Abstract
DELLA proteins are transcriptional regulators present in all land plants which have been shown to modulate the activity of over 100 transcription factors in Arabidopsis, involved in multiple physiological and developmental processes. It has been proposed that DELLAs transduce environmental information to pre-wired transcriptional circuits because their stability is regulated by gibberellins (GAs), whose homeostasis largely depends on environmental signals. The ability of GAs to promote DELLA degradation coincides with the origin of vascular plants, but the presence of DELLAs in other land plants poses at least two questions: what regulatory properties have DELLAs provided to the behavior of transcriptional networks in land plants, and how has the recruitment of DELLAs by GA signaling affected this regulation. To address these issues, we have constructed gene co-expression networks of four different organisms within the green lineage with different properties regarding DELLAs: Arabidopsis thaliana and Solanum lycopersicum (both with GA-regulated DELLA proteins), Physcomitrella patens (with GA-independent DELLA proteins) and Chlamydomonas reinhardtii (a green alga without DELLA), and we have examined the relative evolution of the subnetworks containing the potential DELLA-dependent transcriptomes. Network analysis indicates a relative increase in parameters associated with the degree of interconnectivity in the DELLA-associated subnetworks of land plants, with a stronger effect in species with GA-regulated DELLA proteins. These results suggest that DELLAs may have played a role in the coordination of multiple transcriptional programs along evolution, and the function of DELLAs as regulatory ‘hubs’ became further consolidated after their recruitment by GA signaling in higher plants.
Introduction
Higher plants are characterized by a particularly flexible capacity to adapt to multiple environmental conditions. In other words, environmental signals are very efficient modulators of plant developmental decisions. This ability is generally assumed to be based on at least two mechanistic features: the presence of an extensive and sensitive repertoire of elements that perceive environmental signals (such as light photoreceptors covering a wide range of wavelengths), and the high degree of interconnectivity between the different signaling pathways to allow cellular integration of variable information (Casal et al., 2004).
Evidence has accumulated in recent years about the important role that plant hormones play in the translation of environmental signals into developmental decisions. On one hand, it has become evident that hormone pathways share common components with the pathways that transduce light and other environmental signals (Jaillais and Chory, 2010); and, on the other hand, hormones have been shown to participate in the regulation of developmental processes all throughout a plant’s life cycle (Alabadi et al., 2009). In this context, gibberellins (GAs) and DELLA proteins are a paradigmatic example of the mechanisms that allow environmental signal integration. DELLA proteins constitute a small clade within the GRAS family of loosely defined plant specific nuclear proteins (Vera-Sirera et al., 2015). Their name was coined on the basis of a short stretch of amino acids (D-E-L-L-A) in their N-terminal region, which is tightly conserved among all higher plant species. They also present additional conserved motifs, such as the VHYNP domain, two leucine heptad repeats which may mediate protein–protein interactions, a putative nuclear localization signal, and a putative SH2 phosphotyrosine-binding domain, among others (Vera-Sirera et al., 2015). It has been shown in Arabidopsis thaliana and rice that recognition of GAs by their GID1 receptor allows physical interaction with DELLA proteins and promotes their degradation via the proteasome. In A. thaliana, loss of DELLA function mimics the phenotype of plants treated with an excess of GAs, both anatomically and also at the transcriptional level (Schwechheimer, 2011; Locascio et al., 2013b). Work in the past few years has established that DELLAs regulate transcription through the interaction with more than 100 transcription factors (TFs) in A. thaliana (de Lucas et al., 2008; Feng et al., 2008; Crocco et al., 2010; Hou et al., 2010; Gallego-Bartolomé et al., 2012; Daviere et al., 2014; Marin-de la Rosa et al., 2014, 2015; Resentini et al., 2015). In some cases, interaction with the TF inhibits its ability to bind DNA, while in other cases DELLAs seem to act as co-activators (Locascio et al., 2013b; Daviere and Achard, 2016). For all the cases examined in detail, the DELLA region responsible for the interaction with the TFs is the C-terminal region of the protein, the GRAS domain. Given that GA levels are strongly regulated by environmental signals such as light, temperature and photoperiod (Hedden and Thomas, 2012; Colebrook et al., 2014), cellular DELLA levels seem to be a proxy for the environmental status faced by plants (Claeys et al., 2014). Changes in DELLA levels could in turn differentially modulate distinct sets of TFs and their target genes in various developmental contexts. The promiscuous interaction with TFs, and the observation that A. thaliana dellaKO mutants display constitutive growth even under stress, and suffer from increased sensitivity to several types of stress factors such as salinity, cold, or fungal attacks (Alabadí et al., 2004; Achard et al., 2006, 2007, 2008a,b; Cheminant et al., 2011) suggests that DELLAs are potentially important ‘hubs’ in the transcriptional network that regulates the balance between growth and stress tolerance in higher plants.
Previous interest in the evolution of DELLA proteins is restricted to the question on how they were recruited to mediate cellular signaling by GAs. Based on phylogenetic analyses and shallow molecular analysis with fern and moss orthologs, it seems that the GA/GID1/DELLA module originated with early diverging tracheophytes (Wang and Deng, 2014). For instance, the Selaginella genus possesses the ability to synthesize GAs, a GID1 GA receptor, and a DELLA protein (Wang and Deng, 2014), which is sensitive to GA-induced degradation, even when introduced in an angiosperm, such as A. thaliana (Hirano et al., 2007; Yasumura et al., 2007). On the other hand, the DELLA proteins that existed in other land plants before the emergence of vascular plants were not involved in GA signaling. First, there are no bona-fide DELLA genes in algae and, second, the genomes of bryophytes like Physcomitrella patens encode DELLA proteins that lack the canonical ‘DELLA motif’ (Wang and Deng, 2014), and PpDELLAs are not sensitive to GAs when introduced in A. thaliana (Yasumura et al., 2007). However, the ability of DELLA proteins to modulate transcriptional programs relies on the GRAS domain through which interactions with TFs occur, and the evolution of this activity has not been addressed before.
In an attempt to identify the possible function of ancestral DELLAs and to delineate how evolution has shaped the functions of the GA/DELLA module in higher plants, we have addressed the analysis of the transcriptional networks potentially regulated by DELLAs in several species. For this reason, we have used gene co-expression networks, in which genes are represented as nodes, and if two genes exhibit a significant correlation value for co-expression, the corresponding nodes are joined by an edge. Importantly, if a node is a TF, first neighbors can be confidently taken as targets for that particular TF (Franco-Zorrilla et al., 2014). Therefore, the analysis of topological parameters of a gene co-expression network is an interesting tool that may reveal information about the function and evolutionary history of transcriptional programs (Aoki et al., 2007; Usadel et al., 2009).
Here we have investigated the properties of networks formed by DELLA-interacting TFs and their co-expressing genes in A. thaliana, and compared them with the orthologous networks in three other plant species: (i) Solanum lycopersicum (possessing a fully operative GA/DELLA module); (ii) P. patens (possessing GA-independent DELLA functions); and (iii) Chlamydomonas reinhardtii (without GA perception or DELLAs) (Figure 1A). All the parameters examined suggest that the functions regulated by DELLA-interacting TFs (and thus DELLAs themselves) have increased their level of coordination along evolution.
FIGURE 1
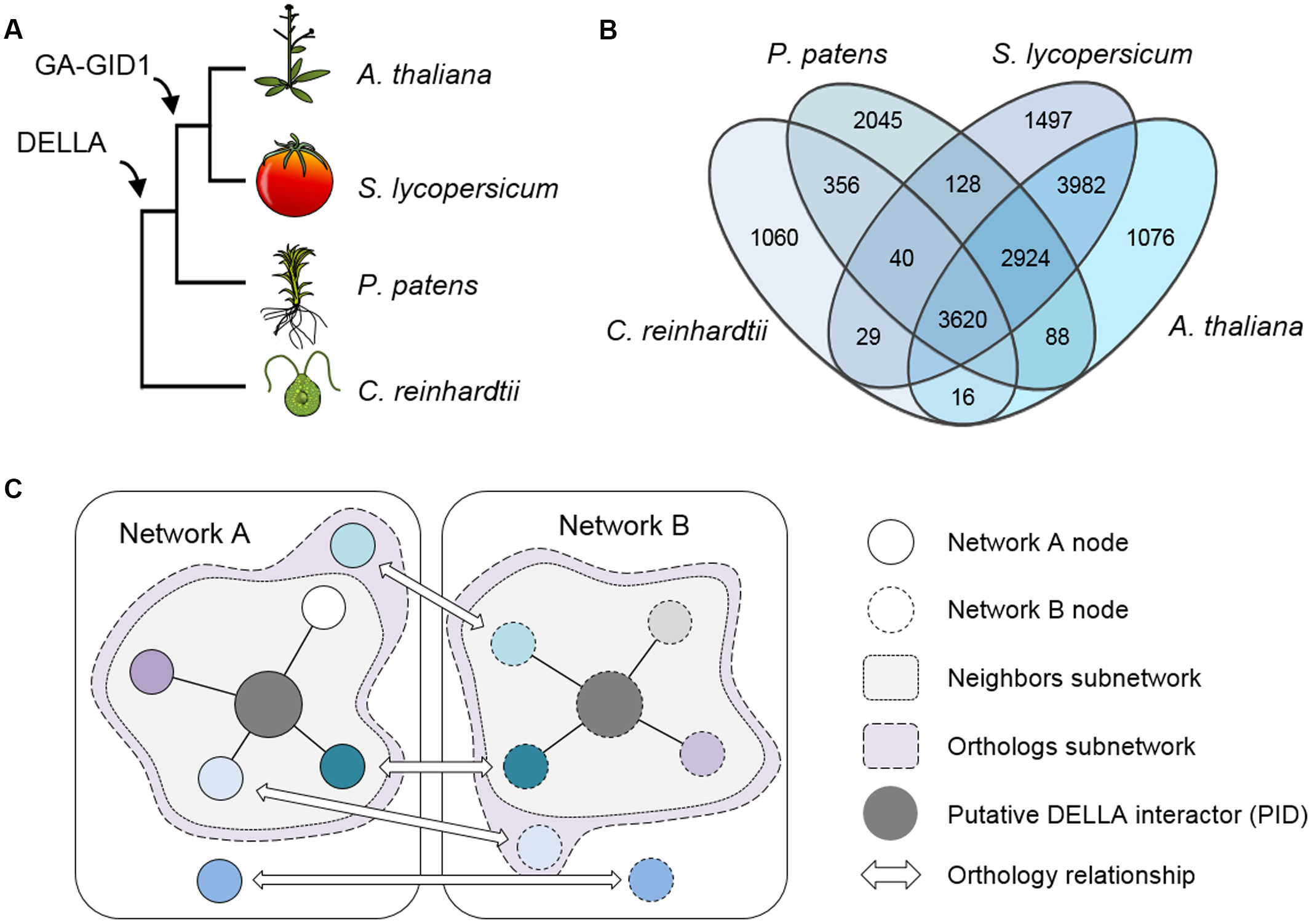
Phylogenetic relationships between the chosen species. (A) Representation of the species tree indicating the origin of key elements related to the gibberellin signaling pathway. (B) Venn’s diagram showing the number of OrthoMCL groups in which genes of each species are present. (C) Schematic representation of the basis for subnetwork design. Nodes in different networks with the same color indicate an orthologous relationship.
Results and Discussion
Construction of Networks and Subnetworks
Gene expression data from RNA sequencing (RNA-seq) experiments in A. thaliana, S. lycopersicum, P. patens, and C. reinhardtii were obtained from the Gene Expression Omnibus, and gene co-expression networks were inferred for each species from transcriptomic data as described in section “Materials and Methods.” All four networks are scale-free networks (Supplementary Figure S1) (Romero-Campero et al., 2013, 2016) and have comparable sizes in terms of number of nodes, but there are remarkable differences in the way they are connected (Table 1). The A. thaliana network contains more than twice as many edges than the others, the average degree of its nodes (average number of connections) is one order of magnitude higher and its average shortest path length (average number of nodes between two random nodes) is lower. Even though the number of genes of each species represented in the networks is similar, in some species they are more connected, possibly due to differences in their endogenous regulation and the availability of experimental data. For that reason, we decided to do every comparative analysis between the different species in relative terms.
Table 1
|
C. reinhardtii
|
P. patens
|
S. lycopersicum
|
A. thaliana
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Full | Neigh | Ortho | Full | Neigh | Ortho | Full | Neigh | Ortho | Full | Neigh | Ortho | |
| Nodes | 8652 | 48 | 658 | 8564 | 448 | 1503 | 7851 | 1314 | 2885 | 5663 | 2070 | 2949 |
| Edges | 145903 | 78 | 1173 | 295317 | 15078 | 19828 | 287409 | 153396 | 169171 | 593730 | 460951 | 512042 |
| Average degree | 33.73 | 3.25 | 3.57 | 68.97 | 67.31 | 26.38 | 73.22 | 233.48 | 117.28 | 209.69 | 445.36 | 347.26 |
| Average shortest path length | 7.37 | 1.91 | 8.71 | 13.11 | 1.39 | 12.01 | 13.78 | 1.67 | 5.63 | 4.28 | 2.15 | 3.09 |
| Diameter | 23 | 4 | 24 | 46 | 4 | 41 | 44 | 6 | 25 | 20 | 9 | 12 |
General parameters in co-expression networks.
Parameters of networks and subnetworks used in this study. Full, full gene co-expression network; Neigh, first neighbors subnetwork; Ortho, orthologs subnetwork; C. reinhardtii, Chlamydomonas reinhardtii; P. patens, Physcomitrella patens; S. lycopersicum, Solanum lycopersicum; A. thaliana, Arabidopsis thaliana.
To be able to compare the co-expression networks of the different species, we first identified the orthologous nodes in each of them using the OrthoMCL method (Li et al., 2003). Up to 17,053 groups of genes were obtained. Genes in the same group were considered orthologs or paralogs if they belonged to different or the same species, respectively. The four species were represented unequally, as both A. thaliana and S. lycopersicum genes were present in ca. 70% of the groups, while P. patens genes were found in little more than 50% of them, and only ca. 30% of the groups contained genes from C. reinhardtii (Figure 1B). This was already expected, given the evolutionary distance among these species and the genomic complexity of each one.
To assess the possible contribution of DELLA proteins to co-expression networks architecture, we created subnetworks based on reported DELLA interactors known to act as transcriptional regulators. First, we compiled a list of all published DELLA interactors (Supplementary Table S1), obtained their orthologs for the four species, and localized them in their respective networks. Since most of the interactions have been described for A. thaliana, the corresponding orthologs in the other species are only “putative interactors of the DELLA proteins” (PIDs), and the first neighbors of AtDELLA interactors and PIDs are their putative expression targets. Second, we built two different subnetworks using this information. The first one, called “Neighbors” subnetwork (abbreviated as AtNeigh, SlNeigh, PpNeigh, and CrNeigh), is composed of the DELLA interactors (or the corresponding PIDs) and their first neighbors (Figure 1C and Supplementary Table S2). The second one, called “Orthologs” subnetwork (abbreviated as AtOrtho, SlOrtho, PpOrtho, and CrOrtho), contains the orthologs of all the first neighbors of PIDs in all the species (Figure 1C and Supplementary Table S3). For a given species, the “Neighbors” subnetwork provides a good approximation to its actual DELLA-dependent transcriptome, while the “Orthologs” subnetwork represents the full landscape of potential transcriptional targets for DELLAs, since it includes orthologs of genes that are DELLA transcriptional targets in other species (Figure 2).
FIGURE 2

Gene co-expression networks. Full Chlamydomonas reinhardtii(A,E), Physcomitrella patens(B,F), Solanum lycopersicum(C,G), and Arabidopsis thaliana(D,H) gene co-expression networks. Neighbors subnetworks are comprised of yellow-marked nodes in A-D. Orthologs subnetworks are comprised of yellow-marked nodes in (E–H).
DELLA-Associated Subnetworks Reflect Increased Relevance of DELLAs after Being Recruited by GA Signaling
It is important to take into account a circumstance that affects the construction of subnetworks: OrthoMCL does not always retrieve orthologs for some of the genes, because either they do not exist in the other species, or the method does not provide high-confidence results. This results in a particular bias toward smaller subnetwork sizes with increasing phylogenetic distance (Table 1). However, the impact of this bias can be disregarded when analyzing relative parameters. Hence, regardless of the absolute sizes, we observed that the average degree in the Neighbor subnetworks increased dramatically in SlNeigh and AtNeigh with respect to their full networks (more than threefold and twofold, respectively), while this parameter did not change in PpNeigh, and it actually decreased in CrNeigh (Table 1). Similarly, the Orthologs subnetworks displayed an equivalent behavior as the Neighbors subnetworks: their diameter and average shortest path length decreased considerably more in SlOrtho and AtOrtho with respect to the full networks; and the same happened with the increase of the average degree. In summary, both subnetworks showed a higher compaction and interconnection of nodes in relative terms in the case of S. lycopersicum and A. thaliana compared with P. patens and C. reinhardtii, indicating that the putative interactors and targets of the DELLAs become more connected in those species presenting GA-regulated DELLAs.
A confirmation of the impact of GA regulation on the relevance of DELLA function is found in the analysis of neighborhood conservation. Figure 3A shows the percentage of genes with a significantly overlapping neighborhood in each comparison (see Materials and Methods). When comparing P. patens with the other species, there are no substantial differences between the full network and the Orthologs subnetwork. On the contrary, SlOrtho and AtOrtho contain a considerably higher proportion of genes with conserved neighborhood than their corresponding full networks (15% vs. 10%). Between S. lycopersicum and A. thaliana, the regulation of the putative DELLA targets is more conserved than for the network in general, so this group of genes seems to have a cohesive element in the two species.
FIGURE 3
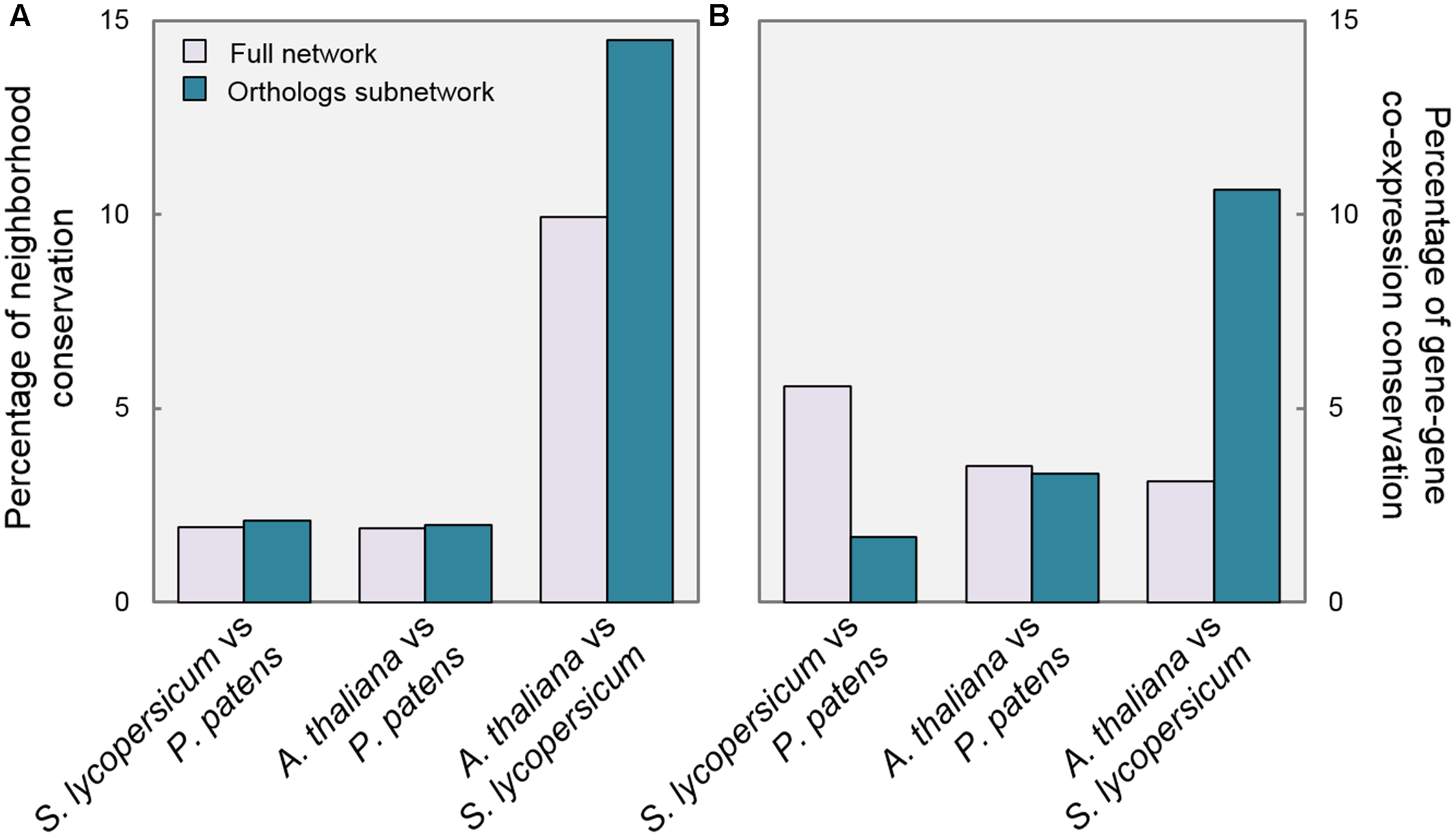
Gene connections are more conserved in species with GA-regulated DELLAs. Pairwise comparisons of P. patens, S. lycopersicum, and A. thaliana Full networks and Ortho subnetworks regarding: (A) Percentage of genes with significantly overlapping neighborhoods; (B) Percentage of conserved gene–gene links.
Furthermore, we examined gene–gene co-expression values, as a measure of the conservation of individual edges. For every pair of linked genes in one species, if the corresponding orthologs are also linked in a second species, it is considered that gene–gene co-expression is conserved. Therefore, the calculation of conserved links between two subnetworks is a measure of functional conservation of a regulatory module. Interestingly, we observed that gene links between PpOrtho and SlOrtho were less conserved than in the full networks, and almost unaltered between PpOrtho and AtOrtho (Figure 3B). However, the gene–gene co-expression was three times more conserved between SlOrtho and AtOrtho than between their full networks (11% vs. 3.5%). In other words, these data are compatible with the proposition that the presence of GA-regulated DELLAs (in S. lycopersicum and A. thaliana) provides stronger links between transcriptional programs, not detected in an organism with GA-independent DELLAs (P. patens).
Efficiency of Transcriptional Regulation Is a DELLA-Associated Parameter
The efficiency of a transcriptional regulatory mechanism can be evaluated through two additional parameters in gene co-expression networks: shortest path length distribution and motif frequency. In network theory, average shortest-path length is defined as the average number of steps along the shortest paths for all possible pairs of network nodes. It is a measure of the efficiency of information propagation on a network, with a shorter average path length being more efficient (Vragovic et al., 2005). When we compared the distribution of shortest path lengths in full and Orthologs subnetworks, we observed a clear tendency toward shorter path lengths in the Orthologs subnetworks of organisms possessing DELLAs (S. lycopersicum, A. thaliana, and P. patens) compared with the situation in an organism without DELLAs (C. reinhardtii) (Figure 4).
FIGURE 4
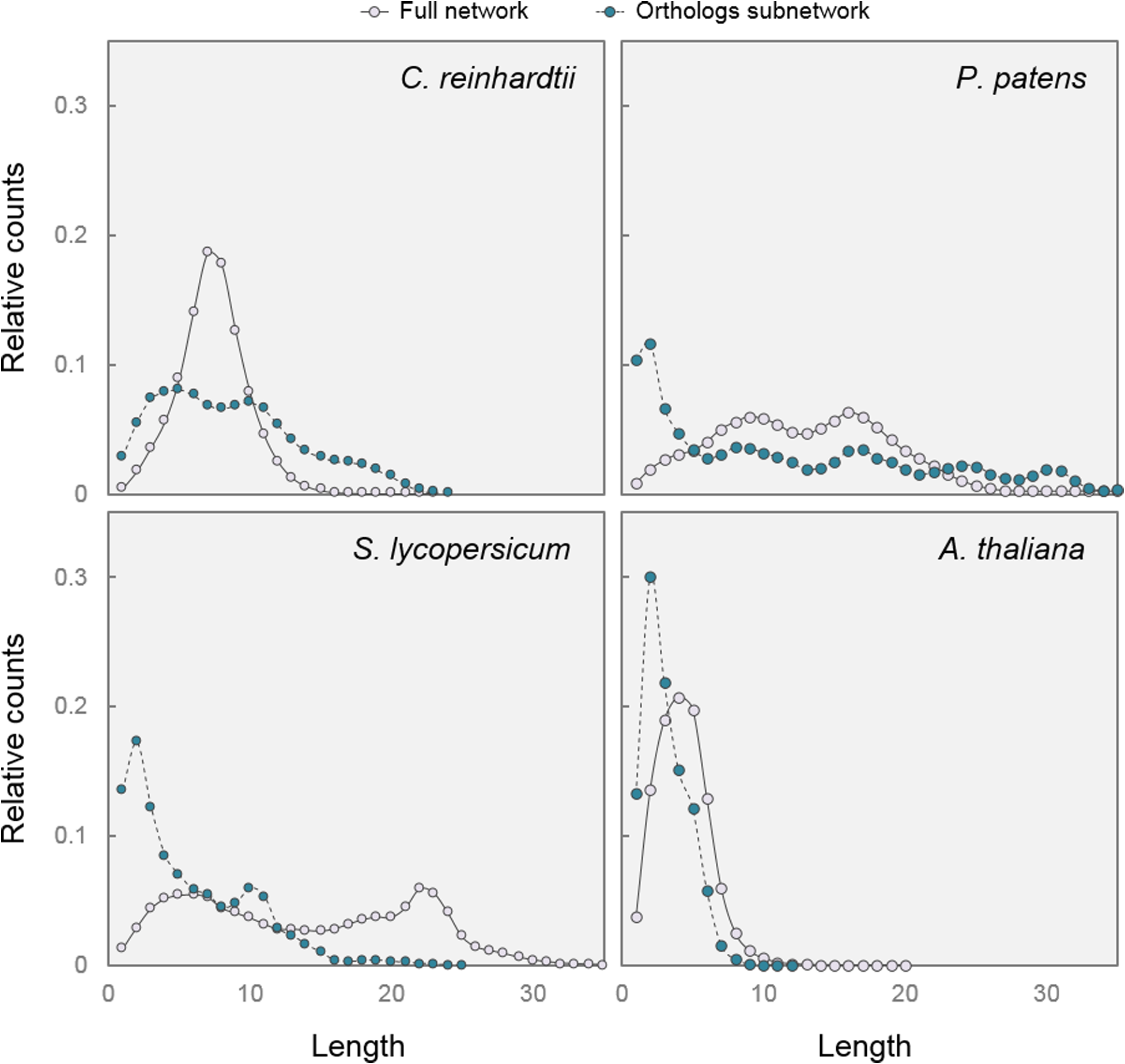
Paths are shorter in DELLA-associated subnetworks. Shortest path length distribution in Full networks and Orthologs subnetworks from the four species. The graphs represent the relative number of nodes (y-axis) joined by a given number of intermediate nodes (x-axis).
Network motifs are small recurring patterns involving a few nodes that appear more frequently in biological networks than in randomized ones. They consist of a certain level of regulation which connects small sets of nodes with a particular topology. Motifs characterize a network, as some of them are useful for the regulation of determined functions, and thus conserved along evolution (Kashtan and Alon, 2005). After measuring the frequency of the eight common motifs composed of three and four nodes in the full networks, we found that there was no relative enrichment of any particular motif between species when comparing the full networks or the Orthologs subnetworks (Figure 5A). However, the AtOrtho, SlOrtho, and PpOrtho subnetworks displayed a clear enrichment in virtually every motif, compared with their respective full networks (Figure 5B). Given that the function of this sort of motifs is to allow coordinated expression of a group of genes with shared function (Alon, 2007), the increase in the proportion of small regulatory patterns among all the putative DELLA targets in species that do contain DELLAs indicates an increase in the complexity of gene regulation, in which DELLAs might mediate the coordination of transcriptional programs.
FIGURE 5
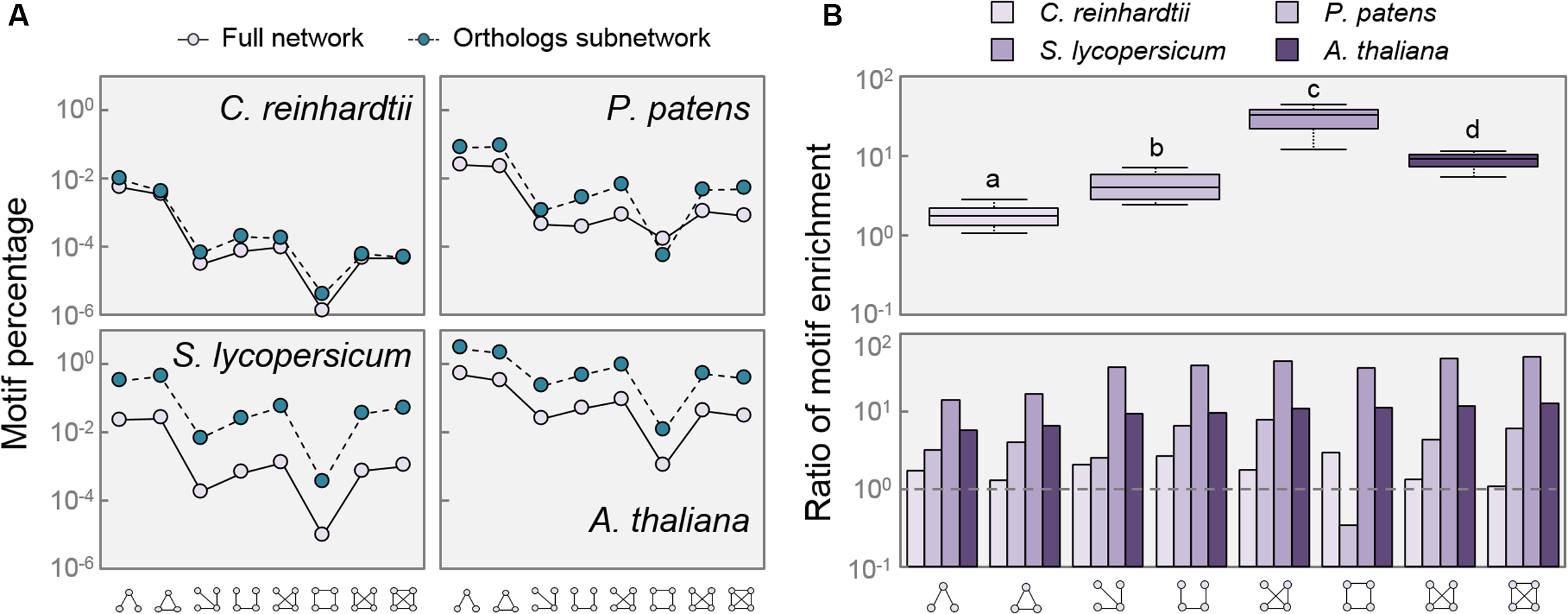
Network motifs are enriched in DELLA related networks. (A) Percentage of motifs found in each network compared to possible combinations of three and four nodes. (B) Ratio of motif enrichment comparing Orthologs subnetworks to Full networks per species (upper panel), and per motif (lower panel). Dashed lines in (B) mark a ratio of 1. Motifs are as depicted in X-axis. Letters indicate significant differences between groups, p < 0.01 (One way ANOVA, Tukey HSD Post Hoc test). Box-plot whiskers are Tukey-defined (extended 1.5 times the IQR from the box edges).
The Regulation of the Stress Response: A Likely Role of Ancestral DELLA Proteins
The results shown above suggest that the origin of DELLAs in land plants would be associated to an increase in the co-expression between genes that are putative targets of DELLA-interacting TFs, both in terms of size of the gene set and degree of the co-expression value. Therefore, DELLAs would have helped in the coordination of certain transcriptional circuits, and their recruitment to mediate GA signaling later in development would have further expanded their coordination capacity. To reveal the most likely functions ultimately regulated by DELLAs in the common ancestor of land plants, we carried out Gene Ontology (GO) analyses on each of the Neighbor subnetworks, with the idea that the terms shared by those in S. lycopersicum, A. thaliana, and P. patens could represent likely functions regulated by the ancestral DELLA proteins.
Not surprisingly, given the larger size of AtNeigh (Table 1), GO analysis rendered a much larger number of terms significantly enriched in this subnetwork, compared to those from the other three organisms (Supplementary Table S4). Terms referring to chloroplast function, such as plastid organization, photosynthesis, or pigment biosynthesis (including chlorophyll) were specifically enriched among the putative DELLA targets in A. thaliana only (Figure 6). This result might reflect functions whose regulation by DELLA has been acquired more recently, or it could simply be a bias of the analysis, caused by the big difference in size of the analyzed sets in the different species. On the contrary, the finding that terms comprised under general ‘response to stress’ were significantly over-represented in the subnetworks of the three land plants, but not C. reinhardtii, suggests that this function might have been the primary target of the regulation by ancestral DELLAs through their interaction with specific TFs.
FIGURE 6
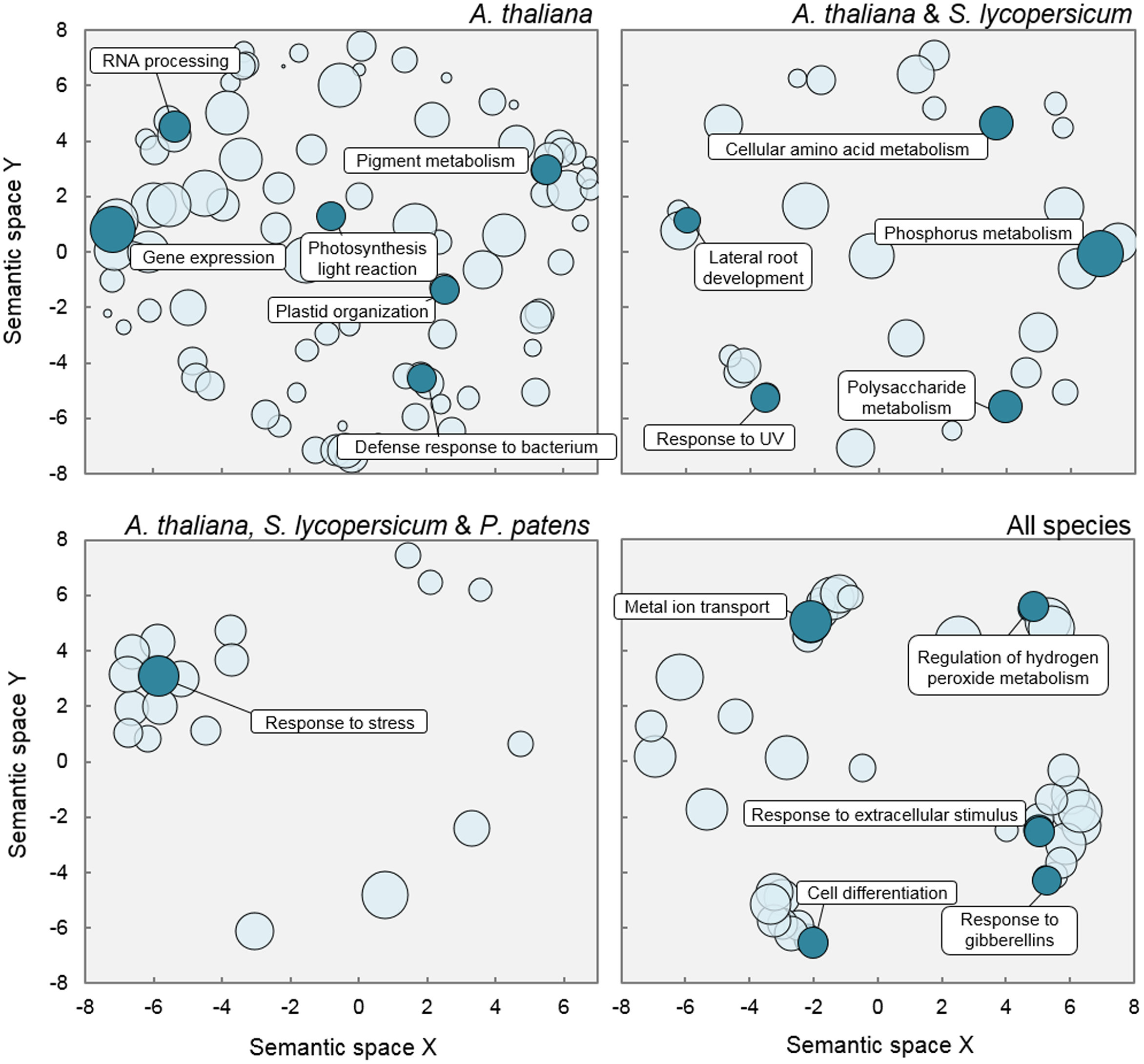
Gene Ontology terms enriched in Neighbors subnetworks. Scatterplots show cluster representatives after redundancy reduction in a two dimensional space derived by applying multidimensional scaling to a matrix of the GO categories semantic similarities. Bubble size is proportional to p-value significance of GO enrichment.
Conclusion
Our analysis suggests that DELLAs may have contributed to the acquisition of an increasing degree of coordination between transcriptional programs during plant evolution. Although these results are consistent with the current view of DELLAs as ‘hubs’ in transcriptional programs in higher plants, and provide a plausible evolutionary scenario, it is important to remark that further experimental work is required to validate most of the conclusions from in silico network analysis. In fact, several reasonable assumptions have been made that would be relatively easy to confirm. For instance, actual transcriptomic data of dellaKO mutants in the different species, coupled to comparative analysis would help establish the role of ancestral DELLAs. Moreover, our current analysis would be strengthened by the experimentally obtained information of which PIDs are in fact bona-fide DELLA interactors in the different species. Finally, the conclusion that DELLAs have probably contributed to the establishment of new co-regulatory circuits during land-plant evolution does not explain the molecular mechanism that supports this progressive acquisition, and it can be generated by changes in DELLA proteins, in their interactors, or in both.
Materials and Methods
Gene Co-expression Network Inference
The C. reinhardtii and A. thaliana networks were downloaded from the web resources of previous work (Romero-Campero et al., 2013, 2016). For the new networks, RNA-seq data were selected from equivalent experiments involving comparable tissues and environmental situations (Supplementary Table S5). The P. patens gene co-expression network was inferred from the RNA-seq data freely available from the Gene Expression Omnibus identified with accession numbers GSE19824, GSE33279, GSE36274, and GSE25237. The S. lycopersicum network was constructed based on the RNA-seq data identified with the accession numbers GSE45774, GSE64665, GSE64981, GSE68018, and GSE77340 in the Gene Expression Omnibus. In both cases, RNA-seq data was processed using the Tuxedo protocol (Trapnell et al., 2012) to obtain gene expression levels measured as FPKM. Briefly, short reads were mapped to the corresponding reference genome using Tophat, transcripts were assembled using Cufflinks and expression levels were computed using Cuffdiff. The Bioconductor R package cummeRbund (Goff et al., 2013) was used for subsequent analysis of the results generated by the Tuxedo protocol. In order to reduce noise in our analysis only genes that were detected as differentially expressed in at least one of the studies integrated in this work were considered. Differentially expressed genes were determined comparing each condition with the corresponding control within each study using a fold-change threshold of two. For each species, a matrix containing the expression levels of the selected genes was extracted. The Pearson correlation coefficient between every pair of gene expression profiles was computed using the cor function from the stats R package to generate a correlation matrix. Two genes were assumed to be co-expressed when the Pearson correlation coefficient between their expression profiles over the analyzed conditions was greater than 0.95. Following this criterion, the corresponding adjacency matrix was generated from the correlation matrix. Using the R package igraph1 (Csardi and Nepusz, 2006), each network was constructed from its adjacency matrix and exported in gml formal for subsequent analysis.
Data Compilation and Processing
The reference proteomes from A. thaliana TAIR10, S. lycopersicum iTAGv2.3, C. reinhardtii v5.5, and P. patens v3.3 were downloaded from Phytozome (Goodstein et al., 2012). From all the possible proteins from each locus tag only the longest protein was kept and assigned to its locus tag. These files were used to identify the orthologs among the four species with OrthoMCL (Li et al., 2003).
The networks were converted to SIF format and processed using the package igraph1 (Csardi and Nepusz, 2006) made with R2 (R Core Team, 2016). Only the edges between two non-identical nodes were conserved. If a given node was not identified in the proteome files, it was removed from the network. Afterward, components with fewer than seven elements were removed from the network to generate the complete network for each species. The orthologs for the set of manually curated DELLA interactors from A. thaliana were identified, and these nodes were selected from the complete networks. The first neighbors for all the selected nodes were identified and used to build a subnetwork. Finally, the orthologs on each species for all the genes in the previous subnetworks were identified and used to generate a new subnetwork for each species.
Network Analysis and Visualization
All networks were imported into the software package Cytoscape (Smoot et al., 2011) for their visualization using the Prefuse Force Directed layout.
The measures of network topology were calculated using both predefined and custom made functions. The gene–gene co-expression and neighborhood conservation were determined following the approach described by Netotea et al. (2014), using Fisher exact tests to check for statistical significance.
Gene Ontology analysis on Neigh subnetworks was made with AgriGO (Du et al., 2010), and represented with ReviGO (Supek et al., 2011).
Statements
Author contributions
AB-M, JH-G and MB conceived and designed the work. FR-C, JR, and FV constructed the co-expression networks. AB-M, CV-C, and JH-G performed network analyses. AB-M and MB wrote the first draft of the manuscript, to which all authors contributed.
Funding
. Work in the laboratories was funded by grants BFU2016-80621-P and BIO2014-52425-P of the Spanish Ministry of Economy, Industry and Competitiveness, and H2020-MSCA-RISE-2014-644435 of the European Union. AB-M and JH-G hold Fellowships of the Spanish Ministry of Education, Culture and Sport FPU14/01941 and FPU15/01756, respectively.
Acknowledgments
We thank the members of the Hormone Signaling and Plasticity Lab at IBMCP (http://www.ibmcp.upv.es/BlazquezAlabadiLab/) for useful discussions and suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00626/full#supplementary-material
TABLE S1Compilation of DELLA interactors used in this study.
TABLE S2Genes included in the ‘Neighbors’ subnetworks.
TABLE S3Genes included in the ‘Orthologs’ subnetworks.
TABLE S4Gene Onthology categories enriched in the ‘Neighbors’ subnetworks.
TABLE S4Gene Onthology categories enriched in the ‘Neighbors’ subnetworks.
References
1
Achard P. Cheng H. De Grauwe L. Decat J. Schoutteten H. Moritz T. et al (2006). Integration of plant responses to environmentally activated phytohormonal signals.Science31191–94. 10.1126/science.1118642
2
Achard P. Gong F. Cheminant S. Alioua M. Hedden P. Genschik P. (2008a). The cold-inducible CBF1 factor-dependent signaling pathway modulates the accumulation of the growth-repressing DELLA proteins via its effect on gibberellin metabolism.Plant Cell202117–2129. 10.1105/tpc.108.058941
3
Achard P. Liao L. Jiang C. Desnos T. Bartlett J. Fu X. et al (2007). DELLAs contribute to plant photomorphogenesis.Plant Physiol.1431163–1172.10.1104/pp.106.092254
4
Achard P. Renou J. P. Berthome R. Harberd N. P. Genschik P. (2008b). Plant DELLAs restrain growth and promote survival of adversity by reducing the levels of reactive oxygen species.Curr. Biol.18656–660. 10.1016/j.cub.2008.04.034
5
Alabadi D. Blazquez M. A. Carbonell J. Ferrandiz C. Perez-Amador M. A. (2009). Instructive roles for hormones in plant development.Int. J. Dev. Biol.531597–1608. 10.1387/ijdb.072423da
6
Alabadí D. Gil J. Blázquez M. A. García-Martínez J. L. (2004). Gibberellins repress photomorphogenesis in darkness.Plant Physiol.1341050–1057.10.1104/pp.103.035451
7
Alon U. (2007). Network motifs: theory and experimental approaches.Nat. Rev. Genet.8450–461. 10.1038/nrg2102
8
Aoki K. Ogata Y. Shibata D. (2007). Approaches for extracting practical information from gene co-expression networks in plant biology.Plant Cell Physiol.48381–390. 10.1093/pcp/pcm013
9
Arnaud N. Girin T. Sorefan K. Fuentes S. Wood T. A. Lawrenson T. et al (2010). Gibberellins control fruit patterning in Arabidopsis thaliana.Genes Dev.242127–2132. 10.1101/gad.593410
10
Bai M. Y. Shang J. X. Oh E. Fan M. Bai Y. Zentella R. et al (2012). Brassinosteroid, gibberellin and phytochrome impinge on a common transcription module in Arabidopsis.Nat. Cell Biol.14810–817. 10.1038/ncb2546
11
Casal J. J. Fankhauser C. Coupland G. Blázquez M. A. (2004). Signalling for developmental plasticity.Trends Plant Sci.9309–314. 10.1016/j.tplants.2004.04.007
12
Cheminant S. Wild M. Bouvier F. Pelletier S. Renou J. P. Erhardt M. et al (2011). DELLAs regulate chlorophyll and carotenoid biosynthesis to prevent photooxidative damage during seedling deetiolation in Arabidopsis.Plant Cell231849–1860. 10.1105/tpc.111.085233
13
Claeys H. De Bodt S. Inze D. (2014). Gibberellins and DELLAs: central nodes in growth regulatory networks.Trends Plant Sci.19231–239.10.1016/j.tplants.2013.10.001
14
Colebrook E. H. Thomas S. G. Phillips A. L. Hedden P. (2014). The role of gibberellin signalling in plant responses to abiotic stress.J. Exp. Biol.21767–75. 10.1242/jeb.089938
15
Crocco C. D. Holm M. Yanovsky M. J. Botto J. F. (2010). AtBBX21 and COP1 genetically interact in the regulation of shade avoidance.Plant J.64551–562. 10.1111/j.1365-313X.2010.04360.x
16
Csardi G. Nepusz T. (2006). The igraph software package for complex network research.InterJournal Complex Syst. 1695.
17
Daviere J. M. Achard P. (2016). A pivotal role of DELLAs in regulating multiple hormone signals.Mol. Plant910–20. 10.1016/j.molp.2015.09.011
18
Daviere J. M. Wild M. Regnault T. Baumberger N. Eisler H. Genschik P. et al (2014). Class I TCP-DELLA interactions in inflorescence shoot apex determine plant height.Curr. Biol.241923–1928. 10.1016/j.cub.2014.07.012
19
de Lucas M. Davière J. M. Rodríguez-Falcón M. Pontin M. Iglesias-Pedraz J. M. Lorrain S. et al (2008). A molecular framework for light and gibberellin control of cell elongation.Nature451480–484. 10.1038/nature06520
20
Du Z. Zhou X. Ling Y. Zhang Z. Su Z. (2010). agriGO: a GO analysis toolkit for the agricultural community.Nucleic Acids Res.38W64–W70.10.1093/nar/gkq310
21
Feng S. Martinez C. Gusmaroli G. Wang Y. Zhou J. Wang F. et al (2008). Coordinated regulation of Arabidopsis thaliana development by light and gibberellins.Nature451475–479. 10.1038/nature06448
22
Feurtado J. A. Huang D. Wicki-Stordeur L. Hemstock L. E. Potentier M. S. Tsang E. W. et al (2011). The Arabidopsis C2H2 zinc finger INDETERMINATE DOMAIN1/ENHYDROUS promotes the transition to germination by regulating light and hormonal signaling during seed maturation.Plant Cell231772–1794. 10.1105/tpc.111.085134
23
Fonouni-Farde C. Tan S. Baudin M. Brault M. Wen J. Mysore K. S. et al (2016). DELLA-mediated gibberellin signalling regulates Nod factor signalling and rhizobial infection.Nat. Commun.7:12636. 10.1038/ncomms12636
24
Franco-Zorrilla J. M. Lopez-Vidriero I. Carrasco J. L. Godoy M. Vera P. Solano R. (2014). DNA-binding specificities of plant transcription factors and their potential to define target genes.Proc. Natl. Acad. Sci. U.S.A.1112367–2372. 10.1073/pnas.1316278111
25
Fukazawa J. Teramura H. Murakoshi S. Nasuno K. Nishida N. Ito T. et al (2014). DELLAs function as coactivators of GAI-ASSOCIATED FACTOR1 in regulation of gibberellin homeostasis and signaling in Arabidopsis.Plant Cell262920–2938. 10.1105/tpc.114.125690
26
Gallego-Bartolomé J. Arana M. V. Vandenbussche F. Zadnikova P. Minguet E. G. Guardiola V. et al (2011). Hierarchy of hormone action controlling apical hook development in Arabidopsis.Plant J.67622–634. 10.1111/j.1365-313X.2011.04621.x
27
Gallego-Bartolomé J. Minguet E. G. Grau-Enguix F. Abbas M. Locascio A. Thomas S. G. et al (2012). Molecular mechanism for the interaction between gibberellin and brassinosteroid signaling pathways in Arabidopsis.Proc. Natl. Acad. Sci. U.S.A.10913446–13451. 10.1073/pnas.1119992109
28
Gallego-Bartolomé J. Minguet E. G. Marín J. A. Prat S. Blázquez M. A. Alabadí D. (2010). Transcriptional diversification and functional conservation between DELLA proteins in Arabidopsis.Mol. Biol. Evol.271247–1256.10.1093/molbev/msq012
29
Goff L. Trapnell C. Kelley D. (2013). cummeRbund: Analysis, Exploration, Manipulation, and Visualization of Cufflinks High-Throughput Sequencing Data in: R Package Version 2.16.0. Burlington, MA: ScienceOpen, Inc.
30
Goodstein D. M. Shu S. Howson R. Neupane R. Hayes R. D. Fazo J. et al (2012). Phytozome: a comparative platform for green plant genomics.Nucleic Acids Res.40D1178–D1186. 10.1093/nar/gkr944
31
Heck C. Kuhn H. Heidt S. Walter S. Rieger N. Requena N. (2016). Symbiotic fungi control plant root cortex development through the novel GRAS transcription factor MIG1.Curr. Biol.262770–2778. 10.1016/j.cub.2016.07.059
32
Hedden P. Thomas S. G. (2012). Gibberellin biosynthesis and its regulation.Biochem. J.44411–25. 10.1042/BJ20120245
33
Hirano K. Nakajima M. Asano K. Nishiyama T. Sakakibara H. Kojima M. et al (2007). The GID1-mediated gibberellin perception mechanism is conserved in the lycophyte Selaginella moellendorffii but not in the bryophyte Physcomitrella patens.Plant Cell193058–3079. 10.1105/tpc.107.051524
34
Hong G. J. Xue X. Y. Mao Y. B. Wang L. J. Chen X. Y. (2012). Arabidopsis MYC2 interacts with DELLA proteins in regulating sesquiterpene synthase gene expression.Plant Cell242635–2648. 10.1105/tpc.112.098749
35
Hou X. Lee L. Y. Xia K. Yan Y. Yu H. (2010). DELLAs modulate jasmonate signaling via competitive binding to JAZs.Dev. Cell19884–894. 10.1016/j.devcel.2010.10.024
36
Hou X. Zhou J. Liu C. Liu L. Shen L. Yu H. (2014). Nuclear factor Y-mediated H3K27me3 demethylation of the SOC1 locus orchestrates flowering responses of Arabidopsis.Nat. Commun.5:4601. 10.1038/ncomms5601
37
Huang D. Wang S. Zhang B. Shang-Guan K. Shi Y. Zhang D. et al (2015). A gibberellin-mediated DELLA-NAC signaling cascade regulates cellulose synthesis in rice.Plant Cell271681–1696. 10.1105/tpc.15.00015
38
Hyun Y. Richter R. Vincent C. Martinez-Gallegos R. Porri A. Coupland G. (2016). Multi-layered regulation of SPL15 and cooperation with SOC1 integrate endogenous flowering pathways at the Arabidopsis shoot meristem.Dev. Cell37254–266. 10.1016/j.devcel.2016.04.001
39
Itoh H. Ueguchi-Tanaka M. Sato Y. Ashikari M. Matsuoka M. (2002). The gibberellin signaling pathway is regulated by the appearance and disappearance of SLENDER RICE1 in nuclei.Plant Cell1457–70. 10.1105/tpc.010319
40
Jaillais Y. Chory J. (2010). Unraveling the paradoxes of plant hormone signaling integration.Nat. Struct. Mol. Biol.17642–645. 10.1038/nsmb0610-642
41
Jin Y. Liu H. Luo D. Yu N. Dong W. Wang C. et al (2016). DELLA proteins are common components of symbiotic rhizobial and mycorrhizal signalling pathways.Nat. Commun.7:12433. 10.1038/ncomms12433
42
Josse E. M. Gan Y. Bou-Torrent J. Stewart K. L. Gilday A. D. Jeffree C. E. et al (2011). A DELLA in disguise: SPATULA restrains the growth of the developing Arabidopsis seedling.Plant Cell231337–1351. 10.1105/tpc.110.082594
43
Kashtan N. Alon U. (2005). Spontaneous evolution of modularity and network motifs.Proc. Natl. Acad. Sci. U.S.A.10213773–13778. 10.1073/pnas.0503610102
44
Li L. Stoeckert C. J. Jr. Roos D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes.Genome Res.132178–2189.10.1101/gr.1224503
45
Li M. An F. Li W. Ma M. Feng Y. Zhang X. et al (2016). DELLA proteins interact with FLC to repress flowering transition.J. Integr. Plant Biol.58642–655. 10.1111/jipb.12451
46
Li Q. F. Wang C. Jiang L. Li S. Sun S. S. He J. X. (2012). An interaction between BZR1 and DELLAs mediates direct signaling crosstalk between brassinosteroids and gibberellins in Arabidopsis.Sci. Signal.5 ra72. 10.1126/scisignal.2002908
47
Lim S. Park J. Lee N. Jeong J. Toh S. Watanabe A. et al (2013). ABA-INSENSITIVE3, ABA-INSENSITIVE5, and DELLAs interact to activate the expression of SOMNUS and other high-temperature-inducible genes in imbibed seeds in Arabidopsis.Plant Cell254863–4878. 10.1105/tpc.113.118604
48
Locascio A. Blazquez M. A. Alabadi D. (2013a). Dynamic regulation of cortical microtubule organization through prefoldin-DELLA interaction.Curr. Biol.23804–809. 10.1016/j.cub.2013.03.053
49
Locascio A. Blazquez M. A. Alabadi D. (2013b). Genomic analysis of della protein activity.Plant Cell Physiol.541229–1237. 10.1093/pcp/pct082
50
Ma Z. Hu X. Cai W. Huang W. Zhou X. Luo Q. et al (2014). Arabidopsis miR171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions.PLoS Genet.10:e1004519. 10.1371/journal.pgen.1004519
51
Marin-de la Rosa N. Pfeiffer A. Hill K. Locascio A. Bhalerao R. P. Miskolczi P. et al (2015). Genome wide binding site analysis reveals transcriptional coactivation of cytokinin-responsive genes by DELLA proteins.PLoS Genet.11:e1005337. 10.1371/journal.pgen.1005337
52
Marin-de la Rosa N. Sotillo B. Miskolczi P. Gibbs D. J. Vicente J. Carbonero P. et al (2014). Large-scale identification of gibberellin-related transcription factors defines Group VII ETHYLENE RESPONSE FACTORS as functional DELLA partners.Plant Physiol.1661022–1032. 10.1104/pp.114.244723
53
Netotea S. Sundell D. Street N. R. Hvidsten T. R. (2014). ComPlEx: conservation and divergence of co-expression networks in A. thaliana, Populus and O. sativa.BMC Genomics15:106. 10.1186/1471-2164-15-106
54
Oh E. Zhu J. Y. Bai M. Y. Arenhart R. A. Sun Y. Wang Z. Y. (2014). Cell elongation is regulated through a central circuit of interacting transcription factors in the Arabidopsis hypocotyl.Elife3:e03031. 10.7554/eLife.03031
55
Park J. Nguyen K. T. Park E. Jeon J. S. Choi G. (2013). DELLA proteins and their interacting RING finger proteins repress gibberellin responses by binding to the promoters of a subset of gibberellin-responsive genes in Arabidopsis.Plant Cell25927–943. 10.1105/tpc.112.108951
56
Pimprikar P. Carbonnel S. Paries M. Katzer K. Klingl V. Bohmer M. J. et al (2016). A CCaMK-CYCLOPS-DELLA complex activates transcription of RAM1 to regulate arbuscule branching.Curr. Biol.26987–998. 10.1016/j.cub.2016.01.069
57
Qi T. Huang H. Wu D. Yan J. Qi Y. Song S. et al (2014). Arabidopsis DELLA and JAZ proteins bind the WD-repeat/bHLH/MYB complex to modulate gibberellin and jasmonate signaling synergy.Plant Cell261118–1133.10.1105/tpc.113.121731
58
R Core Team (2016). R: A Language and Environment for Statistical Computing.Vienna: R Foundation for Statistical Computing.
59
Resentini F. Felipo-Benavent A. Colombo L. Blazquez M. A. Alabadi D. Masiero S. (2015). TCP14 and TCP15 mediate the promotion of seed germination by gibberellins in Arabidopsis thaliana.Mol. Plant8482–485. 10.1016/j.molp.2014.11.018
60
Rombolá-Caldentey B. Rueda-Romero P. Iglesias-Fernandez R. Carbonero P. Onate-Sanchez L. (2014). Arabidopsis DELLA and two HD-ZIP transcription factors regulate GA signaling in the epidermis through the L1 box cis-element.Plant Cell262905–2919. 10.1105/tpc.114.127647
61
Romero-Campero F. J. Lucas-Reina E. Said F. E. Romero J. M. Valverde F. (2013). A contribution to the study of plant development evolution based on gene co-expression networks.Front. Plant Sci.4:291. 10.3389/fpls.2013.00291
62
Romero-Campero F. J. Perez-Hurtado I. Lucas-Reina E. Romero J. M. Valverde F. (2016). ChlamyNET: a Chlamydomonas gene co-expression network reveals global properties of the transcriptome and the early setup of key co-expression patterns in the green lineage.BMC Genomics17:227.10.1186/s12864-016-2564-y
63
Sarnowska E. A. Rolicka A. T. Bucior E. Cwiek P. Tohge T. Fernie A. R. et al (2013). DELLA-interacting SWI3C core subunit of switch/sucrose nonfermenting chromatin remodeling complex modulates gibberellin responses and hormonal cross talk in Arabidopsis.Plant Physiol.163305–317. 10.1104/pp.113.223933
64
Schwechheimer C. (2011). Gibberellin signaling in plants – the extended version.Front. Plant Sci.2:107. 10.3389/fpls.2011.00107
65
Shen Q. Cui J. Fu X. Q. Yan T. X. Tang K. X. (2015). Cloning and characterization of DELLA genes in Artemisia annua.Genet. Mol. Res.1410037–10049. 10.4238/2015.August.21.10
66
Smoot M. E. Ono K. Ruscheinski J. Wang P. L. Ideker T. (2011). Cytoscape 2.8: new features for data integration and network visualization.Bioinformatics27431–432. 10.1093/bioinformatics/btq675
67
Supek F. Bosnjak M. Skunca N. Smuc T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms.PLoS ONE6:e21800.10.1371/journal.pone.0021800
68
Trapnell C. Roberts A. Goff L. Pertea G. Kim D. Kelley D. R. et al (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks.Nat. Protoc.7562–578. 10.1038/nprot.2012.016
69
Usadel B. Obayashi T. Mutwil M. Giorgi F. M. Bassel G. W. Tanimoto M. et al (2009). Co-expression tools for plant biology: opportunities for hypothesis generation and caveats.Plant Cell Environ.321633–1651. 10.1111/j.1365-3040.2009.02040.x
70
Vera-Sirera F. Gomez M. D. Perez-Amador M. A. (2015). “DELLA proteins, a group of GRAS transcription regulators that mediate gibberellin signaling,” inPlant Transcription Factors; Evolutionary, Structural and Functional Aspects, ed.GonzálezD. H. (San Diego, CA: Elsevier), 313–328.
71
Vragovic I. Louis E. Diaz-Guilera A. (2005). Efficiency of informational transfer in regular and complex networks.Phys. Rev. E Stat. Nonlin. Soft Matter Phys.71:036122. 10.1103/physreve.71.036122
72
Wang Y. Deng D. (2014). Molecular basis and evolutionary pattern of GA-GID1-DELLA regulatory module.Mol. Genet. Genomics2891–9. 10.1007/s00438-013-0797-x
73
Wild M. Daviere J. M. Cheminant S. Regnault T. Baumberger N. Heintz D. et al (2012). The Arabidopsis DELLA RGA-LIKE3 is a direct target of MYC2 and modulates jasmonate signaling responses.Plant Cell243307–3319.10.1105/tpc.112.101428
74
Xie Y. Tan H. Ma Z. Huang J. (2016). DELLA proteins promote anthocyanin biosynthesis via sequestering MYBL2 and JAZ suppressors of the MYB/bHLH/WD40 complex in Arabidopsis thaliana.Mol. Plant9711–721. 10.1016/j.molp.2016.01.014
75
Xu F. Li T. Xu P. B. Li L. Du S. S. Lian H. L. et al (2016). DELLA proteins physically interact with CONSTANS to regulate flowering under long days in Arabidopsis.FEBS Lett.590541–549. 10.1002/1873-3468.12076
76
Yamaguchi N. Winter C. M. Wu M. F. Kanno Y. Yamaguchi A. Seo M. et al (2014). Gibberellin acts positively then negatively to control onset of flower formation in Arabidopsis.Science344638–641. 10.1126/science.1250498
77
Yang D. L. Yao J. Mei C. S. Tong X. H. Zeng L. J. Li Q. et al (2012). Plant hormone jasmonate prioritizes defense over growth by interfering with gibberellin signaling cascade.Proc. Natl. Acad. Sci. U.S.A.109E1192–E1200. 10.1073/pnas.1201616109
78
Yasumura Y. Crumpton-Taylor M. Fuentes S. Harberd N. P. (2007). Step-by-step acquisition of the gibberellin-DELLA growth-regulatory mechanism during land-plant evolution.Curr. Biol.171225–1230. 10.1016/j.cub.2007.06.037
79
Ye Y. Liu B. Zhao M. Wu K. Cheng W. Chen X. et al (2015). CEF1/OsMYB103L is involved in GA-mediated regulation of secondary wall biosynthesis in rice.Plant Mol. Biol.89385–401. 10.1007/s11103-015-0376-0
80
Yoshida H. Hirano K. Sato T. Mitsuda N. Nomoto M. Maeo K. et al (2014). DELLA protein functions as a transcriptional activator through the DNA binding of the INDETERMINATE DOMAIN family proteins.Proc. Natl. Acad. Sci. U.S.A.1117861–7866. 10.1073/pnas.1321669111
81
Yu N. Luo D. Zhang X. Liu J. Wang W. Jin Y. et al (2014). A DELLA protein complex controls the arbuscular mycorrhizal symbiosis in plants.Cell Res.24130–133. 10.1038/cr.2013.167
82
Yu S. Galvao V. C. Zhang Y. C. Horrer D. Zhang T. Q. Hao Y. H. et al (2012). Gibberellin regulates the Arabidopsis floral transition through miR156-targeted SQUAMOSA PROMOTER BINDING–LIKE transcription factors.Plant Cell243320–3332. 10.1105/tpc.112.101014
83
Zhang Z. L. Ogawa M. Fleet C. M. Zentella R. Hu J. Heo J. O. et al (2011). Scarecrow-like 3 promotes gibberellin signaling by antagonizing master growth repressor DELLA in Arabidopsis.Proc. Natl. Acad. Sci. U.S.A.1082160–2165. 10.1073/pnas.1012232108
84
Zhou X. Zhang Z. L. Park J. Tyler L. Yusuke J. Qiu K. et al (2016). The ERF11 transcription factor promotes internode elongation by activating gibberellin biosynthesis and signaling.Plant Physiol.1712760–2770. 10.1104/pp.16.00154
Summary
Keywords
gene co-expression networks, integrative molecular systems biology, evo–devo, transcriptional regulation, plant signaling
Citation
Briones-Moreno A, Hernández-García J, Vargas-Chávez C, Romero-Campero FJ, Romero JM, Valverde F and Blázquez MA (2017) Evolutionary Analysis of DELLA-Associated Transcriptional Networks. Front. Plant Sci. 8:626. doi: 10.3389/fpls.2017.00626
Received
04 March 2017
Accepted
07 April 2017
Published
25 April 2017
Volume
8 - 2017
Edited by
Stefan de Folter, Center for Advanced Research, The National Polytechnic Institute, Cinvestav-IPN, Mexico
Reviewed by
Marie Monniaux, Max Planck Institute for Plant Breeding Research (MPG), Germany; Frank Wellmer, Trinity College, Dublin, Ireland; Jean-Michel Davière, UPR2357 Institut de Biologie Moléculaire des Plantes (IBMP), France
Updates
Copyright
© 2017 Briones-Moreno, Hernández-García, Vargas-Chávez, Romero-Campero, Romero, Valverde and Blázquez.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel A. Blázquez, mblazquez@ibmcp.upv.es
This article was submitted to Plant Evolution and Development, a section of the journal Frontiers in Plant Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.