 Nelson Garcia
Nelson Garcia Joachim Messing
Joachim Messing- Waksman Institute of Microbiology, Rutgers University, Piscataway, NJ, United States
The TEL2, TTI1, and TTI2 proteins are co-chaperones for heat shock protein 90 (HSP90) to regulate the protein folding and maturation of phosphatidylinositol 3-kinase-related kinases (PIKKs). Referred to as the TTT complex, the genes that encode them are highly conserved from man to maize. TTT complex and PIKK genes exist mostly as single copy genes in organisms where they have been characterized. Members of this interacting protein network in maize were identified and synteny analyses were performed to study their evolution. Similar to other species, there is only one copy of each of these genes in maize which was due to a loss of the duplicated copy created by ancient allotetraploidy. Moreover, the retained copies of the TTT complex and the PIKK genes tolerated extensive retrotransposon insertion in their introns that resulted in increased gene lengths and gene body methylation, without apparent effect in normal gene expression and function. The results raise an interesting question on whether the reversion to single copy was due to selection against deleterious unbalanced gene duplications between members of the complex as predicted by the gene balance hypothesis, or due to neutral loss of extra copies. Uneven alteration of dosage either by adding extra copies or modulating gene expression of complex members is being proposed as a means to investigate whether the data supports the gene balance hypothesis or not.
Introduction
Maize has undergone a whole-genome duplication event due to allotetraploidization approximately 4.8 million years ago (Swigonova et al., 2004) and was domesticated only about 10,000 years ago (Doebley, 2004). The whole-genome duplication event created 20 pairs of chromosomes, which were later reduced to 10 after diploidization, mostly by chromosome fusions and deletions of centromeres (Wei et al., 2007; Salse et al., 2008). The maize genome also underwent extensive retrotransposition, gene movement (Lai et al., 2004), chromosome contraction and fractionation, and the loss of homoeologous gene copies (Bruggmann et al., 2006; Schnable et al., 2011).
Several hypotheses explain this fractionation process. The gene balance hypothesis calls for an ideal range of stoichiometric balance between members of protein complexes because a disruption due to imbalance in concentrations of the components can have harmful effects (Birchler and Newton, 1981; Veitia, 2002; Papp et al., 2003). This dosage-dependent function can also apply to the interaction of positive and negative regulatory effectors (Birchler et al., 2005), and involved many genes of different functions (Birchler et al., 2001). Indeed, genes that are thought to be dose-sensitive such as those encoding components of proteasome/protein modification complexes, signal transduction machinery, ribosomes, and transcription factor complexes mendelized in Arabidopsis after diploidization from its tetraploid ancestor (Thomas et al., 2006). The same study used the term “connected genes” to describe loci that seemed to be co-regulated and dependent on each other. In addition, there seems to be preferential removal of genes from one of the homoeologs, the same as in maize (Woodhouse et al., 2010). This process is probably a common occurrence in eukaryotes with whole-genome duplication histories (Birchler et al., 2001; Sankoff et al., 2010). Preferential removal of genes from one homoeolog also explains the uneven expansion and contraction of syntenic blocks in maize (Bruggmann et al., 2006). On the other hand, loss of a duplicated copy could simply be due to neutral loss of an extra copy over evolutionary time.
Although genome-wide studies support the fractionation bias of connected genes, we thought to investigate this at the level of specific examples of interacting genes that are parts of functional units during development. We recently described the first TTT co-chaperone complex in plants (Garcia et al., 2017). The TTT complex is composed of Telomere maintenance 2 (Tel2), Tel2-interacting protein 1 (Tti1), and Tel2-interacting protein 2 (Tti2) and functions as co-chaperones for maturation and stability of phosphatidylinositol 3-kinase-related kinases (PIKKs) (Hurov et al., 2010; Takai et al., 2010). The PIKK family on the other hand is involved in cell signaling related to growth in response to nutrients (TOR), DNA damage response (ATM, ATR, and DNA-PKcs), epigenetic transcriptional regulation (TRRAP), and nonsense-mediated RNA decay (SMG-1) (Abraham, 2004; Lovejoy and Cortez, 2009). Mutations in the TTT complex and PIKK members are lethal in many organisms (Brown and Baltimore, 2000; Benard et al., 2001; Menand et al., 2002; Takai et al., 2007; Stirling et al., 2011; Yamamoto et al., 2012). Deregulated expression has been implicated in many diseases including cancer (Populo et al., 2012; Weber and Ryan, 2015; Rodina et al., 2016), which underscores the essential function of these proteins.
Materials and Methods
The human TEL2 (UniProt Q9Y4R8), TTI1 (UniProt O43156), and TTI2 (UniProt Q6NXR4) protein sequences were used to identify orthologs in maize (version 3 assembly) and other animal and fungal species using BLASTP at default settings. These proteins are well-characterized in yeast and mammals and have unique conserved domains that can be used to identify orthologs. The selected maize sequences were then used to identify other plant TEL2, TTI1, and TTI2 homologs using a BLASTP search in the Phytozome database. Sequences from representative organisms were then selected and aligned using ClustalW. A maximum likelihood phylogenetic tree was then created using the JTT model as implemented in the software package MEGA 6 (Tamura et al., 2007), with 500 bootstrap replications to test for clade confidence. The accession numbers for the sequences used are listed in Table 1. For the PIKKs, Arabidopsis TOR (UniProt Q9FR53), ATM (UniProt Q9M3G7), ATR (UniProt Q9FKS4), and human DNA-PKcs (UniProt P78527), SMG-1 (UniProt Q96Q15), and TRRAP (UniProt Q9Y4A5) were used to find their orthologs in maize, sorghum, and rice using BLASTP at default settings. Maize syntenic homologs and their subgenome assignments were obtained from the sorghum-referenced Pan-Grass Syntenic Gene Set (Schnable et al., 2012, 2016). Transposable element (TE) insertions in introns were identified based on RepeatMasker annotations. Previous whole genome DNA methylation studies in B73 from West et al. (2014) were used to identify the CG, CHG, and CHH methylations of these TEs.
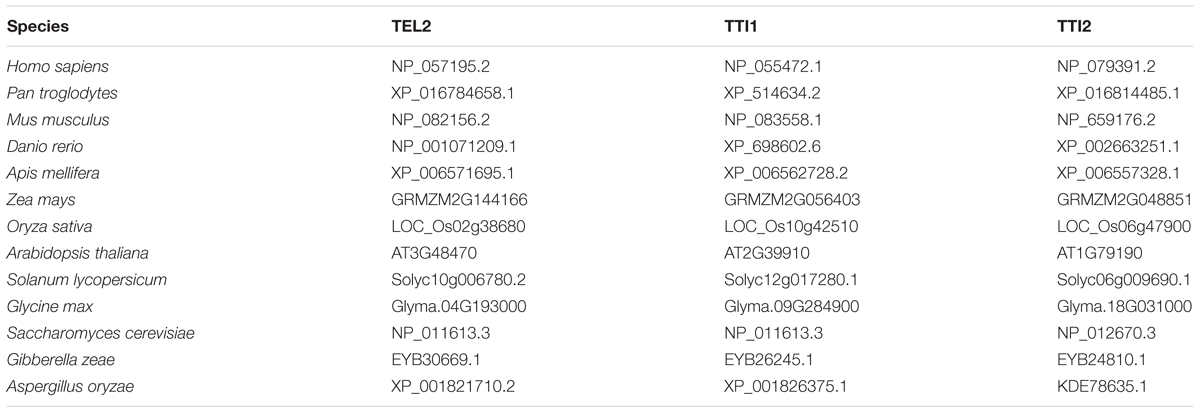
TABLE 1. Accession numbers used in phylogenetic analysis of TTT complex members.
To estimate the insertion time of the LTR-retrotransposons in maize, we used RepeatMasker to retrieve the left and right LTR sequences and aligned them using CLUSTALW. Nucleotide substitution rates between the two LTRs were then calculated using MEGA 6 software using the Distance Estimation analysis option with the Kimura 2-parameter method. Uniform rates were assumed among sites and gaps were deleted from the analysis. Insertion time was then calculated using the reported estimate for LTR nucleotide substitution rate of 1.3 × 10-8 per site per year (Ma and Bennetzen, 2004).
Results and Discussion
Our analysis of the genomes of several animals, fungi, and plants indicate that Tel2, Tti1, and Tti2 are single copy genes as they returned single BLASTP hits. Phylogenetic analyses of their sequences conform to the predicted evolutionary relationships between the species (Figure 1). Likewise, the PIKK genes in maize and rice are single copy (Supplementary Table 1) just like in Arabidopsis (Templeton and Moorhead, 2005) and humans (Bosotti et al., 2000). Synteny analysis in maize using sorghum as a reference confirmed that PIKK and TTT complex genes became single copy because of the removal of the duplicated copy from one of the homoeologs (Figure 2). Dataset from a previous study identified the two maize subgenomes as remnants of the two progenitors of maize, termed maize1 and maize2 (Schnable et al., 2011). In this study, it was hypothesized that fractionation in maize is based on a pattern of overexpression of genes of maize1 over the maize2 subgenome, referred to as genome dominance. It was further suggested that the copy from maize1 was favorably retained because it contributed more to total gene expression relative to its duplicate pair. However, here we can show that in case of the TTT complex Tti1 is located on maize2, whereas the rest of the PIKK and TTT complex genes had copies retained on maize1. Such exceptions would indicate that selection for retention of a gene copy rather depends on the local pattern of transposition events than a particular subgenome. Indeed, the local chromosomal structural organization appeared to be required for the removal of gene copies because of historic homologous recombination events via unequal crossing over as shown for maize and foxtail millet compared to sorghum (Xu et al., 2012).
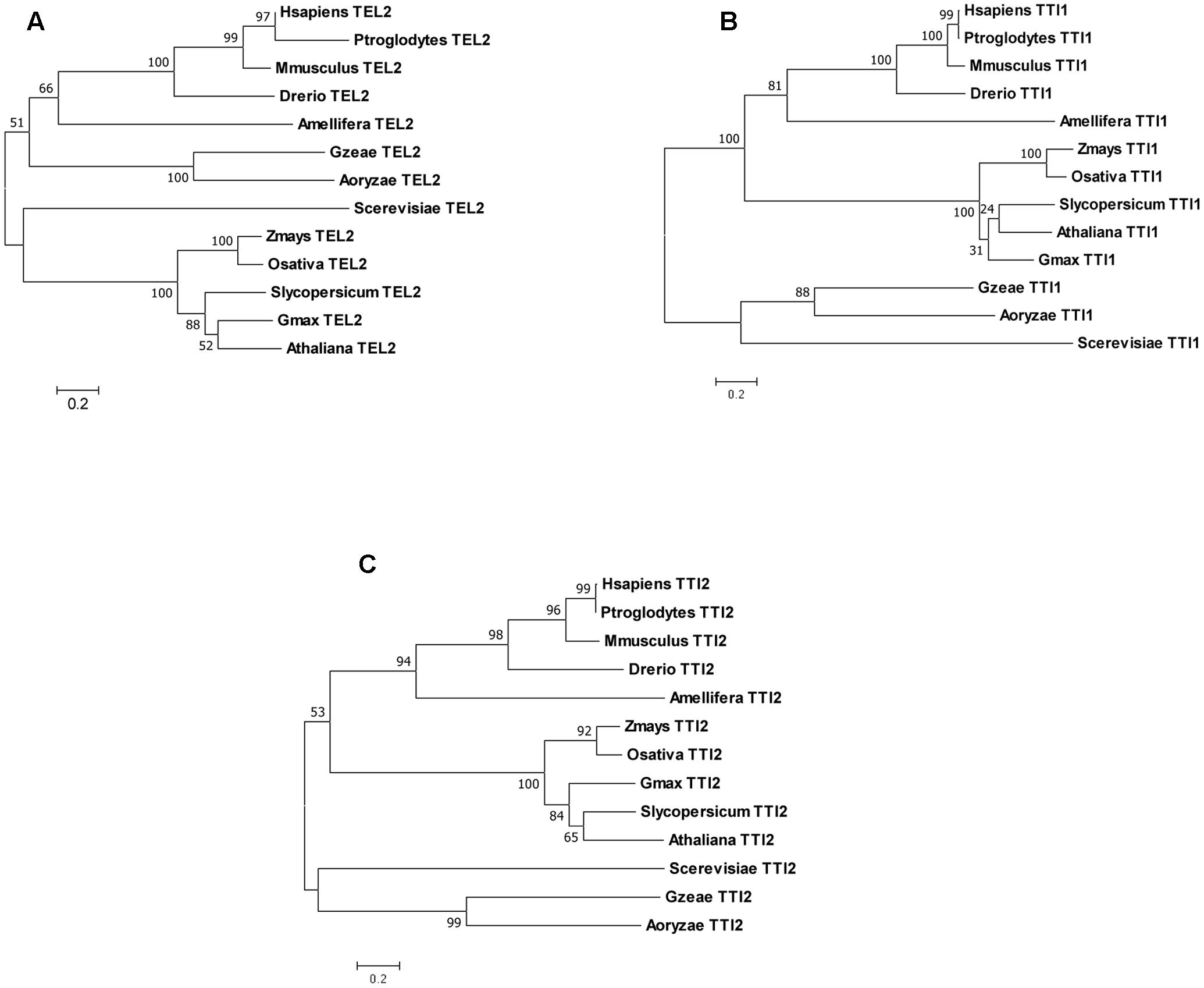
FIGURE 1. Phylogenetic analyses of TEL2 (A), TTI1 (B), and TTI2 (C) from animal, plant, and fungal species. The trees with the highest likelihoods are shown, with nodes indicating bootstrap support values (500 replicates). The bars below indicate distance scale in amino acid substitution per site.
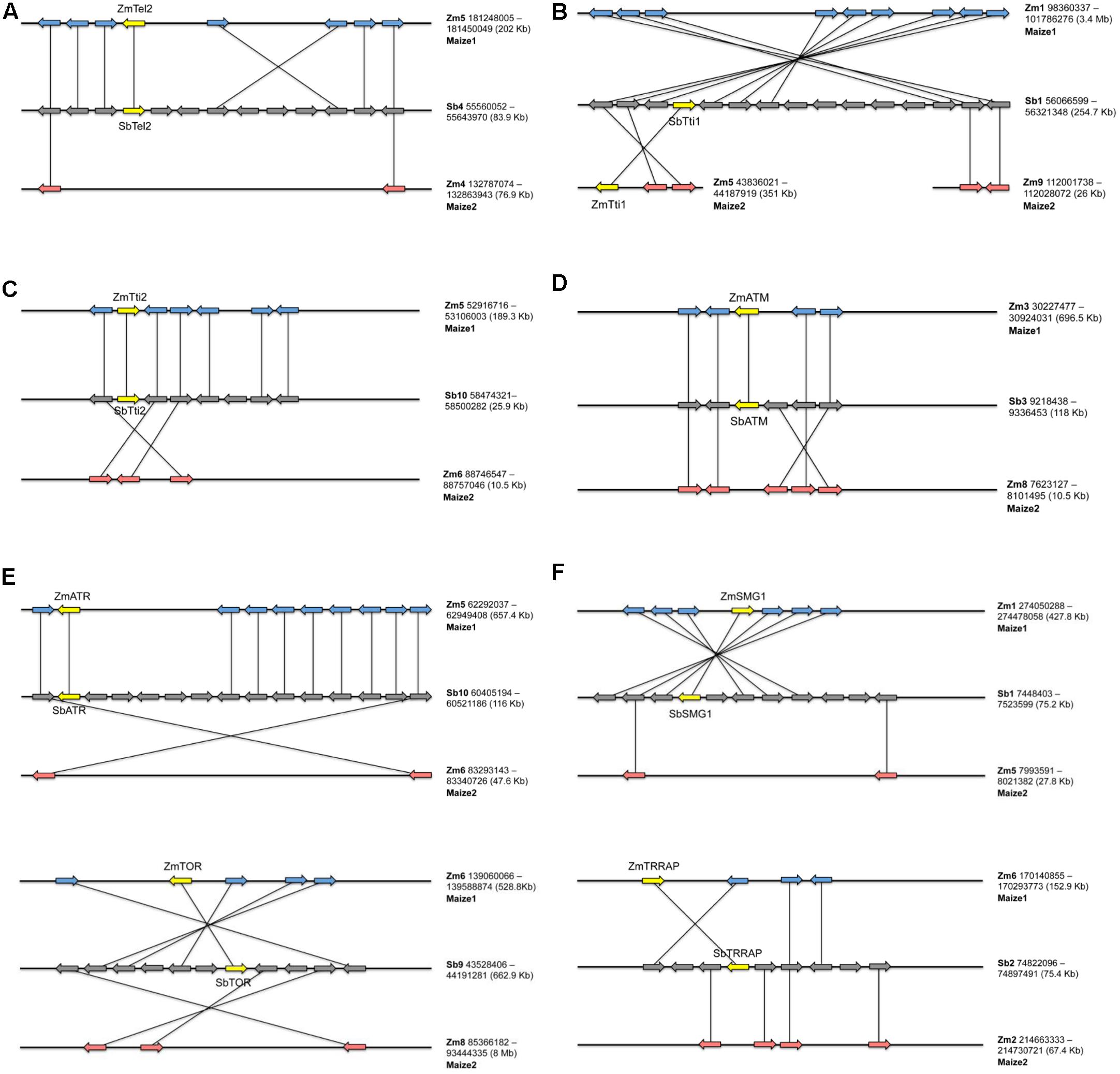
FIGURE 2. Synteny analysis of genes encoding members of TTT co-chaperone complex (A–C) and genes encoding their PIKK client proteins (D–F) in maize. Yellow genes indicate the copy that was retained after polyploidization in maize, and the orthologous gene in sorghum. Sorghum was used as a reference to align the maize syntenic regions (middle genomic segment with gray genes). The top (blue genes) and bottom (pink genes) genomic segments indicate syntenic regions from maize subgenomes 1 and 2 respectively. The coordinates and sizes of the syntenic regions are indicated to the right of the alignments.
Therefore, it is not so much the dominance of one subgenome over the other one, but rather the mendelization of critical functions, which could explain the reversion to single copy of the genes investigated here. The interaction of TTT co-chaperones amongst themselves, as well as their interaction with PIKK proteins as co-chaperones, could be dosage-sensitive. Therefore, the genes coding for these proteins needed to evolve together as a unit (i.e., either all remain duplicated or all lose one duplicate) to attain the best stoichiometric balance needed to maintain fitness. However, it is also important to point out that none of the TTT complex and PIKK mutants displayed haplo-insufficiency, as heterozygotes seemed to be normal. It is therefore possible that expressions of these genes are subject to dosage compensation. If normal phenotypes in heterozygotes are conditional, they would display increased sensitivity compared to wild type, when subjected to certain levels of stress. For example, downregulation of Tti2 expression in yeast is sufficient for normal growth. However, this downregulation strain was more sensitive to PIKK-related stresses compared to wild type (Hoffman et al., 2016). Another example is the Steroidogenic factor-1 (Sf-1) gene in mouse wherein the heterozygote displayed mutant phenotypes only when subjected to stress (Bland et al., 2000). Aside from their role in protein folding, the TTT complex is also needed for the assembly of PIKK complexes (Horejsi et al., 2010; Kaizuka et al., 2010; Takai et al., 2010). Therefore, we extended our investigation to some components of TOR and ATR complexes, which were well-characterized in several organisms. Two proteins called RAPTOR (also known as KOG1) and LST8 (also known as GβL) are integral components of the TOR complex (Loewith et al., 2002; Kim et al., 2003). ATR, on the other hand, closely associates with ATRIP (LCD1 in yeast) (Cortez et al., 2001). Like the TTT complex and PIKKs, RAPTOR, LST8, and ATRIP are conserved in many eukaryotes and are also encoded by single copy genes (Cortez et al., 2001; Loewith et al., 2002). In addition, all three display haplo-insufficient phenotypes in yeast, which is evidence of their dosage-sensitive nature (Pir et al., 2012; Shimada et al., 2013). Homologs of these three genes have been cloned in Arabidopsis and in contrast to humans and yeast, both RAPTOR and LST8 exist as two copies (Deprost et al., 2005; Moreau et al., 2012). However, their investigations revealed that one of the copies was barely expressed, and that mutation in the other copy is enough to cause a mutant phenotype. Moreover, disruption of the barely expressed copy of RAPTOR did not result in a mutant phenotype. Therefore, this is still consistent with the gene balance hypothesis. Analysis of many presence-absence variations (PAVs) in a diverse set of maize and teosinte lines suggests that fractionation might still occur in maize, and that it remains biased (Schnable et al., 2011). Possibly, epigenetic gene silencing is a prelude to the deletion of a gene copy and could reflect an ongoing fractionation in Arabidopsis.
Given the potential critical dosage dependence of members of the TTT complex, their coding regions are sensitive to random transposon insertions that have taken place in maize, which also leads to epigenetic silencing. Based on a high confidence gene set, it was previously estimated that 11.6% of genes in maize contain TEs in their introns (Haberer et al., 2005). This estimate is close to the >10% estimate made by West et al. (2014) by surveying gene bodies for CHG DNA methylation and accompanying H3K9Me2 epigenetic modifications that mark the TEs. We found that six of the eight genes in our study contain TEs in introns (Table 2 and Supplementary Figure 1). For example, there is a 6 Kb Gypsy retrotransposon in intron 3 of ZmTti2, and a 2 Kb hAT element in intron 12 of ZmTti1, which are absent in their putative sorghum orthologs. Estimation of the insertion times of five LTR retrotransposons indicated that three of them transposed less than 4.8 million years ago (Supplementary Table 2). This signifies that some of these TEs are recent transpositions that occurred after the split of maize and sorghum (Swigonova et al., 2004). The maize PIKKs ZmATM, ZmATR, ZmTOR, and ZmSMG1 also have many TE insertions in their introns that expanded the gene size relative to their sorghum counterparts (Table 2 and Supplementary Figure 1). For example, ZmATM genic region expanded to about 131 Kb relative to 71.5 Kb in its sorghum ortholog. However, this is not to imply that the TE insertions were selected for in these genes – they are most likely results of random transposon insertions. Because TEs are silenced by DNA methylation and associated histone modifications to prevent their expression (Slotkin and Martienssen, 2007), we investigated the methylation states of these TEs using datasets from a previous DNA methylation studies in maize (West et al., 2014). As expected, the TEs in the introns are heavily methylated in the CG and CHG contexts, as shown for ZmTti2 and ZmATM in Figure 3. Nevertheless, these genes are still well-expressed in many tissues and at many times during development as shown by the gene expression database available in MaizeGDB. It has even been shown that insertion of a TE into an exon can permit proper gene expression as long as splicing is not affected (Wessler et al., 1987). To ensure that proper splicing occurred, we experimentally validated the expression of ZmTti1 and ZmTti2, enabling us to clone their full-length coding sequences using RT-PCR (Garcia et al., 2017). All the data are indications of strong expression of these genes despite TE insertions in introns. In a genome-wide study, West et al. (2014) observed that about 10% of maize genes have CHG methylation in gene bodies that can be partially attributed retrotransposon insertions in the introns. The CG and CHG methylations are also mostly confined to introns where the TEs are located, indicating a mechanism to stop the spread of methylation to exons. This very accurate marking of boundaries between TE and genic sequences likely enable the genes to be expressed at the right dosage. Methylation in TEs promotes the formation of heterochromatin to suppress transposition (Cedar and Bergman, 2009; Zemach et al., 2013) and methylation in promoters is correlated with transcriptional repression (Weber and Schubeler, 2007). In contrast, the role of gene body methylation in the regulation of gene expression is still not resolved, although some studies found that gene body methylation is associated with transcriptional activation (Zhang et al., 2006; Wang et al., 2015), others dispute this (Bewick et al., 2016). Nevertheless, future studies in maize that probe the role of gene body methylation in gene expression, whether because of TE insertions in introns or not, will help determine whether it has a role in changing the expression level of a “connected” gene, and hence its potential impact on dosage-sensitive functions.
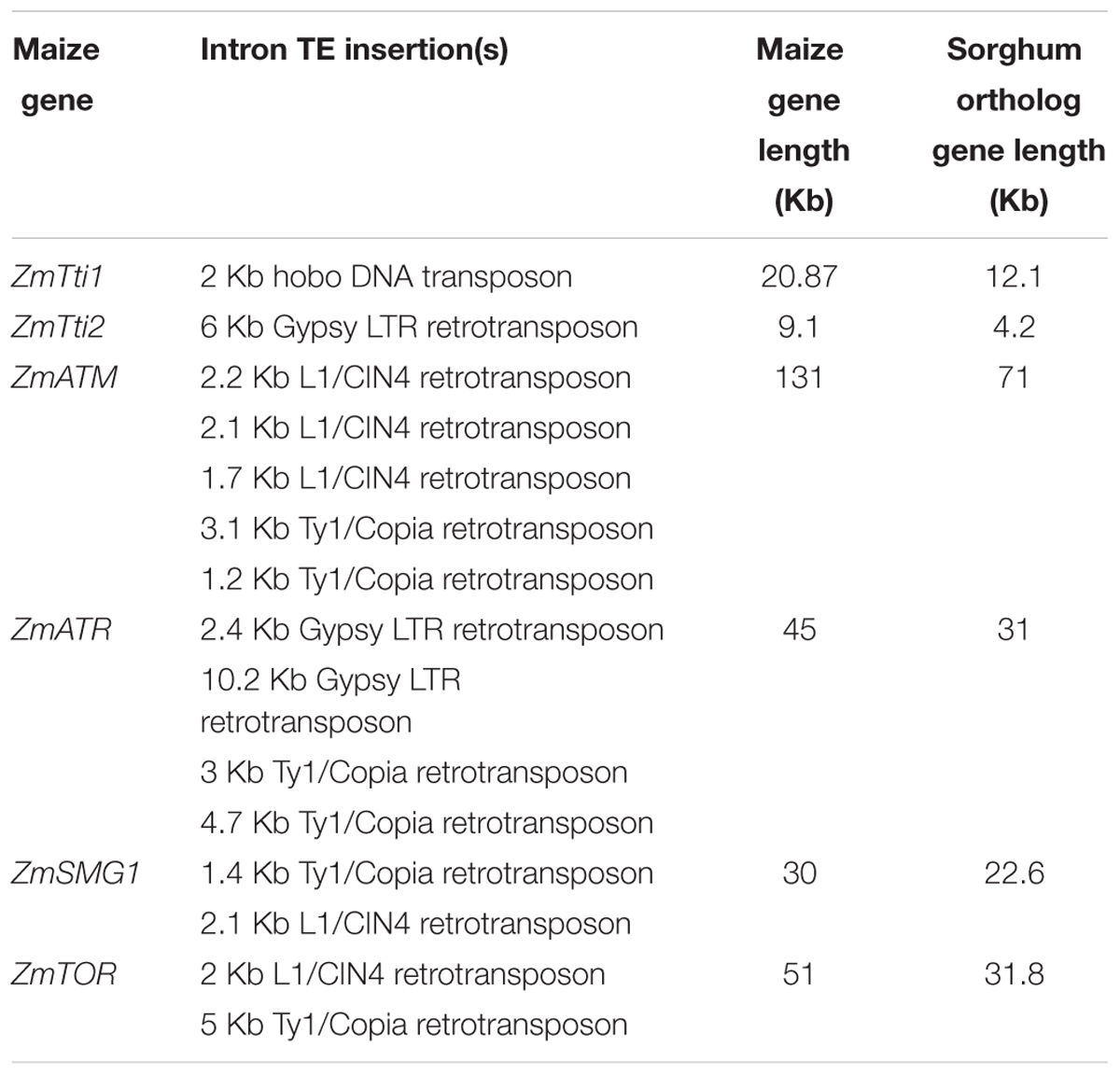
TABLE 2. Examples of TE insertions in introns of maize TTT complex and PIKK genes.
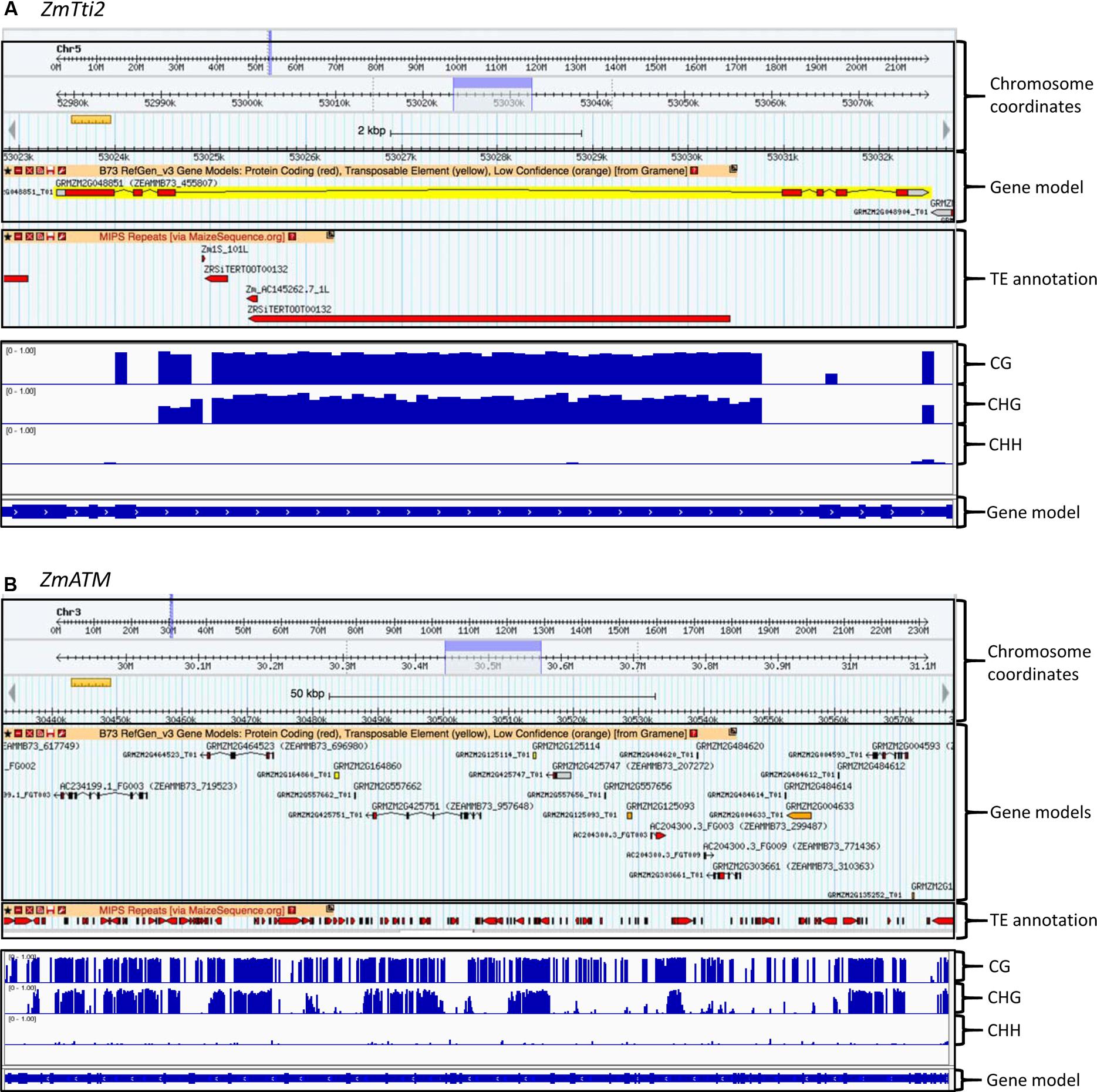
FIGURE 3. Examples of intronic TE insertions and gene body methylation found in some TTT complex and PIKK genes. The top panels show the TE insertion, while the bottom panels show DNA methylation in CG, CHG, and CHH contexts. (A) TE insertion and methylation in the third intron of ZmTti2. (B) Extensive TE insertions and methylation in many introns of ZmATM. Note that the ZmATM gene was misannotated and split into several gene models.
The reversion to single gene copy after genome duplication could be due to neutral loss of extra copies, or selection based on unbalanced gene duplications. It is likely that both mechanisms were involved in the diploidization of maize from its allotetraploid ancestor, which could be based on whether the genes belong to a dosage sensitive protein-protein network or not. Although our data on the TTT complex and PIKK genes is consistent with the gene balance hypothesis, the alternative hypothesis of neutral loss of extra copies cannot be discounted. To test this, uneven alterations in the gene copy number (or uneven levels of gene expression) should be done to see if it results in protein complex destabilization and thus potential negative fitness effects. This approach can be extended to other genes in maize to see if unbalanced duplications in members of protein complexes are deleterious in general.
Author Contributions
NG designed the experiments, performed the analysis, and wrote the manuscript. JM designed the experiments and wrote the manuscript.
Funding
This work was supported with the Selman A. Waksman Chair in Molecular Genetics to JM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2017.01723/full#supplementary-material
References
Abraham, R. T. (2004). PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair 3, 883–887. doi: 10.1016/j.dnarep.2004.04.002
Benard, C., McCright, B., Zhang, Y., Felkai, S., Lakowski, B., and Hekimi, S. (2001). The C. elegans maternal-effect gene clk-2 is essential for embryonic development, encodes a protein homologous to yeast Tel2p and affects telomere length. Development 128, 4045–4055.
Bewick, A. J., Ji, L., Niederhuth, C. E., Willing, E. M., Hofmeister, B. T., Shi, X., et al. (2016). On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. U.S.A. 113, 9111–9116. doi: 10.1073/pnas.1604666113
Birchler, J. A., Bhadra, U., Bhadra, M. P., and Auger, D. L. (2001). Dosage-dependent gene regulation in multicellular eukaryotes: implications for dosage compensation, aneuploid syndromes, and quantitative traits. Dev. Biol. 234, 275–288. doi: 10.1006/dbio.2001.0262
Birchler, J. A., and Newton, K. J. (1981). Modulation of protein levels in chromosomal dosage series of maize: the biochemical basis of aneuploid syndromes. Genetics 99, 247–266.
Birchler, J. A., Riddle, N. C., Auger, D. L., and Veitia, R. A. (2005). Dosage balance in gene regulation: biological implications. Trends Genet. 21, 219–226. doi: 10.1016/j.tig.2005.02.010
Bland, M. L., Jamieson, C. A. M., Akana, S. F., Bornstein, S. R., Eisenhofer, G., Dallman, M. F., et al. (2000). Haploinsufficiency of steroidogenic factor-1 in mice disrupts adrenal development leading to an impaired stress response. Proc. Natl. Acad. Sci. U.S.A. 97, 14488–14493. doi: 10.1073/pnas.97.26.14488
Bosotti, R., Isacchi, A., and Sonnhammer, E. L. (2000). FAT: a novel domain in PIK-related kinases. Trends Biochem. Sci. 25, 225–227. doi: 10.1016/S0968-0004(00)01563-2
Brown, E. J., and Baltimore, D. (2000). ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 14, 397–402.
Bruggmann, R., Bharti, A. K., Gundlach, H., Lai, J., Young, S., Pontaroli, A. C., et al. (2006). Uneven chromosome contraction and expansion in the maize genome. Genome Res. 16, 1241–1251. doi: 10.1101/gr.5338906
Cedar, H., and Bergman, Y. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304. doi: 10.1038/nrg2540
Cortez, D., Guntuku, S., Qin, J., and Elledge, S. J. (2001). ATR and ATRIP - partners in checkpoint signaling. Science 294, 1713–1716. doi: 10.1126/science.1065521
Deprost, D., Truong, H. N., Robaglia, C., and Meyer, C. (2005). An Arabidopsis homolog of RAPTOR/KOG1 is essential for early embryo development. Biochem. Biophys. Res. Commun. 326, 844–850. doi: 10.1016/j.bbrc.2004.11.117
Doebley, J. (2004). The genetics of maize evolution. Annu. Rev. Genet. 38, 37–59. doi: 10.1146/annurev.genet.38.072902.092425
Garcia, N., Li, Y., Dooner, H. K., and Messing, J. (2017). A maize defective kernel mutant generated by insertion of a Ds element in a gene encoding a highly conserved TTI2 co-chaperone. Proc. Natl. Acad. Sci. U.S.A. 114, 5165–5170. doi: 10.1073/pnas.1703498114
Haberer, G., Young, S., Bharti, A. K., Gundlach, H., Raymond, C., Fuks, G., et al. (2005). Structure and architecture of the maize genome. Plant Physiol. 139, 1612–1624. doi: 10.1104/pp.105.068718
Hoffman, K. S., Duennwald, M. L., Karagiannis, J., Genereaux, J., McCarton, A. S., and Brandl, C. J. (2016). Saccharomyces cerevisiae Tti2 regulates PIKK proteins and stress response. G3 6, 1649–1659. doi: 10.1534/g3.116.029520
Horejsi, Z., Takai, H., Adelman, C. A., Collis, S. J., Flynn, H., Maslen, S., et al. (2010). CK2 phospho-dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol. Cell 39, 839–850. doi: 10.1016/j.molcel.2010.08.037
Hurov, K. E., Cotta-Ramusino, C., and Elledge, S. J. (2010). A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev. 24, 1939–1950. doi: 10.1101/gad.1934210
Kaizuka, T., Hara, T., Oshiro, N., Kikkawa, U., Yonezawa, K., Takehana, K., et al. (2010). Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J. Biol. Chem. 285, 20109–20116. doi: 10.1074/jbc.M110.121699
Kim, D.-H., Sarbassov, D. D., Ali, S. J., Latek, R. R., Guntur, K. V. P., Erdjument-Bromage, H., et al. (2003). G βL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 11, 895–904. doi: 10.1016/S1097-2765(03)00114-X
Lai, J., Ma, J., Swigonova, Z., Ramakrishna, W., Linton, E., Llaca, V., et al. (2004). Gene loss and movement in the maize genome. Genome Res. 14, 1924–1931. doi: 10.1101/gr.2701104
Loewith, R., Jacinto, E., Wullschleger, S., Lorberg, A., Crespo, J. L., Bonenfant, D., et al. (2002). Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 10, 457–468. doi: 10.1016/S1097-2765(02)00636-6
Lovejoy, C. A., and Cortez, D. (2009). Common mechanisms of PIKK regulation. DNA Repair 8, 1004–1008. doi: 10.1016/j.dnarep.2009.04.006
Ma, J., and Bennetzen, J. L. (2004). Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. U.S.A. 101, 12404–12410. doi: 10.1073/pnas.0403715101
Menand, B., Desnos, T., Nussaume, L., Berger, F., Bouchez, D., Meyer, C., et al. (2002). Expression and disruption of the Arabidopsis TOR (target of rapamycin) gene. Proc. Natl. Acad. Sci. U.S.A. 99, 6422–6427. doi: 10.1073/pnas.092141899
Moreau, M., Azzopardi, M., Clement, G., Dobrenel, T., Marchive, C., Renne, C., et al. (2012). Mutations in the Arabidopsis homolog of LST8/GbetaL, a partner of the target of Rapamycin kinase, impair plant growth, flowering, and metabolic adaptation to long days. Plant Cell 24, 463–481. doi: 10.1105/tpc.111.091306
Papp, B., Pal, C., and Hurst, L. D. (2003). Dosage sensitivity and the evolution of gene families in yeast. Nature 424, 194–197. doi: 10.1038/nature01771
Pir, P., Gutteridge, A., Wu, J., Rash, B., Kell, D. B., Zhang, N., et al. (2012). The genetic control of growth rate: a systems biology study in yeast. BMC Syst. Biol. 6:4. doi: 10.1186/1752-0509-6-4
Populo, H., Lopes, J. M., and Soares, P. (2012). The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 13, 1886–1918. doi: 10.3390/ijms13021886
Rodina, A., Wang, T., Yan, P., Gomes, E. D., Dunphy, M. P., Pillarsetty, N., et al. (2016). The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538, 397–401. doi: 10.1038/nature19807
Salse, J., Bolot, S., Throude, M., Jouffe, V., Piegu, B., Quraishi, U. M., et al. (2008). Identification and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell 20, 11–24. doi: 10.1105/tpc.107.056309
Sankoff, D., Zheng, C., and Zhu, Q. (2010). The collapse of gene complement following whole genome duplication. BMC Genomics 11:313. doi: 10.1186/1471-2164-11-313
Schnable, J., Zang, Y., and Ngu, D. W. C. (2016). Pan-grass syntenic gene set (sorghum referenced). doi: 10.6084/m9.figshare.3113488.v1
Schnable, J. C., Freeling, M., and Lyons, E. (2012). Genome-wide analysis of syntenic gene deletion in the grasses. Genome Biol. Evol. 4, 265–277. doi: 10.1093/gbe/evs009
Schnable, J. C., Springer, N. M., and Freeling, M. (2011). Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. U.S.A. 108, 4069–4074. doi: 10.1073/pnas.1101368108
Shimada, K., Filipuzzi, I., Stahl, M., Helliwell, S. B., Studer, C., Hoepfner, D., et al. (2013). TORC2 signaling pathway guarantees genome stability in the face of DNA strand breaks. Mol. Cell 51, 829–839. doi: 10.1016/j.molcel.2013.08.019
Slotkin, R. K., and Martienssen, R. (2007). Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–285. doi: 10.1038/nrg2072
Stirling, P. C., Bloom, M. S., Solanki-Patil, T., Smith, S., Sipahimalani, P., Li, Z., et al. (2011). The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLOS Genet. 7:e1002057. doi: 10.1371/journal.pgen.1002057
Swigonova, Z., Lai, J., Ma, J., Ramakrishna, W., Llaca, V., Bennetzen, J. L., et al. (2004). Close split of sorghum and maize genome progenitors. Genome Res. 14, 1916–1923. doi: 10.1101/gr.2332504
Takai, H., Wang, R. C., Takai, K. K., Yang, H., and de Lange, T. (2007). Tel2 regulates the stability of PI3K-related protein kinases. Cell 131, 1248–1259. doi: 10.1016/j.cell.2007.10.052
Takai, H., Xie, Y., de Lange, T., and Pavletich, N. P. (2010). Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev. 24, 2019–2030. doi: 10.1101/gad.1956410
Tamura, K., Dudley, J., Nei, M., and Kumar, S. (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. doi: 10.1093/molbev/msm092
Templeton, G. W., and Moorhead, G. B. G. (2005). The phosphoinositide-3-OH-kinase-related kinases of Arabidopsis thaliana. EMBO Rep. 6, 723–728. doi: 10.1038/sj.embor.7400479
Thomas, B. C., Pedersen, B., and Freeling, M. (2006). Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res. 16, 934–946. doi: 10.1101/gr.4708406
Veitia, R. A. (2002). Exploring the etiology of haploinsufficiency. Bioessays 24, 175–184. doi: 10.1002/bies.10023
Wang, H., Beyene, G., Zhai, J., Feng, S., Fahlgren, N., Taylor, N. J., et al. (2015). CG gene body DNA methylation changes and evolution of duplicated genes in cassava. Proc. Natl. Acad. Sci. U.S.A. 112, 13729–13734. doi: 10.1073/pnas.1519067112
Weber, A. M., and Ryan, A. J. (2015). ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 149, 124–138. doi: 10.1016/j.pharmthera.2014.12.001
Weber, M., and Schubeler, D. (2007). Genomic patterns of DNA methylation: targets and function of an epigenetic mark. Curr. Opin. Cell Biol. 19, 273–280. doi: 10.1016/j.ceb.2007.04.011
Wei, F., Coe, E., Nelson, W., Bharti, A. K., Engler, F., Butler, E., et al. (2007). Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLOS Genet. 3:e123. doi: 10.1371/journal.pgen.0030123
Wessler, S. R., Baran, G., and Varagona, M. (1987). The maize transposable element Ds is spliced from RNA. Science 237, 916–918. doi: 10.1126/science.3039661
West, P. T., Li, Q., Ji, L., Eichten, S. R., Song, J., Vaughn, M. W., et al. (2014). Genomic distribution of H3K9me2 and DNA methylation in a maize genome. PLOS ONE 9:e105267. doi: 10.1371/journal.pone.0105267
Woodhouse, M. R., Schnable, J. C., Pedersen, B. S., Lyons, E., Lisch, D., Subramaniam, S., et al. (2010). Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLOS Biol. 8:e1000409. doi: 10.1371/journal.pbio.1000409
Xu, J. H., Bennetzen, J. L., and Messing, J. (2012). Dynamic gene copy number variation in collinear regions of grass genomes. Mol. Biol. Evol. 126, 861–871. doi: 10.1093/molbev/msr261
Yamamoto, K., Wang, Y., Jiang, W., Liu, X., Dubois, R. L., Lin, C. S., et al. (2012). Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. J. Cell Biol. 198, 305–313. doi: 10.1083/jcb.201204098
Zemach, A., Kim, M. Y., Hsieh, P. H., Coleman-Derr, D., Eshed-Williams, L., Thao, K., et al. (2013). The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153, 193–205. doi: 10.1016/j.cell.2013.02.033
Keywords: gene balance hypothesis, TTT complex, PIKK, genome fractionation, gene body methylation
Citation: Garcia N and Messing J (2017) TTT and PIKK Complex Genes Reverted to Single Copy Following Polyploidization and Retain Function Despite Massive Retrotransposition in Maize. Front. Plant Sci. 8:1723. doi: 10.3389/fpls.2017.01723
Received: 24 May 2017; Accepted: 20 September 2017;
Published: 07 November 2017.
Edited by:
Badri Padhukasahasram, Illumina, United StatesReviewed by:
James A. Birchler, University of Missouri, United StatesRichard John Edwards, University of New South Wales, Australia
Copyright © 2017 Garcia and Messing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joachim Messing, bWVzc2luZ0B3YWtzbWFuLnJ1dGdlcnMuZWR1
†Present address: Nelson Garcia, Department of Horticultural Science, University of Minnesota, Saint Paul, MN, United States