 Shengnan Huang
Shengnan Huang Zhiyong Liu†
Zhiyong Liu† Hui Feng
Hui Feng- Department of Horticulture, Shenyang Agricultural University, Shenyang, China
Female-sterile mutants are ideal materials for studying pistil development in plants. Here, we identified a female-sterile mutant fsm in Chinese cabbage. This mutant, which exhibited stable inheritance, was derived from Chinese cabbage DH line ‘FT’ using a combination of isolated microspore culture and ethyl methanesulfonate mutagenesis. Compared with the wild-type line ‘FT,’ the fsm plants exhibited pistil abortion, and floral organs were also relatively smaller. Genetic analysis indicated that the phenotype of fsm is controlled by a single recessive nuclear gene. Morphological observations revealed that the presence of abnormal ovules in fsm likely influenced normal fertilization process, ultimately leading to female sterility. Comparative transcriptome analysis on the flower buds of ‘FT’ and fsm using RNA-Seq revealed a total of 1,872 differentially expressed genes (DEGs). Of these, a number of genes involved in pistil development were identified, such as PRETTY FEW SEEDS 2 (PFS2), temperature-induced lipocalin (TIL), AGAMOUS-LIKE (AGL), and HECATE (HEC). Furthermore, GO and KEGG pathway enrichment analyses of the DEGs suggested that a variety of biological processes and metabolic pathways are significantly enriched during pistil development. In addition, the expression patterns of 16 DEGs, including four pistil development-related genes and 12 floral organ development-related genes, were analyzed using qRT-PCR. A total of 31,272 single nucleotide polymorphisms were specifically detected in fsm. These results contribute to shed light on the regulatory mechanisms underlying pistil development in Chinese cabbage.
Introduction
Floral organ development is the most obvious characteristic of the reproductive stage of flowering plants. The flowers of typical dicotyledonous plants are composed of four wheel-like structures (whorls). The first whorl (from outside to inside) contains the sepals, the second contains the petals, the third contains the stamens, and the innermost whorl contains the carpels, which are the female organs. In recent years, a variety of floral organ mutants have been characterized, leading to the isolation of a series of floral development- and morphogenesis-related genes using various techniques, and the expression patterns and functions of these genes have also been analyzed (Hu et al., 2003; Szécsi et al., 2006; Koyama et al., 2007; Nag et al., 2009; Sarvepalli and Nat, 2011; Lu et al., 2012; Huang et al., 2015). Studies on the commonality and characteristics of floral development in different species can help elucidate the origins and evolution of flowers. Such information would lay the foundation for altering the process of floral development, which might be used to control the flowering process and fertility in plants.
Female sterility has been identified in crops such as wheat (Triticum aestivum) (Dou et al., 2001), rice (Oryza sativa) (Tang et al., 2002), soybean (Glycine max) (Pereira et al., 1997), pearl millet (Pennisetum glaucum) (Arthur et al., 1993), ramie (Boehmeria nivea) (Zhou, 1996), and rapeseed (Brassica napus) (Chen et al., 2003). Studies have been performed on the biological characteristics, development, genetic methods, and potential applications of female sterility (Brown and Bingham, 1984; Luo et al., 1996; Daskalov and Mihailov, 1998; Xiao et al., 1998; Zhong et al., 1998; Li and Zheng, 2002; Liu et al., 2003; Sun et al., 2009). Female-sterile mutants have been used to identify regulatory genes that influence ovule and female gametophyte development. These regulatory genes are involved in nucellar and integument cell development (Schiefthaler et al., 1999; Balasubramanian and Schneitz, 2000, 2002), the regulation of megasporogenesis (Schneitz et al., 1997; Singh et al., 2011; Chevalier et al., 2013), metabolism, division and differentiation, and the developmental regulation of embryo sac cells (Pagnussat et al., 2005; Portereiko et al., 2006; Johnston et al., 2007; Jones-Rhoades et al., 2007; Colombo et al., 2008; Moll et al., 2008; Punwani et al., 2008).
The female-sterile mutants (with abnormal ovule and embryo sac development) have recently been produced through mutagenesis, including chemical mutagenesis, transposon mutagenesis, and T-DNA insertion mutagenesis (Bao et al., 2005; Venkatesan and Monica, 2010). Mutation of genes controlling female organ development, such as carpel development gene DROPPING LEAF (DL) and ovule development genes FLORAL BINDING PROTEIN 7 (FBP7) and SHATTERPROOF 1 (SHP1) (Cheng et al., 2000; Ferrándiz et al., 2000; Yamaguchi et al., 2004), can lead to female sterility. In addition, such mutations can lead to abnormal ovule or endosperm development, thereby affecting the function of female gametophytes as well as megasporogenesis (Huang and Sheridan, 1996; Siddiqi et al., 2000; Shi et al., 2005). Luo et al. (2001) found that the abnormal pistil development trait in rice mutant dl(t) is controlled by a single recessive gene, whose function may be similar to that of SUPERMAN (SUP), which regulates floral organ development in Arabidopsis thaliana (Sakai et al., 1995). The SUP inhibits the expression of floral organ development genes in the pistil, functioning as a cadastral gene of stamen and pistil genes (Jacobsen and Meyerowitz, 1997; Nibau et al., 2011). These mutants would be useful for studying female sterility-related proteins, cloning the related genes, and further analyzing their structures and functions, shedding light on the molecular mechanisms underlying sex differentiation and development in floral organs.
The male-sterile line has played an important role in the plant hybrid breeding. The research showed that the female-sterile line can also be used as the pollinator, the pollination distance between the both parents can be reduced, thus improving the hybrid seed yield. Therefore, further studies of female-sterile mutants not only help uncover additional information about floral organ development, but also may provide important basic materials for hybrid breeding (Maruyama et al., 1991; Dou et al., 2001).
Transcriptome analysis, i.e., investigating transcription and the regulation of all genes in an organism at the genome-wide level, is an important component of functional genomics research (Qi et al., 2011; Li et al., 2013). Transcriptome analysis can uncover the global expression patterns of genes and provide information about gene-protein interactions in plants (Filichkin et al., 2010; Montgomery et al., 2010; Faulconnier et al., 2011; Li et al., 2012; Ren et al., 2012; Zhang et al., 2012; Huang et al., 2015). The recent transcriptional profiling studies on the female gametophyte development have demonstrated that the female gametophyte formation is a complicated process, and numerous genes are involved in the regulation of female gametophyte formation in several plant species (Pagnussat et al., 2005; Anderson et al., 2013; Chettoor et al., 2014). Therefore, the female-sterile mutants are ideal for revealing the molecular mechanism of pistil development, and further analyses of the gene expression changes during pistil development are very necessary.
Chinese cabbage (Brassica campestris ssp. pekinensis [Lour] Olsson), an economically and nutritionally important vegetable crop, is widely cultivated in Northeast Asia. With the completion of genome sequencing of Chinese cabbage (Wang et al., 2011), transcriptome analysis of this crop has become an important field of study. Considering that the female-sterile plants are a powerful tool to study genes involved in the pistil development and investigate gene functions (Arthur et al., 1993; Rosellini et al., 1998, 2003), and thus the female-sterile mutants may be useful for the study of the reproductive system of Chinese cabbage.
In this study, we identified a female-sterile mutant (fsm) in Chinese cabbage. To help elucidate the molecular mechanism underlying pistil development, we conducted comparative transcriptome analysis using the Illumina sequencing platform HiSeqTM 2000 to characterize the gene expression profiles in flower buds of fsm and the corresponding wild-type line ‘FT’ on a global level. The main objective of this study was to identify differentially expressed genes (DEGs) and potential candidate genes related to pistil development. Our results provide a comprehensive view of the transcriptome of Chinese cabbage, which contributes to increase our understanding of the regulatory mechanisms underlying pistil development in this crop.
Materials and Methods
Plant Materials and Mutagenic Treatment
The fsm mutant was derived from a Chinese cabbage doubled-haploid (DH) line ‘FT’ via a combination of isolated microspore culture and ethyl methanesulfonate (EMS) mutagenesis. Based on our parallel study (Huang et al., 2016a), the isolated microspores were treated with 0.08% EMS solution for 10 min. The microspore regenerated plants were transplanted to pots and cultivated in the greenhouse for further growth and development.
It is thought that the genetic background between ‘FT’ and fsm is highly consistent, with the difference mainly occurring at the mutation sites.
Morphological Observation
In December 2014, the seeds of ‘FT’ and fsm were sown in a greenhouse at Shenyang Agricultural University, China. In March 2015, morphological analyses of ‘FT’ and fsm plants were carried out at the full-bloom stage.
Floral Organs Observation
Four-wheeled floral organs were directly observed and photographed under a dissecting microscope (Nikon SMZ800, Japan).
Identification of Female Sterility
Three ‘FT’ and fsm plants were respectively selected, and bagged for 3 days before pollination. Twenty flower buds were randomly selected from each plant, and the artificial self-pollination of ‘FT’ and fsm was carried out. Additionally, 20 flower buds were randomly selected from each plant again, and a reciprocal cross between ‘FT’ and fsm was performed. After the seeds were mature, the seed setting rates were recorded.
Pollen Viability Detection
The anthers were removed from the stamens and the pollen from each anther was extruded onto a slide. The pollen was immersed in 0.1% TTC (2,3,5-triphenyltetrazolium chloride) dye solution, covered with a cover slip, and dyed for 15–20 min at 35–37°C in an incubator. Pollen viability was observed under an optical microscope (Nikon ECLIPSE 80i, Japan).
Ovary and Ovule Development Observation
Ten flower buds were randomly selected from ‘FT’ and fsm plants, respectively. Flowers on the first flowering day of ‘FT’ and fsm were respectively marked and artificially pollinated. The length and width of the ovaries were measured every other day; ovaries were measured six times, and each measurement was performed in three independent experiments. The ovules in the ovary of ‘FT’ and fsm on the first flowering day and the 5th day after pollination were respectively observed and compared under a dissecting microscope (Nikon SMZ800, Japan); Accordingly, the pistils on the first flowering day and the 5th day after pollination were fixed using FAA (Formalin-Aceto-Alcohol) fixative and then dyed using safranine and fast green. The detailed procedures of paraffin section were performed according to the traditional method of Li (1996), and the ovule development was observed under an optical microscope (Nikon ECLIPSE 80i, Japan).
Genetic Analysis
To investigate the inheritance of fsm, ‘FT’ (P1) and fsm (P2) were used as the parents to develop the F1, BC1, and F2 populations. Phenotypic data were obtained for each plant of the P1, P2, F1, BC1, and F2 populations, and the segregation ratios of the BC1 and F2 populations were analyzed by a Chi-square (χ2) test.
RNA Extraction
In March 2015, developing flower buds were collected from ‘FT’ and fsm at the full-bloom stage and used as transcriptome sequencing materials. All samples were immediately frozen in liquid nitrogen and stored at -80°C.
At the full-bloom stage, five ‘FT’ and fsm plants were respectively selected, and three inflorescences were randomly selected from each plant. All the flower buds from these inflorescences collected from five ‘FT’ and fsm plants were respectively mixed, and the mixed samples were used as a single biological replicate; three independent biological replicates were performed for ‘FT’ and fsm.
Total RNA from six samples of ‘FT’ and fsm (with three independent biological replicates) was respectively extracted using TRIzol reagent (Invitrogen, USA) following the manufacturer’s instructions, and the DNase treatment was applied during RNA extraction to reduce DNA contamination. The quality and integrity of all RNA samples were assessed with a 2100 Bioanalyzer (Agilent Technologies, USA) and by electrophoresis on 1.0% agarose gels.
cDNA Library Construction and Illumina Sequencing
To construct six cDNA libraries, equal amounts of total RNA from the three independent biological replicates of ‘FT’ and fsm were pooled for RNA-Seq library construction, which were designated F1, F2, F4, M1, M2, and M3, respectively. Oligo (dT)-coated magnetic beads were used to isolate mRNA, which was broken into small fragments by the addition of fragmentation buffer. First-strand cDNA was synthesized with these short fragments serving as templates, and second-strand cDNA was synthesized using the reaction system. The short fragments were purified and subjected to end repair and the addition of sequencing adapters. Following agarose gel electrophoresis, suitable fragments were selected as templates for PCR amplification. Quantification and quality analysis of the constructed libraries were conducted using an Agilent 2100 Bioanalyzer and an ABI StepOnePlus Real-Time PCR System (Li et al., 2011; Huang et al., 2016b). The cDNA libraries were then sequenced on Illumina sequencing platform HiSeqTM 2000 with a paired-end sequencing strategy at Beijing Genomics of Institute (BGI), Shenzhen, China.
Mapping of Reads to the Reference Genome
Prior to bioinformatic analysis, the raw image data were transformed into sequence data by base calling. Clean reads were obtained by removing reads containing adaptors, reads with more than 10% unknown nucleotides, and low-quality reads with more than 50% bases with a quality value < 20. Clean reads were mapped to the reference genome (Version 1.5)1 with SOAPaligner/SOAP2 (Li et al., 2009b), allowing no more than five base mismatches in the alignment.
Identification of SNPs
The genotype of three biological replicates of ‘FT’ and fsm were identical. Therefore, RNA-Seq data from the three biological replicates of ‘FT’ and fsm were combined for single nucleotide polymorphism (SNP) identification, respectively. In this study, SNPs were identified using SOAPsnp software (Li et al., 2009a). The SOAPsnp program is a resequencing utility that can detect the consensus sequence for the transcriptome of a sequencing individual based on the alignment of the sequencing reads on the known reference sequence. The SNPs can then be identified on the consensus sequence through the comparison with the reference sequence.
Assessment of Differential Gene Expression
The expression levels of genes determined by RNA-Seq were normalized by the RPKM (reads per kb per million mapped reads) method, thereby limiting the effects of different gene lengths and sequencing levels on the calculation of gene expression level (Mortazavi et al., 2008). DESeq was applied to identify DEGs based on the RPKM-derived baseMean for each gene between samples (Anders and Huber, 2010). The false discovery rate (FDR) was used as the threshold of P-value in multiple tests (Benjamini and Hochberg, 1995). The combination of FDR ≤ 0.001 and the absolute value of log2Ratio ≥ 1 were used as the threshold for judging the significance of differences in gene expression (Benjamini and Yekutieli, 2001). More stringent criteria, including smaller FDR and larger fold-change values, can be used to identify DEGs. In the current study, genes with FDR ≤ 0.001 and the absolute value of log2 Ratio ≥ 4 were defined as DEGs. In addition, the specifically expressed genes (SEGs) were detected, i.e., genes that were not expressed in one library but had baseMean values ≥ 11 in the other library (Tao et al., 2012).
Functional Enrichment Analysis of DEGs
To characterize the biological functions and metabolic pathways of the DEGs, the DEGs were subjected to Gene Ontology (GO)2 functional analysis (Ashburner et al., 2000) and Kyoto Encyclopedia of Genes and Genomes (KEGG)3 pathway enrichment analysis (Kanehisa et al., 2008). Compared to the genome background, the significantly enriched GO terms and KEGG pathways for the DEGs were determined using hypergeometric tests, with the Bonferroni-corrected P-value ≤ 0.05 and Q value ≤ 0.05 as the thresholds, respectively (Abdi, 2007).
Quantitative Real-time PCR (qRT-PCR) Analysis
Total RNA was extracted from the same plant samples of ‘FT’ and fsm as those used for RNA-Seq using TRIzol reagent (Invitrogen, USA), and cDNA was synthesized using a FastQuant First-strand cDNA Synthesis kit (Tiangen, Beijing, China) according to the manufacturer’s instructions. The Actin and 18S rRNA were used as internal reference controls (Huang et al., 2015; Chen et al., 2016) and gene-specific primers were designed using Primer Premier 5.0 software. The qRT-PCR analysis was carried out using SYBR Green as a fluorescent detection dye (Tiangen, Beijing, China) and performed on a Bio-Rad IQ5 real time PCR detection system (Bio-Rad, USA). Each reaction contained 9 μl 2.5× Real MasterMix/20× SYBR solution, 2 μL (2 μmol L-1) of each forward and reverse primers, 2 μl of diluted cDNA (50 ng), and 5 μl ddH2O to a final volume of 20 μL. The qRT-PCR program was performed in 96-well plates under the following cycling conditions: initial activation at 95°C for 3 min, followed by 40 cycles of 95°C for 30 s, 58°C for 30 s, and 68°C for 15 s. This procedure was followed by melting curve analysis from 55 to 95°C to check the specificity of PCR amplification. The 2-ΔΔCt method was employed to calculate the relative expression levels of the target genes (Livak and Schmittgen, 2001). All reactions were performed with three biological and technical replicates, respectively. Differences in gene expression were analyzed using Bio-Rad IQ5 Manager software.
Results
Identification and Genetic Analysis of fsm
A large number of microspore regenerated plants (M0 generation) were obtained using the isolated microspore culture combined with EMS mutagenesis treatment, and the double haploid plants were screened for investigating botanical characteristics. The variant plants were observed and all double haploid plants were selfed in the M0 generation. In the M1 generation, the variant traits and genetic stability of mutants were further identified.
The fsm mutant exhibited the same visible phenotype as wild-type line ‘FT’ in the M0 generation. After selfing, segregation of characters appeared in the M1 generation (segregation ratio of 196: 54): of the 250 plants, 54 plants showed pistil abortion; other plants with the same phenotype as ‘FT’ were further selfed, revealing that character segregation continued in the offspring. These results suggest that the mutation may have occurred during the spontaneous diploid period rather than during the haploid period during the process of microspore culture and that the mutant gene fsm is recessive.
Therefore, to further investigate the inheritance of fsm, ‘FT’ (P1) and fsm (P2) were used as the parents. As shown in Table 1, the ‘FT’: fsm ratio among the BC1 progenies produced from the F1 × fsm backcross was approximately 1: 1 (χ2 = 2.07 < χ20.05,1 = 3.84). Of the 226 F2 plants, 178 and 48 individuals showed the ‘FT’ and fsm phenotypes, respectively, which represents a segregation ratio of 3.71: 1. The segregation ratios in the F2 population conformed to the expected ratio of 3: 1 (χ2 = 1.81 < χ20.05,1 = 3.84). These results indicate that the phenotype of fsm, which exhibiting stable inheritance, is controlled by a single recessive nuclear gene.
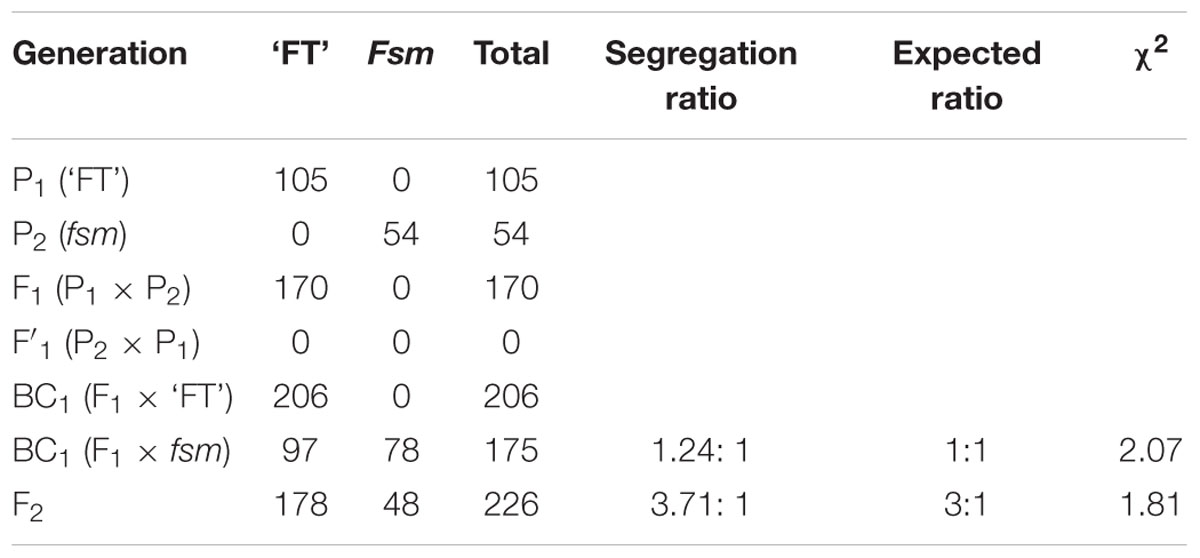
TABLE 1. Genetic analysis of fsm and crosses between fsm and wild-type line ‘FT.’
Morphological Characteristics of Floral Organs in ‘FT’ and fsm
Compared to the wild-type line ‘FT,’ the fsm plants exhibited pistil abortion, and the four-wheeled floral organs were also relatively smaller (Figure 1). As shown in Figure 1E, the pistil parts in fsm were significantly thinner and shorter, especially the ovaries.

FIGURE 1. Morphological characteristics of floral organs from fsm and wild-type line ‘FT.’ (A) Flowers of ‘FT’ (left) and fsm (right); (B) sepals of ‘FT’ (left) and fsm (right); (C) petals of ‘FT’ (left) and fsm (right); (D) stamens of ‘FT’ (left) and fsm (right); (E) pistils of ‘FT’ (left) and fsm (right). Scale bar: 1 mm.
Female Sterility Analysis of fsm
As shown in Table 2, compared to the wild-type line ‘FT,’ the fsm plants exhibited pistil abortion. Whether the fsm mutant was self-pollinated or used as the female parent to accept foreign pollen (wild-type line ‘FT’), the seed setting rates of fsm were both zero. The results showed that the female sterility of fsm was stable.

TABLE 2. The seed setting rates of self-pollination and reciprocal crosses between fsm and wild-type line ‘FT.’
Pollen Viability Observation of fsm
The pistils of fsm were completely sterile; however, there were small amounts of pollen in the stamens. As shown in Figure 2, the pollen of fsm was viable. In accordance with the results of Table 2, therefore, the fertility of fsm stamens was normal.
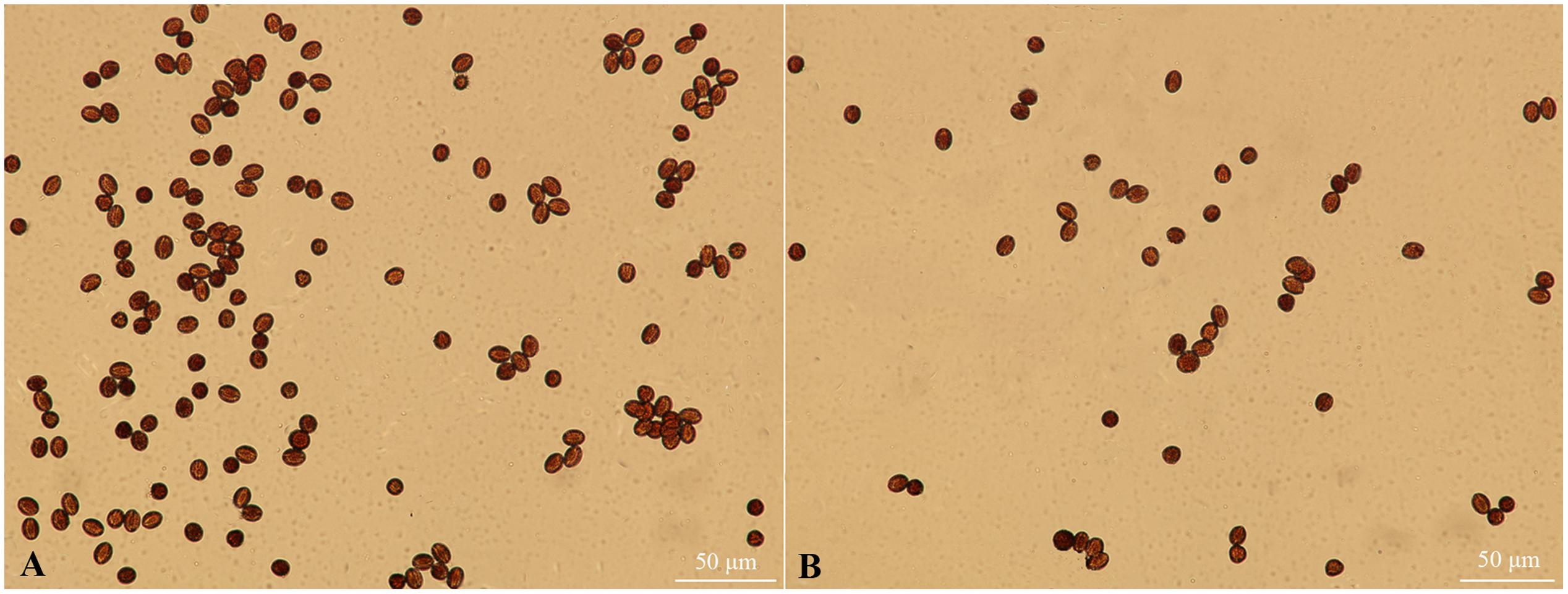
FIGURE 2. Pollen viability observation of fsm and wild-type line ‘FT.’ (A) Mature pollen microspores of ‘FT’; (B) mature pollen microspores of fsm. Scale bar: 50 μm.
Morphological Comparison of Ovary and Ovule Development in ‘FT’ and fsm
In plants, the development of female reproductive organs meant the ovary development, mainly including the development of ovule and formation of embryo sac. To further investigate the female sterility phenotype of fsm, the ovary and ovule development in ‘FT’ vs. fsm were observed and compared. As shown in Figure 3, under artificial pollination conditions, ovary development in fsm stopped at the end of flowering. The ovaries gradually became atrophied and yellow, ultimately leading to abscission. By contrast, the ovaries of ‘FT’ were elongated and widened at the end of flowering, and they developed rapidly.
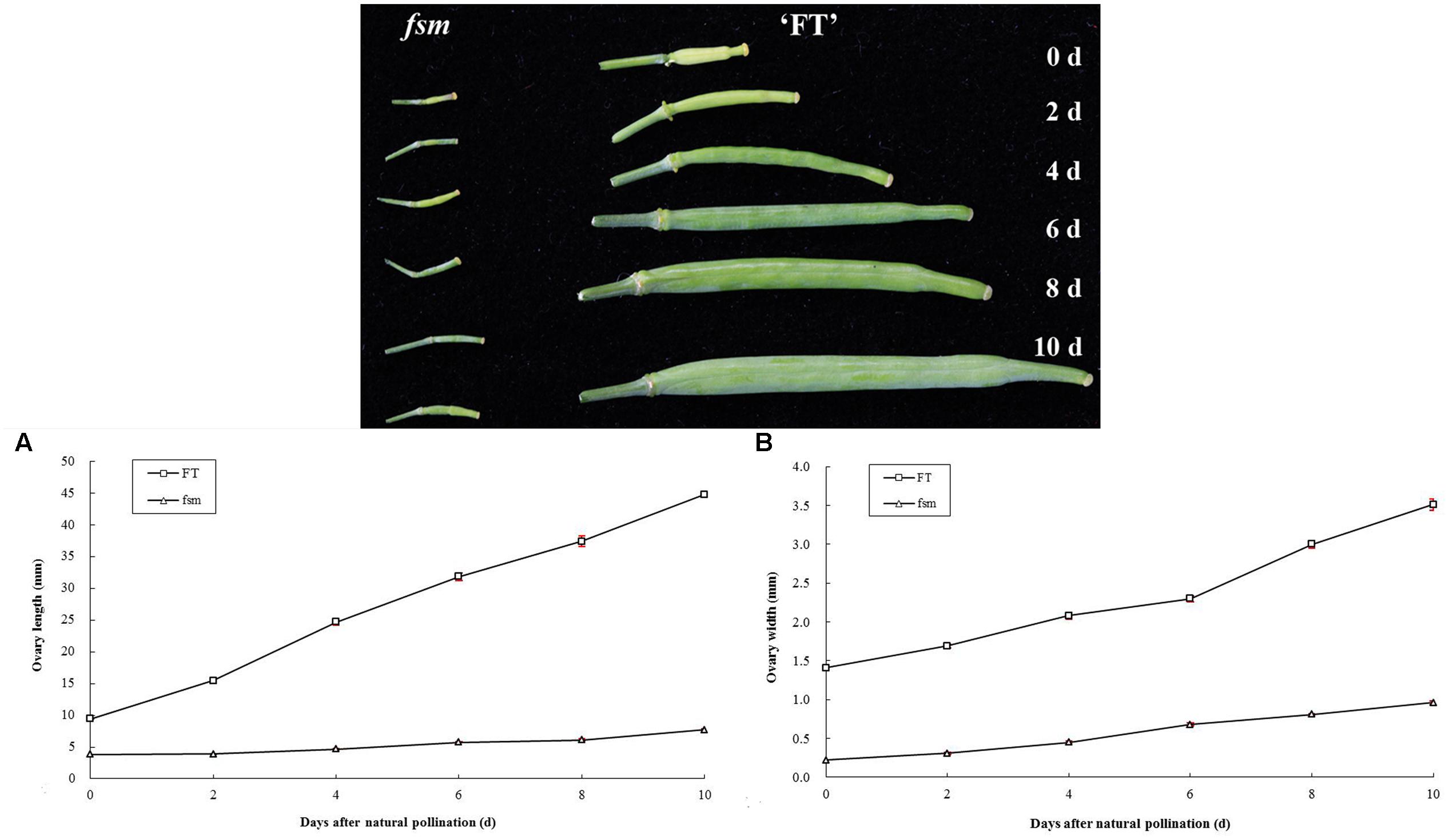
FIGURE 3. Ovary development in fsm and wild-type line ‘FT’ on different days after artificial pollination. (A) Dynamic changes of ovary length; (B) dynamic changes of ovary width. Each value is the mean of three independent experiments. The error bars represent standard error (SE) of the means.
Also, the structural characteristics of ovule development were observed and compared in ‘FT’ and fsm. As shown in Figure 4, compared with ‘FT,’ the ovules of fsm were abnormal, fewer, and smaller. After artificial pollination, the ovules of ‘FT’ were expanded and eventually developed into seeds, whereas the ovules of fsm were shriveled and did not develop into seeds. The observation results were in accordance with the results of paraffin section (Figure 5).

FIGURE 4. Structural characteristics of ovules in fsm and wild-type line ‘FT.’ (A) Ovules on the first flowering day in fsm (above) and ‘FT’ (below); (B) ovules of the 5th day after pollination in fsm (above) and ‘FT’ (below). Scale bar: 1 mm.
FIGURE 5. Morphological comparison of ovule development in fsm and wild-type line ‘FT.’ (A) Ovules on the first flowering day in ‘FT’; (B) ovules on the first flowering day in fsm; (C) ovules of the 5th day after pollination in ‘FT’; (D) ovules of the 5th day after pollination in fsm. Scale bar: 50 μm.
Illumina Sequencing and Mapping Reads to the Reference Genome
To help increase our understanding of the regulatory mechanisms underlying pistil development at the molecular level, we performed comparative floral transcriptome analysis of ‘FT’ and fsm using RNA-Seq technology.
Based on Illumina sequencing, a total of 130,216,688 and 134,955,118 clean reads were generated from the three biological replicates of ‘FT’ and fsm, respectively. Of the total clean reads, the number of reads that could be mapped to the reference genome ranged from 23.3 to 28.3 million, and the percentage of cleans reads ranged from 53.95 to 61.52% in the six libraries. As shown in Table 3, the overwhelming majority of these mapped reads were matched to unique genomic locations. The uniquely matched reads were used for gene expression analysis between ‘FT’ and fsm. These transcriptomes provide valuable resources for further analysis.
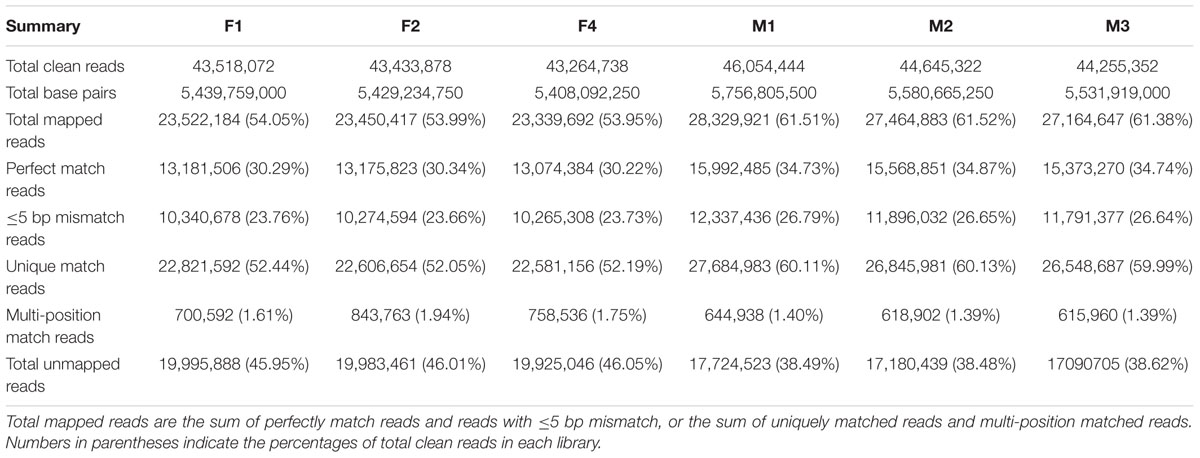
TABLE 3. Reads statistics based on RNA-Seq data of six libraries from fsm and wild-type line ‘FT.’
A total of 36,120 genes were detected in ‘FT’ and fsm. Among these, 32,843 (F1), 32,853 (F2), 32,837 (F4), 32,675 (M1), 32,640 (M2), and 32,665 (M3) expressed genes were identified from the six libraries, respectively (Supplementary Table S1). To further evaluate the RNA-Seq data, we analyzed the distribution of gene coverage in each library, representing the percentage of a gene covered by reads. As shown in Figure 6, genes with coverage > 90% were the most abundant category, accounting for 65–69% of the total number of genes. The second most abundant category was gene coverage of 80–90%, while the percentages of gene coverage for the remaining eight categories were similar.
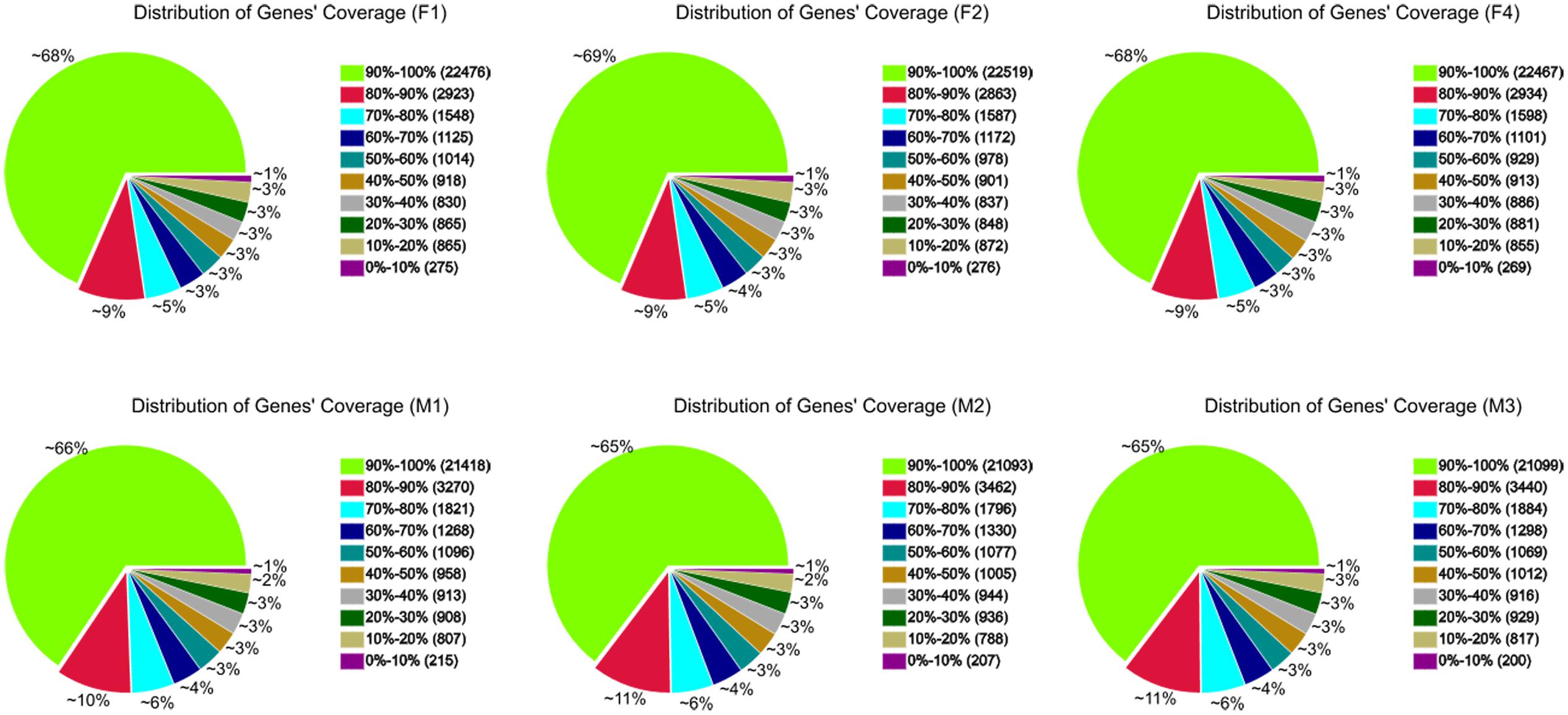
FIGURE 6. Distribution of gene coverage in fsm and wild-type line ‘FT.’
Identification of SNPs
A total of 167,837 and 155,448 SNPs were identified in ‘FT’ and fsm, respectively (Supplementary Tables S2, S3). As shown in Table 4, the most common base substitutions were A/G and C/T, and the least common was C/G. In addition, the SNPs between ‘FT’ and fsm were further analyzed and compared, among these SNPs, 43,661 and 31,272 SNPs were specifically detected in ‘FT’ and fsm, respectively (Supplementary Tables S4, S5).
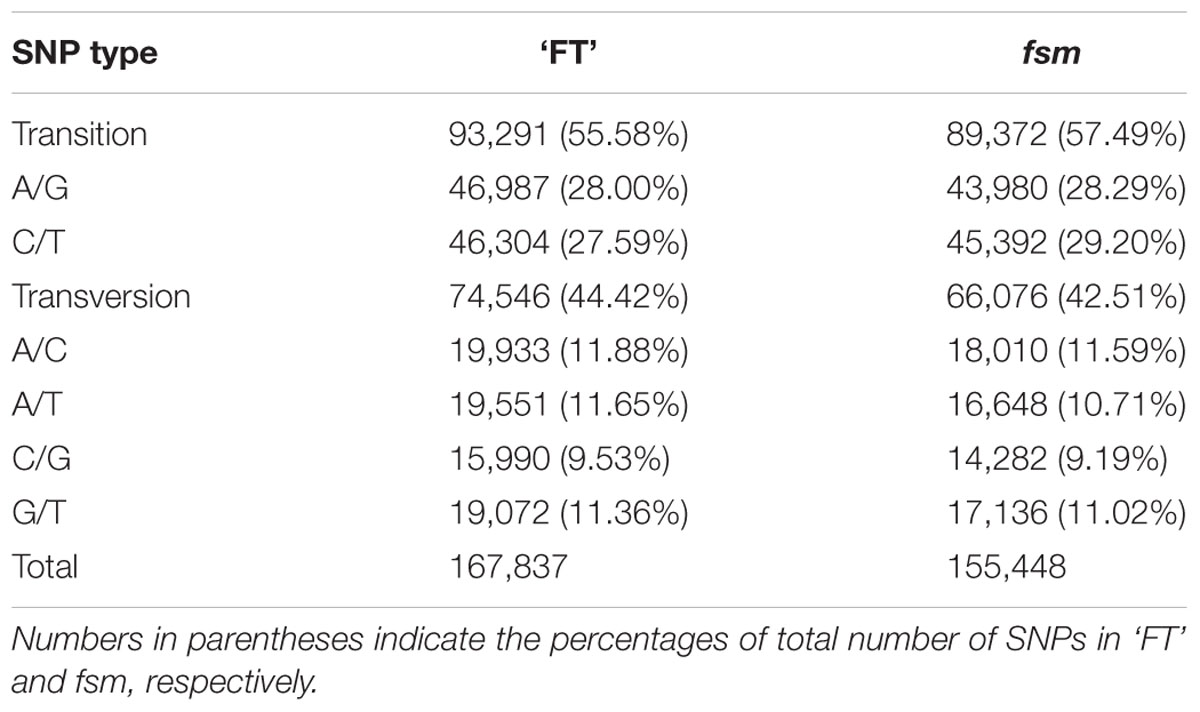
TABLE 4. Summary of single nucleotide polymorphism (SNP) types identified in fsm and wild-type line ‘FT.’
Global Analysis of Differential Gene Expression
Comparative analysis of the gene expression profiles between ‘FT’ and fsm were conducted to identify DEGs. A total of 1,872 DEGs were detected, including 1,021 up-regulated and 851 down-regulated genes in the fsm vs. ‘FT’ comparison, respectively. Therefore, the number of up-regulated DEGs in fsm is higher than the number of down-regulated DEGs (Supplementary Table S6).
We also detected a number of SEGs in this study. A total of 178 SEGs were identified between ‘FT’ and fsm, including 49 SEGs in ‘FT’ and 129 in fsm (Supplementary Table S7).
DEGs Related to Pistil Development
The morphological characterization suggested that the presence of the mutant gene fsm likely influenced pistil development (especially the presence of abnormal ovules), and thus the fertilization process cannot be accomplished, ultimately leading to female sterility.
To identify potential genes related to pistil development, we compared the gene expression profiles of ‘FT’ and fsm. Among the DEGs, a number of pistil development-related genes were identified, including genes for PRETTY FEW SEEDS 2 (PFS2; Bra026791), temperature-induced lipocalin (TIL; Bra020391), AGAMOUS-LIKE (AGL; Bra029154), and HECATE (HEC; Bra012128). In the fsm vs. ‘FT’ comparison, these genes were up-regulated, with relatively high expression levels (Supplementary Table S6), providing clues about the molecular mechanisms underlying female sterility.
The fsm mutant not only exhibited pistil abortion, but the floral organs were also relatively smaller compared to the wild-type line ‘FT’ (Figure 1). Consequently, numerous DEGs involved in floral organ development were also identified in this study, such as genes encoding F-box family protein (Bra001764, Bra004091, Bra027182, and so on), JASMONATE-ZIM-DOMAIN PROTEIN (JAZ; Bra022981, Bra025713, and Bra031065), VANGUARD 1 (VGD 1; Bra000438 and Bra040474), FLOWERING LOCUS T (FT; Bra022475), polygalacturonase (PG; Bra001268, Bra001269, Bra025631, and so on), MYB family transcription factors (Bra012579, Bra013526, and Bra028717), Arabinogalactan proteins (AGPs; Bra003296, Bra014611, Bra016902, and so on), EARLY FLOWERING 4-LIKE 1 (EFL1; Bra000468), pectinesterase family protein (Bra009264, Bra009921, Bra034960, and so on), BROTHER OF FT AND TFL1 (TERMINAL FLOWER 1) protein (BFT; Bra010052), especially for Auxin-Regulated Gene Involved In Organ Size (ARGOS; Bra007491), which can regulate the floral organ size in Arabidopsis thaliana (Hu et al., 2003). Most of these genes were highly expressed in the fsm vs. ‘FT’ comparison (Supplementary Table S6).
Overall, these genes may play important roles in floral organ development in Chinese cabbage. Further investigating possible pistil development-related genes would help elucidate the gene expression patterns and regulatory mechanisms involved in the female sterility phenotype of fsm.
Functional Enrichment Analysis of DEGs Using GO Classification and KEGG Pathway Analysis
To gain insight into the biological functions of the DEGs, all DEGs in the fsm vs. ‘FT’ comparison were mapped to GO terms using GO functional category analysis. For the three main GO categories, DEGs assigned to “biological process” (1,084, 57.9%) accounted for the majority of genes, followed by “molecular function” (1,077, 57.5%) and “cellular component” (960, 51.3%). Among these, the terms “cellular process” (GO: 0009987) and “metabolic process” (GO: 0008152), with 648 genes (59.8%) and 669 genes (61.7%), respectively, were dominant in the biological process category. In the molecular function category, the terms “binding” (GO: 0005488; 677, 62.9%) and “catalytic activity” (GO: 0003824; 638, 59.2%) were the most highly represented. In the cellular component category, “cell” (GO: 0005623; 780, 81.2%), “cell part” (GO: 0044464; 780, 81.2%), and “intracellular” (GO: 0005622; 606, 63.1%) were the most abundant groups (Figure 7).
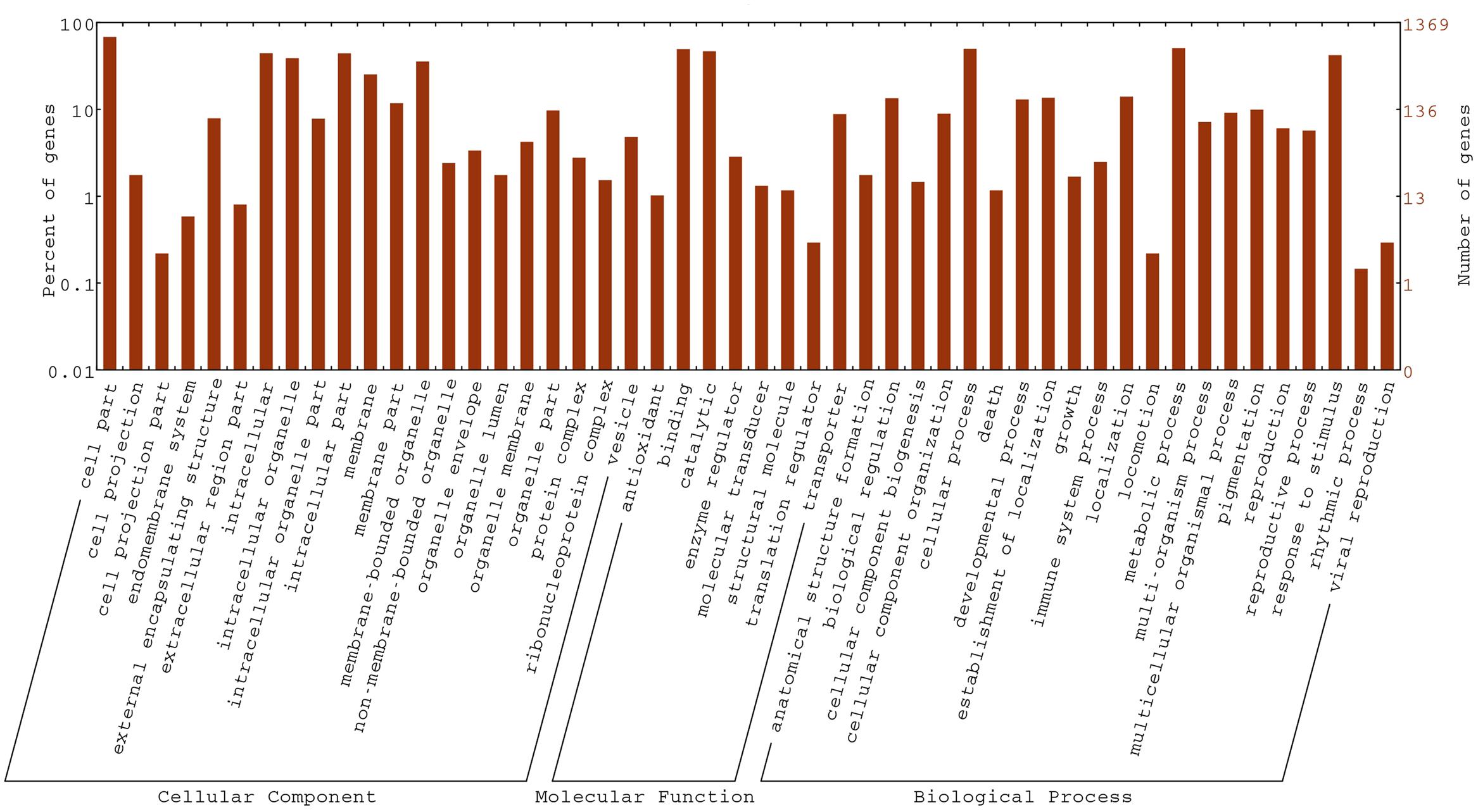
FIGURE 7. Gene ontology (GO) functional classification of DEGs in the fsm vs. ‘FT’ comparison.
We also performed GO term enrichment analysis (corrected P-value ≤ 0.05). The significantly enriched GO terms were shown in Supplementary Table S8. In addition, GO analysis revealed a number of GO terms related to floral organ development, including gynoecium development (Bra012128 and Bra000979), pollen tube growth (Bra037213 and Bra034771), floral organ morphogenesis (Bra012128, Bra000979 and Bra026791), pollen tube development (Bra012579, Bra037213, and Bra034771), floral organ formation (Bra012128 and Bra000979), floral organ development (Bra012128, Bra001005, Bra000979, Bra026791, Bra033931, and Bra012639), floral whorl development (Bra012128 and Bra000979), pollen development (Bra037213, Bra003255, and Bra020195), and flower development (Bra012639, Bra012128, Bra001005, Bra000979, Bra026791, and Bra033931, and so on).
To identify genes involved in metabolic or signal transduction pathways, a total of 949 DEGs were mapped to 226 KEGG pathways. Metabolic pathways (ko01100; 218, 22.97%) was the largest category, which was significantly larger than other pathways, followed by biosynthesis of secondary metabolites (ko01110; 131, 13.80%), plant-pathogen interaction (ko04626; 126, 13.28%), plant hormone signal transduction (ko04075; 103, 10.85%), and protein processing in endoplasmic reticulum (ko04141; 51, 5.37%). In addition, the KEGG enrichment analysis of DEGs was performed in this study. As shown in Table 5, a total of eight KEGG pathways were significantly enriched. These results indicated that a variety of genetic and active metabolic pathways were involved in pistil development, which laid the foundation for further investigating specific processes, functions, and pathways underlying female sterility in Chinese cabbage.
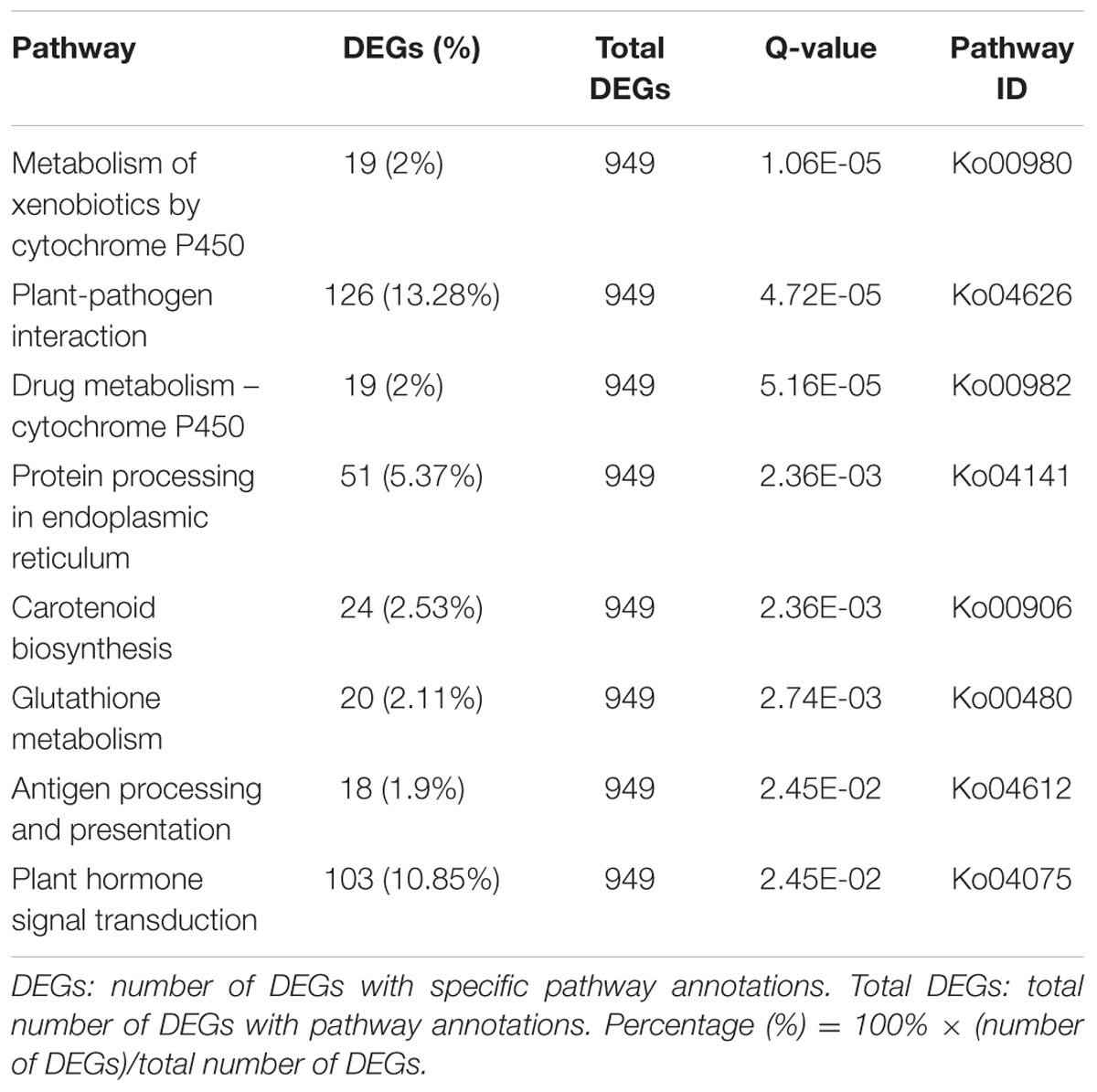
TABLE 5. Significantly enriched KEGG pathways of DEGs in fsm vs. ‘FT.’
Analysis of the Gene Expression Patterns by qRT-PCR
To help confirm the differential expression patterns of the DEGs detected by RNA-Seq, we performed qRT-PCR analysis of various DEGs. A total of 16 DEGs, including four pistil development-related genes (Bra026791, Bra020391, Bra029154 and Bra012128) and 12 floral organ development-related genes (Bra001764, Bra031065, Bra022475, Bra000468, Bra010052, Bra007491, Bra000438, Bra001268, Bra019903, Bra009921, Bra003296, and Bra021235) were selected for qRT-PCR analysis (Supplementary Table S9). As shown in Figure 8, the gene expression patterns obtained by qRT-PCR showed the similar trends as those of RNA-Seq data, thus supporting the reliability of our transcriptome analysis.
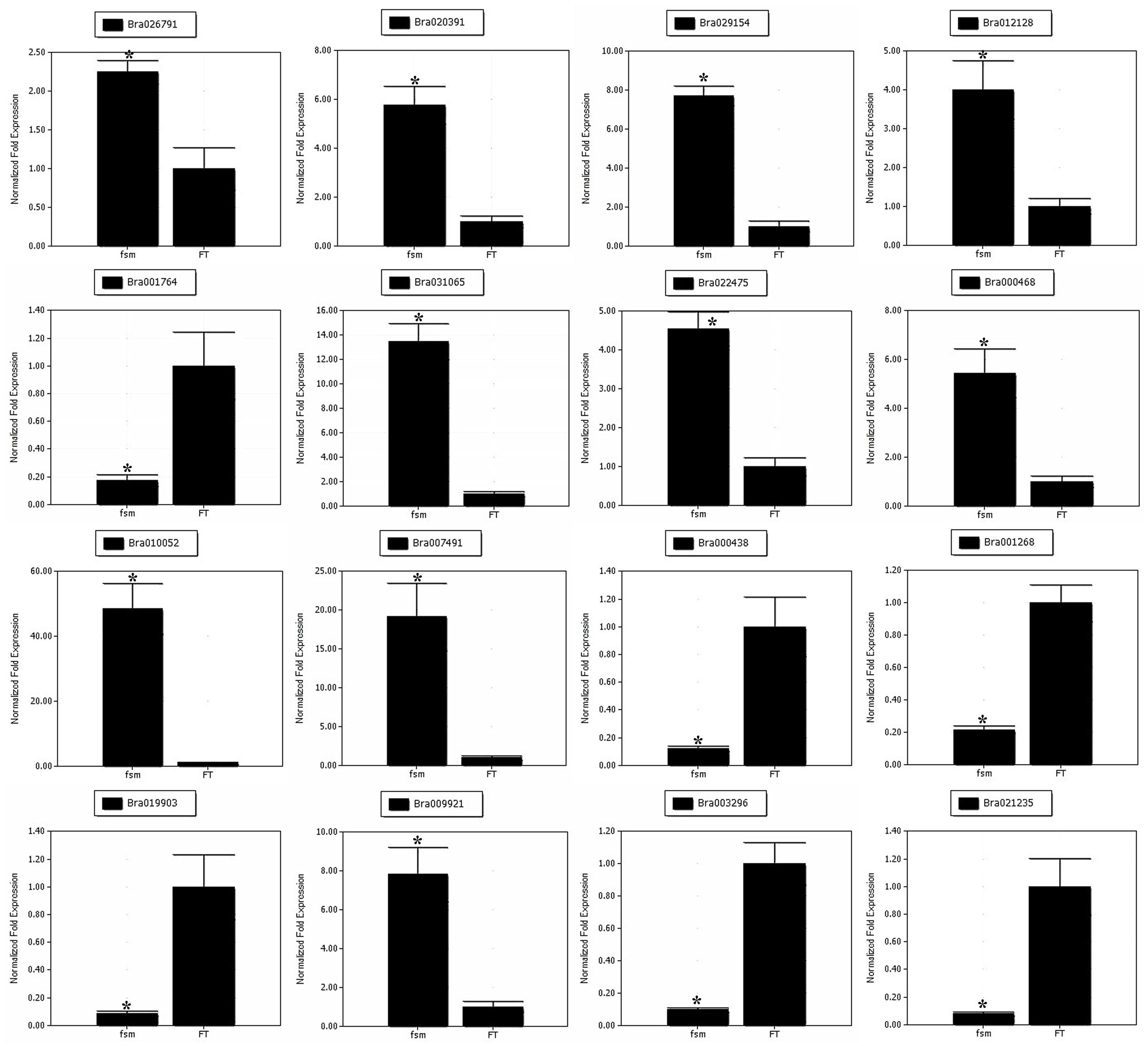
FIGURE 8. Quantitative real-time polymerase chain reaction (qRT-PCR) analysis of gene expression patterns. The relative expression levels of 16 DEGs identified by RNA-Seq analysis are shown. The gene expression analysis was performed based on three biological and technical replicates, respectively. ∗Significantly different at a level of 0.05 by t-test, and the statistical analysis was conducted using SPSS16.0 (Chicago, IL, USA).
Discussion
In this study, we identified the fsm mutant in Chinese cabbage, which exhibited stable inheritance. Based on a comparison with ‘FT,’ we speculate that the mutant gene in fsm likely influences ovule development, which further affects normal fertilization process, eventually leading to female sterility. Comparative transcriptome analysis of ‘FT’ and fsm showed that a number of DEGs are related to pistil development, and numerous DEGs involved in floral organ development were also identified. Further investigating these DEGs may increase our understanding of the regulatory mechanisms underlying female sterility.
The previous study has reported that the microspore culture enjoyed the characteristic of spontaneous doubling, requiring no artificial doubling treatment, and a high percentage of spontaneous diploids were found in Brassica campestris (Zhang and Ting Guan, 1993). The research on the combination of isolated microspore culture and EMS mutagenesis indicated that the frequency of spontaneous diploids were approximately 76.4% in Chinese cabbage (Huang et al., 2016a). In this study, the fsm mutant is different from previously reported mutants (Huang et al., 2015, 2016b). In general, the mutation occurred during the haploid microspore period, however, the mutation of the fsm mutant occurred during the spontaneous diploid period, and the mutant gene fsm was recessive. Given this, the fsm mutant exhibited the same visible phenotype as wild-type line ‘FT’ in the M0 generation, and segregation of characters appeared in the M1 generation, which exhibiting the mutant character.
The female-sterile mutants represent important materials for exploring floral organ-specific gene regulation and function. In the present study, genetic analysis indicated that the mutant phenotype of fsm is controlled by a single recessive nuclear gene, however, fine mapping of fsm remains to be performed. The results of gene mapping and our transcriptome analysis could be combined to further investigate candidate genes in fsm, especially the DEGs identified between ‘FT’ and fsm, as well as SNPs specifically detected in fsm based on the RNA-Seq results. The DEGs with most dramatically reduced expression levels in the fsm mutant could be candidates of the mutant gene, which would be responsible for the pistil developmental phenotype in fsm. Further investigating the fsm gene may help reveal the regulatory mechanisms underlying pistil development in Chinese cabbage. In addition, the developed SNPs markers represent a rich source of valuable molecular markers, which are widely used for genetic mapping and genetic diversity analysis in plants (Blair et al., 2013; Frascaroli et al., 2013). In this study, finding SNPs between ‘FT’ and fsm is especially useful, as some SNPs specifically detected in fsm may be directly related to the mutant phenotype.
Morphological observations suggested that the abnormal ovules in fsm likely influenced normal fertilization process, ultimately leading to female sterility. Therefore, identifying genes involved in pistil development would facilitate the analysis of female sterility. PRETTY FEW SEEDS2 (PFS2) is primarily expressed in developing primordia, and its transcripts are most abundant in developing ovules. PFS2 encodes a homeodomain protein that plays a prominent role during ovule patterning by regulating the differentiation of megaspore mother cells and cell proliferation of maternal integuments (Park et al., 2004). AGAMOUS (AG) can regulate ovule development and floral development (Becker and Theissen, 2003), and the research has showed that the ovule development is closely related to the level of PFS2 activity, which can repress AG expression in Arabidopsis thaliana (Park et al., 2005). In the present study, the up-regulation of PFS2 (Bra026791) detected in fsm vs. ‘FT’ may inhibit the AG expression, and thus affect ovule development of fsm. The female gametophyte development is a complicated process, and numerous genes are involved in its regulation in Arabidopsis thaliana (Wang et al., 2012). The temperature-induced lipocalin (TIL), which is mainly expressed in the embryo sacs of ovules, plays an essential role in female gametophyte development. Mutation of TIL causes ovule abortion and sometimes seed abortion, ultimately leading to low seed set (Chang et al., 2014). In the MADS-box gene family, AGAMOUS (AG) gene plays an important role in regulating floral carpel and ovule development (Alvarez-Buylla et al., 2000; Becker and Theissen, 2003). The related studies indicated that the AGAMOUS-LIKE 6 (AGL6) gene played an essential role in the floral development (Rijpkema et al., 2009). In Arabidopsis thaliana, AGL6 gene was mainly expressed in the ovule and not expressed in the stamens (Schauer et al., 2009), however, the AGL13 gene was expressed in both ovule and stamens, and had the function of regulating stamen and pistil development (Hsu et al., 2014). In addition, the AGL23 gene had an effect on the female gametophyte development (Colombo et al., 2008). The gene function of AGL62 was similar to AGL61, which interacted with AGL80, and they were jointly associated with the differentiation of central cells in the female gametophyte (Portereiko et al., 2006; Bemer et al., 2008; Kang et al., 2008). The basic helix-loop-helix (bHLH) transcription factors, HECATE 1 (HEC1), HEC2 and HEC3 genes are involved in the transmitting tract formation and stigma development, and the HEC activity is very necessary in the developing gynoecium in Arabidopsis thaliana (Gremski et al., 2007). In addition, the HEC genes can regulate both auxin and cytokinine signaling during gynoecium development (Schuster et al., 2015). Therefore, the interaction of these DEGs related to pistil development may influence ovule development, ultimately resulting in the pistil abortion phenotype of fsm.
In this study, the fsm plants not only exhibited pistil abortion, but the floral organs were also relatively smaller compared to the wild-type line ‘FT.’ The floral organ size is closely related to the floral morphology and function in plants (Horiguchi et al., 2006). The two cellular processes, cell proliferation and cell growth, can control the final size of floral organs during the process of floral development (Horiguchi et al., 2006; Krizek and Anderson, 2013). The research has shown that the ARGOS gene can control the floral organ size in Arabidopsis thaliana, with the up-regulation or down-regulation of ARGOS gene, the size of floral organs would be increased or decreased accordingly (Hu et al., 2003). The homologous gene of ARGOS has been isolated from Chinese cabbage, and the overexpression of ARGOS gene can make a significant increase in the leaves and floral organs in Arabidopsis thaliana (Hu et al., 2003, 2006; Feng et al., 2011). Based on the results presented here, the ARGOS gene was up-regulated in fsm vs. ‘FT’ and specifically expressed in fsm, however, the floral organs were relatively smaller in fsm. The result was opposite to the previous studies (Hu et al., 2003, 2006). In this study, except for the ARGOS gene, other DEGs involved in the floral development were also found, such as F-box family protein, FT and MYB family transcription factors. Therefore, we speculated that the interaction and regulation of these DEGs may result in the differences between ‘FT’ and fsm in floral organ sizes.
Among the genes differed in expression between ‘FT’ and fsm, approximately one third of genes which were annotated as unknown function. These genes might be the potential candidates to be involved in the pistil development, especially for the SEGs. The genes expressed exclusively in fsm may lead to the female sterility by inhibiting the pistil development. On the other hand, the genes only expressed in ‘FT’ were detected, which may have a role in the pistil development. Therefore, the actual functions of these genes remain to be further studied.
The previous studies indicated that heat or cold stress response-related proteins were associated with the pistil development in plants (Zinn et al., 2010). In Arabidopsis thaliana, heat-stress treatment can increase the number of aborted ovules and reduce the ovule numbers (Whittle et al., 2009). The related studies showed cold stress can reduce ovule fertilization and ovule viability, and thus affect the pistil function (Srinivasan et al., 1999; Thomashow, 1999). In our study, a number of DEGs related with heat or cold temperature stress were identified, such as heat shock proteins (HSP; Bra018216, Bra006697, Bra002539, and Bra020295) and late embryogenesis abundant (LEA) proteins (Bra022950, Bra027219, and Bra030494). Besides, the GO term enrichment analysis showed the GO term “response to heat” (GO: 0009408; 16 DEGs, 1.5%) was detected. Therefore, we speculated that these genes associated with hot or cold temperature stress may play important roles in the pistil abortion in fsm.
The KEGG pathway analysis revealed that a total of eight KEGG pathways were significantly enriched, of which, 103 (10.85%) DEGs were involved in the Plant hormone signal transduction pathway (ko04075). In plants, flower development was strongly influenced by hormonal regulation (Rudich et al., 1972). The previous studies indicated that the genes involved in the hormone signaling may play important roles in plant sex determination (Chandler, 2011). In recent years, several different transcriptome analyses showed that numerous hormone-related genes were differently expressed between different flower types, which further indicated hormones had roles in the sexual differentiation and development of floral organ (Wu et al., 2010; Ramos et al., 2014; Rocheta et al., 2014; Sobral et al., 2016). For example, auxin enjoyed a critically regulatory function in the process of floral growth and development in plants (Aloni et al., 2006; Cheng and Zhao, 2007). The recent studies indicated that cytokinine facilitates cell proliferation in early reproductive tract development and regulates reproductive meristems and ovule formation (Bartrina et al., 2011; Marsch-Martínez et al., 2012a). The interaction between auxin and cytokinine has been demonstrated to have a role in the gynoecium morphogenesis (Marsch-Martínez et al., 2012b). Another hormone, ethylene, was strongly relevant to the sex determination (Byers et al., 1972). The ethylene was thought to be essential for the process of sex determination in several species, such as cucumber and melon (Guo et al., 2010; Gao et al., 2015). In this study, the genes involved in the auxin signaling, cytokinine signaling and ethylene signaling were differentially expressed and had relatively high expression levels. Further study of these genes related to the hormone signal transduction may contribute to elucidate the female organ determination in Chinese cabbage.
Conclusion
We performed a systematic morphological investigation of fsm, followed by comparative transcriptome analysis between ‘FT’ and fsm. The results provide a comprehensive view of the expression profiles of genes involved in pistil development, which may help uncover the molecular mechanisms determining the phenotypic differences between these lines. Further studies of the functions of DEGs involved in pistil development should increase our understanding of female sterility. Our results provide a solid foundation for the further functional characterization of genes associated with the pistil development in Chinese cabbage.
Database Linking
The transcriptome sequencing data were deposited in the NCBI Gene Expression Omnibus (GEO) Database under accession number GSE76917.
Author Contributions
HF and ZL conceived and designed the research. SH, CL, DL, and RY performed the research. SH, LH, XL, and WL analyzed the data. SH wrote the manuscript. HF revised the manuscript. All authors discussed the results and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (31672144) and the earmarked fund for China Agriculture Research System (CARS-25-A-03).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00546/full#supplementary-material
TABLE S1 | List of all genes detected in the ‘FT’ and fsm libraries (XLS).
TABLE S2 | Summary of SNPs detected in the ‘FT’ library (XLSX).
TABLE S3 | Summary of SNPs detected in the fsm library (XLSX).
TABLE S4 | The SNPs specifically detected in the ‘FT’ library (XLS).
TABLE S5 | The SNPs specifically detected in the fsm library (XLS).
TABLE S6 | List of DEGs identified in the fsm vs. ‘FT’ comparison (XLS). Gene IDs and expression levels of DEGs are shown.
TABLE S7 | List of SEGs identified in the fsm vs. ‘FT’ comparison (XLS).
TABLE S8 | Significantly enriched GO terms identified in the fsm vs. ‘FT’ comparison (XLS).
TABLE S9 | Primer sequences used for qRT-PCR expression analysis (XLS).
Footnotes
References
Abdi, H. (2007). “Bonferroni and Šidák corrections for multiple comparisons,” in Encyclopedia of Measurement and Statistics, ed. N. Salkind (Thousand Oaks, CA: Sage), 103–107.
Aloni, R., Aloni, E., Langhans, M., and Ullrich, C. L. (2006). Role of auxin in regulating Arabidopsis flower development. Planta 223, 315–328. doi: 10.1007/s00425-005-0088-9
Alvarez-Buylla, E. R., Liljegren, S. J., Palaz, S., Gold, S. E., Burgeff, C., Ditta, G. S., et al. (2000). MADS-box gene evolution beyond flowers: expression in pollen, endosperm, guard cells, roots and trichomes. Plant J. 24, 457–466. doi: 10.1111/j.1365-313X.2000.00891.x
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. doi: 10.1038/npre.2010.4282.1
Anderson, S. N., Johnson, C. S., Jones, D. S., Conrad, L. J., Gou, X. P., Russell, S. D., et al. (2013). Transcriptomes of isolated Oryza sativa gametes characterized by deep sequencing: evidence for distinct sex-dependent chromatin and epigenetic states before fertilization. Plant J. 76, 729–741. doi: 10.1111/tpj.12336
Arthur, L., Ozias-Akins, P., and Hanna, W. W. (1993). Female sterile mutant in pearl millet: evidence for initiation of apospory. J. Hered. 84, 112–115. doi: 10.1093/oxfordjournals.jhered.a111290
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Balasubramanian, S., and Schneitz, K. (2000). NOZZLE regulates proximal-distal pattern formation, cell proliferation and early sporogenesis during ovule development in Arabidopsis thaliana. Development 127, 4227–4238.
Balasubramanian, S., and Schneitz, K. (2002). NOZZLE links proximal-distal and adaxial-abaxial pattern formation during ovule development in Arabidopsis thaliana. Development 129, 291–300.
Bao, R. Y., Jiang, C. N., Zheng, C. X., and Ding, K. S. (2005). Molecular mechanism of the regulation of female gametophyte development in plants. J. Beijing For. Univ. 27, 90–96.
Bartrina, I., Otto, E., Strnad, M., Werner, T., and Schmulling, T. (2011). Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. Plant Cell 23, 69–80. doi: 10.1105/tpc.110.079079
Becker, A., and Theissen, G. (2003). The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 29, 464–489. doi: 10.1016/S1055-7903(03)00207-0
Bemer, M., Wolters-Arts, M., Grossniklaus, U., and Angenent, G. C. (2008). The MADS domain protein DIANA acts together with AGAMOUS-LIKE80 to specify the central cell in Arabidopsis ovules. Plant Cell 20, 2088–2101. doi: 10.1105/tpc.108.058958
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300. doi: 10.2307/2346101
Benjamini, Y., and Yekutieli, D. (2001). The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 1165–1188. doi: 10.1214/aos/1013699998
Blair, M. W., Cortes, A. J., Penmetsa, R. V., Farmer, A., Carrasquilla-Garcia, N., and Cook, D. R. (2013). A high-throughput SNP marker system for parental polymorphism screening, and diversity analysis in common bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 126, 535–548. doi: 10.1007/s00122-012-1999-z
Brown, D. E., and Bingham, E. T. (1984). Hybrid alfalfa seed production using a female-sterile pollenizer. Crop Sci. 24, 1207–1208. doi: 10.2135/cropsci1984.0011183X002400060047x
Byers, R. E., Baker, L. R., Sell, H. M., Herner, R. C., and Dilley, D. R. (1972). Ethylene: a natural regulator of sex expression of Cucumismelo L. Proc. Natl. Acad. Sci. U.S.A. 69, 717–720. doi: 10.1073/pnas.69.3.717
Chandler, J. W. (2011). The hormonal regulation of flower development. J. Plant Growth Regul. 30, 242–254. doi: 10.1007/s00344-010-9180-x
Chang, X. Y., Zhao, L. H., Fu, F. L., and Qin, Y. (2014). Temperature-induced lipocalin TIL1 is required for female gametophyte development in Arabidopsis. Plant Physiol. J. 50, 253–262.
Chen, J. J., Pang, W. X., Chen, B., Zhang, C. Y., and Piao, Z. Y. (2016). Transcriptome analysis of Brassica rapa near-isogenic lines carrying clubroot-resistant and –susceptible alleles in response to Plasmodiophora brassicae during early infection. Front. Plant Sci. 6:1183. doi: 10.3389/fpls.2015.01183
Chen, X. J., Qi, C. K., Zhang, J. F., Pu, H. M., Gao, J. Q., and Fu, S. Z. (2003). A primary study on biological characteristics of female sterile mutant FS-M1 of rapeseed (Brassica napus L.). Chin. J. Oil Crop Sci. 25, 12–15.
Cheng, X. F., Wittich, P. E., Kieft, H., Angenent, G., XuHan, X., and van Lammeren, A. A. M. (2000). Temporal and spatial expression of MADS box genes, FBP7 and FBP11, during initiation and early development of ovules in wild type and mutant Petunia hybrida. Plant Biol. 2, 693–701. doi: 10.1055/s-2000-16640
Cheng, Y., and Zhao, Y. (2007). A role for auxin in flower development. J. Integr. Plant Biol. 49, 99–104. doi: 10.1111/j.1744-7909.2006.00412.x
Chettoor, A., Givan, S., Cole, R., Coker, C., Unger-Wallace, E., Vejlupkova, Z., et al. (2014). Discovery of novel transcripts and gametophytic functions via RNA-seq analysis of maize gametophytic transcriptomes. Genome Biol. 15, 414. doi: 10.1186/s13059-014-0414-2
Chevalier, E., Loubert-Hudon, A., and Matton, D. P. (2013). ScRALF3, a secreted RALF-like peptide involved in cell-cell communication between the sporophyte and the female gametophyte in a solanaceous species. Plant J. 73, 1019–1033. doi: 10.1111/tpj.12096
Colombo, M., Masiero, S., Vanzulli, S., Lardelli, P., Kater, M. M., and Colombo, L. (2008). AGL23, a type I MADS-box gene that controls female gametophyte and embryo development in Arabidopsis. Plant J. 54, 1037–1048. doi: 10.1111/j.1365-313X.2008.03485.x
Daskalov, S., and Mihailov, L. (1998). A new method for hybrid seed production based on cytoplasmic male sterility combined with a lethal gene and a female sterile pollenizer in Capsicum annuum L. Theor. Appl. Genet. 76, 530–532. doi: 10.1007/BF00260902
Dou, B. D., Zhang, X. L., Ma, L., Feng, D. J., and Sun, Q. X. (2001). A preliminary study on female sterility in wheat. Acta Agron. Sin. 27, 1013–1016. doi: 10.3321/j.issn:0496-3490.2001.06.053
Faulconnier, Y., Chilliard, Y., Torbati, M. B. M., and Leroux, C. (2011). The transcriptomic profiles of adipose tissues are modified by feed deprivation in lactating goats. Comp. Biochem. Physiol. D 6, 139–149. doi: 10.1016/j.cbd.2010.12.002
Feng, G. P., Qin, Z. X., Yan, J. Z., Zhang, X. R., and Hu, Y. X. (2011). Arabidopsis ORGAN SIZE RELATED1 regulates organ growth and final organ size in orchestration with ARGOS and ARL. New Phytol. 191, 635–646. doi: 10.1111/j.1469-8137.2011.03710.x
Ferrándiz, C., Liljegren, S. J., and Yanofsky, M. F. (2000). Negative regulation of the SHATTERPROOF genes by FRUITFULL during Arabidopsis fruit development. Science 289, 436–438. doi: 10.1126/science.289.5478.436
Filichkin, S. A., Priest, H. D., Givan, S. A., Shen, R. K., Bryant, D. W., Fox, S. E., et al. (2010). Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 20, 45–58. doi: 10.1101/gr.093302.109
Frascaroli, E., Schrag, T. A., and Melchinger, A. E. (2013). Genetic diversity analysis of elite European maize (Zea mays L.) inbred lines using AFLP, SSR, and SNP markers reveals ascertainment bias for a subset of SNPs. Theor. Appl. Genet. 126, 133–141. doi: 10.1007/s00122-012-1968-6
Gao, P., Sheng, Y., Luan, F., Ma, H., and Liu, S. (2015). RNA-seq transcriptome profiling reveals differentially expressed genes involved in sex expression in melon. Crop Sci. 55, 1686–1695. doi: 10.2135/cropsci2014.06.0444
Gremski, K., Ditta, G., and Yanofsky, M. F. (2007). The HECATE genes regulate female reproductive tract development in Arabidopsis thaliana. Development 134, 3593–3601. doi: 10.1242/dev.011510
Guo, S. G., Zheng, Y., Joung, J.-G., Liu, S. Q., Zhang, Z. H., Crasta, O. R., et al. (2010). Transcriptome sequencing and comparative analysis of cucumber flowers with different sex types. BMC Genomics 11:384. doi: 10.1186/1471-2164-11-384
Horiguchi, G., Ferjani, A., Fujikura, U., and Tsukaya, H. (2006). Coordination of cell proliferation and cell expansion in the control of leaf size in Arabidopsis thaliana. J. Plant Res. 119, 37–42. doi: 10.1007/s10265-005-0232-4
Hsu, W. H., Yeh, T. J., Huang, K. Y., Li, J. Y., Chen, H. Y., and Yang, C. H. (2014). AGAMOUS-LIKE13, a putative ancestor for the E functional genes, specifies male and female gametophyte morphogenesis. Plant J. 77, 1–15. doi: 10.1111/tpj.12363
Hu, Y. X., Poh, H. M., and Chua, N. H. (2006). The Arabidopsis ARGOS-LIKE gene regulates cell expansion during organ growth. Plant J. 47, 1–9. doi: 10.1111/j.1365-313X.2006.02750.x
Hu, Y. X., Xie, Q., and Chua, N. H. (2003). The Arabidopsis auxin-inducible gene ARGOS controls lateral organ size. Plant Cell 15, 1951–1961. doi: 10.1105/tpc.013557
Huang, B. Q., and Sheridan, W. F. (1996). Embryo sac development in the maize indeterminate gametophyte mutant: abnormal nuclear behavior and defective microtubule organization. Plant Cell 8, 1391–1407. doi: 10.1046/j.1365-2044.2000.01798-3.x
Huang, S. N., Liu, Z. Y., Li, D. Y., Yao, R. P., and Feng, H. (2016a). A new method for generation and screening of Chinese cabbage mutants using isolated microspore culturing and EMS mutagenesis. Euphytica 207, 23–33. doi: 10.1007/s10681-015-1473-5
Huang, S. N., Liu, Z. Y., Li, D. Y., Yao, R. P., Hou, L., Li, X., et al. (2016b). Physiological characterization and comparative transcriptome analysis of a slow-growing reduced-thylakoid mutant of Chinese cabbage (Brassica campestris ssp. pekinensis). Front. Plant Sci. 7:3. doi: 10.3389/fpls.2016.00003
Huang, S. N., Liu, Z. Y., Yao, R. P., Li, D. Y., and Feng, H. (2015). Comparative transcriptome analysis of the petal degeneration mutant pdm in Chinese cabbage (Brassica campestris ssp. pekinensis) using RNA-seq. Mol. Genet. Genomics 290, 1833–1847. doi: 10.1007/s00438-015-1041-7
Jacobsen, S. E., and Meyerowitz, E. M. (1997). Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science 277, 1100–1103. doi: 10.1126/science.277.5329.1100
Johnston, A. J., Meier, P., Gheyselinck, J., Wuest, S. E. J., Federer, M., Schlagenhauf, E., et al. (2007). Genetic subtraction profiling identifies genes essential for Arabidopsis reproduction and reveals interaction between the female gametophyte and the maternal sporophyte. Genome Biol. 8:R204. doi: 10.1186/gb-2007-8-10-r204
Jones-Rhoades, M. W., Borevitz, J. O., and Preuss, D. (2007). Genome-wide expression profiling of the Arabidopsis female gametophyte identifies families of small, secreted proteins. PLoS Genet. 3:e171. doi: 10.1371/journal.pgen.0030171
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. doi: 10.1093/nar/gkm882
Kang, I. H., Steffen, J. G., Portereiko, M. F., Lloyd, A., and Drews, G. N. (2008). The AGL62 MADS domain protein regulates cellularization during endosperm development in Arabidopsis. Plant Cell 20, 635–647. doi: 10.1105/tpc.107.055137
Koyama, T., Furutani, M., Tasaka, M., and Ohme-Takagi, M. (2007). TCP transcription factors control the morphology of shoot lateral organs via negative regulation of the expression of boundary specific genes in Arabidopsis. Plant Cell 19, 473–484. doi: 10.1105/tpc.106.044792
Krizek, B. A., and Anderson, J. T. (2013). Control of flower size. J. Exp. Bot. 64, 1427–1437. doi: 10.1093/jxb/ert025
Li, M. L., and Zheng, C. X. (2002). RAPD analysis in female sterility (Clone 28) of Pinus tabulaeformis Carr. J. Beijing For. Univ. 24, 42–44.
Li, R. Q., Li, Y. R., Fang, X. D., Yang, H. M., Wang, J., Kristiansen, K., et al. (2009a). SNP detection for massively parallel whole-genome resequencing. Genome Res. 19, 1124–1132. doi: 10.1101/gr.088013.108
Li, R. Q., Yu, C., Li, Y. R., Lam, T. W., Yiu, S. M., Kristiansen, K., et al. (2009b). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967. doi: 10.1093/bioinformatics/btp336
Li, X. B., Xiang, L., Luo, J., Hu, B. L., Tian, S. P., Xie, M., et al. (2013). The strategy of RNA-seq, application and development of molecular marker derived from RNA-seq. Chin. J. Cell Biol. 35, 720–726.
Li, X. L., Bai, B., Wu, J., Deng, Q. Y., and Zhou, B. (2012). Transcriptome analysis of early interaction between rice and Magnaporthe oryzae using next-generation sequencing technology. Hereditas (Beijing) 34, 102–112. doi: 10.3724/SP.J.1005.2012.00102
Li, Z., Zhang, Z., Yan, P., Huang, S., Fei, Z., and Lin, K. (2011). RNA-Seq improves annotation of protein-coding genes in the cucumber genome. BMC Genomics 12:540. doi: 10.1186/1471-2164-12-540
Liu, H. W., Zhou, R. Y., and Xu, L. (2003). Studies on photoperiodicity of photoperiod-sensitive female sterility in ramie [Boehmeria nivea (L.) Gaud.]. Acta Agron. Sin. 29, 222–224. doi: 10.3321/j.issn:0496-3490.2003.02.010
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, Y. H., Arnaud, D., Belcram, H., Falentin, C., Rouault, P., Piel, N., et al. (2012). A dominant point mutation in a RINGv E3 ubiquitin ligase homoeologous gene leads to cleistogamy in Brassica napus. Plant Cell 24, 4875–4891. doi: 10.1105/tpc.112.104315
Luo, C. M., Liu, X. D., Lu, Y. G., and Feng, J. H. (1996). A preliminary studies on photoperiod-thermo sensitive female sterility of T23fs in rice. J. Yunnan Univ. 21, 179.
Luo, Q., Zhou, K. D., Wang, W. M., Wang, X. D., Xiao, H., Wang, X. H., et al. (2001). The genetic analysis and gene molecular mapping of a new leaf and abnormal pistil development mutant in rice. Chin. Sci. Bull. 46, 1277–1280. doi: 10.3321/j.issn:0023-074X.2001.15.012
Marsch-Martínez, N., Ramos-Cruz, D., Irepan Reyes-Olalde, J., Lozano-Sotomayor, P., Zuìnϸiga-Mayo, V. M., and de Folter, S. (2012a). The role of cytokinin during Arabidopsis gynoecia and fruit morphogenesis and patterning. Plant J. 72, 222–234. doi: 10.1111/j.1365-313X.2012.05062.x
Marsch-Martínez, N., Reyes-Olalde, J. I., Ramos-Cruz, D., Lozano-Sotomayor, P., Zuìnϸiga-Mayo, V. M., and de Folter, S. (2012b). Hormones talking: does hormonal cross-talk shape the Arabidopsis gynoecium? Plant Signal. Behav. 7, 1698–1701. doi: 10.4161/psb.22422
Maruyama, K., Kato, H., and Araki, H. (1991). Mechanized production of F1 seeds in rice by mixed planting. Jpn. Agric. Res. Q. 24, 243–252.
Moll, C., von Lyncker, L., Zimmermann, S., Kgi, C., Baumann, N., Twell, D., et al. (2008). CLO/GFA1 and ATO are novel regulators of gametic cell fate in plants. Plant J. 56, 913–921. doi: 10.1111/j.1365-313X.2008.03650.x
Montgomery, S. B., Sammeth, M., Gutierrez-Arcelus, M., Lach, R. P., Ingle, C., Nisbett, J., et al. (2010). Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464, 773–777. doi: 10.1038/nature08903
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Nag, A., King, S., and Jack, T. (2009). miR319a targeting of TCP4 is critical for petal growth and development in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 106, 22534–22539. doi: 10.1073/pnas.0908718106
Nibau, C., Di Stilio, V. S., Wu, H. M., and Cheung, A. Y. (2011). Arabidopsis and Tobacco SUPERMAN regulate hormone signalling and mediate cell proliferation and differentiation. J. Exp. Bot. 62, 949–961. doi: 10.1093/jxb/erq325
Pagnussat, G. C., Yu, H. J., Ngo, Q. A., Rajani, S., Mayalagu, S., Johnson, C. S., et al. (2005). Genetic and molecular identification of genes required for female gametophyte development and function in Arabidopsis. Development 132, 603–614. doi: 10.1242/dev.01595
Park, S. O., Hwang, S., and Hauser, B. A. (2004). The phenotype of Arabidopsis ovule mutants mimics the morphology of primitive seed plants. Proc. R. Soc. Lond. B Biol. Sci. 271, 311–316. doi: 10.1098/rspb.2003.2544
Park, S. O., Zheng, Z. G., Oppenheimer, D. G., and Hauser, B. A. (2005). The PRETTY FEW SEEDS2 gene encodes an Arabidopsis homeodomain protein that regulates ovule development. Development 132, 841–849. doi: 10.1242/dev.01654
Pereira, T. N. S., Lersten, N. R., and Palmer, R. G. (1997). Genetic and cytological analyses of a partial-female-sterile mutant (PS1) in soybean (Glycine max; Leguminosae). Am. J. Bot. 84, 781–791. doi: 10.2307/2445814
Portereiko, M. F., Lloyd, A., Steffen, J. G., Punwani, J. A., Otsuga, D., and Drews, G. N. (2006). AGL80 is required for central cell and endosperm development in Arabidopsis. Plant Cell 18, 1862–1872. doi: 10.1105/tpc.106.040824
Punwani, J. A., Rabiger, D. S., Lloyd, A., and Drews, G. N. (2008). The MYB98 subcircuit of the synergid gene regulatory network includes genes directly and indirectly regulated by MYB98. Plant J. 55, 406–414. doi: 10.1111/j.1365-313X.2008.03514.x
Qi, Y. X., Liu, Y. B., and Rong, W. S. (2011). RNA-Seq and its applications: a new technology for transcriptomics. Hereditas (Beijing) 33, 1191–1202. doi: 10.3724/SP.J.1005.2011.01191
Ramos, M. J. N., Coito, J. L., Silva, H. G., Cunha, J., Costa, M. M. R., and Rocheta, M. (2014). Flower development and sex specification in wild grapevine. BMC Genomics 15:1095. doi: 10.1186/1471-2164-15-1095
Ren, S. C., Peng, Z. Y., Mao, J. H., Yu, Y. W., Yin, C. J., Cao, X., et al. (2012). RNA-Seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noneoding RNAs and aberrant alternative splicing. Cell Res. 22, 806–821. doi: 10.1038/cr.2012.30
Rijpkema, A. S., Zethof, J., Gerats, T., and Vandenbussche, M. (2009). The petunia AGL6 gene has a SEPALLATA-like function in floral patterning. Plant J. 60, 1–9. doi: 10.1111/j.1365-313X.2009.03917.x
Rocheta, M., Sobral, R., Magalhães, J., Amorim, M. I., Ribeiro, T., Pinheiro, M., et al. (2014). Comparative transcriptomic analysis of male and female flowers of monoecious Quercussuber. Front. Plant Sci. 5:599. doi: 10.3389/fpls.2014.00599
Rosellini, D., Ferranti, F., Barone, P. F., and Veronesi, F. (2003). Expression of female sterility in alfalfa (Medicago sativa L.). Sex. Plant Reprod. 15, 271–279. doi: 10.1007/s00497-003-0163-y
Rosellini, D., Lorenzetti, F., and Bingham, E. T. (1998). Quantitative ovule sterility in Medicago sativa. Theor. Appl. Genet. 97, 1289–1295. doi: 10.1007/s001220051021
Rudich, J., Halevy, A. H., and Kedar, N. (1972). Ethylene evolution from cucumber plants as related to sex expression. Plant Physiol. 49, 998–999. doi: 10.1104/pp.49.6.998
Sakai, H., Medrano, L. J., and Meyerowitz, E. M. (1995). Role of SUPERMAN in maintaining Arabidopsis floral whorl boundaries. Nature 378, 199–203. doi: 10.1038/378199a0
Sarvepalli, K., and Nat, U. (2011). Hyper-activation of the TCP4 transcription factor in Arabidopsis thaliana accelerates multiple aspects of plant maturation. Plant J. 67, 595–607. doi: 10.1111/j.1365-313X.2011.04616.x
Schauer, S. E., Schlüter, P. M., Baskar, R., Gheyselinck, J., Bolaños, A., Curtis, M. D., et al. (2009). Intronic regulatory elements determine the divergent expression patterns of AGAMOUS-LIKE6 subfamily members in Arabidopsis. Plant J. 59, 987–1000. doi: 10.1111/j.1365-313X.2009.03928.x
Schiefthaler, U., Balasubramanian, S., Sieber, P., Chevalier, D., Wisman, E., and Schneitz, K. (1999). Molecular analysis of NOZZLE, a gene involved in pattern formation and early sporogenesis during sex organ development in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 96, 11664–11669. doi: 10.1073/pnas.96.20.11664
Schneitz, K., Hülskamp, M., Kopczak, S. D., and Pruitt, R. E. (1997). Dissection of sexual organ ontogenesis: a genetic analysis of ovule development in Arabidopsis thaliana. Development 124, 1367–1376.
Schuster, C., Gaillochet, C., and Lohmann, J. U. (2015). Arabidopsis HECATE genes function in phytohormone control during gynoecium development. Development 142, 3343–3350. doi: 10.1242/dev.120444
Shi, D. Q., Liu, J., Xiang, Y. H., Ye, D., Sundaresan, V., and Yang, W. C. (2005). SLOW WALKER1, essential for gametogenesis in Arabidopsis, encodes a WD40 protein involved in 18S ribosomal RNA biogenesis. Plant Cell 17, 2340–2354. doi: 10.1105/tpc.105.033563
Siddiqi, I., Ganesh, G., Grossniklaus, U., and Subbiah, V. (2000). The dyad gene is required for progression through female meiosis in Arabidopsis. Development 127, 197–207.
Singh, M., Goel, S., Meeley, R. B., Dantec, C., Parrinello, H., Michaud, C., et al. (2011). Production of viable gametes without meiosis in maize deficient for an ARGONAUTE protein. Plant Cell 23, 443–458. doi: 10.1105/tpc.110.079020
Sobral, R., Silva, H. G., Morais-Cecílio, L., and Costa, M. M. R. (2016). The quest for molecular regulation underlying unisexual flower development. Front. Plant Sci. 7:160. doi: 10.3389/fpls.2016.00160
Srinivasan, A., Saxena, N., and Johansen, C. (1999). Cold tolerance during early reproductive growth of chickpea (Cicer arietinum L.): genetic variation in gamete development and function. Field Crops Res. 60, 209–222. doi: 10.1016/S0378-4290(98)00126-9
Sun, H., Zhang, J. Y., Wang, Y. M., and Zhao, L. M. (2009). A review of utilization of heterosis in three legume crops of pigeonpea, alfalfa and soybean. Sci. Agric. Sin. 42, 1528–1539. doi: 10.3864/j.issn.0578-1752.2009.05.004
Szécsi, J., Joly, C., Bordji, K., Varaud, E., Cock, J. M., Dumas, C., et al. (2006). BIGPETALp, a bHLH transcription factor is involved in the control of Arabidopsis petal size. EMBO J. 25, 3912–3920. doi: 10.1038/sj.emboj.7601270
Tang, J. J., Chen, X., Hu, Q. D., Kato, M., Shimizu, K., and Yokoo, M. (2002). A comparatively histological observation on the megagametophytic abortion of female-sterile rice FS-1 and its maternal parent Fujisaka 5. Acta Biol. Exp. Sin. 35, 313–318. doi: 10.3321/j.issn:1673-520X.2002.04.011
Tao, X., Gu, Y. H., Wang, H. Y., Zheng, W., Li, X., Zhao, C. W., et al. (2012). Digital gene expression analysis based on integrated De Novo transcriptome assembly of sweet potato [Ipomoea batatas (L.) Lam.]. PLoS ONE 7:e36234. doi: 10.1371/journal.pone.0036234
Thomashow, M. F. (1999). Plant cold acclimation: freezing tolerance genes and regulatory mechanisms. Annu. Rev. Plant Biol. 50, 571–599. doi: 10.1146/annurev.arplant.50.1.571
Venkatesan, S., and Monica, A. S. (2010). Pattern formation in miniature: the female gametophyte of flowering plants. Development 137, 179–189. doi: 10.1242/dev.030346
Wang, X., Gou, X. X., and Wang, J. J. (2012). Review on molecular mechanism and genetic regulation of female gametophyte development in plant. Mol. Plant Breed. 10, 1067–1079. doi: 10.5376/mpb.cn.2012.10.0009
Wang, X. W., Wang, H. Z., Wang, J., Sun, R. F., Wu, J., Liu, S. Y., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039. doi: 10.1038/ng.919
Whittle, C. A., Otto, S. P., Johnston, M. O., and Krochko, J. E. (2009). Adaptive epigenetic memory of ancestral temperature regime in Arabidopsis thaliana. Botany 87, 650–657. doi: 10.1139/B09-030
Wu, T., Qin, Z. W., Zhou, X. Y., Feng, Z., and Du, Y. L. (2010). Transcriptome profile analysis of floral sex determination in cucumber. J. Plant Physiol. 167, 905–913. doi: 10.1016/j.jplph.2010.02.004
Xiao, F. M., Zhong, R., Gao, F. Y., and Wu, B. J. (1998). RAPD analysis of indica-japonica rice hybrid female sterilit mutant 91fs and its parents. China J. Appl. Environ. Biol. 4, 235–237.
Yamaguchi, T., Nagasawa, N., Kawasaki, S., Matsuoka, M., Nagato, Y., and Hirano, H. Y. (2004). The YABBY gene DROOPING LEAF regulates carpel specification and midrib development in Oryza sativa. Plant Cell 16, 500–509. doi: 10.1105/tpc.018044
Zhang, C. L., Qin, Z. J., Wang, G. Z., Ji, Z. B., and Wang, J. M. (2012). Transcriptome and RNA-Seq technology. Biotech. Bull. 12, 51–56.
Zhang, L., and Ting Guan, J. J. (1993). The discussion on the spontaneous doubling frequency of microspore regenerated plants in Chinese cabbage (Brassica campestris ssp. pekinensis). Beijing Agric. Sci. 11, 23–25.
Zhong, R., Xiao, F. M., Gao, F. Y., and Wu, B. J. (1998). The research on female sterility in plant. Explor. Nat. 17, 75–79.
Zhou, R. Y. (1996). The discovery of female sterile individuals in ramie [Boehmeria nivea (L.) Gaud.]. Sci. Agric. Sin. 29, 96.
Keywords: Chinese cabbage, female-sterile mutant, pistil development, RNA-Seq technology, DEGs
Citation: Huang S, Liu Z, Li C, Yao R, Li D, Hou L, Li X, Liu W and Feng H (2017) Transcriptome Analysis of a Female-sterile Mutant (fsm) in Chinese Cabbage (Brassica campestris ssp. pekinensis). Front. Plant Sci. 8:546. doi: 10.3389/fpls.2017.00546
Received: 27 January 2017; Accepted: 27 March 2017;
Published: 10 April 2017.
Edited by:
Jacqueline Batley, University of Western Australia, AustraliaCopyright © 2017 Huang, Liu, Li, Yao, Li, Hou, Li, Liu and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Feng, ZmVuZ2h1aWFhYUAyNjMubmV0
†These authors have contributed equally to this work and share the first authorship.