




- 1 Plant Biology, Department of Biology, University of Fribourg, Fribourg, Switzerland
- 2 Institute of Plant Biology, Zurich–Basel Plant Science Center, University of Zurich, Zurich, Switzerland
- 3 Department of Horticulture and Landscape Architecture, Purdue University, West Lafayette, IN, USA
In contrast to mammalian ABCB1 proteins, narrow substrate specificity has been extensively documented for plant orthologs shown to catalyze the transport of the plant hormone, auxin. Using the crystal structures of the multidrug exporters Sav1866 and MmABCB1 as templates, we have developed structural models of plant ABCB proteins with a common architecture. Comparisons of these structures identified kingdom-specific candidate substrate-binding regions within the translocation chamber formed by the transmembrane domains of ABCBs from the model plant Arabidopsis. These results suggest an early evolutionary divergence of plant and mammalian ABCBs. Validation of these models becomes a priority for efforts to elucidate ABCB function and manipulate this class of transporters to enhance plant productivity and quality.
Introduction
Members of the ATP-binding cassette (ABC) transphyletic protein superfamily are predominantly transporters that function in movement of a wide variety of substrates across cellular membranes. The B subclass of this superfamily comprises the eukaryotic P-glycoproteins (PGPs; Hrycyna and Gottesman, 1998; Higgins, 2001). The most notorious ABCB transporter is human P-glycoprotein/multiple drug resistance1 (P-GP, MDR1, HsABCB1) due to its contribution to cellular resistance toward multiple cytotoxic chemotherapeutic agents when overexpressed in tumor cells (O’Connor et al., 2007). Elucidation of the mechanistic basis of ABCB multi-substrate specificity has been a longstanding biomedical research priority (Borowski et al., 2005) and has been accelerated by the publication of a high-resolution crystal structure of murine ABCB1 complexed with a cyclic tetrapeptide inhibitor (3G5U, 3G60, 3G61) in a ligand-binding conformation (Aller et al., 2009). Publication of detailed structures of the bacterial ABC transporters Sav1866, MsbA, and MalGFK (Dawson and Locher, 2006, 2007; Dawson et al., 2007; Oldham et al., 2007; Ward et al., 2007; Aller et al., 2009; Oldham and Chen, 2011a) have provided important insights into the conserved mechanisms of ATP hydrolysis, domain organization, and membrane interactions in ABC transporters. The crystal structures of MmABCB1 and Sav1866 represent the putative ligand-binding and ligand-releasing states of the transport cycle, thus allowing modeling of both states in the context of the commonly accepted ABCB export mechanism (Li et al., 2010). Increasingly, these crystal structures are utilized by researchers to inform experimental analyses of specific details of hydrophobic substrate translocation in mammalian ABCBs.
Plant genomes contain an expanded family of ABC-transporter genes: over 120, compared to 50–60 in other organisms of equivalent genome size (Theodoulou, 2000; Sanchez-Fernandez et al., 2001a,b; Martinoia et al., 2002; Jasinski et al., 2003). Despite their high numerous representation, only a few ABCB orthologs have been extensively characterized in plants and shown to catalyze the transport of structurally diverse substrates, such as phytohormones, secondary metabolites, and xenobiotics (Geisler et al., 2005; Sugiyama et al., 2006; Knoller et al., 2010). However, the most extensive analysis of ABCB function has taken place in the model plant Arabidopsis and has focused on AtABCB1 (PGP1) and AtABCB19 (PGP19/MDR1; Geisler et al., 2005; Geisler and Murphy, 2006). Lesions in the genes encoding these proteins result in reductions in long distance transport of the phytohormone auxin and consequent dwarfism in mutant plants (Figure A1 in Appendix). Most notably, the agriculturally important brachytic2 and dwarf3 mutants in maize and sorghum were shown to result from loss-of-function mutations in ABCB1 genes (Figures A1A,B in Appendix; Multani et al., 2003). AtABCB1 and AtABCB19 do not transport standard HsABCB1 substrates, even when overexpressed in human HeLa cells, but directly transport the natural auxin indole-3-acetic acid (IAA) and the artificial auxin 1-NAA (AtABCB1 transports some additional artificial auxins, and, to a lesser extent, auxin-like compounds; Noh et al., 2001; Geisler et al., 2003, 2005; Bouchard et al., 2006; Bailly et al., 2008). A third ABCB transporter, AtABCB4, is a conditional auxin im/exporter with specificity for auxins similar to ABCB1 (Santelia et al., 2005; Terasaka et al., 2005; Kubes et al., 2011). AtABCB14 that is included in this study as non-auxin transporting ABCB is a malate/citrate transporter functioning in Arabidopsis guard cells (Lee et al., 2008).
The relative substrate specificity of these plant transporters in comparison to mammalian orthologs suggests that plant transporters of the ABCB1/19 subgroup contain either very different amino acid compositions in substrate-binding sites identified in HsABCB1 or additional sites that dictate narrower substrate specificity (Shapiro and Ling, 1997). However, amino acid sequences variation associated with these differential specificities are not readily deduced from phylogenetic sequence comparisons alone (Knoller et al., 2010). Robust comparative homology modeling and sequence analysis of a variety of plant and mammalian ABCB1 proteins with dissimilar substrate specificities is therefore a strategy of choice to predict kingdom-specific ligand-recognition patterns within the protein subfamily. The results presented here support evolutionary divergence within the recently proposed substrate-binding domains and indicate that electrostatic changes in surface residues within the translocation chamber dictate substrate specificity.
Materials and Methods
Homology Modeling of ABCB Structures
The high-resolution P-glycoprotein Mus musculus ABCB1 (MmABCB1) structure (3G5U), representing the ligand-binding competent conformation, was utilized as a homology modeling template for Arabidopsis ABCB1, 19, 4, and 14 transporters (GenBank accession numbers: ABCB1:NP_181228, ABCB4: NP_182223, ABCB14: NP_174122, ABCB19: NP_189528). The sequences of AtABCBs and MmABCB1 were used to generate a multiple alignment with MultAlin (Corpet, 1988). The alignments used to build the models are shown in Table S2 in Supplementary Material. The Modeller9v7 was used to generate the AtABCB models based on the alignments and MmABCB1 crystal structure (Sali and Blundell, 1993; Eswar et al., 2006). The N-terminal and linker regions of AtABCBs connecting nucleotide-binding domain (NBD) 1 with transmembrane domain (TMD) 2 were removed since these regions in MmABCB1 are missing in the crystal structure. A second set of homology models was generated using the nucleotide-bound state Sav1866 crystal structure as template (2HYD). AtABCB4, 14, and 19 models were described in Yang and Murphy (2009). Each half of ABCBs were aligned with Sav1866 using BLAST21 and MultAlin (Corpet, 1988). The N-terminal and linker regions were used to blast protein sequence by selecting Protein Data Bank proteins as database. The most similar structures were chosen for templates for N-terminal and linker regions. As an example, the complete template alignments for HsABCB1 are shown in Table S3 in Supplementary Material in two runs of modeling. For each ABCB model, five models were generated and the ones with best molecular probability density function (molpdf) and discrete optimized protein energy (DOPE) scores were selected. The evaluation of the selected models was carried out by analyses of ERRAT (Colovos and Yeates, 1993), Qmean (Benkert et al., 2008), PROCHECK (Laskowski et al., 1993), and WHAT_CHECK (Hooft et al., 1996). The TMDs and NBDs regions in both Sav1866- and MmABCB1-based models showed good scores in assessment with these analyses while the N-terminal, linker, and C-terminal regions in Sav1866-based models showed less satisfied scores in these analyses. These regions were not modeled in MmABCB1-based models due to difficulties to model these regions without disturbing the flexible NBDs. All ABCB models from this study are available as PDB files as listed in the Supplementary Material information.
Alignments and Structure Display
Multiple sequence alignments of the primary polypeptide sequences of identified HsABCB1 and AtABCB1 orthologs (listed in Table S1 in Supplementary Material) and the generation of bootstrapped N-J trees were performed using ClustalX v2.1 (Larkin et al., 2007) in its default settings. In order to map the relative degree of residue conservation onto protein model surfaces, the ConSurf server tool2 (Ashkenazy et al., 2010) has been next used with the relevant PDB templates or generated models and guiding trees, employing the JTT evolutionary substitution model and the Bayesian calculation method. Output figures have been generated using PyMOL v1.33.
Substrate Docking to ABCB TMDs
For the docking process 35 different auxins and auxin-related compounds have been selected according to their different structure–effect relationships in hormonal activity (Ferro et al., 2006, 2010) and were docked into both homology models. Thousand poses for IAA and 100 poses for each of the other compounds were generated using the PyMOL embedded AutoDock Vina toolset (Seeliger and de Groot, 2010). In order to avoid any bias, the binding site was defined either as the whole molecule or the complete transmembrane region for each model conformer. This region in AtABCB1 shows patterns of polar and apolar residues suitable for IAA binding (Figure 2; Tan et al., 2007; Ferro et al., 2010) and, since there is indication that the protein’s translocation chamber is water filled during the catalytic cycle (Gutmann et al., 2010), the ligands were docked in their ionized state.
Electrostatics
Electrostatic surfaces were generated for both inward- and outward-facing conformations of the structural models using the APBS Tools2 plugin (Baker et al., 2001) implemented within PyMOL. The solving of the non-linear Poisson–Boltzmann equation had been performed with the package default settings and molecular surfaces were colored by the potential on solvent accessible surface using a temperature spectrum ranging from −10 to +10 kT.
Results
Predictions from Structural Modeling
ABCB proteins share a common architecture
ATP-binding cassette transporters share a common architecture consisting of two TMDs and two cytosolic NBDs (constituted by a dimer in the “half transporter” subgroup). Models derived from high-resolution crystal structures of MmABCB1, Sav1866, and MalGFK2 are shown in Figures 1A–C (Dawson and Locher, 2006, 2007; Dawson et al., 2007; Oldham et al., 2007; Aller et al., 2009; Oldham and Chen, 2011a,b). Prokaryotic ABC importers contain a fifth component, a periplasmic or cell-surface-associated binding protein that binds specific solutes with high affinity as shown in maltose transporter in both pretranslocation intermediate state (Figure 1C left) and translocation intermediate state (Figure 1C right; Oldham et al., 2007; Oldham and Chen, 2011a,b).
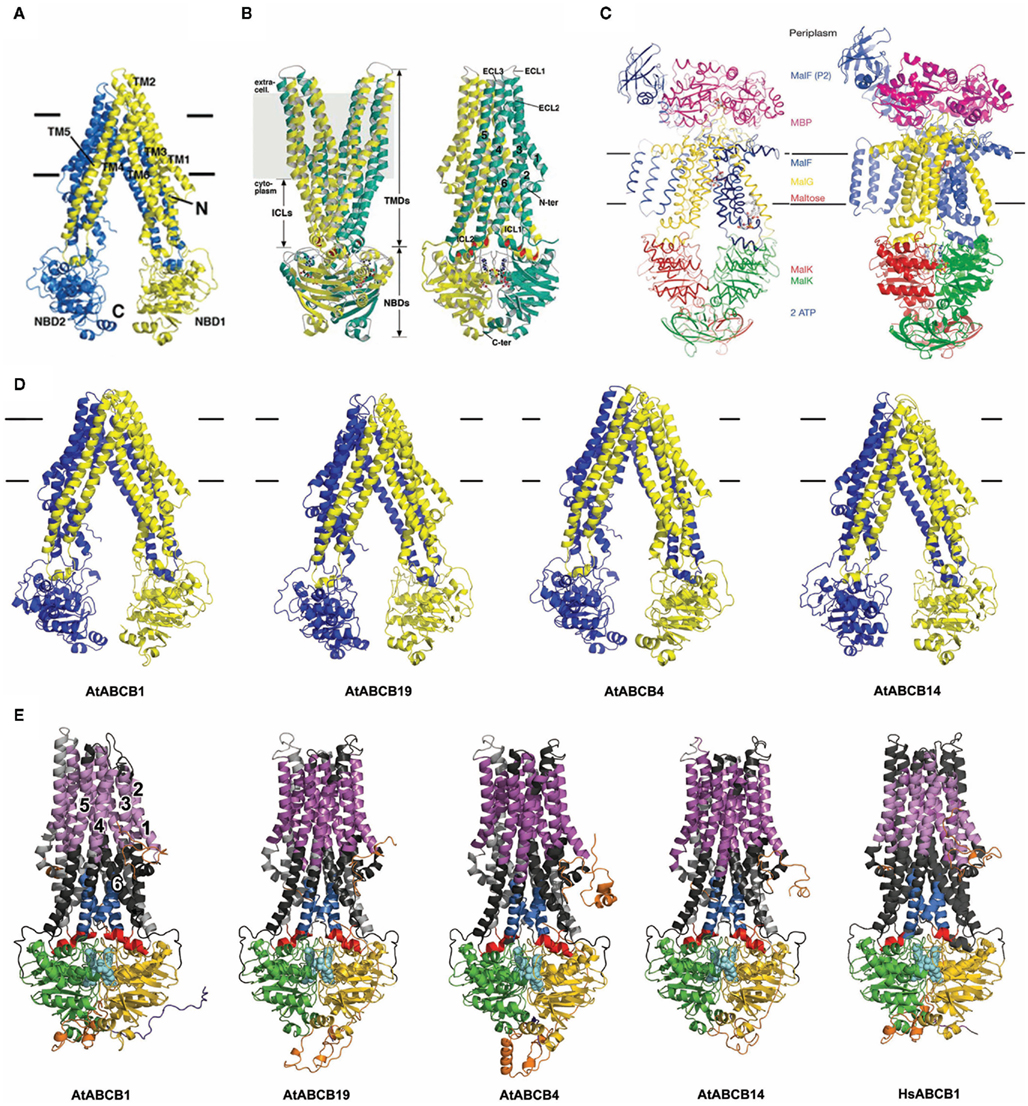
Figure 1. ABC structures and Arabidopsis ABCB models. (A) MmABCB1 (Aller et al., 2009). (B) Sav1866 (Dawson and Locher, 2006). (C) Maltose transporter: left, pretranslocation intermediate state (Oldham and Chen, 2011a), right, translocation intermediate state (Oldham et al., 2007). (D) Arabidopsis ABCB structural models based on MmABCB1. (E) ABCB models based on Sav1866 including AtABCB19 and AtABCB4 models (Yang and Murphy, 2009).
Arabidopsis ABCB transporters, ABCB1, ABCB4, and ABCB19 that were independently and unambiguously demonstrated to function as auxin transporters (Geisler et al., 2005; Santelia et al., 2005; Terasaka et al., 2005; Bouchard et al., 2006; Cho et al., 2007) were chosen for our analyses. Moreover, AtABCB14 as non-auxin transporter (Lee et al., 2008) was included in this study (see Introduction for details). Models of the Arabidopsis unidirectional exporters ABCB1 and ABCB19 and conditional exporters ABCB4 and 14 were threaded onto theMmABCB1 and Sav1866 crystal structures (Figures 1D,E). AtABCB4/14 aligned better with exporters MmABCB1 and Sav1866 than ABC importers with reported crystal structures, suggesting that all Arabidopsis full-length ABCB transporters share ABC exporter-like architectures and are dissimilar to prokaryotic importers. Instead, import activity observed in some plant ABCB transporters appears to be determined by N-terminal linker regions and additional substrate binding or regulatory sites (Yang and Murphy, 2009; Kubes et al., 2011). The linker domains of ABCBs were constructed in outward-facing models based on structures searched from PDB database (described in Materials and Methods; Figure 1E), and these domains may function to stabilize the NBDs as in the maltose transporter MalGFK2 (Oldham et al., 2007).
The canonical structure of ABCB-type proteins can be extended to all generated models and adopts a pseudo-symmetric arrangement of the two TMD–NBD modules (Figures 1D,E). This forms a large internal cavity exposed to the cytosol with separated NBDs in the nucleotide-free conformation, and an asymmetric distribution in the nucleotide-bound state that reveals the conduction chamber to the outer medium (Rosenberg et al., 2005; Dawson and Locher, 2007). In the commonly accepted ABC transport mechanism, helices from each TMD participate to the binding site(s), with a predominant role for TMH6 and TMH12 (Martin et al., 2001; Pleban et al., 2005) and form the translocation pathway for the substrate, while both NBDs transmit the necessary ATP-dependent “power stroke” to perform a complete transport cycle (Hopfner et al., 2000; Hollenstein et al., 2007; Linton and Higgins, 2007). The overall Sav1866 and MmABCB1 architectures and the subsequent structure-based allosteric mechanisms proposed in Hollenstein et al. (2007) are therefore relevant for all close ABCB-type exporters (Dawson and Locher, 2006; Hollenstein et al., 2007). As such, these computational predictions suggest that substrate specificity in plant ABCBs is determined primarily by characteristics of the TMDs. Moreover, biochemical and structural evidences designate the top of the inward-facing translocation chamber as the paradigm for the competent ligand-binding region that could provide access to either lipophilic or cytosolic substrates with minimum energies (Gutmann et al., 2010). For simplicity, further analyses concentrated on comparison of ABCB1 with other characterized plant ABCB transporters and MmABCB1 and Sav1866.
Plant and Mammalian ABCB1 Proteins Display Kingdom-Specific and Substrate-Specific Translocation Chambers
Most of the divergences observed in ABCB substrate recognition can be attributed to residues facing the translocation chamber space. However the differences between poly- and mono-specific transporters can only be identified by analyzing the 3-D feature of the translocation chamber space. The combination of homology modeling and surface mapping of conservation scores employed in this study stands as an initial effort to pinpoint substrate specificity in large transmembrane proteins to domains composed by the dynamic arrangement of multiple transmembrane helices. Alignment and projection of plant ABCB transporter sequences onto the protein model surfaces of MmABCB1 and Sav1866 allowed us to assess the conservation of amino acid residues facing the chamber cavity. Not surprisingly, plant and animal proteins share a high degree of conservation between functional and structural domains such as nucleotide-binding folds and coupling helices, but low degree of conservation in the residues exposed to the transport cavity (Figure 2). Although precursors of plant and animal ABCB genes appear to have diverged during early plant evolution (Rea, 2007; Knoller et al., 2010; De Smet et al., 2011), comparisons of plant and animal sequences indicated considerable conservation within three regions within the translocation chamber and a fourth region exhibiting conservation to a lesser extent (Figure 2). Comparison of mammalian ABCB1 sequences showed a strong and large spatial conservation of the chamber residues that correlates with the poly-specific drug-binding model suggested for HsABCB1 based on published drug-binding data (Aller et al., 2009; Klepsch et al., 2011). This conservation reflects the relatively recent evolution of multi-substrate specificity in mammalian ABCBs and the earlier evolution of plant ABCB auxin transporters (Knoller et al., 2010). Although only a small numbers of ABCB transporters have been characterized in planta (reviewed in Knoller et al., 2010; Zazimalova et al., 2010), the respective auxin transport functions of AtABCB1 and 19 have been conserved in monocots and dicots (Knoller et al., 2010), suggesting that the selective specificity of ABCB auxin transporters appeared early in plant evolution.
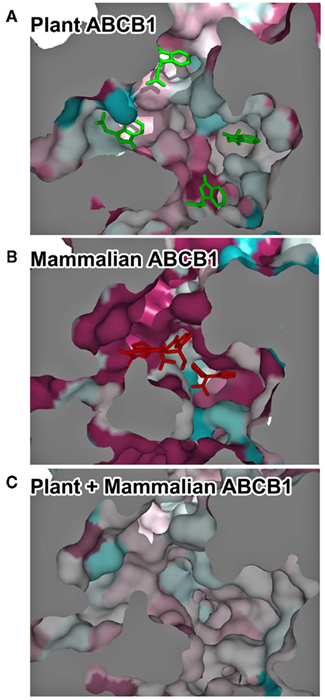
Figure 2. Kingdom-specific conservation of the translocation chamber suggests early specialization of the transporters. (A–C) Temperature-coded residue conservation between plant ABCB1 orthologs (A), mammalian ABCB1 orthologs (B), and all ABCB1 sequences (C). Dark-blue, lowest conservation score; dark red, highest conservation score. Bottom view of the inward-facing translocation chamber. For illustration purposes, representative IAA docking poses are displayed as green sticks and QZ59 crystallographic coordinates are displayed in red sticks.
Docking of Indole-3-Acetic Acid to AtABCB1 Structural Models
The generation of 1000 IAA docking runs into the entire inward- and outward-facing structural models of AtABCB1 and AtABCB19 resulted in homologous clusters of docking poses concentrated within the translocation chambers of the transporters (Figures 3A,B). The poses generated for the outward-facing conformer (Figure 3B) targeted the intracellular loops (ICLs) region and the inner leaflet regions of the transporter, which are similar to the sites predicted in AtABCB19 (Yang and Murphy, 2009). Four binding regions were predicted in inward-facing AtABCB1 model with two regions (1 and 2) at the position of inner leaflet membrane, region 3 at the outer leaflet of membrane and region 4 out of the cell is not an entry binding sites (Figure 3A). The large majority of the resulting poses in outward-facing conformer was distributed at the region 3 (Figure 3A), which overlaps with the binding region of the cyclic peptide QZ59 in the co-crystal structures of MmABCB1 published by Aller et al. (2009). Analysis of the surface residues surrounding the region 3 showed that residues primarily located on TM helices 5, 6, 7, 11, and 12 were involved in binding (Table 1). The importance of a large majority of these residues was formerly assessed in different studies (Loo and Clarke, 2000, 2001; Loo et al., 2006a,b; Pajeva et al., 2009; Bessadok et al., 2011). Numerous residues involved have been previously shown to influence HsABCB1 transport activity (see Table 1). Interestingly, our analysis indicates that most of the identified residues listed in Table 1 are indeed evolutionary conserved among ABCB1 orthologs taken from Table S1 in Supplementary Material (results not shown). This holds also true for Asp974, shown to be highly critical for transport activity of AtABCB1 functionally expressed in yeast (see below).
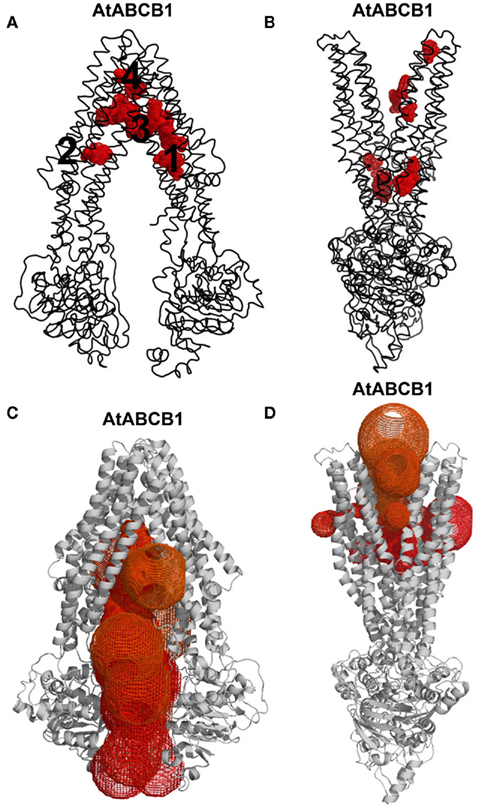
Figure 3. Exhausting in silico docking revealed auxin-binding hotspots in AtABCB1. (A,B) Thousand IAA docking poses (red meshes) segregated in discrete regions of the transporter (black ribbon) mainly located at the bottom of the funnel-shaped translocation chambers in the inward- (A) and outward-facing (B) conformations. (C,D) MOLE-computed access routes (colored meshes) to the bottom of the translocation chambers in the inward- (C) and outward-facing (D) conformations of the transporter (gray cartoon).
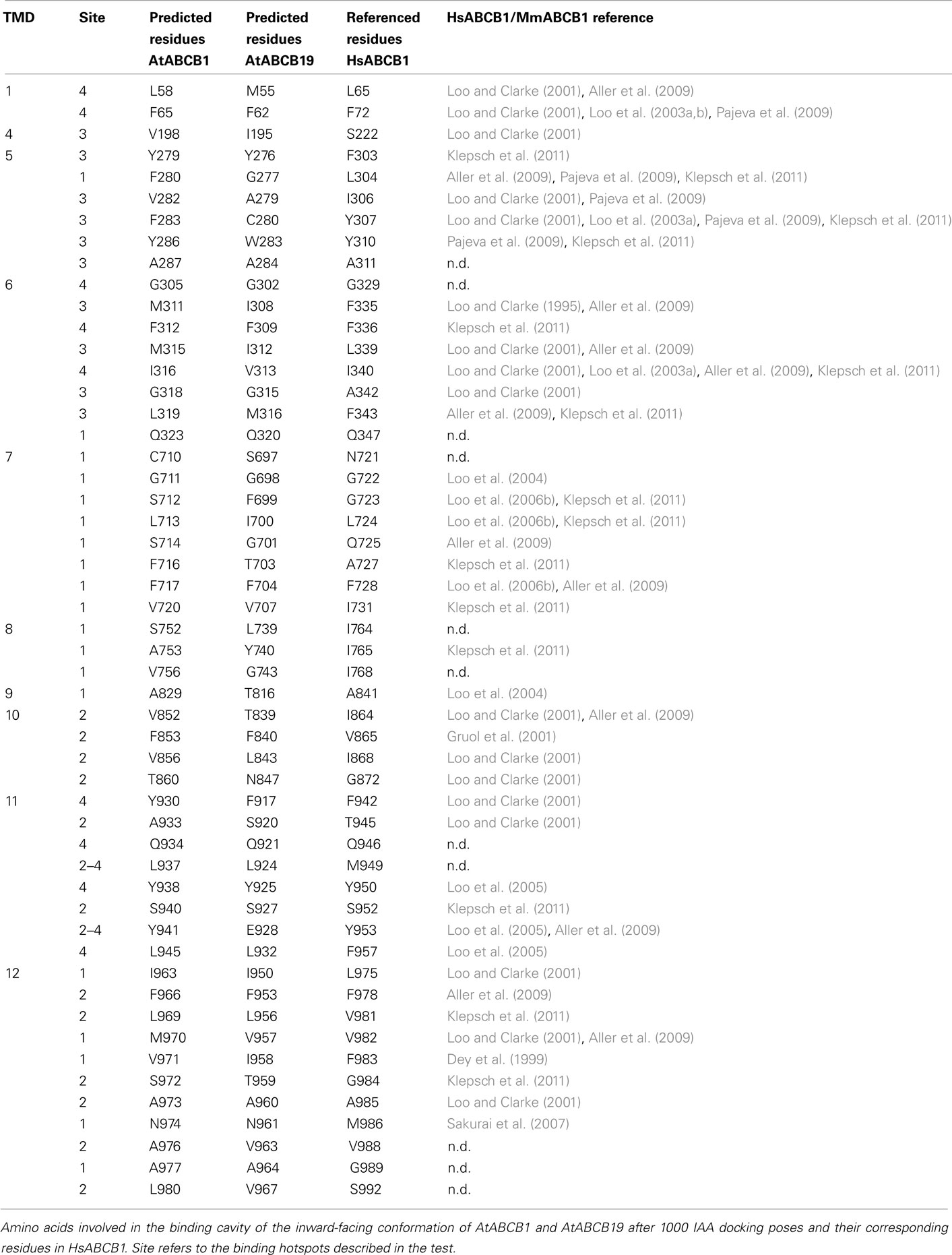
Table 1. ABCB1–ligand interaction.
Putative AtABCB1 Auxin-Binding Sites Are Consistent with the Efflux Process
The transport of the major natural auxin, IAA, across biological membranes has been extensively studied (reviewed in Petrasek and Friml, 2009). Due to the acidic pH in the apoplastic space (around pH 5.5), a part of IAA can as protonated IAAH readily diffuse over the plasma membrane. When IAA reaches the neutral cytosolic compartment, the IAA− anion then prevails consequently trapping the hormone inside the cell (Rubery and Sheldrake, 1974; Raven, 1975). Dedicated plasma membrane transporters, such as AtABCB1/19 or members of the PIN-FORMED family (Petrasek and Friml, 2009) are then needed to control auxin efflux from the cell. This process infers that AtABCB1 recognition sites for the polar IAA− are required to face the cytosolic space. The AtABCB1 inward-facing structure model therefore represents the auxin-binding competent state of the exporter. The two predicted binding sites associated with the inner leaflet are likely to bind IAA− from the cytosolic side of the membrane and are predicted by docking analyses to be more specific. The third predicted region corresponding to the position of QZ59 binding sites of MmABCB1 may be the site for exclusion of IAAH in the lipid bilayer, which is less specific as proposed in Aller et al. (2009): substrates wrapped by lipids enter the central binding sites (site 3) and are flipped out to the outer leaflet of the membrane during the change to outward-facing conformation. Another possibility is that the third region functions as an intermediate site for IAA translocation rather than as an entry site. Apolar localization of AtABCB1 and AtABCB19 suggests that, unlike PINs, AtABCB1, and AtABCB19 (Blakeslee et al., 2007; Wu et al., 2007; Mravec et al., 2008) function primarily in exclusion of IAAH from cellular membranes via site 3. However, the higher substrate specificity observed in the plant ABCB1/19 transporters compared to MmABCB1 favors interactions at the inner leaflet sites in region 1 and 2 (Figure 3A).
Unfortunately, due to the limitations of the docking algorithm it was technically not possible to run the simulations with IAA− as AutoDock Vina ignores the user-supplied partial charges. On the other side, we would like to stress that at the generally assumed apoplastic pH of around 5.5, the majority of IAA (pKa = 4.75) would be in its deprotonated form (roughly 83%, Zazimalova et al., 2010).
In AtABCB1, binding region 3 contains four apparent hotspots. Interestingly, top-scoring IAA docking poses in region 3 cluster in a homologous, if not identical, area in AtABCB19. Mutations in HsABCB1 residues equivalent to AtABCB1 M970 and N974 (V982 and M986, respectively) have biochemically been associated with substrate binding in the human transporter (Loo and Clarke, 2000, 2001; Loo et al., 2003a,b, 2006a,b). An Asp974 to Ala mutation in AtABCB1 functionally expressed in Saccharomyces cerevisiae resulted in a substantial decrease in the auxin efflux activity (Bailly and Geisler, unpublished data). The functional conservation of IAA-binding sites tends to argue for a paradigm of auxin-binding modus preserved in plants under the evolution pressure. Subsequent structure analysis using MOLE (Petrek et al., 2007) revealed that the center cavity of AtABCB1 in inward-facing conformation is only accessible from the cytosol and the inner leaflet of the lipid bilayer, thus allowing the separation between the intra- and extracellular compartments (Figure 3C). In the outward-facing AtABCB1 conformer, upon the large rearrangement of the transmembrane domain and especially demonstrated TMH6 movements (Loo and Clarke, 1997; Rothnie et al., 2004; Oldham et al., 2007; Storm et al., 2008; Becker et al., 2010), important residues for ligand binding to the inward-facing conformation move away from the binding cavity and face the apoplastic space, get buried in the TMD or face the lipid bilayer (Figure 3D). Therefore all putative binding sites are destroyed and favorable protein–ligand interactions are likely to be disrupted, thus representing the step for IAA release outside of the cell (Oldham et al., 2007; Gutmann et al., 2010). Indeed, substrate paths generated by MOLE in the outward-facing conformers exit the hotspots binding region to the extracellular space and outer plasma membrane leaflet (Figure 3D). Furthermore, in our simulations, IAA hardly docked in the upper parts of the transmembrane region when challenged against the outward-facing conformer (Figure 3B). Taken together, these results infer a conserved translocation mechanism in which IAA binds to the high-affinity internal transporter chamber from the cytosol in the closed conformation and is subsequently excluded from the low-affinity, opened binding sites to the outer space in the course of TMD reorganization (Becker et al., 2010).
AtABCB1 Can Distinguish Dissimilar Auxins
The Arabidopsis ABCB1 and ABCB19 proteins have been recently extensively characterized and a large body of physiological, cellular, and biochemical data confirmed their function as auxin-transport components specialized in the export of the hormone across the plasma membrane (Geisler et al., 2005; Wang and Lin, 2005). However, investigators have focused their efforts on few well-studied auxins, thus leaving old questions about other auxin-related compounds unanswered. Therefore we here took the opportunity to challenge our structural models with 34 auxin-related compounds distributed within three classes categorized as Class 1 (active auxins), Class 2 (weak auxins and competitive inhibitors), and Class 3 (inactive, but structurally related compounds; Table 2; Ferro et al., 2006). Best docking results showed IAA poses mainly distributed in hotspots 1, 2, and 3, with a close-fitting clustering in hotspot 1 (Figure 4A). Remarkably, Class 1 molecules followed the same docking pattern as IAA and most of the docking poses were in the same magnitude of computed docking energy as IAA (Table 2) and preferentially targeted hotspots 2, 3, and 1, in this order respectively (Figure 4B). More interestingly, docking results from Class 2 and Class 3 showed a significantly different patterning: although these inactive or transport-inhibiting compounds share close structures with the active auxins, they exhibited binding to hotspots 2 and 3 sites, but almost excluded hotspot 1 (Figure 4C). This suggests a dominant role for hotspot 1 in auxin recognition and this motif may hold the ability to specifically select active molecules while the other binding regions may exhibit less selectivity. Moreover, Class 2 molecules binding energies scored generally lower and Class 3 higher than IAA, thus suggesting that the behavior of those chemicals is linked to the affinity to their binding site(s) (Table 2). Remarkable examples are illustrated by 1-naphtalene acetic acid (1-NAA), 2-napthalene acetic acid (2-NAA), and 2,3,5-triiodobenzoic acid (TIBA; Figure 4D; Figure A2 in Appendix).
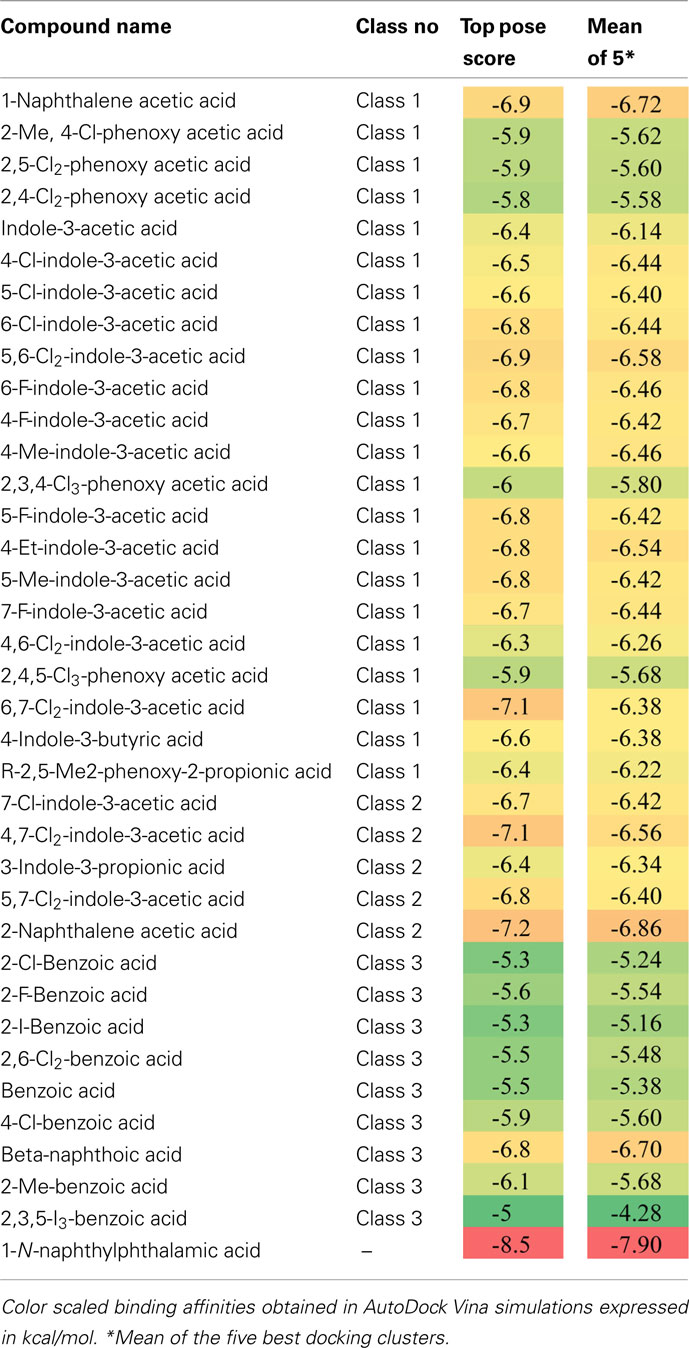
Table 2. Docking scores of auxin-related compounds used in this study.
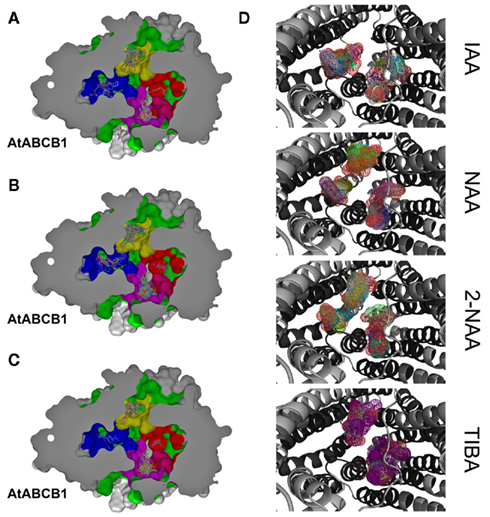
Figure 4. Docking of auxinic compounds to AtABCB1 suggests functional substrate selectivity. (A–C) Docking poses of IAA (A), active (B), and inactive (C) auxinic compounds in the inward-facing cavity. IAA-binding hotspots are colored on the protein surface as 1, blue; 2, magenta; 3, yellow; 4, red. (D) Close-up views of docking poses (shown as colored meshes) of commonly used auxin-related compounds. AtABCB1 is shown as a gray cartoon and predicted transmembrane residues are colored in black.
Synthetic 1-NAA acts as an active lipophilic auxin and mimics IAA docking pattern by occupying the four hotspots (Figure 4D) with lower binding energies than IAA, probably due to the stronger hydrophobicity generated by the naphthalene ring compared to the indole ring in absence of an obvious hydrogen-binding partner for the indole amine (Table 2). Surprisingly, its close enantiomer, the anti-auxin, 2-NAA, an inactive auxin analog, docks to the same regions in AtABCB1 and AtABCB19 but with higher affinities (Table 2). This is further supported by analogous behaviors from auxinic competitors in early radiolabeled auxin-binding protein studies (Lobler and Klambt, 1985). Expression of AtABCB1 in yeast and human heterologous systems lead to enhanced export capacities of IAA and NAA but not 2-NAA (Geisler et al., 2005), inferring that the inefficient transport might lie in its high affinity to the binding pocket. Class 2 compounds displayed the highest binding scoring consistent with the role of transport inhibitors by competing with active auxins for substrate-binding sites (Table 2).
Despite its clearly different ring structure, the weak auxin, TIBA, is thought to act as auxin-transport inhibitor by mimicry of auxin in planta (Keitt and Baker, 1966). However, in computed dockings against AtABCB1 and AtABCB19, TIBA docks to hotspots 2, 3, and 4 with very low scores in the same range as benzoic acid and its derivatives (Figure 4D; Table 2). Moreover, TIBA is excluded from the first IAA-binding hotspot, suggesting that the transporters probably do not recognize the molecule as a potent substrate (Figure 4D). This suggests that the inhibitory effect of TIBA on AtABCB1 activity might be conferred not by direct interference with the substrate-binding site of ABCB1 as found for allosteric inhibitors or by indirect events.
Following the idea that ABCB-mediated polar auxin-transport inhibitors may directly interact with the auxin-binding sites, we included NPA (1-N-naphthylphthalamic acid) in our analysis. Interestingly, in addition to the docking poses in the TMD–TMD interface previously reported (Kim et al., 2010), NPA docked into hotspot 1 and 3 with the highest binding score observed in this study (Table 2), which is in line with its reported striking inhibitory effect on ABCB1-mediated auxin transport (Wang and Lin, 2005; Bailly et al., 2008; Nagashima et al., 2008) and NPA binding studies (Sussman and Gardner, 1980; Muday et al. 1993). These data however also support the concept that ABCBs are primary NPA targets (Rojas-Pierce et al., 2007; Kim et al., 2010).
Surface Electrostatics of Translocation Chambers Reflect Substrate Specificity
In order to understand the functional influence of evolution on the translocation pathways of ABCB proteins and its consequence on substrate specificity, electrostatic potentials were computed for a choice of related plant and mammalian sequences from different species and were mapped to each corresponding chamber surface. Further, the overall distribution of apolar residues and polar residues on both the Arabidopsis and murine ABCB1 binding chambers (Figure 5) is not sufficient to clarify the observed discrepancies in substrate specificity. Elements of answer may come from the analysis of the electrostatic potentials computed for both translocation chambers.
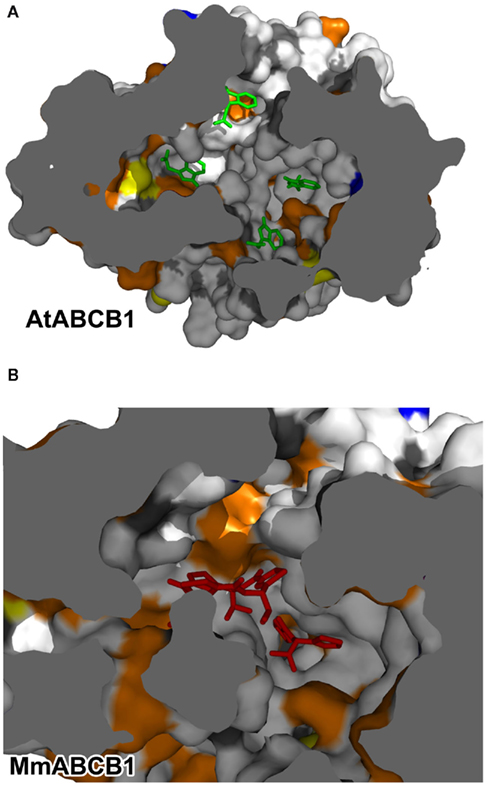
Figure 5. Surface distribution of residue properties in the ABCB translocation chamber. (A) AtABCB1; representative IAA docking poses are depicted as green sticks. (B) MmABCB1; Co-crystallized compounds coordinates are shown as red sticks. Residue property color code is as follow: white, apolar; orange, polar; red, acidic; blue, alkaline; yellow, cysteine.
In its inward-facing modeling, AtABCB1 displayed predominantly lateral negative potentials near the entrance of the cavity that weakens to a more neutral environment in the region where the binding hotspots were described (Figure 4A). In a greater magnitude, AtABCB19 retained the same electrostatic distribution with a solvent accessible surface that remained principally negative, with values >10 kT near the putative binding pocket (Figure 6A). This conserved pattern seems to be functionally important as other models of plant ABCB1 orthologs, despite their divergence in residues aligned in the translocation chamber, kept the same negative to neutral distribution of the surface electrostatic potential (data not shown). Strikingly, AtABCB4 and AtABCB14, described to function as auxin and malate importers (Terasaka et al., 2005; Oldham and Chen, 2011b), respectively, displayed a common neutral to positive electrostatic surface throughout the whole chamber (Figure 6A), arguing for a functional evolution of the plant ABCB subfamily toward a specialization of their transport activities.
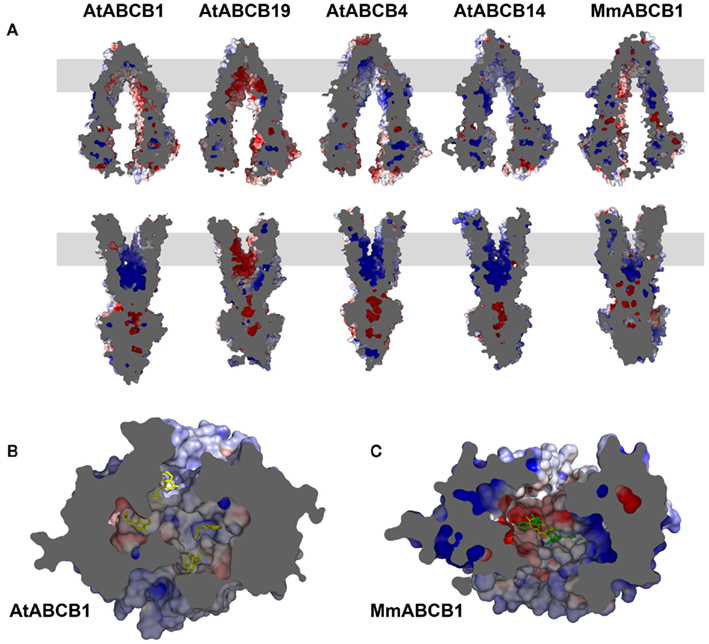
Figure 6. Surface electrostatic potential of the ABCB translocation chamber reflects its substrate specificity. (A) Cut views of Arabidopsis (At) and mouse (Mm) ABCB proteins’ surface electrostatics in both inward- (up) and outward-facing conformers (down). The gray box represents the estimated position of the lipid bilayer. (B) Bottom view of the AtABCB1 inward-facing translocation chamber. Representative IAA docking poses are shown as yellow sticks. (C) Bottom view of the MmABCB1 inward-facing translocation chamber. Co-crystallized compound coordinates are shown as yellow and green sticks. Scale is red −10 kT, blue +10 kT.
Human and murine B1 showed a less homogenous electrostatic potential ranging from strong negative and positive stretches toward the substrate entrance of the translocation pathway to a more neutral electrostatic character with small patches of strong charges closer to the substrate-binding site (Figure 6A). Remarkably, the electrostatic surface for the mammalian ABCB1 seems to mirror the various physico-chemical properties of the lipophilic substrates described for this transporter (Figure A2 in Appendix, see Sharom, 2008) in comparison to the hydrophilic IAA−.
It is worth mentioning that the transmembrane region of the TMDs concentrates most of the differences observed between the different categories of transporters included in this study. Furthermore, the observed neutral to charged environments described for the ligand-accepting conformers are tremendously altered with the TMDs conformational rearrangements associated with ATP binding and the exposition of the cavity to the extracellular space. Indeed, all molecules tested here switched to fully charged surfaces in their outward-facing conformers (Figure 6A). These changes of electrostatic properties may represent an important component of the ligand release step by presenting charged and polar residues to the cavity that were previously buried in the protein and thus creating an incompatible drug-binding situation.
To summarize, computation of surface electrostatics revealed a great level of divergence in electrostatic properties that correlated with the substrate specificity of the transporters. While the mammalian B1 substrate promiscuity is reflected by a broad range distribution of charges, narrow-specific plant transporters show a high level of functional conservation in surface electrostatic potentials.
The network of electrostatic interactions between the surface-facing residues is expected to provide a basis for the distinction between discrete sites or common functional regions. Plant ABCB models clearly show a neutral setting with weak patches of charges in the putative auxin-binding region (Figure 6B). The weak static electric fields thus represent the ideal environment for the transient interaction of such small molecules. Surface electrostatic potentials in the crystal structure of MmABCB1 correlate with the binding of the cyclic peptides QZ59-RRR and QZ59-SSS where patches of negative, neutral, and positive fields provide accommodation to these complex molecules that build hydrophobic and aromatic interaction within the chamber (Figure 6C; Aller et al., 2009). It is worth mentioning that the increased negative potential in AtABCB19 is correlating with increased IAA transport rates reflected finally by the more severe null mutant phenotype (Geisler et al., 2005; Wang and Lin, 2005; Geisler and Murphy, 2006). On the contrary, the strong negative potential concentrated at the top of the MmABCB1 translocation chamber is surrounded by a more neutral environment that may accommodate its large and lipophilic substrates. Taken with the dominantly apolar residues lying in its cavity (Figure 4), the electrostatic properties of the mammalian transporter may account for the diversity in ligands reported so far. The size of the binding region is large enough to receive bulky or long-chained compounds and the flexibility of the amino acid side chains facing the cavity is confidently sufficient to build the binding plasticity or adaptability that forms the canonical multisite previously hypothesized (Corpet, 1988; Garrigos et al., 1997; Knoller et al., 2010). Though Bachas et al. (2011) have proposed a model for multidrug recognition based on a single rigid binding pocket in the Bacillus subtilis BmrR, such a scenario seems unlikely to be true in mammalian ABCB molecules where the different structural and biochemical evidences point to a larger platform for recognition of dissimilar molecules (Lee et al., 2008; Newberry et al., 2008).
Although it represents a loose cavity when compared to enzymatic or receptor’s binding pockets, from all 4 putative binding hotspots proposed in this work, only hotspot 1 forms a close-fitting site for IAA recognition (Figure 4A). It is therefore difficult to sustain that information obtained in silico may stay valid when challenged by in vivo experimentation. However, our first attempts to use the knowledge gathered via this analysis has been the successful site-directed mutagenesis of site 1 by exchanging I963 and N974 to alanines that led to drastic decreases in AtABCB1-mediated IAA efflux from S. cerevisiae cells (not shown). Those regions may therefore form bona fide binding sites congruent with the transport cycle (Oldham et al., 2007) as each pocket described in the inward-facing conformer vanishes in the outward-facing conformer along with the large transmembrane helices movements. However, the molecular dynamics of the ABC-type cavity and its remodeling during the catalytic cycle remain difficult to predict and, although simulations have been recently performed (Oldham et al., 2007; Becker et al., 2010).
Obviously, the long evolutionary period separating plant and mammalian ABCB1 transporters has resulted in different electrostatic properties, which are likely to reflect differences in the recognition and transport of their respective substrate(s).
Discussion
Despite diverse substrate specialization, bacterial, mammalian, and plant ABCB transporters share a close common architecture (Locher, 2004; Hollenstein et al., 2007), underlining the essential function of pumping specific drugs in or out cellular compartments. Our analysis identified candidate substrate regions in the transporter’s transmembrane domains that appear to describe a paradigm for substrate recognition and subsequent translocation. Comparisons of plant and animal ABCB central binding sites suggest that the multi-substrate specificity associated with this region is not due to a randomized distribution of amino acid side chains and is functionally conserved. However, the putative IAA-binding sites and translocation surfaces of the plant ABCBs examined herein are relatively conserved, although they lack a high degree of sequence identity (Figure 4). This suggests that evolutionary pressure may have favored a plastic, multi-site substrate-binding environment over a rigid substrate-binding pocket as found for the auxin receptor (Tan et al., 2007).
The kingdom-specific differences presented here raise the question of how the large ABCB binding pocket can selectively recognize and transport molecules as large as hydrophobic HsABCB1 substrates and as small as auxins? Indeed, IAA binding may not generate sufficient energy to cause the expected “induced-fit” sterical shift by itself. Kimura et al. (2007) proposed a model for human ABC transporters in which sterols or phospholipids would be needed to fill the remaining space and play a role in small substrate recognition. This “cholesterol fill-in” concept is an attractive alternative to a water-filled cavity and is not incompatible with auxin transport but lacks biochemical evidence for plant ABCBs. In support of this concept, activity and membrane stability of Arabidopsis ABCB19 have been shown to be positively modulated by structural sterols (Rojas-Pierce et al., 2007; Titapiwatanakun and Murphy, 2009).
Despite the large space of the translocation chamber, most of the auxinic compounds tested in this analysis share the same binding sites (Figure 4) with different binding energies (Table 2). Most of these compounds display a common structural basis, comprising the ring system and the carboxylic function, but differ in their substituents, inferring that the protein would recognize rather a family of ligands than a definite molecule. The docking scores obtained here are weak and within a close range, probably because of the small size and chemical similarity of auxins, but all three compound classes showed distinct behaviors: Class 1 active molecules displayed midrange scores, Class 2 anti-auxins seemed to have the highest binding affinities and Class 3 inactive auxins had the lowest scores (Table 2). Those properties could partially explain the physiological effects of auxin-like molecules since active compounds bind preferentially to site 1 while inactive and anti-auxins do not (Figure 4). One might speculate that hotspot 1 may represent an active and selective site for active auxins where inhibitors with high affinity could block the translocation mechanism. Interestingly the efficient polar auxin-transport inhibitor, NPA, targeted hotspot 1 and 3 in our docking simulations, with the highest scores obtained in this study (Table 2). The modeling presented here suggests that the ABCB1 chamber could accommodate multiple auxin molecules at the same time. As functional cooperation between multiple sites with different affinities has been observed in HsABCB1 (Shapiro and Ling, 1997), this prediction is a priority target for experimental testing.
Conclusion and Further Direction
The solution of the Sav1886, MsbA, and MmABCB1 crystal structures and an avalanche of sequence information derived from genome sequencing projects have provided the basis for sophisticated structural models of plant ABCB transporters that can be rigorously tested. As plant genomes contain an expanded family of ABC-transporter genes (Theodoulou, 2000; Sanchez-Fernandez et al., 2001a,b; Martinoia et al., 2002; Jasinski et al., 2003), multiple narrow-specificity transporters in these sessile organisms reflects an apparent need (Geisler and Murphy, 2006). Structurally and functionally, ABCB substrate specialization seems to be recent when compared to the ancient origin of ABC proteins. Some genetic studies from water- to land-plants indicate that the complexity of the ABC-transporter family arose with the emergence of auxin-controlled development (Kelch et al., 2004), although this expansion requires further validation as more plant genomes are sequenced. The task at hand is the experimental testing of the models shown here and the identification of the cofactors required for efficient plant ABCB transport.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like thank Jue Chen and her lab members for technical advices, fruitful discussion, and comments on the manuscript. This work was supported by grants from the Division of Energy Biosciences, US Department of Energy (DE-FG02-06ER15804 to Angus S. Murphy and Haibing Yang), from the Forschungskredit of the University of Zurich (to Aurélien Bailly), and from the Swiss National Funds (to Markus Geisler).
Supplementary Material
The Supplementary Material (Tables and zipped folder of homology models in PDB format) for this article can be found online at http://www.frontiersin.org/plant_traffic_and_transport/10.3389/fpls.2011.00108/abstract
Table S1. ABCB1/PGP1 orthologs used in this study. All protein sequences used in the present work have been obtained from BLAST analyzes (blastp, psi-blast, phi-blast, and tblastn) using HsABCB1 and AtABCB1 translated cDNAs and a scoring threshold of 1000 points, 90% coverage, and e-value <0.01.
Table S2. Alignments of AtABCBs and MmABCB1 sequences that were used to build homology models for this study. For details, see Section “Material and Methods.”
Table S3. Complete template alignments for HsABCB1 are shown in two runs of HsABCB1 modeling based on Sav1866. For details, see Section “Material and Methods.”
Footnotes
Abbreviations
ABCB, ATP-binding cassette protein subfamily B; ADP, adenosine-5′-diphosphate; ATP, adenosine-5′-triphosphate; IAA, indole-3-acetic acid; ICL, intracellular loop; MDR, multidrug resistance; NMD, nucleotide-binding domain; PGP, P-glycoprotein; TM, transmembrane; TMD, transmembrane domain.
References
Aller, S. G., Yu, J., Ward, A., Weng, Y., Chittaboina, S., Zhuo, R., Harrell, P. M., Trinh, Y. T., Zhang, Q., Urbatsch, I. L., and Chang, G. (2009). Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722.
Ashkenazy, H., Erez, E., Martz, E., Pupko, T., and Ben-Tal, N. (2010). ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533.
Bachas, S., Eginton, C., Gunio, D., and Wade, H. (2011). Structural contributions to multidrug recognition in the multidrug resistance (MDR) gene regulator, BmrR. Proc. Natl. Acad. Sci. U.S.A. 108, 11046–11051.
Bailly, A., Sovero, V., Vincenzetti, V., Santelia, D., Bartnik, D., Koenig, B. W., Mancuso, S., Martinoia, E., and Geisler, M. (2008). Modulation of P-glycoproteins by auxin transport inhibitors is mediated by interaction with immunophilins. J. Biol. Chem. 283, 21817–21826.
Baker, N. A., Sept, D., Joseph, S., Holst, M. J., and Mccammon, J. A. (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041.
Becker, J. P., Van Bambeke, F., Tulkens, P. M., and Prevost, M. (2010). Dynamics and structural changes induced by ATP binding in SAV1866, a bacterial ABC exporter. J. Phys. Chem. B 114, 15948–15957.
Benkert, P., Tosatto, S. C., and Schomburg, D. (2008). QMEAN: a comprehensive scoring function for model quality assessment. Proteins 71, 261–277.
Bessadok, A., Garcia, E., Jacquet, H., Martin, S., Garrigues, A., Loiseau, N., Andre, F., Orlowski, S., and Vivaudou, M. (2011). Recognition of sulfonylurea receptor (ABCC8/9) ligands by the multidrug resistance transporter P-glycoprotein (ABCB1): functional similarities based on common structural features between two multispecific ABC proteins. J. Biol. Chem. 286, 3552–3569.
Blakeslee, J. J., Bandyopadhyay, A., Lee, O. R., Mravec, J., Titapiwatanakun, B., Sauer, M., Makam, S. N., Cheng, Y., Bouchard, R., Adamec, J., Geisler, M., Nagashima, A., Sakai, T., Martinoia, E., Friml, J., Peer, W. A., and Murphy, A. S. (2007). Interactions among PIN-FORMED and P-glycoprotein auxin transporters in Arabidopsis. Plant Cell 19, 131–147.
Borowski, E., Bontemps-Gracz, M. M., and Piwkowska, A. (2005). Strategies for overcoming ABC-transporters-mediated multidrug resistance (MDR) of tumor cells. Acta Biochim. Pol. 52, 609–627.
Bouchard, R., Bailly, A., Blakeslee, J. J., Oehring, S. C., Vincenzetti, V., Lee, O. R., Paponov, I., Palme, K., Mancuso, S., Murphy, A. S., Schulz, B., and Geisler, M. (2006). Immunophilin-like TWISTED DWARF1 modulates auxin efflux activities of Arabidopsis P-glycoproteins. J. Biol. Chem. 281, 30603–30612.
Cho, M., Lee, S. H., and Cho, H. T. (2007). P-glycoprotein4 displays auxin efflux transporter-like action in Arabidopsis root hair cells and tobacco cells. Plant Cell 19, 3930–3943.
Colovos, C., and Yeates, T. O. (1993). Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 2, 1511–1519.
Corpet, F. (1988). Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890.
Dawson, R. J., Hollenstein, K., and Locher, K. P. (2007). Uptake or extrusion: crystal structures of full ABC transporters suggest a common mechanism. Mol. Microbiol. 65, 250–257.
Dawson, R. J., and Locher, K. P. (2006). Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185.
Dawson, R. J., and Locher, K. P. (2007). Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581, 935–938.
De Smet, I., Voss, U., Lau, S., Wilson, M., Shao, N., Timme, R. E., Swarup, R., Kerr, I., Hodgman, C., Bock, R., Bennett, M., Jurgens, G., and Beeckman, T. (2011). Unraveling the evolution of auxin signaling. Plant Physiol. 155, 209–221.
Dey, S., Hafkemeyer, P., Pastan, I., and Gottesman, M. M. (1999). A single amino acid residue contributes to distinct mechanisms of inhibition of the human multidrug transporter by stereoisomers of the dopamine receptor antagonist flupentixol. Biochemistry 38, 6630–6639.
Eswar, N., Webb, B., Marti-Renom, M. A., Madhusudhan, M. S., Eramian, D., Shen, M. Y., Pieper, U., and Sali, A. (2006). Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics Chap. 5, Unit 5.6.1–5.6.30.
Ferro, N., Bredow, T., Jacobsen, H. J., and Reinard, T. (2010). Route to novel auxin: auxin chemical space toward biological correlation carriers. Chem. Rev. 110, 4690–4708.
Ferro, N., Gallegos, A., Bultinck, P., Jacobsen, H. J., Carbo-Dorca, R., and Reinard, T. (2006). Coulomb and overlap self-similarities: a comparative selectivity analysis of structure-function relationships for auxin-like molecules. J. Chem. Inf. Model. 46, 1751–1762.
Garrigos, M., Mir, L. M., and Orlowski, S. (1997). Competitive and non-competitive inhibition of the multidrug-resistance-associated P-glycoprotein ATPase – further experimental evidence for a multisite model. Eur. J. Biochem. 244, 664–673.
Geisler, M., Blakeslee, J. J., Bouchard, R., Lee, O. R., Vincenzetti, V., Bandyopadhyay, A., Titapiwatanakun, B., Peer, W. A., Bailly, A., Richards, E. L., Ejendal, K. F., Smith, A. P., Baroux, C., Grossniklaus, U., Muller, A., Hrycyna, C. A., Dudler, R., Murphy, A. S., and Martinoia, E. (2005). Cellular efflux of auxin catalyzed by the Arabidopsis MDR/PGP transporter AtPGP1. Plant J. 44, 179–194.
Geisler, M., Kolukisaoglu, H. U., Bouchard, R., Billion, K., Berger, J., Saal, B., Frangne, N., Koncz-Kalman, Z., Koncz, C., Dudler, R., Blakeslee, J. J., Murphy, A. S., Martinoia, E., and Schulz, B. (2003). TWISTED DWARF1, a unique plasma membrane-anchored immunophilin-like protein, interacts with Arabidopsis multidrug resistance-like transporters AtPGP1 and AtPGP19. Mol. Biol. Cell 14, 4238–4249.
Geisler, M., and Murphy, A. S. (2006). The ABC of auxin transport: the role of p-glycoproteins in plant development. FEBS Lett. 580, 1094–1102.
Gruol, D. J., Bernd, J., Phippard, A. E., Ojima, I., and Bernacki, R. J. (2001). The use of a novel taxane-based P-glycoprotein inhibitor to identify mutations that alter the interaction of the protein with paclitaxel. Mol. Pharmacol. 60, 104–113.
Gutmann, D. A., Ward, A., Urbatsch, I. L., Chang, G., and Van Veen, H. W. (2010). Understanding polyspecificity of multidrug ABC transporters: closing in on the gaps in ABCB1. Trends Biochem. Sci. 35, 36–42.
Higgins, C. F. (2001). ABC transporters: physiology, structure and mechanism – an overview. Res. Microbiol. 152, 205–210.
Hollenstein, K., Dawson, R. J., and Locher, K. P. (2007). Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 17, 412–418.
Hooft, R. W., Vriend, G., Sander, C., and Abola, E. E. (1996). Errors in protein structures. Nature 381, 272.
Hopfner, K. P., Karcher, A., Shin, D. S., Craig, L., Arthur, L. M., Carney, J. P., and Tainer, J. A. (2000). Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 101, 789–800.
Hrycyna, C. A., and Gottesman, M. M. (1998). Multidrug ABC transporters from bacteria to man: an emerging hypothesis for the universality of molecular mechanism and function. Drug Resist. Updat. 1, 81–83.
Jasinski, M., Ducos, E., Martinoia, E., and Boutry, M. (2003). The ATP-binding cassette transporters: structure, function, and gene family comparison between rice and Arabidopsis. Plant Physiol. 131, 1169–1177.
Keitt, G. W., and Baker, R. A. (1966). Auxin activity of substituted benzoic acids and their effect on polar auxin transport. Plant Physiol. 41, 1561.
Kelch, D. G., Driskell, A., and Mishler, B. (2004). “Inferring phylogeny using genomic characters: a case study using land plant plastomes,” Molecular Systematics of Bryophytes, eds V. H. B. Goffinet and R. Magill (St. Louis, MO: Missouri Botanical Garden Press), 3–12.
Kim, J. Y., Henrichs, S., Bailly, A., Vincenzetti, V., Sovero, V., Mancuso, S., Pollmann, S., Kim, D., Geisler, M., and Nam, H. G. (2010). Identification of an ABCB/P-glycoprotein-specific inhibitor of auxin transport by chemical genomics. J. Biol. Chem. 285, 23309–23317.
Kimura, Y., Kodan, A., Matsuo, M., and Ueda, K. (2007). Cholesterol fill-in model: mechanism for substrate recognition by ABC proteins. J. Bioenerg. Biomembr. 39, 447–452.
Klepsch, F., Chiba, P., and Ecker, G. F. (2011). Exhaustive sampling of docking poses reveals binding hypotheses for propafenone type inhibitors of P-glycoprotein. PLoS Comput. Biol. 7, e1002036. doi:10.1371/journal.pcbi.1002036
Knoller, A. S., Blakeslee, J. J., Richards, E. L., Peer, W. A., and Murphy, A. S. (2010). Brachytic2/ZmABCB1 functions in IAA export from intercalary meristems. J. Exp. Bot. 61, 3689–3696.
Kubes, M., Yang, H., Richter, G. L., Cheng, Y., Mlodzinska, E., Wang, X., Blakeslee, J., Carraro, N., Petrasek, J., Zazimalova, E., Hoyerova, K., Peer, W. A., and Murphy, A. S. (2011). The Arabidopsis concentration-dependent influx/efflux transporter ABCB4 regulates cellular auxin levels in the root epidermis. Plant J. doi: 10.1111/j.1365-313X.2011.04818.x
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. A., Mcwilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., and Higgins, D. G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948.
Laskowski, R. A., Macarthur, M. W., Moss, D. S., and Thornton, J. M. (1993). Procheck – a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291.
Lee, M., Choi, Y., Burla, B., Kim, Y. Y., Jeon, B., Maeshima, M., Yoo, J. Y., Martinoia, E., and Lee, Y. (2008). The ABC transporter AtABCB14 is a malate importer and modulates stomatal response to CO2. Nat. Cell Biol. 10, 1217–1223.
Li, Y., Yuan, H., Yang, K., Xu, W., Tang, W., and Li, X. (2010). The structure and functions of P-glycoprotein. Curr. Med. Chem. 17, 786–800.
Linton, K. J., and Higgins, C. F. (2007). Structure and function of ABC transporters: the ATP switch provides flexible control. Pflugers Arch. 453, 555–567.
Lobler, M., and Klambt, D. (1985). Auxin-binding protein from coleoptile membranes of corn (Zea mays-L).10.1. Purification by immunological methods and characterization. J. Biol. Chem. 260, 9848–9853.
Locher, K. P. (2004). Structure and mechanism of ABC transporters. Curr. Opin. Struct. Biol. 14, 426–431.
Loo, T. W., Bartlett, M. C., and Clarke, D. M. (2003a). Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J. Biol. Chem. 278, 1575–1578.
Loo, T. W., Bartlett, M. C., and Clarke, D. M. (2003b). Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J. Biol. Chem. 278, 13603–13606.
Loo, T. W., Bartlett, M. C., and Clarke, D. M. (2004). Processing mutations located throughout the human multidrug resistance P-glycoprotein disrupt interactions between the nucleotide binding domains. J. Biol. Chem. 279, 38395–38401.
Loo, T. W., Bartlett, M. C., and Clarke, D. M. (2005). ATP hydrolysis promotes interactions between the extracellular ends of transmembrane segments 1 and 11 of human multidrug resistance P-glycoprotein. Biochemistry 44, 10250–10258.
Loo, T. W., Bartlett, M. C., and Clarke, D. M. (2006a). Transmembrane segment 1 of human P-glycoprotein contributes to the drug-binding pocket. Biochem. J. 396, 537–545.
Loo, T. W., Bartlett, M. C., and Clarke, D. M. (2006b). Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochem. J. 399, 351–359.
Loo, T. W., and Clarke, D. M. (1995). Rapid purification of human P-glycoprotein mutants expressed transiently in HEK 293 cells by nickel-chelate chromatography and characterization of their drug-stimulated ATPase activities. J. Biol. Chem. 270, 21449–21452.
Loo, T. W., and Clarke, D. M. (1997). Drug-stimulated ATPase activity of human P-glycoprotein requires movement between transmembrane segments 6 and 12. J. Biol. Chem. 272, 20986–20989.
Loo, T. W., and Clarke, D. M. (2000). Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J. Biol. Chem. 275, 39272–39278.
Loo, T. W., and Clarke, D. M. (2001). Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J. Biol. Chem. 276, 14972–14979.
Martin, C., Higgins, C. F., and Callaghan, R. (2001). The vinblastine binding site adopts high- and low-affinity conformations during a transport cycle of P-glycoprotein. Biochemistry 40, 15733–15742.
Martinoia, E., Klein, M., Geisler, M., Bovet, L., Forestier, C., Kolukisaoglu, U., Muller-Rober, B., and Schulz, B. (2002). Multifunctionality of plant ABC transporters – more than just detoxifiers. Planta 214, 345–355.
Mravec, J., Kubes, M., Bielach, A., Gaykova, V., Petrasek, J., Skupa, P., Chand, S., Benkova, E., Zazimalova, E., and Friml, J. (2008). Interaction of PIN and PGP transport mechanisms in auxin distribution-dependent development. Development 135, 3345–3354.
Muday, G. K., Brunn, S. A., Haworth, P., and Subramanian, M. (1993). Evidence for a single naphthylphthalamic acid binding site on the zucchini plasma membrane. Plant Physiol. 103, 449–456.
Multani, D. S., Briggs, S. P., Chamberlin, M. A., Blakeslee, J. J., Murphy, A. S., and Johal, G. S. (2003). Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science 302, 81–84.
Nagashima, A., Uehara, Y., and Sakai, T. (2008). The ABC subfamily B auxin transporter AtABCB19 is involved in the inhibitory effects of N-1-naphthyphthalamic acid on the phototropic and gravitropic responses of Arabidopsis hypocotyls. Plant Cell Physiol. 49, 1250–1255.
Newberry, K. J., Huffman, J. L., Miller, M. C., Vazquez-Laslop, N., Neyfakh, A. A., and Brennan, R. G. (2008). Structures of BmrR-drug complexes reveal a rigid multidrug binding pocket and transcription activation through tyrosine expulsion. J. Biol. Chem. 283, 26795–26804.
Noh, B., Murphy, A. S., and Spalding, E. P. (2001). Multidrug resistance-like genes of Arabidopsis required for auxin transport and auxin-mediated development. Plant Cell 13, 2441–2454.
O’Connor, R., Clynes, M., Dowling, P., O’donovan, N., and O’driscoll, L. (2007). Drug resistance in cancer – searching for mechanisms, markers and therapeutic agents. Expert Opin. Drug Metab. Toxicol. 3, 805–817.
Oldham, M. L., and Chen, J. (2011a). Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science 332, 1202–1205.
Oldham, M. L., and Chen, J. (2011b). Snapshots of the maltose transporter during ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 108, 15152–15156.
Oldham, M. L., Khare, D., Quiocho, F. A., Davidson, A. L., and Chen, J. (2007). Crystal structure of a catalytic intermediate of the maltose transporter. Nature 450, 515–521.
Pajeva, I. K., Globisch, C., and Wiese, M. (2009). Comparison of the inward- and outward-open homology models and ligand binding of human P-glycoprotein. FEBS J. 276, 7016–7026.
Petrasek, J., and Friml, J. (2009). Auxin transport routes in plant development. Development 136, 2675–2688.
Petrek, M., Kosinova, P., Koca, J., and Otyepka, M. (2007). MOLE: a Voronoi diagram-based explorer of molecular channels, pores, and tunnels. Structure 15, 1357–1363.
Pleban, K., Kopp, S., Csaszar, E., Peer, M., Hrebicek, T., Rizzi, A., Ecker, G. F., and Chiba, P. (2005). P-glycoprotein substrate binding domains are located at the transmembrane domain/transmembrane domain interfaces: a combined photoaffinity labeling-protein homology modeling approach. Mol. Pharmacol. 67, 365–374.
Raven, J. A. (1975). Transport of indoleacetic-acid in plant-cells in relation to Ph and electrical potential gradients, and its significance for polar IAA transport. New Phytol. 74, 163–172.
Rojas-Pierce, M., Titapiwatanakun, B., Sohn, E. J., Fang, F., Larive, C. K., Blakeslee, J., Cheng, Y., Cutler, S. R., Peer, W. A., Murphy, A. S., and Raikhel, N. V. (2007). Arabidopsis P-glycoprotein19 participates in the inhibition of gravitropism by gravacin. Chem. Biol. 14, 1366–1376.
Rosenberg, M. F., Callaghan, R., Modok, S., Higgins, C. F., and Ford, R. C. (2005). Three-dimensional structure of P-glycoprotein: the transmembrane regions adopt an asymmetric configuration in the nucleotide-bound state. J. Biol. Chem. 280, 2857–2862.
Rothnie, A., Storm, J., Campbell, J., Linton, K. J., Kerr, I. D., and Callaghan, R. (2004). The topography of transmembrane segment six is altered during the catalytic cycle of P-glycoprotein. J. Biol. Chem. 279, 34913–34921.
Sakurai, A., Onishi, Y., Hirano, H., Seigneuret, M., Obanayama, K., Kim, G., Liew, E. L., Sakaeda, T., Yoshiura, K., Niikawa, N., Sakurai, M., and Ishikawa, T. (2007). Quantitative structure – activity relationship analysis and molecular dynamics simulation to functionally validate nonsynonymous polymorphisms of human ABC transporter ABCB1 (P-glycoprotein/MDR1). Biochemistry 46, 7678–7693.
Sali, A., and Blundell, T. L. (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815.
Sanchez-Fernandez, R., Davies, T. G., Coleman, J. O., and Rea, P. A. (2001a). The Arabidopsis thaliana ABC protein superfamily, a complete inventory. J. Biol. Chem. 276, 30231–30244.
Sanchez-Fernandez, R., Rea, P. A., Davies, T. G., and Coleman, J. O. (2001b). Do plants have more genes than humans? Yes, when it comes to ABC proteins. Trends Plant Sci. 6, 347–348.
Santelia, D., Vincenzetti, V., Azzarello, E., Bovet, L., Fukao, Y., Duchtig, P., Mancuso, S., Martinoia, E., and Geisler, M. (2005). MDR-like ABC transporter AtPGP4 is involved in auxin-mediated lateral root and root hair development. FEBS Lett. 579, 5399–5406.
Seeliger, D., and de Groot, B. L. (2010). Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 24, 417–422.
Shapiro, A. B., and Ling, V. (1997). Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem. 250, 130–137.
Sharom, F. J. (2008). ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 9, 105–127.
Storm, J., Modok, S., O’mara, M. L., Tieleman, D. P., Kerr, I. D., and Callaghan, R. (2008). Cytosolic region of TM6 in P-glycoprotein: topographical analysis and functional perturbation by site directed labeling. Biochemistry 47, 3615–3624.
Sugiyama, A., Shitan, N., Sato, S., Nakamura, Y., Tabata, S., and Yazaki, K. (2006). Genome-wide analysis of ATP-binding cassette (ABC) proteins in a model legume plant, Lotus japonicus: comparison with Arabidopsis ABC protein family. DNA Res. 13, 205–228.
Sussman, M. R., and Gardner, G. (1980). Solubilization of the receptor for N-1-naphthylphthalamic acid. Plant Physiol. 66, 1074–1078.
Tan, X., Calderon-Villalobos, L. I., Sharon, M., Zheng, C., Robinson, C. V., Estelle, M., and Zheng, N. (2007). Mechanism of auxin perception by the TIR1 ubiquitin ligase. Nature 446, 640–645.
Terasaka, K., Blakeslee, J. J., Titapiwatanakun, B., Peer, W. A., Bandyopadhyay, A., Makam, S. N., Lee, O. R., Richards, E. L., Murphy, A. S., Sato, F., and Yazaki, K. (2005). PGP4, an ATP binding cassette P-glycoprotein, catalyzes auxin transport in Arabidopsis thaliana roots. Plant Cell 17, 2922–2939.
Titapiwatanakun, B., and Murphy, A. S. (2009). Post-transcriptional regulation of auxin transport proteins: cellular trafficking, protein phosphorylation, protein maturation, ubiquitination, and membrane composition. J. Exp. Bot. 60, 1093–1107.
Wang, H. Y., and Lin, R. C. (2005). Two homologous ATP-binding cassette transporter proteins, AtMDR1 and AtPGP1, regulate Arabidopsis photomorphogenesis and root development by mediating polar auxin transport. Plant Physiol. 138, 949–964.
Ward, A., Reyes, C. L., Yu, J., Roth, C. B., and Chang, G. (2007). Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010.
Wu, G., Lewis, D. R., and Spalding, E. P. (2007). Mutations in Arabidopsis multidrug resistance-like ABC transporters separate the roles of acropetal and basipetal auxin transport in lateral root development. Plant Cell 19, 1826–1837.
Yang, H., and Murphy, A. S. (2009). Functional expression and characterization of Arabidopsis ABCB, AUX 1 and PIN auxin transporters in Schizosaccharomyces pombe. Plant J. 59, 179–191.
Zazimalova, E., Murphy, A. S., Yang, H., Hoyerova, K., and Hosek, P. (2010). Auxin transporters – why so many? Cold Spring Harb. Perspect. Biol. 2, a001552.
Appendix

Figure A1. Mutations in ABCB1 orthologs result in reduced basipetal auxin transport and dwarfism. (A,B) Compressed stalk internodes in maize br2 (brachytic2/zmpgp1) and sorghum dw3 (dwarf3/sbpgp1) mutants (Multani et al., 2003). (C) Drastic dwarfing phenotype in Arabidopsisabcb1 abcb19 double mutant (Geisler et al., 2005). Wt, wild type.
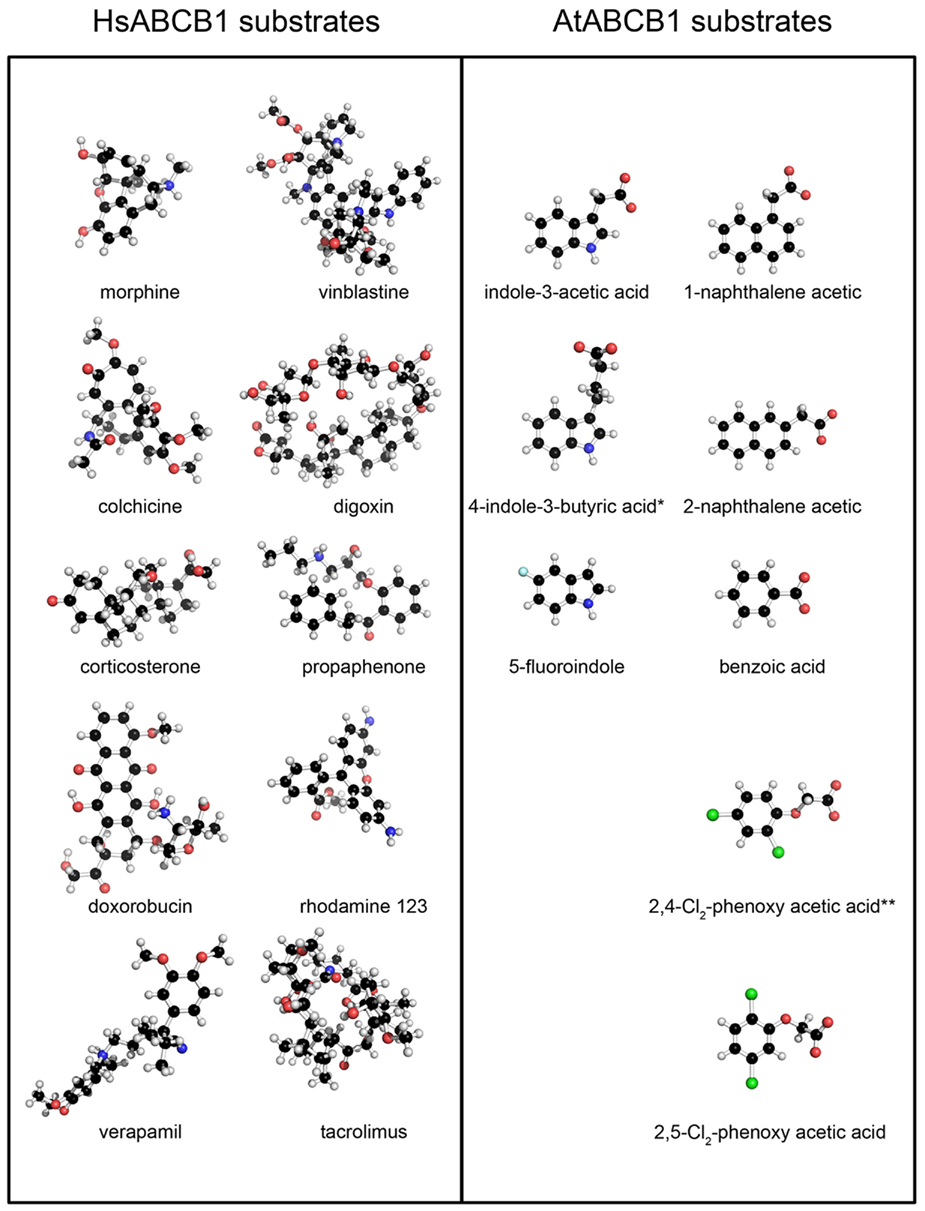
Figure A2. Mammalian and plant ABCB1 proteins show divergent substrate selectivities. Structural representations of discrete HsABCB1 and AtABCB1 substrates evidence the extreme variance in the reported mammalian substrates when compared to the similarity of AtABCB1 substrates. *No experimental evidence for those compounds has been reported to date, although their biological activity in polar auxin transport has been extensively documented.
Keywords: ABCB exporter, ABCB importer, structural modeling, auxin, substrate docking
Citation: Bailly A, Yang H, Martinoia E, Geisler M and Murphy AS (2012) Plant lessons: exploring ABCB functionality through structural modeling. Front. Plant Sci. 2:108. doi: 10.3389/fpls.2011.00108
Received: 13 October 2011;
Accepted: 17 December 2011;
Published online: 05 January 2012.
Edited by:
Heven Sze, University of Maryland, USAReviewed by:
Fatima Cvrckova, Charles University in Prague, Czech RepublicFrantisek Baluska, University of Bonn, Germany
Copyright: © 2012 Bailly, Yang, Martinoia, Geisler and Murphy. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Markus Geisler, Plant Biology, Department of Biology, University of Fribourg, Chemin du Musée 10, CH-1700 Fribourg, Switzerland. e-mail:bWFya3VzLmdlaXNsZXJAdW5pZnIuY2g=