approach for a given dataset. Consequently, there is an increasing demand for tools that present data intuitively, providing context, accuracy estimates, and clear explanations. The central motivation behind these AI-driven approaches is to gain a deeper understanding of organismal evolution while managing the complexities of working with noisy or incomplete data.
7 Challenges and future directions
•While the integration of AI into plant tissue culture and genome editing holds immense potential, several challenges must be addressed. In plant tissue culture, major hurdles include genotype dependence, contamination risks, somaclonal variation, and the high cost and labor-intensive nature of maintaining sterile environments. These limitations hinder the consistent and scalable propagation of plant species. AI, particularly through machine learning models, has shown promise in optimizing media compositions, predicting shoot and root regeneration, and reducing empirical trial-and-error. Future directions in this area include the development of generalized predictive models that can adapt across species, the integration of multi-omics and sensor data for dynamic culture optimization, and the deployment of robotic systems for high-throughput automation (Williamson et al., 2021). However, the success of AI in tissue culture relies heavily on the availability of diverse, high-quality data and the development of user-friendly interfaces accessible to non-experts (Zhang et al., 2025).
•The AI driven methodologies would provide an opportunity to build end-to-end, data-driven pipelines that reduce the time and cost associated with plant transformation and trait development. However, challenges such as data heterogeneity, lack of standardization, high implementation costs, and regulatory uncertainties must be addressed. Developing AI models that can generalize across diverse genotypes, supporting open data sharing, and building interdisciplinary teams of plant scientists, engineers, and data experts are essential steps forward (Farrell et al., 2025; Finzel, 2025). Additionally, ethical concerns related to gene editing, especially in food crops, require transparent regulatory frameworks and public engagement (Harfouche et al., 2021).
•In CRISPR-based genome editing, AI contributes to guide RNA (gRNA) design, prediction of off-target effects, and understanding editing outcomes, all of which are crucial for enhancing editing precision and minimizing unintended changes (Han et al., 2025). Despite this, the effectiveness of CRISPR in plants is often limited by inefficient delivery systems, tissue culture-dependent regeneration processes, and the complexity of polygenic traits (Sebiani-Calvo et al., 2024; Rafiq et al., 2025). AI can accelerate trait discovery by analyzing multi-omics and phenotypic data to identify target genes and regulatory networks. Moreover, future innovations aim to reduce or eliminate the reliance on tissue culture altogether, enabling direct in planta editing. This would not only streamline workflows but also expand editing capabilities to recalcitrant species. Integration of AI across the editing pipeline, from gRNA design to phenotypic evaluation, can significantly enhance efficiency, especially when coupled with high-throughput imaging and omics-based phenotyping platforms (Kim et al., 2025).
•Looking ahead, future research directions include the development of explainable AI models tailored for tissue culture, transfer-learning or few-shot learning approaches to allow models to generalize between plant species, synthetic data and augmentation to overcome limited datasets. Tighter integration of AI with sensor/automation systems and Internet of Things (IoT) environments for real-time feedback in culture systems. The field is also moving toward cloud-based platforms for data/model sharing, which shall facilitate collaborative development and validation of AI-driven tissue culture workflows (Holzinger et al., 2023).
•There is limited research translating AI-driven innovations from controlled laboratory settings to real-world field applications, particularly under variable environmental conditions. Moreover, interdisciplinary collaboration between AI, plant physiology, and molecular biology is still limited, constraining system-level advancements. Finally, ethical and regulatory considerations surrounding AI in plant biotechnology such as societal impacts, biosafety, and governance, remain underexplored, highlighting the need for a more comprehensive and responsible approach to AI-driven plant sciences.
8 Conclusion
Most studies have focused on optimizing experimental conditions and improving automation and efficiency through AI-based models that refine media composition, growth parameters, and editing targets. AI can significantly enhance plant tissue culture methods by optimizing growth conditions, making the process more efficient and scalable. Given the complex and non-deterministic nature of tissue culture, ML and optimization algorithms have been effectively applied to fine-tune key parameters (Hesami and Jones, 2020b). Future research may focus on ML-based virtual simulations to reduce experimental time and accelerate crop improvement (Ray, 2019). However, challenges such as data availability, scalability, and regulatory frameworks must be addressed to fully realize AI’s potential in plant biotechnology. Automated AI-powered systems have further to be streamlined for monitoring and quality control, reducing human error and conserving resources. Gene editing has become increasingly accessible, and advancements in computing power and ML continue to simplify the process (Ran et al., 2013). ML/DL/ANNs have helped overcome key challenges in the CRISPR/Cas system by improving the accuracy of outcome predictions and reducing the need for extensive experimental optimization (Chuai et al., 2017). Enhanced protein structure and functional residue predictions have also lowered the resources required to develop more effective gene-editing tools (Huang et al., 2023).
Author contributions
MN: Writing – original draft, Writing – review & editing. AR: Funding acquisition, Resources, Supervision, Writing – review & editing. BP: Writing – review & editing. HY: Writing – review & editing. PB: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Publication fees were covered by the National Research Council of Canada and Anandi Botanicals (through Result Driven Agriculture Research and Sustainable Canadian Agriculture Partnership).
Acknowledgments
The authors wish to express their sincere gratitude to Devasis Dasgupta of Anandi Botanicals Inc., Lethbridge, Alberta, and Ali Salehpour, Research Officer at the National Research Council of Canada (NRC), Saskatoon, for their critical review and insightful comments on the manuscript. The authors also acknowledge Leah Knickle and Shengjian Ye of the NRC for their valuable assistance in preparing the tables.
Conflict of interest
Author AR was employed by the company Anandi Botanicals Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aasim, M., Ali, S. A., Altaf, M. T., Ali, A., Nadeem, M. A., Baloch, F. S., et al. (2023). Artificial neural network and decision tree facilitated prediction and validation of cytokinin-auxin induced in vitro organogenesis of sorghum (Sorghum bicolor L.). Plant Cell Tiss. Org. Cult. 153, 611–624. doi: 10.1007/s11240-023-02498-3
Aasim, M., Katırcı, R., Akgur, O., Yildirim, B., Mustafa, Z., Nadeem, M. A., et al. (2022). Machine learning (ML) algorithms and artificial neural network for optimizing in vitro germination and growth indices of industrial hemp (Cannabis sativa L.). Ind. Crops Prod. 181. doi: 10.1016/j.indcrop.2022.114801
Abadi, S., Yan, W. X., Amar, D., and Mayrose, I. (2017). A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PloS Comput. Biol. 13, e1005807. doi: 10.1371/journal.pcbi.1005807
Akin, M., Eyduran, E., and Reed, B. M. (2017). Use of RSM and CHAID data mining algorithm for predicting mineral nutrition of hazelnut. Plant Cell Tiss. Org. Cult. 128, 303–316. doi: 10.1007/s11240-016-1110-6
Akin, M., Hand, C., Eyduran, E., and Reed, B. M. (2018). Predicting minor nutrient requirements of hazelnut shoot cultures using regression trees. Plant Cell Tiss. Org. Cult. 132, 545–559. doi: 10.1007/s11240-017-1353-x
Alanagh, E. N., Garoosi, G. A., Haddad, R., Maleki, S., Landín, M., and Gallego, P. P. (2014). Design of tissue culture media for efficient Prunus rootstock micropropagation using artificial intelligence models. Plant Cell Tiss. Org. Cult. 117, 349–359. doi: 10.1007/s11240-014-0444-1
Alanagh, E. N., Garoosi, G. A., Maleki, S., Landín, M., and Gallego, P. P. (2017). Predicting optimal in vitro culture medium for Pistacia vera micropropagation using neural networks models. Plant Cell Tiss. Org. Cult. 129, 19–33. doi: 10.1007/s11240-016-1152-9
Ali, S. A. and Aasim, M. (2024). Response surface methodology and artificial intelligence modeling for in vitro regeneration of Brazilian micro sword (Lilaeopsis brasiliensis). Plant Cell Tiss Org. Cult. 157, 10. doi: 10.1007/s11240-024-02734-4
Allen, F., Crepaldi, L., Alsinet, C., Strong, A. J., Kleshchevnikov, V., De Angeli, P., et al. (2018). Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol 37, 64–72. doi: 10.1038/nbt.4317
Alloghani, M., Al-Jumeily, D., Mustafina, J., Hussain, A., and Aljaaf, A. J. (2020). “A systematic review on supervised and unsupervised machine learning algorithms for data science,” in Supervised and unsupervised learning for data Science (Switzerland: springer), 3–21. doi: 10.1007/978-3-030-22475-2_1
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. doi: 10.1038/s41586-019-1711-4
Ashworth, J., Havranek, J. J., Duarte, C. M., Sussman, D., Monnat, R. J., Jr., Stoddard, B. L., et al. (2006). Computational redesign of endonuclease DNA binding and cleavage specificity. Nature 441, 656–659. doi: 10.1038/nature04818
Awty-Carroll, D., Clifton-Brown, J., and Robson, P. (2018). Using k-NN to analyse images of diverse germination phenotypes and detect single seed germination in Miscanthus sinensis. Plant Methods 14, 5. doi: 10.1186/s13007-018-0272-0
Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876. doi: 10.1126/science.abj8754
Balderas, J., Chen, D., Huang, Y., Wang, L., and Li, R. (2025). A comparative study of deep reinforcement learning for crop production management. Smart Agricultural Technology 10, 100853. doi: 10.48550/arXiv.2411.04106
Bao, C. and Liu, F. (2024). Prediction of CRISPR on-target effects via deep learning. ArXiv abs/2409.05938. doi: 10.48550/arXiv.2409.05938
Barone, J. O. (2019). Use of multiple regression analysis and artificial neural networks to model the effect of nitrogen in the organogenesis of Pinus taeda L. Plant Cell Tiss. Org. Cult. 137, 455–464. doi: 10.1007/s11240-019-01581-y
Brown, D. C. and Thorpe, T. A. (1995). Crop improvement through tissue culture. World J. Microbiol. Biotechnol. 11, 409–415. doi: 10.1007/BF00364616
Bukhari, S. A. S., Mushtaq, M. F., Akram, U., and Ahmad, M. A. (2025). A comprehensive review of novel AI techniques and applications in bioinformatics. Tech. J. 30, 35–46.
Champati, B. B., Padhiari, B. M., Ray, A., Halder, T., Jena, S., Sahoo, A., et al. (2022). Application of a multilayer perceptron artificial neural network for the prediction and optimization of the Andrographolide Content in Andrographis paniculata. Molecules 27, 2765. doi: 10.3390/molecules27092765
Chao, R. and Fei, J. (2023). Off-target effects of CRISPR/Cas9 and their solutions. Highlights Sci. Engin. Technol. 45, 296–301. doi: 10.54097/hset.v45i.7444
Charlier, J., Nadon, R., and Makarenkov, V. (2021). Accurate deep learning off-target prediction with novel sgRNA-DNA sequence encoding in CRISPR-Cas9 gene editing. Bioinformatics 37, 2299–2307. doi: 10.1093/bioinformatics/btab112
Chen, S., Liu, Z., Lai, L., and Li, Z. (2022). Efficient C-to-G base editing with improved target compatibility using engineered deaminase-nCas9 fusions. CRISPR J. 5, 389–396. doi: 10.1089/crispr.2021.0124
Chen, W., McKenna, A., Schreiber, J., Haeussler, M., Yin, Y., Agarwal, V., et al. (2019). Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 47, 7989–8003. doi: 10.1093/nar/gkz487
Cho, H., Kim, Y., Lee, E., Choi, D., Lee, Y., and Rhee, W. (2020). Basic enhancement strategies when using bayesian optimization for hyperparameter tuning of deep neural networks. IEEE Access 8, 52588–52608. doi: 10.1109/ACCESS.2020.2981072
Chou, Y., Moreira, C., Bruza, P., Ouyang, C., and Jorge, J. (2021). Counterfactuals and causability in explainable artificial intelligence: theory, algorithms, and applications. Inf. Fusion 81, 59–83. doi: 10.48550/arXiv.2103.04244
Christian, M., Cermak, T., Doyle, E. L., Schmidt, C., Zhang, F., Hummel, A., et al. (2010). Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186, 757–761. doi: 10.1534/genetics.110.120717
Christopher, M. B. (1995). Neural networks for pattern recognition (United Kingdom: Oxford academic). doi: 10.1093/oso/9780198538493.001.0001
Chuai, G., Ma, H., Yan, J., Chen, M., Hong, N., and Xue, D. (2018). DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Biol. 19, 80. doi: 10.1186/s13059-018-1459-4
Chuai, G. H., Wang, Q. L., and Liu, Q. (2017). In silico meets in vivo: towards computational CRISPR-based sgRNA design. Trends Biotechnol. 35, 12–21. doi: 10.1016/j.tibtech.2016.06.008
Compton, M. E. (2024). Use of statistics in plant cell tissue culture. Methods Mol. Biol. 2827, 15–34. doi: 10.1007/978-1-0716-3954-2_2
Dhanjal, J. K., Dammalapati, S., Pal, S., and Sundar, D. (2020). Evaluation of off-targets predicted by sgRNA design tools. Genomics 112, 3609–3614. doi: 10.1016/j.ygeno.2020.04.024
Dickinson, E., Rusilowicz, M. J., Dickinson, M., Charlton, A. J., Bechtold, U., Mullineaux, P. M., et al. (2018). Integrating transcriptomic techniques and k-means clustering in metabolomics to identify markers of abiotic and biotic stress in Medicago truncatula. Metabolomics 14, 126. doi: 10.1007/s11306-018-1424-y
Dimauro, G., Colagrande, P., Carlucci, R., Ventura, M., Bevilacqua, V., and Caivano, D. (2019). CRISPRLearner: A deep learning-based system to predict CRISPR/Cas9 sgRNA on-target cleavage efficiency. Electronics 8, 1478. doi: 10.3390/electronics8121478
Ding, S., Zheng, J., and Jia, C. (2025). DeepMEns: an ensemble model for predicting sgRNA on-target activity based on multiple features. Brief. Funct. Genomics 24, elae043. doi: 10.1093/bfgp/elae043
Doench, J. G., Fusi, N., Sullender, M., Hegde, M., Vaimberg, E. W., Donovan, K. F., et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191. doi: 10.1038/nbt.3437
Doench, J. G., Hartenian, E., Graham, D. B., Tothova, Z., Hegde, M., Smith, I., et al. (2014). Rational design of highly active sgRNAs for crispr-cas9–mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267. doi: 10.1038/nbt.3026
Douglass, S., Hsu, S. W., Cokus, S., Goldberg, R. B., Harada, J. J., and Pellegrini, M. (2016). A naïve bayesian classifier for identifying plant microRNAs. Plant J. 86, 481–492. doi: 10.1111/tpj.13180
Elkayam, S., Tziony, I., and Orenstein, Y. (2024). DeepCRISTL: deep transfer learning to predict CRISPR/Cas9 on-target editing efficiency in specific cellular contexts. Bioinformatics. 40, btae481. doi: 10.1093/bioinformatics/btae481
Eraslan, G., Avsec, Ž., Gagneur, J., and Theis, F. J. (2019). Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 20, 389–403. doi: 10.1038/s41576-019-0122-6
Fakhrzad, F., Jowkar, A., and Hosseinzadeh, J. (2022). Mathematical modeling and optimizing the in vitro shoot proliferation of wallflower using multilayer perceptron non-dominated sorting genetic algorithm-II (MLP-NSGAII). PloS One 17. doi: 10.1371/jopurnal.pone.0273009
Fallah Ziarani, M., Tohidfar, M., and Navvabi, M. (2022). Modeling and optimizing in vitro percentage and speed callus induction of carrot via multilayer perceptron-single point discrete GA and radial basis function. BMC Biotechnol. 22, 34. doi: 10.1186/s12896-022-00764-4
Farhadi, S., Salehi, M., Moieni, A., Safaie, N., and Sabet, M. S. (2020). Modeling of paclitaxel biosynthesis elicitation in Corylus avellana cell culture using adaptive neuro-fuzzy inference system-genetic algorithm (ANFIS-GA) and multiple regression methods. PloS One 15, e0237478. doi: 10.1371/journal.pone.023747
Farrell, G., Adamidi, E., Buono, R. A., Anton, M., Omar Abdelghani Attafi, O. A., Gutierrez, S. C., et al. (2025). Open and sustainable AI: challenges, opportunities and the road ahead in the life sciences. arXiv preprint arXiv:2505.16619. doi: 10.48550/arXiv.2505.16619
Feng, Z., Zhang, B., Ding, W., Liu, X., Yang, D. L., Wei, P., et al. (2013). Efficient genome editing in plants using a CRISPR/Cas system. Cell Res. 23, 1229–1232. doi: 10.1038/cr.2013.114
Finzel, B. (2025). Current methods in explainable artificial intelligence and future prospects for integrative physiology. Pflugers Archiv: Eur. J. Physiol. 477, 513–529. doi: 10.1007/s00424-025-03067-7
Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. J., Kii, E., and Alt, F. W. (2015). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 33, 179–186. doi: 10.1038/nbt.3101
Gago, J., Landín, M., and Gallego, P. P. (2010). A neurofuzzy logic approach for modeling plant processes: A practical case of in vitro direct rooting and acclimatization of Vitis vinifera L. Plant Sci. 179, 241–249. doi: 10.1016/j.plantsci.2010.05.009
Gago, J., Pérez-Tornero, O., Landín, M., Burgos, L., and Gallego, P. P. (2011). Improving knowledge of plant tissue culture and media formulation by neurofuzzy logic: a practical case of data mining using apricot databases. J. Plant Physiol. 168, 1858–1865. doi: 10.1016/j.jplph.2011.04.008
Garneau, J. E., Dupuis, M.È., Villion, M., Romero, D. A., Barrangou, R., Boyaval, P., et al. (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. doi: 10.1038/nature09523
Goell, J. H. and Hilton, I. B. (2021). CRISPR/Cas-based epigenome editing: advances, applications, and clinical utility. Trends Biotechnol. 39, 678–691. doi: 10.1016/j.tibtech.2020.10.012
Goodfellow, I., Bengio, Y., and Courville, A. (2016). Deep Learning (cambridge, USA: The MIT press), 485–716.
Goodfellow, I., Pouget-Abadie, J., Mirza, M., Xu, B., Farley, D. W., Ozair, S., et al. (2020). Generative adversarial networks. Commun. ACM. 63, 139–144. doi: 10.1145/3422622
Grizot, S., Smith, J., Daboussi, F., Prieto, J., Redondo, P., Merino, N., et al. (2009). Efficient targeting of a SCID gene by an engineered single-chain homing endonuclease. Nucleic Acids Res. 37, 5405–5419. doi: 10.1093/nar/gkp548
Guo, X., Qiu, Y., Nettleton, D., Yeh, C. T., Zheng, Z., Hey, S., et al. (2021). KAT4IA: K-means assisted training for image analysis of field-grown plant phenotypes. Plant phenomics 3, 9805489. doi: 10.34133/2021/9805489
Haeussler, M., Schönig, K., Eckert, H., Eschstruth, A., Mianné, J., Renaud, J. B., et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148. doi: 10.1186/s13059-016-1012-2
Hameg, R., Arteta, T. A., Landin, M., Gallego, P. P., and Barreal, M. E. (2020). Modeling and optimizing culture medium mineral composition for in vitro propagation of Actinidia arguta. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.554905
Han, X., Deng, Z., Liu, H., and Ji, X. (2025). Current advancement and future prospects in simplified transformation-based plant genome editing. Plants 14, 889. doi: 10.3390/plants14060889
Harfouche, A. L., Petousi, V., Meilan, R., Sweet, J., Twardowski, T., and Altman, A. (2021). Promoting ethically responsible use of agricultural biotechnology. Trends Plant Sci. 26, 546–559. doi: 10.1016/j.tplants.2020.12.015
Hastie, T., Tibshirani, R., and Friedman, J. (2009a). “Overview of supervised learning,” in The elements of statistical learning (New York: Springer), 9–41. doi: 10.1007/978-0-387-84858-7_2
Hastie, T., Tibshirani, R., and Friedman, J. (2009b). The elements of statistical learning (New York: Springer). doi: 10.1007/978-0-387-84858-7
He, W., Wang, H., Wei, Y., Jiang, Z., Tang, Y., Chen, Y., et al. (2021). GuidePro: a multi-source ensemble predictor for prioritizing sgRNAs in CRISPR/Cas9 protein knockouts. Bioinformatics 37, 134–136. doi: 10.1093/bioinformatics/btaa1068
Heenkenda, A. C., Nadaraja, S., Hettiarachchi, R., Dias, N., Rathnayake, S., and Vidhanaarachchi, S. (2024). Advanced convolutional neural networks for detecting and quantifying infections in coconut tree tissue cultures. Int. Conf. Advan. Comput., 420–425. doi: 10.1109/ICAC64487.2024.10851098
Hesami, M., Condori-Apfata, J. A., Valderrama Valencia, M., and Mohammadi, M. (2020c). Application of artificial neural network for modeling and studying in vitro genotype-independent shoot regeneration in wheat. Appl. Sci. 10, 5370. doi: 10.3390/app10155370
Hesami, M. and Jones, A. M. P. (2020b). Application of artificial intelligence models and optimization algorithms in plant cell and tissue culture. Appl. Microbiol. Biotechnol. 104, 9449–9485. doi: 10.1007/s00253-020-10888-2
Hesami, M. and Jones, A. M. P. (2021). Modeling and optimizing callus growth and development in Cannabis sativa using random forest and support vector machine in combination with a genetic algorithm. Appl. Microbiol. Biotechnol. 105, 5201–5212. doi: 10.1007/s00253-021-11375-y
Hesami, M., Naderi, R., and Tohidfar, M. (2019). Modeling and optimizing medium composition for shoot regeneration of chrysanthemum via radial basis function-non-dominated sorting genetic algorithm-II (RBF-NSGAII). Sci. Rep. 9, 18237. doi: 10.1038/s41598-019-54257-0
Hesami, M., Naderi, R., Tohidfar, M., and Yoosefzadeh-Najafabadi, M. (2020a). Development of support vector machine-based model and comparative analysis with artificial neural network for modeling the plant tissue culture procedures: effect of plant growth regulators on somatic embryogenesis of chrysanthemum, as a case study. Plant Methods 16, 112. doi: 10.1186/s13007-020-00655-9
Hiranniramol, K., Chen, Y., Liu, W., and Wang, X. (2020). Generalizable sgRNA design for improved CRISPR/Cas9 editing efficiency. Bioinformatics 36, 2684–2689. doi: 10.1093/bioinformatics/btaa041
Holzinger, A., Keiblinger, K., Holub, P., Zatloukal, K., and Müller, H. (2023). AI for life: trends in artificial intelligence for biotechnology. New Biotechnol. 74, 16–24. doi: 10.1016/j.nbt.2023.02.001
Hu, J. H., Miller, S. M., Geurts, M. H., Tang, W., Chen, L., Sun, N., et al. (2018). Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63. doi: 10.1038/nature26155
Hua, K., Tao, X., and Zhu, J. K. (2019). Expanding the base editing scope in rice by using Cas9 variants. Plant Biotechnol. J. 17, 499–504. doi: 10.1111/pbi.12993
Huang, J., Lin, Q., Fei, H., He, Z., Xu, H., Li, Y., et al. (2023). Discovery of deaminase functions by structure-based protein clustering. Cell 186, 3182–3195.e14. doi: 10.1016/j.cell.2023.05.041
Ibrahim, N., Adedamola, A. M., Ibrahim, B., Ahmed, R. T., Raji, I. D., and Bello-Salau, H. (2023). Survey of machine learning and optimization algorithms in plant tissue culture. Eng. Proc. 56, 122. doi: 10.3390/ASEC2023-15259
Isak, M. A., Bozkurt, T., Tütüncü, M., Dönmez, D., İzgü, T., and Şimşek, Ö. (2024). Leveraging machine learning to unravel the impact of cadmium stress on goji berry micropropagation. PloS One 13, 19(6), e0305111. doi: 10.1371/journal.pone.0305111
Ishibuchi, H. (1996). Development of fuzzy neural networks. Fuzzy Model. (Boston: Springer) 185–202. doi: 10.1007/978-1-4613-1365-6_9
Ivashchuk, O. A., Fedorova, V., Shcherbinina, N. V., Maslova, E. V., and Shamraeva, E. (2018). Microclonal propagation of plant process modeling and optimization of its parameters based on neural network. Drug Invent Today 10, p.3170–p.3175.
Jafari, M. and Daneshvar, M. H. (2023). Prediction and optimization of indirect shoot regeneration of Passiflora caerulea using machine learning and optimization algorithms. BMC Biotechnol. 23, 27. doi: 10.1186/s12896-023-00796-4
James, G., Witten, D., Hastie, T., and Tibshirani, R. (2021). “Unsupervised learning,” in An introduction to statistical learning (New York: Springer), 497–552. doi: 10.1007/978-1-0716-1418-1_12
Ji, Y., Zhou, Z., Liu, H., and Davuluri, R. V. (2021). DNABERT: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics 37, 2112–2120. doi: 10.1093/bioinformatics/btab083
Jones, J. E. (2012). Method of Classifying Plant Embryos Using Penalized Logistic Regression U.S. Patent 8,321,191 (Washington, DC: U.S. Patent and Trademark Office), 27.
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
Kaushik, P., Rani, M., Khurana, N., Pandey, P., Payal, and Kapoor, S. (2025). Revolutionizing plant tissue culture: harnessing artificial intelligence for precision propagation and optimization. Nat. Prod. J. 15. doi: 10.2174/0122103155302871240527094915
Kim, D., Bae, S., Park, J., Kim, E., Kim, S., Yu, H. R., et al. (2015). Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 12, 237–243. doi: 10.1038/nmeth.3284
Kim, Y. G., Cha, J., and Chandrasegaran, S. (1996). Hybrid restriction enzymes: zinc finger fusions to FokI cleavage domain. Proc. Natl. Acad. Sci. U.S.A 93, 1156–1160. doi: 10.1073/pnas.93.3.1156
Kim, N., Choi, S., Kim, S., Song, M., Seo, J. H., Min, S., et al. (2024). Deep learning models to predict the editing efficiencies and outcomes of diverse base editors. Nat. Biotechnol. 42, 484–497. doi: 10.1038/s41587-023-01792-x
Kim, M., Go, M. J., Kang, S. H., Jeong, S. H., and Lim, K. (2025). Revolutionizing CRISPR technology with artificial intelligence. Exp. Mol. Med. 57, 1419–1431. doi: 10.1038/s12276-025-01462-9
Kim, D., Kim, S., Kim, S., Park, J., and Kim, J. S. (2016). Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res. 26, 406–415. doi: 10.1101/gr.199588.115
Kim, H. K., Yu, G., Park, J., Min, S., Lee, S., Yoon, S., et al. (2021). Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol. 39, 198–206. doi: 10.1038/s41587-020-0677-y
Kleinstiver, B. P., Prew, M. S., Tsai, S. Q., Topkar, V. V., Nguyen, N. T., Zheng, Z., et al. (2015). Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485. doi: 10.1038/nature14592
Koblan, L. W., Arbab, M., Shen, M. W., Hussmann, J. A., Anzalone, A. V., Doman, J. L. Z., et al. (2021). Efficient C•G-to-G•C base editors developed using CRISPRi screens, target-library analysis, and machine learning. Nat. Biotechnol. 39, 1414–1425. doi: 10.1038/s41587-021-00938-z
Koeppel, J., Weller, J., Peets, E. M., Pallaseni, A., Kuzmin, I., Raudvere, U., et al. (2023). Prediction of prime editing insertion efficiencies using sequence features and DNA repair determinants. Nat. Biotechnol. 41, 1446–1456. doi: 10.1038/s41587-023-01678-y
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. doi: 10.1038/nature17946
Konstantakos, V., Nentidis, A., Krithara, A., and Paliouras, G. (2022). CRISPRedict: a CRISPR-Cas9 web tool for interpretable efficiency predictions. Nucleic Acids Res. 50, W191–W198. doi: 10.1093/nar/gkac466
Kovalchuk, I. Y., Mukhitdinova, Z., Turdiyev, T., Madiyeva, G., Akin, M., Eyduran, E., et al. (2017). Modeling some mineral nutrient requirements for micropropagated wild apricot shoot cultures. Plant Cell Tiss. Org. Cult. 129, 325–335. doi: 10.1007/s11240-017-1180-0
Lackner, M., Helmbrecht, N., Pääbo, S., and Riesenberg, S. (2023). Detection of unintended on-target effects in CRISPR genome editing by DNA donors carrying diagnostic substitutions. Nucleic Acids Res. 51, e26. doi: 10.1093/nar/gkac1254
Lazzarotto, C. R., Malinin, N. L., Li, Y., Zhang, R., Yang, Y., Lee, G., et al. (2020). CHANGE-seq reveals genetic and epigenetic effects on CRISPR-Cas9 genome-wide activity. Nat. Biotechnol. 38, 1317–1327. doi: 10.1038/s41587-020-0555-7
Lee, K.-H., Samsuzzaman, Reza, M. N., Islam, S., Ahmed, S., Cho, Y. J., et al. (2025). Evaluation of machine learning models for stress symptom classification of cucumber seedlings grown in a controlled environment. Agronomy 15, 90. doi: 10.3390/agronomy15010090
Leenay, R. T., Aghazadeh, A., Hiatt, J., Tse, D., Roth, T. L., Apathy, R., et al. (2019). Large dataset enables prediction of repair after CRISPR-Cas9 editing in primary T cells. Nat. Biotechnol. 37, 1034–1037. doi: 10.1038/s41587-019-0203-2
Li, S., An, J., Li, Y., Zhu, X., Zhao, D., Wang, L., et al. (2022). Automated high-throughput genome editing platform with an AI learning in situ prediction model. Nat. Commun. 13, 7386. doi: 10.1038/s41467-022-35056-0
Li, Y. and Chao, X. (2021). Semi-supervised few-shot learning approach for plant diseases recognition. Plant Methods 17, 68. doi: 10.1186/s13007-021-00770-1
Li, J. F., Norville, J. E., Aach, J., McCormack, M., Zhang, D., Bush, J., et al. (2013). Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 31, 688–691. doi: 10.1038/nbt.2654
Li, J., Xu, R., Qin, R., Liu, X., Kong, F., and Wei, P. (2021b). Genome editing mediated by SpCas9 variants with broad non-canonical PAM compatibility in plants. Mol. Plant 14, 352–360. doi: 10.1016/j.molp.2020.12.017
Li, V. R., Zhang, Z., and Troyanskaya, O. G. (2021a). CROTON: an automated and variant-aware deep learning framework for predicting CRISPR/Cas9 editing outcomes. Bioinformatics 37, i342–i348. doi: 10.1093/bioinformatics/btab268
Li, J., Zhu, C., Yang, C., Zheng, Q., Wang, B., Tu, J., et al. (2025). DM_CorrMatch: a semi-supervised semantic segmentation framework for rapeseed flower coverage estimation using UAV imagery. Plant Methods 21, 54. doi: 10.1186/s13007-025-01373-w
Lin, J. and Wong, K. C. (2018). Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics 34, i656–i663. doi: 10.1093/bioinformatics/bty554
Lin, J., Zhang, Z., Zhang, S., Chen, J., and Wong, K. (2020c). CRISPR-Net: A recurrent convolutional network quantifies CRISPR off-target activities with mismatches and indels. Adv. Sci. 7, 1903562. doi: 10.1002/advs.201903562
Lin, J., Zhao, Y., Watson, D., and Chen, C. (2020a). The radial basis funcion differential quadrature method with ghost points. Math. Comput. Simul. 173, 105–114. doi: 10.1016/j.matcom.2020.01.006
Lin, Q., Zong, Y., Xue, C., Wang, S., Jin, S., Zhu, Z., et al. (2020b). Prime genome editing in rice and wheat. Nat. Biotechnol. 38, 582–585. doi: 10.1038/s41587-020-0455-x
Listgarten, J., Weinstein, M., Kleinstiver, B. P., Sousa, A. A., Joung, J. K., Crawford, J., et al. (2018). Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng. 2, 38–47. doi: 10.1038/s41551-017-0178-6
Liu, Q., Cheng, X., Liu, G., Li, B., and Liu, X. (2020a). Deep learning improves the ability of sgRNA off-target propensity prediction. BMC Bioinf. 21, 51. doi: 10.1186/s12859-020-3395-z
Liu, Q., He, D., and Xie, L. (2019). Prediction of off-target specificity and cell-specific fitness of CRISPR-Cas System using attention boosted deep learning and network-based gene feature. PloS Comput. Biol. 15, e1007480. doi: 10.1371/journal.pcbi.1007480
Liu, F., Huang, S., Hu, J., Chen, X., Song, Z., Dong, J., et al. (2023). Design of prime-editing guide RNAs with deep transfer learning. Nat. Mach. Intell. 5, 1261–1274. doi: 10.1038/s42256-023-00739-w
Liu, G., Lin, Q., Jin, S., and Gao, C. (2022a). The CRISPR-Cas toolbox and gene editing technologies. Mol. Cell. 82, 333–347. doi: 10.1016/j.molcel.2021.12.002
Liu, X., Wang, S., and Ai, D. (2022b). Predicting CRISPR/Cas9 Repair Outcomes by attention-based deep learning framework. Cells 11, 1847. doi: 10.3390/cells11111847
Liu, X., Yang, Y., Qiu, Y., Reyad-Ul-Ferdous, M., Ding, Q., and Wang, Y. (2020b). SeqCor: correct the effect of guide RNA sequences in clustered regularly interspaced short palindromic repeats/Cas9 screening by machine learning algorithm. J. Genet. Genomics 47, 672–680. doi: 10.1016/j.jgg.2020.10.007
Loberant, B. and Altman, A. (2010). “Micropropagation of plants,” in Encyclopedia of industrial biotechnology: bioprocess, bioseparation, and cell technology (New York: Wiley), 3499–3515. doi: 10.1002/9780470054581.eib442
Loyola-Vargas, V. M. and Ochoa-Alejo, N. (2018). An introduction to plant tissue culture: advances and perspectives. Methods Mol. Biol. 1815, 3–13. doi: 10.1007/978-1-4939-8594-4_1
Lundberg, S. M., Erion, G. G., and Lee, S. (2018). Consistent individualized feature attribution for tree ensembles. ArXiv abs/1802.03888. doi: 10.48550/arXiv.1802.03888
Lundberg, S. M. and Lee, S. (2017). A unified approach to interpreting model predictions. Neu. Infor. Process. Syst. 30. doi: 10.48550/arXiv.1705.07874
Ma, E., Chen, K., Shi, H., Stahl, E. C., Adler, B., Trinidad, M., et al. (2022). Improved genome editing by an engineered CRISPR-Cas12a. Nucleic Acids Res. 50, 12689–12701. doi: 10.1093/nar/gkac1192
Machnicka, M. A. and Wilczynski, B. (2020). “Machine learning and deep learning for the advancement of epigenomics,” in Epigenetics of the Immune System. Eds. Kabelitz, D. and Bhat, J. (United States: Academic Press), 217–237.
Madsen, S. L., Dyrmann, M., Jørgensen, R. N., and Karstoft, H. (2019). Generating artificial images of plant seedlings using generative adversarial networks. Biosyst. Eng. 187, 147–159. doi: 10.1016/j.biosystemseng.2019.09.005
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J., Charpentier, E., Horvath, P., et al. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477. doi: 10.1038/nrmicro2577
Malangsa, R. D. and Maravillas, E. A. (2017). Performance comparison of naïve bayes and K-NN algorithms on contamination grading for Abaca tissue culture (in vitro). Int. J. Comput. Sci. Inf. Technol. 5, 5–10.
Marquart, K. F., Allam, A., Janjuha, S., Sintsova, A., Villiger, L., Frey, N., et al. (2021). Predicting base editing outcomes with an attention-based deep learning algorithm trained on high-throughput target library screens. Nat. Commun. 12, 5114. doi: 10.1038/s41467-021-25375-z
Mathis, N., Allam, A., Kissling, L., Marquart, K. F., Schmidheini, L., Solari, C., et al. (2023). Predicting prime editing efficiency and product purity by deep learning. Nat. Biotechnol. 41, 1151–1159. doi: 10.1038/s41587-022-01613-7
Mazoure, B., Mazoure, A., Bédard, J., and Makarenkov, V. (2022). DUNEScan: a web server for uncertainty estimation in skin cancer detection with deep neural networks. Sci. Rep. 12, 179. doi: 10.1038/s41598-021-03889-2
Mikolov, T., Sutskever, I., Chen, K., Corrado, G. S., and Dean, J. (2013). Distributed representations of words and phrases and their compositionality. Neu. Inform. Process. Syst. 26. doi: 10.48550/arXiv.1310.4546
Mucherino, A., Papajorgji, P. J., and Pardalos, P. M. (2009). “k-Nearest Neighbor Classification,” in Data mining in agriculture (New York: Springer), 83–106. doi: 10.1007/978-0-387-88615-2_4
Muhammad Rafid, A. H., Toufikuzzaman, M., Rahman, M. S., and Rahman, M. S. (2020). CRISPRpred(SEQ): a sequence-based method for sgRNA on target activity prediction using traditional machine learning. BMC Bioinf. 21, 223. doi: 10.1186/s12859-020-3531-9
Najafian, K., Ghanbari, A., Sabet Kish, M., Eramian, M., Shirdel, G. H., Stavness, I., et al. (2023). Semi-self-supervised learning for semantic segmentation in images with dense patterns. Plant phenomics 5, 25. doi: 10.34133/plantphenomics.0025
Nasrabadi, N. M. (2007). Pattern recognition and machine learning. J. Electron. Imag. 16. doi: 10.1117/1.2819119
Nasti, L., Vecchiato, G., Heuret, P., Rowe, N. P., Palladino, M., and Marcati, P. (2024). A reinforcement learning approach to study climbing plant behaviour. Sci. Rep. 14, 18222. doi: 10.1038/s41598-024-62147-3
Nekrasov, V., Staskawicz, B., Weigel, D., Jones, J. D., and Kamoun, S. (2013). Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 691–693. doi: 10.1038/nbt.2655
Ng, P. (2017). dna2vec: Consistent vector representations of variable-length k-mers. ArXiv abs/1701.06279. doi: 10.48550/arXiv.1701.06279
Niazian, M. and Niedbała, G. (2020). Machine learning for plant breeding and biotechnology. Agriculture 10, 436. doi: 10.3390/agriculture10100436
Nishimasu, H., Shi, X., Ishiguro, S., Gao, L., Hirano, S., Okazaki, S., et al. (2018). Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262. doi: 10.1126/science.aas9129
Niu, R., Peng, J., Zhang, Z., and Shang, X. (2021). R-CRISPR: A deep learning network to predict off-target activities with mismatch, insertion and deletion in CRISPR-Cas9 system. Genes 12, 1878. doi: 10.3390/genes12121878
Onay, A., Jeffree, C., Theobald, C., and Yeoman, M. M. (2000). Analysis of the effects of maturation treatments on the probabilities of somatic embryo germination and plantlet regeneration in pistachio using a linear logistic method. Plant Cell Tiss. Org. Cult. 60, 121–129. doi: 10.1023/A:1006464505072
Osama, K., Mishra, B. N., and Somvanshi, P. (2015). “Machine learning techniques in plant biology,” in The omics of plant science (New York: Springer), 731–754. doi: 10.1007/978-81-322-2172-2_26
Özcan, E., Atar, H. H., Ali, S. A., and Aasim, M. (2023). Artificial neural network and decision tree–based models for prediction and validation of in vitro organogenesis of two hydrophytes—Hemianthus callitrichoides and Riccia fluitans. In Vitro Cell. Dev. Biol.-Plant 59, 547–562. doi: 10.1007/s11627-023-10367-z
Pallaseni, A., Peets, E. M., Koeppel, J., Weller, J., Vanderstichele, T., Ho, U. L., et al. (2022). Predicting base editing outcomes using position-specific sequence determinants. Nucleic Acids Res. 50, 3551–3564. doi: 10.1093/nar/gkac161
Palvadi, S. K. and Kadiravan, G. (2025). “Future Trends in Bioinformatics AI Integration,” in Multimodal Data Fusion for Bioinformatics Artificial Intelligence. Eds. Lilhore, U. K., Kumar, A., Vyas, N., Simaiya, S., and Dutt, V (USA: Scrivener Publishing, LLC). doi: 10.1002/9781394269969.ch13
Park, J. and Kim, H. K. (2023). Prediction of base editing efficiencies and outcomes using DeepABE and DeepCBE. Methods Mol. Bio., 23–32. doi: 10.1007/978-1-0716-2879-9_3
Peng, H., Zheng, Y., Zhao, Z., Liu, T., and Li, J. (2018). Recognition of CRISPR/Cas9 off-target sites through ensemble learning of uneven mismatch distributions. Bioinformatics 34, i757–i765. doi: 10.1093/bioinformatics/bty558
Phillips, G. C. and Garda, M. (2019). Plant tissue culture media and practices: an overview. In Vitro Cell. Dev. Biol.-Plant 55, 242–257. doi: 10.1007/s11627-019-09983-5
Prasad, V. and Gupta, S. D. (2008). Applications and potentials of artificial neural networks in plant tissue culture. Plant Tiss. Cult. Eng. 6, 47–67. doi: 10.1007/978-1-4020-3694-1_3
Rafiq, S., Macha, M. A., and Assad, A. (2025). Machine learning and deep learning for genomic data: a data-centric approach to CRISPR/Cas9 gene editing. Int. J. Data Sci. Anal. 20, 5037–5066. doi: 10.1007/s41060-025-00773-4
Rahman, M. K. and Rahman, M. S. (2017). CRISPRpred: A flexible and efficient tool for sgRNAs on-target activity prediction in CRISPR/Cas9 systems. PloS One 12, e0181943. doi: 10.1371/journal.pone.0181943
Rajapaksha, H. M. S. N., Thiwanka, W. A. D. A., Tissera, W., Vidhanaarachchi, S., Kishara, J., and Vidhanaarachchi, V. R. M. (2024). Integrating deep learning for plant growth monitoring and browning disease severity identification in coconut tissue culture. Int. Conf. Advan. Comput., 1–6. doi: 10.1109/ICAC64487.2024.10851150
Ran, F. A., Cong, L., Yan, W. X., Scott, D. A., Gootenberg, J. S., Kriz, A. J., et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191. doi: 10.1038/nature14299
Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. protocol. 8, 2281–2308. doi: 10.1038/nprot.2013.143
Rauschert, S., Raubenheimer, K., Melton, P. E., and Huang, R. C. (2020). Machine learning and clinical epigenetics: a review of challenges for diagnosis and classification. Clin. Epigenet. 12, 51. doi: 10.1186/s13148-020-00842-4
Ray, S. (2019). “A quick review of machine learning algorithms,” in Int. conf. on machine learning, big data, cloud and parallel computing (COMITCon). (India: IEEE) 35–39. doi: 10.1109/COMITCon.2019.8862451
Rezaei, H., Mirzaie-Asl, A., Abdollahi, M. R., and Tohidfar, M. (2023a). Enhancing petunia tissue culture efficiency with machine learning: A pathway to improved callogenesis. PloS One 18, e0293754. doi: 10.1371/journal.pone.0293754
Rezaei, H., Mirzaie-Asl, A., Abdollahi, M. R., and Tohidfar, M. (2023b). Comparative analysis of different artificial neural networks for predicting and optimizing in vitro seed germination and sterilization of petunia. PloS One 18, e0285657. doi: 10.1371/journal.pone.0285657
Şimşek, Ö. (2024). Machine learning offers insights into the impact of in vitro drought stress on strawberry cultivars. Agriculture 14, 294. doi: 10.3390/agriculture14020294
Sadat-Hosseini, M., Arab, M. M., Soltani, M., Eftekhari, M., Soleimani, A., and Vahdati, K. (2022). Predictive modeling of persian walnut (Juglans regia L.) in vitro proliferation media using machine learning approaches: a comparative study of ANN, KNN and GEP models. Plant Methods 18, 48. doi: 10.1186/s13007-022-00871-5
Salehi, M., Farhadi, S., Moieni, A., Safaie, N., and Hesami, M. (2021). A hybrid model based on general regression neural network and fruit fly optimization algorithm for forecasting and optimizing paclitaxel biosynthesis in Corylus avellana cell culture. Plant Methods 17, 13. doi: 10.1186/s13007-021-00714-9
Sarker, I. H. (2022). AI-based modeling: techniques, applications and research issues towards automation, intelligent and smart systems. SN Comput. Sci. 3, 158. doi: 10.1007/s42979-022-01043-x
Schmid-Burgk, J. L., Gao, L., Li, D., Gardner, Z., Strecker, J., Lash, B., et al. (2020). Highly parallel profiling of cas9 variant specificity. Mol. Cell 78, 794–800.e8. doi: 10.1016/j.molcel.2020.02.023
Schoonenberg, V. A. C., Cole, M. A., Yao, Q., Macias-Treviño, C., Sher, F., Schupp, P. G., et al. (2018). CRISPRO: identification of functional protein coding sequences based on genome editing dense mutagenesis. Genome Biol. 19, 169. doi: 10.1186/s13059-018-1563-5
Sebiani-Calvo, A., Hernández-Soto, A., Hensel, G., and Gatica-Arias, A. (2024). Crop genome editing through tissue-culture-independent transformation methods. Front. Genome editing 6. doi: 10.3389/fgeed.2024.1490295
Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., et al. (2020). Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710. doi: 10.1038/s41586-019-1923-7
Shavindi, H. D. M., Kumarasiri, D. K. N., Tissera, W., Vidhanaarachchi, S., Joseph, J., and Vidhanaarachchi, V. (2024). Effectiveness of deep learning and IoT for disease classification and characterizing tissue culture calli. Int. Conf. Advan. Comput., 522–527. doi: 10.1109/ICAC64487.2024.10851116
Shen, M. W., Arbab, M., Hsu, J. Y., Worstell, D., Culbertson, S. J., Krabbe, O., et al. (2018). Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651. doi: 10.1038/s41586-018-0686-x
Shrawgi, H. and Sisodia, D. S. (2019). Convolution neural network model for predicting single guide RNA efficiency in CRISPR/Cas9 system. Chemom. Intell. Lab. Syst. 189, 149–154. doi: 10.1016/j.chemolab.2019.04.008
Simonyan, K. and Zisserman, A. (2014). Very deep convolutional networks for large-scale image recognition. CoRR abs/1409, 1556. doi: 10.48550/arXiv.1409.1556
Singh, R., Kuscu, C., Quinlan, A., Qi, Y., and Adli, M. (2015). Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 43, e118. doi: 10.1093/nar/gkv575
Slaymaker, I. M., Gao, L., Zetsche, B., Scott, D. A., Yan, W. X., and Zhang, F. (2016). Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88. doi: 10.1126/science.aad5227
Song, M., Kim, H. K., Lee, S., Kim, Y., Seo, S. Y., Park, J., et al. (2020). Sequence-specific prediction of the efficiencies of adenine and cytosine base editors. Nat. Biotechnol. 38, 1037–1043. doi: 10.1038/s41587-020-0573-5
Specht, D. F. (1991). A general regression neural network. IEEE T. Neur. Net. 2, 568–576. doi: 10.1109/72.97934
Sperandei, S. (2014). Understanding logistic regression analysis. Biochem. Med. 24, 12–18. doi: 10.11613/BM.2014.003
Stemmer, M., Thumberger, T., Del Sol Keyer, M., Wittbrodt, J., and Mateo, J. L. (2015). CCTop: An intuitive, flexible and Rrliable CRISPR/Cas9 target prediction tool. PloS One 10, e0124633. doi: 10.1371/journal.pone.0124633
Störtz, F. and Minary, P. (2021). crisprSQL: a novel database platform for CRISPR/Cas off-target cleavage assays. Nucleic Acids Res. 49, D855–D861. doi: 10.1093/nar/gkaa885
Sudheer, W. N., Praveen, N., Al-Khayri, J. M., and Jain, S. M. (2022). “Role of plant tissue culture medium components,” in Adv. in plant tissue culture (United Kingdom: Academic Press) 51–83. doi: 10.1016/B978-0-323-90795-8.00012-6
Sun, J., Guo, J., and Liu, J. (2024). CRISPR-M: Predicting sgRNA off-target effect using a multi-view deep learning network. PloS Computat. Biol. 20, e1011972. doi: 10.1371/journal.pcbi.1011972
Szepesvári, C. (2010). “Value prediction problems,” in Algorithms for reinforcement learning (New York: Springer), 11–36. doi: 10.1007/978-3-031-01551-9_2
Talpur, N., Abdulkadir, S. J., Alhussian, H., Hasan, M. H., Aziz, N., and Bamhdi, A. (2023). Deep neuro-fuzzy system application trends, challenges, and future perspectives: a systematic survey. Artif. Intell. Rev. 56, 865–913. doi: 10.1007/s10462-022-10188-3
Tan, F., Gao, X., Cang, H., Wu, N., Di, R., Yan, J., et al. (2025). Early detection of verticillium wilt in cotton by using hyperspectral imaging combined with recurrence plots. Agronomy 15, 213. doi: 10.3390/agronomy15010213
The ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Thomas, T., Vijayaraghavan, P. A., and Emmanuel, S. (2020). “Applications of decision trees,” in Machine learning approaches in cyber security analytics (New York: Springer), 157–184. doi: 10.1007/978-981-15-1706-8_9
Tipping, M. E. and Bishop, C. M. (1999). Probabilistic principal component analysis. J. R. Statist. Soc B 61, 611–622. doi: 10.1111/1467-9868.00196
Tsai, S. Q., Nguyen, N. T., Malagon-Lopez, J., Topkar, V. V., Aryee, M. J., and Joung, J. K. (2017). CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods 14, 607–614. doi: 10.1038/nmeth.4278
Tsai, S. Q., Zheng, Z., Nguyen, N. T., Liebers, M., Topkar, V. V., Thapar, V., et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197. doi: 10.1038/nbt.3117
van Bemmelen van der Plaat, A., van Treuren, R., and van Hintum, T. J. L. (2021). Reliable genomic strategies for species classification of plant genetic resources. BMC Bioinf. 22, 173. doi: 10.1186/s12859-021-04018-6
Vapnik, V. N. (2000). “Introduction: Four periods in the research of the learning problem,” in The nature of statistical learning theory (New York: Springer), 1–15. doi: 10.1007/978-1-4757-3264-1_1
Venkadesh, P., Divya, S. V., Mathumitha, V., Nels, S. N., Balaji, J., and Ramesh, A. (2025). “AI in bioinformatics unlocking the secrets of life,” in The Role of Artificial Intelligence in Advancing Applied Life Sciences (USA: IGI Global Scientific Publishing Platform) 119–148. doi: 10.4018/979-8-3693-9208-9.ch004
Vilone, G. and Longo, L. (2021). Notions of explainability and evaluation approaches for explainable artificial intelligence. Inf. Fusion 76, 89–106. doi: 10.1016/j.inffus.2021.05.009
Vinodkumar, P. K., Ozcinar, C., and Anbarjafari, G. (2021). Prediction of sgRNA off-Target activity in CRISPR/Cas9 gene editing using graph convolution network. Entropy 23, 608. doi: 10.3390/e23050608
Walton, R. T., Christie, K. A., Whittaker, M. N., and Kleinstiver, B. P. (2020). Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Sci. 368, 290–296. doi: 10.1126/science.aba8853
Wang, T., Wei, J. J., Sabatini, D. M., and Lander, E. S. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Sci. 343, 80–84. doi: 10.1126/science.1246981
Wang, J., Xiang, X., Bolund, L., Zhang, X., Cheng, L., and Luo, Y. (2020). GNL-Scorer: a generalized model for predicting CRISPR on-target activity by machine learning and featurization. J. Mol. Cell Biol. 12, 909–911. doi: 10.1093/jmcb/mjz116
Wang, D., Zhang, C., Wang, B., Li, B., Wang, Q., Liu, D., et al. (2019). Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 10, 4284. doi: 10.1038/s41467-019-12281-8
Watkins, C. J. C. H. and Dayan, P. (1992). Q- learning. Mach. Learn. 8, 279–292. doi: 10.1007/BF00992698
Webb, G. I. (2011). “Naïve Bayes,” in Encyclopedia of machine learning (New York: Springer), 713–714. doi: 10.1007/978-0-387-30164-8_576
Williamson, H. F., Brettschneider, J., Caccamo, M., Davey, R. P., Goble, C., Kersey, P. J., et al. (2021). Data management challenges for artificial intelligence in plant and agricultural research. F1000research 10, 324. doi: 10.12688/f1000research.52204.2
Xiang, X., Corsi, G. I., Anthon, C., Qu, K., Pan, X., Liang, X., et al. (2021). Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat. Commun. 12, 3238. doi: 10.1038/s41467-021-23576-0
Xiao, L. M., Wan, Y. Q., and Jiang, Z. R. (2021). AttCRISPR: a spacetime interpretable model for prediction of sgRNA on-target activity. BMC Bioinf. 22, 589. doi: 10.1186/s12859-021-04509-6
Xu, H., Xiao, T., Chen, C. H., Li, W., Meyer, C. A., Wu, Q., et al. (2015). Sequence determinants of improved CRISPR sgRNA design. Genome Res. 25, 1147–1157. doi: 10.1101/gr.191452.115
Xu, C., Zhou, Y., Xiao, Q., He, B., Geng, G., Wang, Z., et al. (2021). Programmable RNA editing with compact CRISPR-Cas13 systems from uncultivated microbes. Nat. Methods 18, 499–506. doi: 10.1038/s41592-021-01124-4
Xue, L., Tang, B., Chen, W., and Luo, J. (2019). Prediction of CRISPR sgRNA activity using a deep convolutional neural network. J. Chem. Inf. Model. 59, 615–624. doi: 10.1021/acs.jcim.8b00368
Yang, Q., Wu, L., Meng, J., Ma, L., Zuo, E., and Sun, Y. (2022). EpiCas-DL: Predicting sgRNA activity for CRISPR-mediated epigenome editing by deep learning. Comput. Struct. Biotechnol. J. 21, 202–211. doi: 10.1016/j.csbj.2022.11.034
Younis, O. G., Corinzia, L., Athanasiadis, I. N., Krause, A., Buhmann, J. M., and Turchetta, M. (2024). Breeding programs optimization with reinforcement learning. ArXiv abs/2406.03932. doi: 10.48550/arXiv.2406.03932
Yu, G., Kim, H. K., Park, J., Kwak, H., Cheong, Y., Kim, D., et al. (2023). Prediction of efficiencies for diverse prime editing systems in multiple cell types. Cell 186, 2256–2272.e23. doi: 10.1016/j.cell.2023.03.034
Yuan, T., Wu, L., Li, S., Zheng, J., Li, N., Xiao, X., et al. (2024). Deep learning models incorporating endogenous factors beyond DNA sequences improve the prediction accuracy of base editing outcomes. Cell Discov. 10, 20. doi: 10.1038/s41421-023-00624-1
Zarate, O. A., Yang, Y., Wang, X., and Wang, J. P. (2022). BoostMEC: predicting CRISPR-Cas9 cleavage efficiency through boosting models. BMC Bioinf. 23, 446. doi: 10.1186/s12859-022-04998-z
Zarbakhsh, S., Shahsavar, A. R., and Soltani, M. (2024). Optimizing PGRs for in vitro shoot proliferation of pomegranate with bayesian-tuned ensemble stacking regression and NSGA-II: a comparative evaluation of machine learning models. Plant Methods 20, 82. doi: 10.1186/s13007-024-01211-5
Zhang, G., Dai, Z., and Dai, X. (2020a). C-RNNCrispr: Prediction of CRISPR/Cas9 sgRNA activity using convolutional and recurrent neural networks. Comput. Struct. Biotechnol. J. 18, 344–354. doi: 10.1016/j.csbj.2020.01.013
Zhang, Z. R. and Jiang, Z. R. (2022). Effective use of sequence information to predict CRISPR-Cas9 off-target. Comput. Struct. Biotechnol. J. 20, 650–661. doi: 10.1016/j.csbj.2022.01.006
Zhang, Z., Lamson, A. R., Shelley, M., and Troyanskaya, O. (2023). Interpretable neural architecture search and transfer learning for understanding CRISPR-Cas9 off-target enzymatic reactions. Nat. Comput. Sci. 3, 1056–1066. doi: 10.1038/s43588-023-00569-1
Zhang, S., Li, X., Lin, Q., and Wong, K. C. (2019). Synergizing CRISPR/Cas9 off-target predictions for ensemble insights and practical applications. Bioinformatics 35, 1108–1115. doi: 10.1093/bioinformatics/bty748
Zhang, Y., Long, Y., Yin, R., and Kwoh, C. K. (2020b). DL-CRISPR: A deep learning method for off-target activity prediction in CRISPR/Cas9 with data augmentation. IEEE Access 8, 76610–76617. doi: 10.1109/ACCESS.2020.2989454
Zhang, G., Luo, Y., Xie, H., and Dai, Z. (2024). Crispr-SGRU: Prediction of CRISPR/Cas9 off-target activities with mismatches and indels using stacked BiGRU. Int. J. Mol. Sci. 25, 10945. doi: 10.3390/ijms252010945
Zhang, D., Xu, F., Wang, F., Le, L., and Pu, L. (2025). Synthetic biology and artificial intelligence in crop improvement. Plant Commun. 6. doi: 10.1016/j.xplc.2024.101220
Zhang, G., Zeng, T., Dai, Z., and Dai, X. (2021). Prediction of CRISPR/Cas9 single guide RNA cleavage efficiency and specificity by attention-based convolutional neural networks. Comput. Struct. Biotechnol. J. 19, 1445–1457. doi: 10.1016/j.csbj.2021.03.001
Zhong, Z., Liu, G., Tang, Z., Xiang, S., Yang, L., Huang, L., et al. (2023). Efficient plant genome engineering using a probiotic sourced CRISPR-Cas9 system. Nat. Commun. 14, 6102. doi: 10.1038/s41467-023-41802-9
Keywords: artificial intelligence, plant tissue culture, genome editing, machine learning, deep learning, CRISPR
Citation: Narra M, Ray A, Polley B, Yang H and Bhowmik PK (2025) AI-driven advances in plant biotechnology: sharpening the edge of plant tissue culture and genome editing. Front. Plant Sci. 16:1718810. doi: 10.3389/fpls.2025.1718810
Received: 04 October 2025; Accepted: 20 November 2025; Revised: 17 November 2025;
Published: 10 December 2025.
Edited by:
Marcos Egea-Cortines, Polytechnic University of Cartagena, SpainReviewed by:
Mostafa Farajpour, Mazandaran Agricultural and Natural Resources Research and Education Center, IranPoulomi Sen, Sister Nivedita University, India
Copyright © 2025 Narra, Polley, Yang and Bhowmik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pankaj K. Bhowmik, UGFua2FqLkJob3dtaWtAbnJjLWNucmMuZ2MuY2E=