 Marco Loehrer1
Marco Loehrer1 Alexander Vogel2
Alexander Vogel2 Bruno Huettel3
Bruno Huettel3 Richard Reinhardt3
Richard Reinhardt3 Vladimir Benes4
Vladimir Benes4 Sébastien Duplessis5,6
Sébastien Duplessis5,6 Björn Usadel2,7
Björn Usadel2,7 Ulrich Schaffrath1*
Ulrich Schaffrath1*- 1Department of Plant Physiology, Rheinisch-Westfälische Technische Hochschule Aachen University, Aachen, Germany
- 2Institute for Botany and Molecular Genetics, Institute for Biology I, Rheinisch-Westfälische Technische Hochschule Aachen University, Aachen, Germany
- 3Max Planck Institute for Plant Breeding Research, Köln, Germany
- 4Genomics Core Facility, European Molecular Biology Laboratory, Heidelberg, Germany
- 5Institut National de la Recherche Agronomique, Interactions Arbres/Microorganismes, UMR 1136, Champenoux, France
- 6Université de Lorraine, Interactions Arbres/Microorganismes, UMR 1136, Vandoeuvre-lès-Nancy, France
- 7Institute of Bio- and Geosciences-2 Plant Sciences, Institute for Bio- and Geosciences, Forschungszentrum Jülich, Jülich, Germany
Recent advances in the field of sequencing technologies and bioinformatics allow a more rapid access to genomes of non-model organisms at sinking costs. Accordingly, draft genomes of several economically important cereal rust fungi have been released in the last 3 years. Aside from the very recent flax rust and poplar rust draft assemblies there are no genomic data available for other dicot-infecting rust fungi. In this article we outline rust fungus sequencing efforts and comment on the current status of Phakopsora pachyrhizi (Asian soybean rust) genome sequencing.
Sequencing of fungal genomes represented a significant milestone in the emerging era of “genomics.” In fact, the first eukaryotic genome ever sequenced was that of baker’s yeast, Saccharomyces cerevisiae, which consequently strengthened its position as a fungal model organism after the release of the 12 Mb genome with approximately 6000 genes in 1996 (Goffeau et al., 1996). Some time thereafter the genomes of the fission yeast S. pombe (14 Mb) and the filamentous ascomycete Neurospora crassa (40 Mb) were released in Wood et al. (2002) and Galagan et al. (2003), respectively. Accelerated progress in sequencing technology from early clone-by-clone approaches through Sanger-based whole-genome shotgun sequencing (WGS) to today’s next-generation sequencing (NGS) shortened the periods between releases of novel genomes considerably (Grigoriev, 2014). This paved the way for comparative genomics which opened new possibilities for people working in the field of agriculture and biotechnology or combating human, animal or plant diseases (Vebø et al., 2009; Manning et al., 2013; Bolger et al., 2014).
In the latter field, the sequencing of the genome of the ascomycete Magnaporthe oryzae was achieved by Dean et al. (2005). Along with the genome of rice (Goff et al., 2002), the M. oryzae host plant, an understanding of the plant–pathogen interaction became possible at the genome level. Since then, several plant-pathogenic fungi were sequenced; however, a group of pathogens that exclusively feed from living plant tissue, so-called obligate biotrophs, remained recalcitrant. This was disappointing particularly because some of the most economically serious threats to human nutrition, such as powdery mildew fungi and rust fungi, are among this group.
Rust fungi have long been in the focus of plant pathologists. Already in the 19th century, Anton de Bary, who is considered as a founder of plant pathology, picked up Puccinia graminis with its various formae speciales that are specialized for parasitism on particular cereal hosts, as subject for his groundbreaking studies. Later Harold Henry Flor developed the famous “gene-for-gene” concept based on his work on the interaction of flax rust (Melampsora lini) with its host plant flax (Linum usitatissimum; Flor, 1955). Despite considerable interest, sequencing of rust genomes was not achieved until most recently. Thus, the 101 Mb genome of Melampsora larici-populina and the 89 Mb draft genome of Puccinia graminis f. sp. tritici were sequenced in a common effort by the Joint Genome Institute and the Broad Institute, respectively, and published in Duplessis et al. (2011). Following, more or less advanced draft genomes of other rust fungi were sequenced and published by the community, such as several Puccinia striiformis f. sp. tritici races (56–110 Mb) and the flax rust genome M. lini (Cantu et al., 2011, 2013; Zheng et al., 2013; Nemri et al., 2014; see Table 1). Although Pucciniales is an order with a lesser coverage compared to other fungi1, more genomic resources are becoming accessible. A major drawback encountered during sequencing efforts of rust genomes was their unexpected large sizes, a fact that also hampered attempts of sequencing the genome of the Asian soybean rust fungus Phakopsora pachyrhizi, an economically important threat to soybean cultivation. The following commentary is written to give an overview on the current status of P. pachyrhizi genome sequencing and is intended to initiate combined activities toward this goal.
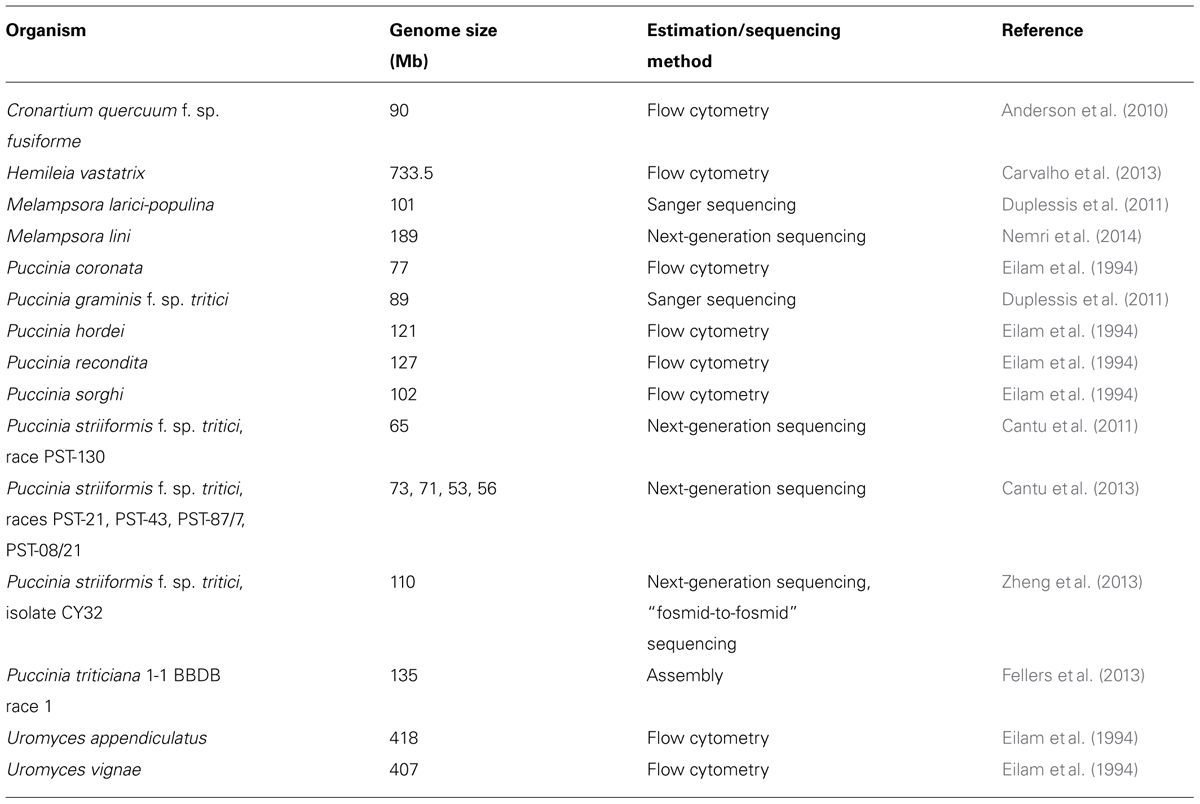
TABLE 1. Published rust fungi draft genomes and genome size estimations in alphabetical order.
What makes P. pachyrhizi so interesting? For sure it is a devastating fungal disease of the important crop plant soybean. The origin of the pathogen can be traced back to Asia and most likely it spread alongside with the propagation of soybean cultivation. P. pachyrhizi is able to infect more than 31 species from 17 genera of legumes, which is a rather unusual feature for rust fungi that usually are highly specialized for particular hosts (Goellner et al., 2010). P. pachyrhizi differs in a further important aspect from the majority of rusts: it directly penetrates leaf cells rather than entering the leaf via stomata at the uredinial stage. On the contrary, most rust fungi use stomata to get inside the host tissues at this stage and a direct penetration is only observed for some rust fungi when basidiospores infect the aecial host at later stages of the rust life cycle (Heath, 1997). Recent studies imply that generation of high turgor pressure of around 5 MPa in the non-melanized appressoria supports penetration (Loehrer et al., 2014). Penetrated epidermal cells undergo a cell death response, again an unexpected property for a biotrophic pathogen. Experiments with non-host plants such as barley and Arabidopsis showed that during penetration and concomitant epidermal cell death, marker genes associated with responses to necrotrophic pathogens are switched on and that cell death suppression had a negative influence on infection success of P. pachyrhizi (Loehrer et al., 2008; Hoefle et al., 2009). Regarding its lifestyle, P. pachyrhizi which forms so far only a single spore type in the wild, i.e. urediospores, is a minimalist compared to, e.g., Puccinia graminis f. sp. tritici which has five distinct spore types and performs a host jump (Leonard and Szabo, 2005). Despite the unknown or missing sexual life cycle the genetic diversity of P. pachyrhizi seems not to be impaired. This may be explained by parasexual nuclear recombination occurring between different isolates after germ tube fusion or hyphal anastomosis, a feature also reported for cereal rusts (Wang and McCallum, 2009; Vittal et al., 2012).
Public information about the P. pachyrhizi genome sequencing project is rare. In the DoE JGI Community Sequencing Program of 2004, a project was launched to sequence the genome of P. pachyrhizi (isolate Taiwan 72-1) based on a fosmid shotgun sequencing approach. The genome size prediction with 50 Mb at that time was much underestimated. The sequencing project has now a “permanent draft” status at the JGI2. Besides the recently released mitochondrial genome sequence (Stone et al., 2010), information on assembly attempts of the nuclear genome have not been published. The major drawback for progress in P. pachyrhizi genome sequencing seems to be its huge size. An update on this topic was given at the National Soybean Rust Symposium 2005 in Nashville (TN, USA). Genome size estimations ranged from 300 to 950 Mb depending on the analysis method used (Posada-Buitrago et al., 2005). A similar statement was provided by Igor Grigoriev (Head of the JGI Fungal Program) suggesting a genome size above 850 Mb (Duplessis et al., 2012). Besides, other general features of rust fungi genomes unraveled since then, such as expanded multigene families and very large amount of transposable elements (>45%), pose serious problems for proper genome assembly.
We started our own efforts toward uncovering the genome size of P. pachyrhizi by using our lab isolate (Brazil 05-1) and we followed a strategy based on k-mer analysis. By breaking down the reads obtained by Illumina sequencing into short nucleotide sequences of defined length k (k-mers), several characteristics of genomes, like size, heterozygosity and repeat content, can be analyzed, that would normally require a complete de novo assembly. As basis for our analysis, DNA was generated from urediospores of the P. pachyrhizi isolate Brazil 05-1. A total of 47 Gb Illumina whole-genome sequencing data (100 bp paired-end reads) were then subjected to analysis using the program JELLYFISH (Marçais and Kingsford, 2011). In the 17-mer distribution depicted in Figure 1, two peaks could be differentiated at a depth of 37 and 75. This can be explained by the dikaryotic nature of the urediospores of rust fungi, which means that these organisms maintain two haploid nuclei separately during prolonged stages of their lifecycle. The two peaks in the k-mer histogram point to a high degree of heterozygosity between the two nuclei or to largely heterozygotic regions within the haploid nuclei. Similar results were also observed by Zheng et al. (2013) in the case of the wheat stripe rust fungus.
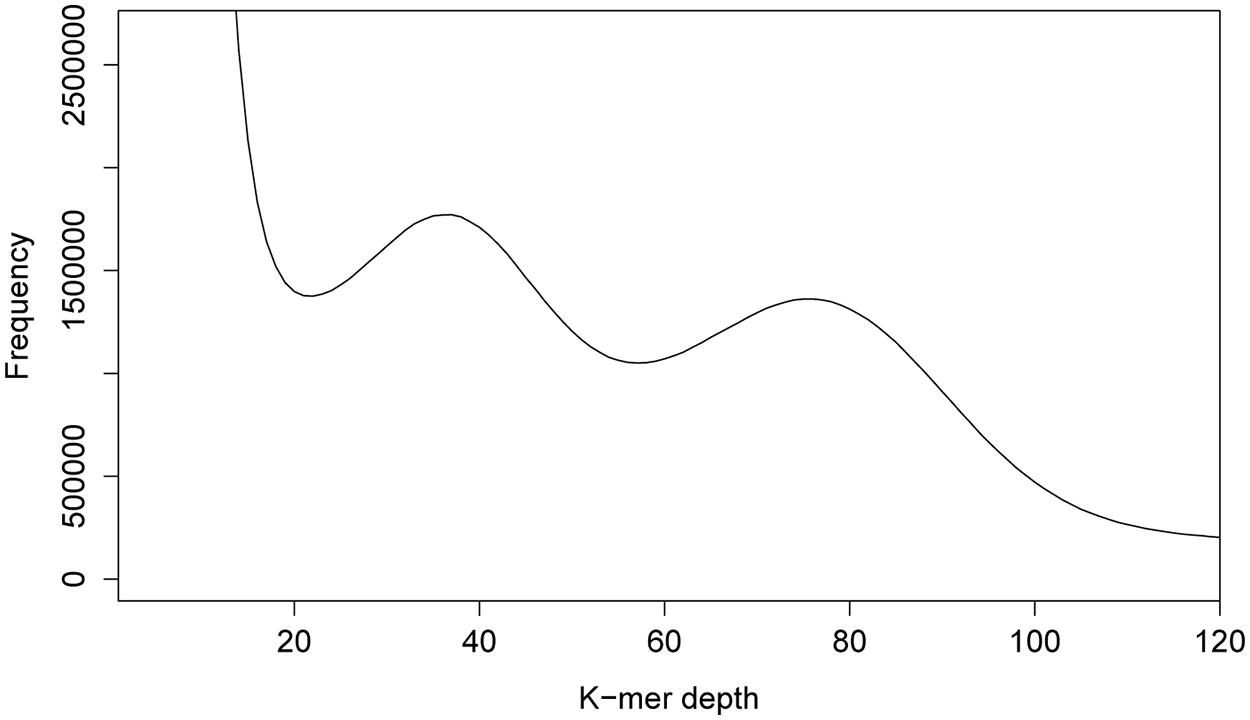
FIGURE 1. K-mer analysis for P. pachyrhizi whole-genome sequencing data. The 17-mer distribution for 47 Gb of 100 bp paired-end Illumina whole-genome sequencing data indicates two peaks at a depth of 37 and 75. These findings point to a possibly highly repetitive genome with a high degree of heterozygosity between the genomes of the two haploid nuclei.
By adding up the products of k-mer depth coverage and frequency for each pair of values in Figure 1, divided by the depth coverage of the first peak (=37), the size of the genome in bp was computed, similarly as in (Li et al., 2010). Values, smaller than the first minimum in Figure 1, were considered noise caused by sequencing errors and were excluded from the calculation. Based on this analysis, the overall size of the dikaryotic genome of P. pachyrhizi is at most around 1 Gb. However, due to the unknown degree of heterozygosity between or within the genomes of both nuclei, this might be an overestimation (see above). The minimal size of the haploid genome can be estimated to be around 500 Mb, based on the second peak in the k-mer analysis (Figure 1). This would place the genome of the Asian soybean fungus in the same range as published rust genomes, e.g., Hemileia vastatrix (733.5 Mb) and Uromyces spp. (420 Mb; Table 1). It should be noted, however, that the analysis method might considerably influence the outcome of such genome size estimations. The genome size of H. vastatrix, e.g., was estimated by DNA-staining in combination with flow cytometry which itself is prone to errors but has the advantage of not being sequencing-dependent (Bainard et al., 2010). Phenomena related to the partial heterozygosity of the P. pachyrhizi genome are only detectable by assembly or k-mer analysis as described above. Since we did not use a large insert size sequencing approach for genome size estimation, we obtained a N50 value of 569 bp after assembly and scaffolding with SOAPdenovo. This allowed no prediction on gene number or length. In future studies a combined BAC- and third generation sequencing approach hopefully will increase the assembly quality to a point at which comprehensive gene predictions become possible.
Working with organisms, whose genome has been sequenced provides many advantages over working with non-sequenced species. Besides the comprehensive prediction of all genes, intra-genomic structural analyses or comparative genome analyses between different species become possible. An alternative to genomic-based approaches in large-scale analyses of plant-pathogen-interactions, however, is the use of transcriptomics, proteomics, or metabolomics (Tan et al., 2009). Up to now, only limited information is available on P. pachyrhizi transcriptomics, though very recent publications have broadened the view on particular aspects of the infection process of P. pachyrhizi (Tremblay et al., 2010, 2012, 2013; Link et al., 2013). For instance, Illumina-based transcriptome profiling at several stages of soybean leaf infection has led to the identification of nearly 19,000 transcripts not previously identified in other rust fungi (Tremblay et al., 2013). This would imply a much larger gene complement in the soybean rust than in other rust fungi. So far, the numbers of genes reported in rust fungi are between 15,000 and 20,000 genes (Duplessis et al., 2014). Although biases in the RNA-Seq approach can not be excluded, it is possible that the P. pachyrhizi genome has experienced a high level of gene duplication during its evolution along with important transposable element activity that could explain the huge genome size predicted for this species. There is an urgent need for genome sequences as prerequisite for accurate large scale expression analysis and more RNA-seq efforts are needed. Without a genome, transcript reads have to be assembled first and not only RNA quality and sequencing technique used will influence the resulting assembly quality but also the algorithms used for assembly. And even if these problems could be sufficiently solved, the resulting contigs are much smaller than transcribed ORFs, limiting for example predictions of putatively secreted proteins. Also, redundancy within gene families could be better resolved when compared to a reference genome sequence.
Hopefully in the near future, the development of novel sequencing and assembly strategies, together with dropping costs for NGS, will make the sequencing of large and complex genomes more affordable and will help to unravel the secrets of the genome of P. pachyrhizi.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ralph Panstruga (Biology I, RWTH Aachen) for helpful discussions. We thank Anthony Bolger (Biology I, RWTH Aachen University) for helpful advice on k-mer analysis. Sébastien Duplessis acknowledges the ANR “Investissements d’Avenir” program (ANR-11-LABX-0002-01, Lab of Excellence ARBRE).
Footnotes
- ^http://genome.jgi.doe.gov/programs/fungi/1000fungalgenomes.jsf
- ^http://genome.jgi.doe.gov/genome-projects/pages/project-status.jsf?projectId=16847
References
Anderson, C. L., Kubisiak, T. L., Nelson, C. D., Smith, J. A., and Davis, J. M. (2010). Genome size variation in the pine fusiform rust pathogen Cronartium quercuum f.sp. fusiforme as determined by flow cytometry. Mycologia 102, 1295–1302. doi: 10.3852/10-040
Bainard, J. D., Fazekas, A. J., and Newmaster, S. G. (2010). Methodology significantly affects genome size estimates: quantitative evidence using bryophytes. Cytometry 77, 725–732. doi: 10.1002/cyto.a.20902
Bolger, M. E., Weisshaar, B., Scholz, U., Stein, N., Usadel, B., and Mayer, K. F. (2014). Plant genome sequencing — applications for crop improvement. Curr. Opin. Biotechnol. 26, 31–37. doi: 10.1016/j.copbio.2013.08.019
Cantu, D., Govindarajulu, M., Kozik, A., Wang, M., Chen, X., Kojima, K. K.,et al. (2011). Next generation sequencing provides rapid access to the genome of Puccinia striiformis f. sp. tritici, the causal agent of wheat stripe rust. PLoS ONE 6:e24230. doi: 10.1371/journal.pone.0024230
Cantu, D., Segovia, V., MacLean, D., Bayles, R., Chen, X., Kamoun, S.,et al. (2013). Genome analyses of the wheat yellow (stripe) rust pathogen Puccinia striiformis f. sp. tritici reveal polymorphic and haustorial expressed secreted proteins as candidate effectors. BMC Genomics 14:270. doi: 10.1186/1471-2164-14-270
Carvalho, G. M. A., Carvalho, C. R., Barreto, R. W., and Evans, H. C. (2013). Coffee rust genome measured using flow cytometry: does size matter? Plant Pathol. doi: 10.1111/ppa.12175
Dean, R. A., Talbot, N. J., Ebbole, D. J., Farman, M. L., Mitchell, T. K., Orbach, M. J.,et al. (2005). The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434, 980–986. doi: 10.1038/nature03449
Duplessis, S., Bakkeren, G., and Hamelin, R. (2014). “Advancing knowledge on biology of rust fungi through genomics,” in Advances in Botanical Research, 1st Edn, Vol. 70, ed. F. Martin (London: Elsevier), 173–209.
Duplessis, S., Cuomo, C. A., Lin, Y.-C., Aerts, A., Tisserant, E., Veneault-Fourrey, C.,et al. (2011). Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. U.S.A. 108, 9166–9171. doi: 10.1073/pnas.1019315108
Duplessis, S., Joly, D. J., and Dodds, P. N. (2012). “Rust Effectors,” in Effectors in Plant-Microbe Interactions, 1st Edn, eds F. Martin and S. Kamoun (Chichester: John Wiley and Sons, Ltd), 155–193.
Eilam, T., Bushnell, W. R., and Anikster, Y. (1994). Relative nuclear DNA content of rust fungi estimated by flow cytometry of propidium iodide-stained pycniospores. Phytopathology 84, 728–735. doi: 10.1094/Phyto-84-728
Fellers, J. P., Soltani, B. M., Bruce, M., Linning, R., Cuomo, C. A, Szabo, L. J.,et al. (2013). Conserved loci of leaf and stem rust fungi of wheat share synteny interrupted by lineage-specific influx of repeat elements. BMC Genomics 14:60. doi: 10.1186/1471-2164-14-60
Flor, H. H. (1955). Host-parasite interaction in flax rust - its genetics and other implications. Phytopathology 45, 680–685.
Galagan, J. E., Calvo, S. E., Borkovich, K. A., Selker, E. U., Read, N. D., Jaffe, D.,et al. (2003). The genome sequence of the filamentous fungus Neurospora crassa. Nature 422, 859–868. doi: 10.1038/nature01554
Goellner, K., Loehrer, M., Langenbach, C., Conrath, U., Koch, E., and Schaffrath, U. (2010). Phakopsora pachyrhizi, the causal agent of Asian soybean rust. Mol. Plant Pathol. 11, 169–177. doi: 10.1111/j.1364-3703.2009.00589.x
Goff, S. A., Ricke, D., Lan, T.-H., Presting, G., Wang, R., Dunn, M.,et al. (2002). A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92–100. doi: 10.1126/science.1068275
Goffeau, A., Barrell, B. G., Bussey, H., Davis, R. W., Dujon, B., Feldmann, H.,et al. (1996). Life with 6000 genes. Science 274, 546–567. doi: 10.1126/science.274.5287.546
Grigoriev, I. V. (2014). “A changing landscape of fungal genomics,” in Ecological Genomics of the Fungi, 1st Edn, ed. F. Martin (Chichester: John Wiley and Sons, Ltd), 3–20.
Heath, M. C. (1997). Signalling between pathogenic rust fungi and resistant or susceptible host plants. Ann. Bot. 80, 713–720. doi: 10.1006/anbo.1997.0507
Hoefle, C., Loehrer, M., Schaffrath, U., Frank, M., Schultheiss, H., and Hückelhoven, R. (2009). Transgenic suppression of cell death limits penetration success of the soybean rust fungus Phakopsora pachyrhizi into epidermal cells of barley. Phytopathology 99, 220–226. doi: 10.1094/PHYTO-99-3-0220
Leonard, K. J., and Szabo, L. J. (2005). Stem rust of small grains and grasses caused by Puccinia graminis. Mol. Plant Pathol. 6, 99–111. doi: 10.1111/j.1364-3703.2005.00273.x
Li, R., Fan, W., Tian, G., Zhu, H., He, L., Cai, J.,et al. (2010). The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317. doi: 10.1038/nature08696
Link, T. I., Lang, P., Scheffler, B. E., Duke, M. V., Graham, M. A., Cooper, B.,et al. (2013). The haustorial transcriptomes of Uromyces appendiculatus and Phakopsora pachyrhizi and their candidate effector families. Mol. Plant Pathol. 15, 379–393. doi: 10.1111/mpp.12099
Loehrer, M., Botterweck, J., Jahnke, J., Mahlmann, D. M., Gaetgens, J., Oldiges, M.,et al. (2014). In vivo assessment of the invasive force exerted by the Asian soybean rust fungus by mach-zehnder double-beam interferometry. New Phytol. 203, 620–631. doi: 10.1111/nph.12784
Loehrer, M., Langenbach, C., Goellner, K., Conrath, U., and Schaffrath, U. (2008). Characterization of nonhost resistance of Arabidopsis to the Asian soybean rust. Mol. Plant Microbe Interact. 21, 1421–1430. doi: 10.1094/MPMI-21-11-1421
Manning, V. A., Pandelova, I., Dhillon, B., Wilhelm, L. J., Goodwin, S. B., Berlin, A. M.,et al. (2013). Comparative genomics of a plant-pathogenic fungus, Pyrenophora tritici-repentis, reveals transduplication and the impact of repeat elements on pathogenicity and population divergence. G3 (Bethesda) 3, 41–63. doi: 10.1534/g3.112.004044
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Nemri, A., Saunders, D. G. O., Anderson, C., Upadhyaya, N. M., Win, J., Lawrence, G. J.,et al. (2014). The genome sequence and effector complement of the flax rust pathogen Melampsora lini. Front. Plant Sci. 5:98. doi: 10.3389/fpls.2014.00098
Posada-Buitrago, M. L., Boore, J. L., and Frederick, R. D. (2005). “Soybean Rust Genome Sequencing Project,” in Proceedings of the National Soybean Rust Symposium, Nashville, TN. Available at: http://www.plantmanagementnetwork.org/infocenter/topic/soybeanrust/symposium/posters/3.pdf [accessed November 14–16, 2005].
Stone, C. L., Posada-Buitrago, M. L., Boore, J. L., and Frederick, R. D. (2010). Analysis of the complete mitochondrial genome sequences of the soybean rust pathogens Phakopsora pachyrhizi and P. meibomiae. Mycologia 102, 887–897. doi: 10.3852/09-198
Tan, K.-C., Ipcho, S. V. S., Trengove, R. D., Oliver, R. P., and Solomon, P. S. (2009). Assessing the impact of transcriptomics, proteomics and metabolomics on fungal phytopathology. Mol. Plant Pathol. 10, 703–715. doi: 10.1111/j.1364-3703.2009.00565.x
Tremblay, A., Hosseini, P., Alkharouf, N. W., Li, S., and Matthews, B. F. (2010). Transcriptome analysis of a compatible response by Glycine max to Phakopsora pachyrhizi infection. Plant Sci. 179, 183–193. doi: 10.1016/j.plantsci.2010.04.011
Tremblay, A., Hosseini, P., Li, S., Alkharouf, N. W., and Matthews, B. F. (2012). Identification of genes expressed by Phakopsora pachyrhizi, the pathogen causing soybean rust, at a late stage of infection of susceptible soybean leaves. Plant Pathol. 61, 773–786. doi: 10.1111/j.1365-3059.2011.02550.x
Tremblay, A., Hosseini, P., Li, S., Alkharouf, N. W., and Matthews, B. F. (2013). Analysis of Phakopsora pachyrhizi transcript abundance in critical pathways at four time-points during infection of a susceptible soybean cultivar using deep sequencing. BMC Genomics 14:614. doi: 10.1186/1471-2164-14-614
Vebø, H. C., Snipen, L., Nes, I. F., and Brede, D. A. (2009). The transcriptome of the nosocomial pathogen Enterococcus faecalis V583 reveals adaptive responses to growth in blood. PLoS ONE 4:e7660. doi: 10.1371/journal.pone.0007660
Vittal, R., Yang, H.-C., and Hartman, G. L. (2012). Anastomosis of germ tubes and migration of nuclei in germ tube networks of the soybean rust pathogen, Phakopsora pachyrhizi. Eur. J. Plant Pathol. 132, 163–167. doi: 10.1007/s10658-011-9872-5
Wang, X., and McCallum, B. (2009). Fusion body formation, germ tube anastomosis, and nuclear migration during the germination of urediniospores of the wheat leaf rust fungus, Puccinia triticina. Phytopathology 99, 1355–1364. doi: 10.1094/PHYTO-99-12-1355
Wood, V., Gwilliam, R., Rajandream, M.-A., Lyne, M., Lyne, R., Stewart, A.,et al. (2002). The genome sequence of Schizosaccharomyces pombe. Nature 415, 871–880. doi: 10.1038/nature724
Keywords: fungal genomics, rust fungi, Asian soybean rust, next-generation sequencing, herterozygosity, genome size, k-mer analysis
Received: 31 May 2014; Accepted: 14 July 2014;
Published online: 27 August 2014.
Citation: Loehrer M, Vogel A, Huettel B, Reinhardt R, Benes V, Duplessis S, Usadel B and Schaffrath U (2014) On the current status of Phakopsora pachyrhizi genome sequencing. Front. Plant Sci. 5:377. doi: 10.3389/fpls.2014.00377
Edited by:
David L. Joly, Université de Moncton, CanadaReviewed by:
John Fellers, United States Department of Agriculture – Agricultural Research Service, USARalf Thomas Voegele, Universität Hohenheim, Germany
Copyright © 2014 Loehrer, Vogel, Huettel, Reinhardt, Benes, Duplessis, Usadel and Schaffrath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ulrich Schaffrath, Department of Plant Physiology, RWTH Aachen University, Worringerweg 1, 52056 Aachen, Germany e-mail:c2NoYWZmcmF0aEBiaW8zLnJ3dGgtYWFjaGVuLmRl