

- Division of Life Science, The Hong Kong University of Science and Technology, Hong Kong, China
Protein phosphorylation is one of the most important post-translational modifications (PTMs) as it participates in regulating various cellular processes and biological functions. It is therefore crucial to identify phosphorylated proteins to construct a phosphor-relay network, and eventually to understand the underlying molecular regulatory mechanism in response to both internal and external stimuli. The changes in phosphorylation status at these novel phosphosites can be accurately measured using a 15N-stable isotopic labeling in Arabidopsis (SILIA) quantitative proteomic approach in a high-throughput manner. One of the unique characteristics of the SILIA quantitative phosphoproteomic approach is the preservation of native PTM status on protein during the entire peptide preparation procedure. Evolved from SILIA is another quantitative PTM proteomic approach, AQUIP (absolute quantitation of isoforms of post-translationally modified proteins), which was developed by combining the advantages of targeted proteomics with SILIA. Bioinformatics-based phosphorylation site prediction coupled with an MS-based in vitro kinase assay is an additional way to extend the capability of phosphosite identification from the total cellular protein. The combined use of SILIA and AQUIP provides a novel strategy for molecular systems biological study and for investigation of in vivo biological functions of these phosphoprotein isoforms and combinatorial codes of PTMs.
Introduction
As one of hundreds of known post-translational modifications (PTM), protein phosphorylation participates in almost every regulatory event known to us in cellular process and biological function (Krishna and Wold, 1993; Hunter, 2000). It is therefore regarded as one of the most important regulatory PTM events. Understanding of the regulation of protein phosphorylation in response to environmental cues/stimuli provides novel insights into how phosphor-relay-mediated signals are transduced and regulated within a plant cell. Mass spectrometry-based phosphoproteomics has emerged as a powerful and high-throughput approach in profiling phosphoproteins participating in cellular processes and signaling regulation (Schmelzle and White, 2006; Schulze, 2010). Recently, various quantitative proteomics strategies have been developed to gain insight into the phosphoprotein dynamics, including label-free, chemical or stable-isotope labeling and corresponding statistical assessment (Schulze and Usadel, 2010). In vivo metabolic incorporation of stable isotopes, particularly the heavy nitrogen (15N), is firstly described in 1999 (Oda et al., 1999) and has emerged as one of the favorite strategies given the autotrophic nature of plants (Dunkley et al., 2004; Gevaert et al., 2008; Gouw et al., 2010; Guo and Li, 2011; Arsova et al., 2012). Examples are the stable isotope labeling by/with amino acids in cell culture (SILAC; Ong et al., 2002) and the stable isotope labeling in planta (SILIP; Schaff et al., 2008). Based on these protocols, treated and untreated plants are differentially labeled with 14N- or 15N-coded salt, respectively, with reciprocal repeats. Consequently, proteins from each group of plants are differentially labeled and are mixed at ratios of 1:1–1.5 (14N:15N) depending on the actual incorporation rate before a mixture of peptides are processed (Guo and Li, 2011). This early protein mixing step is supposed to exclude the variation resulting from peptide preparation, separation, and MS analysis processes (Gevaert et al., 2008). The absolute quantification of proteins (AQUA; Gerber et al., 2003) was introduced in 2003 by Steven Gygi’s group. Heavy isotope-labeled peptides are used by AQUA method as the internal standard, added preferentially as early as possible in the analytical process. Multiplexed absolute quantification was achieved by constructing a recombinant gene that concatenates different tryptic peptides to be quantified (Beynon et al., 2005; Pratt et al., 2006).
As there are already a number of excellent reviews covering the developments and perspectives in quantitative proteomics (Aebersold and Mann, 2003; Ong and Mann, 2005; Bantscheff et al., 2007; Schulze and Usadel, 2010) as well as plant proteomics (Gerber et al., 2003; Chen and Harmon, 2006; Thelen and Peck, 2007; Kota and Goshe, 2011; Arsova et al., 2012), this review will focus on the application of 15N stable isotope-based quantitative and differential PTM proteomics in identification of important PTM protein components during cellular processes and on the investigation of their in vivo functions in plant. The 15N-stable isotope labeling in Arabidopsis (SILIA)-based quantitative proteomic protocol is first applied onto Arabidopsis grown on a solid-medium (Guo and Li, 2011). Consequently, the successfully identified phosphosites are further investigated by bioinformatics-prediction and in vitro kinase assays in combination with mutant kinase extracts and quantitative methods such as the isobaric tags for relative and absolute quantitation (iTRAQ; Ross et al., 2004) so that these phosphopeptides can be first validated and a subgroup of highly interesting phosphorylation sites are selected for the following in vivo quantitation using the absolute quantitation of isoforms of post-translationally modified proteins (AQUIP) approach (Li et al., 2012). AQUIP was developed from the targeted and quantitative proteomics by combining the advantages of AQUA (Gerber et al., 2003) and the protein standard absolute quantification strategy (PSAQ; Brun et al., 2007; Lebert et al., 2011) with SILIA. Finally, at the end of the integrated in vitro and in vivo quantitative PTM proteomics, the biological function studies are performed on the group of highly selected phosphosites and phosphoproteins to unravel their molecular, cellular, and biological roles in plants.
Silia-Based Quantitative Phosphoproteomics
Previous metabolic labeling experiments were mostly performed in aqueous solutions in the form of cell suspension cultures (SILAC; Engelsberger et al., 2006; Benschop et al., 2007) or seedlings suspended in liquid media (Dunkley et al., 2004; Huttlin et al., 2007; Nelson et al., 2007). The labeling was further performed on plants either grown in hydroponic solutions (HILEP, the hydroponic isotope labeling of entire plants; Bindschedler et al., 2008; Hebeler et al., 2008) or in soil (SILIP; Schaff et al., 2008). In the case of SILIA, a general metabolic-labeling strategy was designed for agar-based plant growth, which is frequently used in molecular genetic screens and/or physiological studies. SILIA-medium was designed to allow the 15N as the only nitrogen source for Arabidopsis growth and to maximize the incorporation rate (Guo and Li, 2011). Another major improvement in SILIA-based quantitative PTM proteomics is the use of urea-based protein denaturing buffer (UEB), which provides a fully denaturing condition to inhibit protease-mediated protein degradation during protein extraction/separation processes and, especially, to prevent non-specific phosphorylation/dephosphorylation events from occurring in protein extracts. A detailed workflow of the SILIA-based quantitative proteomics has been described in an earlier publication (Guo and Li, 2011). As data generated by quantitative proteomics methods (e.g., iTRAQ and SILIA) usually show relative changes (i.e., fold change) in phosphorylation among different conditions, statistical methods are needed to confirm that there is a significant difference between the degrees of phosphorylation of two conditions (de la Fuente van Bentem et al., 2008). Both forward and reciprocal experiments are usually repeated two to four times to obtain a set of satisfactory quantitative phosphoprotein-profiling data.
In Vitro Kinase Assay to Validate the MS-Identified and Bioinformatics-Predicted Phosphosites
Non-biased phosphoprotein profiling performed by SILIA provides valuable quantitative information about protein phosphorylation changes in response to external or internal stimuli in plants. As MS-identification of phosphopeptides is routinely evaluated with a false discovery rate (FDR), in vitro phosphorylation validation is usually required to be performed on a group of randomly selected phosphopeptides before they are chosen as candidates for further in vivo quantitation and a long-term functional study. Secondly, although substantial advancements have been made in MS technology and bioinformatics software during the discovery of phosphosites, the number of MS-identified phosphopeptides is still far below the theoretically predicted number (de la Fuente van Bentem et al., 2008; Heazlewood et al., 2008; Durek et al., 2010). Thus, prediction of putative phosphosites from inducible phosphorylation sites using bioinformatics is expected to increase the probability of identifying additional phosphosites.
The amino acid sequence around a phosphosite serves as a structural feature for kinase’s substrate recognition (Jorgensen and Linding, 2008; Mok et al., 2010). Because the amino acid sequence surrounding the phosphosite may be highly conserved among various substrates, a short stretch (9–21 amino acid-long) oligopeptide deduced from the primary sequence of an identified phosphosite, is employed to BLAST against the non-redundant plant protein sequence database (Arabidopsis in this case; Li et al., 2009). The alignment leads to the identification of phosphorylation motif. Synthetic peptides are then made according to the predicted phosphosites and fused to a hexa-His tag at N-terminus to facilitate substrate peptide purification following kinase assay. Huber and colleagues have demonstrated a case, in which a highly conserved phosphorylation site was established among ACC synthase (a key enzyme in ethylene biosynthesis) isomers using in vitro kinase assay and the calmodulin-dependent protein kinase (CDPK) was found to be responsible for the phosphorylation of this site (Hernandez et al., 2004).
To place the newly identified phosphosites in the context of a cellular process or a specific signal regulation, a general approach has been developed using quantitative proteomics in combination with kinase extracts of both the treated and mutant plants (Figure 1; Li et al., 2009, 2012; Zhu et al., 2012). The in vitro kinase assay is then performed using the synthetic peptides mixed with plant kinase extracts in the presence of phosphatase inhibitors. The chemical labeling quantitative proteomic protocol, such as iTRAQ, is then applied to investigate the differentially regulated phosphorylation events. A routine iTRAQ-based in vitro phosphosite quantitation requires at least two sets of forward and reciprocal experiments. Results on MS-derived and bioinformatics-derived synthetic peptides can provide information on the differential regulation of phosphosites in response to the internal and external stimuli.
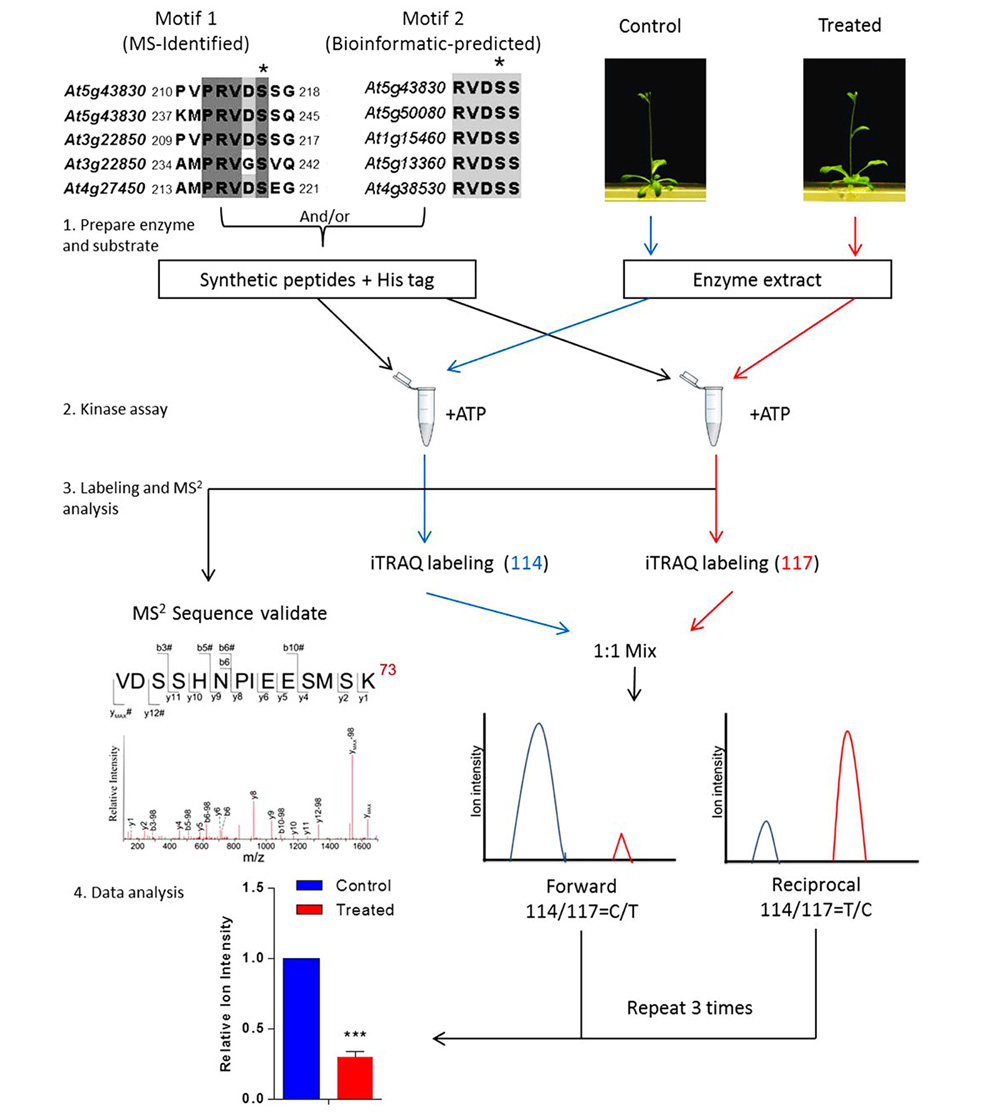
FIGURE 1. Workflow of in vitro assay for signal-regulated phosphorylation. Upper left panel shows two alignments of conserved sequences of phosphorylation motif generated with CLUSTAL W, which was reported in a previous study (Li et al., 2009). In this case, 9–21 amino acid-long oligopeptide sequences deduced from the primary sequence of an aluminum-induced protein covering the entire phosphorylation motif was employed to search against the non-redundant protein sequence database (organism: Arabidopsis thaliana, taxid: 3702). Candidates with a homology of more than 55.5% are considered to contain the phosphorylation motif. The most conserved candidates are aligned to obtain the motif sequence. The phosphorylation site Ser(S) is marked with a black asterisk and TAIR IDs of the genes are shown on the left. To validate such a prediction, peptides including those amino acid sequences flanking the predicted phosphosite motif are synthesized and used as substrates for in vitro kinase assay. The control or treated means the plant without or with particular treatment of interest, respectively. The quantitation is performed at MS2 spectra by directly comparing the reporter ion intensity. Plant extracts from the control or the treated plants are used separately. Peptides assayed are reciprocally labeled with iTRAQ reagents. Three sets of reciprocal labelings are required before a statistical analysis is performed.
Kinase/Phosphatase and Phosphosite-Containing Peptide Interactomics
The highly conserved amino acid sequence or phosphosite motif is useful when one wants to isolate the phosphosite-containing peptide-binding proteins. To achieve that, synthetic peptides that have been validated by in vitro kinase assay will be used in combination with the 15N-stable isotope labeled plants. By this approach, the validated phosphopeptides together with their corresponding non-phosphorylated peptide cognates will be synthesized chemically and conjugated with biotin according to the method described previously (Christofk et al., 2011). Each set of biotinylated peptides will be immobilized on beads and packed into a chromatography column. Light nitrogen-labeled proteins will be passed through a column immobilized with the phosphorylated peptides, whereas the heavy nitrogen-labeled proteins will be passed through a column immobilized with its corresponding non-phosphopeptide. As a result, 15N-coded putative kinases and phosphosites-binding proteins are presumably enriched on the column with non-phosphorylated synthetic peptide containing a phosphosite or motif sequence. The two protein passages will be mixed together at 1:1 ratio (w/w) and resolved on SDS-PAGE gel, which will then be tryptic digested and subject to MS analysis as described before (Li et al., 2009; Guo and Li, 2011). Both phosphosite-containing peptides (non-phosphorylated and phosphorylated)-binding proteins will be identified differentially due to a difference in between the heavy and light isotopolog envelopes of their peptide ions.
In Vivo Quantitation of PTM Occupancy
Once phosphosites are identified from SILIA-based phosphoproteomics and confirmed with in vitro kinase assays, an in planta validation and measurement of these phosphosites and signal-specific phosphorylation level will be performed using a transgenic approach. A transgene encoding the target protein will be transferred into plant to measure the alteration of a particular phosphorylation in response to various treatments, cell types, or disease models (Havlis and Shevchenko, 2004; Steen et al., 2008). The term occupancy (Raqu or Risf) defines the ratio of the amount of a protein PTM isomer (or PTM peptide) over the total amount of the corresponding protein (or the corresponding PTM site peptide; Li et al., 2012). A signal-induced alteration in occupancy on a phosphosite usually measures the change in phosphorylation level elicited by both internal and external signals. It is therefore that Raqu or Risf are used to describe signal-specific phosphorylation change. To determine Raqu (or Risf) of a phosphosite, 14N-coded peptide standards are established (Figure 2) against the 15N-coded recombinant protein expressed in the transgenic plant. One is the synthetic phosphopeptide that contains targeted phosphosite, whereas the other is its unmodified cognate. A series of synthetic peptide samples with a wide range of concentrations are then made, in the meantime, an aliquot of known amount of highly purified recombinant protein expressed in transgenic plant is resolved on SDS-PAGE and undergo in-gel tryptic digestion. Once the peptides are produced from the 15N-coded recombinant protein, 14N-coded standard phosphopeptides (both phosphorylated and non-phosphorylated) of various amounts are added into aliquots of the recombinant protein-derived peptides and analyzed using MS. The resulting data is used to establish two individual standard curves to calculate the molar amount of phosphopeptide Paqu and its non-phosphorylated cognate NPaqu. The Raqu is determined using Eq. 1:
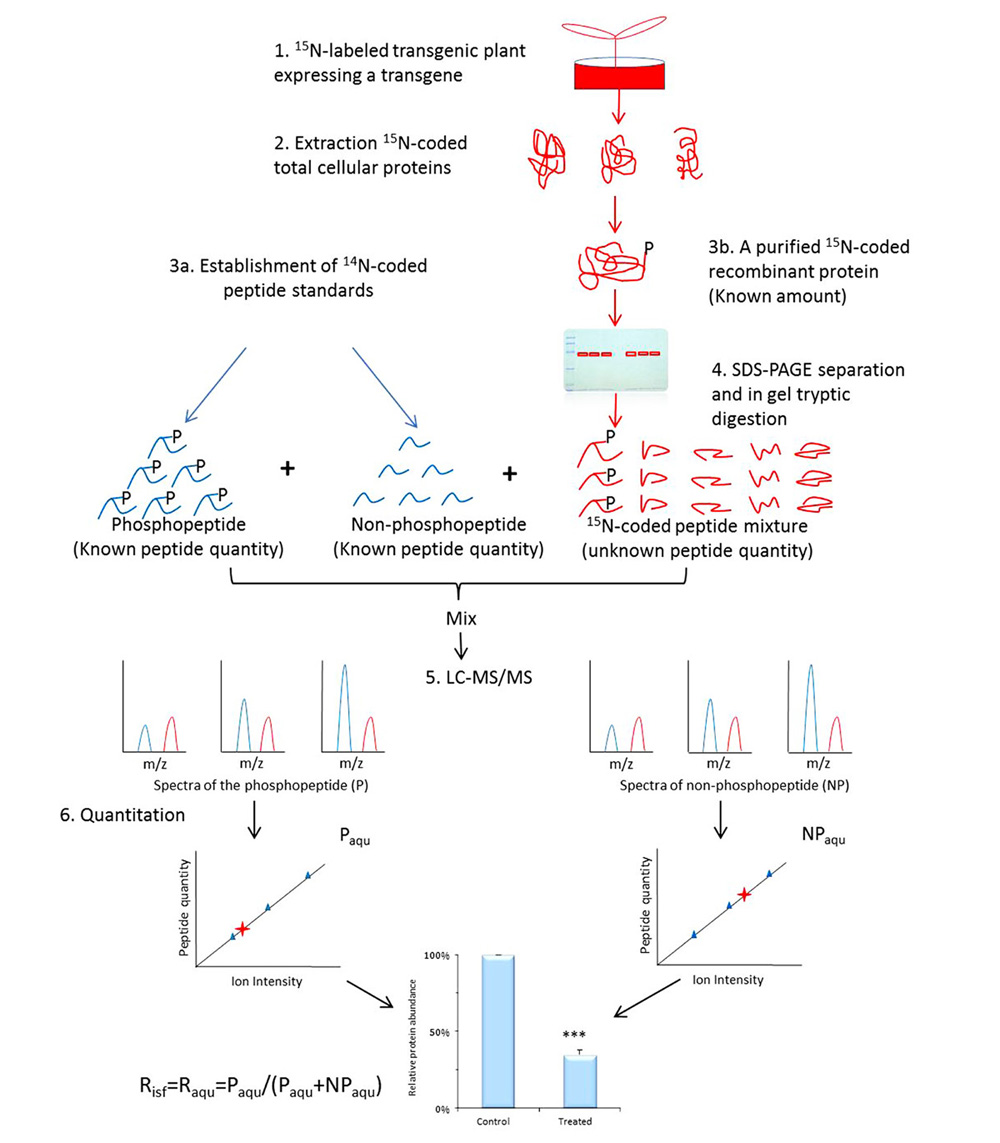
FIGURE 2. Workflow of determination of occupancy (Raqu or Risf) using AQUIP. A known quantity of 15N-coded recombinant proteins, including both the target phosphorylated isoform and all of its cognates of different types of PTMs, are highly purified from 15N-labeled transgenic plant (either the control or the treated plants) and divided into six aliquots and resolved on SDS-PAGE gel, followed by in-gel trypsin digestion. The 14N-coded synthetic phosphopeptide and its non-phosphorylated cognate are mixed together with a known concentration and spiked into 6 aliquots of 15N-coded peptide samples. After the oxidization of methionine residues on the peptides and LC-MS/MS analysis, two standard curves are built using a method between the ion intensity ratios of peptide P and NP standards and the molar ratios of peptide standards. The molar amount of phosphorylated peptide (Paqu) and its non-phosphorylated cognate (NPaqu), which are derived from the highly purified and 15N-coded protein isoforms, is determined according to these two standard curves separately. The percentage of target-phosphorylated isoform among all isoforms in the total cellular proteins is defined as the site-specific phosphorylation occupancy Risf, which is equivalent to Raqu when the ratio of P and NP peptide amount is in concern (Li et al., 2012). Error values indicate standard deviations. The Risf obtained from either the control or the treated plants is used to calculate the relative abundance of the phosphorylation event and subject to statistical assessment, as shown at the bottom of the figure. ***Means significant difference.
Because it is believed that the fold increase from a partial to a full phosphorylation status may lead to different cellular effects (Steen et al., 2008; Wu et al., 2011), a number of MS-based methods have been invented for quantitation of phosphorylated isoform. Using stable isotope-labeled synthetic phosphopeptides and artificial concatemer of standard peptides (Gerber et al., 2003; Mayya et al., 2006; Wu et al., 2011) and QconCAT (Beynon et al., 2005; Anderson and Hunter, 2006; Pratt et al., 2006; Rivers, et al., 2007) approaches are frequently used for quantitative analysis of protein phosphorylation. Since peptide digestion may be incomplete or proteins of interest requires isolation (Brun et al., 2007), protein standard absolute quantification approach, which uses the full-length stable isotope-labeled protein as standard, becomes popular in quantitation of non-PTM proteins (Brun et al., 2007; Lebert et al., 2011). To address the difficulty in measuring abundance of a phosphorylated isoform, AQUIP (Figure 2; Li et al., 2012) strategy, which combines these advantageous features with a recombinant HisTag fusion protein (His8-BCCP-His8), was invented to achieve absolute quantitation for the phosphorylation level of a particular phosphosite in vivo. In short, in AQUIP: (1) both 14N synthetic peptide standards and purified 15N-coded protein of interest are used to determine the total amount of the target protein (Tisf); (2) a pair of synthetic peptides of phosphosites are used to calculate the phosphosite occupancy (Raqu or Risf), and (3) the absolute amount of phosphorylated isoform Pisf is calculated via an equation: Pisf = Tisf ∙ Raqu, where Tisf stands for the molar amount of the whole recombinant protein isolated from plant cell lysate.
To determine the absolute amount of the recombinant protein in the cell lysate, the plant is first labeled via SILIA. The 15N-coded total cellular protein is extracted and resolved on SDS-PAGE. The gel slice that contains the targeted protein is excised out to perform the in-gel trypsin digestion and then divided into seven equal fractions. Two gradients of 14N-peptide standards of known concentrations are mixed with the target protein sample to establish two standard curves between the peptide concentration and the ion intensity of the whole isotopic envelope. These standard curves established above are used to calculate the molar amount of the 15N-coded target peptide from recombinant fusion protein sample. Because the digestion efficiency and recovery rate of each peptide varies during peptide preparation, peptide yield (km) of each peptide needs to be determined. To obtain km, a highly purified target protein with known amount is obtained via tandem affinity purification (Li et al., 2012). The purified targeted recombinant protein is divided into two fractions with high or low molar amount and mixed with equal amount of standard peptides mixtures. The two mixtures of 15N-coded target proteins and 14N-coded peptide standards are then subjected to protease digestion separately and subjected to MS analysis to build standard curves. These MS results are used to determine the peptide yield km. Finally, Tisf of the targeted protein could be obtained follow Eq. 2:
where k represents peptide yield and m represents number of peptides used for quantitation. The absolute molar amount of the PTM isoform is determined as the following Eq. 3:
This method has been applied in Arabidopsis to measure phosphorylation event on transcriptional factor ERF110 in both air-treated and ethylene-treated transgenic plants, and confirmed an ethylene down-regulating phosphorylation event on ERF110 Ser62 in the ein2-5 mutant background (Li et al., 2012). Its application could be extend to different treatments and plant species, and not limited to quantitation of phosphorylated isoforms. The absolute amount of PTM isoforms could also serve as biomarkers that correlate with phenotypic changes, which could be applied to the clinical aspects (Schrohl et al., 2003; Barnidge et al., 2004).
In Vivo Validation of Biological Function of a Phosphosite or a Phosphoprotein
To study the biological function of a phosphoprotein identified from quantitative PTM proteomics, loss of function lines of these phosphoproteins can be obtained by both T-DNA insertion (Krysan et al., 1999) and RNA interference (Klink and Wolniak, 2000) to study the function of these candidate genes in vivo. Alternatively, the candidate phosphoprotein gene can be expressed ectopically to monitor the gain-of-function effect of these genes. To further confirm the biological function of phosphosites, phosphorylation- and dephosphorylation-mimetic mutants are introduced to plants. A successful example is that phosphorylation of Ser (S) can be functionally substituted by Asp (D) in a rat liver kinase, resulting in an identical substrate dependency. In contrast, Ala (A) mutant showed exactly the same kinetic property of its de-phosphorylation form (Kurland et al., 1992). These molecular genetic methods have been successfully applied in a wide number of organisms, which include, for example, determination of the phosphorylation-dependent kinase activity of eukaryotic elongation factor 2 kinase (eEF-2K; Tavares et al., 2012), alteration of conformation of human oncogene stathmin by phosphorylation (Curmi et al., 1994) as well as the phosphorylation-dependent accumulation/degradation of both ACC synthase (Liu and Zhang, 2004) and signaling component EIN3 (Yoo et al., 2008) in Arabidopsis. The commonly used amino acid substitution that are mimetic of the dephosphorylation status at phosphosite are amino acid S (or T) mutated to A (or I) and Y to F, respectively. In contrast, the phosphosite (S) mimicking the phosphorylated status is usually mutated to D or E. The mutant genes of site-directed point-mutations are then transformed into a plant to verify its biological function in vivo using the wild-type gene as a control. Phenotypes of all three transgenic plants expressing the mutants and wild-type gene reveal the possible role of the phosphosites in plant. By this strategy, the Ser62 phosphosite of ERF110 has been found to play a role in the organ development in Arabidopsis (Li et al., 2012; Zhu et al., 2012).
Conclusion
In conclusion, we have summarized an integrated quantitative and functional phosphoproteomics approach, which starts from a large-scale identification of phosphopeptides, to bioinformatics-prediction, to in vitro validation of phosphosites, protein and phosphopeptide interactomics, to in planta quantitation of phosphosites and finally to the investigation of biological functions of these phosphosites in vivo. Knowledge generated via such a strategy might ultimately be integrated to interpret the combinatorial codes of different PTMs (including phosphorylation) events and those PTM changes in response to developmental cues and environmental stimuli. During application of this quantitative PTM proteomics approach to a special biological problem, one may have to pay special attention to the amount of labor involved, the cost for synthesis of peptides and computational skills used in analysis of these large set of MS data.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The research was supported by grants CAS10SC01, CAS10SC01-M1, NMESL11SC01, W0298, CGPL08/09.SC01, and GMGS11SC02 awarded to Ning Li. The authors want to express sincere gratitude to Mr. Guang Yu GUO and Dr. Zhu YANG for their useful suggestion during the preparation of this manuscript.
References
Anderson, L., and Hunter, C. L. (2006). Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 5, 573–588.
Arsova, B., Kierszniowska, S., and Schulze, W. X. (2012). The use of heavy nitrogen in quantitative proteomics experiments in plants. Trends Plant Sci. 17, 102–112.
Bantscheff, M., Schirle, M., Sweetman, G., Rick, J., and Kuster, B. (2007). Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031.
Barnidge, D. R., Goodmanson, M. K., Klee, G. G., and Muddiman, D. C. (2004). Absolute quantification of the model biomarker prostate-specific antigen in serum by LC-Ms/MS using protein cleavage and isotope dilution mass spectrometry. J. Proteome Res. 3, 644–652.
Benschop, J. J., Mohammed, S., O’Flaherty, M., Heck, A. J., Slijper, M., and Menke, F. L. (2007). Quantitative phosphoproteomics of early elicitor signaling in Arabidopsis. Mol. Cell. Proteomics 6, 1198–1214.
Beynon, R. J., Doherty, M. K., Pratt, J. M., and Gaskell, S. J. (2005). Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat. Methods 2, 587–589.
Bindschedler, L. V., Palmblada, M., and Cramera, R. (2008). Hydroponic isotope labelling of entire plants (HILEP) for quantitative plant proteomics: an oxidative stress case study. Phytochemistry 69, 1962–1972.
Brun, V., Dupuis, A., Adrait, A., Marcellin, M., Thomas, D., Court, M., et al. (2007). Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol. Cell. Proteomics 6, 2139–2149.
Christofk, H. R., Wu, N., Cantley, L. C., and Asara, J. M. (2011). Proteomic screening method for phosphopeptide motif binding proteins using peptide libraries. J. Proteome Res. 10, 4158–4164.
Curmi, P. A., Maucuer, A., Asselin, S., Lecourtois, M., Chaffotte, A., Schmitter, J. M., et al. (1994). Molecular characterization of human stathmin expressed in Escherichia coli site-directed mutagenesis of 2 phosphorylatable serines (Ser-25 and Ser-63). Biochem. J. 300, 331–338.
de la Fuente van Bentem, S., Mentzen, W. I., de la Fuente, A., and Hirt, H. (2008). Towards functional phosphoproteomics by mapping differential phosphorylation events in signaling networks. Proteomics 8, 4453–4465.
Dunkley, T. P., Watson, R., Griffin, J. L., Dupree, P., and Lilley, K. S. (2004). Localization of organelle proteins by isotope tagging (LOPIT). Mol. Cell. Proteomics 3, 1128–1134.
Durek, P., Schmidt, R., Heazlewood, J. L., Jones, A., MacLean, D., Nagel, A., et al. (2010). PhosPhAt: the Arabidopsis thaliana phosphorylation site database. An update. Nucleic Acids Res. 38, D828–D834.
Engelsberger, W. R., Erban, A., Kopka, J., and Schulze, W. X. (2006). Metabolic labeling of plant cell cultures with K(15)NO3 as a tool for quantitative analysis of proteins and metabolites. Plant Methods 2, 14.
Gerber, S. A., Rush, J., Stemman, O., Kirschner, M. W., and Gygi, S. P. (2003). Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 100, 6940–6945.
Gevaert, K., Impens, F., Ghesquiere, B., Van Damme, P., Lambrechts, A., and Vandekerckhove, J. (2008). Stable isotopic labeling in proteomics. Proteomics 8, 4873–4885.
Gouw, J. W., Krijgsveld, J., and Heck, A. J. (2010). Quantitative proteomics by metabolic labeling of model organisms. Mol. Cell. Proteomics 9, 11–24.
Guo, G., and Li, N. (2011). Relative and accurate measurement of protein abundance using 15N stable isotope labeling in Arabidopsis (SILIA). Phytochemistry 72, 1028–1039.
Havlis, J., and Shevchenko, A. (2004). Absolute quantification of proteins in solutions and in polyacrylamide gels by mass spectrometry. Anal. Chem. 76, 3029–3036.
Heazlewood, J. L., Durek, P., Hummel, J., Selbig, J., Weckwerth, W., Walther, D., et al. (2008). PhosPhAt: a database of phosphorylation sites in Arabidopsis thaliana and a plant-specific phosphorylation site predictor. Nucleic Acids Res. 36, D1015–D1021.
Hebeler, R., Oeljeklaus, S., Reidegeld, K. A., Eisenacher, M., Stephan, C., Sitek, B., et al. (2008). Study of early leaf senescence in Arabidopsis thaliana by quantitative proteomics using reciprocal 14N/15N labeling and difference gel electrophoresis. Mol. Cell. Proteomics 7, 108–120.
Hernandez, S. C., Hardin, S. C., Clouse, S. D., Kieber, J. J., and Huber, S. C. (2004). Identification of a new motif for CDPK phosphorylation in vitro that suggests ACC synthase may be a CDPK substrate. Arch. Biochem. Biophys. 428, 81–91.
Huttlin, E. L., Hegeman, A. D., Harms, A. C., and Sussman, M. R. (2007). Comparison of full versus partial metabolic labeling for quantitative proteomics analysis in Arabidopsis thaliana. Mol. Cell. Proteomics 6, 860–881.
Jorgensen, C., and Linding, R. (2008). Directional and quantitative phosphorylation networks. Brief. Funct. Genomics Proteomics 7, 17–26.
Klink, V. P., and Wolniak, S. M. (2000). The efficacy of RNAi in the study of the plant cytoskeleton. J. Plant Growth Regul. 19, 371–384.
Kota, U., and Goshe, M. B. (2011). Advances in qualitative and quantitative plant membrane proteomics. Phytochemistry 72, 1040–1060.
Krishna, R. G., and Wold, F. (1993). Post-translational modification of proteins. Adv. Enzymol. Relat. Areas Mol. Biol. 67, 265–298.
Krysan, P. J., Young, J. C., and Sussman, M. R. (1999). T-DNA as an insertional mutagen in Arabidopsis. Plant Cell 11, 2283–2290.
Kurland, I. J., Elmaghrabi, M. R., Correia, J. J., and Pilkis, S. J. (1992). Rat-liver 6-phosphofructo-2-kinase fructose-2,6-bisphosphatase – properties of phospho-forms and dephospho-forms and of 2 mutants in which Ser32 has been changed by site-directed mutagenesis. J. Biol. Chem. 267, 4416–4423.
Lebert, D., Dupuis, A., Garin, J., Bruley, C., and Brun, V. (2011). Production and use of stable isotope-labeled proteins for absolute quantitative proteomics. Methods Mol. Biol. 753, 93–115.
Li, H., Wong, W. S., Zhu, L., Guo, H. W., Ecker, J., and Li, N. (2009). Phosphoproteomic analysis of ethylene-regulated protein phosphorylation in etiolated seedlings of Arabidopsis mutant ein2 using two-dimensional separations coupled with a hybrid quadrupole time-of-flight mass spectrometer. Proteomics 9, 1646–1661.
Li, Y., Shu, Y., Peng, C., Zhu, L., Guo, G., and Li, N. (2012). AQUIP: absolute quantitation of isoforms of post-translationally modified proteins in transgenic organism. Mol. Cell. Proteomics 11,272–285.
Liu, Y. D., and Zhang, S. Q. (2004). Phosphorylation of 1-aminocyclopropane-1-carboxylic acid synthase by MPK6, a stress-responsive mitogen-activated protein kinase, induces ethylene biosynthesis in Arabidopsis. Plant Cell 16, 3386–3399.
Mayya, V., Rezual, K., Wu, L., Fong, M. B., and Han, D. K. (2006). Absolute quantification of multisite phosphorylation by selective reaction monitoring mass spectrometry: determination of inhibitory phosphorylation status of cyclin-dependent kinases. Mol. Cell. Proteomics 5, 1146–1157.
Mok, J., Kim, P. M., Lam, H. Y., Piccirillo, S., Zhou, X., Jeschke, G. R., et al. (2010). Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signal. 3, ra12.
Nelson, C. J., Huttlin, E. L., Hegeman, A. D., Harms, A. C., and Sussman, M. R. (2007). Implications of 15N-metabolic labeling for automated peptide identification in Arabidopsis thaliana. Proteomics 7, 1279–1292.
Oda, Y., Huang, K., Cross, F. R., Cowburn, D., and Chait, B. T. (1999). Accurate quantitation of protein expression and site-specific phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 96, 6591–6596.
Ong, S. E., Blagoev, B., Kratchmarova, I., Kristensen, D. B., Steen, H., Pandey, A., et al. (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386.
Ong, S. E., and Mann, M. (2005). Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 1, 252–262.
Pratt, J. M., Simpson, D. M., Doherty, M. K., Rivers, J., Gaskell, S. J., and Beynon, R. J. (2006). Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat. Protoc. 1, 1029–1043.
Rivers, J., Simpson, D. M., Robertson, D. H., Gaskell, S. J., and Beynon, R. J. (2007). Absolute multiplexed quantitative analysis of protein expression during muscle development using QconCAT. Mol. Cell. Proteomics 6, 1416–1427.
Ross, P. L., Huang, Y. N., Marchese, J. N., Williamson, B., Parker, K., Hattan, S., et al. (2004). Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169.
Schaff, J. E., Mbeunkui, F., Blackburn, K., Bird, D. M., and Goshe, M. B. (2008). SILIP: a novel stable isotope labeling method for in planta quantitative proteomic analysis. Plant J. 56, 840–854.
Schmelzle, K., and White, F. M. (2006). Phosphoproteomic approaches to elucidate cellular signaling networks. Curr. Opin. Biotechnol. 17, 406–414.
Schrohl, A. S., Holten-Andersen, M., Sweep, F., Schmitt, M., Harbeck, N., Foekens, J., et al. (2003). Tumor markers: from laboratory to clinical utility. Mol. Cell. Proteomics 2, 378–387.
Schulze, W. X. (2010). Proteomics approaches to understand protein phosphorylation in pathway modulation. Curr. Opin. Plant Biol. 13, 280–287.
Schulze, W. X., and Usadel, B. (2010). Quantitation in mass-spectrometry-based proteomics. Annu. Rev. Plant Biol. 61, 491–516.
Steen, J. A., Steen, H., Georgi, A., Parker, K., Springer, M., Kirchner, M., et al. (2008). Different phosphorylation states of the anaphase promoting complex in response to antimitotic drugs: a quantitative proteomic analysis. Proc. Natl. Acad. Sci. U.S.A. 105, 6069–6074.
Tavares, C. D. J., O’Brien, J. P., Abramczyk, O., Devkota, A. K., Shores, K. S., Ferguson, S. B., et al. (2012). Calcium/calmodulin stimulates the autophosphorylation of elongation factor 2 kinase on Thr-348 and Ser-500 to regulate its activity and calcium dependence. Biochemistry 51, 2232–2245.
Thelen, J. J., and Peck, S. C. (2007). Quantitative proteomics in plants: choices in abundance. Plant Cell 19, 3339–3346.
Wu, R., Haas, W., Dephoure, N., Huttlin, E. L., Zhai, B., Sowa, M. E., et al. (2011). A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat. Methods 8, 677–683.
Yoo, S. D., Cho, Y. H., Tena, G., Xiong, Y., and Sheen, J. (2008). Dual control of nuclear EIN3 by bifurcate MAPK cascades in C2H4 signalling. Nature 451, U789–U781.
Keywords: SILIA, AQUIP, plant, quantitative proteomics, post-translational modification, cell signaling and regulation, mass spectrometry-based interactomics
Citation: Zhu L and Li N (2013) Quantitation, networking, and function of protein phosphorylation in plant cell. Front. Plant Sci. 3:302. doi: 10.3389/fpls.2012.00302
Received: 27 September 2012; Accepted: 16 December 2012;
Published online: 08 January 2013.
Edited by:
Waltraud X Schulze, Max Planck Institute for Plant Physiology, GermanyReviewed by:
Stefanie Wienkoop, University of Vienna, AustriaBorjana Arsova, Heinrich-Heine University, Germany
Copyright: © 2013 Zhu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Lin Zhu, Division of Life Science, The Hong Kong University of Science and Technology, Clear Water Bay, Hong Kong, China. e-mail:bGluekB1c3QuaGs=;Ym9uaW5nbGlAdXN0Lmhr