 Graham J. King1,2,3*
Graham J. King1,2,3*- 1Southern Cross Plant Science, Southern Cross University, Lismore, NSW, Australia
- 2National Key Laboratory for Crop Genetic Improvement, Huazhong Agricultural University, Wuhan, China
- 3Crops for the Future, Biotechnology and Breeding Systems, Semenyih, Malaysia
Crop plants encounter thermal environments which fluctuate on a diurnal and seasonal basis. Future climate resilient cultivars will need to respond to thermal profiles reflecting more variable conditions, and harness plasticity that involves regulation of epigenetic processes and complex genomic regulatory networks. Compartmentalization within plant cells insulates the genomic central processing unit within the interphase nucleus. This review addresses the properties of the chromatin hardware in which the genome is embedded, focusing on the biophysical and thermodynamic properties of DNA, histones and nucleosomes. It explores the consequences of thermal and ionic variation on the biophysical behavior of epigenetic marks such as DNA cytosine methylation (5mC), and histone variants such as H2A.Z, and how these contribute to maintenance of chromatin integrity in the nucleus, while enabling specific subsets of genes to be regulated. Information is drawn from theoretical molecular in vitro studies as well as model and crop plants and incorporates recent insights into the role epigenetic processes play in mediating between environmental signals and genomic regulation. A preliminary speculative framework is outlined, based on the evidence of what appears to be a cohesive set of interactions at molecular, biophysical and electrostatic level between the various components contributing to chromatin conformation and dynamics. It proposes that within plant nuclei, general and localized ionic homeostasis plays an important role in maintaining chromatin conformation, whilst maintaining complex genomic regulation that involves specific patterns of epigenetic marks. More generally, reversible changes in DNA methylation appear to be consistent with the ability of nuclear chromatin to manage variation in external ionic and temperature environment. Whilst tentative, this framework provides scope to develop experimental approaches to understand in greater detail the internal environment of plant nuclei. It is hoped that this will generate a deeper understanding of the molecular mechanisms underlying genotype × environment interactions that may be beneficial for long-term improvement of crop performance in less predictable climates.
Introduction
Crop plants are sessile autotrophs, represented by relatively few monocotyledon and dicotyledon angiosperm species which lack the internal thermoregulation of hot blooded animals. Modern breeding programs have contributed to increases in yield, with major advances made during a period of relative climate stability. However, the planet has entered a period of climate variability, in which higher global temperatures also increase amplitude and temporal variance of climate parameters, and temperature accounts for over 30% of global crop yield variability (Porter and Semenov, 2005; Ray et al., 2015). These effects are compounded by the progressive salinization of many available arable soils (Pimental et al., 2004).
Such issues require a deeper understanding of the molecular mechanisms underlying plant responses to the environment (Baulcombe and Dean, 2014). Crop performance, yield and quality are sensitive to interactions between genotype and environment (GxE), with built-in phenotypic plasticity required for crop cultivars to cope with variable environments (Bloomfield et al., 2014). This is particularly critical where management of the internal thermal and ionic environment affects growth rates and developmental phase transitions. The internal ionic status of a plant is strongly dependent on external nutrient availability, with mineral fertilizers a major cost for crop production (Timilsena et al., 2014). In particular, the major macronutrient potassium plays a key role in metabolic adjustment during plant development, affecting yield and responses to salinity, drought and cold.
The detection of temperature by plants is required for appropriate responses on multiplexed timescales covering periods from seconds to years (Blanchard and Runkle, 2011; Way and Pearcy, 2012), with supply of mineral ions varying on an intermediary timescale (Le Bot et al., 1998). Crop yields are sensitive to the pattern of diurnal variation in air and soil temperature that affects the rate of growth and development (Schlenker and Roberts, 2009; Lobell and Gourdji, 2012; Gourdji et al., 2013). Productivity is dependent both on the ability to perceive minor fluctuations in ambient temperature, as low as ±1°C (Porter and Semenov, 2005; Hüve et al., 2006; Blanchard and Runkle, 2011), and plastic responses that involve keeping tally of accumulated thermal history during specific developmental phases (Muchow et al., 1990; McMaster and Wilhelm, 1997; Tan et al., 2000). Thus fluctuations in thermal environment that perturb the ontogenetic timeline have potential for a significant impact on crop yield and quality (Craufurd and Wheeler, 2009; Bloomfield et al., 2014; Ray et al., 2015).
Although many crop traits and developmental phase changes are dependent upon thermal and ionic signals, conventional genetic models have not provided a complete understanding of the relevant signal transduction pathways and behavior (Hammond et al., 2011; McClung and Davis, 2010). More recently it has become apparent that epigenetic marks play a significant role and are able to provide a mechanistic framework in the context of chromatin dynamics.
Environment is detected in a number of ways, including via effector proteins, small RNAs (Mirouze and Paszkowski, 2011) and directly by chromatin (Kumar and Wigge, 2010). As we will see, sophistication and ruggedness in crop plasticity depends to a great extent upon epigenetic feedback loops that contribute to genomic regulation, with crosstalk between physiological and sub-cellular systems (Bloomfield et al., 2014; Kissoudis et al., 2014; Kulcheski et al., 2015). Whilst many of these mechanisms involve specific gene networks and epigenetic marks, at the molecular level within the plant nucleus the relationship between temperature and electrostatic interactions mediated by ion concentration merits investigation.
Plant growth and development progress within biophysical and thermodynamic constraints imposed by the molecular composition of cells (Lintilhac, 2014; Wolfe, 2015). In eukaryotes, sub-cellular compartmentalization reduces the impact of environment on key sub-systems (Millar et al., 2009), and helps preserve the integrity of enzymes and other informational macromolecules. In the nucleus, the dynamic composition and status of chromatin plays a central role in genomic regulation, and is sensitive to local ionic and thermal environment (Gan and Schlick, 2010; Woodcock and Ghosh, 2010; Allahverdi et al., 2015). Management of temperature and ionic homeostasis represents a major energetic and organizational overhead (Jones and Rotenberg, 2001; Alekseeva et al., 2007; Watling et al., 2008), and involves complex signal transduction systems that are fine-tuned to generate appropriate physiological and developmental responses (Wilson, 2013). These systems include epigenetic processes that provide an environmental memory heritable through mitosis, and in some situations through meiosis (Heard and Martienssen, 2014). Thus it has become clear that RNA-mediated epigenetic mechanisms, along with DNA methylation and histone protein epigenetic marks, significantly extend the adaptive responses of plants (Bräutigam et al., 2013; Pikaard and Scheid, 2014). More complex crop plant genomes, with a high load of repetitive sequences and associated pool of epigenetic marks, may offer greater opportunities for regulation of phenotypic plasticity (Bloomfield et al., 2014).
The performance potential of crop plants relies on maximizing harvest index, the ratio of harvestable to total biomass (Unkovich et al., 2010). This is dependent upon the timely transition between distinct phases of development, where genomic and phenotypic plasticity enables this to be orchestrated in the context of variation in the cultivation environment and crop management practices (Bloomfield et al., 2014). It is recognized that coping with the more extreme environmental fluctuations during a crop cycle is likely to rely on secondary systems (Cramer et al., 2011), as well as gene neo-functionalization, with novel genetic loci conferring distinct regulation or function in order to maintain plant homeostasis (Mickelbart et al., 2015).
This review describes the macromolecular components of the chromatin hardware in which plant genomes are embedded. It explores their biophysical behavior in the context of the ionic and thermal environment of the nucleus, and how this is affected by the local distribution of DNA and histone epigenetic marks. Evidence from in vitro and molecular modeling studies is placed where possible in the context of in vivo observations for model species and crop plants. The contribution of ions to mediating electrostatic interactions of chromatin and epigenetic marks is placed in the context of ionic variation at whole plant level. Recent advances in understanding how specific epigenetic marks mediate plant thermosensory signaling and other responses to abiotic environment are placed in the context of chromatin dynamics and biophysics.
A preliminary speculative framework is outlined, based on the evidence of what appears a cohesive set of interactions at molecular, biophysical and electrostatic level between the various components contributing to chromatin conformation and dynamics (Figure 1). It proposes that within plant nuclei, general and localized ionic homeostasis plays an important role in maintaining chromatin conformation, whilst maintaining complex genomic regulation that involve specific patterns of epigenetic marks. More generally, reversible changes in DNA methylation appear to be consistent with the ability of nuclear chromatin to manage variation in external ionic and temperature environment. Whilst tentative, this framework provides scope to develop experimental approaches to understand in greater detail the internal environment of plant nuclei. It is hoped that this will generate a deeper understanding of the molecular mechanisms underlying genotype × environment interactions that may be beneficial for long-term improvement of crop performance in less predictable climates.
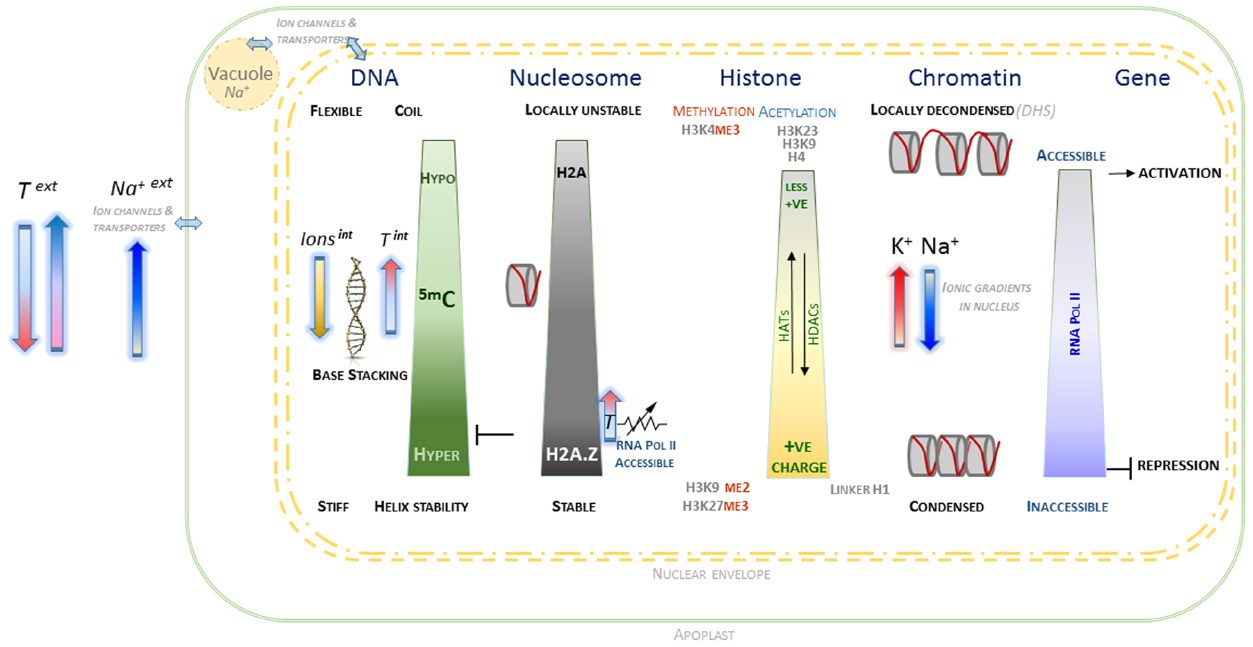
Figure 1. Schematic overview of interactions associated with chromatin and component macromolecules within the electrostatic environment of the plant nucleus. The contrasting states of DNA, histones, nucleosomes and chromatin as affected by epigenetic marks and localized nuclear ionic environment are indicated. The arrows indicating external temperature and salt are oriented in relation to their observed effects on DNA methylation and H2A status. Internal cation concentrations contribute to level of chromatin condensation, along with more complex electrostatic interactions, including those involving histone modifications and divalent cations. The contribution of histone acetylases (HAT) and deacetylases (HDAC) to H4 and H3 acetylation are indicated, which along with the interplay with histone methylation is able to provide a complex chromatin code. The nuclear envelope is represented by dashed double yellow lines, with a very simplified representation of interactions between the cytoplasm, vacuole and external apoplast.
The Genome at Home in the Nucleus
Crop plants have derived from taxa that represent different levels of genome complexity (King, 2002). Some are well adapted to the relatively uniform annual environments of the tropics and subtropics (Gepts, 2008), while others must manage variability in length of temperate seasons and severity of cold winter periods (Rosenzweig and Liverman, 1992; Craufurd and Wheeler, 2009). Compared with the condensed genome of Arabidopsis (125 Mbp), crop genome sizes vary over 60-fold, from around 265 Mbp (peach, Prunus persica) to the larger cereal genomes (barley, 5.1 Gbp; wheat: 17 Gbp; Michael and Jackson, 2013). In common with all eukaryotes, this variation is directly proportional to nuclear volume (Cavalier-Smith, 1985), which suggests strong selective pressure to maintain an optimal dense nuclear environment, where crowding due to proteins, DNA and RNA leads to a macromolecular concentration calculated at over 100 mg ml–1 (Hancock and Hadj-Sahraoui, 2009; Hancock, 2012a), considerably higher than those typically used for many in vitro experiments (Hancock, 2012a). This greatly reduced effective solvent volume also means that the equivalent molar concentrations of mono- and divalent ions may be considerably different from those regarded as cytoplasmic or physiological. At present few reliable estimates of nuclear water content exist and, as already noted, it appears that a significant proportion of ions are bound to chromatin and other macromolecules (Garner, 2002; Hancock and Hadj-Sahraoui, 2009).
Compared with the cytoplasm, the rate of molecular mobility, expressed as diffusion constants, is about five times lower in the nucleus, and 10-fold lower in the nucleolus (Bancaud et al., 2009). Despite this, regulatory factors appear still able to locate genomic targets rapidly, due to the presence of electrochemical gradients generated by ion distributions in the remaining space, which have been modeled as channels between chromatin fibers represented by percolation clusters (Wedemeier et al., 2007; Fritsch and Langowski, 2010; Bancaud et al., 2012). Such an environment may also encourage diffusiophoresis (Hancock, 2012a), a process that involves dispersed particles moving spontaneously in a fluid induced by a diffusion or concentration gradient. Indeed, in vivo imaging of HeLa nuclei indicates a “mesh spacing” of chromatin fibers on the order of 63 nm, significantly larger than the typical size of diffusing protein complexes (Weidemann et al., 2003). From this and other in vivo studies, these authors concluded that all nuclear locations are accessible for diffusing protein complexes (Weidemann et al., 2003). Thus signal transduction via diffusion of regulatory factors to the genome does not appear to be a time-limiting factor for regulatory systems involving rapid transcription, or epigenetic response to external environment. At 7–19 mg ml–1, the 30 nM chromatin fiber represents about 10% of the nuclear volume (Strickfaden et al., 2012) and so the time for a non-interacting protein to explore the whole nucleus is of the order of a few seconds, and four times faster than in water (Strickfaden et al., 2012). For example, GFP (27 kDa) may move 12 μM sec–1, a similar rate as between the cytoplasm and nucleus (Pack et al., 2006). The extent of this molecular crowding within the nucleus has led to the suggestion that entropic forces, such as those associated with polymer elasticity, may be more significant for chromatin structure and dynamics than some of the electrostatic forces observed under typical in vitro conditions (Hancock, 2012b).
Chromatin Architecture and Dynamics
Eukaryote nuclear chromosomes represent the complex macromolecular structures formed of chromatin, in which genomic DNA is embedded along with highly charged proteins and varying amounts of RNA. Nucleosomes represent the building blocks of chromatin, with ∼147 bp of DNA wrapped around core nucleosome particles, each of which consists of a H3-H3-H4-H4 histone tetramer and two H2A–H2B dimers. Within this octamer, the H2A-H2B dimers occupy the peripheral 2 × ∼30 bp DNA, and the H3–H4 tetramer the inner region, with tails that interact both within and between nucleosomes and contribute to accessibility for transcriptional apparatus (Iwasaki et al., 2013). Post-translational changes to histones, including acetylation and methylation, are able to generate a combinatorial code of epigenetic marks (Lothrop et al., 2013), along with variants such as H2A.Z.
Based on in vitro and modeling studies, electrostatic interactions appear to be the dominant factor affecting overall nucleosome stability (Fenley et al., 2010), with higher ionic concentration a major source for destabilization and disruption (Gansen et al., 2009). The wrapping and unwrapping of DNA around the histone core is sensitive to the charge state of the globular core (Fenley et al., 2010), with in vivo post-translational acetylation of a single lysine residue in H3 or H4 able to decrease the charge and decrease the strength of binding to DNA. This reduces the tendency of chromatin to fold into highly compact structures, making it more accessible to transcription factors (TFs) and other interacting proteins, as well as having greater sensitivity to DNase I (Garcia-Ramirez et al., 1995; Tse et al., 1998). This epigenetic switch is mediated by histone acetyltransferases (HATs) that neutralize the positive charge on lysine, and histone deacetylases (HDACs) which play an inverse role, by providing a more positive net charge (Eberharter and Becker, 2002).
Nucleosome Positioning and Epigenetic Marks Define Chromatin State
Nucleosome positioning with respect to genomic sequence is sensitive to many intrinsic and external factors (Tsankov et al., 2011; West et al., 2014), especially in euchromatin, where positioning is dynamic and responsive to cellular identity and internal nuclear environment (Hughes et al., 2012). Gene activation is facilitated by DNA and nucleosome thermodynamics, the nucleosomal surface and chromatin higher order structure (Dechassa and Luger, 2012). Thus initiation and progress of transcription is dependent upon RNA polymerase II (RNA Pol II) gaining access to DNA wrapped around nucleosomes, by harnessing fluctuations that locally unwrap DNA, rather than unwrapping nucleosomes (Mack et al., 2012). Targeted protein and snRNA regulatory factors are able to effect rapid and reversible changes in transcription as a result of this highly dynamic behavior (Ivashuta et al., 2011; Chen and Rajewsky, 2014; Franco-Zorrilla et al., 2014), which is mediated by the epigenetic marks of histone and DNA modifications (Pikaard and Scheid, 2014). The detection of an association between nucleosome phasing, introns, and RNA splicing (Schwartz et al., 2009a; Tilgner et al., 2009; Chodavarapu et al., 2010) also highlights the central role nucleosomes play in managing RNA Pol II transcription in complex eukaryote genomes.
The distribution of epigenetic marks is an important contributor to the organization of nucleosomes in promoter regions, with histone modifications able to mediate very specific access to DNA to enable gene activation (Bannister and Kouzarides, 2011). This can result in distinct and dynamic histone landscapes associated with specific plant processes, such as de-etiolation in Arabidopsis (Charron et al., 2009). Experimental data and statistical mechanics thermodynamic predictions both indicate that nucleosomes are able to block the binding of many TFs by competing with common binding sites, as well as contributing to cooperative binding between TFs (Raveh-Sadka et al., 2009). In general, core histones and variant forms tend to be stably bound to DNA on a timescale of hours (Rippe, 2012), whilst the half-life turnover of histone acetylation is on the order of minutes (Nightingale et al., 2007). This confers a combination of high thermodynamic stability whilst being sensitive to factors that allow rapid access to DNA when required.
Chromatin Accessibility
Decondensed or open chromatin is detectable by the presence of DNAse I hypersensitive sites (DHSs), which provide an accurate experimental indication of where the DNA is exposed and accessible (Wu et al., 2014). For example, in Arabidopsis ∼90% of the binding sites of the MADS-box TFs APETALA1 and SEPALLATA3 are covered with DHSs (Zhang et al., 2012), indicating that these form a barrier for nucleosome formation. However, in general only a subset of nucleosomes are reproducibly positioned, with phasing associated with flanking of transcription start sites (TSS) of active genes in Arabidopsis (Zhang et al., 2012). In rice, DHSs are associated with regions flanked by strongly phased nucleosome arrays (Wu et al., 2014). This is consistent with a barrier model, where intergenic and other regions in which regulatory proteins may be bound to the genomic DNA to provide a barrier that facilitates phased nucleosome arrays to organize either side (Mobius and Gerland, 2010). Rice DHSs may also span a single phased nucleosome (Wu et al., 2014). For promoters of constitutively transcribed genes a DHS detectable barrier may be permanent (Wu et al., 2014), while for binding sites of TFs associated with tissue-specific, organ-specific or environmentally induced gene expression such barriers may be transient, thus allowing nucleosome rearrangement to facilitate transcription (Zhang et al., 2012).
Epigenetic Modifications Modify Chromatin Architecture
Although many components of chromatin and epigenetic marks are conserved between plants and animals, it is important to be aware of a number significant plant-specific features. These include 5′-methylcytosine (5mC) occurring in all context in plants compared with solely CG in animals, the presence of hydroxymethylated cytosine (5 hmC) in animals, the distinct plant methyltransferases CMT3, DRM1/2, and MET (Jang et al., 2014) and defined distribution of methylation with respect to cis-regulatory and gene body sequences (Cokus et al., 2008). In addition, plants display a characteristic pattern of nucleosome distribution (Chodavarapu et al., 2010), and tighter distribution of intron length (Wu et al., 2013) compared with animals. Plants and animals also appear to have evolved distinct DNA demethylation systems, with the DEMETER (DME) family DNA glycosylases able to remove 5mC efficiently in plants, resulting in DNA demethylation and transcriptional activation of target genes (Jang et al., 2014).
Management of DNA Methylation
Various experimental approaches have shown that DNA cytosine methylation contributes to regulating higher order chromatin structure in plants (Tariq and Paszkowski, 2004), primarily through interactions with histones, and ultimately affecting nucleosome positioning. There is extensive evidence that gene silencing and repression of active euchromatin is associated with hypermethylation of DNA in plants (Vaillant and Paszkowski, 2007). Although there is less evidence for direct involvement of 5mC in condensation of heterochromatin (Gilbert et al., 2007), at the primary chromatin level DNA methylation has been shown to have a strong interaction with nucleosome formation (Pennings et al., 2005), and particularly in plants (Chodavarapu et al., 2010). These phenomenological observations appear to be consistent with what is known of the underlying properties of the component molecules and their interactions.
5mC has been shown to shift the preferred rotational position of nucleosomes in vitro by 3 bp (Buttinelli et al., 1998). In mammals, the stabilizing effect of 5mC on DNA duplexes is able to be reversed by hydroxymethylation to 5 hmC (Rodriguez Lopez et al., 2010; Thalhammer et al., 2011). However, the 5 hmC modification appears absent in plant chromatin (Erdmann et al., 2014), and at present it is unclear whether functional analogs for regulating chromatin dynamics exist.
In plants, the involvement of DNA cytosine methylation (5mC) in regulation of gene expression makes a significant contribution to definition of cellular identity and coordination of ontogeny (Vaillant and Paszkowski, 2007; Heard and Martienssen, 2014). The specific molecular attributes of 5mC compared with unmethylated C, and the dynamic nature of DNA methylation, are critical in providing a “toggle switch” mechanism. Thus 5mC provides a versatile heritable epigenetic mark able to define tissue specific expression patterns, and mediate responses to the environment (Zhang et al., 2011; Bloomfield et al., 2014; Widman et al., 2014). The higher density of 5mCG observed within plant genes compared with promoters (Cokus et al., 2008) appears to have a greater effect on transcription due to inhibition of elongation (Chodavarapu et al., 2010; Gelfman et al., 2013). Within the rice genome, the methylation map indicates single peaks close to start codons (Li et al., 2008). In Arabidopsis transcriptional units, 5mC appears to be enriched over the first nucleosome in a transcription unit, with strong periodicity of ∼180 bp in methylation over subsequent nucleosomes (Chodavarapu et al., 2010), and a strong signal associated with positioning in exons.
The processes underlying hypomethylation that are associated with reprogramming, particularly in establishing pluripotency and imprinting effects in plant and animal systems, are now starting to be uncovered (Feng et al., 2010). Dynamic control of DNA methylation involves a cyclic enzyme cascade that consists of cytosine methylation, iterative oxidation of the methyl group by TET (ten eleven translocation) dioxygenases which act as 5mC oxidase, and replacement with unmodified cytosine (Zhao and Chen, 2014). In plants, this latter step of active DNA demethylation is primarily carried out by a small group of bifunctional DNA glycosylases that include ROS1, DME, DML2, and DML3 (Gong and Zhu, 2011). These remove the methylated cytosine base and create an abasic site, with the gap refilled by an unmethylated cytosine through a base-excision-repair pathway (Gong and Zhu, 2011).
RNA Directed DNA Methylation
The RNA-directed DNA methylation (RdDM) epigenetic pathway is the primary mechanism by which plants mediate responses involving small RNAs, and is dependent upon the RNA polymerases Pol IV and Pol V, which are specific to plants, along with various accessory proteins currently being characterized (Matzke and Mosher, 2014). There is increasing evidence for involvement of RdDM in a wide range of developmental and physiological processes that include stress responses, pathogen defense as well as reproductive development (Boyko et al., 2010; Gutierrez-Marcos and Dickinson, 2012; Matzke and Mosher, 2014). This is in addition to the major role played in repression of subsets of transposons as well as protein coding genes, and the interplay between these in complex crop genomes has yet to be fully explored.
Within the nucleus snRNAs operate to repress epigenetic modifications such as 5mC and histone methylation directly at specific target sites, resulting in transcriptional gene silencing (TGS; Simon and Meyers, 2011; Matzke and Mosher, 2014). This involves processing of Pol IV transcripts within the nucleus and cytoplasm, and re-introduction into the nucleus, where siRNAs are able to facilitate targeting of Pol V nascent transcripts (Simon and Meyers, 2011). Recruitment of methyltransferase leads to de novo methylation of cytosines in each of the CG, CHG, CHH contexts, and although Pol V-mediated RdDM operates over many genomic regions, there appears to be a preference toward euchromatin, more recently acquired intergenic TEs, and genes containing TEs or other repeat sequences in their promoters and introns (Matzke and Mosher, 2014). A large proportion of RdDM targets are also modified by modified histone H3K9me, which can provide a feedback loop with DNA methylation to reinforce TGS.
Biophysical Properties of 5mC
The methylation of cytosine (5mC) affects a wide range of DNA biophysical properties, with variation in the localized patterns of steric and conformational energy, as well as hydrophobic modifications to polarity (Hausheer et al., 1989; Wanunu et al., 2011). Together with electrostatic alterations that affect internal base pair dynamics, and variation in base stacking energy, these lead to variation in DNA flexibility (increased flexibility or bending propensity = decreased stiffness) and duplex stability. In particular, stacking energies between neighboring dinucleotides in DNA are represented by elastic force constants that contribute both to DNA flexibility and helical opening (Severin et al., 2011). In 5mC, these effects appear to be specifically associated with molecular polarizability of the pyrimidine, which increases the base stacking energy and reduced flexibility (Norberg and Vihinen, 2001; Acosta-Silva et al., 2010). This is due in part to the protrusion of the hydrophobic methyl group into the major groove, which alters the steric arrangement and local charge environment (Song et al., 2013). The base stacking interactions can generate local distortions in DNA (Acosta-Silva et al., 2010; Yusufaly et al., 2013) and inhibit CG:CG step overtwisting, which in turn decreases flexibility (Yusufaly et al., 2013).
In addition to DNA flexibility, stacking energies also contribute to the cooperative melting associated with the DNA helical-coil transition that is observed both in naked form (Anselmi et al., 2000) as well as within protein complexes (Perez et al., 2004), with helical stability also proportional to the local cation environment (Yakovchuk et al., 2006). Atomic force experiments and molecular dynamics simulations suggest that the contribution of 5mC to increased cooperative DNA helical stability may also depend on methylation level and sequence context, with perhaps more significant effects on mechanical stability and relative stiffness (Severin et al., 2011). Independent experimental evidence based on high resolution melting has also shown that 5mC confers increased helical stability compared with unmodified C (Rodriguez Lopez et al., 2010; Wanunu et al., 2011).
These contributions of 5mC to increased DNA duplex stability and reduced flexibility also appear to affect some aspects of nucleosome positioning, as well as the ability of nucleotide sequences to wrap around the histone complex (Dantas Machado et al., 2015). A picture that is emerging from recent plant whole genome methylome and nucleosome positioning studies (Pennings et al., 2005; Chodavarapu et al., 2010; Gelfman et al., 2013) suggests a discontinuous variation of 5mC in nucleosomal regions.
H3.3 Distribution
Histone H3 is a substrate that provides considerable molecular complexity in terms of epigenetic marks for most eukaryotes, having two major variants, H3.1 and H3.3, as well as accommodating a range of post-translational modifications in the N-terminal amino acid residues. In Arabidopsis, H3.1 is enriched in silent areas of the genome, including those with the H3K27me3 and H3K9me3 modifications that contribute to transcriptional repression, as well as with DNA methylation (Stroud et al., 2012). In contrast, H3.3 has been shown to play a role in maintaining accessible chromatin (Jin and Felsenfeld, 2007), and is enriched in actively transcribed regions, particularly in the 3′ of Arabidopsis genes, where it is correlated with H3K4me3 and H3B ubiquitylation (Stroud et al., 2012).
Histone H2A.Z Distribution
The histone variant H2A.Z is evolutionarily conserved, and often ∼60% identical to canonical H2A within a species, while being ∼80% conserved between species (Zlatanova and Thakar, 2008). The variant plays an important role in marking the epigenetic state of nucleosomes (To and Kim, 2013), and is preferentially localized toward the 5′ of genes in Arabidopsis, where it has been shown to be excluded from sites of heavily methylated DNA within actively transcribed genes (Zilberman et al., 2008). This inverse relationship between the H2A.Z and 5mC has been interpreted as providing a mechanism whereby H2A.Z protects DNA from cytosine methylation in euchromatic regions (Meneghini et al., 2003; Zilberman et al., 2008).
Increasing the wrapping of DNA around the core of H2A.Z containing nucleosomes can reduce the intrinsic fluctuations in DNA accessibility which facilitate transcription (Bowman and Poirier, 2015). Thus H2A.Z marked nucleosomes are often found in regions flanking TSS, and these provide a “molecular rheostat” for initiation of RNA Pol II transcription (Weber et al., 2014; Subramanian et al., 2015).
H1 Linker
H1 histones interact with the linker DNA between adjacent nucleosomes, and cooperatively contribute to formation of the stable and compact 30 nm fiber (McBryant et al., 2010). Although the linker histones ensure compaction and stabilization of higher order chromatin, the variant forms also mediate variation in conformation and accessibility (Wong et al., 2007). It should be noted that linker H1 facilitates self-association of chromatin fibers at salt concentrations considerably lower than for nucleosomal arrays lacking H1 (McBryant et al., 2010). The stoichiometrical relationship between H1 and core nucleosomes has been shown to range from 0.5 to 1 in different tissues (Woodcock et al., 2006), with linker length conventionally described as a diagnostic feature of chromatin from different taxa and/or tissues (Woodcock et al., 2006).
Transient binding of H1 determines the trajectory of DNA entering and exiting the nucleosome (Bednar et al., 1998) by asymmetric binding of an entry or exit linker with the dyad axis (Brown et al., 2006), and constrains an additional 19–20 bp beyond the nucleosome core (Noll and Kornberg, 1977; Simpson, 1978). This is achieved primarily by neutralizing the negative charge of linker DNA, with the binding of the H1 C-terminal domain contributing to chromatin condensation (McBryant et al., 2010). More recently, additional roles for H1 histones have been uncovered, with the C terminal ends associated with molecular “hubs” that recruit proteins involved in accessing and modifying the chromatin fiber (McBryant et al., 2010).
Plants appear to have a wider range of H1 variants than animals (Over and Michaels, 2014), with many monocot and dicot species having at least one shorter variant that may be induced under drought stress (Jerzmanowski et al., 2000). For example, in Arabidopsis, H1.3 is drought inducible and has greater binding to chromatin (Ascenzi and Gantt, 1999). However, it should be noted that not all “drought inducible” H1 variants are associated with drought, and may contribute other functions during development (Over and Michaels, 2014). Depletion of the variants H1.1 and H1.2, along with removal of H2A.Z, is consistent with the global pattern of chromatin decondensation observed in Arabidopsis female megaspore mother cells (She et al., 2013).
Various lines of evidence have suggested a close interaction between linker H1 and ordered DNA methylation in plants (Wierzbicki and Jerzmanowski, 2005). For example, knockdown of H1 in ddm1 mutants of Arabidopsis can lead to restoration of DNA methylation by RdDM (Zemach et al., 2013), suggesting that DDM1 is able to remove H1 to facilitate access to the DNA methylation machinery (Over and Michaels, 2014). Additional evidence has come from analysis of parent-of-origin imprinted loci in the MEDEA (MEA): DEMETER (DME) system, where DME acts as a 5mC demethylase and physically interacts with H1.2 (Rea et al., 2012). The same study has shown that H1 mutants increase DNA methylation in maternal copies of MEA and FWA promoters in Arabidopsis endosperm. More recently, analysis of h1.3 mutants has indicated that the absence of H1.3 can lead to significantly reduced stress-related DNA methylation, with this being most evident in the CHH context (Rutowicz et al., 2015). The requirement of H1.3 for a significant proportion of the DNA methylation associated with environmental stress suggests that this linker histone variation may facilitate chromatin accessibility in direct competition with the primary variants H1.1 and H1.2 (Rutowicz et al., 2015). More generally H1 depletion and DNA hypomethylation, along with H3K27me3 demethylation, appear to be key contributors to pluripotency that is facilitated by chromatin decondensation (Alatzas et al., 2008; He et al., 2012).
Biophysical Properties of Histone Modifications
The complimentary roles of 5mC and H2A.Z associated with nucleosome stability may also be based on their respective biophysical and thermodynamic properties. H2A.Z has been shown to contribute to increased nucleosome stability compared with the canonical H2A, with structural and thermodynamic evidence for a more stable interface via the extended acidic path of the H2A.Z dimer and the charged tails of the (H3–H4)2 tetramer (Dechassa and Luger, 2012). These differences in electrostatic potential and size affect the interface with neighboring nucleosomes and other nuclear proteins (Chakravarthy et al., 2005), and can also contribute to compaction of the 30 nM fiber (Fan et al., 2002).
Sequence analysis of human H2A.Z and H2A-containing nucleosomes has also indicated a prominent association with DNA flexibility at nucleosome boundaries (Gervais and Gaudreau, 2009), with H2A.Z being slightly more rigid than corresponding H2A sequences. Moreover, a DNA flexibility model is able to predict the presence of H2A.Z bordering TSS (Gervais and Gaudreau, 2009). Biophysical studies have also indicated a decreased sensitivity of H2A.Z to ionic strength, with reduced organization of only ∼118 bp of core nucleosomal DNA compared with the canonical 147 bp (Doyen et al., 2006).
By adding a negative charge, phosphorylation of H1 generally has the effect of weakening the electrostatic interaction between H1 and DNA, thus increasing H1 mobility (Roque et al., 2008; Over and Michaels, 2014). Although the precise arrangement of H1 in relation to linker and nucleosome is still unclear (Woodcock et al., 2006), recent models suggest increased bending of DNA at the ends of the nucleosome core (Cui and Zhurkin, 2009).
Nuclear Ionic Status and Chromatin Dynamics
The Ionic Environment of the Nucleus
The nuclear envelope provides a boundary within which the genome resides and benefits from a distinct ionic environment buffered from the cytoplasm (Mekhail and Moazed, 2010; Van de Vosse et al., 2011). The two membranes of the nuclear envelope provide an interface, with discrete functions serving the nucleus and cytoplasm. In plants, the vacuole provides the primary store for inorganic ions (Seidel et al., 2013). However, it has become apparent that in all eukaryotes the perinuclear space between inner and outer nuclear envelope provides a store for calcium and other inorganic ions (Matzke et al., 2010), which may contribute to intracellular signaling (Draguhn et al., 1997), including rapid responses for maintaining selective homeostasis of ions such as K+ to sustain nuclear function (Wyn Jones and Lunt, 1967).
Although nuclear pores are not freely permeable to Na+ and K+ the outer membrane of the nuclear envelope contains distinct ion channel classes (Franco-Obregon et al., 2000), including K+ channels, which contribute to the Na+/K+ gradients between the perinuclear lumen, the nucleus and cytoplasm both in animals (Garner, 2002) and plants (Charpentier et al., 2008). Early X-ray microanalysis of oocytes demonstrated that only a portion of the K+ in interphase nuclei is in free ionic state, with the remainder being absorbed to the nuclear macromolecules, including DNA and histones (Cameron, 1985). As we shall see, the conformation of chromatin can be modulated by the electrostatic interaction mediated by ions such as K+. More generally, it is recognized that monovalent cations that are actively transported through nuclear channels are likely to play an important role in modulation of chromatin structure and gene expression (Garner, 2002).
Chromatin Sensitivity to Cation Environment
The dynamic state of chromatin is subject to variations in the immediate thermal and ionic environment (Spadafora et al., 1979; Caño et al., 2006; Arya and Schlick, 2009). As we have seen, the genome exists in a crowded nuclear environment, embedded in chromatin and serviced by an array of RNA and protein molecules, with access to the read-only transcriptional capability being affected by thermodynamic and biophysical properties of the constituent macromolecules. The ionic environment of chromatin has significant effects on higher level chromatin conformation, with salt-dependent chain folding indicated by in vitro (Bertin et al., 2007) and electron microscope (Thoma et al., 1979) studies. As well as being guided by genomic sequence and distribution of epigenetic marks, global aspects of nucleosome assembly and disassembly appears to be dependent upon salt concentration, with the internal (H3–H4)2 tetramer of the nucleosome binding DNA more often at higher ionic strengths than the H2A–H2B dimer (Dechassa and Luger, 2012).
Evidence from molecular combing experiments, which generate a uniformly stretched array of DNA, has suggested that both Na+ and K+ inhibit binding of histone to DNA, whilst divalent cations significantly enhance binding, with Mn++ inducing condensation and aggregation of histone-DNA complexes in vitro (Liu et al., 2005). Thus increasing ionic strength is able to condense the 10 nm nucleosome fiber to form the 30 nm chromatin fiber as part of a reversible process arising from electrostatic repulsion overcoming nucleosome stacking interactions (Poirer et al., 2002). While an increase in monovalent cations above normal range may result in destabilization of interphase chromatin, low concentrations (10 mM) of divalent cations are able to condense chromatin (Visvanathan et al., 2013), possibly as a result of Mg++ mediating attraction between single negative charges along chromatin (Poirer et al., 2002).
Large scale sensitivity to the ionic environment is also apparent from the fact that attractive electrostatic interactions in chromatin can be screened by a high ion density with salt concentrations >100 mM (Poirer et al., 2002). In terms of visible phenotype, this has been found to lead to unfolding and expansion of chromosomes in Notophthalmus viridescens (eastern newt; Poirer et al., 2002). This may also account for the early observation that increasing the external supply of K+ up to 0.3 M in Lolium temulentum (ryegrass) had the effect of increasing meiotic chiasmata frequency at 30°C, although with little effect at 20°C (Law, 1963), and with little effect from Ca+.
Of more general significance, different lines of evidence from molecular modeling and in vitro studies now suggest that K+ and Na+ ions have distinct roles in condensation of DNA and chromatin, with recent in vitro evidence indicating that Na+ promotes the folding into 30 nm fibers in the presence of Mg++, whereas K+ limits this effect (Allahverdi et al., 2015). This appears to be due to the different binding behaviors of each ion to DNA, with K+ binding to the electronegative sites of DNA bases in the major and minor grooves, and Na+ interacting preferentially with the phosphate groups (Cheng et al., 2006). Moreover, there appears to be greater variation in the mobility of both water and ions in the K-DNA system than the Na-DNA system (Allahverdi et al., 2015). The consequences of this phenomenon for transcription, as well as the maintenance of Na:K ratios within the plant nucleus, have yet to be fully explored, but may have extensive ramifications for our understanding of how plant genomes harness and respond to the complex electrostatic environment within the nucleus.
Ionic Variation in Plants
Cellular organisms expend a substantial proportion of their energy ensuring that the biochemical and other components within the cell are able to operate within boundaries of a relatively consistent ionic environment (Alekseeva et al., 2007). The internal concentration of specific mono- and divalent cations in plants appears to be under strong selection, with considerable variation observed across plant taxa based on data derived from tissue level assessment (Thompson et al., 1997; Broadley et al., 2004; Harada and Leigh, 2006). Whilst internal K, N, and P concentrations have been found to vary sixfold to ninefold between species of related angiosperm taxa (Thompson et al., 1997), concentrations of the divalent cations Mg++ and Ca++ appear much more variable, with a 49-fold variation in Ca++ and a 24-fold variation in Mg++. Most of this variance is allocated between monocot and dicot species (Thompson et al., 1997), and compares with P, where most of the variance is found at or below the species level.
Potassium (K+) is the most abundant inorganic cation in plants, representing up to 10% of dry weight (Watanabe et al., 2007), which is significantly more than required for optimal growth. Since the greater proportion is sequestered into the vacuole, most research attention has focused on its role as an osmoticant in the vacuole and cytosol, as well as an enzyme activator (Leigh and Wyn Jones, 1984), rather than its role in the nucleus. Much of the intra-specific variation appears to be under genetic control, with a greater than twofold variation in shoot K observed in Brassica oleracea, and quantitative trait loci (QTL) analysis suggesting a significant role for variation in K+ transporters (White et al., 2009). In the same species, levels of shoot Ca and Mg vary two and twofold to threefold respectively (Broadley et al., 2008), with a range of genes contributing to the uptake and homeostasis in leaf tissue (Hammond et al., 2011; Graham et al., 2014).
In general, plant species are able to exclude most of the salt (NaCl) present in soil solution, allowing about 2% to be transported in the xylem to shoots (Munns et al., 2006). Na+ severely inhibits most enzymes at levels >100 mM, and since more than 50 enzymes require K+ as a co-factor these are sensitive to Na+ and high Na+/K+ ratios (Tester and Davenport, 2003; Munns et al., 2006). Thus, while halophytic plants continue to grow at >250 mM, a number of crops, including rice, are compromised and die if soil salinity rises to 100 mM NaCl (Munns et al., 2006). Na+ toxicity is strongly associated with a plant’s ability to maintain uptake of K+, as well as the within plant distribution (Kader and Lindberg, 2005). Rice appears to have evolved in a low salt environment with plentiful supply of fresh water (Zong et al., 2007), and so yields start to decline at 30 mM Na+, compared with wheat at between 60 and 80 mM (Munns et al., 2006).
External Ionic Conditioning and Epigenetic Variation
An early study using Medicago callus cultures indicated that while short term exposure to NaCl in had no effect on histone variant composition, it did lead to major increases in acetylation of H3.1, H3.3, and H4 (Waterborg et al., 1989). This was interpreted as an altered intra-nuclear ionic environment in the presence of salt, and possibly also representing an adaptive response in chromatin structure to permit chromatin function as Na+ increases (Waterborg et al., 1989). Since this study, a number of studies have detected an interaction between salt stress and histone modifications including both acetylation and methylation (Kim et al., 2015).
Although global DNA methylation is not significantly different between Arabidopsis shoots and roots, those regions of the genome that are differentially methylated tend to be preferentially (1.85×) hypermethylated in shoots (Widman et al., 2014), consistent with findings in rice (Karan et al., 2012). Within Arabidopsis, these sites in genes are primarily in the CG context, with a higher proportion at transcription initiation and termination boundaries. This also corresponds to a higher nucleosome density in these regions for the differentially transcribed genes, with the corresponding gene body being less nucleated. Overall it appears that roots tend to have a higher nucleosome density over genic regions and a more marked periodicity of DNA methylation (Widman et al., 2014). Moreover, genes with >10 × higher level of transcription in root tissue are more nucleosome-rich in the boundary regions compared with shoot tissue. The relationship between such observations and the prevailing intracellular thermal and ionic environments has yet to be explored. However, based on the findings in rice it has been suggested that the relative DNA hypomethylation observed in roots provides greater plasticity or preparedness for salt response genes (Karan et al., 2012).
DNA methylation has been shown to provide levels of environmental responsiveness in plant phenotypes, while providing some evolutionary flexibility in terms of heritability (Heard and Martienssen, 2014). Previous reviews have addressed a broad range of genetic and some epigenetic responses to extreme temperature or salt stress events (Madlung and Comai, 2004; Atkinson and Urwin, 2012; Mickelbart et al., 2015). Initial evidence that epigenetic mechanisms are more extensively involved in a range of plant responses to abiotic stress has come from reactivation of transgenes silenced by DNA methylation (Ito et al., 2011; Lang-Mladek et al., 2010). Thus, in tobacco the elevation of cold, salt, and metal ions all lead to hypomethylation of coding regions (Choi and Sano, 2007). The wider contribution of 5mC to management of stress responses has been revealed by the identification of large numbers of differentially methylated genomic regions, many with associated transcriptional changes, as a result of induction by stresses including bacteria and abiotic factors (Dowen et al., 2012). Although transposons may occur in these differentially methylated regions, such responses appear to be accompanied by up-regulation of 21-nt siRNAs, with many coupled to changes in transcription of the transposon itself and/or nearby genes (Dowen et al., 2012).
In soybean, salt stress has also been shown to induce hypomethylation, along with transcriptional activation of salt stress-induced TFs (Song et al., 2012). From evidence in rice, it appears that remodeling of DNA methylation may play a more general role in conditioning salt tolerance. For example, two salt tolerant genotypes have been found to have a significant level of hypermethylation compared with hypomethylation in two salt-sensitive genotypes (Feng et al., 2012). Although an independent study (Karan et al., 2012) did not find any specific methylation pattern associated with salt tolerant or susceptible genotypes under salt stress, there was a significant association with level of methylation and salt treatment in the shoot of four genotypes and in the root of two others. The authors concluded that many methylation changes associated with salinity were not directed (Karan et al., 2012), which may suggest a more generalized effect on the genome, particularly in the context of 5mCG gene-body methylation. This would be consistent with chromatin being in a more repressed (condensed) state, as well as the observation that stress leads to hypermethylation in satellite DNA at (non-genic) CHG sites within the halophyte Mesembryanthemum crystallinum (Dyachenko et al., 2006). It also appears consistent with the observation that mutations in components of the HDAC complexes reduce the ability of Arabidopsis to cope with salt and cold (Zhu et al., 2008; Chen et al., 2010), where more condensed chromatin may provide some protection against these stresses.
Thermal Physiology and Contributions to Chromatin Conformation
Thermal Physiology of Crop Plants
A wide range of crop phenotypic traits are affected by thermal environment over multiplexed time-scales, from transitory responses through diurnal, circadian, and annual cycles (Bita and Gerats, 2013). Progress through the sequential developmental phase transitions associated with the detection, initiation, onset and progression of inflorescence development is co-ordinated by integration of environmental signals (Baurle and Dean, 2006; Huijser and Schmid, 2011; Figure 2). Detection of temperature is particularly important for the regulation and integration of signals contributing to onset of flowering, including the vernalization and photoperiod pathways (Huijser and Schmid, 2011). Many annual and perennial plants have evolved to fine-tune the sensing and integration of thermal signals (Patel and Franklin, 2009), with the ability to integrate periods of prolonged exposure below critical temperatures, together with thermal responsiveness, directly affecting crop performance (Luo, 2011; Robertson et al., 2013).
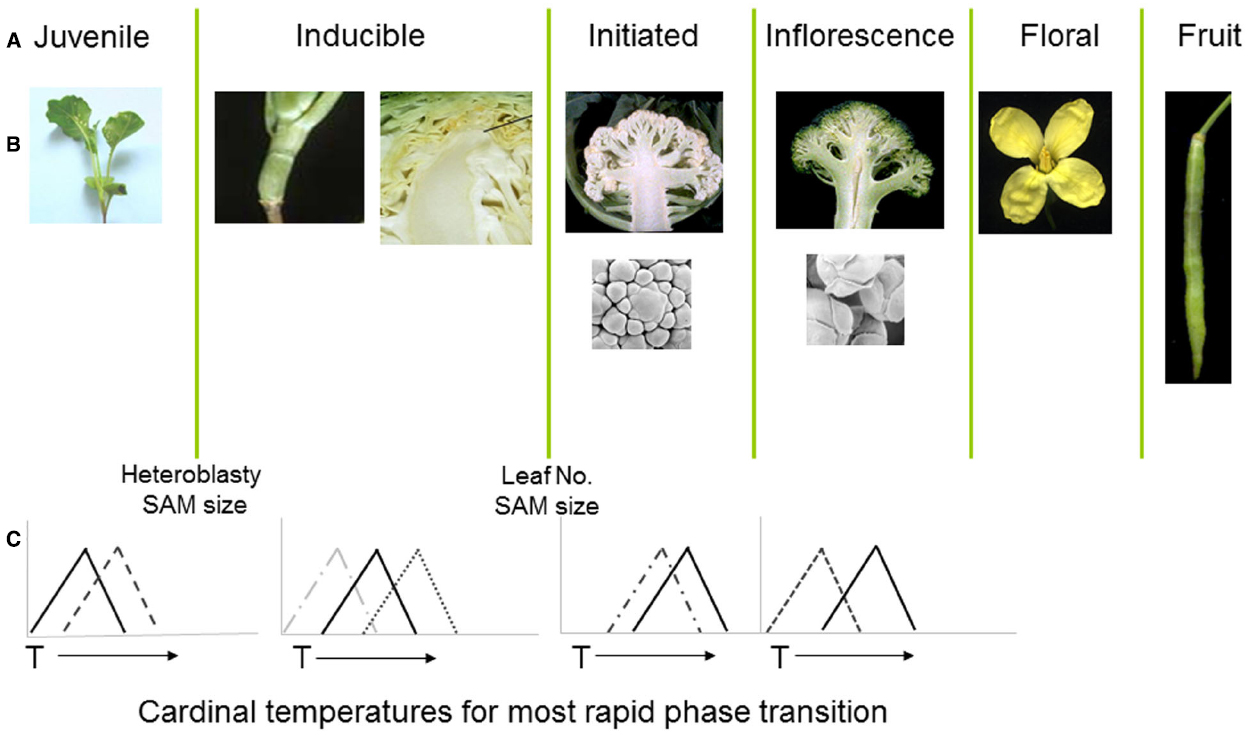
Figure 2. (A,B) Distinct developmental phases and phase transitions are clearly delineated in different genotypes of Brassica oleracea. Following floral initiation in cauliflower (var. botrytis) arrested development leads to proliferation of vegetative and inflorescence meristems, whereas in broccoli (var. italica) development is arrested at the later floral bud stage. (C) Each developmental phase may have a distinct cardinal temperature associated with an optimal rate of growth leading to the subsequent transition. X axes represent arbitrary temperature scale; Y axis relative growth rate.
The cauliflower crop (B. oleracea var. botrytis) provides a useful demonstration of the consequences of temperature variation at different phases of development, with each phase transition typically sensitive to a genotype-specific optimal temperature range (Figure 2). Following seedling emergence, the plant remains in a vegetative juvenile phase during which it is unable to detect signals to initiate floral development (Wurr et al., 1994; Guo et al., 2004). The duration of this phase is sensitive to ambient temperatures (Wurr et al., 1995) with considerable variation dependent upon genotype (Fellows et al., 1999; Wurr et al., 2004). In most cultivars, including non-winter types, the plant undergoes a vernalization phase during which the vegetative apical meristem is responsive to accumulated thermal units in a range around a “cardinal” temperature optimum, with considerable genotypic variation also associated with this cardinal temperature (Wurr et al., 2004).
The developmental program in cauliflower leads to a proliferation of vegetative and inflorescence meristems to form the harvested curd (Smith and King, 2000), which is also sensitive to temperature variation (Wurr et al., 1990; Rahman et al., 2007). Radiation of the cauliflower and related broccoli crops from their centre of diversity in Italy (Massie et al., 1999) has resulted in cultivars containing alleles able to provide distinct cardinal temperature optima for length of juvenile period, vernalization, floral initiation, curd, and inflorescence initiation (Irwin et al., 2012). For some genotypes, high temperatures at the curd arrest stage may lead to the development of bracts, with a reduction in marketable quality (Kop et al., 2003), whilst others grown at temperatures below an optimal range may develop an undesirable “ricey” phenotype (Fujime and Okuda, 1996), where the arrested floral meristems progress to a later stage in development. This diversity of responses and phenotypic consequences are indicative of sophisticated gene regulatory mechanisms that are able to manage complex thermal signals in the context of distinct phases of development.
Chromatin Responses to Thermal Variation
A range of properties associated with the chromatin macromolecular complex are affected by thermal variation. Thermal fluctuations are able to induce partial unwrapping of DNA from nucleosomes in vitro, and introduce twist or loop defects into the DNA wrapped round the core particle, resulting in repositioning in relation to the DNA (Blossey and Schiessel, 2011). Increased affinity (free energy, or enthalpy) due to association of histone proteins with DNA may also contribute to nucleosome translational or rotational positioning (Lowary and Widom, 1997), while relatively small quantities of free energy appear sufficient to precipitate association/disassociation with histone H1 (Rippe, 2012). In the absence of ATP-dependent DNA remodeling complexes, translational sliding of nucleosomes along DNA is temperature dependent, with repositioning taking place relatively slowly, at a rate of a few hours per 200 bp (Blossey and Schiessel, 2011). More generally, a range of histone modifications have been found to be associated with the heat stress response (Kim et al., 2015).
H2A.Z occupancy, especially at the TSS+1 nucleosomes of temperature-induced genes, has been shown in Arabidopsis to decrease with temperature, independent of transcription (Kumar and Wigge, 2010), so that when H2A.Z deposition is prevented, plants have a constitutive warm temperature response. Thus the canonical H2A nucleosomes do not contribute to unwrapping of DNA from the nucleosome in response to temperature, while H2A.Z nucleosomes become increasingly accessible to RNA Pol II as temperature rises. Where gene transcription decreases with temperature this may be due to H2A.Z providing greater access to binding of repressors at these loci, or by facilitating de novo DNA methylation (Kumar and Wigge, 2010).
Involvement of H2A.Z in Thermosensory Flowering
The autonomous flowering pathway was conventionally regarded as being independent of environmental signals such as photoperiod. However, mutant analysis has demonstrated that genes of this pathway are also directly involved in mediating the effects of ambient temperature (Blázquez et al., 2003), with consequent effects on the expression of FLOWERING LOCUS T (FT), the mobile integrator gene of the floral pathway.
The mechanism by which the FT locus mediates the thermosensory flowering pathway (Halliday et al., 2003; Balasubramanian et al., 2006) is now being unraveled. It appears that while H2A.Z is enriched in the promoter region of FT, it is depleted at higher temperature, providing an explanation for acceleration of flowering in Arabidopsis arp6 mutants deficient in H2A.Z (Kumar and Wigge, 2010). The chromatin modification that results from the heat-induced removal of H2A.Z from nucleosomes provides access to the FT promoter by the bHLH TF PIF4 (Kumar et al., 2012). In terms of control logic, H2A.Z is able to provide a genome-wide mechanism that is directly and rapidly coupled to temperature, and thus facilitate fine-tuning of phenotypic plasticity in response to environment.
This generic mechanism appears conserved in Brachypodium, a close relative of the major Pooideae grain crops, where H2A.Z nucleosomes appear responsible for the observed increase in thermal sensitivity of endosperm compared with vegetative tissue in the major monocot grain crops (Boden et al., 2013). Notably, H2A.Z nucleosome occupancy was more responsive to increases in ambient temperature in grain reproductive tissues, and correlated with the sensitivity to increased ambient temperature during early maturity. Thus the genomic organization of H2A.Z in Brachypodium results in limited impact of temperature on the phase transition from vegetative to reproductive stage, whilst retaining sensitivity at grain filling—a major contributor to yield in temperate grain crops. Perturbing the deposition of H2A.Z was found to be sufficient to mimic the effects of a warm temperature environment on grain development (Boden et al., 2013).
Temperature Effects on RdDM
Temperature also appears to play a role in mediating RNA silencing (Szittya et al., 2003; Romon et al., 2013; Zhong et al., 2013; Liu et al., 2015). Post transcriptional gene silencing (PTGS) is characterized by accumulation of snRNAs, targeted degradation of mRNAs and DNA methylation of target genes. This can be inhibited in Arabidopsis by increasing growth temperature from 22 to 30°C (Zhong et al., 2013), and inherited through meiosis, affecting DNA methylation status as a result of exposure to higher temperatures in the previous generation. The release of PTGS appears to be due to a reduction in the formation of dsRNA required for production of siRNAs in the RNA silencing pathway, where the temperature increase reduces abundance of SUPPRESSOR OF GENE SILENCING 3 (SGS3). When over-expressed, SGS3 can reverse the warmth-induced inhibition of siRNA biogenesis and so reduce the transgenerational epigenetic memory (Zhong et al., 2013). Moreover, temperature induced release of sense transgene-mediated PTGS is dose dependent and stochastic between 24 and 28°C, but becomes deterministic at 30°C, with associated variation in warmth-induced DNA methylation within the target transgenes.
Basal heat tolerance in Arabidopsis also involves the RdDM pathway (Popova et al., 2013), with consequences for transcription and epigenetic regulation of transposons. Arabidopsis plants defective in either NRPD2, a subunit of RNA Pol IV and V, or in HDA6, an Rpd3-type histone acetylase, are hypersensitive to heat exposure, and these genes have independent roles in transcriptional reprogramming in response to temperature stress (Popova et al., 2013).
Thermal Memory
Detection of accumulated thermal units or growing day degrees above or below a threshold has been resolved to major QTLs (Sadok et al., 2007; Bogard et al., 2014; Sánchez-Pérez et al., 2014), even in complex crop genomes such as Brassica napus (canola, rapeseed), where candidate genes at such loci appear to account for differences between over-wintering and summer crop types (Wang et al., 2012; Nelson et al., 2014). Natural flowering responses in Arabidopsis have also been localized to the cis-regulatory regions of the FT locus (Schwartz et al., 2009b). In B. napus, six copies of FT appear to have contributed to more complex mechanisms of floral regulation and niche adaptation compared to Arabidopsis (Wang et al., 2012) with promoter analysis (Wang et al., 2012) indicating that one copy (FT-C2) has been repressed by transposon insertion, with high levels of 5mC in both B. napus and the ancestral B. oleracea. Meanwhile the FT-A7/C6 homologs are specifically silenced in winter type B. napus, but abundantly expressed in spring type cultivars under vernalization-free conditions.
In Arabidopsis, dissection of the molecular mechanisms underlying vernalization has uncovered the role of epigenetic marks, particularly the polycomb-mediated additive effect of histone modifications, which under cold conditions regulate silencing of the flowering repressor FLOWERING LOCUS C (FLC, Sheldon et al., 2009; Romera-Branchat et al., 2014). Subsequently, it has been found that long non-coding RNAs (lncRNAs) COOLAIR (Swiezewski et al., 2009) and COLDAIR (Heo and Sung, 2011) are embedded within the FLC locus, and also induced during vernalization in Arabidopsis by periods of cold. COLDAIR has been proposed to recruit the H3K27me3 mark to the FLC gene, thus contributing to FLC repression (Heo and Sung, 2011), whereas some alternatively spliced isoforms of COOLAIR may contribute to activation of FLC (Csorba et al., 2014; Romera-Branchat et al., 2014).
Temperature Conditioning and Epigenetic Variation
An early study by Burn et al. (1993) found that cold temperatures lead to hypomethylation in Arabidopsis and a Nicotiana cell line. Subsequently Finnegan et al. (1998) studied the effect of imbibing Arabidopsis C24 seed for 4 weeks at 8°C, and detected a large (86%) albeit transitory effect of hypomethylation in mature leaves compared with untreated controls at the same stage of development. These phenomena appeared to be reversible, as after 7 days growth at 22°C DNA methylation in seedlings developed from the vernalized controls was comparable to those of control seedlings. Cold stress has since been found to lead to genome-wide demethylation in maize seedlings (Steward et al., 2002), while growth of Antirrhinum majus in low-temperature conditions results in hypomethylation of the transposon Tam3 (Hashida et al., 2006). A related phenomenon is also observed with heat-stress, where inducible alterations in endogenous loci generally lead to hypomethylation of retro-elements, with depression of transcription along with transient changes in nucleosome density (Lang-Mladek et al., 2010; Pecinka et al., 2010; Ito et al., 2011).
Histone methylation at H3K27me3 has also been shown to decrease gradually during cold exposure in two Arabidopsis cold-responsive genes COR15A and ATGOLS3 (Kwon et al., 2009). It appears that in this case gene activation leads to removal of H3K27me3 and that this mark is able to be inherited quantitatively, providing a memory of recent transcriptional activity.
A Speculative Framework: Electrostatic and Epigenetic Interactions within the Plant Nucleus
Our current understanding of the direct involvement of ionic and temperature variation on chromatin structure and transcription is fragmentary. However, it is clear that some key biophysical properties of chromatin components and epigenetic marks are affected by electrostatic and thermal interactions, and these more fundamental observations are starting to align with observations at cellular and whole organism level. In order to unravel these relationships, experimental approaches need to distinguish between direct effects and those mediated by signal transduction pathways that sense variation in external ionic or temperature environment and are then apparent at the level of epigenetic modifications.
Few studies have systematically evaluated the ensemble of interactions that place the ionic and thermal environment, the biophysical attributes of 5mC, H2A.Z and other epigenetic marks in the context of chromatin dynamics and genomic regulation (McClung and Davis, 2010). Thus, at present there is no cohesive model that takes into account the contribution made by each of the distinct epigenetic marks to chromatin conformation, and the ability of plants to maintain complex genomic regulation under fluctuating external environmental conditions. However, from the information presented in this review it is clear that temperature and ionic conditions both play an important role in determining the biophysical behavior of histone and DNA macromolecules, their interaction in forming nucleosomes, and in higher order chromatin conformation.
Taken together, the various lines of evidence outlined here appear to be internally consistent in describing contributions to accessible versus inaccessible chromatin. The framework that emerges is based on what appears to be a cohesive set of interactions at molecular, biophysical and electrostatic level between the various components that affect chromatin conformation and dynamics. This is represented in a simple schematic (Figure 1) that outlines the behavior of the key components of DNA, histones, and nucleosomes in the context of epigenetic marks and ionic environment within the plant nucleus. From this set of interactions, it is possible to speculate that within plant nuclei, general and localized ionic homeostasis plays a significant important role in maintaining chromatin conformation, whilst maintaining complex genomic regulation involving specific patterns of epigenetic marks.
The contributions of 5mC to local DNA stability and reduced flexibility appear to be consistent with the association of 5mC with stable, more ordered nucleosomes and localized transcription, with denser methylation leading to tighter chromatin and gene repression. The complementary contributions of 5mC and H2A.Z to less accessible chromatin is consistent with their observed relative mutual exclusivity in chromatin (Zilberman et al., 2008), and H2A.Z providing the transcriptionally responsive mark in response to external temperature (Kumar and Wigge, 2010).
Complex electrostatic interactions within the nucleus contribute to the condensation state of chromatin, with the localized net charge state of the interaction between DNA and histone affecting position and stability of nucleosomes. Post-translational histone modifications play a major role, although it is currently unclear how these may be affected by the proposed differential roles played by Na+ and K+ in chromatin condensation (Allahverdi et al., 2015). The latter findings have yet to be incorporated into our understanding of ionic variation in plants, and the dearth of knowledge about the ionic environment of plant nuclei, although Na+ and K+ gradients are observed within some eukaryote nuclei (Garner, 2002). Given the huge resources devoted to K fertilizer use of around 30 mt per annum (Timilsena et al., 2014) and concerns about salinization of cultivated land, it would seem timely to explore these phenomena in more detail at a molecular level to understand mineral ion availability in the nuclei of crop plants.
Whilst tentative, this framework provides scope to develop experimental approaches to understanding in greater detail the internal environment of plant nuclei. It is hoped that this will generate a deeper understanding of the molecular mechanisms underlying genotype × environment interactions that may be beneficial for long-term improvement of crop performance in less predictable climates.
Concluding Remarks
A high proportion of crop traits exhibit quantitative inheritance, many with relatively low penetrance. For example, in rapeseed hundreds of significant environment-specific QTL have been identified for yield and other traits in a “fixed” segregating population of homozygous lines grown in 10 environments, with relatively few coinciding in multiple environments (Shi et al., 2009). In many cases the extensive genotype × environment interactions associated with crop yield traits has limited the ability to identify underlying genes. However, it is now apparent that QTL may also be accounted for by changes in DNA methylation status, with, e.g., 60–90% of heritability for the complex traits of flowering time and primary root length being detected in epiRILs of Arabidopsis (Cortijo et al., 2014). It is worth noting that in Arabidopsis, spontaneous transgenerational epiallelic variation can occur at a rate 103 times higher than the genetic mutation rate (Becker et al., 2011), with hypermethylated alleles associated with siRNA production and TGS (Schmitz et al., 2011). This stochastic generation of epialleles has the potential to alter transcriptional behavior and generate novel phenotypic variation subject to selection. Thus formation of random epialleles mediated by RdDM may be of more significance than genetic variation (Matzke and Mosher, 2014). For crop breeding, there are clear indications that such variation needs to be under active selection, and attention given to maintenance of germplasm to ensure that epigenetic plasticity is hard-wired into new cultivars.
At present we have only a partial understanding of how the various epigenetic components confer dynamic functional information content in the context of the ionic and thermal environment of the nucleus. It is becoming clear that most regions of complex eukaryotic genomes play some role in gene regulation (Haudry et al., 2013). However, there is a need for systematic analysis of the relationships between plant genome complexity, the known taxonomic variation in ionic composition, the distribution of epigenetic marks and measures of genome ruggedness or plasticity. For example, does the relatively compact genome of rice, with less scope for redundancy in epigenetic regulation, also contribute to its inability to manage more extreme and complex abiotic stresses?
The ability of plants to accommodate fluctuations in thermal and ionic environment is an essential fitness attribute and a key determinant for crop performance, and requires a deeper understanding of the interactions at intra-cellular and intra-nuclear level, including those with epigenetic marks and processes. The availability of comprehensive tissue-specific epi-genome, nucleosome and snRNA datasets will contribute to more comprehensive models of interactions between genome organization, chromatin dynamics, and epigenetic signaling systems. These can help provide new tools and approaches for breeding selection and agronomic management of crops able to perform in changeable environments.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by Southern Cross University, Australia and Hubei Province Chutian Scholar programme, China.
References
Acosta-Silva, C., Branchadell, V., Bertran, J., and Oliva, A. (2010). Mutual relationship between stacking and hydrogen bonding in DNA. Theoretical study of guanine-cytosine, guanine-5-methylcytosine, and their dimers. J. Phys. Chem. B 114, 10217–10227. doi: 10.1021/jp103850h
Alatzas, A., Srebreva, L., and Foundouli, A. (2008). Distribution of linker histone variants during plant cell differentiation in the developmental zones of the maize root, dedifferentiation in callus culture after auxin treatment. Biol. Res. 41, 205–215. doi: 10.4067/s0716-97602008000200010
Alekseeva, V. T., Tsentsevitsky, A. N., Loseva, N. L., Gordon, L. K., and Rakhimova, G. G. (2007). Energy expenditure induced by changes in ion homeostasis of wheat root cells. Thermoch. Acta. 458, 97–100. doi: 10.1016/j.tca.2007.03.005
Allahverdi, A., Chen, Q., Korolev, N., and Nordenskold, L. (2015). Chromatin compaction under mixed salt conditions, opposite effects of sodium and potassium ions on nucleosome array folding. Sci. Rep. 5, 8512. doi: 10.1038/srep08512
Anselmi, C., Bocchinfuso, G., Santis, P. D., Savino, M., and Scipioni, A. (2000). A theoretical model for the prediction of sequence-dependent nucleosome thermodynamic stability. Biophys. J. 7, 601–613. doi: 10.1016/S0006-3495(00)76319-3
Arya, G., and Schlick, T. (2009). A tale of tails: how histone tails mediate chromatin compaction in different salt and linker histone environments. J. Phys. Chem. A 113, 4045–59. doi: 10.1021/jp810375d
Ascenzi, R., and Gantt, J. S. (1999). Molecular genetic analysis of the drought-inducible linker histone variant in Arabidopsis thaliana. Plant Mol. Biol. 41, 159–169. doi: 10.1023/A:1006302330879
Atkinson, N. J., and Urwin, P. E. (2012). The interaction of plant biotic and abiotic stresses, from genes to the field. J. Exp. Bot. 63, 3523–3543. doi: 10.1093/jxb/ers100
Balasubramanian, S., Sureshkumar, S., Lempe, J., and Weigel, D. (2006). Potent induction of Arabidopsis thaliana flowering by elevated growth temperature. PLoS Genet. 2:e106. doi: 10.1371/journal.pgen.0020106
Bancaud, A., Huet, S., Daigle, N., Mozziconacci, J., Beaudouin, J., and Ellenberg, J. (2009). Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organisation of chromatin. EMBO J. 28, 3785–3798. doi: 10.1038/emboj.2009.340
Bancaud, A., Lavelle, C., Huet, S., and Ellenberg, J. A. (2012). Fractal model for nuclear organization: current evidence and biological implications. Nucleic Acids Res. 40, 8783–8792. doi: 10.1093/nar/gks586
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modification. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Baulcombe, D. C., and Dean, C. (2014). Epigenetic regulation in plant responses to the environment. Cold Spring Harb. Perspect. Biol. 6, a019471. doi: 10.1101/cshperspect.a019471
Baurle, I., and Dean, C. (2006). The timing of developmental transitions in plants. Cell 125, 655–664. doi: 10.1016/j.cell.2006.05.005
Becker, C., Hagmann, J., Müller, J., Koenig, D., Stegle, O., Borgwardt, K., et al. (2011). Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480, 245–249. doi: 10.1038/nature10555
Bednar, J., Horowitz, R. A., Grigoryev, S. A., Carruthers, L. M., Hansen, J. C., Koster, A. J., et al. (1998). Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. U.S.A. 95, 14173–14178. doi: 10.1073/pnas.95.24.14173
Bertin, A., Mangenot, S., Renouard, M., and Livolant, F. (2007). Structure and phase diagram of nucleo, some core particles aggregated by multivalent cations. Biophys. J. 93, 3652–3653. doi: 10.1529/biophysj.107.108365
Bita, C. E., and Gerats, T. (2013). Plant tolerance to high temperature in a changing environment, scientific fundamentals and production of heat stress-tolerant crops. Front. Plant Sci. 4:273. doi: 10.3389/fpls.2013.00273
Blanchard, M. G., and Runkle, E. S. (2011). The influence of day and night temperature fluctuations on growth and flowering of annual bedding plants and greenhouse heating cost predictions. Hortscience 46, 599–603.
Blázquez, M. A., Ahn, J. H., and Weigel, D. (2003). A thermosensory pathway controlling flowering time in Arabidopsis thaliana. Nat. Genet. 33, 168–171. doi: 10.1038/ng1085
Bloomfield, J., Rose, T. J., and King, G. J. (2014). Sustainable harvest: managing plasticity for resilient crops. Plant Biotech. J. 12, 517–533. doi: 10.1111/pbi.12198
Blossey, R., and Schiessel, H. (2011). The dynamics of the nucleosome, thermal effects, external forces and ATP. FEBS J. 278, 3619–3632. doi: 10.1111/j.1742-4658.2011.08283.x
Boden, S. A., Kavanová, M., Finnegan, E. J., and Wigge, P. A. (2013). Thermal stress effects on grain yield in Brachypodium distachyon occur via H2A.Z-nucleosomes. Genome Biol. 14, R65. doi: 10.1186/gb-2013-14-6-r65
Bogard, M., Ravel, C., Paux, E., Bordes, J., Balfourier, F., Chapman, S. C., et al. (2014). Predictions of heading date in bread wheat (Triticum aestivum L.) using QTL-based parameters of an ecophysiological model. J. Exp. Bot. 65, 5849–5865. doi: 10.1093/jxb/eru328
Bowman, G. D., and Poirier, M. G. (2015). Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev. 115, 2274–2295. doi: 10.1021/cr500350x
Boyko, A., Blevins, T., Yao, Y., Golubov, A., Bilichak, A., Ilnytskyy, Y., et al. (2010). Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS ONE 5:e9514. doi: 10.1371/journal.pone.0009514
Bräutigam, K., Vining, K. J., Lafon-Placette, C., Fossdal, C. G., Mirouze, M., Marcos, J. G., et al. (2013). Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol. Evol. 3, 399–415. doi: 10.1002/ece3.461
Broadley, M. R., Bowen, H. C., Cotterill, H. L., Hammond, J. P., Meacham, M. C., Mead, A., et al. (2004). Phylogenetic variation in the shoot mineral concentration of angiosperms. J. Exp. Bot. 55, 321–336. doi: 10.1093/jxb/erh002
Broadley, M. R., Hammond, J. P., King, G. J., Astley, D., Bowen, H. C., Meacham, M. C., et al. (2008). Shoot calcium (Ca) and magnesium (Mg) concentrations differ between subtaxa, are highly heritable, and associate with potentially pleiotropic loci in Brassica oleracea. Plant Physiol. 146, 1707–1720. doi: 10.1104/pp.107.114645
Brown, D. T., Izard, T., and Misteli, T. (2006). Mapping the interaction surface of linker histone H1 with the nucleosome of native chromosome in vivo. Nat. Struct. Mol. Biol. 13, 250–255. doi: 10.1038/nsmb1050
Burn, J. E., Bagnall, D. J., Metzger, J. D., Dennis, E. S., and Peacock, W. J. (1993). DNA methylation, vernalization, and the initiation of flowering. Proc. Natl. Acad. Sci. U.S.A. 90, 287–291. doi: 10.1073/pnas.90.1.287
Buttinelli, M., Minnock, A., Panetta, G., Waring, M., and Travers, A. (1998). The exocyclic groups of DNA modulate the affinity and positioning of the histone octamer. Proc. Natl. Acad. Sci. U.S.A. 95, 8544. doi: 10.1073/pnas.95.15.8544
Cameron, I. L. (1985). Electron probe x-ray microanalysis studies on the ionic environment of nuclei and the maintenance of chromatin structure. Prof. Clin. Biol. Res. 196, 223–239.
Caño, S., Caravaca, J. M., Martín, M., and Daban, J. R. (2006). Highly compact folding of chromatin induced by cellular cation concentrations. Evidence from atomic force microscopy studies in aqueous solution. Eur. Biophys. J. 35, 495–501. doi: 10.1007/s00249-006-0057-7
Cavalier-Smith, T. (1985). “Cell volume and the evolution of eukaryotic genome size,” in The Evolution of Genome Size, ed. T. Cavalier-Smith (Chichester: John Wiley & Sons Ltd.), 105–184.
Chakravarthy, S., Gundimella, S. K., Caron, C., Perche, P. Y., Pehrson, J. R., Khochbin, S., et al. (2005). Structural characterisations of the histone variant macro-H2A preferentially forms “hybrid nucleosomes”. J. Biol. Chem. 281, 25522–25531. doi: 10.1074/jbc.M602258200
Charpentier, M., Bredemeier, R., Wanner, G., Takeda, N., Schleiff, E., and Parniske, M. (2008). Lotus japonicus CASTOR and POLLUX are ion channels essential for perinuclear calcium spiking in legume root endosymbiosis. Plant Cell 20, 3467–3479. doi: 10.1105/tpc.108.063255
Charron, J.-B. F., He, H., Elling, A. A., and Deng, X. W. (2009). Dynamic landscapes of four histone modifications during deetiolation in Arabidopsis. Plant Cell 21, 3732–3748. doi: 10.1105/tpc.109.066845
Chen, K., and Rajewsky, N. (2014). The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 8, 93–103. doi: 10.1038/nrg1990
Chen, L. T., Luo, M., Wang, Y. Y., and Wu, K. (2010). Involvement of Arabidopsis histone deacetylase HDA6 in ABA and salt stress response. J. Exp. Bot. 61, 3345–3353. doi: 10.1093/jxb/erq154
Cheng, Y., Korolev, N., and Nordenskiold, L. (2006). Similarities and differences in interaction of K+ and Na+ with condensed ordered DNA. A molecular dynamics computer simulation study. Nucleic Acids Res. 34, 686–696. doi: 10.1093/nar/gkj434
Chodavarapu, R. K., Feng, S., Bernatavichute, Y. V., Chen, P. Y., Stroud, H., Yu, Y., et al. (2010). Relationship between nucleosome positioning and DNA methylation. Nature 466, 388–392. doi: 10.1038/nature09147
Choi, C. S., and Sano, H. (2007). Abiotic-stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase-like protein in tobacco plants. Mol. Genet. Genomics 277, 689–600. doi: 10.1007/s00438-007-0209-1
Cokus, S. J., Feng, S., Zhang, X., Chen, Z., Merriman, B., Haudenschild, C. D., et al. (2008). Shotgun bisulfite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219. doi: 10.1038/nature06745
Cortijo, S., Wardenaar, R., Colomé-Tatché, M., Gilly, A., Etcheverry, M., Labadie, K., et al. (2014). Mapping the epigenetic basis of complex traits. Science 343, 1145–1148. doi: 10.1126/science.1248127
Cramer, G. R., Urano, K., Delrot, S., Pezzotti, M., and Shinozaki, K. (2011). Effects of abiotic stress on plants, a systems biology perspective. BMC Plant Biol. 11:163. doi: 10.1186/1471-2229-11-163
Craufurd, P. Q., and Wheeler, T. R. (2009). Climate change and the flowering time of annual crops. J. Exp. Bot. 60, 2529–2539. doi: 10.1093/jxb/erp196
Csorba, T., Questa, J. I., Sun, Q., and Dean, C. (2014). Antisense COOLAIR mediates the co-ordinated switching of chromatin states at FLC during vernalization. Proc. Natl. Acad. Sci. U.S.A. 111, 16160–16165. doi: 10.1073/pnas.1419030111
Cui, F., and Zhurkin, V. B. (2009). Distinctive sequence patterns in metazoan and yeast nucleosomes: implications for linker histone binding to AT-rich and methylated DNA. Nucleic Acids Res. 37, 2818–2829. doi: 10.1093/nar/gkp113
Dantas Machado, A. C., Zhou, T., Rao, S., Goel, P., Rastogi, C., Lazarovici, A., et al. (2015). Evolving insights on how cytosine methylation affects protein-DNA binding. Brief. Funct. Genomics 14, 61–73. doi: 10.1093/bfgp/elu040
Dechassa, M. L., and Luger, K. (2012). “Nucleosomes as control elements for accessing the genome,” in Genome Organisation and Function in the Cell Nucleus, ed. K. Rippe (Heidelberg: Wiley-VCH), 55–75.
Dowen, R. H., Pelizzola, M., Schmitz, R. J., Lister, R., Dowen, J. M., Nery, J. R., et al. (2012). Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. U.S.A. 109, E2183–E2191. doi: 10.1073/pnas.1209329109
Doyen, C. M., Montel, F., Gautier, T., Menoni, H., Claudet, C., Delacour-Larose, M., et al. (2006). Dissection of the unusual structure and functional properties of the variant H2A.Bbd nucleosome. EMBO J. 25, 4234–4244. doi: 10.1038/sj.emboj.7601310
Draguhn, A., Borner, G., Beckman, R., Buchner, K., Heinmann, U., and Hucho, F. (1997). Large conductance cation channels in the envelope of nuclei from rat cerebral cortex. J. Membr. Biol. 158, 159–166. doi: 10.1007/s002329900253
Dyachenko, O. V., Zakharchenko, N. S., Shevchuk, T. V., Bohnert, H. J., Cushman, J. C., and Buryanov, Y. I. (2006). Effect of hypermethylation of CCWGG sequences in DNA of Mesembryanthemum crystallinum plants on their adaptation to salt stress. Biochemistry (Mosc.) 71, 461–465. doi: 10.1134/S000629790604016X
Eberharter, A., and Becker, P. B. (2002). Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 3, 224–229. doi: 10.1093/embo-reports/kvf053
Erdmann, R. M., Souza, A. L., Clish, C. B., and Gehring, M. (2014). 5-hydroxymethylcytosine is not present in appreciable quantities in Arabidopsis DNA. G3 (Bethesda) 5, 1–8. doi: 10.1534/g3.114.014670
Fan, J. Y., Gordon, F., Luger, K., Hansen, J. C., and Tremethick, D. J. (2002). The essential histone variant H2A.Z regulates the equilibrium between different chromatin conformational states. Nat. Struct. Biol. 9, 172–176. doi: 10.1038/nsb0402-316b
Fellows, J. R., Wurr, D. C. E., Phelps, K., and Reader, R. J. (1999). Initiation of early summer cauliflowers in response to temperature. J. Hort. Sci. Biotechnol. 74, 328–336.
Feng, Q. Z., Yang, C. W., and Lin, X. Y. (2012). Salt and alkaline stress induced transgenerational alteration in DNA methylation of rice (Oryza sativa). Aust. J. Crop Sci. 6, 877–883.
Feng, S., Jacobsen, S. E., and Reik, W. (2010). Epigenetic reprogramming in plant and animal development. Science 330, 622–627. doi: 10.1126/science.1190614
Fenley, A. T., Adams, D. A., and Onufriev, A. V. (2010). Charge state of the globular histone core controls stability of the nucleosome. Biophys. J. 99, 1577–1585. doi: 10.1016/j.bpj.2010.06.046
Finnegan, E. J., Genger, R. K., Kovac, K., Peacock, W. J., and Dennis, E. S. (1998). DNA methylation and the promotion of flowering by vernalization. Proc. Natl. Acad. Sci. U.S.A. 95, 5824–5829. doi: 10.1073/pnas.95.10.5824
Franco-Obregon, A., Wang, H.-W., and Clapham, D. E. (2000). Distinct ion channel classes are expressed on the outer nuclear envelope of T- and B-lymphocyte cell lines. Biophys. J. 79, 202–214. doi: 10.1016/S0006-3495(00)76284-9
Franco-Zorrilla, J. M., López-Vidriero, I., Carrasco, J. L., Godoy, M., Vera, P., and Solano, R. (2014). DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc. Natl. Acad. Sci. U.S.A.111, 2367–2372. doi: 10.1073/pnas.1316278111
Fritsch, C. C., and Langowski, J. (2010). Anomalous diffusion in the interphase cell nucleus: the effect of spatial correlations of chromatin. J. Chem. Phys. 133, 025101. doi: 10.1063/1.3435345
Fujime, N., and Okuda, N. (1996). The physiology of flowering in Brassicas, especially about cauliflower and broccoli. Acta Hortic. 407, 247–254. doi: 10.17660/ActaHortic.1996.407.30
Gan, H. H., and Schlick, T. (2010). Chromatin ionic atmosphere analyzed by a mesoscale electrostatic approach. Biophys. J. 99, 2587–2596. doi: 10.1016/j.bpj.2010.08.023
Gansen, A., Tóth, K., Schwarz, N., and Langowski, J. (2009). Structural variability of nucleosomes detected by single-pair Förster resonance energy transfer, histone acetylation, sequence variation, and salt effects. J. Phys. Chem. B 113, 2604–2613. doi: 10.1021/jp7114737
Garcia-Ramirez, M., Rocchini, C., and Ausio, J. (1995). Modulation of chromatin folding by histone acetylation. J. Biol. Chem. 270, 17923–17928. doi: 10.1074/jbc.270.30.17923
Garner, M. H. (2002). Na, K-ATPase in the nuclear envelope regulates Na+, K+ gradients in hepatocyte nuclei. J. Membr. Biol. 187, 97–115. doi: 10.1007/s00232-001-0155-5
Gelfman, S., Cohen, N., Yearim, A., and Ast, G. (2013). DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res. 23, 789–799. doi: 10.1101/gr.143503.112
Gepts, P. (2008). “Tropical environments, biodiversity, and the origin of crops,” in Genomics of Tropical Crop Plants, eds P. H. Moore, and R. Ming (New York: Springer-Verlag), 1–20.
Gervais, A. L., and Gaudreau, L. (2009). Discriminating nucleosomes containing histone H2A.Z or H2A based on genetic and epigenetic information. BMC Mol. Biol. 10:18. doi: 10.1186/1471-2199-10-18
Gilbert, N., Thomson, I., Boyle, S., Sllan, J., Ramsahoye, B., and Bickmore, W. A. (2007). DNA methylation affects nuclear organisation, histone modifications and linker histone binding but not chromatin compaction. J. Cell Biol. 177, 401–411. doi: 10.1083/jcb.200607133
Gong, Z., and Zhu, J.-K. (2011). Active DNA demethylation by oxidation and repair. Cell Res. 21, 1649–1651. doi: 10.1038/cr.2011.140
Gourdji, S. M., Mathews, K. L., Reynolds, M. P., Crossa, J., and Lobell, D. B. (2013). An assessment of wheat yield sensitivity and breeding gains in hot environments. Proc. Biol. Sci. 280, 20122190. doi: 10.1098/rspb.2012.2190
Graham, N. S., Hammond, J. P., Lysenko, A., Mayes, S., O Lochlainn, S., Blasco, B., et al. (2014). Genetical and comparative genomics of Brassica under altered Ca supply identifies Arabidopsis Ca-transporter orthologues. Plant Cell 26, 2818–2830. doi: 10.1105/tpc.114.128603
Guo, D. P., Shah, G. A., Zheng, G. W., and Zheng, S. J. (2004). The interaction of plant growth regulators and vernalization on the growth and flowering of cauliflower (Brassica oleracea var. botrytis). Plant Growth Reg. 43, 163–171. doi: 10.1023/B:GROW.0000040114.84122.11
Gutierrez-Marcos, J. F., and Dickinson, H. G. (2012). Epigenetic reprogramming in plant reproductive lineages. Plant Cell Physiol. 53, 817–823. doi: 10.1093/pcp/pcs052
Halliday, K. J., Salter, M. G., Thingnaes, E., and Whitelam, G. C. (2003). Phytochrome control of flowering is temperature sensitive and correlates with expression of the floral integrator FT. Plant J. 33, 875–885. doi: 10.1046/j.1365-313X.2003.01674.x
Hammond, J. P., Mayes, S., Bowen, H. C., Graham, N. S., Hayden, R. M., Love, C. G., et al. (2011). Regulatory hotspots are associated with plant gene expression under varying soil phosphorus (P). supply in Brassica rapa. Plant Physiol. 156, 1230–1241. doi: 10.1104/pp.111.175612
Hancock, R. (2012a). “The crowded environment of the genome,” in Genome Organisation and Function in the Cell Nucleus, ed. K. Rippe (Heidelberg: Wiley-VCH), 169–184.
Hancock, R. (2012b). Structure of metaphase chromosomes: a role for effects of macromolecular crowding. PLoS ONE 7:e36045. doi: 10.1371/journal.pone.0036045
Hancock, R., and Hadj-Sahraoui, Y. (2009). Isolation of cell nuclei using inert macromolecules to mimic the crowded cytoplasm. PLoS ONE 4:e7560. doi: 10.1371/journal.pone.0007560
Harada, H., and Leigh, R. A. (2006). Genetic mapping of natural variation in potassium concentrations in shoots of Arabidopsis thaliana. J. Exp. Bot. 57, 953–960. doi: 10.1093/jxb/erj081
Hashida, S. N., Uchiyama, T., Martin, C., Kishima, Y., Sano, Y., and Mikami, T. (2006). The temperature-dependent change in methylation of the Antirrhinum transposon Tam3 is controlled by the activity of its tranposase. Plant Cell 18, 104–118. doi: 10.1105/tpc.105.037655
Haudry, A., Platts, A. E., Vello, E., Hoen, D. R., Leclercq, M., Williamson, R. J., et al. (2013). An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat. Genet. 45, 891–898. doi: 10.1038/ng.2684
Hausheer, F. H., Rao, S. N., Gamcsik, M. P., Kollman, P. A., Colvin, O. M., Saxe, J. D., et al. (1989). Computational analysis of structural and energetic consequences of DNA methylation. Carcinogenesis 10, 1131–1137. doi: 10.1093/carcin/10.6.1131
He, C., Chen, X., Huang, H., and Xu, L. (2012). Reprogramming of H3K27me3 is critical for acquisition of pluripotency from cultured Arabidopsis tissues. PLoS Genet. 8:e1002911. doi: 10.1371/journal.pgen.1002911
Heard, E., and Martienssen, R. A. (2014). Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157, 95–109. doi: 10.1016/j.cell.2014.02.045
Heo, J. B., and Sung, S. (2011). Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 331, 76–79. doi: 10.1126/science.1197349
Hughes, A. L., Jin, Y., Rando, O. J., and Struhl, K. (2012). A functional evolutionary approach to identify determinants of nucleosome positioning: a unifying model for establishing the genome-wide pattern. Mol. Cell 48, 5–15. doi: 10.1016/j.molcel.2012.07.003
Huijser, P., and Schmid, M. (2011). The control of developmental phase transitions in plants. Development 138, 4117–4129. doi: 10.1242/dev.063511
Hüve, K., Bichele, I., Tobias, M., and Niinemets, U. (2006). Heat sensitivity of photosynthetic electron transport varies during the day due to changes in sugars and osmotic potential. Plant Cell Environ. 29, 212–228. doi: 10.1111/j.1365-3040.2005.01414.x
Irwin, J. A., Lister, C., Soumpourou, E., Zhang, Y., Howell, E. C., Teakle, G., et al. (2012). Functional alleles of the flowering time regulator FRIGIDA in the Brassica oleracea genome. BMC Plant Biol. 12:21. doi: 10.1186/1471-2229-12-21
Ito, H., Gaubert, H., Bucher, E., Mirouze, M., Vaillant, I., and Paszkowski, J. (2011). An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 472, 115–119. doi: 10.1038/nature09861
Ivashuta, S., Banks, I. R., Wiggins, B. E., Zhang, Y., Ziegler, T. E., Roberts, J. K., et al. (2011). Regulation of gene expression in plants through miRNA inactivation. PLoS ONE 6:e21330. doi: 10.1371/journal.pone.0021330
Iwasaki, W., Miya, Y., Horikoshi, N., Osakabe, A., Taguchi, H., Tachiwana, H., et al. (2013). Contribution of histone N-terminal tails to the structure and stability of nucleosomes. FEBS Open Biol. 3, 363–369. doi: 10.1016/j.fob.2013.08.007
Jang, H., Shin, H., Eichman, B. F., and Huh, J. H. (2014). Excision of 5-hydroxymethylcytosine by DEMETER family DNA glycosylases. Biochem. Biophys. Res. Commun. 446, 1067–1072. doi: 10.1016/j.bbrc.2014.03.060
Jerzmanowski, A., Przewłoka, M., and Grasser, K. D. (2000). Linker histones and HMG1 proteins of higher plants. Plant Biol. 2, 586–597. doi: 10.1055/s-2000-16648
Jin, C., and Felsenfeld, G. (2007). Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 21, 1519–1529. doi: 10.1101/gad.1547707
Jones, H. G., and Rotenberg, E. (2001). “Energy, radiation and temperature regulation in plants,” in eLS (Chichester: John Wiley & Sons Ltd.), 1–8. doi: 10.1002/9780470015902.a0003199.pub2
Kader, M. A., and Lindberg, S. (2005). Uptake of sodium in protoplasts of salt-sensitive and salt-tolerant cultivars of rice, Oryza sativa L., determined by the fluorescent dye SBFI. J. Exp. Bot. 56, 3149–3158. doi: 10.1093/jxb/eri312
Karan, R., DeLeon, T., Biradar, H., and Subudh, P. K. (2012). Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS ONE 7:e40203. doi: 10.1371/journal.pone.0040203
Kim, J. M., Sasaki, T., Ueda, M., Sako, K., and Seki, M. (2015). Chromatin changes in response to drought, salinity, heat, and cold stresses in plants. Front. Plant Sci. 6:114. doi: 10.3389/fpls.2015.00114
King, G. J. (2002). Through a genome, darkly: comparative analysis of plant chromosomal DNA. Plant Mol. Biol. 48, 5–20. doi: 10.1023/A:1013705704925
Kissoudis, C., van de Wiel, C., Visser, R. G., and van der Linden, G. (2014). Enhancing crop resilience to combined abiotic and biotic stress through the dissection of physiological and molecular crosstalk. Front. Plant Sci. 5:207. doi: 10.3389/fpls.2014.00207
Kop, E. P., Teakle, G. R., McClenaghan, E. R., Lynn, J., and King, G. J. (2003). Molecular genetic analysis of the bracting trait in cauliflower and broccoli. Plant Sci. 164, 803–808. doi: 10.1016/S0168-9452(03)00068-2
Kulcheski, F. R., Côrrea, R., Gomes, I. A., de Lima, J. C., and Margis, R. (2015). NPK macronutrients and microRNA homeostasis. Front. Plant Sci. 16:451. doi: 10.3389/fpls.2015.00451
Kumar, S. V., Lucyshyn, D., Jaeger, K. E., Alós, E., Alvey, E., Harberd, N. P., et al. (2012). Transcription factor PIF4 controls the thermosensory activation of flowering. Nature 484, 242–245. doi: 10.1038/nature10928
Kumar, S. V., and Wigge, P. A. (2010). H2A.Z-containing nucleosomes mediate the thermosensory response in Arabidopsis. Cell 140, 136–147. doi: 10.1016/j.cell.2009.11.006
Kwon, C. S., Lee, D., Choi, G., and Ching, W. (2009). Histone occupancy-dependent and independent removal of H3K27 trimethylation at cold-responsive genes in Arabidopsis. Plant J. 60, 112–121. doi: 10.1111/j.1365-313X.2009.03938.x
Lang-Mladek, C., Popova, O., Klok, K., Berlinger, M., Rakic, B., Aufstatz, W., et al. (2010). Transgenerational inheritance and resetting of stress-induced loss of epigenetic gene silencing in Arabidopsis. Mol. Plant 3, 594–602. doi: 10.1093/mp/ssq014
Law, C. N. (1963). An effect of potassium on chiasma frequency and recombination. Genetics 33, 313–329. doi: 10.1007/bf01725768
Le Bot, J., Adamowicz, S., and Robin, P. (1998). Modelling plant nutrition of horticultural crops: a review. Sci. Hortic. 74, 47–82. doi: 10.1016/S0304-4238(98)00082-X
Leigh, R. A., and Wyn Jones, R. G. (1984). A hypothesis relating critical potassium concentrations for growth to the distribution and functions of this ion in the plant cell. New Phytol. 97, 1–13. doi: 10.1111/j.1469-8137.1984.tb04103.x
Li, X., Wang, X., He, K., Ma, Y., Su, N., and He, H. (2008). High resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell 20, 259–276. doi: 10.1105/tpc.107.056879
Lintilhac, P. M. (2014). The problem of morphogenesis: unscripted biophysical control systems in plants. Protoplasma 251, 25–36. doi: 10.1007/s00709-013-0522-y
Liu, J., Feng, L., Li, J., and He, Z. (2015). Genetic and epigenetic control of plant heat responses. Front. Plant Sci. 6:267. doi: 10.3389/fpls.2015.00267
Liu, Y.-Y., Wang, P.-Y., Dou, S.-X., Xie, P., Wang, W.-C., and Yin, H.-W. (2005). Study the effect of metallic ions on the combination of DNA and histones with molecular combing technique. Chin. Sci. Bull. 8, 731–737. doi: 10.1007/BF03183670
Lobell, D. B., and Gourdji, S. M. (2012). The influence of climate change on global crop productivity. Plant Physiol. 160, 1686–1697. doi: 10.1104/pp.112.208298
Lothrop, A. P., Torres, M. P., and Fuchs, S. M. (2013). Deciphering post-translational modification codes. FEBS Lett. 587, 1247–1257. doi: 10.1016/j.febslet.2013.01.047
Lowary, P. T., and Widom, J. (1997). Nucleosome packaging and nucleosome positioning of genomic DNA. Proc. Natl. Acad. Sci. U.S.A. 18, 1183–1188. doi: 10.1073/pnas.94.4.1183
Luo, Q. (2011). Temperature thresholds and crop production: a review. Clim. Change 109, 583–598. doi: 10.1007/s10584-011-0028-6
Mack, A. H., Schlingman, D. J., Ilagan, R. P., Regan, L., and Mochrie, S. G. (2012). Kinetics and thermodynamics of phenotype, unwinding and rewinding the nucleosome. J. Mol. Biol. 423, 687–701. doi: 10.1016/j.jmb.2012.08.021
Madlung, A., and Comai, L. (2004). The effect of stress on genome regulation and structure. Ann. Bot. 94, 481–495. doi: 10.1093/aob/mch172
Massie, I. H., Astley, D., King, G. J., and Ingrouille, M. (1999). “Patterns of ecogeographic variation in the Italian landrace cauliflower and broccoli (Brassica oleracea L. var. botrytis L. and var. italica Plenck),” in Taxonomy of Cultivated Plants, 3rd International Symposium, eds S. Andrews, A. C. Leslie, and C. Alexander (Kew: Royal Botanic Gardens), 443–445.
Matzke, A. J. M., Weiger, T. M., and Matzke, M. (2010). Ion channels at the nucleus, electrophysiology meets the genome. Mol. Plant 3, 642–652. doi: 10.1093/mp/ssq013
Matzke, A. M., and Mosher, R. A. (2014). RNA-directed DNA methylation, an epigenetic pathway of increasing complexity. Nat. Rev. 15, 394–408. doi: 10.1038/nrg3683
McBryant, S. J., and Lu, X., Hansen, J. C. (2010). Multifunctionality of the linker histones: an emerging role for protein-protein interactions. Cell Res. 20, 519–528. doi: 10.1038/cr.2010.35
McClung, C. R., and Davis, S. J. (2010). Ambient thermometers in plants: from physiological outputs towards mechanisms of thermal sensing. Curr. Biol. 20, R1086–R1092. doi: 10.1016/j.cub.2010.10.035
McMaster, G. S., and Wilhelm, W. W. (1997). Growing degree-days: one equation, two interpretations. Agric. For. Meteorol. 87, 291–300. doi: 10.1016/S0168-1923(97)00027-0
Mekhail, K., and Moazed, D. (2010). The nuclear envelope in genome organisation, expression and stability. Nat. Rev. Mol. Cell Biol. 11, 317–328. doi: 10.1038/nrm2894
Meneghini, M. D., Wu, M., and Madhani, H. D. (2003). Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell 112, 725–736. doi: 10.1016/S0092-8674(03)00123-5
Michael, T. P., and Jackson, S. (2013). The first 50 plant genomes. Plant Genome 6, 2. doi: 10.3835/plantgenome2013.03.0001in
Mickelbart, M. V., Hasegawa, P. M., and Bailey-Serres, J. (2015). Genetic mechanisms of abiotic stress tolerance that translate to crop yield stability. Nat. Rev. Genet. 16, 237–251. doi: 10.1038/nrg3901
Millar, A. H., Carrie, C., Pogson, B., and Whelan, J. (2009). Exploring the function-location nexus: using multiple lines of evidence in defining the subcellular location of plant proteins. Plant Cell 21, 1625–1631. doi: 10.1105/tpc.109.066019
Mirouze, M., and Paszkowski, J. (2011). Epigenetic contribution to stress adaptation in plants. Curr. Opin. Plant Biol. 14, 267–274. doi: 10.1016/j.pbi.2011.03.004
Mobius, W., and Gerland, U. (2010). Quantitative test of the barrier nucleosome model for statistical positioning of nucleosomes up- and downstream of transcription start sites. PLoS Comput. Biol. 6:e1000891. doi: 10.1371/journal.pcbi.1000891
Muchow, R. C., Sinclair, T. R., and Bennett, J. M. (1990). Temperature and solar radiation effects on potential maize yield across locations. Agron. J. 82, 338–343. doi: 10.2134/agronj1990.00021962008200020033x
Munns, R., James, R. A., and Lauchli, A. (2006). Approaches to increasing the salt tolerance of wheat and other cereals. J. Exp. Bot. 57, 1025–1043. doi: 10.1093/jxb/erj100
Nelson, M. N., Rajasekaran, R., Smith, A., Chen, S., Beeck, C. P., Siddique, K. H., et al. (2014). Quantitative trait loci for thermal time to flowering and photoperiod responsiveness discovered in summer annual-type Brassica napus L. PLoS ONE 9:e102611. doi: 10.1371/journal.pone.0102611
Nightingale, K. P., Gendreizig, S., White, D. A., Bradbury, C., Hollfelder, F., and Turner, B. M. (2007). Cross-talk between histone modifications in response to histone deacetylate inhibitors, MLL4 links histone H3 acetylation and histone H3K4 methylation. J. Biol. Chem. 282, 4408–4416. doi: 10.1074/jbc.M606773200
Noll, M., and Kornberg, R. D. (1977). Action of micrococcal nuclease on chromatin and the location of histone H1. J. Mol. Biol. 109, 393–404.
Norberg, J., and Vihinen, M. (2001). Molecular dynamics simulation of the effects of cytosine methylation on structure of oligonucleotides. J. Mol. Struct. 546, 51–62. doi: 10.1016/S0166-1280(01)00435-3
Over, R. S., and Michaels, S. D. (2014). Open and closed: the roles of linker histones in plants and animals. Mol. Plant 7, 481–491. doi: 10.1093/mp/sst164
Pack, C., Saito, K., Tamura, M., and Kinjo, M. (2006). Microenvironment and effect of energy depletion in the nucleus analysed by mobility of multiple oligomeric EGFPs. Biophys. J. 91, 3921–3936. doi: 10.1529/biophysj.105.079467
Patel, D., and Franklin, K. A. (2009). Temperature-regulation of plant architecture. Plant Signal. Behav. 4, 577–579. doi: 10.4161/psb.4.7.8849
Pecinka, A., Dinh, H. Q., Baubec, T., Rosa, M., Lettner, N., and Mittelsten Scheid, O. (2010). Epigenetic regulation of repetitive elements is attenuated by prolonged heat stress in Arabidopsis. Plant Cell 22, 3118–3129. doi: 10.1105/tpc.110.078493
Pennings, S., Allan, J., and Davey, C. S. (2005). DNA methylation, nucleosome formation and positioning. Brief. Funct. Genomic Proteomic 3, 351–361. doi: 10.1093/bfgp/3.4.351
Perez, A., Noy, A., Lankas, F., Luque, F. J., and Orozco, M. (2004). The relative flexibility of B-DNA and A-RNA duplexes: database analysis. Nucleic Acids Res. 32, 6144–6151. doi: 10.1093/nar/gkh954
Pikaard, C. S., and Scheid, O. M. (2014). Epigenetic regulation in plants. Cold Spring Harb. Perspect. Biol. 6, a019315. doi: 10.1101/cshperspect.a019315
Pimental, D., Berger, B., Filiberto, D., Newton, M., Wolfe, B., Karabinakis, E., et al. (2004). Water resources: agricultural and environmental issues. Bioscience 54, 909–918. doi: 10.1641/0006-3568(2004)054[0909:WRAAEI]2.0.CO;2
Poirer, M. G., Nonhait, T., and Marko, J. F. (2002). Reversible hypercondensation and decondensation of mitotic chromosomes studied using combined chemical-micromechanical techniques. J. Cell. Biochem. 85, 422–434. doi: 10.1002/jcb.10132
Popova, O. V., Dinh, H. Q., Aufsatz, W., and Jonak, C. (2013). The RdDM pathway is required for basal heat tolerance in Arabidopsis. Mol. Plant 6, 396–410. doi: 10.1093/mp/sst023
Porter, J. R., and Semenov, M. A. (2005). Crop responses to climatic variation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 2021–2035. doi: 10.1098/rstb.2005.1752
Rahman, H. U., Hadley, P., and Pearson, S. (2007). Relationship between temperature and cauliflower (Brassica oleracea L. var. botrytis) growth and development after curd initiation. Plant Growth Regul. 52, 61–72. doi: 10.1007/s10725-007-9177-z
Raveh-Sadka, T., Levo, M., and Segal, E. (2009). Incorporating nucleosomes into thermodynamic models of transcription regulation. Genome Res. 19, 1480–1496. doi: 10.1101/gr.088260.108
Ray, D. K., Gerber, J. S., MacDonald, G. K., and West, P. C. (2015). Climate variation explains a third of global crop yield variability. Nat. Commun. 22, 5989. doi: 10.1038/ncomms6989
Rea, M., Zheng, W., Chen, M., Braud, C., Bhangu, D., Rognan, T. N., et al. (2012). Histone H1 affects gene imprinting and DNA methylation in Arabidopsis. Plant. J. 71, 776–786. doi: 10.1111/j.1365-313X.2012.05028.x
Rippe, K. (2012). “The folding of the nucleosome chain,” in Genome Organisation and Function in the Cell Nucleus, ed. K. Rippe (Heidelberg: Wiley-VCH), 139–167. doi: 10.1002/9783527639991
Robertson, S. M., Jeffrey, S. R., Unterschultz, J. R., and Boxall, P. C. (2013). Estimating yield response to temperature and identifying critical temperatures for annual crops in the Canadian Prairie region. Can. J. Plant Sci. 93, 1237–1247. doi: 10.4141/cjps2013-125
Rodriguez Lopez, C. M., Asenjo, B. G., Lloyd, A. J., and Wilkinson, M. J. (2010). Direct detection and quantification of methylation in nucleic acid sequences using high-resolution melting analysis. Anal. Chem. 82, 9100–9108. doi: 10.1021/ac1024057
Romera-Branchat, M., Andrés, F., and Coupland, G. (2014). Flowering responses to seasonal cues: what’s new? Curr. Opin. Plant Biol. 21, 120–127. doi: 10.1016/j.pbi.2014.07.006
Romon, M., Soustre-Gacougnolle, I., Schmitt, C., Perrin, M., Burdloff, Y., Chevalier, E., et al. (2013). RNA silencing is resistant to low-temperature in grapevine. PLoS ONE 8:e82652. doi: 10.1371/journal.pone.0082652
Rosenzweig, C., and Liverman, D. (1992). “Predicted effects of climate change on agriculture: a comparison of temperate and tropical regions,” in Global Climate Change: Implications, Challenges, and Mitigation Measures, ed. S. K. Majumdar (Philadelphia: The Pennsylvania Academy of Sciences), 342–361.
Roque, A., Ponte, I., Arrondo, J. L., and Suau, P. (2008). Phosphorylation of the carboxy-terminal domain of histone H1: effects on secondary structure and DNA condensation. Nucleic Acids Res. 36, 4719–4726. doi: 10.1093/nar/gkn440
Rutowicz, K., Puzio, M., Halibart-Puzio, J., Lirski, M., Kroteń, M. A., Kotliński, M., et al. (2015). A specialized histone H1 variant is required for adaptive responses to complex abiotic stress and related DNA methylation in Arabidopsis. Plant Physiol. doi: 10.1104/pp.15.00493 [Epub ahead of print].
Sadok, W., Naudin, P., Boussuge, B., Muller, B., Welcker, C., and Tardieu, F. (2007). Leaf growth rate per unit thermal time follows QTL-dependent daily patterns in hundreds of maize lines under naturally fluctuating conditions. Plant Cell Environ. 30, 135–146. doi: 10.1111/j.1365-3040.2006.01611.x
Sánchez-Pérez, R., Del Cueto, J., Dicenta, F., and Martínez-Gómez, P. (2014). Recent advancements to study flowering time in almond and other Prunus species. Front. Plant Sci. 5:334. doi: 10.3389/fpls.2014.00334
Schlenker, W., and Roberts, M. J. (2009). Nonlinear temperature effects indicate severe damages to U.S. crop yields under climate change. Proc. Natl. Acad. Sci. U.S.A. 106, 15594–15598. doi: 10.1073/pnas.0906865106
Schmitz, R. J., Schultz, M. D., Lewsey, M. G., O’Malley, R. C., Urich, M. A., Libiger, O., et al. (2011). Transgenerational epigenetic instability is a source of novel methylation variants. Science 334, 369–373. doi: 10.1126/science.1212959
Schwartz, S., Meshorer, E., and Ast, G. (2009a). Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 16, 990–995. doi: 10.1038/nsmb.1659
Schwartz, C., Balasubramanian, S., Warthmann, N., Michael, T. P., Lempe, J., Sureshkumar, S., et al. (2009b). Cis-regulatory changes at FLOWERING LOCUS T mediate natural variation in flowering responses of Arabidopsis thaliana. Gene. 183, 723–732. doi: 10.1534/genetics.109.104984
Seidel, T., Siek, M., Marg, B., and Dietz, K. J. (2013). Energization of vacuolar transport in plant cells and its significance under stress. Int. Rev. Cell Mol. Biol. 304, 57–131. doi: 10.1016/B978-0-12-407696-9.00002-6
Severin, P. M. D., Zou, X., Gaub, H. E., and Schulten, K. (2011). Cytosine methylation alters DNA mechanical properties. Nucleic Acids Res. 39, 8740–8751. doi: 10.1093/nar/gkr578
She, W., Grimanelli, D., Rutowicz, K., Whitehead, M. W., Puzio, M., Kotlinski, M., et al. (2013). Chromatin reprogramming during the somatic-to-reproductive cell fate transition in plants. Development 140, 4008–4019. doi: 10.1242/dev.095034
Sheldon, C. C., Finnegan, E. J., Peacock, W. J., and Dennis, E. S. (2009). Mechanisms of gene repression by vernalization in Arabidopsis. Plant J. 59, 488–498. doi: 10.1111/j.1365-313X.2009.03883.x
Shi, J., Li, R., Qiu, D., Jiang, C., Long, Y., Morgan, C., et al. (2009). Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 182, 851–861. doi: 10.1534/genetics.109.101642
Simon, S. A., and Meyers, B. C. (2011). Small RNA-mediated epigenetic modifications in plants. Curr. Opin. Plant Biol. 14, 148–155. doi: 10.1016/j.pbi.2010.11.007
Simpson, R. T. (1978). Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochemistry 17, 5524–5531. doi: 10.1021/bi00618a030
Smith, L. B., and King, G. J. (2000). The distribution of BoCAL-a alleles in Brassica oleracea is consistent with a genetic model for curd development and domestication of the Cauliflower. Mol. Breed. 6, 603–613. doi: 10.1023/A:1011370525688
Song, Q., Qiu, Z., Wang, H., Xia, Y., Shen, J., and Zhang, Y. (2013). The effect of methylation on the hydrogen-bonding and stacking interaction of nucleic acid bases. Struct. Chem. 24, 55–65. doi: 10.1007/s11224-012-0027-x
Song, Y., Ji, D., Li, S., Wang, P., Li, Q., and Xiang, F. (2012). The dynamic changes of DNA methylation and histone modifications of salt responsive transcription factor genes in soybean. PLoS ONE 7:e41274. doi: 10.1371/journal.pone.0041274
Spadafora, C., Oudet, P., and Chambon, P. (1979). Rearrangement of chromatin structure induced by increasing ionic strength and temperature. Eur. J. Biochem. 100, 225–235. doi: 10.1111/j.1432-1033.1979.tb02053.x
Steward, N., Ito, M., Yamaguchi, Y., Koizumi, N., and Sano, H. (2002). Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress. J. Biol. Chem. 288, 37741–37746. doi: 10.1074/jbc.M204050200
Strickfaden, H., Cremer, T., and Rippe, K. (2012). “Higher order chromatin organization and dynamics,” in Genome Organisation and Function in the Cell Nucleus, ed. K. Rippe (Winheim: Wiley-VCH), 417–447.
Stroud, H., Otero, S., Desvoyes, B., Ramirez-Parra, E., Jacobsen, S. E., and Gutierrez, C. (2012). Genome-wide analysis of histone H3.1 and H3.3 variants in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 109, 5370–5375. doi: 10.1073/pnas.1203145109
Subramanian, V., Fields, P. A., and Boyer, L. A. (2015). H2A.Z: a molecular rheostat for transcriptional control. F1000Prime Rep. 7, 01. doi: 10.12703/P7-01
Swiezewski, S., Liu, F., Magusin, A., and Dean, C. (2009). Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 462, 799–802. doi: 10.1038/nature08618
Szittya, G., Silhavy, D., Molnár, A., Havelda, Z., Lovas, A., Lakatos, L., et al. (2003). Low temperature inhibits RNA silencing-mediated defence by the control of siRNA generation. EMBO J. 22, 633–640. doi: 10.1093/emboj/cdg74
Tan, D. K. Y., Birch, C. J., Wearing, A. H., and Rickert, K. G. (2000). Predicting broccoli development: I. Development is predominantly determined by temperature rather than photoperiod. Sci. Hortic. 84, 227–243. doi: 10.1016/S0304-4238(99)00139-9
Tariq, M., and Paszkowski, J. (2004). DNA and histone methylation in plants. Trends Genet. 20, 244–251. doi: 10.1016/j.tig.2004.04.005
Tester, M., and Davenport, R. (2003). Na+ tolerance and Na+ transport in higher plants. Ann. Bot. 91, 503–527. doi: 10.1093/aob/mcg058
Thalhammer, A., Hansen, A. S., El-Sagheer, A. H., Brown, T., and Schofield, C. J. (2011). Hydroxylation of methylated CpG dinucleotides reverses stabilisation of DNA duplexes by cytosine t-methylation. Chem. Commun. (Camb.) 47, 5325–5327. doi: 10.1039/c0cc05671e
Thoma, F., Koller, T., and Klug, A. J. (1979). Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. Cell Biol. 83, 403–427. doi: 10.1083/jcb.83.2.403
Thompson, K., Parkinson, J. A., Band, S. R., and Spencer, R. E. (1997). A comparative study of leaf nutrient concentrations in a regional herbaceous flora. New Phytol. 136, 679–689. doi: 10.1046/j.1469-8137.1997.00787.x
Tilgner, H., Nikolaou, C., Althammer, S., Sammeth, M., Beato, M., Valcárcel, J., et al. (2009). Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 16, 996–1001. doi: 10.1038/nsmb.1658
Timilsena, Y. P., Adhikari, R., Casey, P., Muster, T., Gill, H., and Adhikari, B. (2014). Enhanced efficiency fertilisers: a review of formulation and nutrient release patterns. J. Sci. Food Agric. 95, 1131–1142. doi: 10.1002/jsfa.6812
To, K., and Kim, J. M. (2013). Epigenetic regulation of gene responsiveness in Arabidopsis. Front. Plant Sci. 4:548. doi: 10.3389/fpls.2013.00548
Tsankov, A., Yanagisawa, Y., Rhind, N., Regev, A., and Rando, O. J. (2011). Evolutionary divergence of intrinsic and trans-regulated nucleosome positioning sequences reveals plastic rules for chromatin organization. Genome Res. 21, 1851–1862. doi: 10.1101/gr.122267.111
Tse, C., Sera, T., Wolffe, A. P., and Hansen, J. C. (1998). Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol. Cell. Biol. 18, 4629–4638. doi: 10.1128/MCB.18.8.4629
Unkovich, M., Baldock, J., and Forbes, M. (2010). Variability in harvest index of grain crops and potential significance for carbon accounting: examples from Australian agriculture. Adv. Agron. 105, 173–219. doi: 10.1016/S0065-2113(10)05005-4
Vaillant, I., and Paszkowski, J. (2007). Role of histone and DNA methylation in gene regulation. Curr. Opin. Plant Biol. 10, 528–533. doi: 10.1016/j.pbi.2007.06.008
Van de Vosse, D. W., Wan, Y., Wozniak, R. W., and Aitchison, J. D. (2011). Role of the nuclear envelope in genome organization and gene expression. Wiley Interdiscip. Rev. Syst. Biol. Med. 3, 147–166. doi: 10.1002/wsbm.101
Visvanathan, A., Ahmed, K., Even-Faitelson, L., Lleres, D., Bazett-Jones, D. P., and Lamond, A. I. (2013). Modulation of higher order chromatin conformation in mammalian cell nuclei can be mediated by polyamines and divalent cations. PLoS ONE 8:e67689. doi: 10.1371/journal.pone.0067689
Wang, J., Hopkins, C. J., Hou, J., Zou, X., Wang, C., Long, Y., et al. (2012). Promoter variation and transcript divergence in Brassicaceae lineages of FLOWERING LOCUS T. PLoS ONE 7:e47127. doi: 10.1371/journal.pone.0047127
Wanunu, M., Cohen-Karni, D., Johnson, R. R., Fields, L., Benner, J., Peterman, N., et al. (2011). Discrimination of methylcytosine from hydroxymethylcytosine in DNA molecules. J. Am. Chem. Soc. 133, 486–492. doi: 10.1021/ja107836t
Watanabe, T., Broadley, M. R., Jansen, S., White, P. J., Takada, J., Satake, K., et al. (2007). Evolutionary control of leaf element composition in plants. New Phytol. 174, 516–523. doi: 10.1111/j.1469-8137.2007.02078.x
Waterborg, J. H., Harrington, R. E., and Winicov, I. (1989). Differential histone acetylation in alfalfa (Medicago sativa) due to growth in NaCl: responses in salt stressed and salt tolerant callus cultures. Plant Physiol. 90, 237–245. doi: 10.1104/pp.90.1.237
Watling, J. R., Grant, N. M., Miller, R. E., and Robinson, S. A. (2008). Mechanisms of thermoregulation in plants. Plant Signal. Behav. 3, 595–597. doi: 10.4161/psb.3.8.6341
Way, D. A., and Pearcy, R. W. (2012). Sunflecks in trees and forests: from photosynthetic physiology to global change biology. Tree Physiol. 32, 1066–1081. doi: 10.1093/treephys/tps064
Weber, C. M., Ramachandran, S., and Henikoff, S. (2014). Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol. Cell 53, 819–830. doi: 10.1016/j.molcel.2014.02.014
Wedemeier, A., Merlitz, H., Wu, C.-X., and Jörg Langowski, J. (2007). Modeling diffusional transport in the interphase cell nucleus. J. Chem. Phys. 127, 045102. doi: 10.1063/1.2753158
Weidemann, T., Wachsmuth, M., Knoch, T. A., Muller, G., Waldeck, W., and Langowski, J. (2003). Counting nucleosomes in living cells with a combination of fluorescence correlation spectroscopy and confocal imaging. J. Mol. Biol. 334, 229–240. doi: 10.1016/j.jmb.2003.08.063
West, J. A., Cook, A., Alver, B. H., Stadtfeld, M., Deaton, A. M., Hochedlinger, K., et al. (2014). Nucleosomal occupancy changes locally over key regulatory regions during cell differentiation and reprogramming. Nat. Commun. 5, 4719. doi: 10.1038/ncomms5719
White, P. J., Hammond, J. P., King, G. J., Bowen, H. C., Hayden, R. M., Meacham, M. C., et al. (2009). Genetic analysis of potassium use efficiency in Brassica oleracea L. Ann. Bot. 105, 1199–1210. doi: 10.1093/aob/mcp253
Widman, N., Feng, S., Jacobsen, S. E., and Pellegrini, M. (2014). Epigenetic differences between shoots and roots in Arabidopsis reveals tissue-specific regulation. Epigenetics 9, 1–7. doi: 10.4161/epi.26869
Wierzbicki, A. T., and Jerzmanowski, A. (2005). Suppression of histone H1 genes in Arabidopsis results in heritable developmental defects and stochastic changes in DNA methylation. Genetics 169, 997–1008. doi: 10.1534/genetics.104.031997
Wilson, D. F. (2013). Regulation of cellular metabolism: programming and maintaining metabolic homeostasis. J. Appl. Physiol. 115, 1583–1588. doi: 10.1152/japplphysiol.00894.2013
Wolfe, J. (2015). “Cellular thermodynamics: the molecular and macroscopic views,” in eLS (Chichester: John Wiley & Sons, Ltd.), 1–13. doi: 10.1002/9780470015902.a0001363.pub2
Wong, H., Victor, J. M., and Mozziconacci, J. (2007). An all-atom model of the chromatin fibre containing linker histones reveals a versatile structure tuned by nucleosomal repeat length. PLoS ONE 2:e877. doi: 10.1371/journal.pone.0000877
Woodcock, C. L., and Ghosh, R. P. (2010). Chromatin higher-order structure and dynamics. Cold Spring Harb. Perspect. Biol. 2, a000596. doi: 10.1101/cshperspect.a000596
Woodcock, C. L., Skoultchi, A. I., and Fan, Y. (2006). Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 14, 17–25. doi: 10.1007/s10577-005-1024-3
Wu, J., Xiao, J., Wang, L., Zhong, J., Yin, H., Wu, S., et al. (2013). Systematic analysis of intron size and abundance parameters in diverse lineages. Sci. China Life Sci. 56, 968–974. doi: 10.1007/s11427-013-4540-y
Wu, Y., Zhang, W., and Jiang, J. (2014). Genome-wide nucleosome positioning is orchestrated by genomic regions associated with DNase I hypersensitivity in rice. PLoS Genet. 10:e1004378. doi: 10.1371/journal.pgen.1004378
Wurr, D. C. E., Fellows, J. R., and Fuller, M. P. (2004). Simulated effects of climate change on the production pattern of winter cauliflower in the UK. Sci. Hortic. 101, 359–372. doi: 10.1016/j.scienta.2003.11.011
Wurr, D. C. E., Fellows, J. R., Phelps, K., and Reader, R. J. (1994). Testing a vernalization model on field-grown crops of four cauliflower cultivars. J. Hortic. Sci. 69, 251–255.
Wurr, D. C. E., Fellows, J. R., Phelps, K., and Reader, R. J. (1995). Vernalization in calabrese (Brassica oleracea var. italica)—a model for apex development. J. Exp. Bot. 46, 1487–1496. doi: 10.1093/jxb/46.10.1487
Wurr, D. C. E., Fellows, J. R., Sutherland, R. A., and Elphinstone, E. D. (1990). A model of cauliflower curd growth to predict when curds reach a specified size. J. Hortic. Sci. 65, 555–564.
Wyn Jones, R. G., and Lunt, O. R. (1967). The function of calcium in plants. Bot. Rev. 33, 407–426. doi: 10.1007/BF02858743
Yakovchuk, P., Protozanova, E., and Frank-Kamenetskii, M. D. (2006). Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 34, 564–574. doi: 10.1093/nar/gkj454
Yusufaly, W. I., Li, Y., and Olson, W. K. (2013). 5-Methylation of cytosine in CG:CG base-pair steps, a physicochemical mechanism for the epigenetic control of DNA nanomechanics. J. Phys. Chem. B 117, 16436–16442. doi: 10.1021/jp409887t
Zemach, A., Kim, M. Y., Hsieh, P. H., Coleman-Derr, D., Eshed-Williams, L., Thao, K., et al. (2013). The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153, 193–205. doi: 10.1016/j.cell.2013.02.033
Zhang, M., Xu, C., von Wettstein, D., and Liu, B. (2011). Tissue-specific differences in cytosine methylation and their association with differential gene expression in sorghum. Plant Physiol. 156, 1955–1966. doi: 10.1104/pp.111.176842
Zhang, W., Zhang, T., Wu, Y., and Jiang, J. (2012). Genome-wide identification of regulatory DNA elements and protein-binding footprints using signatures of open chromatin in Arabidopsis. Plant Cell 24, 2719–2731. doi: 10.1105/tpc.112.098061
Zhao, H., and Chen, T. (2014). Tet family of 5-methylcytosine dioxygenases in mammalian development. J. Hum. Genet. 58, 421–427. doi: 10.1038/jhg.2013.63
Zhong, S. H., Liu, J. Z., Jin, H., Lin, L., Li, Q., Chen, Y., et al. (2013). Warm temperatures induce transgenerational epigenetic release of DNA silencing by inhibiting siRNA biogenesis in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 110, 9171–9176. doi: 10.1073/pnas.1219655110
Zhu, J., Jeong, J. C., Zhu, Y., Sokolchik, I., Miyazaki, S., Zhu, J. K., et al. (2008). Involvement of HOS15 in histone deacetylation and cold tolerance. Proc. Natl. Acad. Sci. U.S.A. 105, 4945–4950. doi: 10.1073/pnas.0801029105
Zilberman, D., Coleman-Derr, D., Ballinger, T., and Henikoff, S. (2008). Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456, 125–129. doi: 10.1038/nature07324
Zlatanova, J., and Thakar, A. (2008). H2A.Z, view from the top. Structure 16, 166–179. doi: 10.1016/j.str.2007.12.008
Keywords: crop epigenetics, chromatin dynamics, thermal homeostasis, ionic homeostasis, DNA methylation, phenotypic plasticity, G × E interactions
Citation: King GJ (2015) Crop epigenetics and the molecular hardware of genotype × environment interactions. Front. Plant Sci. 6:968. doi: 10.3389/fpls.2015.00968
Received: 18 May 2015; Accepted: 22 October 2015;
Published: 06 November 2015.
Edited by:
Raúl Alvarez-Venegas, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoReviewed by:
Célia Baroux, Zurich-Basel Plant Science Center, SwitzerlandChris Helliwell, Commonwealth Scientific and Industrial Research Organisation, Australia
Copyright © 2015 King. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Graham J. King, Z3JhaGFtLmtpbmdAc2N1LmVkdS5hdQ==