 Florian Walder1*
Florian Walder1* Klaus Schlaeppi1
Klaus Schlaeppi1 Raphaël Wittwer1
Raphaël Wittwer1 Alain Y. Held1
Alain Y. Held1 Susanne Vogelgsang2
Susanne Vogelgsang2 Marcel G. A. van der Heijden1,3,4
Marcel G. A. van der Heijden1,3,4- 1Plant-Soil Interactions, Research Division Agroecology and Environment, Agroscope, Zurich, Switzerland
- 2Ecology of Noxious and Beneficial Organisms, Research Division Plant Protection, Agroscope, Zurich, Switzerland
- 3Department of Evolutionary Biology and Environmental Studies, University of Zürich, Zurich, Switzerland
- 4Plant-Microbe Interactions, Institute of Environmental Biology, Faculty of Science, Utrecht University, Utrecht, Netherlands
Fusarium head blight, caused by fungi from the genus Fusarium, is one of the most harmful cereal diseases, resulting not only in severe yield losses but also in mycotoxin contaminated and health-threatening grains. Fusarium head blight is caused by a diverse set of species that have different host ranges, mycotoxin profiles and responses to agricultural practices. Thus, understanding the composition of Fusarium communities in the field is crucial for estimating their impact and also for the development of effective control measures. Up to now, most molecular tools that monitor Fusarium communities on plants are limited to certain species and do not distinguish other plant associated fungi. To close these gaps, we developed a sequencing-based community profiling methodology for crop-associated fungi with a focus on the genus Fusarium. By analyzing a 1600 bp long amplicon spanning the highly variable segments ITS and D1–D3 of the ribosomal operon by PacBio SMRT sequencing, we were able to robustly quantify Fusarium down to species level through clustering against reference sequences. The newly developed methodology was successfully validated in mock communities and provided similar results as the culture-based assessment of Fusarium communities by seed health tests in grain samples from different crop species. Finally, we exemplified the newly developed methodology in a field experiment with a wheat-maize crop sequence under different cover crop and tillage regimes. We analyzed wheat straw residues, cover crop shoots and maize grains and we could reveal that the cover crop hairy vetch (Vicia villosa) acts as a potent alternative host for Fusarium (OTU F.ave/tri) showing an eightfold higher relative abundance compared with other cover crop treatments. Moreover, as the newly developed methodology also allows to trace other crop-associated fungi, we found that vetch and green fallow hosted further fungal plant pathogens including Zymoseptoria tritici. Thus, besides their beneficial traits, cover crops can also entail phytopathological risks by acting as alternative hosts for Fusarium and other noxious plant pathogens. The newly developed sequencing based methodology is a powerful diagnostic tool to trace Fusarium in combination with other fungi associated to different crop species.
Introduction
Fusarium represents a complex fungal genus inhabiting a vast range of different ecological niches and is often found in associations with plants (Summerell et al., 2010). A set of plant-associated Fusarium species include some of the most important plant pathogens worldwide causing the disease complexes Fusarium ear rot (FER) in maize (Zea mays) and Fusarium head blight (FHB) in several small-grain cereals, such as wheat (Triticum aestivum) and barley (Hordeum vulgare) (Parry et al., 1995; Desjardins, 2003; Schöneberg et al., 2016). The danger of both FER and FHB epidemics is not only due to the severe yield losses they cause but also because of the contamination of grains with harmful mycotoxins, which might devastate entire harvests (Bottalico and Perrone, 2002; Nganje et al., 2004; Reddy et al., 2010; Vogelgsang et al., 2011). Hence, Fusarium constitutes not only a severe threat to plant health but also to food and feed safety and sustainable agricultural production.
For several reasons, it is of great agronomical importance to understand the structure of Fusarium communities. First, the FER and FHB disease complexes are caused by 15–20 different species, where usually more than one species are simultaneously found in individual grain samples (Xu et al., 2005; Xu and Nicholson, 2009). Important causal species of FER in maize are Fusarium graminearum, F. verticillioides, F. proliferatum, and F. subglutinans (Desjardins, 2003; Dorn et al., 2009; Summerell et al., 2010), while the main causal species for FHB include F. graminearum, F. culmorum, F. poae and F. avenaceum (Parry et al., 1995; Xu et al., 2005). Additionally, it is important to note that different Fusarium species differ significantly in their response to diverse environmetal conditions and to particular agricultural practices (Edwards, 2004; Xu and Nicholson, 2009). Finally, disease severity and also the type and amount of mycotoxin production depends on the identity of Fusarium involved, as species vary substantially in their aggressiveness, host range and mycotoxin proflies (Bottalico and Perrone, 2002; Uhlig et al., 2006; Stȩpień et al., 2011). Thus, revealing the composition of the Fusarium community is important to understand its agro-ecological impact and finally to develop effective control measures (Xu and Nicholson, 2009).
The genetic diversity within the Fusarium genus is complex, exhibiting high genetic variablity within morphologically defined species (Leslie et al., 2007; O’Donnell et al., 2009). A comprehensive molecular study revealed that most of the species within this genus could be resolved into monophyletic species complexes that consist of multiple morphological cryptic species (O’Donnell et al., 2013). Hence, the phylogenetic relationship among Fusarium species remains unclear and the construction of a reliable taxonomic system based on the combination of morphological, molecular, toxicological, and biological traits is needed (Leslie and Bowden, 2008; Watanabe, 2013). In the present study, we are using a species definition which is based on the morphological species concept. Thus, Fusarium species are defined in a broader sense and we do not differentiate between cryptic species within given species complexes. We therefore refer in the following to species as sensu lato (s.l.).
Suitable tools to identify and quantify Fusarium communities are limited. The assessment of Fusarium is traditionally performed employing so-called seed health tests (SHTs; Dorn et al., 2011; Vogelgsang et al., 2011; Infantino et al., 2012), which are cultivation-based assays on agar media making use of species-specific morphological characteristics. However, this approach is laborious and only applicable if highly experienced and specialized personnel is available (Summerell and Leslie, 2011). On the other hand, molecular detection of Fusarium is often based on species-specific real-time quantitative polymerase chain reactions (qPCR; Gao et al., 2004; Waalwijk et al., 2004; Hogg et al., 2007; Edwards et al., 2012). qPCR assays allow a precise quantification of Fusarium, but are restricted to a limited number of species. The state-of-the-art technique for molecular identification of fungal communities is high-throughput amplicon sequencing providing relative abundances of operational taxonomical units (OTUs; Lindahl et al., 2013). A major drawback of high throughput sequencing approaches is that they often fail to resolve microbes on species levels (Eren et al., 2013). So far, only one high-throughput sequencing approach is available able to resolve Fusarium fungi down to morphological species level (Karlsson et al., 2016). However, this approach is restricted to the genus Fusarium because it uses taxon-specific primers. The use of universal fungal primers has many benefits over taxa specific approaches because other fungi (including a wide range of fungal pathogens) can be detected at the same time providing important additional information. Also, commensal plant-associated fungal communities can interact with pathogens, thus determining their infection success (Berendsen et al., 2012; Agler et al., 2016). Hence, sequencing tools that simultaneously capture pathogens (e.g., Fusarium) and non-pathogenic fungi can provide additional information and might thus contribute to the search for beneficial plant-associated fungi with potential bio-control activity.
In the present work, we are aiming to develop a sequencing based community profiling methodology enabling the detection of Fusarium down to morphological species level in the frame of a universal fungal approach. Most promising loci for a sequence-based identification of Fusarium taxa are the translation elongation factor 1α, β-tubulin, and the largest subunit of RNA polymerase (Balajee et al., 2009; O’Donnell et al., 2015). However, these loci are less practical for a universal approach as available primers for such markers usually amplify a narrow taxonomic range (Schoch et al., 2012). More suitable for a universal fungal target locus appears to be the nuclear ribosomal operon, particularly its internal transcribed spacer (ITS; Schoch et al., 2012; Lindahl et al., 2013). Although the ITS segment is highly variable, it lacks species-level resolution in some fungal groups including Fusarium (O’Donnell et al., 1998; Yli-Mattila et al., 2004; Santamaria et al., 2009). In addition, paralogous sequences for the ITS segment were found within Fusarium species, with the consequence that the ITS segment is not always reflecting the Fusarium phylogeny (Waalwijk et al., 1996; O’Donnell and Cigelnik, 1997). The segment at the 3′-prime end of the large subunit (LSU) of the ribosomal operon including the highly variable segments D1 and D2 (D1–D2) (Nilsson et al., 2013) provide a valid alternative. The D1–D2 segment was also used to distinguish among Fusarium species (Guadet et al., 1989; Hennequin et al., 1999). However, it has been shown that the D1–D2 segment on its own was not able to resolve Fusarium taxa down to species or isolate level (O’Donnell et al., 2008b). The combination of the highly variable ITS and D1–D2 segments has been tested as well, but showed an inferior performance than other loci (O’Donnell et al., 2008a, 2009).
Nonetheless, as the ribosomal operon offers some indisputable advantages for the sequencing based assessment of fungi over the whole kingdom, we aimed to explore this possibility based on the analysis of a large amplicon spanning the entire ITS and the D1–D2 segment of the ribosomal operon using single molecule real time (SMRT) sequencing methodology. SMRT sequencing presents a valid technology for high-resolution community sequencing of bacteria and fungi (Franzén et al., 2015; Schlaeppi et al., 2016; Schloss et al., 2016; Singer et al., 2016; Tedersoo et al., 2017). We hypothesized that such an approach will permit the description of Fusarium communities down to morphological species level.
The goal of this study was the development of a sequencing-based methodology that robustly quantifies known Fusarium in the phyllosphere, including shoots and grains, of different plant species. To achieve this aim, we first examined the ribosomal operon for a suitable marker region to discriminate Fusarium identities based on a set of morphologically validated species and isolates. We then evaluated four PCR primer combinations to capture the taxonomic breath of Fusarium taxa and how accurately they quantify them. Subsequently, we successfully validated the diagnostic power of the new methodology in several samples of different crops in comparison to sequencing-independent and cultivation-based SHTs. Finally, we applied the new sequencing-based methodology to a test case under field conditions. We monitored Fusarium species in a field experiment investigating the effect of different cover crops and tillage regimes to get insights on the dispersal of FHB causing species along a wheat-maize crop sequence. Both tillage and cover crops can have a substantial impact on plant yield (Wittwer et al., 2017) and on the dispersal of Fusarium species (Dill-Macky and Jones, 2000; Edwards, 2004; Friberg et al., 2009). Thus, the potential agronomic benefits of cover crops and reduced tillage systems can only become effective if their phytopathological risks for crop sequences are thoroughly assessed.
Materials and Methods
Analysis of Ribosomal Operon Sequences of Swiss Fusarium Isolates
Forty-four isolates belonging to 14 different Fusarium species based on morphological characterization reflecting the diversity of FHB causing species in Switzerland were selected to survey the genetic variability of the ribosomal operon (ITS, LSU) among the genus Fusarium (Supplementary Table S1). The single conidia isolates were collected in the frame of a Swiss Fusarium monitoring effort (Dorn et al., 2009; Vogelgsang et al., 2009; Schöneberg et al., 2016). Fungal mycelium was used for DNA extraction (see Supplementary Methods for details). Ribosomal operon sequences of the 44 Fusarium isolates were obtained by amplification with the PCR primer pair ITS1F and LR6 (Supplementary Table S2 for references), spanning the entire ITS region and the highly variable segments D1, D2 and D3 of the LSU, and subsequent Sanger sequencing (Microsynth AG, Balgach, Switzerland; see Supplementary Methods for details).
Based on these Sanger sequences, we evaluated the entire amplicon of ITS1F and LR6 region and additionally the ITS and the D1–D2 segment on its own for their suitability to discriminate the Fusarium isolates at the morphological species level (Figure 1A). The different segments of the ribosomal operon were extracted in silico with flexbar (Dodt et al., 2012) using for the ITS segment the PCR primers ITS5 and ITS4, and for the D1–D2 segment of the LSU region the primers LR0R and LR3 (see Supplementary Table S2 for details and references, Figure 1A). We clustered the sequences for each operon segment and defined OTUs at similarity levels of 100% using USEARCH (see below). For each segment, we report the taxonomic resolution among the 44 isolate sequences (Figure 1B). Utilizing the full-length operon, we found 23 unique sequences among the 44 tested isolates. The sequences determined in this study were deposited in GenBank (Accession Numbers: MG274294-MG274317).
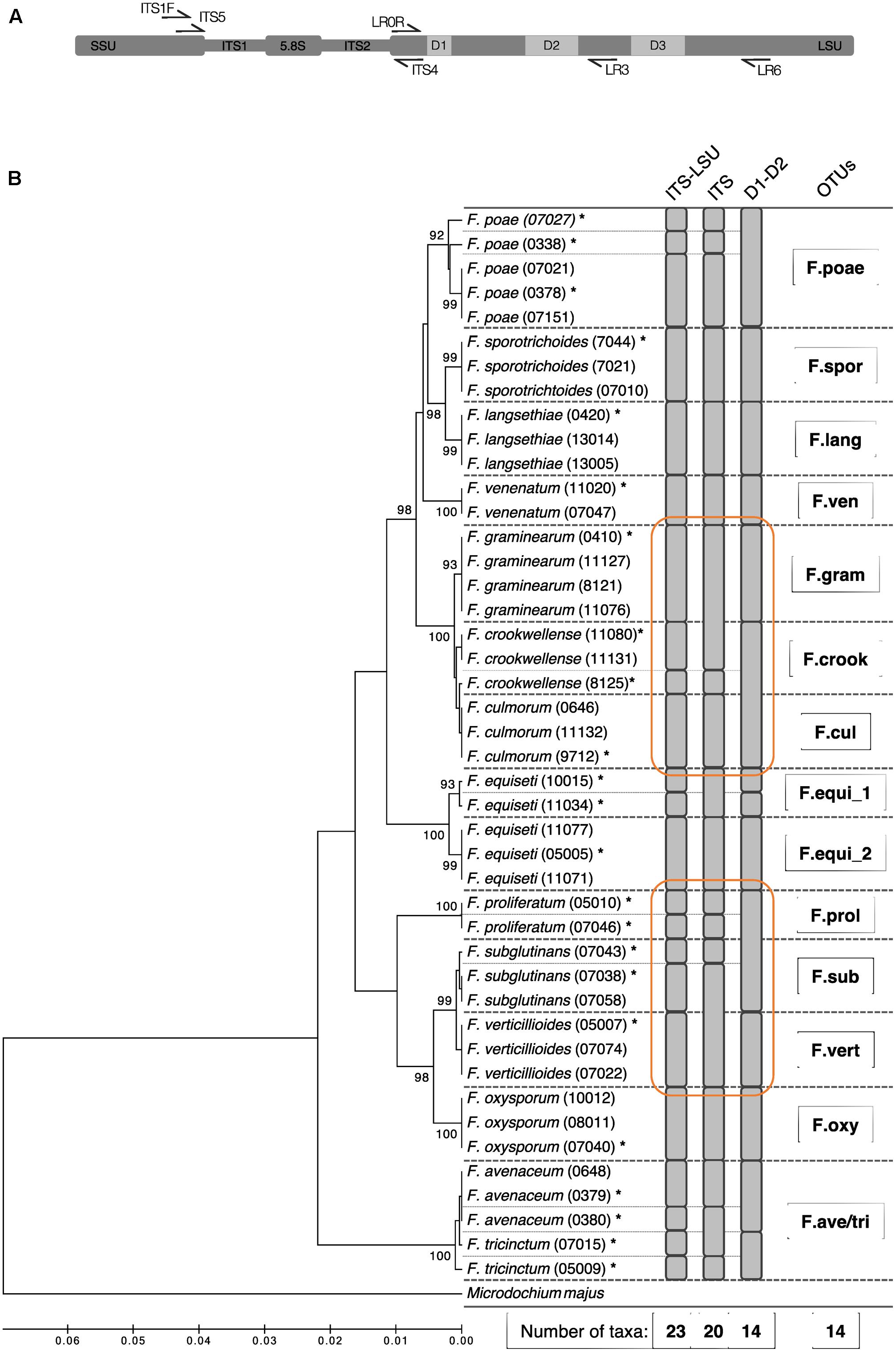
FIGURE 1. Phylogenetic diversity and taxonomic clustering of 44 Swiss Fusarium isolates. (A) Primer binding at the ribosomal operon used to defined different segments: ITS1F and LR6 (used in this study) for the ITS-LSU, ITS5 and ITS4 for the ITS, and LR0R and LR3 for the D1–D2 segment. (B) The tree was constructed using the UPGMA method based on the partial sequences of the ribosomal operon amplified by the primer pair ITS1F and LR6 (∼1.6 kb). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) is shown next to the branches. The distances were computed using the Jukes-Cantor method in MEGA7 (Tamura et al., 2013). Microdochium majus was used as outgroup. Fine dashed lines indicate sequence differences among isolates. Gray bars indicate clusters computed by USEARCH with a similarity threshold of 100% using different segments of the ribosomal operon (ITS-LSU, ITS, LSU). Stars above isolate names mark sequences which were considered for the Fusarium sequence reference dataset. Bold dashed lines indicate isolates binned into the same Fusarium OTU by the method introduced in the present study. Orange circles highlight superior clustering of the ITS-LSU segment compared to ITS and LSU on its own.
Performance of Different Primer Pairs
Initially, different primer pairs were tested for their Fusarium community profiling performance on three different test samples. The three test samples included maize grains infected by Fusarium graminearum, shoot material of winter wheat and a composite sample of co-occurring weeds sampled on the OSCAR field experiment (see below for details). Maize grains were dried with forced air at 32°C for 3 days and ground to a fine powder with a sample mill (Cyclotec, 1093, IG AG, Zurich, Switzerland). Leaf samples were washed after harvest with a PBS-S buffer (130 mM NaCl, 7 mM Na2HPO4, 3 mM NaH2PO4, pH 7.0, 0.02% Tween 20), two times for 20 min on a platform shaker at 120 rpm to remove microbes that are weakly associated. Samples were dried with laboratory grade tissue papers, lyophilized for 48 h and ground in a ball mill (Retsch MM200 mixer mill, Retsch GmbH, Hann, Germany) for 1.5 min with 30 Hz. DNA was extracted from the three different samples using Nucleo spin lysis buffer PL1 for 15 min at 65°C followed by the Nucleo spin plant II extraction kit protocol (Macherey & Nagel, Düren, Germany), and was used as template for PCR with the following primer combinations; ITS1f:LR5, ITS1f:LR6, fITS7:LR5 and fITS7:LR6 (Supplementary Table S2). Amplicons were analyzed with PacBio SMRT sequencing (details are described below).
DNA Extraction, PCR Cycling and SMRT Sequencing for Community Profiling
Fungal or plant material (∼20 mg dry weight) was used for DNA extraction (Nucleo spin plant II extraction kit, Macherey & Nagel, Düren, Germany). The DNA samples were amplified by different primers targeting the ITS region and the 5′-end of the LSU as indicated above. The forward and reverse primers were synthesized with a 5-nucleotide-long padding sequence followed by barcode tags at the 5′-end to allow multiplexing of samples within a single sequencing run. Details of PCR and purification are described in Supplementary Methods. Library preparation and SMRT sequencing were conducted at the Functional Genomics Centre Zurich1 on the PacBio® RS II Instrument (PacBio, San Diego, CA, United States).
Sequence Data Processing
The sequence data processing was mainly conducted following the pipeline established by Schlaeppi et al. (2016) and is described in detail in the Supplementary Methods. In brief, the SMRT Portal was used to extract from the raw data (available from the European Nucleotide Archive, study accession number: PRJEB23144) the circular consensus sequences (CCS) of at least five passes yielding in similar error rates as 454 or MiSeq sequencing platforms (Schlaeppi et al., 2016). Subsequently, the CCS reads were quality filtered in mothur (v.1.35.0; Schloss et al., 2009). Quality reads were demultiplexed based on the barcode-primer sequences using fexbar (Dodt et al., 2012).
QIIME 1.8 (Caporaso et al., 2010) was used for further downstream read processing. De novo chimera detection was performed on quality reads using UCHIME (Edgar et al., 2011). Reads were clustered into OTUs according to the open reference protocol using UCLUST (Edgar, 2010) within QIIME applying a sequence similarity threshold of 98%. This involves first clustering reads against the hand-curated Fusarium reference sequence dataset composed of the 23 unique Fusarium full-length amplicon sequences (see above). The reads clustered to the Fusarium reference dataset were subsequently binned to Fusarium OTUs with a custom R code (Figure 1B; see Supplementary Data). Reads that failed to assign to the reference dataset were clustered de novo into OTUs with UCLUST. OTUs of low abundance (less than 0.1% global abundance and less than 0.5% abundance within a specific sample) were removed from the data set yielding in a data set of abundant community members only. Fusarium and de novo OTUs were classified taxonomically against the UNITE database (Kõljalg et al., 2013) and against the fungal LSU training set of the Ribosomal Database Project (RDP) (Cole et al., 2014). The OTU and taxonomy tables were filtered to exclude OTUs classified as non-fungal. Results of mock communities were used to define optimal settings for quality filtering, demulitplexing and cut-off for low abundant OTUs (Bokulich et al., 2012).
Evaluation of Fusarium Community Profile Performance
The sequence based Fusarium community profiling method was evaluated by three different approaches; (i) by determining artificial Fusarium mock communities, and by comparing sequence-based profiling to the assessment of Fusarium communities with the cultivation-based seed health test (SHT) in different field samples of (ii) Swiss Fusarium monitoring and (iii) of a Fusarium-inoculation field experiment.
Mock communities were composed of ten Fusarium species s.l. and isolates: F. avenaceum (0379), F. crookwellense (0825), F. culmorum (11132), F. equiseti (00505), F. equiseti (10015), F. graminearum (0410), F. langsethiae (0420), F. poae (0338), F. sporotrichioides (07010) and F. verticillioides (05007; Supplementary Table S1). Fusarium isolates were chosen to represent the expected phylogenetic diversity of Fusarium fungi associated to small-grain cereals and maize in Swiss agro-ecosystems (Dorn et al., 2009; Vogelgsang et al., 2011). Additionally, closely related isolates [e.g., F. equiseti (05005) and F. equiseti (10015), or F. crookwellense (0825) and F. culmorum (11132), Figure 1B] were included to test for resolution power. Two different types of mock communities were assembled; (i) all species were added at equal concentrations resulting in the equal mock community, or (ii) species were added in three difference concentration ratios, i.e., F. culmorum (11132) and F. langsethiae (0420) were ten times higher, and F. equiseti (10015) and F. graminearum (0410) were ten times lower concentrated as the remaining six isolates in the staggered mock community.
In SHTs, Fusarium species s.l. from grain samples are determined based on species-specific morphological characteristics (see details in Supplementary Methods). Briefly, 100 surface-sterilized grains were plated on potato dextrose agar, after 7 days of incubation at 19 ± 1°C with a photoperiod of 12 h dark/12 h near-UV light, Fusarium species colonizing the grains were identified using morphological characteristics according to Nelson et al. (1983) and Leslie and Summerell (2008). Results are presented as incidence of each Fusarium species expressed in percentage.
To evaluate the community profile methodology in an experimental environment, we re-assessed Fusarium communities previously profiled with SHT in a winter wheat field experiment where plots were or were not artificially inoculated with F. poae conidia. Treatments consisted of inoculations with a water-based conidial suspension of F. poae with Tween 20® or with Tween 20® alone, serving as the control treatment. Each treatment was replicated four times. Plots were combine-harvested and the grains were passed through a grain cleaning machine. To obtain a representative subsample, grains for the STHs were passed through a mechanical grain divider (detailed information regarding the Fusarium inoculation field experiment is available in the Supplementary Methods).
Finally, we used 15 additional samples of winter wheat, maize and barley to compare the Fusarium communities assessed by SHT to the community profile based on sequencing (Supplementary Table S3). Winter wheat and barley samples originate from commercial samples of Swiss farmers taken in the frame of the Swiss Fusarium monitoring. Maize samples originate from a maize variety trial at Agroscope. Grains of all three crops were passed through a grain-cleaning machine and randomized using a grain divider.
Fusarium Diversity and Distribution along a Wheat-Maize Crop Sequence
The investigations on diversity and distribution of Fusarium along the crop sequence were within an OSCAR field trial during the growing years 2014 and 2015 (details on the OSCAR field trial is available in the Supplementary Methods). The field experiment exhibiting a wheat-maize crop sequence was arranged in a strip-split-plot design with four replicates. On the main plots, two different tillage intensities were applied: (i) conventional inversion tillage by mouldboard plowing (ct) and (ii) no-tillage (nt), both employed before maize sowing in the second year of each experiment. In addition, each main plot was divided into four sub plots where the cover crop treatments were applied: (i) subterranean clover (Trifolium subterraneum) undersown in winter wheat and re-sown after wheat harvest (“clover”), (ii) hairy vetch (Vicia villosa) as legume cover crop (“vetch”), (iii) oilseed radish (Raphanus sativus) as non-legume cover crop (“radish”), and (iv) fallow (“control”).
Winter wheat (Triticum aestivum) was sown either as pure crop (vetch, radish and control cover crop treatments) or intercropped with subterranean clover (clover treatment). After wheat harvest, all three winter cover crops (clover, vetch and radish) were sown. In the next spring (end of April/begin of May), cover crops were either terminated by tillage (ct) or by applying glyphosate in the nt treatment. Maize (Zea mays) was then sowed in the end of May.
For winter wheat residues and cover crop shoots, five subsamples were taken on 16 plots (in a W-scheme) and pooled in April 2015. Cover crop samples were composed of shoot material of the cover species (clover, vetch or radish) or of a composite sample of weeds present on the control plot. All cover crop samples were washed with PBS-S buffer as described above. Residue and cover crop samples were lyophilized for 48 h and ground in a ball mill for 1.5 min with 30 Hz for DNA extraction. Maize grains were harvested in two rows along 7 m length each with an adapted plot-sized combine harvesters. Maize grains were dried at 60°C, grinded and used for DNA extraction. The DNA samples served as templates for PCR with the primer pair ITS1f:LR6 (Supplementary Table S2). Amplicons were subsequently analyzed with SMRT sequencing as described above.
Statistical Analysis
All analyses were performed using R (R Core Team, 2016), and the specific R and Bioconductor packages (as indicated below). Abundance of individual taxa was normalized by the sampling depth of each sample and expressed as percentage relative abundance. The correlation between Fusarium communities determined by sequencing and by SHT was tested using Spearman rank order correlation.
Rarefaction analysis was performed in QIIME on the by abundance filtered OTU table (exported from R for this purpose) and on the original OTU table (Supplementary Figure S1). The analysis of α- and β-diversity along the crop rotation in the OSCAR field trial were performed on rarefied samples using the Bioconductor package phyloseq (McMurdie and Holmes, 2013). Differences in α-diversity measures (OTU richness and Shannon Index) were tested using One- or Two-way ANOVA implementing the factors sample type, cover crop and tillage if applicable for the group of samples (the first only for comparison over all sample types, the latter only for maize grain samples). To quantify the major variance factors of β-diversity along the crop rotation, we performed a multivariate analysis of fungal diversity including a testing for the experimental factors by permutational analysis of variance (PERMANOVA) followed in case of significant effects by a constrained canonical analysis of principal coordinates (CAP; Anderson and Willis, 2003). Finally, a characterization of the taxa responsible for the multivariate patterns based indicator species analysis was performed (De Cáceres and Legendre, 2009). All p-values were adjusted for multiple testing with the FDR correction using the Benjamin–Hochberg method (Benjamini and Hochberg, 1995).
Results
Taxonomic Resolution among Fusarium Species Based on the Ribosomal Operon
In a first step, we evaluated in silico different segments of the ribosomal operon to define the marker region for best possible discrimination among Fusarium isolates at species level. The combined use of ITS and LSU offered a higher resolution compared to single segments (Figure 1B). The ca. 1.6 kb long segment spanning the entire ITS segment and the highly variable segments D1, D2, and D3 of the LSU could separate 23 unique sequences and differentiate 13 out of 14 Fusarium species s.l. tested. Only the species F. avenaceum and F. tricinctum were not discriminated properly and thus clustered into the same Fusarium OTU (referred to as ‘F.ave/tri’). Moreover, we identified two distinct sequence groups in various isolates of F. equiseti that are all classified as one species using morphological characteristics. Overall, the 44 tested Swiss Fusarium isolates clustered into 14 distinct Fusarium OTUs with the new methodology. While the combined ITS-LSU segment discriminated Fusarium species correctly to morphological assessed species boundaries, the single ITS or D1–D2 segments were insufficient to resolve on this level (Figure 1B). The ITS segment failed to separate F. graminearum and F. culmorum, and F. subglutinans and F. verticillioides isolates. The D1–D2 segment failed to discriminate F. crookwellense and F. culmorum, and F. proliferatum and F. subglutinans. Moreover, the ITS segment on its own showed clusters not adherent to morphological assessed species boundaries and thus failed to group all isolates of a single species into the same OTU (e.g., see clustering of F. graminearum and F. crookwellense or of F. subglutinans and F. verticillioides; Figure 1B). Based on these results, we decided to move forward with an amplicon spanning the entire ITS region and highly variable parts of the LSU (D1, D2, and D3) to profile Fusarium communities with the highest resolution based on the ribosomal operon segments considered.
Validation of Fusarium Abundance As Revealed by Community Quantification
We tested four different PCR primer pairs for the development of a sequencing-based method to measure Fusarium communities. We describe the corresponding sequencing effort in Supplementary Table S4 (Library Mthd1). We compared the primers for their specificity to amplify fungi and their coverage of the genus Fusarium on three different sample types; maize grains heavily infected by Fusarium, shoot material of winter wheat and a composite sample of co-occurring weeds. The primer’s specificity to amplify fungal signals was highly dependent on the forward primer and sample origin (Supplementary Figure S2A). In the maize grain and wheat shoot samples, all reads were assigned to the fungal kingdom by both forward primer, however, in the composite weed sample a substantial number of plant sequences were detected. Regardless the reverse primer used, pairs including ITS1F amplified less than 2.5% plant reads, whereas primer pairs including fITS7 amplified up to 44% plant reads (Supplementary Figure S2A). The taxonomic analysis at genus level revealed differences between tested primer pairs in their coverage of the genus Fusarium (dark red; Supplementary Figure S2B). In the weed composite sample, only primer pairs including the reverse primer LR6 were able to detect Fusarium OTUs. Similarly, in the winter wheat sample, only the primer pair ITS1f:LR6 was able to detect Fusarium OTUs (dark red; Supplementary Figure S2B). Overall, the primer pair ITS1f:LR6 showed highest fungal specificity in combination with the best coverage of Fusarium taxa and was therefore chosen for further analysis.
In a next step, we confirmed the method’s technical reproducibility by comparing three replicated profiles of the same sample (see Supplementary Results for details; Supplementary Figure S3) and assessed accuracy in reproducing pre-defined Fusarium mock communities (Figure 2). The artificial mock communities consisted of DNA mixtures of ten Fusarium isolates, which were either assembled in even ratios or with a staggered distribution. In general, the sequencing profiles of the tested mock communities reflected well the expected abundances of the mixed Fusarium (Figure 2). For the even mock community, an average OTU abundance of ten per cent would be expected for each of the ten Fusarium isolates (Figure 2A). The measured mean OTU abundances did not differ more than around twofold from the expected values (rho = 0.7, p = 0.02). The OTU F.gram was consistently overrepresented, whereas the OTUs F.vert, F.poae, F.equi_1, and F.ave/tri tended to be underrepresented. In the staggered mock community, the two most abundant OTUs F.cul and F.lang corresponded to the enriched DNA templates, while only one of the two OTUs (F.equi_2) with a reduced DNA concentration belonged to the two rarest OTUs (rho = 0.61, p = 0.06; Figure 2B). We noted again that the OTU F.gram was overrepresented compared to the expected value and this was also true for the OTU F.crook. Overall, the test revealed a reliable display of the artificial Fusarium communities.
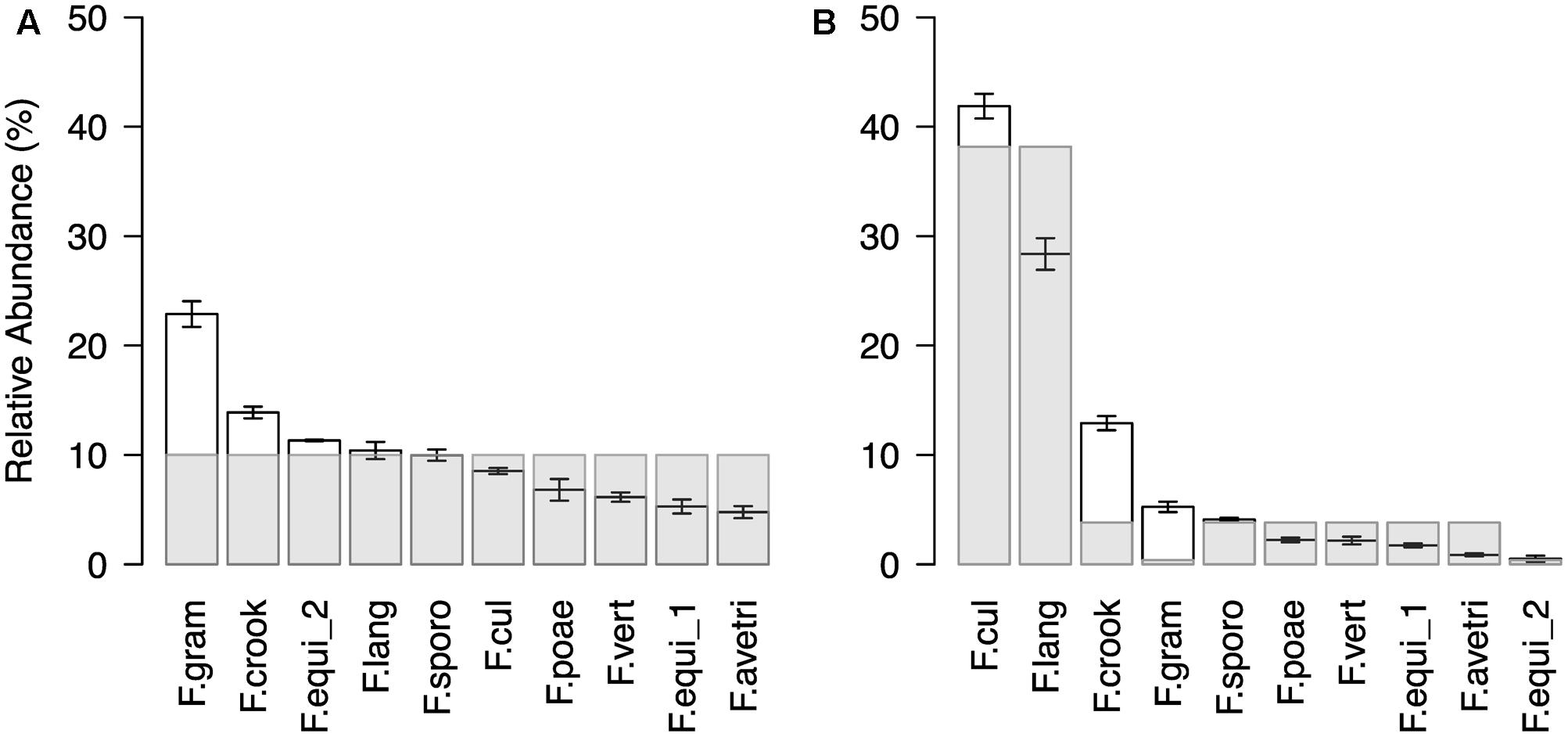
FIGURE 2. Community profile of Fusarium mock communities. Ranked mean OTU abundances (±SE, n = 3) of (A) even and (B) staggered mock communities composed of ten different isolates (F. avenaceum, F. crookwellense, F. culmorum, F. equiseti (two isolates), F. graminearum, F. langsethiae, F. poae, F. sporotrichioides, F. verticillioides; Abbreviation of Fusarium OTUs as described in Figure 1). In the even mock community, the Fusarium isolates were equally represented in the template mixture. In the staggered mock community, the isolate of the species F. culmorum (F.cul) and F. langsethiae (F.lang) were ten times higher concentrated and an isolate of F. equiseti (F.equi_2) and F. graminearum (F.gram) were ten times lower concentrated as the remaining six isolates. Light gray bars indicated expected abundances.
In a further step, we compared the performance of the sequence-based Fusarium profiling with the cultivation-dependent SHT. To this end a total of 23 field samples were assessed with both methods. Eight of these samples originated from a field experiment with wheat, where F. poae was inoculated and we found highly significant correlations between the abundance of Fusarium taxa assessed by the two different methods in control and inoculated treatments (Figures 3A,B). Both methods revealed that in the control treatment F. graminearum was the most abundant Fusarium taxon detected, although its incidence was rather low, and other fungal OTUs showed higher relative abundances (Figure 3A). In the inoculated samples, both methods determined a heavy infection with F. poae as reflected in a high incidence score and high relative abundance of the OTU F.poae (Figure 3B). For the other tested samples, including grain samples from barley, maize and wheat, we also found sound correlations between Fusarium taxa determined by SHT or sequencing with a mean Spearman’s rho of 0.78 ± 0.05 (mean ± SE, n = 15; Figure 3C). Only two of the 15 analyzed monitoring samples exhibited non-significant relationships between the two methods.
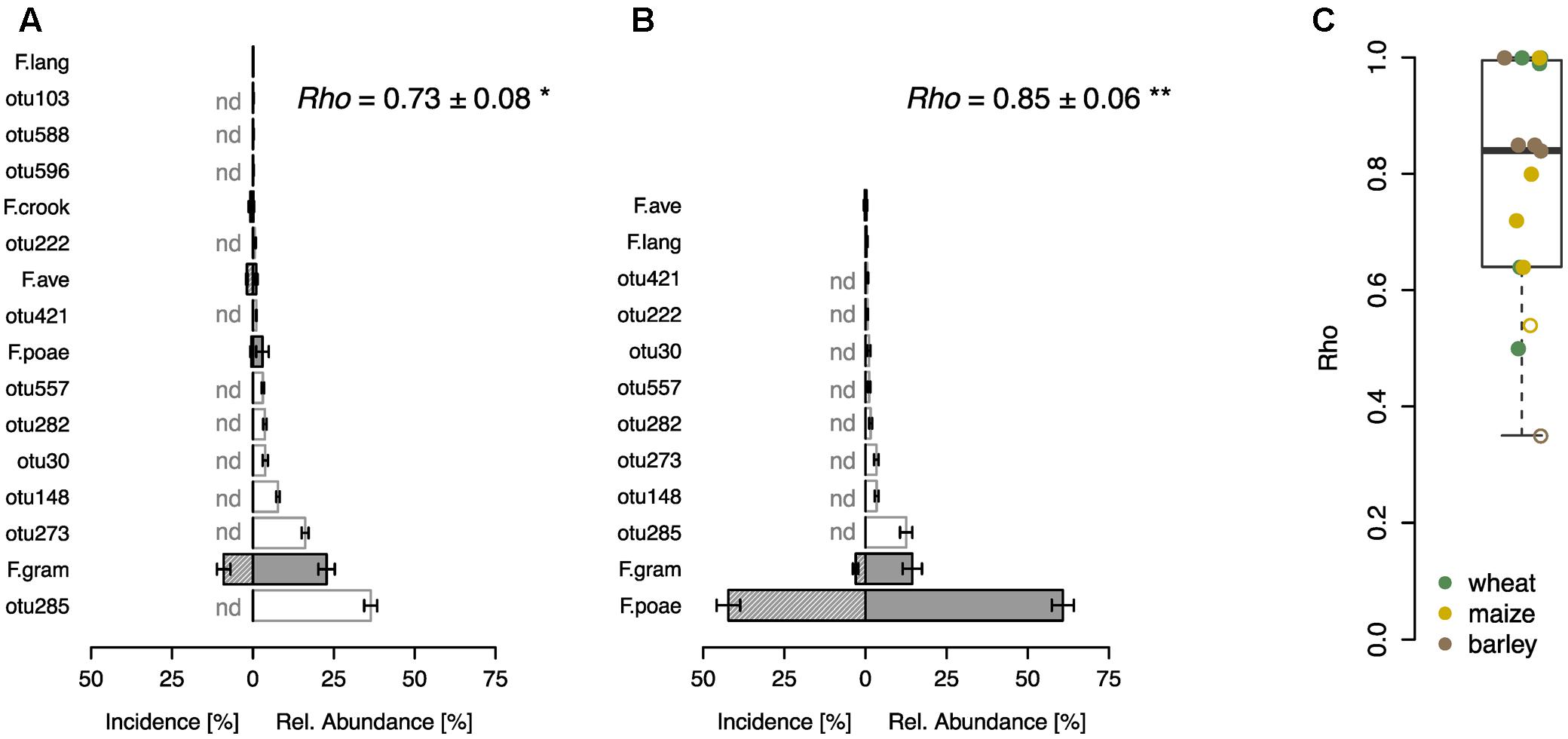
FIGURE 3. Comparison of community profiling based on sequencing and cultivation-based seed health test on different grain samples. Community profiling on a wheat field trial where plots were either not inoculated (A) or inoculated with F. poae (B). Bars represent ranked mean OTU abundances determined by sequencing based community profiling (±SE, n = 4; gray bars indicate Fusarium OTUs abbreviated as described in Figure 1, white bars represent other fungal OTUs). Hatched bars represent incidence of Fusarium species determined by cultivation-based seed health test (±SE, n = 4), other fungal taxa could not be detected (nd). Spearman correlation index (rho-values) are given in the respective upper right corner demonstrating the statistical dependence between the ranking of Fusarium taxa of the two compared methods (∗p < 0.05, ∗∗p < 0.01). (C) Spearman correlation analysis (rho-values) of Fusarium taxa on wheat, maize and barley samples (n = 15). Closed circles indicate a significant correlation (p < 0.05), open circles are not significant.
Fusarium Abundance and Distribution along a Wheat-Maize Crop Sequence
Finally, we applied the newly developed methodology to a field experiment, where we traced the Fusarium community composition in the phyllosphere of different crops along a wheat-maize crop sequence. We investigated how the different cover crop and tillage treatments affected the Fusarium abundance along a wheat-maize sequence to get insights in the dispersal of specific Fusarium taxa (Figure 4A). Fusarium communities were analyzed in the wheat residues and in the different cover crop species before maize was sown, and finally in the grains of the subsequent maize crop. Overall, the relative abundances of Fusarium taxa were low and the fungal communities were dominated by other OTUs (Supplementary Figure S5). In wheat residues and cover crop shoots, only the Fusarium OTU F.ave/tri was detected, whereas in maize nine different Fusarium OTUs were detected of which F.ave/tri, F.prol, F.gram, F.cul, and F.oxy were among the 25 most abundant fungal OTUs (Supplementary Figure S5C). However, no significant difference in Fusarium taxa richness between different treatments (cover crop and tillage) was found within maize grains (data not shown).
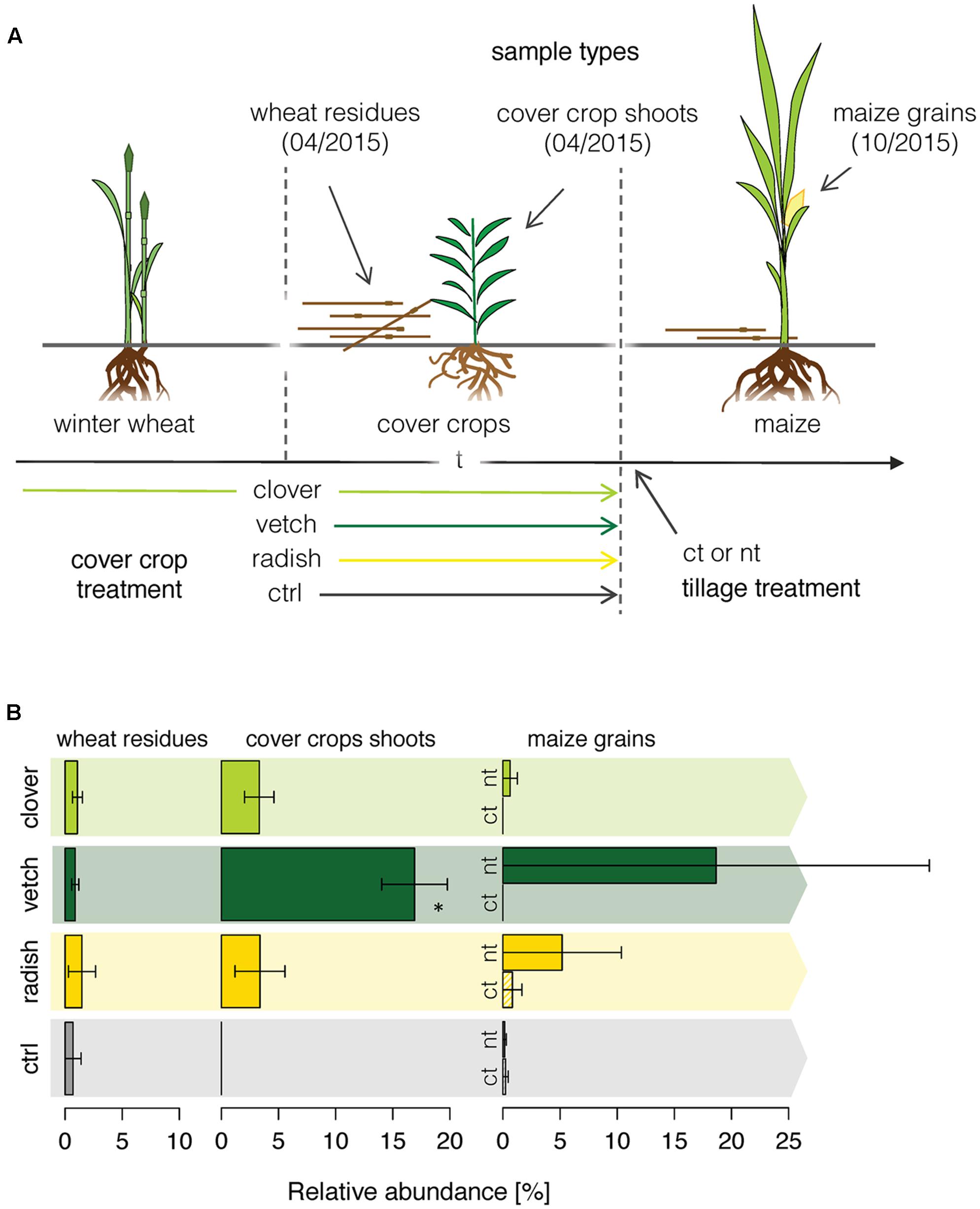
FIGURE 4. OSCAR field experiment investigating the effect of cover crop and tillage treatments on the diversity and dispersal of Fusarium fungi along a wheat-maize crop sequence. (A) Cover crops were applied either as undersown subterranean clover in winter wheat and re-sown after wheat harvest (clover), as leguminous (vetch) or non-leguminous (radish) cover crop both sown after wheat, and green fallow as control (ctrl). Tillage treatments were conventional inversion tillage (ct) and no-tillage (nt) applied before maize in the second year of the experiment. Each treatment was replicated 4 times. (B) Dispersal of the Fusarium OTU F.ave/tri along the crop rotation. Relative abundance of the OTU F.ave/tri (±SE, n = 4) in winter wheat residues, cover crop shoots and maize grains as affected by cover crop and tillage (tillage effect only in maize) treatments. Stars above bars indicate a significant difference within a sample type.
F.ave/tri was the only Fusarium OTU that was detected throughout the wheat-maize crop sequence and thus only its dispersal could be analyzed for effects of cover crop or tillage treatments. While the relative abundance of the OTU F.ave/tri was rather low and similar among all treatments in wheat residues, it was substantially affected by the treatments in cover crops exhibiting a more than eightfold increase in vetch compared to the other treatments (Figure 4B). In maize grains, the abundance of F.ave/tri was highly variable and not even detected in some of the samples, therefore, treatment related patterns could not be statistically confirmed. Nonetheless, means of all no-tillage samples tended to be higher than under plowed conditions, particularly, the combination of no-tillage and vetch as cover crop exhibited the highest mean abundance (Figure 4B).
Fungal Community Dynamics along a Wheat-Maize Crop Sequence
In addition to Fusarium, we assessed the overall composition of fungal communities by means of the universal fungal approach (see Supplementary Results for further information) using SMRT sequencing. We found substantial differences in fungal community composition along the wheat-maize sequence, mainly between the different sample types (wheat residues, cover crop shoots and maize grains; Figure 5A). Further, we investigated the effect of cover crop and tillage treatments (the latter only for maize) on fungal communities separately for the different sample types. In wheat residues collected at the end of cover cropping period, the fungal communities were only slightly affected by cover crop treatments (Figure 5B). In contrast, the analysis of the cover crop shoots revealed species-specific fungal communities in the phyllosphere of different cover crops (Figure 6A). The influence of cover crop treatments to β-diversity was large (CAP revealed 77% of variance is explained by the constrained factor “cover crop treatment”; Figure 6A) and all four treatments harbored significantly different fungal communities based on pair-wise comparison (Supplementary Table S7). Finally, we did not find an effect of the factors cover crop nor tillage to have an influence on the fungal communities in maize grain samples (Figure 5C).
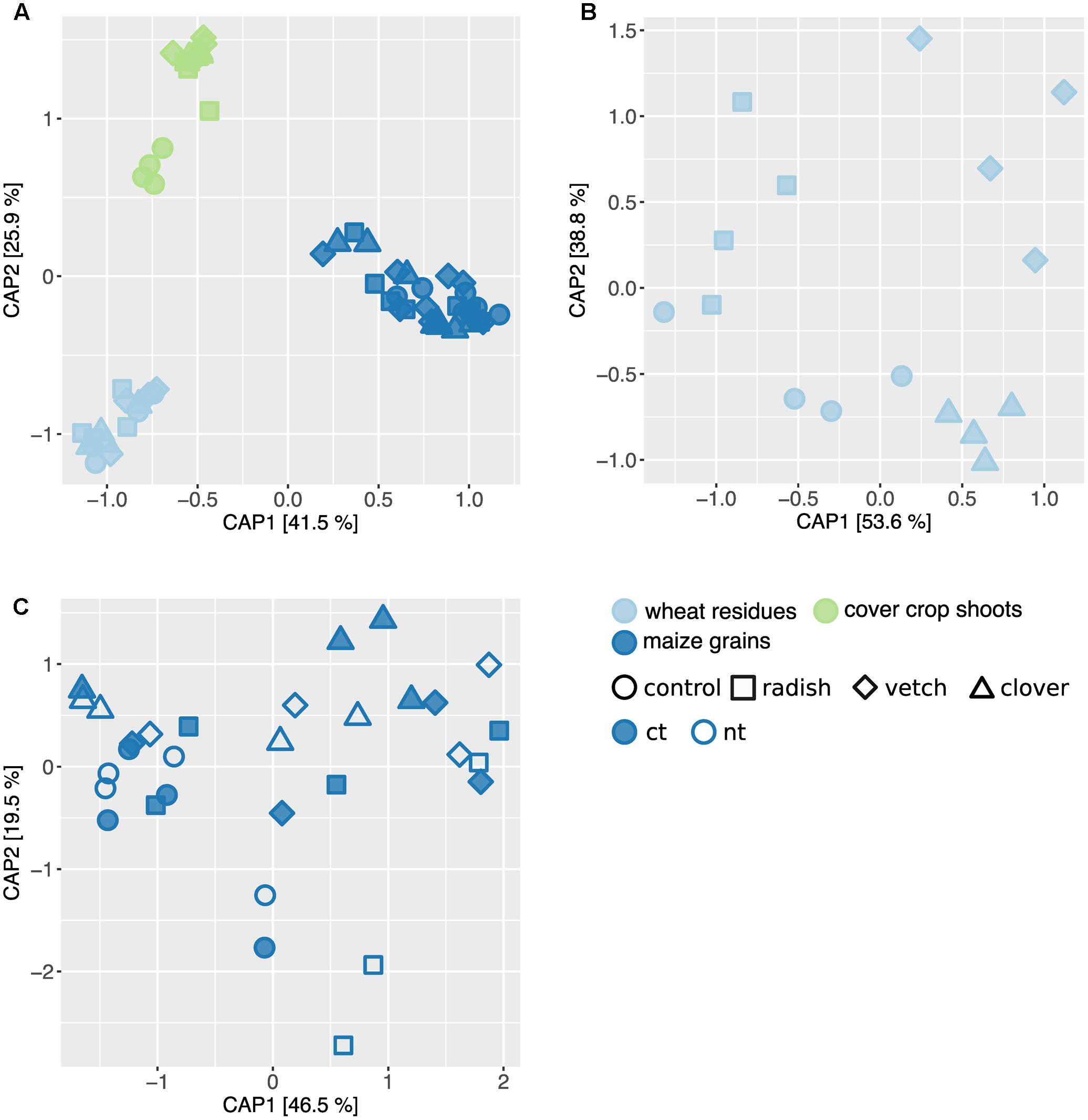
FIGURE 5. Effect of sample type and treatments on fungal community structure along a wheat-maize cropping sequence. CAP ordinations of Bray–Curtis similarities calculated based on relative OTU abundances showing major differences among fungal communities induced by sample types and different treatments. (A) The constrained factors (sample type ∗ cover crop treatment) explain 58% of variance (p = 0.001) between the fungal communities along the cropping sequence. (B) In wheat residues, the constrained factors (cover crop treatment, conditioned for block effect) explain 26% of variance (p = 0.029) between the fungal communities. (C) In maize grains, the constrained factors (cover crop and tillage treatment) explain 23% of variance (p = 0.49) between the fungal communities. Percentages indicated for each axis referring to their contribution to the constrained variation.
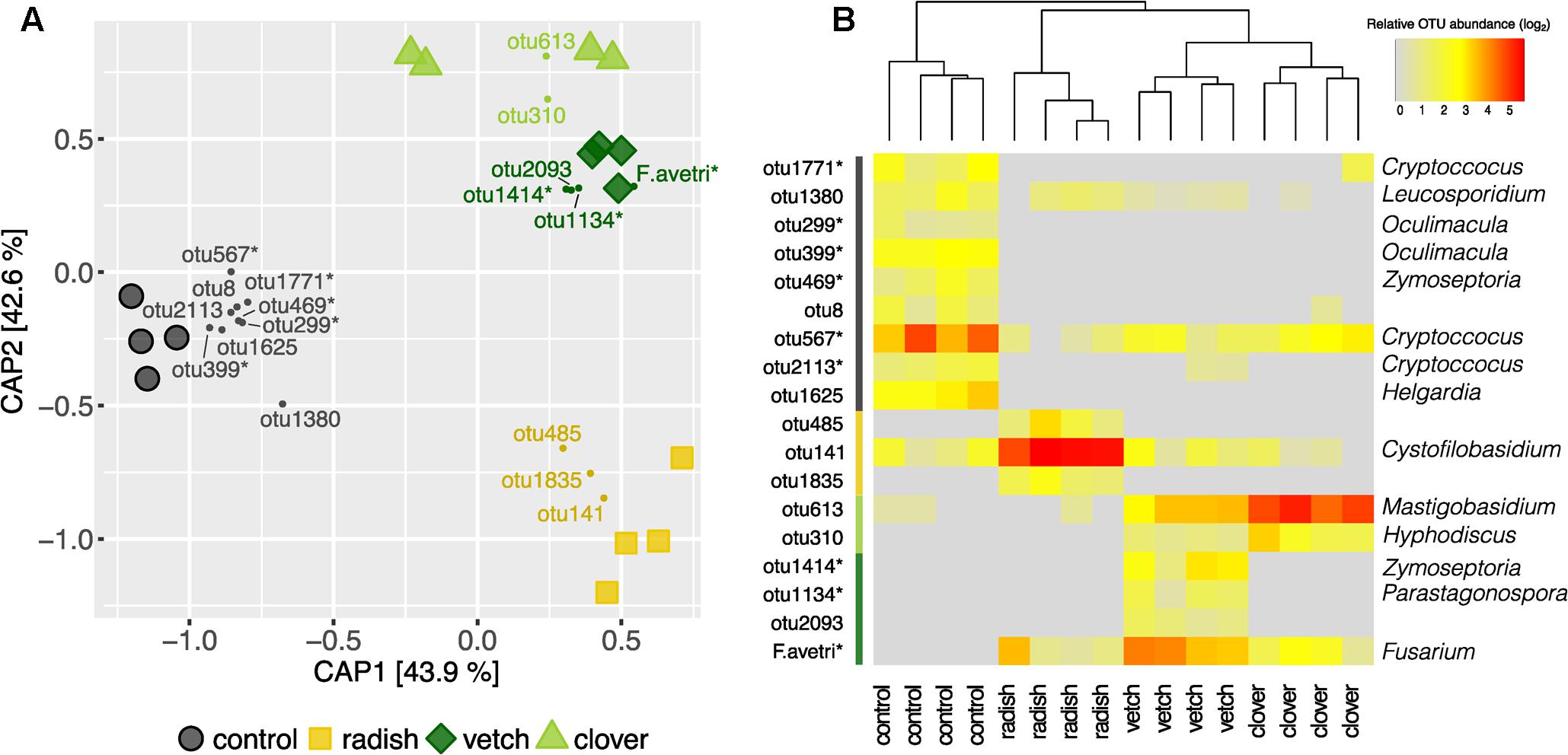
FIGURE 6. Effect of cover crops on its associated fungal communities. (A) CAP ordination of Bray–Curtis similarities calculated based on relative OTU abundances showing major differences among fungal communities induced by different cover crop treatments. The constrained factors (cover crop treatment) explain 77% of variance (p = 0.001) between the fungal communities. Percentages indicated for each axis referring to their contribution to the constrained variation. Cover crop sensitive OTUs are displayed in the corresponding color (clover: light green, vetch: dark green, radish: yellow and control: black). (B) Relative abundances of cover crop sensitive OTUs. Dendrogram is based upon Bray–Curtis distances among fungal communities in cover crop samples. Stars next to OTU names indicate potential plant pathogenic phylogenetical background. Taxonomical assignment (genus) of cover crop sensitive OTUs is indicated on the right if available.
Complementary to the examination of diversity patterns, we also identified treatment-sensitive OTUs based on indicator statistics (see Supplementary Results for further information). However, we found only treatment-sensitive taxa in the cover crop samples (Figure 6B and Table 1). Remarkably, vetch and weeds sampled on the control plots harbored several treatment-sensitive OTUs linked to plant pathogens. One of these is the Fusarium OTU F.ave/tri only characteristically associated to vetch. Moreover, OTUs assigned to the wheat pathogens Zymoseptoria tritici and Parastagonospora nodorum, both causing wheat leaf blotch, were more abundant in vetch samples (Figure 6B and Table 1). In addition, the control plots harbored also several potential plant pathogens including OTUs assigned to Z. tritici and Oculimacula yallundae (Figure 6B and Table 1). Taken together, different cover crop practices exhibited distinct fungal communities of which vetch and weeds on the control plots harbored the highest phytopathological risk.
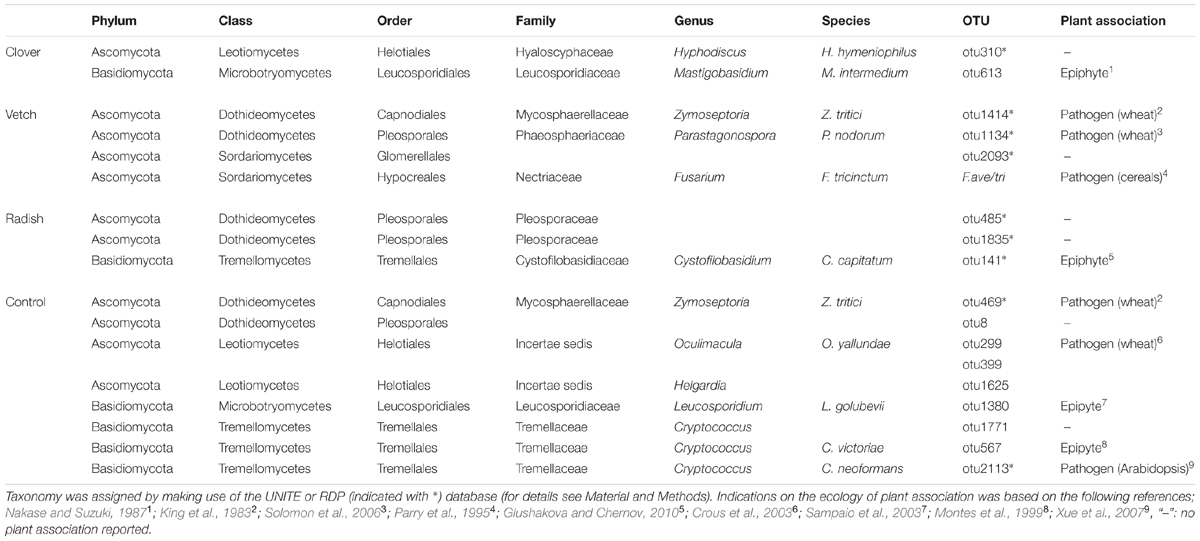
TABLE 1. Taxonomic assignments of treatment sensitive taxa in cover crops.
Discussion
Suitability of the Ribosomal Operon to Discriminate among Fusarium Species
The first objective of this study was the development of a sequencing-based methodology that robustly quantifies Fusarium taxa in shoot and grain samples. In addition, we aimed to develop an approach allowing to focus on Fusarium, but also assessing other fungi associated to crops. By making use of a long amplicon spanning the entire ITS and the 3′-end of the LSU including the highly variable segments D1, D2, and D3 in combination with clustering against a comprehensive reference sequence database, we were able to determine Fusarium beyond the morphological species resolution based on the prime target region for fungal barcoding, the ribosomal operon (Seifert, 2009). Moreover, the present study represents the first attempt to combine the highly variable segments ITS and D1–D3 in a long amplicon for fungal profiling in high resolution, both displaying most favored segments of the ribosomal operon for fungal barcoding (Schoch et al., 2012; Nilsson et al., 2013). The combination of these two segments generates an additive value for fungal community profiling as we could demonstrate for the genus Fusarium. The combined segment allowed to discriminate between the tested Fusarium taxa, while the two segments on their own were less powerful in terms of resolution, and moreover, in terms of species assignments of individual isolates. The D1–D2 segment revealed mainly a poor taxonomic resolution as reported earlier (O’Donnell et al., 2008b). In contrast, the ITS segment on its own revealed weaknesses in clustering isolates to the corresponding Fusarium species. This might be caused by paralogous sequences in the ITS segment known to occur within Fusarium species and making taxonomic assignments based on the ITS segment difficult (Waalwijk et al., 1996; O’Donnell and Cigelnik, 1997).
However, using the sequence combining the ITS and D1–D3 segment, only the two of the tested Fusarium species s.l., F. avenaceum and F. tricinctum, have not been separated properly. This is not surprising as both belong to a large species complex known to be not easily distinguishable with ribosomal tools (Yli-Mattila et al., 2002). In contrast, the species F. equiseti is separated into two clusters based on our analysis. This agrees with previous studies on the phylogeny of F. equiseti revealing high genetic diversity and two distinct clusters within this species (O’Donnell et al., 2009; Marín et al., 2012).
In conclusion, the newly developed methodology is able to identify Fusarium down to morphological species level or even beyond. This ability displays an important feature for a diagnostic tool as conclusions on the phytopathological risk of Fusarium taxa can only be drawn on species level, because pathogenicity as well as mycotoxin production largely differs among Fusarium species and even among isolates (Appel and Gordon, 1996; Bottalico and Perrone, 2002). For instance, among isolates of the species F. graminearum and F. poae, different chemotypes exhibiting distinct mycotoxin profiles have been identified (Jennings et al., 2004; Stenglein, 2009; Stȩpień et al., 2011; Pasquali et al., 2016). Moreover, Marín et al. (2012) found different mycotoxin profiles among two isolate clusters of F. equiseti which may be coherent with the two distinct F. equiseti OTUs we found in the present study (Figure 1B). However, recent development on Fusarium phylogeny has revealed that the morphological species concept misses parts of the phylogenetic diversity of this genus (O’Donnell et al., 2013). Hence, the taxonomical resolution assessed with the methodology introduced in this study might partially underrepresent the Fusarium diversity. Nevertheless, other recent high-throughput approaches did not exceed the taxonomical resolution of our study and also resolve within morphological species boundaries (Karlsson et al., 2016).
Validation of the Diagnostic Power of the Newly Developed Methodology
Besides its resolution, the newly developed methodology showed its diagnostic power in the validation based on mock communities and in the comparison to the assessment of Fusarium communities with the cultivation-based SHT. In the mock communities, we found a twofold difference between the most and the least abundant species in the equal community displaying a similar accuracy as in other sequencing-based Fusarium profiling methods (Karlsson et al., 2016). Particularly, F. graminearum was overrepresented in the mock communities. It is important to note that the mock communities were just equilibrated by mixing equal amounts of genomic DNA, and thus, differences in copy numbers of the ribosomal operon (Rodland and Russell, 1982), and in addition, differences in genome size could have resulted in various ratios of the ribosomal operon to total DNA concentration. For example, the genome of F. graminearum has been estimated to be relatively small compared to F. oxysporum and F. verticillioides (Ma et al., 2010), thus F. graminearum exhibits relative higher number of copies of the genome with equal DNA amount. Overall, however, the quantitative relationship between species was well displayed with the newly developed methodology.
Similarly, the comparison with SHT revealed a high correlation between Fusarium communities assessed with the two different approaches. This is somewhat surprising considering the inherent differences between cultivation- and sequence-based approaches, but also demonstrates the robustness of both methods. More importantly, both approaches were able to detect the epidemic threat of F. poae in the inoculation field experiment, thus demonstrating the usability of both approaches to track taxa-specific FHB causing species in grain samples. In addition, the newly developed methodology allows a perspective beyond Fusarium fungi as other fungal species are also assessed, which can reveal further phytopathological risks as revealed for the OSCAR field experiment in the current study.
A major drawback of the newly developed methodology is its dependency on a suitable reference sequence database to reach highest taxonomical resolution. We established a comprehensive reference sequence database starting with 44 Fusarium isolates collected in Swiss agro-ecosystems. This resulted in a reference sequence database of 23 unique sequences covering a large range of Fusarium species s.l. occurring in Swiss cereal and maize fields (Dorn et al., 2009; Vogelgsang et al., 2011). In the herein presented test case, the OSCAR field experiment, all OTUs assigned to the genus Fusarium were OTUs clustering to the reference sequence database. This illustrates that the Fusarium reference sequence database used covers the major diversity of Fusarium taxa in Swiss cereal and maize fields. Temporarily, the newly developed methodology is relying on hand-curated reference sequence database spanning the ITS and D1–D3. The current fungal sequence databases comprise the ITS (UNITE; Kõljalg et al., 2013), or just the D1-D2 segment alone (RDP; Cole et al., 2014), but there is not yet a sequence database available combining these two potent target regions. Nevertheless, due to the increased use of SMRT sequencing and the improved throughput of Pacific Bioscience with the newly developed ‘SEQUEL’ system2, more reference sequence databases comprising longer sequences will probably be developed in the near future, which will help to overcome the limitation of the newly developed methodology.
Fusarium Diversity and Distribution along the Wheat-Maize Crop Sequence
The focus of the herein introduced methodology lies on Fusarium fungi causing one of the globally most noxious plant diseases, particularly in small grain cereals and maize (Parry et al., 1995; Desjardins, 2003). Overall, the abundance of Fusarium was rather low and other taxa dominated the fungal communities. Only in maize, a considerable Fusarium diversity comprised of nine different species could be detected. This is comparable to the species richness defined in grain samples in another sequence-based Fusarium community study in winter wheat (Karlsson et al., 2017). However, earlier studies have found up to 20 species to be associated with cereals (Xu et al., 2005; Bourdages et al., 2006; Dorn et al., 2009). In contrast to maize, only a single Fusarium OTU, F.ave/tri, was detected in wheat residues and shoots of cover crops and weeds. This very low species richness is unexpected as most of the Fusarium species found in maize grains are also able to colonize crop residues (Munkvold, 2003; Xu and Nicholson, 2009), and have been isolated from a wide range of other plants including different weed species (Jenkinson and Parry, 1994; Lager and Wallenhammar, 2003). Generally, however, it is important to note that the low extent of Fusarium fungi found in the investigated field experiment is probably related to the dry weather in 2014/2015 that resulted in an overall low incidence of FHB in Switzerland (Schöneberg et al., 2016).
The dispersal of only Fusarium OTU F.ave/tri could be analyzed along the crop sequence in this study. This OTU comprises the Fusarium species F. avenaceum, which is along with F. graminearum one of the predominant pathogens of FHB (Parry et al., 1995; Xu and Nicholson, 2009). The abundance of the Fusarium OTU F.ave/tri revealed interesting effects of cover crop and tillage treatments on its dispersal along the crop sequence. While its abundance was not affected by any treatment in wheat residues, a substantial higher abundance of F.ave/tri was found in vetch compared with the other cover crop species and weeds of the control treatment. This interesting pattern was less explicit in maize grains, where the abundance of F.ave/tri was highly variable. Nonetheless, the reported mean abundances of F.ave/tri in maize indicate that the cover crop vetch in combination with no tillage has the potential to increase the dispersal along the crop sequence. This finding is supported by an earlier study showing that F. avenaceum was more frequently associated with cereals in sequence with leguminous crops under reduced tillage (Fernandez et al., 2007). It is generally known that prevalence of Fusarium is greatest for no-tillage and reduced under conventional tillage (Bateman et al., 2007; Dill-Macky, 2008). Up to now, this was mainly attributed to the fact that most of Fusarium species depend on proliferation on crop residues, which serve as inoculum for the following crop (Xu and Nicholson, 2009). However, we demonstrate here that certain cover crops act as alternative hosts of Fusarium and hence could represent another inoculum source for FHB in a crop sequence.
Distinct Fungal Communities along a Wheat-Maize Crop Sequence
The application of the newly developed methodology to track crop-associated fungi along a wheat-maize sequence depicted at first sight that the three sample types, wheat residues, cover crop shoots and maize grains, harbored structurally highly distinct fungal communities. This is not surprising as the sample types represented different substrates, including plant debris and living plants, and additionally originate from different plant species. It has been broadly shown that the species of a plant displays a major driver shaping microbial communities inhabiting its phyllosphere (Redford et al., 2010; Rastogi et al., 2012; Kembel and Mueller, 2014). Moreover, the herein reported impact of sample type has also a temporal component as the sample types have not been harvested at the same time. Temporal effects have also shown to significantly affect microbial community structures in agro-ecosystems (Lukow et al., 2000; Lauber et al., 2013).
While we found substantial differences between sampling types, the impact of the different cropping measures on fungal communities was not apparent throughout the crop sequence. The cover crop treatment was only a significant driver of fungal community structure in cover crop shoot samples. It is important to note that in the sample type cover crop shoots, different plant species have been sampled in the different treatments, which can be a major driver of the microbial community structure of the phyllosphere as we just have observed (see above). The fungal communities of the leguminous cover crops, clover and vetch, clustered closer together compared to the other two treatments hosting phylogenetical more distant plant species. This finding is supported by earlier studies indicating that phylogenetic distance of host plants contributes to diversification of associated microbial communities (Leff and Fierer, 2013; Schlaeppi et al., 2014).
Although we could report substantial differences in the fungal communities of cover crops, this had no impact on the fungal communities in the subsequent crop maize. Fungal communities of maize grains were not affected by cover crop or tillage treatments. Hence, we must conclude that the distinct fungal communities associated to cover crops have not been spread or established in the subsequent maize under the investigated condition and that the phyllosphere of previous crops may not be the main source for the recruitment of the phyllosphere community of the current crop. Indeed, so far it is still unclear how microbial communities in the phyllosphere establish. Further studies have to elucidate how the recruitment via other plants, soil or air contribute to the establishment of phyllosphere communities (Vorholt, 2012).
Phytopathological Risks of Different Cover Crop Treatments
Several cover crop treatment-sensitive fungal taxa were identified. The fungal communities of vetch and weeds sampled in the control treatment were characterized by several OTUs assigned to plant pathogens, while clover and radish treatments did harbor commensal plant epiphytes. Particularly vetch revealed a high potential to act as alternative host for noxious plant pathogens featuring besides the Fusarium OTU F.ave/tri also an OTU assigned to Zymoseptoria tritici, a major fungal plant pathogen (Dean et al., 2012). The weed species of the control treatment hosted also plant pathogens, particularly of wheat (Z. tritici and Oculimacula yallundae). It is important to note that weed samples also include volunteer wheat regrown on the control plots (up to 30% of surface cover; data not shown), which may explain the prominent role of wheat pathogens in the weed sample. In summary, the distinct fungal communities found in cover crops feature diverging phytopathological risks for a crop sequence by acting as alternative host of noxious plant pathogens. However, the phytopathological risks found in cover crops were not apparent in the subsequent crop maize of the current experiment (see above).
Conclusion
We tested and developed a new sequencing based methodology for fungal community profiling that allows to detect fungal pathogens from the genus Fusarium beyond the morphological species level based on the highly variable segments of the ribosomal operon. We demonstrate that the herein introduced method is a powerful diagnostic tool to track Fusarium communities in combination with other fungal pathogens and non-pathogenic fungi associated to crop species. By analyzing a field experiment, we revealed that cover crops differ in their potential to act as alternative hosts for Fusarium, and moreover to host further fungal plant pathogens including Z. tritici. Hence, the choice of cover crops with respect to their phytopathological risk displays an important feature to improve the control of FER and FHB epidemics along a crop sequence.
Author Contributions
FW and MvdH designed the study. SV contributed selected Fusarium isolates and information of the respective cropping histories. FW and RW collected the data. RW was responsible for the OSCAR field experiment. FW and AH performed the molecular work. FW and KS analyzed the data. FW wrote the manuscript and all co-authors contributed substantially to revisions.
Funding
This work was supported by Agroscope and the EU FP7 project OSCAR (no. 289277).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Irene Bänziger, Eveline Jenny, Tomke Musa, and Torsten Schöneberg for providing Fusarium cultures and the assessment of Fusarium incidence by SHT. Special thanks are going to Andrea Patrignani and Giancarlo Russo for running SMRT sequencing including initial read processing at the Functional Genomic Center Zürich. They also thank Kyle Hartman for helpful comments on statistical analysis. Finally, they want to acknowledge Maria Finckh and Peter Baresel, the coordinators of the EU project OSCAR, for their great support and giving us the opportunity to conduct the study presented herein.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2017.02019/full#supplementary-material
Footnotes
References
Agler, M. T., Ruhe, J., Kroll, S., Morhenn, C., Kim, S. T., Weigel, D., et al. (2016). Microbial hub taxa link host and abiotic factors to plant microbiome mariation. PLOS Biol. 14:e1002352. doi: 10.1371/journal.pbio.1002352
Anderson, M. J., and Willis, T. J. (2003). Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology 84, 511–525. doi: 10.1890/0012-9658(2003)084[0511:CAOPCA]2.0.CO;2
Appel, D. J., and Gordon, T. R. (1996). Relationships among pathogenic and nonpathogenic isolates of Fusarium oxysporum based on the partial sequence of the intergenic spacer region of the ribosomal DNA. Mol. Plant Microbe Interact. 9, 125–138. doi: 10.1094/MPMI-9-0125
Balajee, S. A., Borman, A. M., Brandt, M. E., Cano, J., Cuenca-Estrella, M., Dannaoui, E., et al. (2009). Sequence-based identification of Aspergillus, Fusarium, and Mucorales species in the clinical mycology laboratory: where are we and where should we go from here? J. Clin. Microbiol. 47, 877–884. doi: 10.1128/JCM.01685-08
Bateman, G. L., Gutteridge, R. J., Gherbawy, Y., Thomsett, M. A., and Nicholson, P. (2007). Infection of stem bases and grains of winter wheat by Fusarium culmorum and F. graminearum and effects of tillage method and maize-stalk residues. Plant Pathol. 56, 604–615. doi: 10.1111/j.1365-3059.2007.01577.x
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300.
Berendsen, R. L., Pieterse, C. M. J., and Bakker, P. A. H. M. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2012). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Bottalico, A., and Perrone, G. (2002). Toxigenic Fusarium species and mycotoxins associated with head blight in small-grain cereals in Europe. Eur. J. Plant Pathol. 108, 611–624. doi: 10.1023/A:1020635214971
Bourdages, J. V., Marchand, S., Belzile, F. J., and Rioux, S. (2006). Diversity and prevalence of Fusarium species from Quebec barley fields. Can. J. Plant Pathol. 28, 419–425. doi: 10.1080/07060660609507315
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cole, J. R., Wang, Q., Fish, J. A., Chai, B., McGarrell, D. M., Sun, Y., et al. (2014). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, 633–642. doi: 10.1093/nar/gkt1244
Crous, P. W., Groenewald, J. Z. E., and Gams, W. (2003). Eyespot of cereals revisited: ITS phylogeny reveals new species relationships. Eur. J. Plant Pathol. 109, 841–850. doi: 10.1023/A:1026111030426
De Cáceres, M., and Legendre, P. (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. doi: 10.1890/08-1823.1
Dean, R., Van Kan, J. A. L., Pretorius, Z. A., Hammond-Kosack, K. E., Di Pietro, A., Spanu, P. D., et al. (2012). The top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13, 414–430. doi: 10.1111/j.1364-3703.2011.00783.x
Desjardins, A. E. (2003). Giberella from (Avenaceae) to (Zeae). Annu. Rev. Phytopathol. 41, 177–198. doi: 10.1146/annurev.phyto.41.011703.115501
Dill-Macky, R. (2008). Cultural control practices for Fusarium head blight: problems and solutions. Cereal Res. Commun. 36, 653–657. doi: 10.1556/CRC.36.2008.Suppl.B.55
Dill-Macky, R., and Jones, R. K. (2000). The effect of previous crop residues and tillage on Fusarium head blight of wheat. Plant Dis. 84, 71–76. doi: 10.1094/PDIS.2000.84.1.71
Dodt, M., Roehr, J. T., Ahmed, R., and Dieterich, C. (2012). FLEXBAR- Flexible barcode and adapter processing for next-generation sequencing platforms. Biology 1, 895–905. doi: 10.3390/biology1030895
Dorn, B., Forrer, H. R., Jenny, E., Wettstein, F. E., Bucheli, T. D., and Vogelgsang, S. (2011). Fusarium species complex and mycotoxins in grain maize from maize hybrid trials and from grower’s fields. J. Appl. Microbiol. 111, 693–706. doi: 10.1111/j.1365-2672.2011.05091.x
Dorn, B., Forrer, H. R., Schürch, S., and Vogelgsang, S. (2009). Fusarium species complex on maize in Switzerland: occurrence, prevalence, impact and mycotoxins in commercial hybrids under natural infection. Eur. J. Plant Pathol. 125, 51–61. doi: 10.1007/s10658-009-9457-8
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Edwards, S. G. (2004). Influence of agricultural practices on Fusarium infection of cereals and subsequent contamination of grain by trichothecene mycotoxins. Toxicol. Lett. 153, 29–35. doi: 10.1016/j.toxlet.2004.04.022
Edwards, S. G., Imathiu, S. M., Ray, R. V., Back, M., and Hare, M. C. (2012). Molecular studies to identify the Fusarium species responsible for HT-2 and T-2 mycotoxins in UK oats. Int. J. Food Microbiol. 156, 168–175. doi: 10.1016/j.ijfoodmicro.2012.03.020
Eren, A. M., Maignien, L., Sul, W. J., Murphy, L. G., Grim, S. L., Morrison, H. G., et al. (2013). Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. Methods Ecol. Evol. 4, 1111–1119. doi: 10.1111/2041-210X.12114
Fernandez, M. R., Ulrich, D., Sproule, L., Brandt, S. A., Thomas, A. G., Olfert, O., et al. (2007). “Impact of crop management systems on diseases of spring wheat on the Canadian Prairies,” in Wheat Production in Stressed Environments, eds H. T. Buck, J. E. Nisi, and N. Salomon (Berlin: Springer), 265–271.
Franzén, O., Hu, J., Bao, X., Itzkowitz, S. H., Peter, I., and Bashir, A. (2015). Improved OTU-picking using long-read 16S rRNA gene amplicon sequencing and generic hierarchical clustering. Microbiome 3:43. doi: 10.1186/s40168-015-0105-6
Friberg, H., Edel-Hermann, V., Faivre, C., Gautheron, N., Fayolle, L., Faloya, V., et al. (2009). Cause and duration of mustard incorporation effects on soil-borne plant pathogenic fungi. Soil Biol. Biochem. 41, 2075–2084. doi: 10.1016/j.soilbio.2009.07.017
Gao, X., Jackson, T. A., Lambert, K. N., Li, S., and Hartman, G. L. (2004). Detection and quantification of Fusarium solani f. sp. glycines in soybean roots with real-time quantitative polymerase chain reaction. Plant Dis. 88, 1372–1380. doi: 10.1094/PDIS.2004.88.12.1372
Glushakova, A. M., and Chernov, I. Y. (2010). Seasonal dynamics of the structure of epiphytic yeast communities. Microbiology 79, 830–839. doi: 10.1134/S0026261710060160
Guadet, J., Julien, J., Lafay, J. F., and Brygoo, Y. (1989). Phylogeny of some Fusarium species, as determined by large-subunit rRNA sequence comparison. Mol. Biol. Evol. 6, 227–242.
Hennequin, C., Abachin, E., Symoens, F., Lavarde, V., Reboux, G., Nolard, N., et al. (1999). Identification of Fusarium species involved in human infections by 28S rRNA gene sequencing. J. Clin. Microbiol. 37, 3586–3589.
Hogg, A. C., Johnston, R. H., and Dyer, A. T. (2007). Applying real-time quantitative PCR to Fusarium crown rot of wheat. Plant Dis. 91, 1021–1028. doi: 10.1094/PDIS-91-8-1021
Infantino, A., Santori, A., and Shah, D. A. (2012). Community structure of the Fusarium complex on wheat seed in Italy. Eur. J. Plant Pathol. 132, 499–510. doi: 10.1007/s10658-011-9892-1
Jenkinson, P., and Parry, D. W. (1994). Isolation of Fusarium species from common broad-leaved weeds and their pathogenicity to winter wheat. Mycol. Res. 98, 776–780. doi: 10.1016/S0953-7562(09)81054-X
Jennings, P., Coates, M. E., Walsh, K., Turner, J. A., and Nicholson, P. (2004). Determination of deoxynivalenol-and nivalenol-producing chemotypes of Fusarium graminearum isolated from wheat crops in England and Wales. Plant Pathol. 53, 643–652. doi: 10.1111/j.0032-0862.2004.01061.x
Karlsson, I., Edel-Hermann, V., Gautheron, N., Durling, M. B., Kolseth, A. K., Steinberg, C., et al. (2016). Genus-specific primers for study of Fusarium communities in field samples. Appl. Environ. Microbiol. 82, 491–501. doi: 10.1128/AEM.02748-15
Karlsson, I., Friberg, H., Kolseth, A.-K., Steinberg, C., and Persson, P. (2017). Agricultural factors affecting Fusarium communities in wheat kernels. Int. J. Food Microbiol. 252, 53–60. doi: 10.1016/j.ijfoodmicro.2017.04.011
Kembel, S. W., and Mueller, R. C. (2014). Plant traits and taxonomy drive host associations in tropical phyllosphere fungal communities. Botany 92, 303–311. doi: 10.1139/cjb-2013-0194
King, J. E., Cook, R. J., and Melville, S. C. (1983). A review of Septoria diseases of wheat and barley. Ann. Appl. Biol. 103, 345–373. doi: 10.1111/j.1744-7348.1983.tb02773.x
Kõljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F. S., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Lager, J., and Wallenhammar, A.-C. (2003). Crop loss from soil-borne pathogens in white clover stands assessed by chemical treatments. J. Plant Dis. Prot. 110, 120–128.
Lauber, C. L., Ramirez, K. S., Aanderud, Z., Lennon, J., and Fierer, N. (2013). Temporal variability in soil microbial communities across land-use types. ISME J. 7, 1641–1650. doi: 10.1038/ismej.2013.50
Leff, J. W., and Fierer, N. (2013). Bacterial communities associated with the surfaces of fresh fruits and vegetables. PLOS ONE 8:e59310. doi: 10.1371/journal.pone.0059310
Leslie, J., and Bowden, R. (2008). Fusarium graminearum: when species concepts collide. Cereal Res. Commun. 36, 609–615. doi: 10.1556/CRC.36.2008.Suppl.B.50
Leslie, J. F., Anderson, L. L., Bowden, R. L., and Lee, Y. W. (2007). Inter- and intra-specific genetic variation in Fusarium. Int. J. Food Microbiol. 119, 25–32. doi: 10.1016/j.ijfoodmicro.2007.07.059
Leslie, J. F., and Summerell, B. A. (2008). The Fusarium Laboratory Manual. Hoboken, NJ: John Wiley & Sons.
Lindahl, B. D., Nilsson, R. H., Tedersoo, L., Abarenkov, K., Carlsen, T., Kjøller, R., et al. (2013). Fungal community analysis by high-throughput sequencing of amplified markers-a user’s guide. New Phytol. 199, 288–299. doi: 10.1111/nph.12243
Lukow, T., Dunfield, P., and Liesack, W. (2000). Use of the T-RFLP technique to assess spatial and temporal changes in the bacterial community structure within an agricultural soil planted with transgenic. FEMS Microbiol. Ecol. 32, 241–247. doi: 10.1111/j.1574-6941.2000.tb00717.x/full
Ma, L.-J., van der Does, H. C., Borkovich, K. A., Coleman, J. J., Daboussi, M.-J., Di Pietro, A., et al. (2010). Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464, 367–373. doi: 10.1038/nature08850
Marín, P., Moretti, A., Ritieni, A., Jurado, M., Vázquez, C., and González-Jaén, M. T. (2012). Phylogenetic analyses and toxigenic profiles of Fusarium equiseti and Fusarium acuminatum isolated from cereals from Southern Europe. Food Microbiol. 31, 229–237. doi: 10.1016/j.fm.2012.03.014
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLOS ONE 8:e61217. doi: 10.1371/journal.pone.0061217
Montes, M. J., Belloch, C., Galiana, M., Garcia, M. D., Andrés, C., Ferrer, S., et al. (1999). Polyphasic taxonomy of a novel yeast isolated from antarctic environment; description of Cryptococcus victoriae sp. nov. Syst. Appl. Microbiol. 22, 97–105. doi: 10.1016/S0723-2020(99)80032-0
Munkvold, G. P. (2003). Epidemiology of Fusarium diseases and their mycotoxins in maize ears. Eur. J. Plant Pathol. 109, 705–713. doi: 10.1023/A:1026078324268
Nakase, T., and Suzuki, M. (1987). Studies on ballistospore-forming dead leaves of Miscanthus sinensis of the new species Sporobolomyces miscanthi, Sporobolomyces subroseus, and Sporobolomyces weijmani. J. Gen. Appl. Microbiol. 33, 177–196. doi: 10.2323/jgam.33.177
Nelson, P. E., Toussoun, T. A., and Marasas, W. F. O. (1983). Fusarium Species: An Illustrated Manual for Identification. University Park, PA: Penn State University Press.
Nganje, W. E., Bangsund, D. A., Leistritz, F. L., Wilson, W. W., and Tiapo, N. M. (2004). Regional economic impacts of Fusarium head blight in wheat and barley. Rev. Agric. Econ. 26, 332–347. doi: 10.1111/j.1467-9353.2004.00183.x
Nilsson, R. H., Tedersoo, L., Abarenkov, K., Carlsen, T., Pennanen, T., and Stenlid, J. (2013). Methods fungal community analysis by high-throughput sequencing of amplified markers – a user’s guide. New Phytol. 199, 288–299. doi: 10.1111/nph.12243
O’Donnell, K. O., and Cigelnik, E. (1997). Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous. Mol. Phylogenet. Evol. 7, 103–116. doi: 10.1006/mpev.1996.0376
O’Donnell, K. O., Cigelnik, E., and Nirenberg, H. I. (1998). Molecular systematics and phylogeography of the Gibberella fujikuroi species complex. Mycologia 90, 465–493. doi: 10.2307/3761407
O’Donnell, K. O., Rooney, A. P., Proctor, R. H., Brown, D. W., McCormick, S. P., Ward, T. J., et al. (2013). Phylogenetic analyses of RPB1 and RPB2 support a middle Cretaceous origin for a clade comprising all agriculturally and medically important fusaria. Fungal Genet. Biol. 52, 20–31. doi: 10.1016/j.fgb.2012.12.004
O’Donnell, K. O., Sutton, D. A., Fothergill, A., McCarthy, D., Rinaldi, M. G., Brandt, M. E., et al. (2008a). Molecular phylogenetic diversity, multilocus haplotype nomenclature, and in vitro antifungal resistance within the Fusarium solani species complex. J. Clin. Microbiol. 46, 2477–2490. doi: 10.1128/JCM.02371-07
O’Donnell, K. O., Sutton, D. A., Rinaldi, M. G., Gueidan, C., Crous, P. W., and Geiser, D. M. (2009). Novel multilocus sequence typing scheme reveals high genetic diversity of human pathogenic members of the Fusarium incarnatum-F. equiseti and F. chlamydosporum species complexes within the United States. J. Clin. Microbiol. 47, 3851–3861. doi: 10.1128/JCM.01616-09
O’Donnell, K. O., Ward, T. J., Aberra, D., Kistler, H. C., Aoki, T., Orwig, N., et al. (2008b). Multilocus genotyping and molecular phylogenetics resolve a novel head blight pathogen within the Fusarium graminearum species complex from Ethiopia. Fungal Genet. Biol. 45, 1514–1522. doi: 10.1016/j.fgb.2008.09.002
O’Donnell, K. O., Ward, T. J., Robert, V. A. R. G., Crous, P. W., Geiser, D. M., and Kang, S. (2015). DNA sequence-based identification of Fusarium: current status and future directions. Phytoparasitica 43, 583–595. doi: 10.1007/s12600-015-0484-z
Parry, D. W., Jenkinson, P., and McLeod, L. (1995). Fusarium ear blight (scab) in small grain cereals - a review. Plant Pathol. 44, 207–238. doi: 10.1111/j.1365-3059.1995.tb02773.x
Pasquali, M., Beyer, M., Logrieco, A., Audenaert, K., Balmas, V., Basler, R., et al. (2016). A European database of Fusarium graminearum and F. culmorum trichothecene genotypes. Front. Microbiol. 7:406. doi: 10.3389/fmicb.2016.00406
R Core Team (2016). R: A Language and Environment for Statistical Computing. Available at: https://www.r-project.org/
Rastogi, G., Sbodio, A., Tech, J. J., Suslow, T. V., Coaker, G. L., and Leveau, J. H. J. (2012). Leaf microbiota in an agroecosystem: spatiotemporal variation in bacterial community composition on field-grown lettuce. ISME J. 6, 1812–1822. doi: 10.1038/ismej.2012.32
Reddy, K., Salleh, B., Saad, B., Abbas, H., Abel, C., and Shier, W. (2010). An overview of mycotoxin contamination in foods and its implications for human health. Toxin Rev. 29, 3–26. doi: 10.3109/15569541003598553
Redford, A. J., Bowers, R. M., Knight, R., Linhart, Y., and Fierer, N. (2010). The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ. Microbiol. 12, 2885–2893. doi: 10.1111/j.1462-2920.2010.02258.x
Rodland, K. D., and Russell, P. J. (1982). Regulation of ribosomal RNA cistron number in a strain of Neurospora crassa with a duplication of the nucleolus organizer region. Biochim. Biophys. Acta 697, 162–169. doi: 10.1016/0167-4781(82)90072-0
Sampaio, J. P., Gadanho, M., Bauer, R., and Weiß, M. (2003). Taxonomic studies in the Microbotryomycetidae: Leucosporidium golubevii sp. nov., Leucosporidiella gen. nov. and the new orders Leucosporidiales and Sporidiobolales. Mycol. Prog. 2, 53–68. doi: 10.1007/s11557-006-0044-5
Santamaria, M., Vicario, S., Pappadà, G., Scioscia, G., Scazzocchio, C., and Saccone, C. (2009). Towards barcode markers in Fungi: an intron map of Ascomycota mitochondria. BMC Bioinformatics 10(Suppl. 6):S15. doi: 10.1186/1471-2105-10-S6-S15
Schlaeppi, K., Bender, S. F., Mascher, F., Russo, G., Patrignani, A., Camenzind, T., et al. (2016). High-resolution community profiling of arbuscular mycorrhizal fungi. New Phytol. 212, 780–791. doi: 10.1111/nph.14070
Schlaeppi, K., Dombrowski, N., Oter, R. G., Ver Loren van Themaat, E., and Schulze-Lefert, P. (2014). Quantitative divergence of the bacterial root microbiota in Arabidopsis thaliana relatives. Proc. Natl. Acad. Sci. U.S.A. 111, 585–592. doi: 10.1073/pnas.1321597111
Schloss, P. D., Jenior, M. L., Koumpouras, C. C., Westcott, S. L., and Highlander, S. K. (2016). Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 4:e1869. doi: 10.7717/peerj.1869
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U.S.A. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Schöneberg, T., Martin, C., Wettstein, F. E., Bucheli, T. D., Mascher, F., Bertossa, M., et al. (2016). Fusarium and mycotoxin spectra in Swiss barley are affected by various cropping techniques. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 33, 1608–1619. doi: 10.1080/19440049.2016.1219071
Seifert, K. A. (2009). Progress towards DNA barcoding of fungi. Mol. Ecol. Resour. 9, 83–89. doi: 10.1111/j.1755-0998.2009.02635.x
Singer, E., Bushnell, B., Coleman-Derr, D., Bowman, B., Bowers, R. M., Levy, A., et al. (2016). High-resolution phylogenetic microbial community profiling. ISME J. 10, 2020–2032. doi: 10.1038/ismej.2015.249
Solomon, P. S., Lowe, R. G. T., Tan, K., Waters, O. D. C., and Oliver, R. P. (2006). Stagonospora nodorum: cause of Stagonospora nodorum blotch of wheat. Mol. Plant Pathol. 7, 147–156. doi: 10.1111/j.1364-3703.2006.00326.x
Stenglein, S. A. (2009). Fusarium poae: a pathogen that needs more attention. J. Plant Pathol. 91, 25–36.
Stȩpień,Ł, Koczyk, G., and Waśkiewicz, A. (2011). Genetic and phenotypic variation of Fusarium proliferatum isolates from different host species. J. Appl. Genet. 52, 487–496. doi: 10.1007/s13353-011-0059-8
Summerell, B. A., Laurence, M. H., Liew, E. C. Y., and Leslie, J. F. (2010). Biogeography and phylogeography of Fusarium: a review. Fungal Divers 44, 3–13. doi: 10.1007/s13225-010-0060-2
Summerell, B. A., and Leslie, J. F. (2011). Fifty years of Fusarium: how could nine species have ever been enough? Fungal Divers 50, 135–144. doi: 10.1007/s13225-011-0132-y
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tedersoo, L., Tooming-Klunderud, A., and Anslan, S. (2017). PacBio metabarcoding of Fungi and other eukaryotes: errors, biases and perspectives. New Phytol. doi: 10.1111/nph.14776 [Epub ahead of print].
Uhlig, S., Jestoi, M., Knutsen, A. K., and Heier, B. T. (2006). Multiple regression analysis as a tool for the identification of relations between semi-quantitative LC-MS data and cytotoxicity of extracts of the fungus Fusarium avenaceum (syn. F. arthrosporioides). Toxicon 48, 567–579. doi: 10.1016/j.toxicon.2006.07.007
Vogelgsang, S., Hecker, A., Musa, T., Dorn, B., and Forrer, H. R. (2011). On-farm experiments over 5 years in a grain maize/winter wheat rotation: effect of maize residue treatments on Fusarium graminearum infection and deoxynivalenol contamination in wheat. Mycotoxin Res. 27, 81–96. doi: 10.1007/s12550-010-0079-y
Vogelgsang, S., Jenny, E., Hecker, A., Baenziger, I., and Forrer, H.-R. (2009). Fusaria and mycotoxins in wheat-monitoring of harvest samples from growers’ fields. Agrarforschung 16, 238–243.
Vorholt, J. A. (2012). Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828–840. doi: 10.1038/nrmicro2910
Waalwijk, C., Koning, J. R. A., de Baayen, R. P., and Gams, W. (1996). Discordant groupings of Fusarium spp. from sections Elegans, Liseola and Dlaminia based on Ribosomal ITS1 and ITS2 Sequences. Mycologia 88, 361–368. doi: 10.2307/3760877
Waalwijk, C., Van Der Heide, R., De Vries, I., Van Der Lee, T., Schoen, C., Costrel-de Corainville, G., et al. (2004). Quantitative detection of Fusarium species in wheat using TaqMan. Eur. J. Plant Pathol. 110, 481–494. doi: 10.1023/B:EJPP.0000032387.52385.13
Watanabe, M. (2013). Molecular phylogeny and identification of Fusarium species based on nucleotide sequences. Mycotoxins 63, 133–142. doi: 10.2520/myco.63.133
Wittwer, R., Dorn, B., Jossi, W., and Van Der Heijden, M. G. A. (2017). Cover crops support ecological intensification of arable cropping systems. Sci. Rep. 7:41911. doi: 10.1038/srep41911
Xu, X., and Nicholson, P. (2009). Community ecology of fungal pathogens causing wheat head blight. Annu. Rev. Phytopathol. 47, 83–103. doi: 10.1146/annurev-phyto-080508-081737
Xu, X. M., Parry, D. W., Nicholson, P., Thomsett, M. A., Simpson, D., Edwards, S. G., et al. (2005). Predominance and association of pathogenic fungi causing Fusarium ear blightin wheat in four European countries. Eur. J. Plant Pathol. 112, 143–154. doi: 10.1007/s10658-005-2446-7
Xue, C., Tada, Y., Dong, X., and Heitman, J. (2007). The human hungal pathogen Cryptococcus can complete its sexual cycle during a pathogenic association with plants. Cell Host Microbe 1, 263–273. doi: 10.1016/j.chom.2007.05.005
Yli-Mattila, T., Mach, R. L., Alekhina, I. A., Bulat, S. A., Koskinen, S., Kullnig-Gradinger, C. M., et al. (2004). Phylogenetic relationship of Fusarium langsethiae to Fusarium poae and Fusarium sporotrichioides as inferred by IGS, ITS, β-tubulin sequences and UP-PCR hybridization analysis. Int. J. Food Microbiol. 95, 267–285. doi: 10.1016/j.ijfoodmicro.2003.12.006
Yli-Mattila, T., Paavanen-Huhtala, S., Bulat, S. A., Alekhina, I. A., and Nirenberg, H. I. (2002). Molecular, morphological and phylogenetic analysis of the Fusarium avenaceum/F. arthrosporioides/F. tricinctum species complex – a polyphasic approach. Mycol. Res. 106, 655–669. doi: 10.1017/S0953756202006020
Keywords: Fusarium community analysis, plant-associated fungi, single molecule real-time (SMRT) sequencing, PacBio, Mycobiome
Citation: Walder F, Schlaeppi K, Wittwer R, Held AY, Vogelgsang S and van der Heijden MGA (2017) Community Profiling of Fusarium in Combination with Other Plant-Associated Fungi in Different Crop Species Using SMRT Sequencing. Front. Plant Sci. 8:2019. doi: 10.3389/fpls.2017.02019
Received: 06 August 2017; Accepted: 13 November 2017;
Published: 28 November 2017.
Edited by:
Stéphane Hacquard, Max Planck Institute for Plant Breeding Research (MPG), GermanyReviewed by:
Jose Gaspar Maciá-Vicente, Goethe University Frankfurt, GermanySurya Saha, Boyce Thompson Institute, United States
Copyright © 2017 Walder, Schlaeppi, Wittwer, Held, Vogelgsang and van der Heijden. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Florian Walder, ZmxvcmlhbndhbGRlckBnbXguY2g=