 Yanci Yang
Yanci Yang Tao Zhou
Tao Zhou Dong Duan
Dong Duan Guifang Zhao
Guifang Zhao- Key Laboratory of Resource Biology and Biotechnology in Western China (Ministry of Education), College of Life Sciences, Northwest University, Xi’an, China
Quercus is considered economically and ecologically one of the most important genera in the Northern Hemisphere. Oaks are taxonomically perplexing because of shared interspecific morphological traits and intraspecific morphological variation, which are mainly attributed to hybridization. Universal plastid markers cannot provide a sufficient number of variable sites to explore the phylogeny of this genus, and chloroplast genome-scale data have proven to be useful in resolving intractable phylogenetic relationships. In this study, the complete chloroplast genomes of four Quercus species were sequenced, and one published chloroplast genome of Quercus baronii was retrieved for comparative analyses. The five chloroplast genomes ranged from 161,072 bp (Q. baronii) to 161,237 bp (Q. dolicholepis) in length, and their gene organization and order, and GC content, were similar to those of other Fagaceae species. We analyzed nucleotide substitutions, indels, and repeats in the chloroplast genomes, and found 19 relatively highly variable regions that will potentially provide plastid markers for further taxonomic and phylogenetic studies within Quercus. We observed that four genes (ndhA, ndhK, petA, and ycf1) were subject to positive selection. The phylogenetic relationships of the Quercus species inferred from the chloroplast genomes obtained moderate-to-high support, indicating that chloroplast genome data may be useful in resolving relationships in this genus.
Introduction
The genus Quercus (Fagaceae) is distributed throughout the Northern Hemisphere, and consists of approximately 500 species (Nixon, 1993; Manos et al., 1999). Oak taxonomy is perplexing, because of intermediate morphological traits caused by extensive hybridization (Rushton, 1993; Cavender-Bares et al., 2004; Curtu et al., 2007; Burgarella et al., 2009; Moran et al., 2012), introgression, incomplete lineage sorting, and convergent evolution (Kole, 2011). Based on pollen characteristics and nuclear markers, six major intrageneric groups (Cyclobalanopsis, Cerris, Ilex, Lobatae, Protobalanus, and Quercus) have been identified (Oh and Manos, 2008; Denk and Grimm, 2009, 2010; Hubert et al., 2014). In China, Quercus has generally been divided into five sections, based on morphological characteristics (Zhou et al., 1994; Pu et al., 2001; Peng et al., 2007). Among these, Sect. Echinolepides is an intermediate group between evergreen oaks (Sect. Brachylepides and Sect. Engleriana) and deciduous oaks (Sect. Aegilops and Sect. Quercus). However, the phylogenetic relationships among Quercus species are still not fully understood because of incomplete sampling, the use of markers with insufficient phylogenetic signals, and complex evolutionary issues.
Because of their highly conserved structure, general recombination-free, uniparental inheritance, and small effective population sizes (Birky et al., 1983), chloroplast (cp) DNA sequences have been extensively employed to resolve plant phylogenies (Jansen et al., 2007; Moore et al., 2010; Shaw et al., 2014). With the rapid development of next-generation sequencing, it is now cheaper and faster to obtain genomes than by traditional Sanger sequencing (Alkan et al., 2011). Therefore, cp genome-scale data have been increasingly used to infer phylogenetic relationships at high taxonomical levels, and even in lower taxa, great progress has been made (Jansen et al., 2007; Moore et al., 2007, 2010; Parks et al., 2009; Barrett et al., 2013; Ma et al., 2014; Carbonell-Caballero et al., 2015). Most angiosperm cp genomes have a quadripartite circular structure, and are composed of two copies of inverted repeat (IR) regions that are separated by a large single copy (LSC) region and a small single copy (SSC) region (Jansen et al., 2005; Jansen and Ruhlman, 2012). Despite the fact that angiosperm cp genomes exhibit a remarkably conserved gene content and order (Jansen and Ruhlman, 2012), some lineages (such as Campanulaceae, Fabaceae, Geraniaceae, and Oleaceae) exhibit different levels of genomic upheaval, such as gene, intron, or even IR region loss, gene duplications, and large-scale rearrangements (Cosner et al., 2004; Lee et al., 2007; Cai et al., 2008; Guisinger et al., 2010, 2011; Martin et al., 2014).
In the present study, the comparative analysis of five complete Quercus cp genomes was conducted in order to explore the sequences’ molecular evolution. Highly variable regions were identified that could serve as potential markers for phylogenetic analysis or candidate DNA barcoding in future studies.
Materials and Methods
Plant Material and DNA Extraction
The materials used were Q. dolicholepis, Q. variabilis, Q. aliena, and Q. aliena var. acuteserrata. Voucher specimens of these species were deposited in the herbarium of Northwest University, Xi’an, China. Total genomic DNA was isolated from silica-dried leaf material using a modified CTAB method (Doyle, 1987), which was conducted by Biomarker Technologies, Inc. (Beijing, China). The complete cp genome of Quercus baronii (GenBank accession No. KT963087; Yang et al., 2015) was recovered in order to conduct a comparative analysis with these four species.
Illumina Sequencing, Assembly, and Annotation
Total genomic DNA was sequenced using an Illumina Hiseq 2500 platform by Biomarker Technologies, Inc. Firstly, all of the raw reads were trimmed using a CLC Genomics Workbench v7.5 (CLC Bio, Aarhus, Denmark) with the default parameters set. Reference-guided assembly was then used to reconstruct the chloroplast genomes with the program MITObim v1.7 (Hahn et al., 2013; Table 1). In this process, in order to obtain accurate sequences, every species was assembled five times with the reference genomes Q. rubra (JX970937), Q. spinosa (KM841421), Q. aquifolioides (KP340971), Q. aliena (KP301144), and Castanea mollissima (HQ336406). A few gaps in the assembled cp genomes were corrected by Sanger sequencing. Primers were designed using Lasergene 7.1 (DNASTAR, Madison, WI, USA). Primer synthesis, and the sequencing of the polymerase chain reaction products, was conducted by Sangon Biotech (Shanghai, China). The primers and amplifications are shown in Supplementary Table S1. The complete cp genomes were annotated using the program DOGMA (Wyman et al., 2004), and then manually corrected by comparing them with the complete cp genomes of the abovementioned, related species in GENEIOUS R8 (Biomatters, Ltd., Auckland, New Zealand). Circular plastid genome maps were drawn using OGDRAW1 (Lohse et al., 2013).
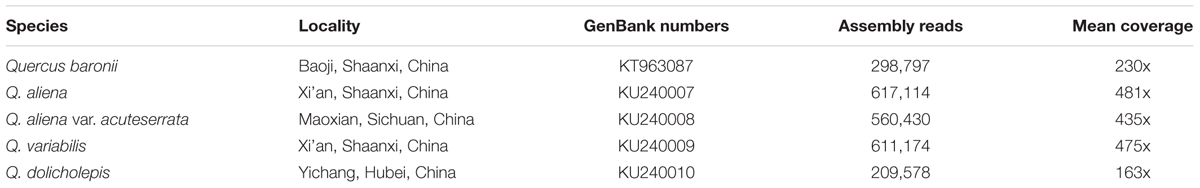
TABLE 1. Assembly information for the five Quercus species.
Repeat Elements Analysis
Tandem repeat sequences (>10 bp in length) were detected using the online program Tandem Repeats Finder (Benson, 1999), with 2, 7, and 7 set for the alignment parameters match, mismatch, and indel, respectively. The minimum alignment score and maximum period size were 80 and 500, respectively. REPuter (Kurtz et al., 2001) was used to find dispersed and palindromic repeats in which the minimal repeat size was 30 bp and the two repeat copies had at least 90% similarity. The gap size between palindromic repeats had a maximum length of 3 kb. All of the repeats found were manually verified and redundant results were removed. The positions and types of simple sequence repeats (SSRs) were ascertained using msatcommander (Faircloth, 2008). The minimum numbers of repeats were 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexanucleotides, respectively.
Sequence Divergence Analysis
The alignments of the five complete chloroplast genome sequences were visualized using mVISTA (Frazer et al., 2004) in order to show interspecific variation. The percentage of variable characters for each coding and non-coding region with an aligned length of more than 200 bp was obtained based on the method of Zhang et al. (2011). Variable sites and parsimony-informative sites across the complete chloroplast genomes and LSC, SSC, and IR regions of the five taxa were calculated using DnaSP v5.0 (Librado and Rozas, 2009). Nucleotide substitutions were counted using MEGA 5.0 (Tamura et al., 2011), and indels were manually detected across the cp genomes. Selective pressures were computed for protein-encoding genes that were located in SC regions and one IR region. Non-synonymous (KA) and synonymous (KS) substitution rates were calculated using PAML with the yn00 program (Yang, 2007). There were 10 pairwise alignments for each gene, which contributed to a total of 790 KA/KS values.
Phylogenetic Analysis
Phylogenetic analysis was conducted based on 10 taxa, including five species in the current study, three other Quercus species (Q. rubra, Q. spinosa, and Q. aquifolioides), and two Fagaceae species (C. mollissima and Castanopsis echidnocarpa) that were used as outgroups. The sequences were aligned using MAFFT (Katoh and Standley, 2013) in GENEIOUS R8 with the default parameters set, and were manually adjusted in MEGA 5.0. Because molecular evolutionary rates differ in different cp genome regions, we constructed the phylogenetic tree using the following datasets: (1) the LSC region; (2) the SSC region; (3) the inverted repeat B (IRB) region; (4) the LSC + SSC regions; (5) the LSC + SSC + IRB regions; and (6) the complete chloroplast genome sequences.
Modeltest 3.7 (Posada and Crandall, 1998) was used to determine the best-fitting model for each dataset based on the Akaike information criterion. Maximum likelihood analysis was performed using RAxML v7.2.8 (Stamatakis, 2006) with 1000 bootstrap replicates. Bayesian inference was performed using the program MrBayes v3.1.2 (Ronquist and Huelsenbeck, 2003). Markov chain Monte Carlo simulations were independently run twice for 2 million generations, and sampling trees every 100 generations. Convergence was determined by examining the average standard deviation of split frequencies (<0.01). The first 25% of trees was discarded as burn-in, and the remaining trees were used to build a majority-rule consensus tree.
Results
Complete Chloroplast Genomes of Quercus Species
The five chloroplast genomes ranged in size from 161,072 bp (Q. baronii) to 161,237 bp (Q. dolicholepis; Figure 1). All of them displayed a typical quadripartite structure, and the same regions were of similar lengths (Table 2). Gene content and order were identical in the five species, and were similar to other published chloroplast genomes in Fagaceae (Jansen et al., 2011; Alexander and Woeste, 2014; Dane et al., 2015; Du et al., 2015; Lu et al., 2015). Although genomic structure and size were highly conserved in the five cp genomes, the IR/SC boundary regions still varied slightly (Figure 2). For example, the distance from the ycf1 5′ end to the junction of IRB/SSC was 43 bp in Q. aliena and Q. aliena var. acuteserrata, 75 bp in Q. baronii and Q. dolicholepis, and 33 bp in Q. variabilis. The assembled cp genomes encoded 134 genes, which consisted of 86 protein-coding genes, 40 transfer RNA (tRNA) genes, and 8 ribosomal RNA (rRNA) genes. Eighteen genes were duplicated in the IR region, including seven protein-coding genes, seven tRNA genes, and four rRNA genes (Table 2). A total of 14 protein-coding genes and 8 tRNA genes contained one or more introns (Supplementary Table S2). The GC content of each analyzed species was very similar in the same region or complete cp genome, but in the IR region it was clearly higher than in the other regions, possibly because of the high GC content of the rRNA (55.5%) that was located in the IR regions (Table 2).
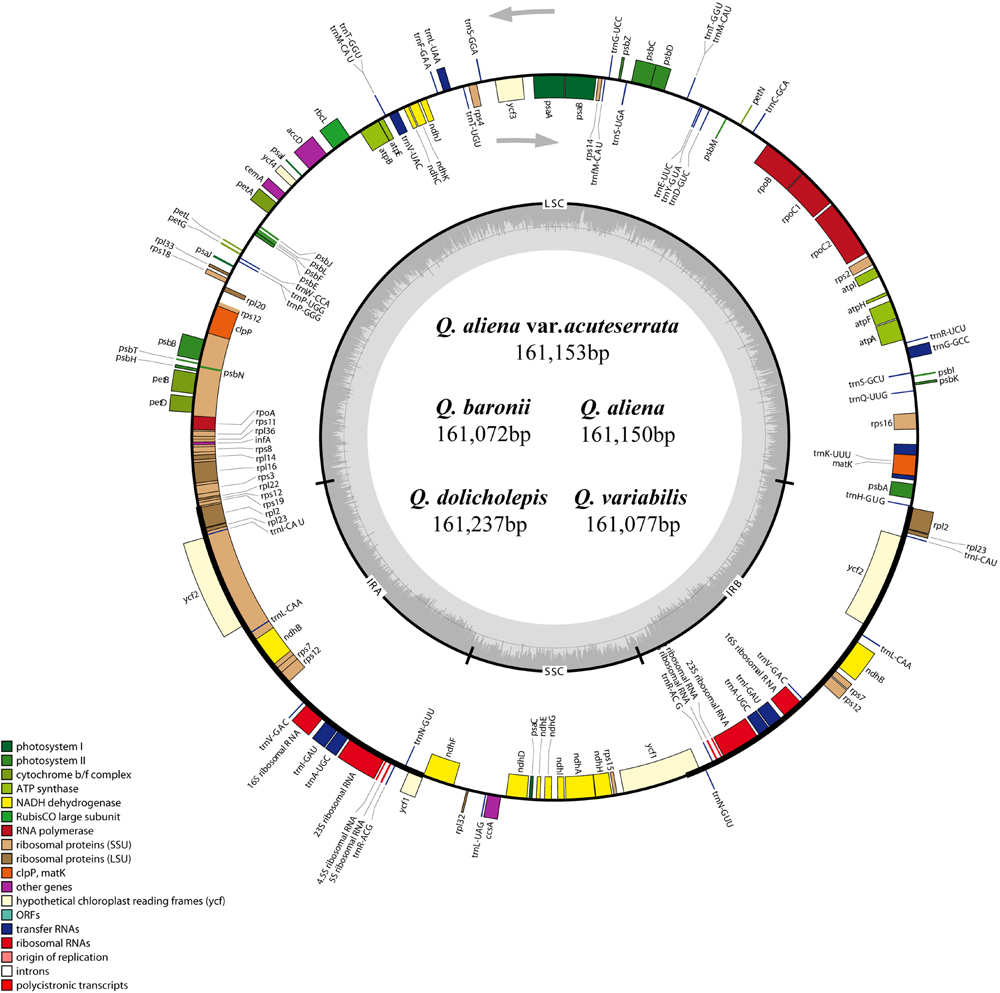
FIGURE 1. Gene map of the five Quercus chloroplast genomes. The genes shown outside of the circle are transcribed clockwise, while those inside are transcribed counterclockwise. Genes belonging to different functional groups are color coded. Dashed area in the inner circle indicates the GC content of the chloroplast genome.
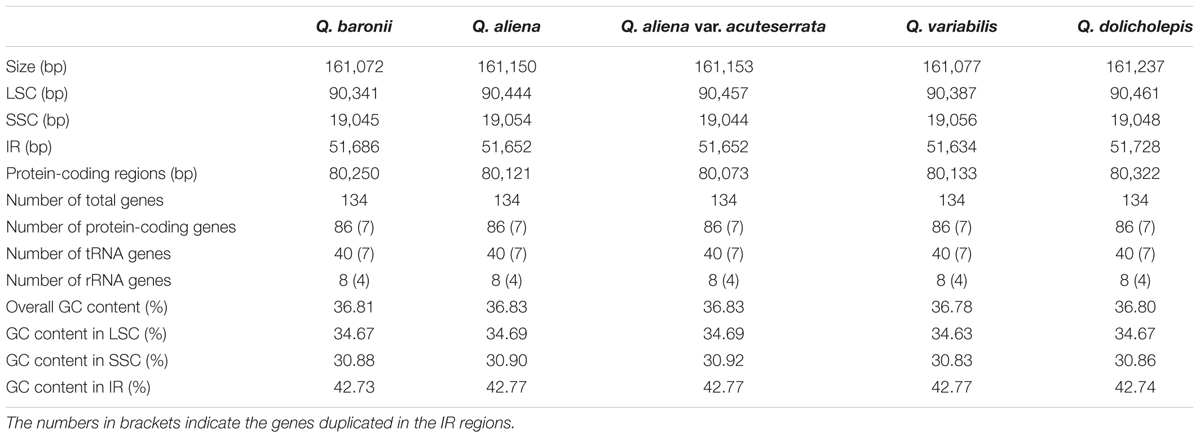
TABLE 2. Characteristics of Quercus chloroplast genomes.
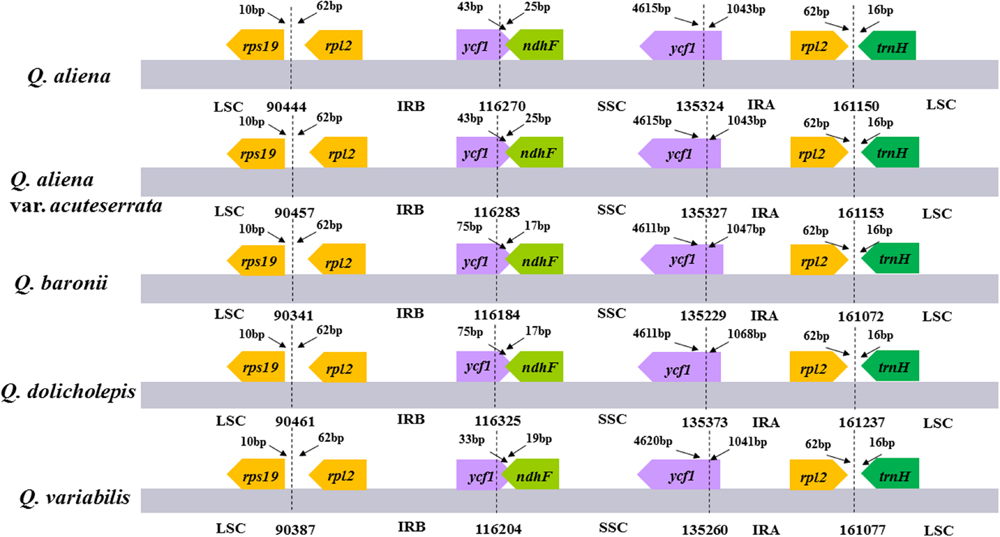
FIGURE 2. The comparison of the LSC, IR, and SSC border regions among the five Quercus chloroplast genomes. Number above the gene features means the distance between the ends of genes and the borders sites. These features are not to scale.
Repeat Elements Analysis
The numbers and distributions of all of the repeat types in the five cp genomes were similar and conserved (Figure 3). There were 132 repeats, which included tandem, dispersed, and palindromic repeats. The lengths of the repeat units ranged from 14 to 40 bp. Most of them were distributed in intergenic or intron regions, and only a minority were located in gene regions (ycf1, ycf2, psaA, psaB, trnS-GCU, trnS-UGA, trnG-GCC, trnG-UCC, trnS-UGA, and trnS-GGA; Supplementary Table S3). We then analyzed the cp genome SSRs, which are often used as genetic markers in population genetics and evolutionary studies. The most abundant were mononucleotide repeats, which accounted for about 80% of the total SSRs, followed by dinucleotides (Table 3). Overall, there were slightly more tetranucleotide repeats than trinucleotide repeats, and penta- and hexanucleotides were very rare across the cp genomes. Protein-coding regions accounted for approximately half of the lengths of the cp genomes but only contained about 13% of the total SSRs, which meant that the SSR distribution was uneven across the cp genomes. SSRs located in the coding DNA sequence (CDS) region were mainly found in rpoC2 and ycf1; rpoB, atpB, accD, ndhF, rpl32, and ndhD contained the remaining SSRs (Supplementary Table S4).
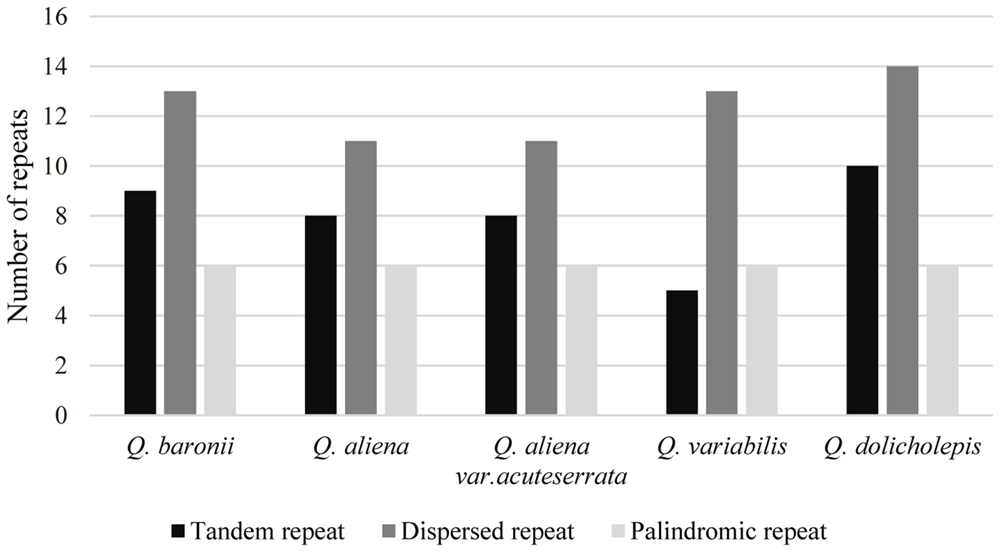
FIGURE 3. Repeat number. Histogram showing the number of three repeat types in five Quercus complete cp genomes.
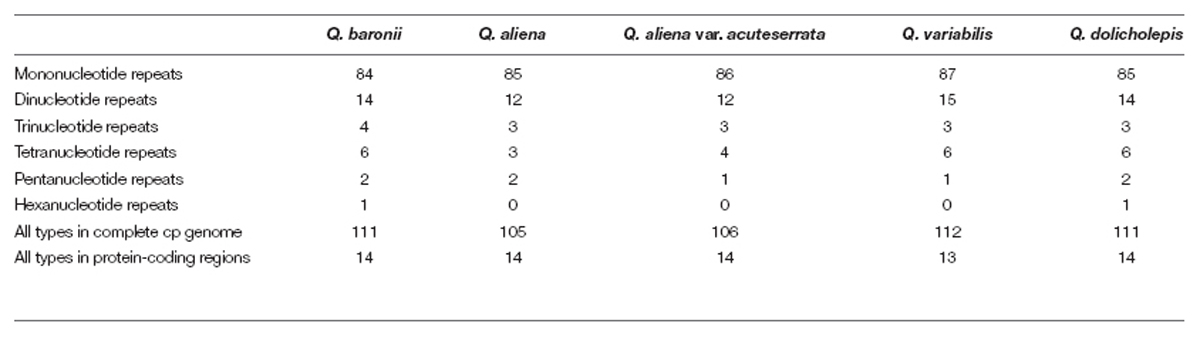
TABLE 3. Types and number of SSRs in cp genomes.
Sequence Divergence Analysis
We used mVISTA to perform a sequence identity analysis, with Q. aliena as a reference (Figure 4). The alignment revealed high sequence similarity across the five cp genomes, which suggests that they are highly conserved. As expected, non-coding and SC regions exhibited higher divergence levels than coding and IR regions, respectively. The percentage of variation in non-coding regions ranged from 0 to 5.00%, with an average of 1.38%, which was threefold higher than that in the coding regions (0.40% on average; Figure 5). In the non-coding regions, the mean percentages of variations in the LSC, SSC, and IR regions were 1.59, 1.87, and 0.23%, respectively, which demonstrated that the IR region had fewer mutations and was highly conserved. However, in the coding regions, there were no significant differences among the regions (0.38, 0.54, and 0.32% for LSC, SSC, and IR regions, respectively), because there was a highly variable gene, ycf1 (2.33%), that was located in the IR region. Genes that were located in SC regions (rps16, rpl20, rpl22, and ndhF) also exhibited higher variability (average value > 1%) than the other genes.
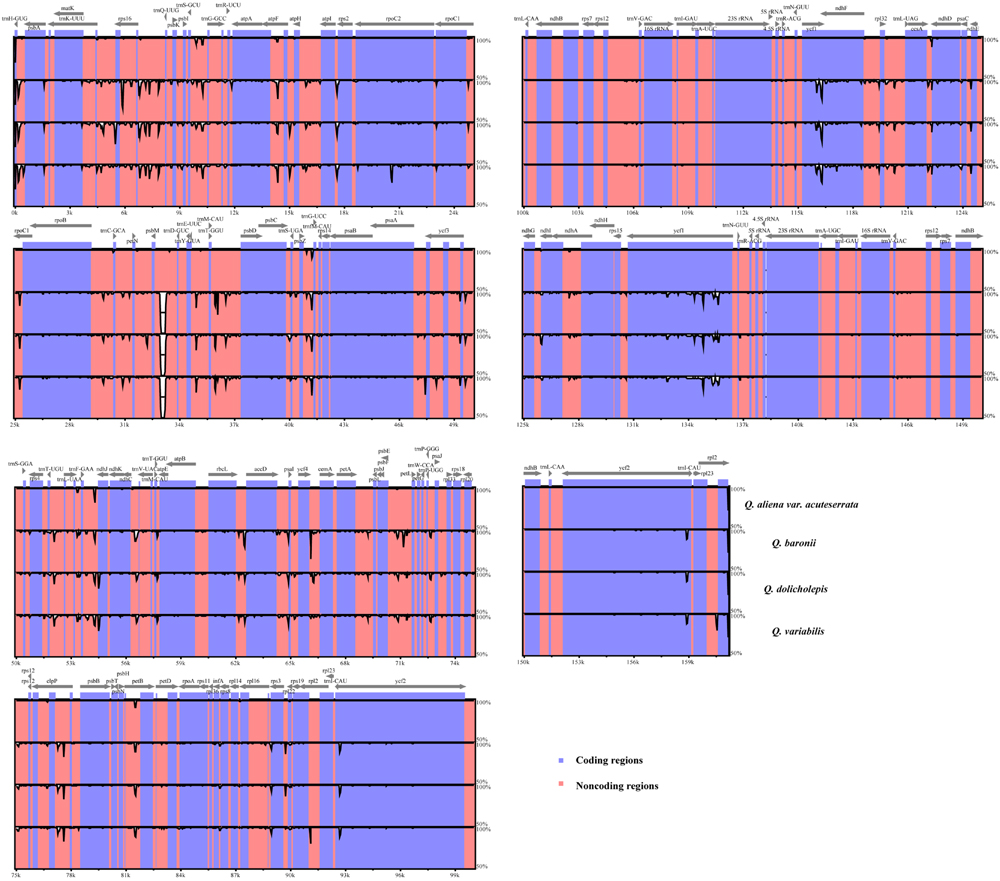
FIGURE 4. Sequence identity plot comparing the five Quercus chloroplast genomes with Q. aliena as a reference by using mVISTA. The y-axis represents % identity ranging from 50 to 100%. Coding and non-coding regions are marked in purple and pink, respectively.
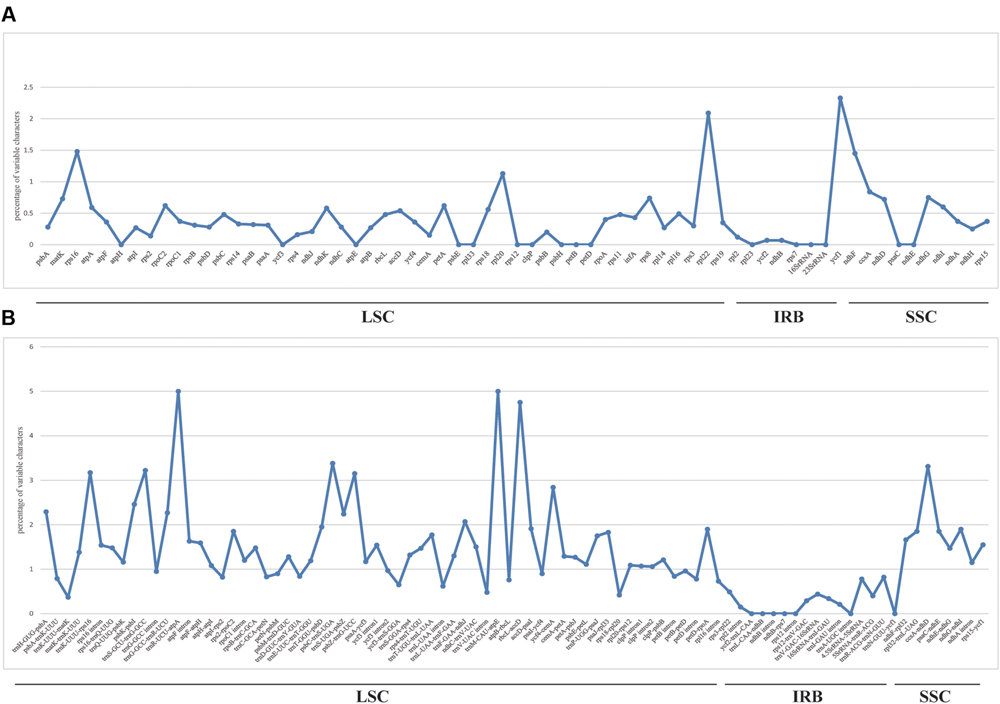
FIGURE 5. Percentage of variable characters in aligned five Quercus chloroplast genomes. (A) Coding region. (B) Non-coding region. These regions are oriented according to their locations in the chloroplast genome.
We then investigated sequence divergence patterns in the five cp genomes. We found 904 single nucleotide variants (SNVs; 0.56%) across the complete cp genomes of the five taxa, including 620 parsimony-informative sites (0.38%). There was a relatively small number of SNVs in the IR regions and coding sequences (Supplementary Table S5). The numbers of nucleotide substitutions and indels varied from 16 to 720 and 4 to 108, respectively (Table 4). There were always fewer transitions than transversions, and there were no transitions between Q. aliena and Q. aliena var. acuteserrata. Among the substitution events in the CDS region, all of the pairwise sequence comparisons showed that there was almost an equal number of synonymous and non-synonymous substitutions (Table 5); however, several NADH genes had more non-synonymous than synonymous substitutions, and most photosynthetic genes had only a few non-synonymous substitutions (Supplementary Tables S6 and S7). Most indels were located in non-coding regions, but some were also detected in psbA, rpoC2, rpl22, ycf1, ycf2, and ndhF. Interestingly, ycf1 had the most number of indels (Supplementary Table S8).

TABLE 4. Numbers of nucleotide substitutions and indels in five complete cp genomes.
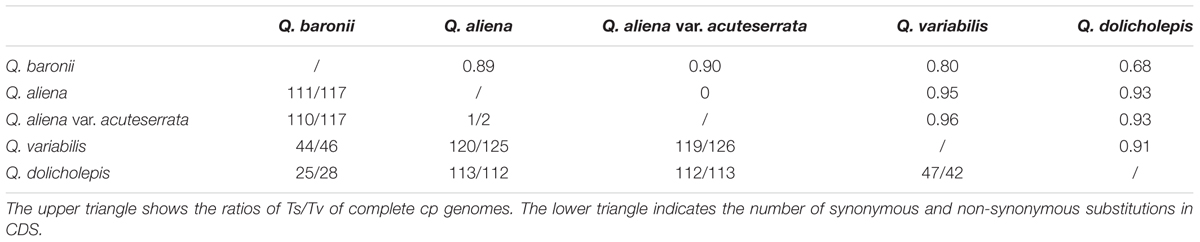
TABLE 5. Numbers of synonymous and non-synonymous substitutions in CDS and ratios of Ts/Tv in complete cp genomes.
To estimate selection pressures, ratios of non-synonymous (KA) versus synonymous (KS) substitutions were calculated for 79 protein-coding genes (Supplementary Table S9), and 293 pairwise comparison results were obtained. The KA/KS ratios of the remaining comparisons could not be calculated due to KS = 0. Four genes (ndhA, ndhK, petA, and ycf1) had KA/KS ratios above 1.0, indicating that these genes are under positive selection.
Phylogenetic Analysis
Six data partitions (Table 6) from the 10 Fagaceae cp genomes were used to construct the phylogenetic trees (Figure 6). All of the six datasets produced similar phylogenetic trees with moderate-to-high support, except for the IRB dataset, which received poor support. All of the datasets indicated that Q. aliena and Q. aliena var. acuteserrata form a monophyletic clade and then cluster with Q. rubra. Another monophyletic branch showed that Q. baronii appeared to be more closely related to Q. dolicholepis than to Q. variabilis. Differences in topological structure mainly involved the placements of Q. spinosa and Q. aquifolioides, which belong to Sect. Brachylepides. For example, in datasets 5 (LSC + SSC + IRB regions) and 6 (complete cp genome sequences), they clustered with Q. rubra; however, in datasets 1 (LSC region) and 4 (LSC + SSC regions), they formed a monophyletic group.
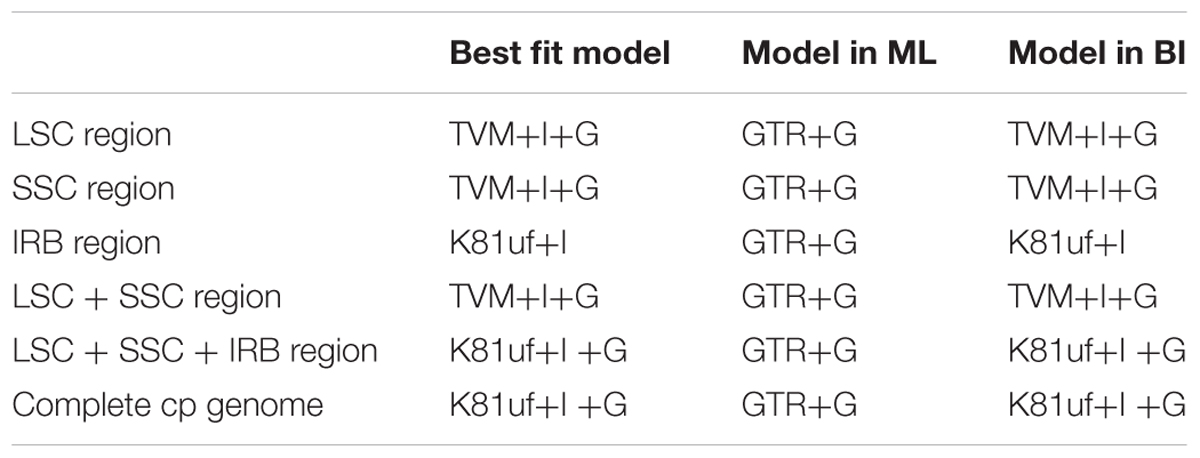
TABLE 6. Model in ML and BI analysis.
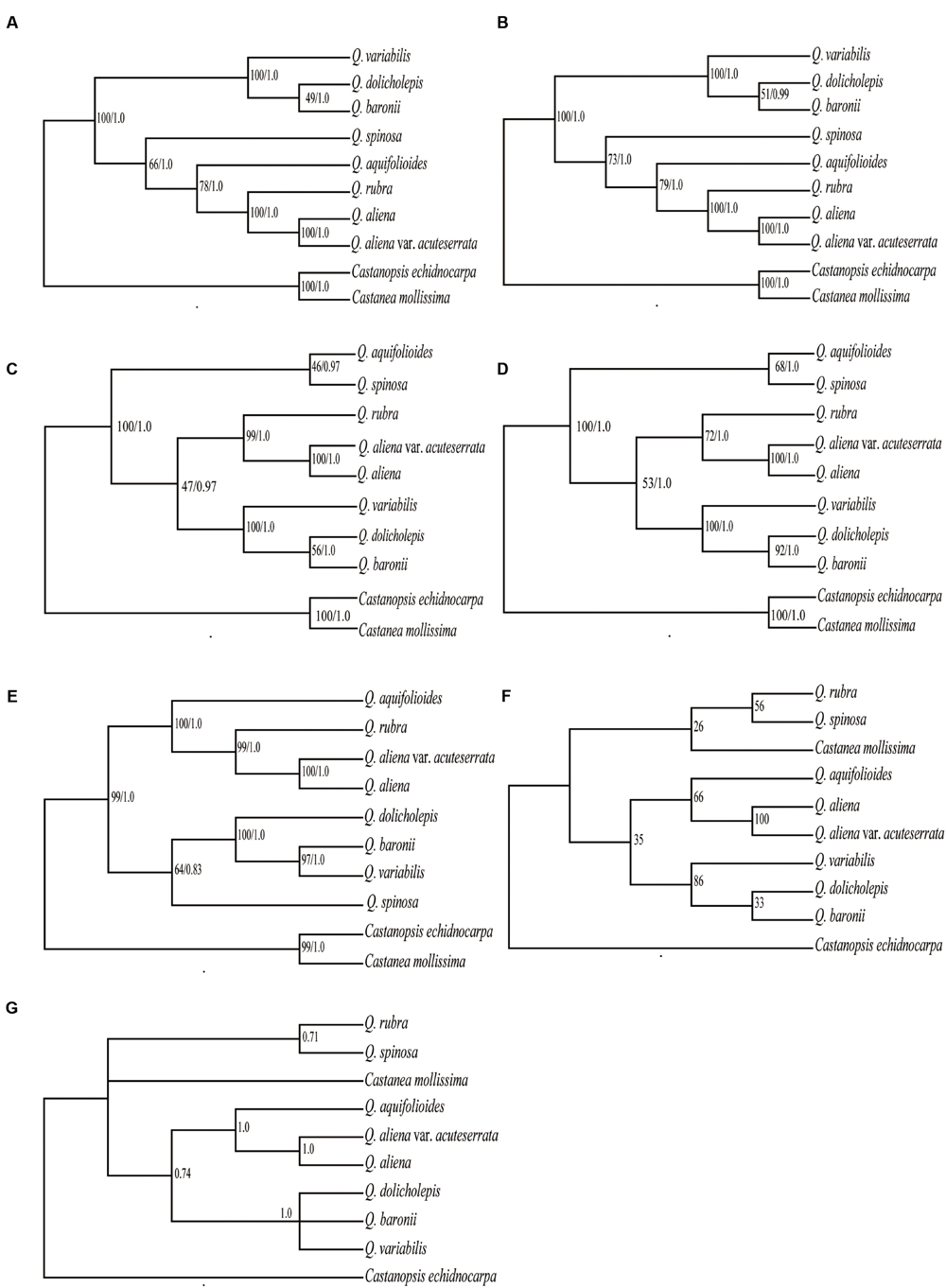
FIGURE 6. Phylogeny of the 10 Fagaceae species inferred from ML and BI analyses of different data partitions. (A) Whole chloroplast genome. (B) LSC+SSC+IRB region. (C) LSC+SSC region. (D) LSC region. (E) SSC region. (F) ML topology of IRB region. (G) BI topology of IRB region. The numbers associated with each node are bootstrap support values and posterior probability values in (A–E). The numbers associated with each node are bootstrap support values and posterior probability values in (F,G), respectively.
Discussion
Chloroplast Sequence Evolution
Although cp genomes are highly conserved in terms of genomic structure and size, the IR/SC junction position change may be caused by the contraction or expansion of the IR region, which is a common evolutionary phenomenon in plants (Kim and Lee, 2004; Hansen et al., 2007; Wang et al., 2008; Huang et al., 2014).
Larger and more complex repeat sequences may play an important role in the rearrangement of cp genomes and sequence divergence (Timme et al., 2007; Weng et al., 2013); therefore, we investigated the numbers and distributions of tandem, dispersed, and palindromic repeats. We found that repeats in different species were usually located in the same genes (ycf1 and ycf2), or genes with similar functions (psaB/psaA, trnS-GCU/trnS-UGA, trnG-GCC/trnG-UCC, and trnS-UGA/trnS-GGA).
Understanding nucleotide substitution rates is of fundamental importance in molecular evolution (Muse and Gaut, 1994), and indels play a significant role in evolutionary processes (Britten et al., 2003). Based on the numbers and distribution of SNVs, indels, and proportions of variability, the IR regions were more conserved than the SC regions. During the process of searching for SNVs and indels, we found that the cp genome sequences of Q. baronii, Q. dolicholepis, and Q. variabilis had similar mutation modes, while the other two species shared another mutation mode. Therefore, the phylogenetic relationships of these species may be affected by different mutation modes. Transitions occur at higher frequencies than transversions in almost all DNA sequences, and transition/transversion bias is a general property of DNA sequence evolution (Yang and Yoder, 1999). However, all of the pairwise sequence comparisons in our study revealed that there was a greater number of transversions than transitions. This has also been found in other taxa (Cai et al., 2015; Song et al., 2015; Kong and Yang, 2016), and may be due to a high AT content in the cp genome; transversion substitutions usually occur in datasets with a high AT content (Morton and Clegg, 1995; Morton et al., 1997). This bias may also be associated with genome content and the genetic characteristics of codons (Yang and Yoder, 1999; Morton, 2003). The estimation of synonymous and non-synonymous substitution rates may play an important role in understanding the dynamics of molecular evolution, and non-synonymous substitutions could be subject to natural selection during the evolutionary process (Yang and Nielsen, 2000; Seo and Kishino, 2008). In this study, the numbers of synonymous and non-synonymous substitutions in the CDS regions were almost equal. However, in several NADH genes, more non-synonymous substitutions than synonymous substitutions were detected, while most photosynthetic genes had only a few non-synonymous substitutions, possibly due to strong selection pressure during cp genome evolution (Matsuoka et al., 2002).
Because of complex evolutionary issues in Quercus, its taxonomy is still difficult to assess. Barcoding is a molecular tool that is used to identify living organisms (Hebert et al., 2003). The loci rbcL, matK, and trnH/psbA, and nuclear ribosomal internal transcribed spacers, are recommended regions for DNA barcoding in plants (CBOL Plant Working Group, 2009; Hollingsworth et al., 2011). In a DNA barcoding study of Quercus, these plastid markers and an extra locus (rpoC1) had extremely low discriminatory power (Piredda et al., 2011; Simeone et al., 2013). Therefore, we chose the five most variable coding regions and 14 most variable non-coding regions that might be regarded as potential molecular markers for Quercus species, with variation percentages that exceeded 1 and 2%, respectively. They were rps16, rpl20, rpl22, ycf1, ndhF, trnH-GUG/psbA, trnK-UUU/rps16, psbK/psbI, trnS-GCU/trnG-GCC, trnG-GCC/trnR-UCU, trnR-UCU/atpA, psbC/trnS-UGA, trnS-UGA/psbZ, psbZ/trnG-UCC, trnF-GAA/ndhJ, trnM-CAU/atpE, rbcL/accD, ycf4/cemA, and ccsA/ndhD. Primers for these regions are shown in Supplementary Table S10. Further work is still necessary to determine whether these highly variable regions could be used in Quercus phylogenetic analyses, or serve as candidate DNA barcodes.
Our analysis indicated that four genes were under positive selection (ndhA, ndhK, petA, and ycf1). Eleven genes (ndhA–ndhK) are found in the cp genomes of most land plants, and encode a NAD(P)H dehydrogenase (NDH) complex that is involved in photosystem I cyclic electron transport and chlororespiration (Kofer et al., 1998; Shikanai et al., 1998). The chloroplast NDH complex is divided into A, B, and membrane and lumen subcomplexes; ndhA is a member of a membrane subunit and ndhK belongs to subcomplex A (Peng et al., 2011). The chloroplast NDH monomer, which is sensitive to strong light intensity, might have changed drastically to develop novel functions for stress resistance (Peng et al., 2011). petA encodes the apoprotein of cytochrome f, which is a membrane component of the cytochrome bf complex and has the function of transferring electrons (Gray, 1992). ycf1 is one of the largest plastid genes, and encodes a protein that is a component of the chloroplast inner envelope membrane protein translocon (Kikuchi et al., 2013). Although this gene appears to be essential for cell survival in tobacco (Drescher et al., 2000), it is a pseudogene or has been lost in various groups, such as rice, maize, palm, and some Geraniaceae species (Maier et al., 1995; Yang et al., 2010; Weng et al., 2013). It has also been shown to be subject to positive selection in many lineages (Greiner et al., 2008; Carbonell-Caballero et al., 2015; Hu et al., 2015).
Phylogenetic Analysis
The phylogenetic trees, which were based on different datasets, produced similar topological structures except for the IRB dataset, possibly because IRB is more conserved and provides fewer variable sites than SC regions. Q. aliena and Q. aliena var. acuteserrata, which belong to Sect. Quercus, had the closest relationship among the species, because Q. aliena var. acuteserrata is considered a variant of Q. aliena (Wu et al., 1994). Q. rubra, which belongs to Sect. Lobatae, always had a close relationship with the above two species, and the phylogenetic trees also showed that Sect. Lobatae is a sister clade of Sect. Quercus (Hubert et al., 2014). Q. dolicholepis is closely related to Q. baronii, and both of them are members of Sect. Echinolepides, which is consistent with their morphological characteristics. A phylogenetic tree that was based on morphological characteristics showed that the two deciduous sections were closely related, and formed a sister clade to an intermediate group (Pu et al., 2001); however, the phylogeny inferred from the cp genomes showed that deciduous oaks should not be clustered in the same clade. Q. variabilis, which belongs to Sect. Aegilops, was clustered with an intermediate group. Furthermore, a phylogenetic reconstruction of 108 Quercus species (including Q. variabilis, Q. aliena, and Q. dolicholepis from China) based on multiple nuclear genes showed that Q. variabilis is more closely related to Q. dolicholepis than to Q. aliena (Hubert et al., 2014). Across the different datasets, the positions of Q. spinosa and Q. aquifolioides were not consistent, and the bootstrap values of the two species were not high enough (<80%), probably because the limited number of species in our study might have influenced the analysis. Therefore, it is necessary to use more species in order to verify the relationships among different sections. Overall, the phylogenetic relationships inferred from the cp genome data obtained high support values and were similar to those indicated by nuclear genes data, which suggests that cp genome data can effectively resolve the phylogenetic relationships of this genus.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by the Ph.D. Programs Foundation of Ministry of Education of China (Grand No. 20136101130001) and National Natural Science Foundation of China (Grand No. 31270364).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00959
Footnotes
References
Alexander, L. W., and Woeste, K. E. (2014). Pyrosequencing of the northern red oak (Quercus rubra L.) chloroplast genome reveals high quality polymorphisms for population management. Tree Genet. Genomes 10, 803–812. doi: 10.1007/s11295-013-0681-1
Alkan, C., Sajjadian, S., and Eichler, E. E. (2011). Limitations of next-generation genome sequence assembly. Nat. Methods 8, 61–65. doi: 10.1038/nmeth.1527
Barrett, C. F., Davis, J. I., Leebens-Mack, J., Conran, J. G., and Stevenson, D. W. (2013). Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics 29, 65–87. doi: 10.1111/j.1096-0031.2012.00418.x
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucl. Acids Res. 27, 573. doi: 10.1093/nar/27.2.573
Birky, C. W., Maruyama, T., and Fuerst, P. (1983). An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics 103, 513–527.
Britten, R. J., Rowen, L., Williams, J., and Cameron, R. A. (2003). Majority of divergence between closely related DNA samples is due to indels. Proc. Natl. Acad. Sci. U.S.A. 100, 4661–4665. doi: 10.1073/pnas.0330964100
Burgarella, C., Lorenzo, Z., Jabbour-Zahab, R., Lumaret, R., Guichoux, E., Petit, R., et al. (2009). Detection of hybrids in nature: application to oaks (Quercus suber and Q. ilex). Heredity 102, 442–452. doi: 10.1038/hdy.2009.8
Cai, J., Ma, P. F., Li, H. T., and Li, D. Z. (2015). Complete plastid genome sequencing of four Tilia species (Malvaceae): a comparative analysis and phylogenetic implications. PLoS ONE 10:e0142705. doi: 10.1371/journal.pone.0142705
Cai, Z., Guisinger, M., Kim, H. G., Ruck, E., Blazier, J. C., McMurtry, V., et al. (2008). Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. J. Mol. Evol. 67, 696–704. doi: 10.1007/s00239-008-9180-7
Carbonell-Caballero, J., Alonso, R., Ibañez, V., Terol, J., Talon, M., and Dopazo, J. (2015). A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 32, 2015–2035. doi: 10.1093/molbev/msv082
Cavender-Bares, J., Ackerly, D., Baum, D., and Bazzaz, F. (2004). Phylogenetic overdispersion in Floridian oak communities. Am. Nat. 163, 823–843. doi: 10.1086/386375
CBOL Plant Working Group. (2009). A DNA barcode for land plants. Proc. Natl. Acad. Sci. U.S.A. 106, 12794–12797. doi: 10.1073/pnas.0905845106
Cosner, M. E., Raubeson, L. A., and Jansen, R. K. (2004). Chloroplast DNA rearrangements in Campanulaceae: phylogenetic utility of highly rearranged genomes. BMC Evol. Biol. 4:1. doi: 10.1186/1471-2148-4-27
Curtu, A. L., Gailing, O., and Finkeldey, R. (2007). Evidence for hybridization and introgression within a species-rich oak (Quercus spp.) community. BMC Evol. Biol. 7:1. doi: 10.1186/1471-2148-7-218
Dane, F., Wang, Z., and Goertzen, L. (2015). Analysis of the complete chloroplast genome of Castanea pumila var. pumila, the Allegheny chinkapin. Tree Genet. Genomes 11, 1–6. doi: 10.1007/s11295-015-0840-7
Denk, T., and Grimm, G. W. (2009). Significance of pollen characteristics for infrageneric classification and phylogeny in Quercus (Fagaceae). Int. J. Plant Sci. 170, 926–940. doi: 10.1086/600134
Denk, T., and Grimm, G. W. (2010). The oaks of western Eurasia: traditional classifications and evidence from two nuclear markers. Taxon 59, 351–366.
Doyle, J. J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19, 11–15.
Drescher, A., Ruf, S., Calsa, T., Carrer, H., and Bock, R. (2000). The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 22, 97–104. doi: 10.1046/j.1365-313x.2000.00722.x
Du, F. K., Lang, T., Lu, S., Wang, Y., Li, J., and Yin, K. (2015). An improved method for chloroplast genome sequencing in non-model forest tree species. Tree Genet. Genomes 11, 1–14. doi: 10.1007/s11295-015-0942-2
Faircloth, B. C. (2008). MSATCOMMANDER: detection of microsatellite repeat arrays and automated, locus-specific primer design. Mol. Ecol. Resour. 8, 92–94. doi: 10.1111/j.1471-8286.2007.01884.x
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Gray, J. C. (1992). Cytochrome f: structure, function and biosynthesis. Photosynth. Res. 34, 359–374. doi: 10.1007/BF00029811
Greiner, S., Wang, X., Herrmann, R. G., Rauwolf, U., Mayer, K., Haberer, G., et al. (2008). The complete nucleotide sequences of the 5 genetically distinct plastid genomes of Oenothera, subsection Oenothera: II. A microevolutionary view using bioinformatics and formal genetic data. Mol. Biol. Evol. 25, 2019–2030. doi: 10.1093/molbev/msn149
Guisinger, M. M., Chumley, T. W., Kuehl, J. V., Boore, J. L., and Jansen, R. K. (2010). Implications of the plastid genome sequence of Typha (Typhaceae, Poales) for understanding genome evolution in Poaceae. J. Mol. Evol. 70, 149–166. doi: 10.1007/s00239-009-9317-3
Guisinger, M. M., Kuehl, J. V., Boore, J. L., and Jansen, R. K. (2011). Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol. Biol. Evol. 28, 583–600. doi: 10.1093/molbev/msq229
Hahn, C., Bachmann, L., and Chevreux, B. (2013). Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads - a baiting and iterative mapping approach. Nucl. Acids Res. 41:e129. doi: 10.1093/nar/gkt371
Hansen, D. R., Dastidar, S. G., Cai, Z., Penaflor, C., Kuehl, J. V., Boore, J. L., et al. (2007). Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae). Mol. Phylogenet. Evol. 45, 547–563. doi: 10.1016/j.ympev.2007.06.004
Hebert, P. D., Ratnasingham, S., and de Waard, J. R. (2003). Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. Lond. B Biol. 270, S96–S99. doi: 10.1098/rsbl.2003.0025
Hollingsworth, P. M., Graham, S. W., and Little, D. P. (2011). Choosing and using a plant DNA barcode. PLoS ONE 6:e19254. doi: 10.1371/journal.pone.0019254
Hu, S., Sablok, G., Wang, B., Qu, D., Barbaro, E., Viola, R., et al. (2015). Plastome organization and evolution of chloroplast genes in Cardamine species adapted to contrasting habitats. BMC Genomics 16:1. doi: 10.1186/s12864-015-1498-0
Huang, H., Shi, C., Liu, Y., Mao, S. Y., and Gao, L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:1. doi: 10.1186/1471-2148-14-151
Hubert, F., Grimm, G. W., Jousselin, E., Berry, V., Franc, A., and Kremer, A. (2014). Multiple nuclear genes stabilize the phylogenetic backbone of the genus Quercus. Syst. Biodivers. 12, 405–423. doi: 10.1080/14772000.2014.941037
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., dePamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U.S.A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Jansen, R. K., Raubeson, L. A., Boore, J. L., dePamphilis, C. W., Chumley, T. W., Haberle, R. C., et al. (2005). Methods for obtaining and analyzing whole chloroplast genome sequences. Method Enzymol. 395, 348–384. doi: 10.1016/S0076-6879(05)95020-9
Jansen, R. K., Saski, C., Lee, S. B., Hansen, A. K., and Daniell, H. (2011). Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 28, 835–847. doi: 10.1093/molbev/msq261
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kikuchi, S., Bédard, J., Hirano, M., Hirabayashi, Y., Oishi, M., Imai, M., et al. (2013). Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 339, 571–574. doi: 10.1126/science.1229262
Kim, K. J., and Lee, H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kofer, W., Koop, H. U., Wanner, G., and Steinmüller, K. (1998). Mutagenesis of the genes encoding subunits A, C, H, I, J and K of the plastid NAD (P) H-plastoquinone-oxidoreductase in tobacco by polyethylene glycol-mediated plastome transformation. Mol. Gen. Genet. 258, 166–173. doi: 10.1007/s004380050719
Kole, C. (2011). Wild Crop Relatives: Genomic and Breeding Resources: Industrial Crops. Berlin: Springer Press.
Kong, W., and Yang, J. (2016). The complete chloroplast genome sequence of Morus mongolica and a comparative analysis within the Fabidae clade. Curr. Genet. 62, 165–172. doi: 10.1007/s00294-015-0507-9
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucl. Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lee, H. L., Jansen, R. K., Chumley, T. W., and Kim, K. J. (2007). Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 24, 1161–1180. doi: 10.1093/molbev/msm036
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucl. Acids Res. 41, 575–581. doi: 10.1093/nar/gkt289
Lu, S., Hou, M., Du, F. K., Li, J., and Yin, K. (2015). Complete chloroplast genome of the Oriental white oak: Quercus aliena Blume. Mitochondrial DNA A DNA MappSeq. Anal. 27, 2802–2804. doi: 10.3109/19401736.2015.1053074
Ma, P. F., Zhang, Y. X., Zeng, C. X., Guo, Z. H., and Li, D. Z. (2014). Chloroplast phylogenomic analysis resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 63, 933–950. doi: 10.1093/sysbio/syu054
Maier, R. M., Neckermann, K., Igloi, G. L., and Kössel, H. (1995). Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 251, 614–628. doi: 10.1006/jmbi.1995.0460
Manos, P. S., Doyle, J. J., and Nixon, K. C. (1999). Phylogeny, biogeography, and processes of molecular differentiation in Quercus subgenus Quercus (Fagaceae). Mol. Phylogenet. Evol. 12, 333–349. doi: 10.1006/mpev.1999.0614
Martin, G. E., Rousseau-Gueutin, M., Cordonnier, S., Lima, O., Michon-Coudouel, S., Naquin, D., et al. (2014). The first complete chloroplast genome of the Genistoid legume Lupinus luteus: evidence for a novel major lineage-specific rearrangement and new insights regarding plastome evolution in the legume family. Ann. Bot. 113, 1197–1210. doi: 10.1093/aob/mcu050
Matsuoka, Y., Yamazaki, Y., Ogihara, Y., and Tsunewaki, K. (2002). Whole chloroplast genome comparison of rice, maize, and wheat: implications for chloroplast gene diversification and phylogeny of cereals. Mol. Biol. Evol. 19, 2084–2091. doi: 10.1093/oxfordjournals.molbev.a004033
Moore, M. J., Bell, C. D., Soltis, P. S., and Soltis, D. E. (2007). Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. U.S.A. 104, 19363–19368. doi: 10.1073/pnas.0708072104
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G., and Soltis, D. E. (2010). Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. U.S.A. 107, 4623–4628. doi: 10.1073/pnas.0907801107
Moran, E. V., Willis, J., and Clark, J. S. (2012). Genetic evidence for hybridization in red oaks (Quercus sect, Lobatae, Fagaceae). Am. J. Bot. 99, 92–100. doi: 10.3732/ajb.1100023
Morton, B. R. (2003). The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J. Mol. Evol. 56, 616–629. doi: 10.1007/s00239-002-2430-1
Morton, B. R., and Clegg, M. T. (1995). Neighboring base composition is strongly correlated with base substitution bias in a region of the chloroplast genome. J. Mol. Evol. 41, 597–603. doi: 10.1007/BF00175818
Morton, B. R., Oberholzer, V. M., and Clegg, M. T. (1997). The influence of specific neighboring bases on substitution bias in noncoding regions of the plant chloroplast genome. J. Mol. Evol. 45, 227–231. doi: 10.1007/PL00006224
Muse, S. V., and Gaut, B. S. (1994). A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol. 11, 715–724.
Nixon, K. C. (1993). Infrageneric classification of Quercus (Fagaceae) and typification of sectional names. Ann. For. Sci. 50, 25s–34s. doi: 10.1051/forest:19930701
Oh, S. H., and Manos, P. S. (2008). Molecular phylogenetics and cupule evolution in Fagaceae as inferred from nuclear CRABS CLAW sequences. Taxon 57, 434–451. doi: 10.2307/25066014
Parks, M., Cronn, R., and Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:1. doi: 10.1186/1741-7007-7-84
Peng, L., Yamamoto, H., and Shikanai, T. (2011). Structure and biogenesis of the chloroplast NAD (P) H dehydrogenase complex. BBA-Bioenergetics 1807, 945–953. doi: 10.1016/j.bbabio.2010.10.015
Peng, Y. S., Chen, L., and Li, J. Q. (2007). Study on numerical taxonomy of Quercus L.(Fagaceae) in China. J. Wuhan Bot. Res. 25, 149–157. doi: 10.1016/j.jcis.2009.12.058
Piredda, R., Simeone, M. C., Attimonelli, M., Bellarosa, R., and Schirone, B. (2011). Prospects of barcoding the Italian wild dendroflora: oaks reveal severe limitations to tracking species identity. Mol. Ecol. Resour. 11, 72–83. doi: 10.1111/j.1755-0998.2010.02900.x
Posada, D., and Crandall, K. A. (1998). Modeltest: testing the model of DNA substitution. Bioinformatics 14, 817–818. doi: 10.1093/bioinformatics/14.9.817
Pu, C. X., Zhou, Z. K., and Luo, Y. (2001). A cladistic analysis of Quercus (Fagaceae) in China based on leaf epidermic and architecture. Acta Bot. Yunnanica 24, 689–698.
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rushton, B. (1993). Natural hybridization within the genus Quercus L. Ann. For. Sci. 50, 73s–90s. doi: 10.1051/forest:19930707
Seo, T. K., and Kishino, H. (2008). Synonymous substitutions substantially improve evolutionary inference from highly diverged proteins. Syst. Biol. 57, 367–377. doi: 10.1080/10635150802158670
Shaw, J., Shafer, H. L., Leonard, O. R., Kovach, M. J., Schorr, M., and Morris, A. B. (2014). Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. Am. J. Bot. 101, 1987–2004. doi: 10.3732/ajb.1400398
Shikanai, T., Endo, T., Hashimoto, T., Yamada, Y., Asada, K., and Yokota, A. (1998). Directed disruption of the tobacco ndhB gene impairs cyclic electron flow around photosystem I. Proc. Natl. Acad. Sci. U.S.A. 95, 9705–9709. doi: 10.1073/pnas.95.16.9705
Simeone, M. C., Piredda, R., Papini, A., Vessella, F., and Schirone, B. (2013). Application of plastid and nuclear markers to DNA barcoding of Euro-Mediterranean oaks (Quercus, Fagaceae): problems, prospects and phylogenetic implications. Bot. J. Linn. Soc. 172, 478–499. doi: 10.1111/boj.12059
Song, Y., Dong, W., Liu, B., Xu, C., Yao, X., Gao, J., et al. (2015). Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machilus balansae in the family Lauraceae. Front. Plant Sci. 6:662. doi: 10.3389/fpls.2015.00662
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analysis with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Timme, R. E., Kuehl, J. V., Boore, J. L., and Jansen, R. K. (2007). A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: identification of divergent regions and categorization of shared repeats. Am. J. Bot. 94, 302–312. doi: 10.3732/ajb.94.3.302
Wang, R. J., Cheng, C. L., Chang, C. C., Wu, C. L., Su, T. M., and Chaw, S. M. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8:1. doi: 10.1186/1471-2148-8-36
Weng, M. L., Blazier, J. C., Govindu, M., and Jansen, R. K. (2013). Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats and nucleotide substitution rates. Mol. Biol. Evol. 31, 645–659. doi: 10.1093/molbev/mst257
Wu, Z.-Y., Raven, P. H., Shehbaz, I. A. A., and Bartholomew, B. (1994). Flora of China. Beijing: Science Press.
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Yang, M., Zhang, X., Liu, G., Yin, Y., Chen, K., Yun, Q., et al. (2010). The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 5:e12762. doi: 10.1371/journal.pone.0012762
Yang, Y. C., Zhou, T., Yang, J., Meng, X., Zhu, J., and Zhao, G. F. (2015). The complete chloroplast genome of Quercus baronii (Quercus L.). Mitochondrial DNA [Epub ahead of print]. doi: 10.3109/19401736.2015.1118084
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., and Nielsen, R. (2000). Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43. doi: 10.1093/oxfordjournals.molbev.a026236
Yang, Z., and Yoder, A. D. (1999). Estimation of the transition/transversion rate bias and species sampling. J. Mol. Evol. 48, 274–283. doi: 10.1007/PL00006470
Zhang, Y. J., Ma, P. F., and Li, D. Z. (2011). High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 6:e20596. doi: 10.1371/journal.pone.0020596
Keywords: Quercus, chloroplast genome, repeat, nucleotide substitution, positive selection, plastid marker, phylogeny
Citation: Yang Y, Zhou T, Duan D, Yang J, Feng L and Zhao G (2016) Comparative Analysis of the Complete Chloroplast Genomes of Five Quercus Species. Front. Plant Sci. 7:959. doi: 10.3389/fpls.2016.00959
Received: 18 March 2016; Accepted: 15 June 2016;
Published: 28 June 2016.
Edited by:
Xiaowu Wang, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Ming Kang, Chinese Academy of Sciences, ChinaTae-Jin Yang, Seoul National University, South Korea
Copyright © 2016 Yang, Zhou, Duan, Yang, Feng and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guifang Zhao, Z2Z6aGFvQG53dS5lZHUuY24=