 Na Yang
Na Yang Wenping Zhou
Wenping Zhou Xiaofan Wang
Xiaofan Wang Xiaoyan Cao
Xiaoyan Cao Zhezhi Wang
Zhezhi Wang- Key Laboratory of the Ministry of Education for Medicinal Resources and Natural Pharmaceutical Chemistry, National Engineering Laboratory for Resource Development of Endangered Crude Drugs in Northwest of China, Shaanxi Normal University, Xi’an, China
MYC2 is a core transcription factor in the plant response to jasmonates. It also functions in secondary metabolism and various processes for growth and development. However, the knowledge about its role in Salvia miltiorrhiza is still very limited. We determined that the biosynthesis of salvianolic acid B (Sal B) was strongly induced in 2-month-old transgenic plants that over-expressed SmMYC2. In the roots of transgenic line 12 that over-expressed SmMYC2 (OEM-12), the Sal B concentration was as high as 5.95 ± 0.07 mg g-1, a level that was 1.88-fold higher than that in control plants that had been transformed with an empty vector. Neither tanshinone IIA nor cryptotanshinone was detected by high-performance liquid chromatography in any of the genotypes. Global transcriptomic analysis using RNA sequencing revealed that most enzyme-encoding genes for the phenylpropanoid biosynthesis pathway were up-regulated in the overexpression lines. Furthermore, both the phenylalanine and tyrosine biosynthesis pathways were activated in those transgenics. Our data demonstrate that overexpression of SmMYC2 promotes the production of phenolic acids by simultaneously activating both primary and secondary pathways for metabolism in S. miltiorrhiza.
Introduction
Salvia miltiorrhiza Bunge is a very useful medicinal plant and has been applied to treat various maladies, especially coronary and cerebrovascular diseases (Chun-Yan et al., 2015). As one of the best-known Chinese traditional herbs, its root has been clinically used for more than 2000 years (Guo et al., 2014). According to pharmacological investigations, active ingredients in S. miltiorrhiza are classified as lipophilic diterpene quinone pigments, generally known as tanshinones (Liao et al., 2009) and hydrophilic phenolic acids, which are mainly rosmarinic acid (RA) and its derivative salvianolic acid B (Sal B) (Huang et al., 2008; Gu et al., 2012). Among the phenolic acids, Sal B is considered the most important active content when S. miltiorrhiza is extracted with water in a traditional method. Designated as a marker component of S. miltiorrhiza in the official Chinese Pharmacopeia, Sal B appears to offer protection against hepatic, neural, and cardiovascular diseases, as well as certain cancers (Ho, 2011). Because of the economic value of Sal B, exogenous transcription factors (TFs) have been used with genetic engineering to enhance its production in S. miltiorrhiza (Wang et al., 2013), the pathways for phenolic acid biosynthesis and relevant TFs are now receiving more research attention (Song and Wang, 2011; Zhang et al., 2013, 2014; Wang et al., 2015; Wang Z.J. et al., 2016; Xu et al., 2016).
Jasmonates (JAs) are integral to various defense responses that lead to the accumulation of secondary metabolites (Browse, 2009). MYC2 is a basic helix-loop-helix (bHLH) TF in the JA signaling pathway. Under stress-free growth conditions, jasmonate ZIM-domain (JAZ) proteins inhibit the function of TFs which are response to JA (Pauwels et al., 2010; Shyu et al., 2012). The JAZ proteins combine with TOPLESS (TPL) and TPL-related proteins to form a repressor complex, through direct or indirect interaction with the adaptor protein Novel Interactor of JAZ (NINJA) (Iván et al., 2013). In response to biotic or abiotic stresses, jasmonoyl-L-isoleucine (JA-Ile) is rapidly synthesized in plant tissues (Wasternack and Kombrink, 2010). The binding of JA-Ile to Coronatine Insensitive 1 (COI1)-JAZ co-receptor complexes results in the removal of JAZ repressors by the 26S proteasome (Sheard et al., 2010; Zhang et al., 2016). Destruction of JAZ repressors liberates MYC2 from suppression. The latter orchestrates the expression of many JA-responsive genes, including several TFs that have an important role in the JA signaling pathway (Kemal and Manners, 2013).
The roles of JA in controlling genes participated in the synthesis of secondary metabolites are well-established. MYC2 positively or negatively regulates secondary metabolism during JA signaling in a species-specific manner. For example, it can positively regulate the biosynthesis of flavonoid (e.g., anthocyanin) but inhibit JA-responsive biosynthesis of Trp-derived indole-glucosinolates in Arabidopsis plants with JA treatment (Dombrecht et al., 2007; Kemal and Manners, 2013). The MYC2 orthologs from tobacco species function as positive regulators of JA activation during nicotine biosynthesis in Nicotiana benthamiana (Todd et al., 2010; Kathleen et al., 2011). CrMYC2 regulates the expression of ORCA genes which are response to JA and the genes have an effect on alkaloid biosynthesis in Catharanthus roseus (Zhang et al., 2011). Results from ectopic RNA interference-mediated knockdown experiments with hairy root cultures of S. miltiorrhiza have suggested that SmMYC2a and SmMYC2b two MYC2 orthologs from that species promote the biosynthesis of tanshinones and Sal B (Zhou Y. et al., 2016). We previously isolated SmMYC2 (GenBank accession number KJ945636) from S. miltiorrhiza. This gene encodes a putative MYC2, and its expression can be induced by methyl jasmonate (MeJA), light, or wounding (Zhou W.P. et al., 2016). However, it has not been clarified the effect of SmMYC2 overexpression on S. miltiorrhiza.
Here, we investigated whether such overexpression might increase plant levels of tanshinones and Sal B. Our goal was to gain a better understanding of the role of MYC2 in S. miltiorrhiza, a valuable medicinal plant. The development of new strategies for genetic manipulation of these plants might be used to enhance the production of bioactive compounds. We conducted genome-wide transcriptional profiling of SmMYC2-overexpression transgenic S. miltiorrhiza and empty vector-transformed control plants, and also performed functional analyses. Roles were identified for MYC2 in the regulation of phenolic acids and tanshinones in that species. Moreover, overexpression of SmMYC2 greatly improved the production of hydrophilic phenolic acids by activating not only the phenylpropanoid biosynthesis pathway but also the pathways for phenylalanine and tyrosine biosynthesis.
Materials and Methods
Experimental Materials
Seeds of S. miltiorrhiza were surface-sterilized as we have described previously (Yan and Wang, 2007). Sterile plants were cultured on an MS basal medium under the conditions presented earlier (Hua et al., 2011). All chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, United States). Solvents were high-performance liquid chromatography (HPLC) grade. The standards (RA, Sal B, tanshinone IIA, and cryptotanshinone) used for HPLC were bought from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). We prepared the standards as stock solutions with methanol and put them at -18°C in a dark environment. The primers are listed in Supplementary Table 1.
Vector Construction and Transformation
To construct SmMYC2-overexpression vectors, we used the pMD19T–SmMYC2 construct already developed in our laboratory (Zhou W.P. et al., 2016). SmMYC2 was amplified from pMD19T–SmMYC2 with primers 207-SmMYC2-F/207-SmMYC2-R, which contain attB1/attB2 sites. Polymerase chain reaction (PCR) products were cloned into entry vector pDONR207, using the BP recombination reaction, and then transferred into destination vector pEarleyGate201 (Earley et al., 2006) through an LR reaction, according to the protocol from the Gateway technology manufacturer (Invitrogen, United States). The resulting pDONR207–SmMYC2 and pEarleyGate201–SmMYC2 were sequenced by Beijing Ao Ke Wei Ye Technologies Co., Ltd. (China).
The pEarleyGate201–SmMYC2 construct was mobilized into Agrobacterium tumefaciens GV3101. Resistant colonies were verified by PCR-amplification and used for transformation experiments (Zhang et al., 2010). Agrobacterium-mediated gene transfer was performed according to the protocol established in our laboratory (Yan and Wang, 2007). The pEarleyGate 201 vector was also transformed by the same method used for control plants (CK) that were transformed with an empty vector.
Transformant Selection and Molecular Characterization
After transformation, explants co-cultured with A. tumefaciens were transferred to an MS selection medium containing 1 mg L-1 naphthalene acetic acid, 10 mg L-1 6-benzyl-aminopurine, 10 mg L-1 glufosinate-ammonium, and 200 mg L-1 cefotaxime. They were transferred to fresh selection media at 10-day intervals. As described previously, developing shoots were excised and placed on a 1/2-MS selection medium supplemented with 10 mg L-1 glufosinate-ammonium and 200 mg L-1 cefotaxime for root induction (Wang et al., 2013). A well-developed root system usually formed within 2 weeks. The rooted plants were cut into intermodal segments and propagated on a 1/2-MS basal medium (Wei et al., 2017).
Molecular characterization of transgenic plants was performed as described before (Wang H.Q. et al., 2016). Transgenic lines of the T1 generation were used for molecular analysis. Genomic DNA was extracted from the leaves of 1-month-old herbicide-resistant plants. Positive pEarleyGate201 transgenic lines and pEarleyGate201–SmMYC2 transgenic lines were detected using 35S-F/R (Supplementary Table 1). All PCR reactions were performed as follows: preheating at 94°C for 3 min; then 32 cycles at 94°C for 30 s, 58°C for 40 s, and 72°C for 1 min; with a final extension at 72°C for 10 min. The target fragments for all transgenic lines were 721 bp long. The pEarleyGate201–SmMYC2 vector served as the positive control, while genomic DNA from wild-type plants was the negative control.
Total RNA was isolated with a Plant RNA Kit (OMEGA, Houston, TX, United States), according to the manufacturer’s protocol, and was converted into cDNA using a primeScript® RT Reagent Kit (TaKaRa, Japan). The reverse-transcription (RT) products were amplified with SmMYC2-RTF/R primers so that we could monitor gene expression via real-time quantitative PCR. A housekeeping gene, SmUbiquitin, served as the control and was amplified with Primers SmUbiquitin-F/R. Quantitative real-time reactions were performed in triplicate under the following conditions: one cycle of pre-denaturation at 95°C for 3 min, then 45 cycles of amplification at 95°C for 10 s and 60°C for 30 s. Relative expression for each gene was calculated by the comparative CT method (Vandesompele et al., 2002).
HPLC Analysis of Phenolic Compounds and Tanshinones
Roots were collected from 2-month-old plants and air-dried at 20 ± 2°C. We used standards of RA, Sal B, tanshinone IIA, and cryptotanshinone in HPLC experiments. Phenolic compounds and tanshinones were extracted as described before (Zhang et al., 2010). Extracts were analyzed with a C18 column (250 × 4.6 mm, 5 μm particle size) on an Agilent 1260 Infinity LC System (United States). The mobile phase comprised 0.4% acetic acid in water (A), acetonitrile (B), and methanol (C). The solvent gradient was as follows: 0–5 min: A 95–90% and B 5–10%; 5–25 min: A 90–67%, B 10–30%, and C 0–3%; 25–40 min: A 67–60%, B 30–5%, and C 3–5%; 40–60 min: A 60–35%, B 35–55%, and C 5–10%; 60–70 min: A 35–25%, B 55–60%, and C 10–15%; 70–75 min: A 25–10%, B 60–70%, and C 15–20%; and 75–80 min: A 10–0%, B 70–80%, and C 20–20%. The flow rate was adjusted to 1.0 mL min-1 and the detection wavelength was 280 nm. All separations were performed at 30°C.
Determination of Total Phenolic and Total Flavonoid Concentrations
The amounts of total phenolics and flavonoids were determined from 2-month-old plants, as we have described previously (Zhang and Wang, 2009). Total phenolics were measured using a modified colorimetric Folin–Ciocalteu method and the absorbance was read at 765 nm. Calculations were based on a calibration curve obtained with gallic acid. The calibration equation for gallic acid was y = 0.0107x + 0.0752 (R2 = 0.9997). Total flavonoid content was determined by using a colorimetric method and the absorbance was measured at 510 nm. Calculations were based on a calibration curve obtained with (-)-epicatechin. The calibration equation for epicatechin was y = 1.113x + 0.0517 (R2 = 0.9971).
Calculation of Jasmonate Concentration
The levels of JA were investigated by culturing samples of fresh leaves from transgenic line OEM-12 and CK plants. After the tissues were ground in liquid nitrogen, 9 mL of PBS buffer (pH 7.4) was added. The extracts were then centrifuged (8000 × g for 30 min at 4°C), and the upper layers were retained for our extractions, which were measured in triplicate. Endogenous JAs were measured in both transgenic and control plants using a Plant Jasmonic acid (JA) ELISA Kit (mlbio, China) according to the manufacturer’s protocol. A set of calibration standards was assayed to produce a standard curve of optical density (OD) versus JA concentration at 0, 125, 250, 500, 1000, and 2000 pmol L-1. The amount of JA in each sample was determined by comparing OD values with the standard curve. The intensity of the final reaction color was measured spectrophotometrically at 450 nm to calculate the final JA concentration.
Transcriptome Analysis
Total RNA was isolated from 2-month-old control and transgenic plants (n = 3 each), using TRIzol® Reagent (Invitrogen, United States) according to the manufacturer’s protocol. Samples from OEM-12 and CK plants were sequenced by Biomarker Technologies Co., Ltd. (Beijing, China). Three biological replicates were used for library preparation and RNAseq. Two samples were used for library construction and sequencing in triplicate. The cDNA library construction and Illumina HiSeq4000 sequencing were performed as described previously (Cao et al., 2016). Our raw data have been submitted to National Center for Biotechnology Information (NCBI) with the accession number of SRP113567.
Identification of Differentially Expressed Genes (DEGs)
Total RNA-Seq reads were mapped to the S. miltiorrhiza genome. Expression levels of various genes were calculated by Cufflinks v2.2.1 (Trapnell et al., 2010); genes with value Q < 0.05, a false discovery rate (FDR) < 0.001, and an estimated absolute log2 fold-change (log2 FC) > 1.0 were selected. Blast X alignments were made (E-values < 0.00001) between the DEGs, using information from the protein databases NR, SwissProt, Pfam, and KEGG. For examining pathway enrichment, all DEGs were mapped to terms in the KEGG database to identify significantly over-represented metabolic pathways or signal transduction pathways as described before (Cao et al., 2016). We primarily focused on differentially regulated pathways closely related to the biosynthesis of salvianolic acids and tanshinones.
Validation of RNA-seq Data by qRT-PCR
To validate the RNA-seq data, we selected 12 genes involved in salvianolic acid production, tanshinone biosynthesis, flavonoids biosynthesis, and the JA signaling pathway for our qRT-PCR experiments. SmUbiquitin served as the reference gene. Quantitative reactions were performed on a LightCycler® 96 real-time PCR detection system (Roche, Switzerland), using SYBR Premix Ex TaqTM (TaKaRa). Relative expression for each gene was calculated by the comparative CT method (Vandesompele et al., 2002). All assays for each gene were performed simultaneously, and in triplicate, under identical conditions.
Statistical Analysis
The statistical evaluation was performed using SPSS version 17.0 software. All data were presented as the means ± standard error (SE) of at least three replicates. Analysis of variance (ANOVA) was followed by Tukey’s pairwise comparison tests. Mean values were considered significantly different at P < 0.05.
Results
Generation of Transgenic Salvia miltiorrhiza Plants
Using Agrobacterium-mediated transformation protocol established in our laboratory (Yan and Wang, 2007), we generated transgenic S. miltiorrhiza plants that over-expressed SmMYC2. After selective culturing on an herbicide medium, those plants were confirmed through PCR amplification to contain an expected 721-bp fragment of the CaMV 35S promoter (Figure 1A). Real-time quantitative PCR demonstrated that SmMYC2 was over-expressed in lines OEM-3, OEM-8, OEM-12, and OEM-14 (Figure 1B). Because expression was significantly higher in OEM-8 and OEM-12 than in the other lines and the CK (plants transformed with the empty vector), we chose them for further analysis.
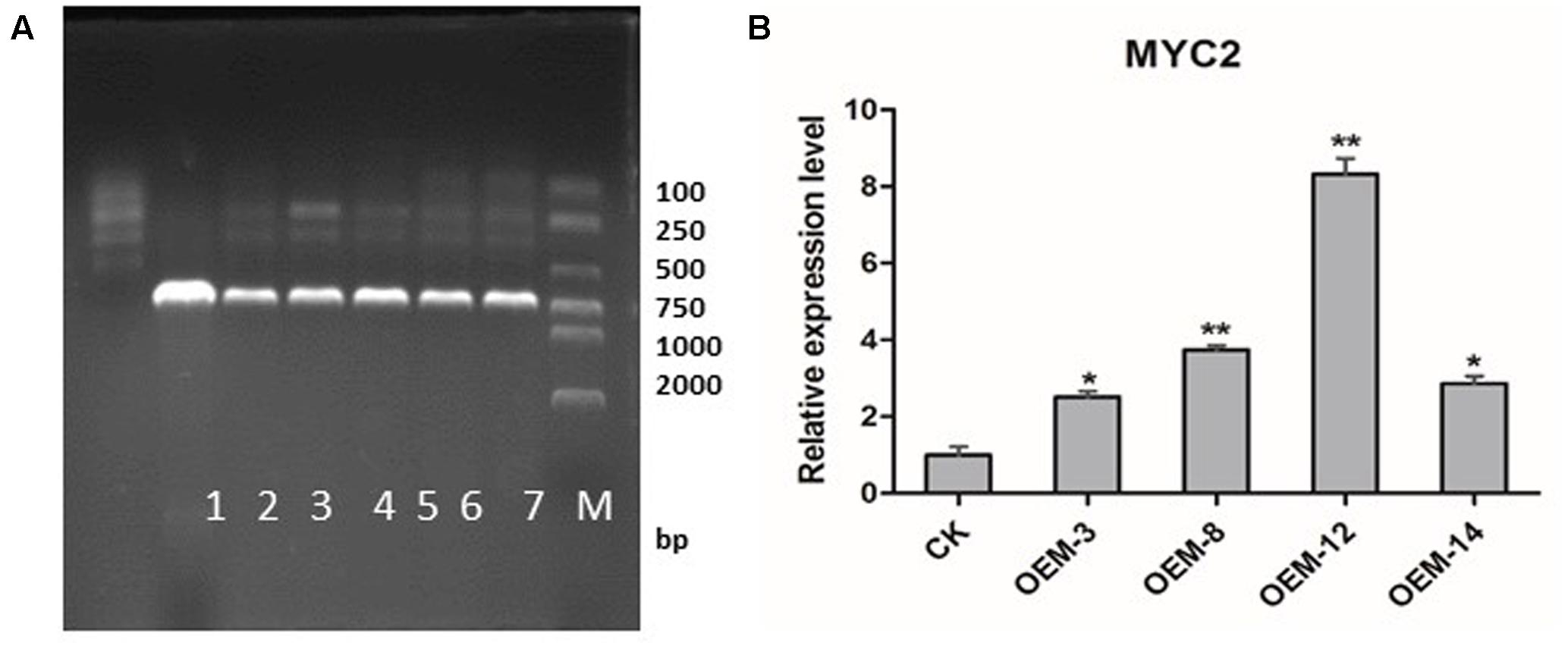
FIGURE 1. Expression of SmMYC2 in control (CK) and overexpression transgenic lines. (A) PCR detection. M, DL2000 DNA marker; band sizes (bp) are shown on right side. Lanes 1, negative control; 2, positive control; 3–7, different transgenic lines. (B) Real-time quantitative PCR analysis; all data are means of three replicates, with error bars indicating SD; ∗ and ∗∗, values are significantly different from CK at P < 0.05 and P < 0.01, respectively.
Overexpression of SmMYC2 Enhances Production of Salvianolic and Rosmarinic Acids in Transgenic S. miltiorrhiza
To characterize how production of phenolic acids and tanshinones is modified in plants over-expressing SmMYC2, we extracted them and subjected them to HPLC separation. Overexpression tended to improve their biosynthesis. In the roots of 2-month-old plants, RA and its dimer Sal B were the major components, showing respective retention times of 24.02 ± 0.02 and 25.60 ± 0.02 min (Figure 2A). Compared with levels in CK samples, the concentrations of both RA and Sal B were increased significantly in the transgenics (P < 0.01) (Figure 2B), particularly in OEM-12, which had mean levels of 6.36 ± 0.21 mg g-1 RA and 5.95 ± 0.07 mg g-1 Sal B. RA and Sal B concentrations in CK roots were 2.59 ± 0.04 and 3.17 ± 0.01 mg g-1, respectively. Compared with CK, the OEM-12 showed a 2.46-fold increase in RA and a 1.88-fold increase in Sal B. In OEM-8, concentrations of RA and Sal B were 6.21 ± 0.03 and 4.80 ± 0.01 mg g-1, respectively, which was approximately 2.40- and 1.51-fold higher than those of the CK.
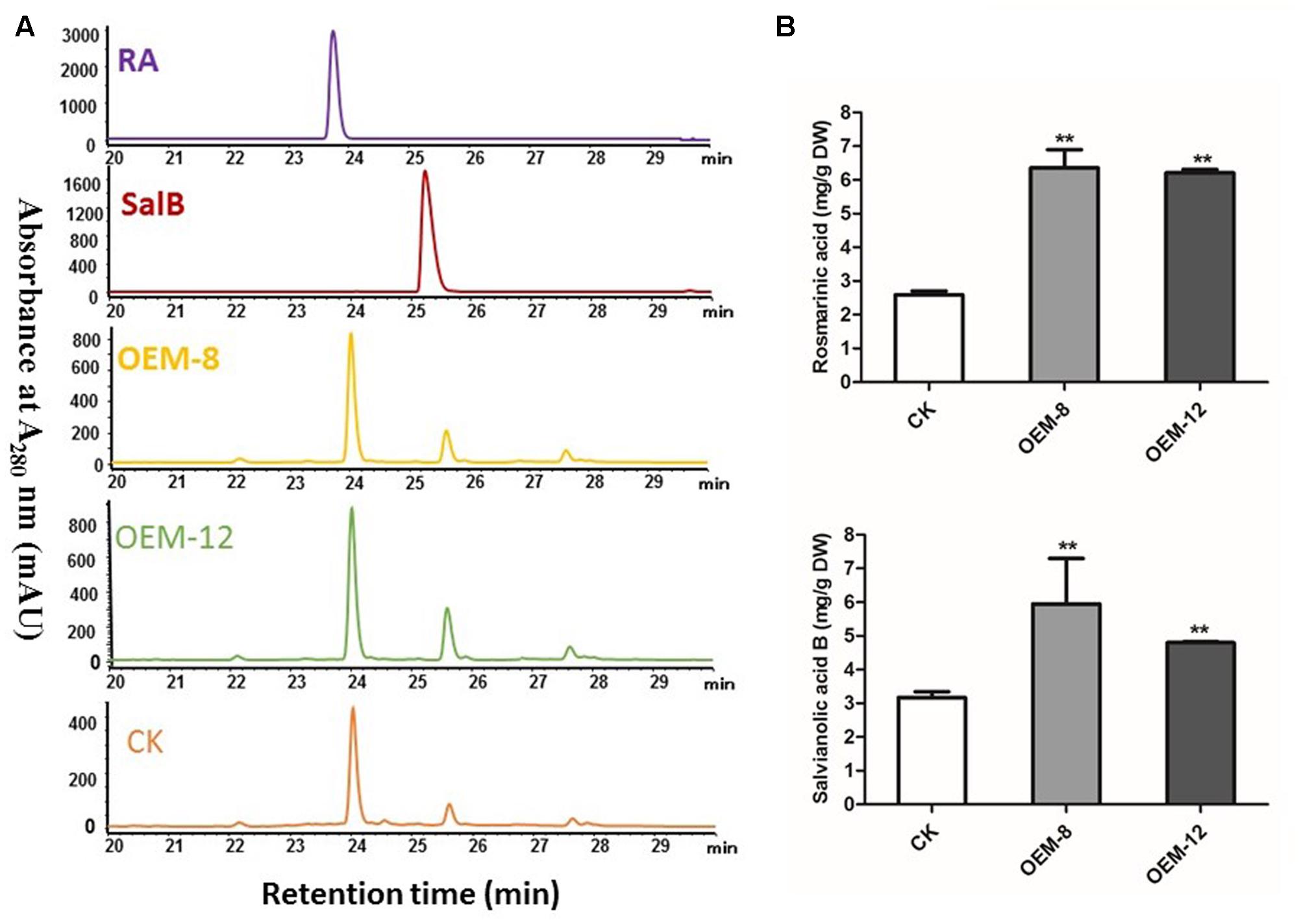
FIGURE 2. Overexpression of SmMYC2 enhances production of rosmarinic acid (RA) and salvianolic acid B (Sal B) in S. miltiorrhiza. (A) Representative HPLCs of 2-month-old plants and standards at the wavelength of 280 nm: RA, standard of RA; Sal B, standard of Sal B; CK, control transformed with empty vector; OEM-8 and OEM-12, positive transgenic lines. (B) Concentrations of RA and Sal B in root extracts from CK and transgenic plants. All data are means of three replicates, with error bars indicating SD. ∗∗Values are significantly different from CK at P < 0.01.
The two tanshinones investigated here are key active ingredients in danshen. Retention times were 75.2 ± 0.2 min for tanshinone IIA and 68.1 ± 0.3 min for cryptotanshinone. However, neither was detected by HPLC in the 2-month-old OEM lines and control plants (Supplementary Figure 1).
Transgenic Plants Show Higher Levels of Total Phenolics and Total Flavonoids
Our results presented above demonstrated that overexpression of SmMYC2 modified the accumulation of two non-flavonoid phenolic acids, RA and Sal B. To evaluate whether this upregulation induced the phenylpropanoid pathway and provided substrates for the biosynthesis of other types of end products, we performed global assays for phenolics and flavonoids which share an upstream core phenylpropanoid metabolism with Sal B. As showed in Figure 3A, morphological differences were apparent among the tested lines. Both types of compounds accumulated at higher levels in the roots of the overexpression lines than in CK samples. Concentrations of total phenolics and total flavonoids were 4.47 ± 0.13 and 32.43 ± 10.31 mg g-1 for CK, 10.61 ± 0.21 and 66.84 ± 8.48 mg g-1 for OEM-8; and 14.98 ± 0.11 and 73.91 ± 12.31 mg g-1 for OEM-12, respectively (Figure 3B).
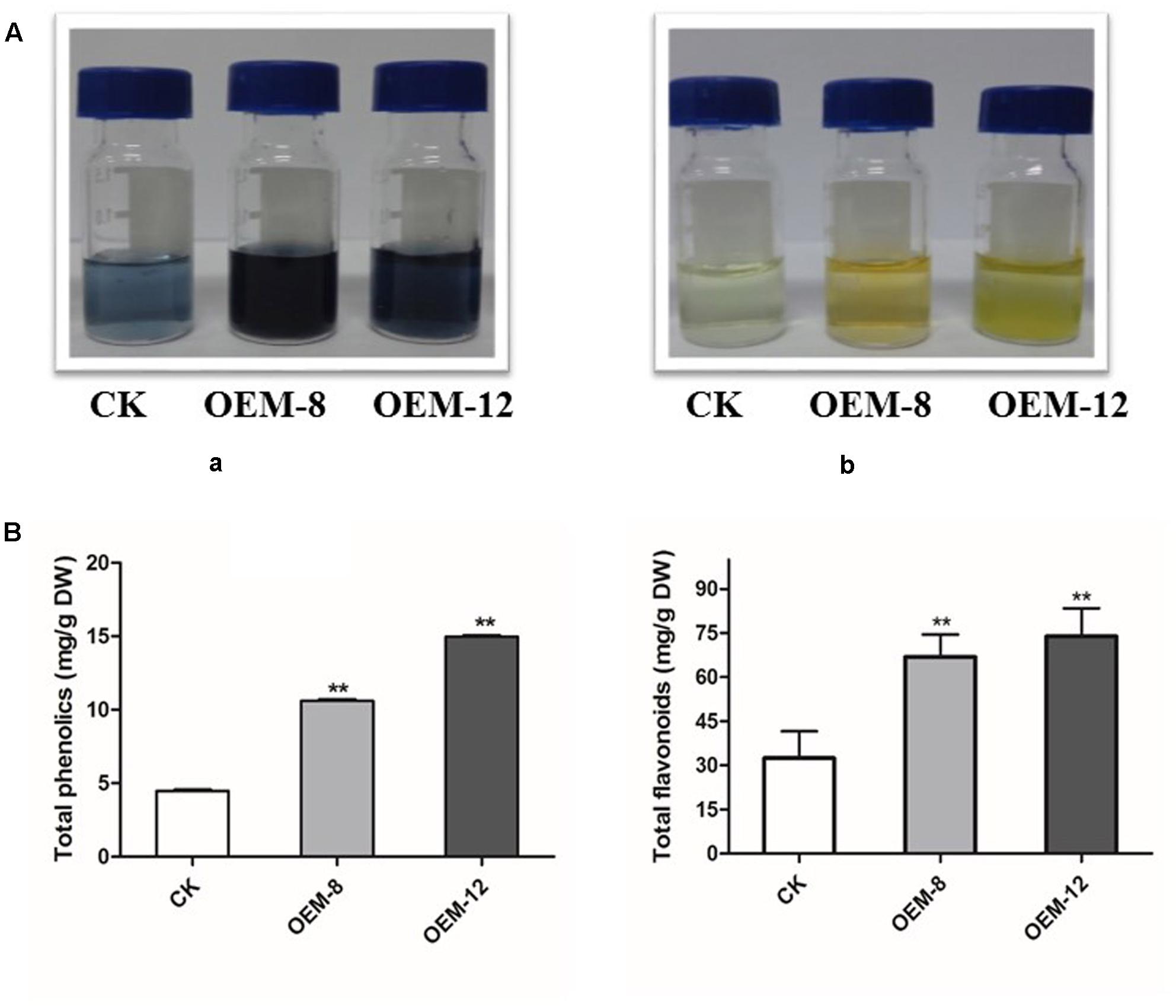
FIGURE 3. Concentrations of total phenolics and total flavonoids in transgenic lines and CK, control. (A) Visible signs of total phenolics (a) and total flavonoids (b). (B) Total accumulations in root extracts. All data are means of three replicates, with error bars indicating SD. ∗∗Values are significantly different from CK at P < 0.01.
Transcriptomic Analysis of SmMYC2-Overexpression and Control Plants of S. miltiorrhiza
For further investigation into the molecular mechanisms involved in the enhancement of salvianolic acid production in SmMYC2-overexpression transgenics, we compared the global expression profiles of OEM-12 versus CK. Two cDNA libraries were sequenced by Illumina deep sequencing to obtain approximately 31.26 and 25.93 million high-quality clean reads for OEM-12 and CK, respectively. Each read averaged 296 bp long. The Q30 value (percentage of sequences with a sequencing error rate < 0.1%) was 93.01% for OEM-12 and 92.86% for CK.
We analyzed the expression of unigenes using Bowtie (Langmead et al., 2009) and RSEM software (Li and Dewey, 2011) and normalized the values by Fragments Per Kilobase of transcript per Million mapped reads (FPKM) (Trapnell et al., 2010). Transcriptome analysis identified 2694 DEGs, with 1540 unigenes being up-regulated and 1154 down-regulated in OEM-12 when compared with expression in CK. The top 20 most up-regulated and most down-regulated genes are listed in Supplementary Tables 2 and 3.
Our KEGG analysis enabled us to identify 20 significantly enriched metabolic pathways for those DEGs (Figure 4). To examine the molecular basis for stimulation of phenolic acid production, we focused on biosynthetic pathways for phenolics, phenylalanine, tyrosine, and JA.
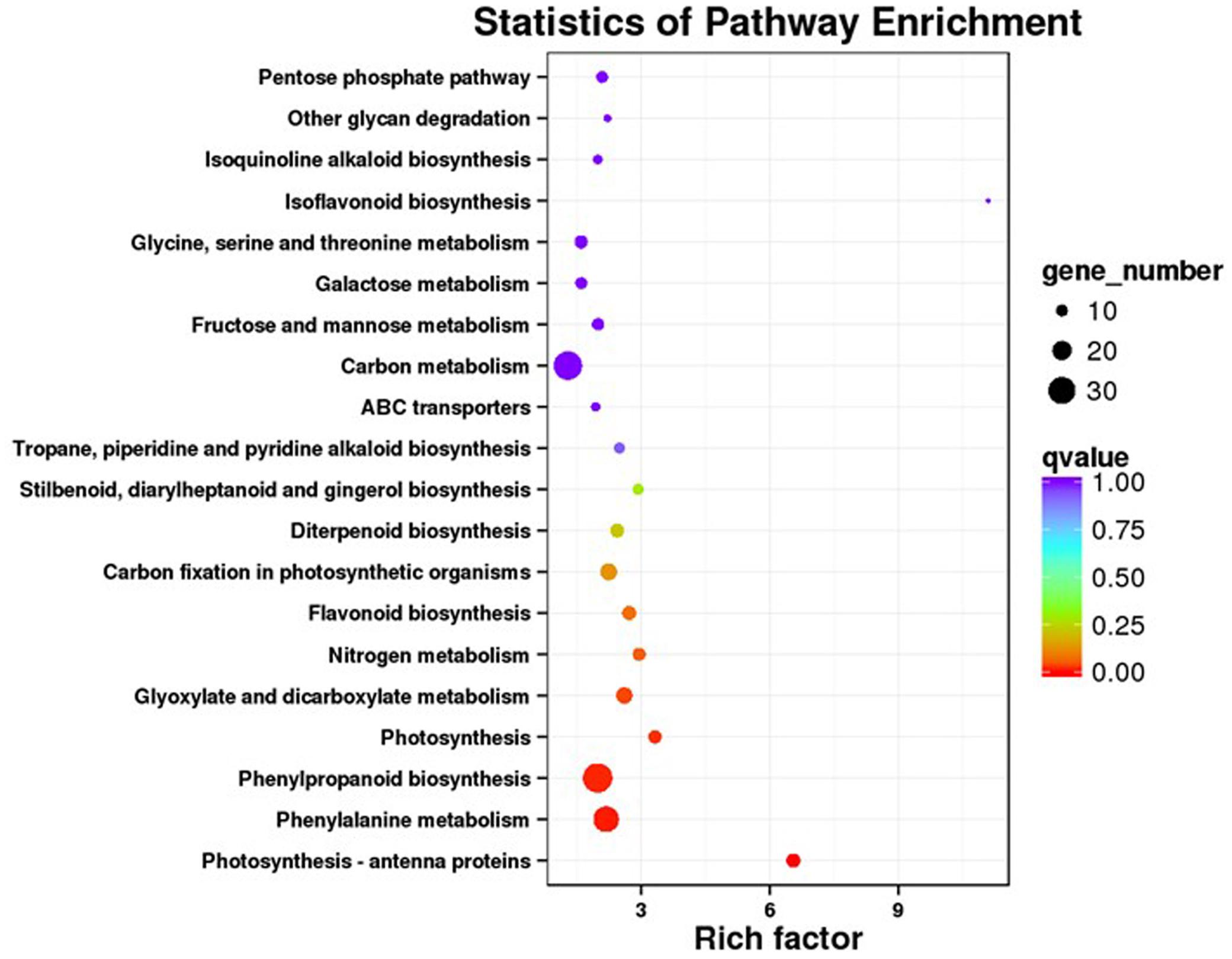
FIGURE 4. KEGG pathway enrichment analysis of DEGs.
DEGs Involved in JA Biosynthesis and Calculation of Endogenous JA Levels
Jasmonates play an integral role in various defense responses, leading to the accumulation of secondary metabolites (Browse, 2009). MYC2, a bHLH TF, acts as a transcriptional activator of JA-responsive genes that encode JA metabolic enzymes such as lipoxygenase (LOX), allene oxide synthase, allene oxide cyclase (AOC), 12-oxophytodienoate reductase (OPR), JA-amido synthetase (also known as JASMONATE RESISTANCE1), and cytochrome P450 monooxygenase 94B3 (CYP94B3) (Lorenzo et al., 2004; Sasaki-Sekimoto et al., 2013). We investigated changes in the expression of genes closely associated with α-linolenic acid metabolism, which finally leads to JA biosynthesis, and found six DEGs for the latter pathway (Figure 5A). Two DEGs encoding putative LOX and AOC showed increased transcript abundance. Among the DEGs encoding putative OPR, two were transcriptionally activated while two were transcriptionally suppressed. We then applied ELISA techniques to determine the concentrations of endogenous JA in fresh leaves from OEM-12 and CK plants. Based on the OD values of our samples, we learned that JA levels were not significantly changed by overexpression of SmMYC2, with values of 2.85 ± 0.23 ng g-1 for Line OEM-12 versus 2.81 ± 0.21 ng g-1 for CK (Figure 5B).
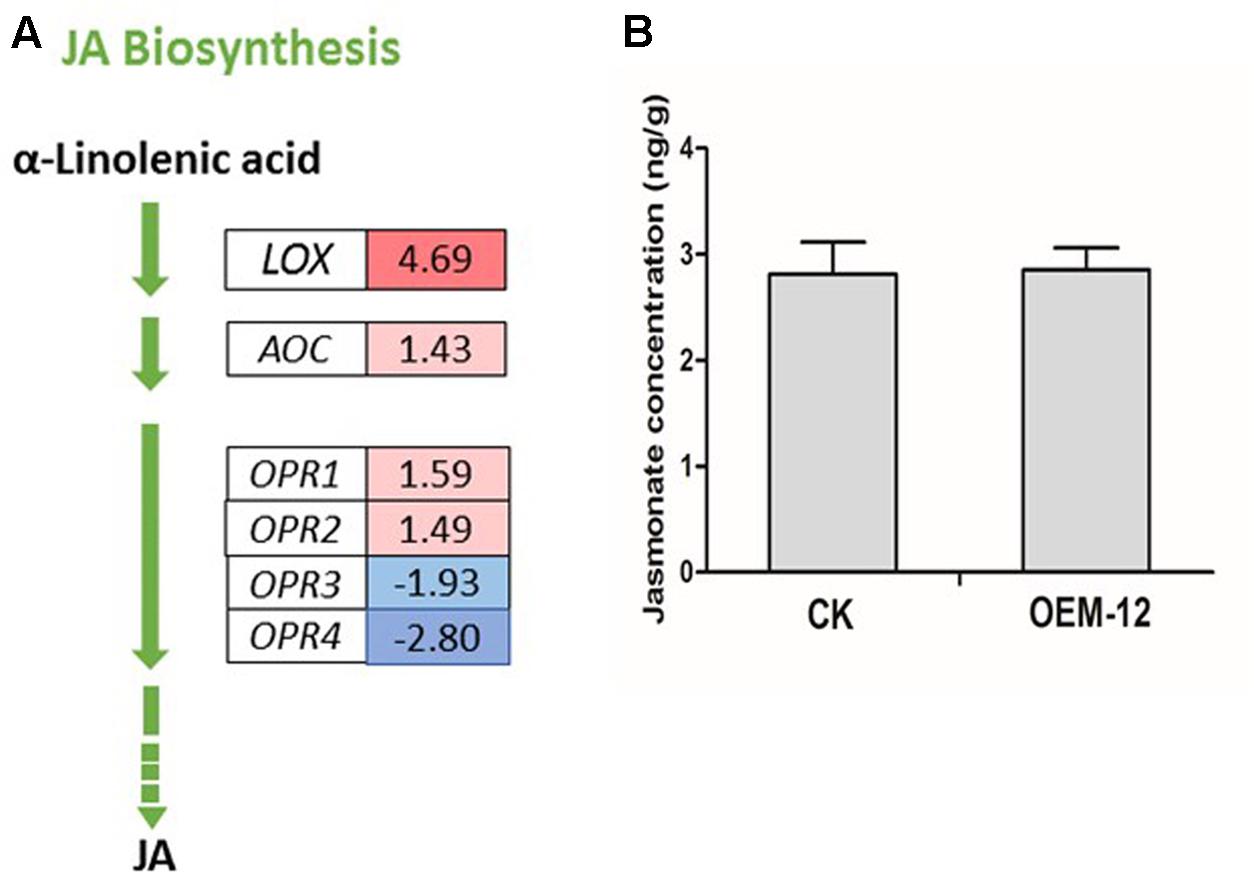
FIGURE 5. Changes in JA concentrations were not significant for transgenic plants over-expressing SmMYC2. (A) MYC2-mediated transcriptional activation of genes for JA metabolic enzymes. Blue boxes, putative encoding genes are down-regulated; red boxes, putative encoding genes are up-regulated. (B) Concentrations of JA in leaf extracts from CK control and transgenic line OEM-12.
DEGs Involved in Pathway for Salvianolic Acid Biosynthesis
To evaluate whether the elevated accumulation of salvianolic acids in transgenic lines was a result of up-regulated expression of SmMYC2, we examined transcript levels for all of the putative enzyme genes in the pathway for salvianolic acid biosynthesis (Figure 6). The transcriptome database revealed 21 genes homologous to SmPAL, SmC4H, Sm4CL, SmTAT, SmHPPR, SmRAS, and SmCYP98A78 (N.B., PAL, phenyl-alanine ammonia-lyase; C4H, cinnamate 4-hydroxylase; 4CL, 4-coumarate: coenzyme A ligase; TAT, tyrosine amino-transferase; HPPR, hydroxyphenylpyruvate reductase; and RA, rosmarinic acid synthase). Among the eight DEGs in the salvianolic acid biosynthesis pathway, five (SmTAT1, SmPAL1, SmC4H1, Sm4CL3, and SmRAS1) were up-regulated in the roots of OEM-12 while three (Sm4CL9, SmRAS2, and SmRAS6) were down-regulated. In that line, transcription was increased by 367.1-fold for SmPAL1, a critical gene for the synthesis of major water-soluble pharmaceutical ingredients within S. miltiorrhiza (Song and Wang, 2011). Expression for most of the other genes that were not differentially expressed slightly increased in OEM-12, according to the transcriptome data. Thus, upregulation of genes in the pathway for salvianolic acid biosynthesis was consistent with the elevated concentrations of salvianolic acids detected in the roots of transgenic plants.
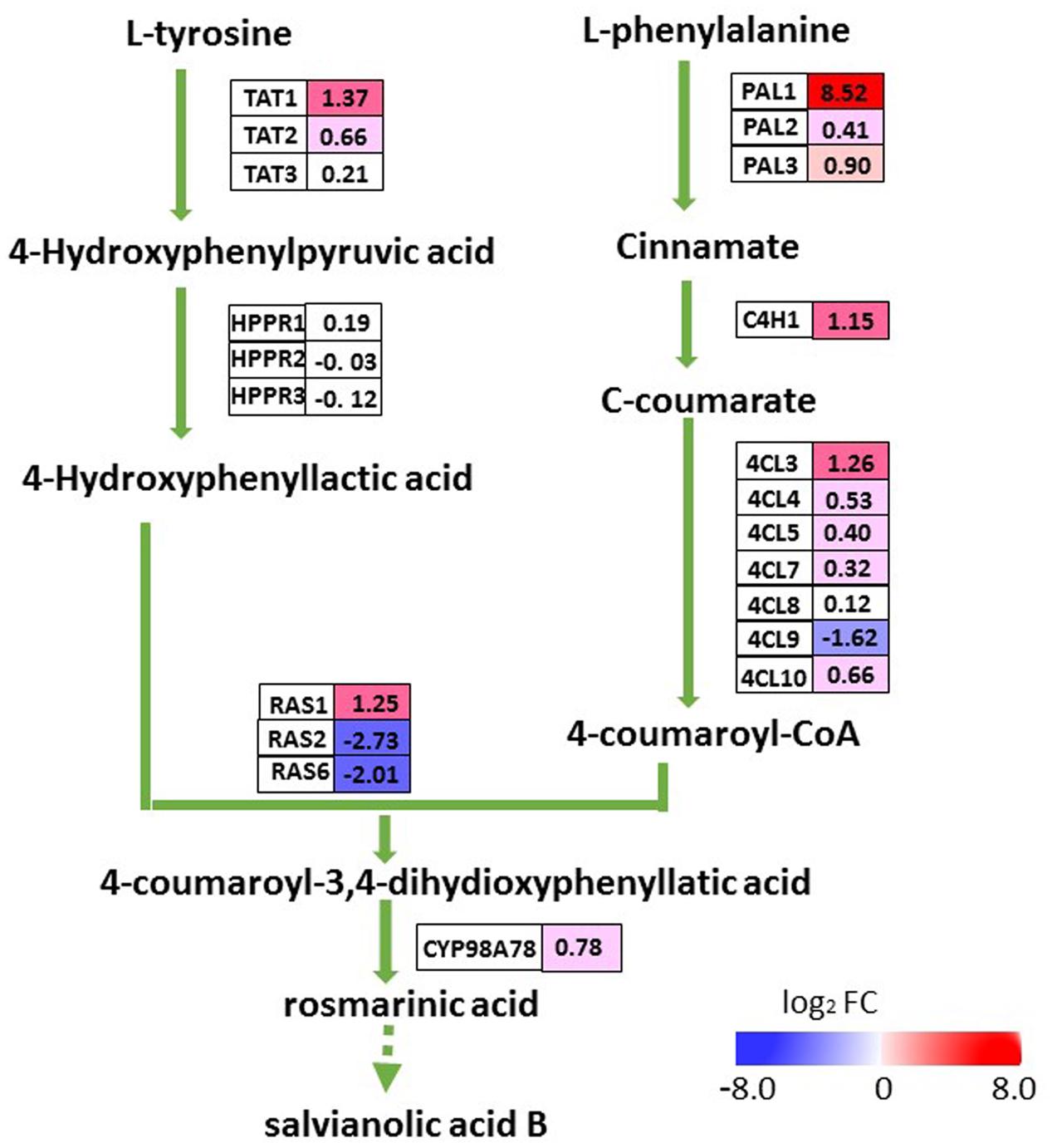
FIGURE 6. DEGs involved in pathway for salvianolic acid biosynthesis. For each gene, relative expression (OEM-12 versus control) is represented in log2 FC. Blue boxes, putative encoding genes are down-regulated; red boxes, putative encoding genes are up-regulated.
DEGs Involved in Phenylalanine and Tyrosine Biosynthesis
Phenolic secondary metabolites and their precursors are synthesized via the pathway of shikimate biosynthesis and its numerous branch points. That pathway is closely interlinked with those of the aromatic amino acids (L-tryptophan, L-phenylalanine, and L-tyrosine) (Tohge et al., 2013). Phenylalanine and tyrosine are precursors of RA and its derivative Sal B. In S. miltiorrhiza, the phenolic acid biosynthetic pathway starts from the general phenylpropanoid pathway and the tyrosine-derived pathway (Peng et al., 2013). Therefore, phenylalanine and tyrosine are of great importance. We found that nine DEGs related to their pathways were up-regulated in OEM-12 (Figure 7). Therefore, activation of the pathways for phenylalanine and tyrosine biosynthesis, which then provides more precursors for phenolic acids, may contribute to the improvement in RA and Sal B concentrations.
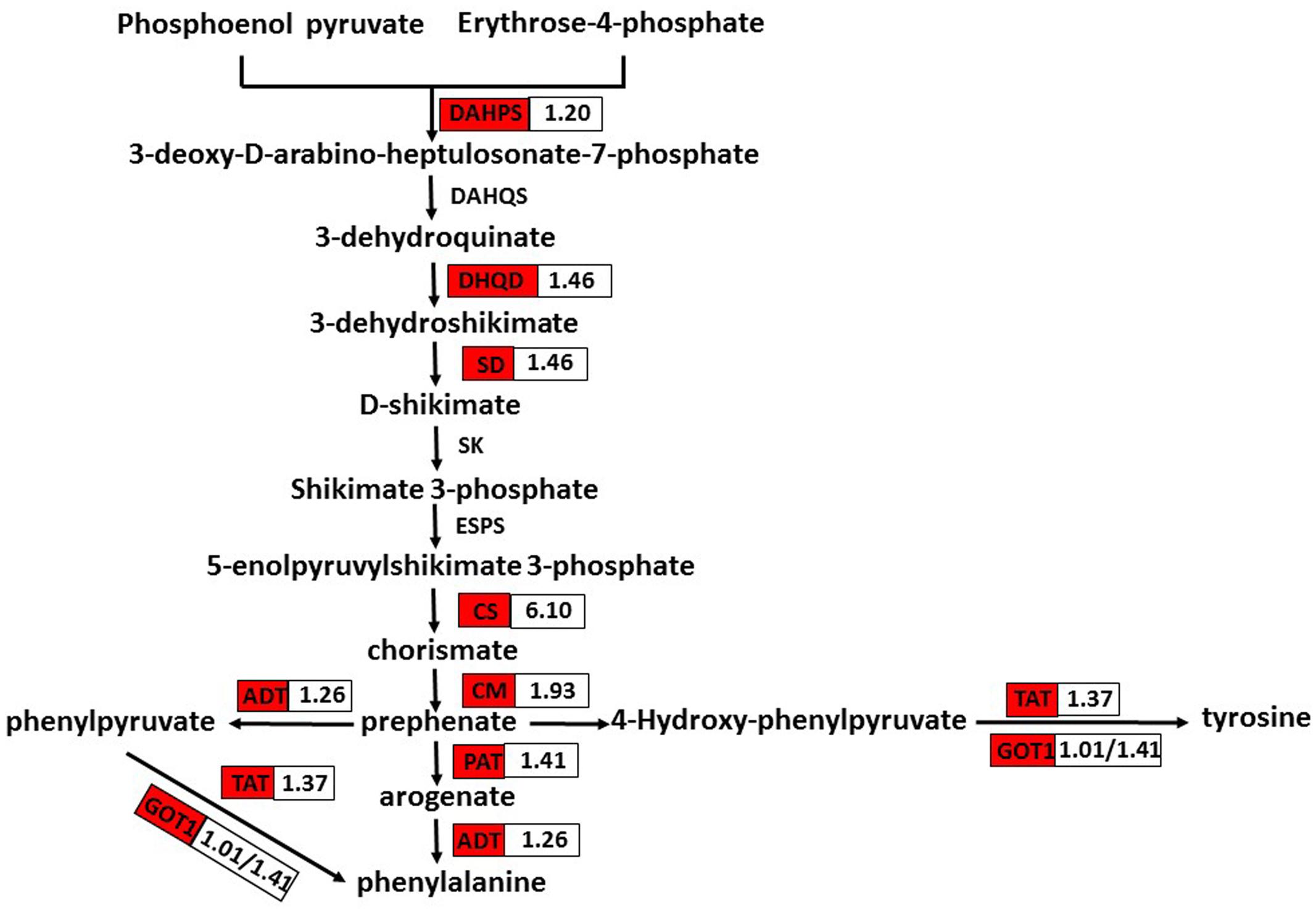
FIGURE 7. DEGs involved in phenylalanine and tyrosine biosynthesis. Red boxes, putative encoding genes are up-regulated. CS, chorismate synthase; CM, chorismate mutase; PAT, bifunctional aspartate aminotransferase and glutamate/aspartate-prephenate aminotransferase; ADT, arogenate/prephenate dehydratase; GOT1, aspartate aminotransferase; and TAT, tyrosine aminotransferase.
DEGs Involved in Tanshinone Biosynthesis
Tanshinones are derived from their precursor isopentenyl pyrophosphate (IPP), which is produced by two biosynthesis pathways: the mevalonate (MVA) pathway in the cytosol and the methylerythritol phosphate (MEP) pathway in the plastids. Although we did not detect tanshinone IIA or cryptotanshinone by HPLC in 2-month-old overexpression lines or control plants, we monitored the transcript levels of putative enzyme genes in the tanshinone biosynthesis pathway, and identified only three DEGs (SmCPS, SmKSL1, and SmMK) there, all of which were up-regulated in OEM-12. We found it interesting that expression for most MEP pathway genes was slightly decreased while most MVA pathway genes were somewhat induced (Figure 8). This implied that the pathway for tanshinone biosynthesis was not obviously activated in the transgenics.
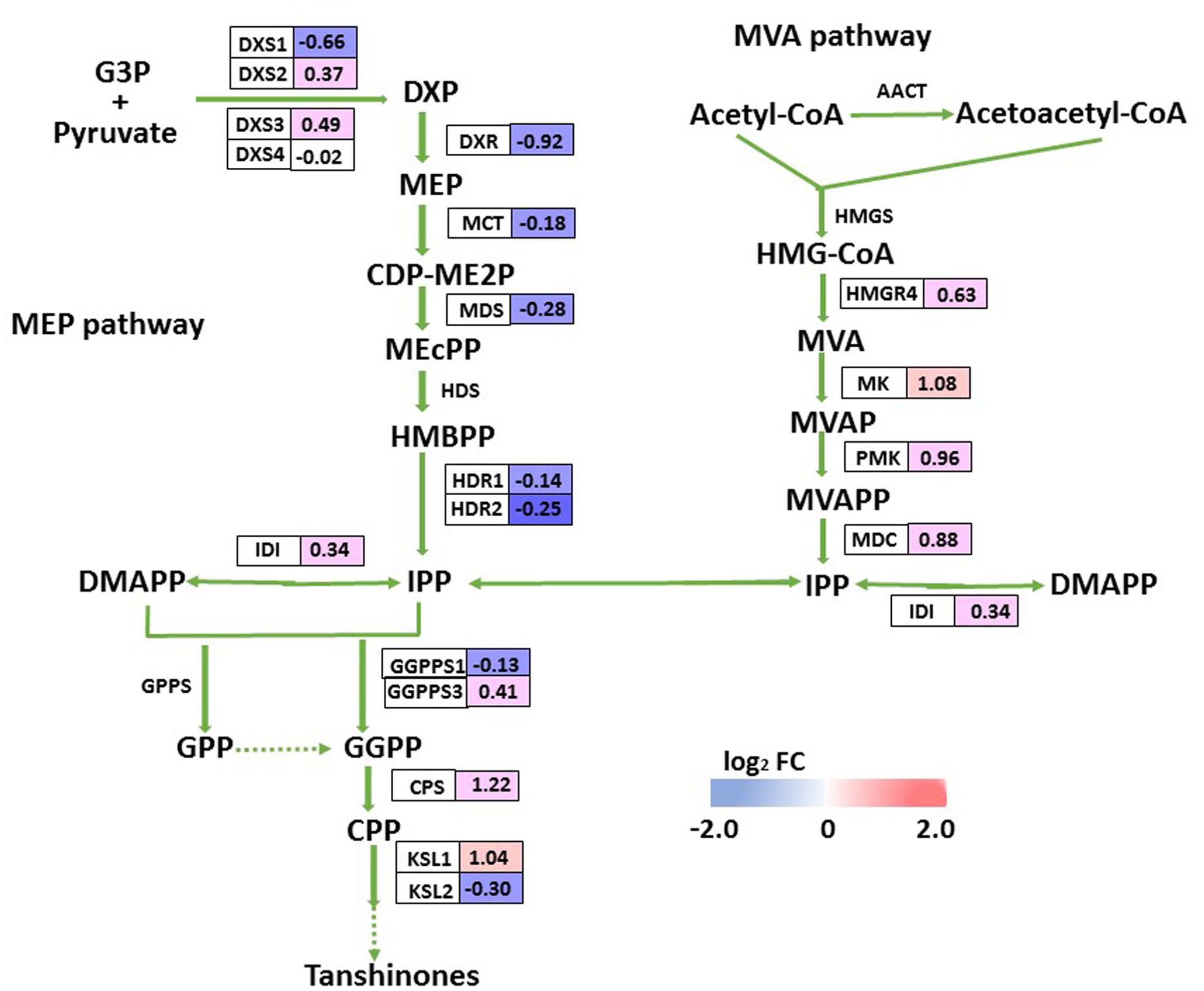
FIGURE 8. DEGs involved in tanshinone biosynthesis. For each gene, relative expression (OEM-12 versus control) is represented in log2 FC. Blue boxes, putative encoding genes are down-regulated; red boxes, putative encoding genes are up-regulated.
G-Box Analysis of Related Genes
MYC2 is both directly and indirectly involved in regulating secondary metabolites. The G-box fragment (5′-CACGTG-3′) is the preferred core binding site of MYC2, followed by the 5′-CACGTT-3′ (∼35% of binding activity of 5′-CACGTG-3′) and 5′-CACATG-3′ (∼20% of binding activity of 5′-CACGTG-3′) sequences (Shoji and Hashimoto, 2011; Kemal and Manners, 2013). We analyzed the promoter sequences of 29 putative genes for enzymes in the pathway for salvianolic acid biosynthesis and found that many, e.g., SmTAT1, SmHPPR1, SmCYP98A78, and SmRAS2, contain G-box sequences in their promoters (Supplementary Table 4). This suggests that MYC2 directly binds to the promoter of each of those genes to regulate their expression. Moreover, SmPAL1 has only a G-box-like sequence (5′-CATCTG-3′) in its promoter, and its transcript levels increased 367.1-fold in our OEM-12 transgenic line. Additional studies, such as chromatin immunoprecipitation (ChIP) sequencing analyses, are needed for genome-wide identification of direct MYC2 targets in S. miltiorrhiza (Kemal and Manners, 2013).
Confirmation of MYC2-Mediated Transcriptional Changes by Quantitative RT-PCR
To verify that these transcriptional changes were induced by MYC2, we examined 12 genes involved in phenolic acid biosynthesis (TAT, PAL, HPPR, C4H, and RAS), the JA signaling pathway (JAZ1 and JAZ3), tanshinone biosynthesis (DXS3, GGPPS1, and HMGR4), and flavonoids biosynthesis (FLS and F3′5′H). Their expression patterns, as detected by qRT-PCR, were consistent with those obtained from the RNA-Seq data (Figure 9). Overall, this qRT-PCR analysis confirmed that the RNA-Seq results were statistically reliable and comparable to transcriptomic data.
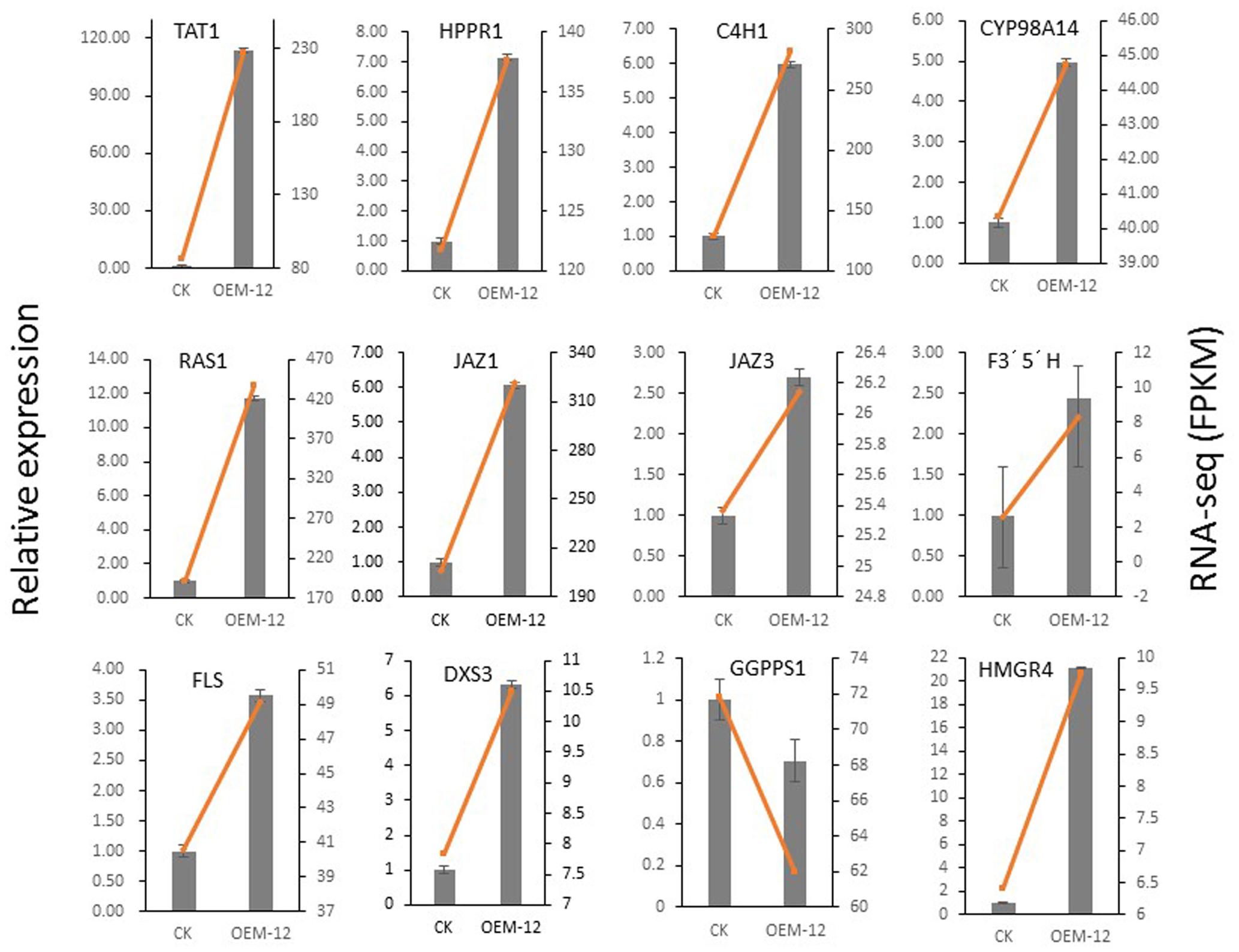
FIGURE 9. Validation by qRT-PCR of 12 genes involved in phenolic acid biosynthesis, JA signaling pathway, and tanshinone biosynthesis in control and transgenic line OEM-12.
Discussion
Jasmonates act as conserved elicitors of plant secondary metabolism. Exogenous application of MeJA triggers an extensive transcriptional reprogramming of metabolism and dramatically increases the biosynthesis of active ingredients in S. miltiorrhiza (Ge et al., 2015). Accordingly, MYC2 serves as a regulatory hub within the JA signaling pathway and regulates secondary metabolism (Kemal and Manners, 2013). AtMYC2 in Arabidopsis thaliana modulates the biosynthesis of glucosinolate, insect activity, and feeding behavior (Schweizer et al., 2013). In Nicotiana tabacum, NtMYC2a and NtMYC2b control multiple JA-inducible steps in nicotine biosynthesis (Zhang et al., 2012; Zhou Y. et al., 2016). Furthermore, MdMYC2 positively regulates flavonoid biosynthesis by modulating the expression of positive regulators in Malus domestica during JA signaling (An et al., 2016). Here, we investigated the function of SmMYC2 in regulating the production of active ingredients by over-expressing SmMYC2 in S. miltiorrhiza, followed by metabolic analysis and RNA-seq.
Positive regulation by SmMYC2 of phenolic acid biosynthesis and overexpression of SmMYC2 significantly improved the concentrations of RA and Sal B calculated in S. miltiorrhiza. Transcriptome analysis showed that such overexpression activated not only the phenolic acid biosynthetic pathway but also the pathways for phenylalanine and tyrosine biosynthesis. Both SmMYC2a and SmMYC2b are positive regulators of multiple genes in the pathways for tanshinone and phenolic acid biosynthesis in S. miltiorrhiz (Zhou Y. et al., 2016). Because we did not detect either tanshinone IIA or cryptotanshinone in any of our tested genotypes, this seemed to indicate that the tanshinone biosynthetic pathway is not obviously activated in OEM-12, based on the results of RNA-seq. Transcript levels for SmMYC2 were 3.77-fold higher in OEM-8 and 8.39-fold higher in OEM-12 when compared with CK. However, that enhancement was not as high as we had expected, possibly because MYC2 negatively regulates its own expression (Dombrecht et al., 2007). Therefore, we might speculate that SmMYC2 prioritizes the phenolic acid biosynthetic pathway over the tanshinone biosynthetic pathway.
Rosmarinic acid is an ester of 3,4-dihydroxyphenyllactic acid and caffeic acid. Those two compounds are synthesized via the tyrosine-derived pathway and the phenylpropanoid pathway, respectively (Petersen et al., 1993; Song and Wang, 2011). The latter pathway includes PAL, C4H, and 4CL, while the former includes TAT and HPPR. Line OEM-12 showed elevated accumulations of RA and SalB, demonstrating the upregulation of most enzyme genes in the pathway for phenolic acid biosynthesis. The first enzyme in that pathway is PAL, and PAL1 plays a major role during the development and metabolic course of S. miltiorrhiza (Song and Wang, 2011). The transcription level of SmPAL1 increased by 367.1-fold in OEM-12 according to the transcriptome data. SmTAT1 was previously identified as being involved in RA biosynthesis in hairy root cultures of S. miltiorrhiza (Xiao et al., 2011). Our qRT-PCR results showed that the transcript level of SmTAT1 was 110-fold higher in OEM-12 than in CK. This strong induction of SmPAL1 and SmTAT1 contributed to the accumulation of phenolic acids. The coupling of products from the phenylpropanoid and tyrosine-derived pathways is catalyzed by RAS, and we identified six SmRASs in the S. miltiorrhiza genome. Based on TBLASTN with amino acid sequences from other plant species, only SmRAS1 has been verified to form the precursor of RA in S. miltiorrhiza (Peng et al., 2013; Wang et al., 2015). We noted here that transcripts of SmRAS1 increased while those of SmRAS2 and SmRAS6 decreased in OEM-12. Although the S. miltiorrhiza genome contains at least 29 genes related to phenolic acid biosynthesis (Wang et al., 2015), most of them, including SmRAS2 and SmRAS6, have not yet been experimentally proven to participate in that process.
It is well known that exogenous JAs can up-regulate the expression of MYC2 and constitutive expression of AtMYC2 in the wild-type background induced an exaggerated response to JA (Lorenzo et al., 2004), but we don’t know if overexpression of MYC2 will influence the endogenous JA biosynthesis. We compared the JA levels between CK and OEM-12. The results showed that overexpression of SmMYC2 did not significantly change the content of endogenous JA. Besides, our transcriptome analysis showed that among the six DEGs in JA biosynthesis pathway, four (LOX, AOC, OPR1, and OPR2) were up-regulated in OEM-12 while two (OPR3 and OPR4) were down-regulated. The transcriptome data suggested that the pathway for JA biosynthesis was vibrated, but not obviously activated in OEM-12, which was consistent with the JA levels.
It was verified that two MYC2 orthologs from S. miltiorrhiza positively regulate the biosynthesis of tanshinones and Sal B by ectopic RNA interference mediated knockdown experiments with hairy root cultures (Zhou Y. et al., 2016), but it did not mean that overexpression of SmMYC2 would certainly increase the contents of tanshinones and phenolic acids in transgenic plantlets. Our results showed that overexpression of SmMYC2 significantly increased the contents of phenolic acids by activating not only the phenylpropanoid biosynthesis pathway but also the pathways for phenylalanine and tyrosine biosynthesis. Overall, although the molecular mechanism by which MYC2 regulates phenolic acid biosynthesis in S. miltiorrhiza is still unknown, our research results indicate that it has an important role in modulating this biosynthesis. It actives pathways for both secondary and primary metabolism. Therefore, our study of SmMYC2 overexpression in S. miltiorrhiza provides a good foundation for elucidating the molecular mechanism regulated by MYC2.
Author Contributions
The experiments were conceived and organized by XC and ZW. NY and WZ performed the experiments. JS, XW, and LL contributed to the data analysis. The paper was written by NY, LW, and XC. All authors discussed and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (31670299), the major Project of Shaanxi Province, China (2017ZDXM-SF-005), and the Fundamental Research Funds for the Central Universities (GK201706004).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2017.01804/full#supplementary-material
References
An, J. P., Li, H. H., Song, L. Q., Ling, S., Xin, L., You, C. X., et al. (2016). The molecular cloning and functional characterization of MdMYC2, a bHLH transcription factor in apple. Plant Physiol. Biochem. 108, 24–31. doi: 10.1016/j.plaphy.2016.06.032
Browse, J. (2009). Jasmonate passes muster: a receptor and targets for the defense hormone. Annu. Rev. Plant Biol. 60, 183–205. doi: 10.1146/annurev.arplant.043008.092007
Cao, X. Y., Guo, X. R., Yang, X. B., Wang, H. Q., Hua, W. P., He, Y. H., et al. (2016). Transcriptional responses and gentiopicroside biosynthesis in methyl jasmonate-treated Gentiana macrophylla seedlings. PLOS ONE 11:e0166493. doi: 10.1371/journal.pone.0166493
Chun-Yan, S. U., Ming, Q. L., Khalid, R., Han, T., and Qin, L. P. (2015). Salvia miltiorrhiza: traditional medicinal uses, chemistry, and pharmacology. Chin. J. Nat. Med. 13, 163–182.
Dombrecht, B., Xue, G. P., Sprague, S. J., Kirkegaard, J. A., Ross, J. J., Reid, J. B., et al. (2007). MYC2 differentially modulates diverse jasmonate-dependent functions in Arabidopsis. Plant Cell 19, 2225–2245. doi: 10.1105/tpc.106.048017
Earley, K. W., Haag, J. R., Pontes, O., Opper, K., Juehne, T., Song, K., et al. (2006). Gateway-compatible vectors for plant functional genomics and proteomics. Plant J. 45, 616–629. doi: 10.1111/j.1365-313X.2005.02617.x
Ge, Q., Zhang, Y., Hua, W. P., Wu, Y. C., Jin, X. X., Song, S. H., et al. (2015). Combination of transcriptomic and metabolomic analyses reveals a JAZ repressor in the jasmonate signaling pathway of Salvia miltiorrhiza. Sci. Rep. 5:14048. doi: 10.1038/srep14048
Gu, X. C., Chen, J. F., Xiao, Y., Di, P., Xuan, H. J., Zhou, X., et al. (2012). Overexpression of allene oxide cyclase promoted tanshinone/phenolic acid production in Salvia miltiorrhiza. Plant Cell Rep. 31, 2247–2259. doi: 10.1007/s00299-012-1334-9
Guo, Y., Li, Y., Xue, L., Severino, R. P., Gao, S., Niu, J., et al. (2014). Salvia miltiorrhiza : an ancient Chinese herbal medicine as a source for anti-osteoporotic drugs. J. Ethnopharmacol. 155, 1401–1416. doi: 10.1016/j.jep.2014.07.058
Ho, H. C. (2011). Salvianolic acids: small compounds with multiple mechanisms for cardiovascular protection. J. Biomed. Sci. 18, 1–5. doi: 10.1186/1423-0127-18-30
Hua, W., Song, J., Li, C., and Wang, Z. (2011). Molecular cloning and characterization of the promoter of SmGGPPs and its expression pattern in Salvia miltiorrhiza. Mol. Biol. Rep. 39, 5775–5783. doi: 10.1007/s11033-011-1388-8
Huang, B., Duan, Y., Yi, B., Sun, L., Lu, B., Yu, X., et al. (2008). Characterization and expression profiling of cinnamate 4-hydroxylase gene from Salvia miltiorrhiza in rosmarinic acid biosynthesis pathway. Russ. J. Plant Physiol. 55, 390–399. doi: 10.1134/S1021443708030163
Iván, F. A., Debora, G., Aurore, C., Stéphanie, S., Luca, S., and Edward, E. F. (2013). Role of NINJA in root jasmonate signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 15473–15478. doi: 10.1073/pnas.1307910110
Kathleen, D. B., Sofie, T., Laurens, P., Robin, V. B., Valerie, D. S., Rudy, V., et al. (2011). APETALA2/ETHYLENE RESPONSE FACTOR and basic helix-loop-helix tobacco transcription factors cooperatively mediate jasmonate-elicited nicotine biosynthesis: ERFs and bHLHs control nicotine biosynthesis. Plant J. 66, 1053–1065. doi: 10.1111/j.1365-313X.2011.04566.x
Kemal, K., and Manners, J. M. (2013). MYC2: the master in action. Mol. Plant 6, 686–703. doi: 10.1093/mp/sss128
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Liao, P., Zhou, W., Zhang, L., Wang, J., Yan, X., Zhang, Y., et al. (2009). Molecular cloning, characterization and expression analysis of a new gene encoding 3-hydroxy-3-methylglutaryl coenzyme A reductase from Salvia miltiorrhiza. Acta Physiol. Plant. 31, 565–572. doi: 10.1016/j.jplph.2010.06.008
Lorenzo, O., Chico, J. M., Sánchezserrano, J. J., and Solano, R. (2004). JASMONATE-INSENSITIVE1 encodes a MYC transcription factor essential to discriminate between different jasmonate-regulated defense responses in Arabidopsis. Plant Cell 16, 1938–1950. doi: 10.1105/tpc.022319
Pauwels, L., Barbero, G. F., Geerinck, J., Tilleman, S., Grunewald, W., Pérez, A. C., et al. (2010). NINJA connects the co-repressor TOPLESS to jasmonate signalling. Nature 464, 788–791. doi: 10.1038/nature08854
Peng, D., Lei, Z., Chen, J., Tan, H., Ying, X., Xin, D., et al. (2013). 13C tracer reveals phenolic acids biosynthesis in hairy root cultures of Salvia miltiorrhiza. ACS Chem. Biol. 8, 1537–1548. doi: 10.1021/cb3006962
Petersen, M., Häusler, E., Karwatzki, B., and Meinhard, J. (1993). Proposed biosynthetic pathway for rosmarinic acid in cell cultures of Coleus blumei Benth. Planta 189, 10–14. doi: 10.1007/BF00201337
Sasaki-Sekimoto, Y., Jikumaru, Y., Obayashi, T., Saito, H., Masuda, S., Kamiya, Y., et al. (2013). Basic helix-loop-helix transcription factors JASMONATE-ASSOCIATED MYC2-LIKE1 (JAM1), JAM2, and JAM3 are negative regulators of jasmonate responses in Arabidopsis. Plant Physiol. 163, 291–304. doi: 10.1104/pp.113.220129
Schweizer, F., Fernándezcalvo, P., Zander, M., Diezdiaz, M., Fonseca, S., Glauser, G., et al. (2013). Arabidopsis basic helix-loop-helix transcription factors MYC2, MYC3, and MYC4 regulate glucosinolate biosynthesis, insect performance, and feeding behavior. Plant Cell 25, 3117–3132. doi: 10.1105/tpc.113.115139
Sheard, L. B., Tan, X., Mao, H., Withers, J., Ben-Nissan, G., Hinds, T. R., et al. (2010). Jasmonate perception by inositol phosphate-potentiated COI1-JAZ co-receptor. Nature 468, 400–405. doi: 10.1038/nature09430
Shoji, T., and Hashimoto, T. (2011). Tobacco MYC2 regulates jasmonate-inducible nicotine biosynthesis genes directly and by way of the NIC2-Locus ERF genes. Plant Cell Physiol. 52, 1117–1130. doi: 10.1093/pcp/pcr063
Shyu, C., Figueroa, P., Depew, C. L., Cooke, T. F., Sheard, L. B., Moreno, J. E., et al. (2012). JAZ8 lacks a canonical degron and has an EAR motif that mediates transcriptional repression of jasmonate responses in Arabidopsis. Plant Cell 24, 536–550. doi: 10.1105/tpc.111.093005
Song, J., and Wang, Z. (2011). RNAi-mediated suppression of the phenylalanine ammonia-lyase gene in Salvia miltiorrhiza causes abnormal phenotypes and a reduction in rosmarinic acid biosynthesis. J. Plant Res. 124, 183–192. doi: 10.1007/s10265-010-0350-5
Todd, A. T., Liu, E., Polvi, S. L., Pammett, R. T., and Page, J. E. (2010). A functional genomics screen identifies diverse transcription factors that regulate alkaloid biosynthesis in Nicotiana benthamiana. Plant J. 62, 589–600. doi: 10.1111/j.1365-313X.2010.04186.x
Tohge, T., Watanabe, M., Hoefgen, R., and Fernie, A. R. (2013). Shikimate and phenylalanine biosynthesis in the green lineage. Front. Plant Sci. 4:62. doi: 10.3389/fpls.2013.00062
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., Baren, M. J. V., et al. (2010). Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Vandesompele, J., Preter, K. D., Pattyn, F., Poppe, B., Roy, N. V., Paepe, A. D., et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, research0034.1–research0034.11. doi: 10.1186/gb-2002-3-7-research0034
Wang, B., Sun, W., Li, Q. S., Li, Y., Luo, H. M., Song, J. G., et al. (2015). Genome-wide identification of phenolic acid biosynthetic genes in Salvia miltiorrhiza. Planta 241, 711–725. doi: 10.1007/s00425-014-2212-1
Wang, D. H., Song, Y., Chen, Y. Q., Yao, W., Li, Z., Liu, W. C., et al. (2013). Metabolic pools of phenolic acids in Salvia miltiorrhiza are enhanced by co-expression of Antirrhinum majus Delila and Rosea1 transcription factors. Biochem. Eng. J. 74, 115–120. doi: 10.1016/j.bej.2013.02.014
Wang, H. Q., Wu, Y. C., Yang, X. B., Guo, X. R., and Cao, X. Y. (2016). SmLEA2, a gene for late embryogenesis abundant protein isolated from Salvia miltiorrhiza, confers tolerance to drought and salt stress in Escherichia coli and S. miltiorrhiza. Protoplasma 254, 685–696. doi: 10.1007/s00709-016-0981-z
Wang, Z. J., Ge, Q., Chen, C., Jin, X. X., Cao, X. Y., and Wang, Z. Z. (2016). Function analysis of Caffeoyl-CoA O-methyltransferase for biosynthesis of lignin and phenolic acid in Salvia miltiorrhiza. Appl. Biochem. Biotechnol. 181, 562–572. doi: 10.1007/s12010-016-2231-4
Wasternack, C., and Kombrink, E. (2010). Jasmonates: structural requirements for lipid-derived signals active in plant stress responses and development. ACS Chem. Biol. 5, 63–77. doi: 10.1021/cb900269u
Wei, T., Deng, K. J., Zhang, Q. X., Gao, Y. H., Liu, Y., Yang, M. L., et al. (2017). Modulating AtDREB1C expression improves drought tolerance in Salvia miltiorrhiza. Front. Plant Sci. 8:52. doi: 10.3389/fpls.2017.00052
Xiao, Y., Zhang, L., Gao, S., Saechao, S., Di, P., Chen, J., et al. (2011). The c4h, tat, hppr and hppd genes prompted engineering of rosmarinic acid biosynthetic pathway in Salvia miltiorrhiza hairy root cultures. PLOS ONE 6:e29713. doi: 10.1371/journal.pone.0029713
Xu, Z., Luo, H., Ji, A., Zhang, X., Song, J., and Chen, S. (2016). Global identification of the full-length transcripts and alternative splicing related to phenolic acid biosynthetic genes in Salvia miltiorrhiza. Front. Plant Sci. 7:100. doi: 10.3389/fpls.2016.00100
Yan, Y. P., and Wang, Z. Z. (2007). Genetic transformation of the medicinal plant Salvia miltiorrhiza by Agrobacterium tumefaciens -mediated method. Plant Cell Tissue Organ Cult. 88, 175–184. doi: 10.1007/s12010-015-1826-5
Zhang, H. B., Bokowiec, M. T., Rushton, P. J., Han, S. C., and Timko, M. P. (2012). Tobacco transcription factors NtMYC2a and NtMYC2b form nuclear complexes with the NtJAZ1 repressor and regulate multiple jasmonate-inducible steps in nicotine biosynthesis. Mol. Plant 5, 73–84. doi: 10.1093/mp/ssr056
Zhang, H. T., Hedhili, S., Montiel, G., Zhang, Y. X., Chatel, G., Pré, M., et al. (2011). The basic helix-loop-helix transcription factor CrMYC2 controls the jasmonate-responsive expression of the ORCA genes that regulate alkaloid biosynthesis in Catharanthus roseus. Plant J. 67, 61–71. doi: 10.1111/j.1365-313X.2011.04575.x
Zhang, L., Junfeng, C., Qing, L., and Wan, W. C. (2016). Transcriptome-wide analysis of basic helix-loop-helix transcription factors in Isatis indigotica and their methyl jasmonate responsive expression profiling. Gene 576, 150–159. doi: 10.1016/j.gene.2015.09.083
Zhang, S., Ma, P., Yang, D., Li, W., Liang, Z., Liu, Y., et al. (2013). Cloning and Characterization of a putative R2R3 MYB transcriptional repressor of the rosmarinic acid biosynthetic pathway from Salvia miltiorrhiza. PLOS ONE 8:e73259. doi: 10.1371/journal.pone.0073259
Zhang, Y., and Wang, Z. Z. (2009). Phenolic composition and antioxidant activities of two Phlomis species: a correlation study. C. R. Biol. 332, 816–826. doi: 10.1016/j.crvi.2009.05.006
Zhang, Y., Yan, Y. P., and Wang, Z. Z. (2010). The Arabidopsis PAP1 transcription factor plays an important role in the enrichment of phenolic acids in Salvia miltiorrhiza. J. Agric. Food Chem. 58, 12168–12175. doi: 10.1021/jf103203e
Zhang, Y., Yan, Y. P., Wu, Y. C., Hua, W. P., Chen, C., Ge, Q., et al. (2014). Pathway engineering for phenolic acid accumulations in Salvia miltiorrhiza by combinational genetic manipulation. Metab. Eng. 21, 71–80. doi: 10.1016/j.ymben.2013.10.009
Zhou, W. P., Wang, H. Q., Guo, X. R., Yang, X. B., Hua, W. P., and Cao, X. Y. (2016). Cloning and expression analysis of a bHLH transcription factor gene from Salvia miltiorrhiza Bunge. Plant Sci. J. 34, 246–254.
Keywords: SmMYC2, Salvia miltiorrhiza, secondary metabolism, salvianolic acid B, tanshinones, transcriptome
Citation: Yang N, Zhou W, Su J, Wang X, Li L, Wang L, Cao X and Wang Z (2017) Overexpression of SmMYC2 Increases the Production of Phenolic Acids in Salvia miltiorrhiza. Front. Plant Sci. 8:1804. doi: 10.3389/fpls.2017.01804
Received: 26 July 2017; Accepted: 04 October 2017;
Published: 18 October 2017.
Edited by:
Angelos K. Kanellis, Aristotle University of Thessaloniki, GreeceReviewed by:
Tatiana Matveeva, Saint Petersburg State University, RussiaKexuan Tang, Shanghai Jiao Tong University, China
Vasiliki Falara, University of Delaware, United States
Copyright © 2017 Yang, Zhou, Su, Wang, Li, Wang, Cao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyan Cao, Y2FveGlhb3lhbkBzbm51LmVkdS5jbg== Zhezhi Wang, enp3YW5nQHNubnUuZWR1LmNu