 Zhenyan Yang
Zhenyan Yang Yunheng Ji*
Yunheng Ji*- Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China
The Arcto-Tertiary relict genera, Camptotheca, Davidia, and Nyssa represent deep lineages in the asterid order Cornales. Recent phylogenetic studies suggested that these genera should be placed in a newly circumscribed family, Nyssaceae. However, because these analyses were based upon a few genes, it is prudent and necessary to examine further evidence before adopting this taxonomic treatment. In this study, we determined the complete chloroplast (cp) genomes of Camptotheca acuminata, Davidia involucrata, and Nyssa sinensis. Their cp genomes ranged from 156,672 to 158,409 bp, which included 115 genes, and their genome features were highly similar to those of other species within the order Cornales. The phylogenetic relationships among the genera Camptotheca, Davidia, Nyssa, and 23 related taxa in the asterids were analyzed based on 73 protein-coding genes from the cp genomes. All of the previously recognized major clades (namely Cornales, Ericales, Campanulids, and Lamiids) in the asterids, as well as their relationships, were recovered with robust support. A clade including the genera Davidia, Nyssa, Camptotheca, and Diplopanax, was resolved as a well-supported monophyletic group, which was fully separated from the family Cornaceae by the family Hydrangeaceae. Our results provide novel evidence to support the acceptance of the family Nyssaceae outlined by the updated Angiosperm Phylogeny Group.
Introduction
The woody dioecious genera, Camptotheca, Davidia, and Nyssa are very likely to be deep branches within the asterid order Cornales (Xiang et al., 2011). Davidia and Camptotheca have, respectively, only one and two extant species native to subtropical China (Qin and Chamlong, 2007), whereas Nyssa (approximately eight species) has a disjunct distribution in the middle latitudes of East Asia and North America (Wen and Stuessy, 1993). However, all three genera have extensive fossil records throughout the northern hemisphere during the Paleocene and Neogene (Eyde, 1997; Manchester, 2002; Manchester et al., 2009, 2015). Their current, relatively narrow distributions may have, in part, resulted from a range contraction triggered by the Neogene climate cooling and the Pleistocene glaciations (Axelrod, 1959; Qian and Ricklefs, 2000; Manchester et al., 2009). The extant species of Camptotheca, Davidia, and Nyssa are thus excellent examples of Arcto-Tertiary relicts. Their phylogenetic profiles would deepen our understanding of the evolution of the Arcto-Tertiary flora in the northern hemisphere.
The phylogenetic position of the genera Camptotheca, Davidia and Nyssa, has long been contentious. Historically, they were placed into either the family Cornaceae (Harms, 1898; Angiosperm Phylogeny Group, 1998, 2003, 2009), or the family Nyssaceae (Wangerin, 1910; Hutchinson, 1967; Cronquist, 1981; Angiosperm Phylogeny Group, 2016), or the families Davidiaceae (Davidia) and Nyssaceae (Camptotheca and Nyssa) (Takhtajan, 1980). The family Nyssaceae outlined by the Angiosperm Phylogeny Group (2016) contains the genera Camptotheca, Davidia, and Nyssa, as well as two other genera (Diplopanax and Mastixia) that were previously placed in the family Cornaceae. This taxonomic treatment was supported by prior phylogenetic analyses based on single or multi-locus DNA sequence data (Xiang et al., 1998, 2002, 2011; Fan and Xiang, 2003). Nonetheless, these studies were based on just a few genes, and the use of a limited number of informative loci may significantly increase the errors in the inferred phylogeny (Rokas and Carroll, 2005; Philippe et al., 2011). It is therefore, necessary to seek further evidence to test the delimitation of the newly circumscribed family Nyssaceae.
Chloroplast (cp) genome sequencing, by providing more genetic information, has proven itself as a method offering great potential for the resolution of historically difficult problems in phylogenetics (Jansen et al., 2007; Moore et al., 2007, 2010; Barrett et al., 2013, 2014; Ma et al., 2014; Stull et al., 2015; Attigala et al., 2016; Huang et al., 2016). Here, we present the complete cp genomes of Davidia involucrata, Nyssa sinensis, and Camptotheca acuminata through Illumina sequencing and a reference-guided assembly of the de novo contigs. The primary aim of this study was to evaluate the circumscription of the family Nyssaceae (Angiosperm Phylogeny Group, 2016) with a cp genome-based dataset. Together with the previously reported cp genome sequences that represent a wide phylogenetic diversity in the asterids, the phylogenetic relationships of the genera Davidia, Nyssa, and Camptotheca with related taxa were investigated.
Materials and Methods
Sample Preparation, DNA Extraction, Sequencing, and Genome Assembly
Fresh leaves of Davidia involucrata, N. sinensis, and C. acuminata were collected from the Botanical Garden of Kunming Institute of Botany, Chinese Academy of Sciences; voucher information is presented in Supplementary Table S1. Total genomic DNA was extracted from 100 mg of fresh leaves using a modified CTAB (cetyltrimethylammonium bromide) method (Doyle and Doyle, 1987), whereby 4% CTAB was used instead of 2% CTAB, and approximately 1% polyvinyl polypyrrolidone and 0.2% DL-dithiothreitol was added. Next, the complete cp genome sequences were amplified by using the nine primer pairs and protocols developed by Yang et al. (2014). Purified DNA (approximately 6 μg) from the resulting PCR products was fragmented and used to construct short-insert (500 bp) libraries according to the manufacturer’s manual (Illumina, San Diego, CA, United States). Paired-end sequencing was performed on the Illumina HiSeq 2000 platform at BGI (Shenzhen, Guangdong, China).
The Illumina raw data were filtered by using the NGS QC Toolkit (Patel and Jain, 2012), with an 80% read length and a cut-off value of 30 for the PHRED quality score. High-quality reads were assembled into contigs by using the software CLC Genomics Workbench v8.0 (CLC Bio), with k-mer = 63 and a minimum length of 1000 bp. Contigs were aligned with a reference cp genome of Diplopanax stachyanthus (NC_029750), which was the most similar genome identified via BLAST1. The assembly of the cp genome of each species was performed in Geneious version 7.0 (Kearse et al., 2012), by using the algorithm MUMmer. The validated complete cp genome sequences were deposited in GenBank (Supplementary Table S2).
Genomic Annotation and Comparison
The annotation of the cp genomes was initially done with the Dual Organellar Genome Annotator database tool (Wyman et al., 2004). Start and stop codons and intron/exon boundaries were manually checked. All tRNAs were further confirmed by tRNA scan-SE 1.21 (Schattner et al., 2005) set to the default parameters. The functional classification of the cp genes was determined by referring to the CpBase2. The graphical maps of the circular cp genomes were drawn using OrganellarGenome DRAW3 (Lohse et al., 2007).
To compare the cp genome structure and sequence divergence among members of the order Cornales, the complete cp genomes of Diplopanax stachyanthus, Hydrangea serrata, and Swida controversa were downloaded from the NCBI GenBank database (Supplementary Table S2). Multiple sequence alignment was performed in the MAFFT software program (Katoh et al., 2002), and manually edited whenever necessary. The boundaries of large single-copy (LSC) regions, inverted repeated (IR) regions, and small single-copy (SSC) regions in the cp genomes were compared among the six species by using Geneious v7.0 (Kearse et al., 2012). The sequence divergence among the six cp genomes was compared by the mVISTA tool (Frazer et al., 2004), for which S. controversa was set as a reference. To identify the single nucleotide polymorphisms (SNPs) across the six species, the Shuffle-LAGAN model in Geneious v7.0 (Kearse et al., 2012) was used with the parameter setting of “Only Find SNPs.” The divergent frequencies of SNPs across these species were calculated manually.
Phylogenomic Analysis
The phylogenetic analysis included six complete Cornales cp genomes, of which three were newly generated in the present study. To investigate the systematic position of the genera Davidia, Nyssa, and Camptotheca, the 23 cp genomes encompassing a wide phylogenetic diversity in the asterids were included in the analyses. Rheum palmatum, from the order Caryophyllales, was set to root the phylogenetic tree. The complete genomes reported for each species were downloaded from the NCBI GenBank database (Supplementary Table S2).
Seventy-three protein-coding genes commonly shared by these 26 taxa were used to reconstruct the phylogeny (Supplementary Table S3). The alignments of these genes were concatenated by the MAFFT software (Katoh et al., 2002). To test the phylogenetic effects of different regions of the cp genome, we defined the following four datasets based on various partition schemes: (1) one partition that had all genes and codons; (2) partitioned by all the first, second, and third codon positions in each gene (i.e., three partitions in total); (3) partitioned by each gene (73 partitions); and (4) partitioned by the first, second, and third codon positions in each gene (219 partitions). The best-fitting partition scheme and nucleotide substitution models were screened in the program PartitionFinder v2.1.1 (Lanfear et al., 2012). For each analysis, the branch lengths were linked, and the models of nucleotides substitution were restricted to those available in either RAxML (Stamatakis et al., 2008; Miller et al., 2010) or MrBayes (Ronquist and Huelsenbeck, 2003) independently; we used the “greedy” search algorithm. The partition that was able to include all genes and codons was selected as the best-fitting scheme.
The phylogenetic analyses were carried out using two approaches: Bayesian inference (BI) and maximum-likelihood analysis (ML). The most suitable nucleotide substitution model for ML and BI analyses suggested by the program PartitionFinder v2.1.1 (Lanfear et al., 2012) was GTR+G. The BI analyses were performed in MrBayes v3.2 (Ronquist and Huelsenbeck, 2003). Four Markov chains, each starting with a random tree, were run simultaneously for one million generations, with trees sampled every 100th generation. Trees from the first 250,000 generations were regarded as “burn in” and discarded. The posterior probability values (PP) were determined from the remaining 750,000 trees. The ML analyses were performed in RAxML-HPC BlackBox v8.1.24 (Stamatakis et al., 2008; Miller et al., 2010); 10 independent ML searches were conducted, and the branch support was determined by computing 1000 non-parametric bootstrap replicates.
Results
Chloroplast Genome Features
The average depths of sequencing coverage were 1154, 1169, and 1123× for N. sinensis, Davidia involucrata, and C. acuminata, respectively. Their complete cp genome sizes were 156,672–15,8409 bp. All three genomes, consisting of a pair of IRs (25,971–25,878 bp) separated by the LSC (86,184–87,611 bp) and SSC (18,260–18,856 bp) regions, showed a typical quadripartite structure that is similar to the majority of land plant cp genomes (Figure 1 and Table 1). The cp genomes of the three relict species contained 115 unique genes (81 protein-coding genes, 30 tRNA, and 4 rRNA) arranged in the same order, of which 18 were duplicated in the IR regions. Among these unique genes, 18 genes contained introns, 12 of which were protein-coding genes (atpF, ndhA, ndhB, petB, petD, rpl16, rpl2, rpoC1, rps12, rps16, clpP, and ycf3) and six were tRNA (trnA-UGC, trnG-GCC, trnI-GAU, trnK-UUU, trnL-UAA, and trnV-UAC). Sixteen of these 18 genes contained a single intron, while the other two had two introns (clpP and ycf3) (Table 2). The ycf1 gene at the IRB/SSC border was identified as a pseudogene in all taxa of the order Cornales. In addition, the ycf15 gene is likely also a pseudogene in Davidia involucrata (Table 2).
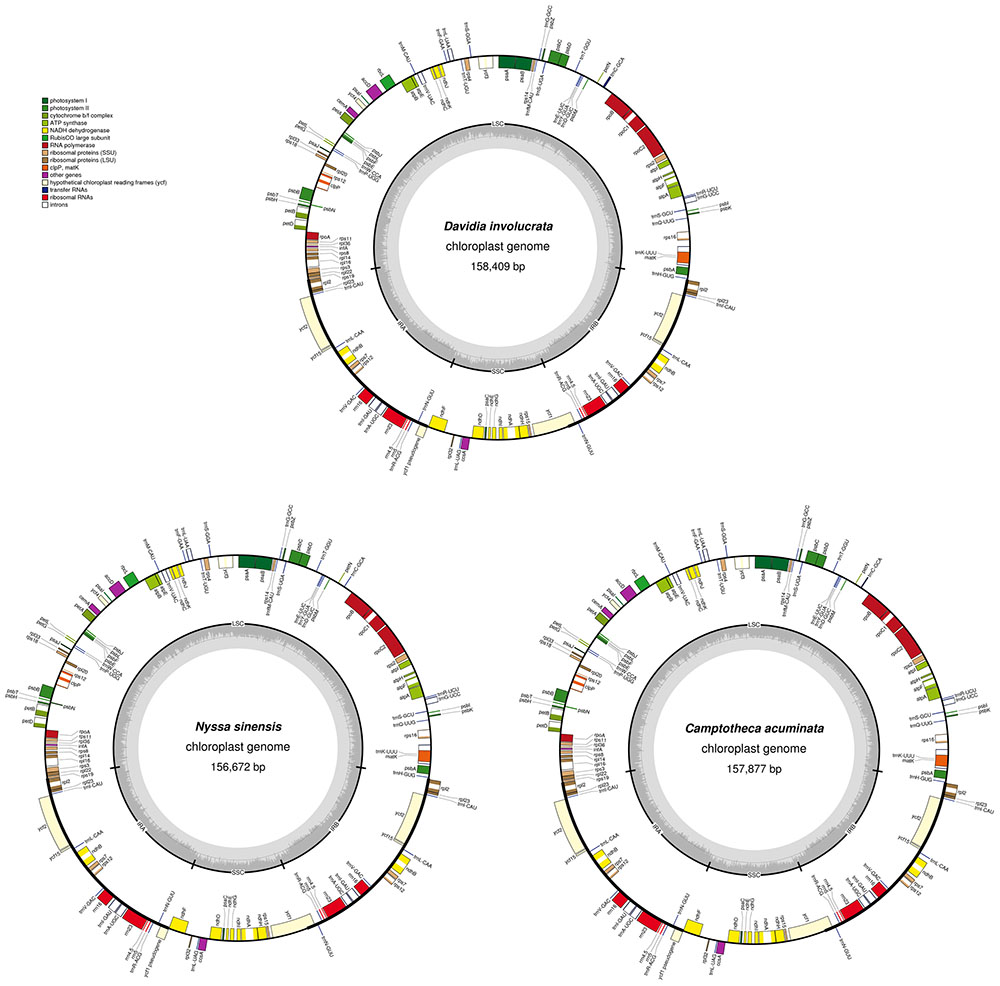
FIGURE 1. Gene map of the Camptotheca acuminata, Davidia involucrata, and Nyssa sinensis chloroplast genomes. Genes shown outside of the outer layer circle are transcribed counterclockwise, whereas genes inside of this circle are transcribed clockwise The colored bars indicate the known protein-coding genes, tRNA, and rRNA. The dashed darker gray area of the inner circle denotes the GC content, while the lighter gray area indicates the AT content of the genome. LSC, large single-copy; SSC, small single-copy; IR, inverted repeat.
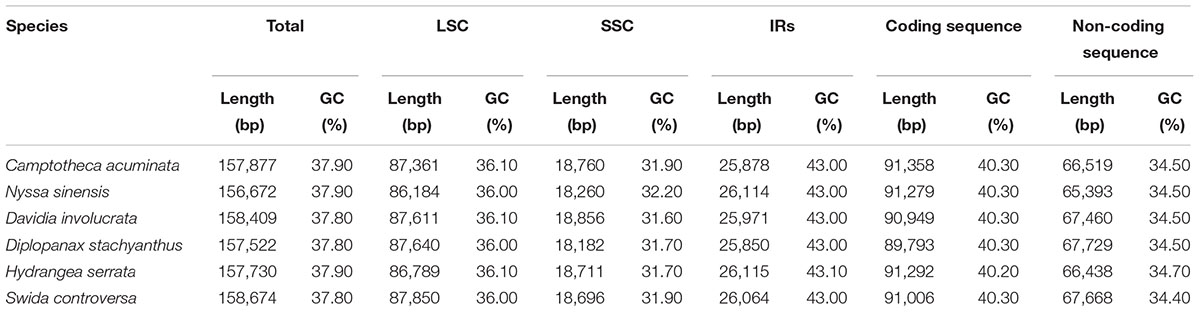
TABLE 1. Features of the Cornales chloroplast genomes.
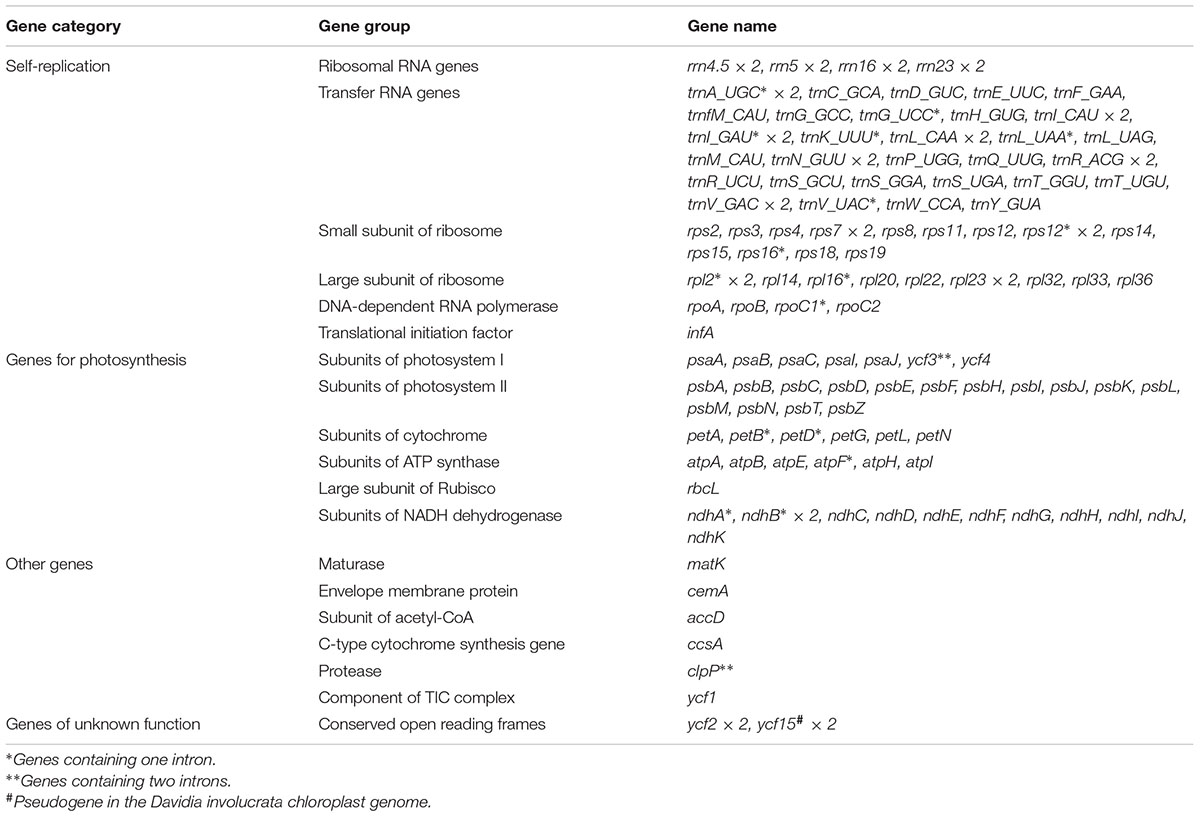
TABLE 2. List of genes identified in the chloroplast genomes of Davidia involucrata, Camptotheca acuminata, and Nyssa sinensis.
The IRA/LSC boundary in all the Cornales cp genomes was located between the rpl2 and trnH genes. Expansion of the IR regions into the rps19 and ycf1 genes at the IRB/LSC and IRA/SSC boundaries was detected, respectively, in all six Cornales species. Although the expansion of the IRB region into the ycf1 pseudogene at the IR/SSC junctions occurred in all species, the overlap between the ycf1 pseudogene and ndhF was only detected in C. acuminata, N. sinensis, and H. serrata (Figure 2).
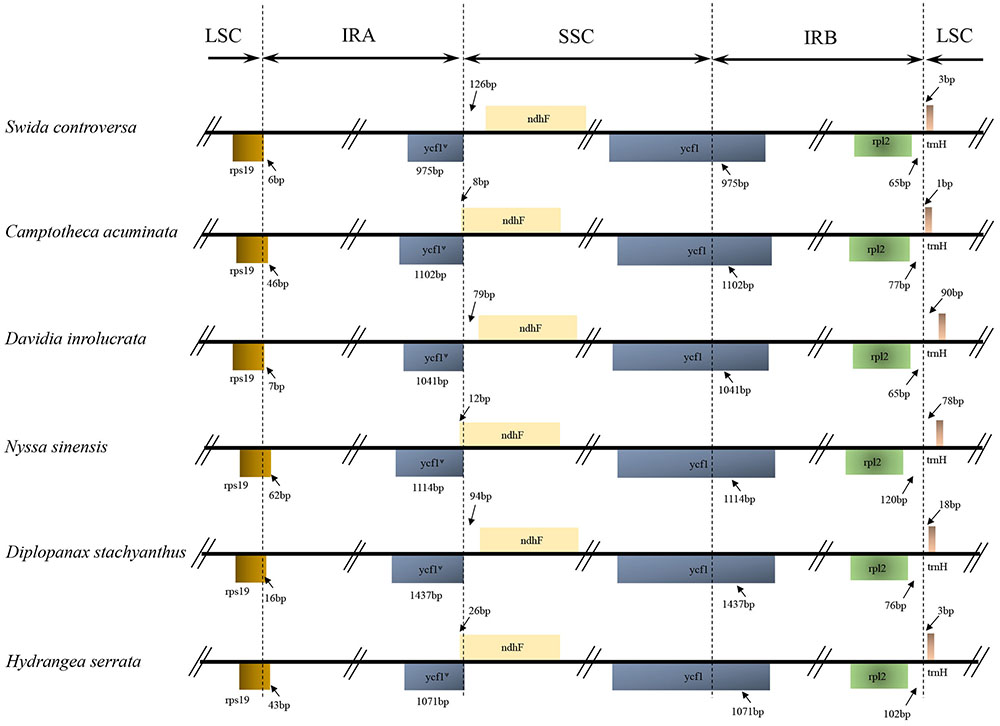
FIGURE 2. Comparison of the borders of the LSC, SSC, and IR regions among the Camptotheca acuminata, Davidia involucrata, Nyssa sinensis, Diplopanax stachyanthus, Hydrangea serrata, and Swida controversa chloroplast genomes. LSC, large single-copy; SSC, small single-copy; IR, inverted repeat.
Sequence Divergence in the Cornales Chloroplast Genomes
Regions containing SNPs were identified by the cp genome-wide comparison (Figure 3). A total of 4,886 SNPs were found in the matrix of the six cp genomes, and the average variant frequency was 3.01%. For all of these SNP mutations, 69.18% of the SNP sites were detected in the LSC region, 21.88% in the SSC region, and 8.94% in the IR region. The corresponding average variant frequency of LSC, SSC, and IR regions was 3.71, 5.08, and 0.87%. In addition, 1994 SNPs (average variant frequency = 2.19%) were detected in the coding regions, while 2,892 SNPs (average variant frequency = 4.05%) were detected in the non-coding regions (Table 3). The divergent frequencies of the exons varied from 0.00 to 6.79% (Supplementary Table S4), whereas those of the non-coding regions varied more, from 0.18 to 11.11% (Supplementary Table S5). According to the sequence divergence analysis, we screened 10 protein-coding regions (rps15, ccsA, rpl22, rps19, ndhG, clpP, ndhD, rps8, psbI, and rps3), with lengths ranging from 250 to 1,500 bp that could be utilized as potential molecular markers to reconstruct the phylogeny in the order Cornales. The percentage of SNPs in these divergence hotspot regions exceeded 3.5%.

FIGURE 3. Visualized alignment of the six Cornales chloroplast genomes. The mVISTA-based identity plots show the sequence identity among the six cp genomes, with S. controversa serving as a reference. Gray arrows indicate the position and direction of each gene. Genome regions are color-coded as protein-coding, rRNA, tRNA, or conserved non-coding regions. Black lines define the regions of sequence identity shared with S. controversa (by using a 50%-identity cutoff).
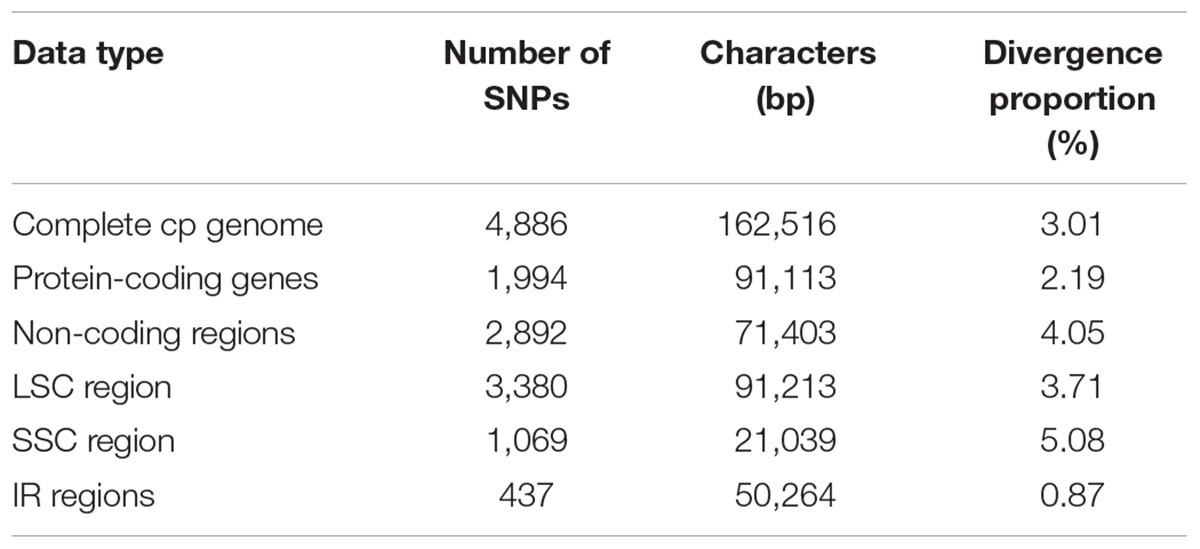
TABLE 3. Summary of the single nucleotide polymorphisms (SNPs) found in the six Cornales cp genomes.
Phylogenetic Analysis
The phylogenetic relationships of the asterids were reconstructed through the BI and ML analyses. The resulting ML and BI tree topologies were identical to each another. Figure 4 shows the phylogenetic tree generated by these BI and ML analyses, including the two types of support values: BI posterior probabilities (PP) and ML bootstrap values (MLBS). The asterids was resolved as four fully supported monophyletic lineages: Cornales, Ericales, Campanulids, and Lamiids. The order Cornales was recovered as the earliest diverged clade in the asterids; the Campanulids and Lamiids formed two sister clades (PP = 1.00, MLBS = 100%), which had diverged from the order Ericales (PP = 1.00, MLBS = 100%). The evolutionary relationships among these clades were consistent with those reported by Stull et al. (2015) and Angiosperm Phylogeny Group (2016).

FIGURE 4. The Bayesian inference (BI, left) and maximum-likelihood (ML, right) trees of 26 taxa reconstructed using 73 chloroplast protein-coding genes. Numbers indicate the posterior probabilities from the BI analyses and bootstrap values from the ML analyses.
Within the order Cornales, the four genera Nyssa, Camptotheca, Davidia, and Diplopanax formed a strongly supported monophyletic group (PP = 1.00, MLBS = 100%). This clade corresponds to the family Nyssaceae that was circumscribed by the Angiosperm Phylogeny Group (2016). Among the four genera, Nyssa is sister to Camptotheca (PP = 1.00, MLBS = 100%), and these two genera, in turn, are sister to Davidia (PP = 1.00, MLBS = 100%); Diplopanax is sister to the Nyssa+Camptotheca+Davidia Clade. In addition, the tree topologies clearly indicated that Nyssaceae circumscribed by the Angiosperm Phylogeny Group (2016) was fully separated from the family Cornaceae by the family Hydrangeaceae (Figure 4).
Discussion
Comparison of Chloroplast Genomes in the Cornales
Although several protein-coding genes (i.e., accD, ycf1, ycf2, rpl22, rps16, rpl23, infA, and ndhF) have been independently lost over the course of angiosperm evolution (e.g., Millen et al., 2001; Jansen et al., 2007), these genes were often detected in the six representatives of the Cornales (Table 2). In addition, no significant structural rearrangements, such as inversions or gene relocations, were observed in any of these six Cornales cp genomes (Figure 1). Taken together, these results suggest that the gene contents and arrangements of the cp genome are likely to be highly conserved in the Cornales.
The pseudogenization or loss of the ycf15 gene has been observed in a wide diversity of lineages in the angiosperms (e.g., Chumley et al., 2006; Raubeson et al., 2007). Previous studies proposed that, in the asterids, this mutation occurred only in the lineages that were diverged later (Chumley et al., 2006; Raubeson et al., 2007; Shi et al., 2013). However, our study indicates that this gene was pseudogenized in Davidia involucrata (Table 2), which is a member of the basally branching order (Cornales) in the asterids. This result suggests that the pseudogenization of ycf15 may have originated independently during the evolution of the asterid lineages; hence, it may not provide relevant phylogenetic information.
The IR expansions often lead to size variations in the angiosperm cp genomes (e.g., Cosner et al., 1997; Plunkett and Downie, 2000; Chumley et al., 2006). For example, a significant expansion of IR regions (ca. 4 kb) may be responsible for the relatively large cp genome of both Tetracentron sinense and Trochodendron aralioides (Sun et al., 2013). The IR/LSC junctions among the six Cornales cp genomes were highly conserved: the IRA/LSC boundaries were located between the rpl2 and trnH genes, while the IRB regions expanded into rps19 at the IRB/LSC junction (Figure 2). It is notable that this type of IR/LSC boundary has not been detected in the other asterid orders (Kim and Lee, 2004; Huang et al., 2014; Downie and Jansen, 2015; Stull et al., 2015; Yao et al., 2016); this suggests it could serve as a potential molecular marker for Cornales. In contrast to the IR/LSC junctions, the IR/SSC boundaries among the six Cornales cp genomes were variable, yet this variability may contribute little to the overall size variations in the chloroplast genomes of these plants. For instance, the largest overall cp genome size among the six Cornales species was observed in S. controversa (Figure 2), but this plant has the shortest expansion of the IR/SSC junction to ycf1 among the six species investigated (975 bp; Figure 2). Although Diplopanax stachyanthus has the longest expansion of the IR/SSC junction to the ycf1 gene (1,437 bp; Figure 2), its cp genome size is notably smaller than that of S. controversa, Davidia involucrata, C. acuminata, and H. serrata.
Phylogenetic Inferences
The key objective of our study was to evaluate the circumscription of the family Nyssaceae (Angiosperm Phylogeny Group, 2016) by using a cp genome-based dataset. Our phylogenomic analyses recovered a fully supported monophyletic clade that included the genera Camptotheca, Nyssa, Davidia, and Diplopanax in the order Cornales, which was separated from the family Cornaceae by the family Hydrangeaceae with substantial empirical support (Figure 4). This result provides additional evidence to accept the newly circumscribed family Nyssaceae (Angiosperm Phylogeny Group, 2016). It is notable that these genera share a distinct morphological similarity: their fruits have germination valves on the fruit stones. This can be the synapomorphy to recognize the family Nyssaceae.
Our analyses also resolved well the evolutionary relationships among the genera Camptotheca, Nyssa, and Davidia (Figure 4), which are consistent with other phylogenetic analyses (Xiang et al., 2002, 2011; Fan and Xiang, 2003). Several lines of evidence support the affinity between Camptotheca and Nyssa. Firstly, the fossil evidence suggests that Camptotheca and Nyssa may be derived from a common ancestor in the Eocene (Eyde, 1997; Manchester et al., 2009). Secondly, the two genera share similar fruit and inflorescence morphologies (Eyde, 1968), as well as wood anatomy (Titman, 1949). Finally, the basal chromosome number of Camptotheca and Nyssa is same (x = 22), whereas that of Davidia is x = 21 (Goldblatt, 1978). This last consideration further suggests that Camptotheca is more closely related to Nyssa than to Davidia. In this respect, it is noteworthy that the earliest fossil record for the Davidia, Camptotheca, and Nyssa belongs to the extinct species, Davidia antique, which occurred in the Paleocene of North America (Manchester, 2002). This is consistent with the basally branching position of Davidia among the three genera in the tree topologies we inferred.
A question that remains unresolved by our study is the phylogenetic position of the genus Mastixia. Previous molecular phylogenetic analyses indicated that this genus is closely related to Diplopanax (Xiang et al., 2002, 2011), and both genera produce flowers with hooked petals that are arranged in paniculate inflorescences (Zhu and Xiang, 1999). However, its basal chromosome number (x = 11) is far lower than that of Camptotheca, Nyssa, and Davidia (Goldblatt, 1978). Since we did not obtain a sample of Mastixia, clarifying its relationship(s) to the other genera in the family Nyssaceae will require further investigation.
Author Contributions
YJ designed the research; ZY collected and analyzed the data; YJ and ZY prepared the manuscript.
Funding
This research was financially supported by the Major Program of National Natural Science Foundation of China (No. 31590823) and the National Natural Science Foundation of China (No. 31070297).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We are grateful to Zhengshan He at the Kunming Institute of Botany, Chinese Academy of Sciences, for his help with the data analyses.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01536/full#supplementary-material
Footnotes
- ^http://blast.ncbi.nlm.nih.gov/
- ^http://chloroplast.ocean.washington.edu/
- ^http://ogdraw.mpimp-golm.mpg.de/
References
Angiosperm Phylogeny Group (1998). An ordinal classification for the families of flowering plants. Ann. Mo. Bot. Gard. 85, 531–553. doi: 10.2307/2992015
Angiosperm Phylogeny Group (2003). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Bot. J. Linn. Soc. 141, 399–436. doi: 10.1046/j.1095-8339.2003.t01-1-00158.x
Angiosperm Phylogeny Group (2009). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 161, 105–121. doi: 10.1111/j.1095-8339.2009.00996.x
Angiosperm Phylogeny Group (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.12385
Attigala, L., Wysocki, W. P., Duvall, M. R., and Clark, L. G. (2016). Phylogenetic estimation and morphological evolution of Arundinarieae (Bambusoideae: Poaceae) based on plastome phylogenomic analysis. Mol. Phylogenet. Evol. 101, 111–121. doi: 10.1016/j.ympev.2016.05.008
Axelrod, D. I. (1959). Poleward migration of early angiosperm flora. Science 130, 203–207. doi: 10.1126/science.130.3369.203
Barrett, C. F., Davis, J. I., Leebens-Mack, J., Conran, J. G., and Stevenson, D. W. (2013). Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics 29, 65–87. doi: 10.1111/j.1096-0031.2012.00418.x
Barrett, C. F., Specht, C. D., Leebens-Mack, J., Stevenson, D. W., Zomlefer, W. B., and Davis, J. I. (2014). Resolving ancient radiations: can complete plastid gene sets elucidate deep relationships among the tropical gingers (Zingiberales)? Ann. Bot. 113, 119–133. doi: 10.1093/aob/mct264
Chumley, T. W., Palmer, J. D., Mower, J. P., Fourcade, H. M., Calie, P. J., Boore, J. L., et al. (2006). The complete chloroplast genome sequence of Pelargonium x hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. doi: 10.1093/molbev/msl089
Cosner, M. E., Jasen, P. K., Palmer, J. D., and Downie, S. R. (1997). The highly rearranged chloroplast genome of Trachelium caeruleum (Campanuceae): insertions/deletions, and several repeat families. Curr. Genet. 31, 419–429. doi: 10.1007/s002940050225
Cronquist, A. (1981). An Integrated System of Classification of Flowering Plants. New York, NY: Columbia University Press.
Downie, S. R., and Jansen, R. K. (2015). A comparative analysis of whole plastid genomes from the Apiales: expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst. Bot. 40, 336–351. doi: 10.1600/036364415X686620
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Eyde, R. H. (1997). Fossil record and ecology of Nyssa (Cornaceae). Bot. Rev. 63, 97–123. doi: 10.1007/BF02935928
Fan, C., and Xiang, Q. Y. J. (2003). Phylogenetic analyses of Cornales based on 26S rRNA and combined 26S rDNA-matK-rbcL sequence data. Am. J. Bot. 90, 1357–1372. doi: 10.3732/ajb.90.9.1357
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Goldblatt, P. (1978). A contribution to cytology in Cornales. Ann. Mo. Bot. Gard. 65, 650–655. doi: 10.2307/2398864
Harms, H. (1898). “Cornaceae,” in Die Natürlichen Pflanzenfamilien, Vol. III-8, eds A. Engler and K. Prantl (Leipzig: Wilhelm Engelmann), 250–270.
Huang, H., Shi, C., Liu, Y., Mao, S. Y., and Gao, L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. doi: 10.1186/1471-2148-14-151
Huang, Y., Li, X., Yang, Z., Yang, C., Yang, J., and Ji, Y. (2016). Analysis of complete chloroplast genome sequences improves phylogenetic resolution in Paris (Melanthiaceae). Front. Plant Sci. 7:1797. doi: 10.3389/fpls.2016.01797
Hutchinson, J. (1967). The Genera of Flowering Plants Angiospermae, Vol. 2. London: Oxford University Press.
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Leebens-Mack, J., Müller, K. F., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U.S.A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Katoh, K., Misawa, K., Kuma, K. I., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, K. J., and Lee, H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Lanfear, R., Calcott, B., Ho, S. Y., and Guindon, S. (2012). PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29, 1695–1701. doi: 10.1093/molbev/mss020
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Ma, P. F., Zhang, Y. X., Zeng, C. X., Guo, Z. H., and Li, D. Z. (2014). Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 63, 933–950. doi: 10.1093/sysbio/syu054
Manchester, S. R. (2002). Leaves and fruits of Davidia (Cornales) from the Paleocene of North America. Syst. Bot. 27, 368–382. doi: 10.1043/0363-6445-27.2.368
Manchester, S. R., Chen, Z. D., Lu, A. M., and Uemura, K. (2009). Eastern Asian endemic seed plant genera and their paleogeographic history throughout the Northern Hemisphere. J. Syst. Evol. 47, 1–42. doi: 10.1111/j.1759-6831.2009.00001.x
Manchester, S. R., Grímsson, F., and Zetter, R. (2015). Assessing the fossil record of Asterids in the context of our current phylogenetic framework. Ann. Mo. Bot. Gard. 100, 329–363. doi: 10.3417/2014033
Millen, R. S., Olmstead, R. G., Adams, K. L., Palmer, J. D., Lao, N. T., Heggie, L., et al. (2001). Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13, 645–658. doi: 10.1105/tpc.13.3.645
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA. doi: 10.1109/GCE.2010.5676129
Moore, M. J., Bell, C. D., Soltis, P. S., and Soltis, D. E. (2007). Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. U.S.A. 104, 19363–19368. doi: 10.1073/pnas.0708072104
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G., and Soltis, D. E. (2010). Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. U.S.A. 107, 4623–4628. doi: 10.1073/pnas.0907801107
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLOS ONE 7:e30619. doi: 10.1371/journal.pone.0030619
Philippe, H., Brinkmann, H., Lavrov, D. V., Littlewood, D. T. J., Manuel, M., Wörheide, G., et al. (2011). Resolving difficult phylogenetic questions: why more sequences are not enough. PLOS Biol. 9:e1000602. doi: 10.1371/journal.pbio.1000602
Plunkett, G. M., and Downie, S. R. (2000). Expansion and contraction of the chloroplast inverted repeat in Apiaceae subfamily Apioideae. Syst. Bot. 25, 648–667. doi: 10.2307/2666726
Qian, H., and Ricklefs, R. E. (2000). Large-scale processes and the Asian bias in species diversity of temperate plants. Nature 407, 180–182. doi: 10.1038/35025052
Qin, H. N., and Chamlong, P. (2007). “Nyssaceae,” in Flora of China 13, eds Z. Y. Wu and P. H. Raren (Beijing: Science Press), 301–304.
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8:174. doi: 10.1186/1471-2164-8-174
Rokas, A., and Carroll, S. B. (2005). More genes or more taxa? The relative contribution of gene number and taxon number to phylogenetic accuracy. Mol. Biol. Evol. 22, 1337–1344. doi: 10.1093/molbev/msi121
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Shi, C., Liu, Y., Huang, H., Xia, E. H., Zhang, H. B., and Gao, L. Z. (2013). Contradiction between plastid gene transcription and function due to complex posttranscriptional splicing: an exemplary study of ycf15 function and evolution in angiosperms. PLOS ONE 8:e59620. doi: 10.1371/journal.pone.0059620
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. doi: 10.1080/10635150802429642
Stull, G. W., de Stefano, R. D., Soltis, D. E., and Soltis, P. S. (2015). Resolving basal lamiid phylogeny and the circumscription of Icacinaceae with a plastome-scale data set. Am. J. Bot. 102, 1794–1813. doi: 10.3732/ajb.1500298
Sun, Y. X., Moore, M. J., Meng, A. P., Soltis, P. S., Soltis, D. E., Li, J. Q., et al. (2013). Complete plastid genome sequencing of Trochodendraceae reveals a significant expansion of the inverted repeat and suggests a Paleogene divergence between the two extant species. PLOS ONE 8:e60429. doi: 10.1371/journal.pone.0060429
Takhtajan, A. L. (1980). Outline of the classification of flowering plants (magnoliophyta). Bot. Rev. 46, 225–359. doi: 10.1007/BF02861558
Titman, P. W. (1949). Studies in the woody anatomy of the family Nyssaceae. J. Elisha Mitchell Sci. Soc. 65, 245–261.
Wangerin, W. (1910). “Cornaceae,” in Das Pflanzenreich, Series IV, Family 229 (Heft 41), ed. A. Engler (Leipzig: W. Engelmann).
Wen, J., and Stuessy, T. F. (1993). The phylogeny and biogeography of Nyssa (Cornaceae). Syst. Bot. 18, 68–79. doi: 10.2307/2419789
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xiang, Q. Y., Moody, M. L., Soltis, D. E., Fan, C. Z., and Soltis, P. S. (2002). Relationships within Cornales and circumscription of Cornaceae-matK and rbcL sequence data and effects of outgroups and long branches. Mol. Phylogenet. Evol. 24, 35–57. doi: 10.1016/S1055-7903(02)00267-1
Xiang, Q. Y., Soltis, D. E., and Soltis, P. S. (1998). Phylogenetic relationships of Cornaceae and close relatives inferred from matK and rbcL sequences. Am. J. Bot. 85, 285–297. doi: 10.2307/2446317
Xiang, Q. Y. J., Thomas, D. T., and Xiang, Q. P. (2011). Resolving and dating the phylogeny of Cornales—effects of taxon sampling, data partitions, and fossil calibrations. Mol. Phylogenet. Evol. 59, 123–138. doi: 10.1016/j.ympev.2011.01.016
Yang, J. B., Li, D. Z., and Li, H. T. (2014). Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol. Ecol. Resour. 14, 1024–1031. doi: 10.1111/1755-0998.12251
Yao, X., Tan, Y. H., Liu, Y. Y., Song, Y., Yang, J. B., and Corlett, R. T. (2016). Chloroplast genome structure in Ilex (Aquifoliaceae). Sci. Rep. 6:28559. doi: 10.1038/srep28559
Keywords: Camptotheca acuminata, chloroplast genome, Cornales, Davidia involucrata, Nyssa sinensis, Nyssaceae, phylogenomics
Citation: Yang Z and Ji Y (2017) Comparative and Phylogenetic Analyses of the Complete Chloroplast Genomes of Three Arcto-Tertiary Relicts: Camptotheca acuminata, Davidia involucrata, and Nyssa sinensis. Front. Plant Sci. 8:1536. doi: 10.3389/fpls.2017.01536
Received: 01 April 2017; Accepted: 22 August 2017;
Published: 11 September 2017.
Edited by:
Federico Luebert, University of Bonn, GermanyReviewed by:
Ming Kang, South China Institute of Botany (CAS), ChinaHengchang Wang, Wuhan Botanical Garden (CAS), China
Copyright © 2017 Yang and Ji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunheng Ji, aml5aEBtYWlsLmtpYi5hYy5jbg==