Abstract
Epigenetic mechanisms, including histone modifications and DNA methylation, mutually regulate chromatin structure, maintain genome integrity, and affect gene expression and transposon mobility. Variations in DNA methylation within plant populations, as well as methylation in response to internal and external factors, are of increasing interest, especially in the crop research field. Methylation Sensitive Amplification Polymorphism (MSAP) is one of the most commonly used methods for assessing DNA methylation changes in plants. This method involves gel-based visualization of PCR fragments from selectively amplified DNA that are cleaved using methylation-sensitive restriction enzymes. In this study, we developed and validated a new method based on the conventional MSAP approach called Methylation Sensitive Amplification Polymorphism Sequencing (MSAP-Seq). We improved the MSAP-based approach by replacing the conventional separation of amplicons on polyacrylamide gels with direct, high-throughput sequencing using Next Generation Sequencing (NGS) and automated data analysis. MSAP-Seq allows for global sequence-based identification of changes in DNA methylation. This technique was validated in Hordeum vulgare. However, MSAP-Seq can be straightforwardly implemented in different plant species, including crops with large, complex and highly repetitive genomes. The incorporation of high-throughput sequencing into MSAP-Seq enables parallel and direct analysis of DNA methylation in hundreds of thousands of sites across the genome. MSAP-Seq provides direct genomic localization of changes and enables quantitative evaluation. We have shown that the MSAP-Seq method specifically targets gene-containing regions and that a single analysis can cover three-quarters of all genes in large genomes. Moreover, MSAP-Seq's simplicity, cost effectiveness, and high-multiplexing capability make this method highly affordable. Therefore, MSAP-Seq can be used for DNA methylation analysis in crop plants with large and complex genomes.
Introduction
DNA methylation is an epigenetic mechanism that influences gene expression, transposon mobility and genome integrity. Additionally, methylation affects the chromatin structure and controls its condensation in the nucleus (Richards and Elgin, 2002). Epigenetic modifications are generally defined as stable changes that do not involve DNA sequence alteration but result from the addition of specific chemical substituents to nucleotide bases in DNA or histone protein tails (Berger et al., 2009). DNA methylation is a modification resulting from the covalent addition of a methyl group to the fifth position of the aromatic ring in cytosine. The addition and removal of DNA methylation can be induced by both internal and external stimuli (Meyer, 2015).
In plants, DNA methylation occurs commonly within three sequence contexts: CG, CHG and CHH (where H is A, C, or T); however, it varies depending on the level and pattern found within different genomic and intergenic regions. The methylome of the model plant Arabidopsis thaliana has been extensively studied, although its DNA methylation levels and patterns are not shared by all plants. This phenomenon is observed because A. thaliana has a small genome size with low repetitive element content in addition to dissimilarities within methylating/demethylating enzymes (Kapazoglou et al., 2013; Yamauchi et al., 2014). One study observed that 24% of CG, 6.7% of CHG and 1.7% of CHH sequences were methylated in A. thaliana using bisulfite treatments coupled with global next generation sequencing (Cokus et al., 2008). Alternatively, in the Oryza sativa genome, which is three times larger, 59% of CG, 21% of CHG, and 2.2% of CHH sequences were methylated (Feng et al., 2010). Additionally, in the 20 times larger Zea mays genome, 86% of CG, 74% of CHG, and 5.4% of CHH sequences were methylated (Gent et al., 2013).
Many techniques have been developed to analyze DNA methylation and its alterations and can be classified as general or specific. General DNA methylation analyses, such as the popular HPLC or ELISA-based methods, determine the total level of DNA methylation within a genome, whereas specific methods identify regional changes within short sequences or even particular cytosines. Specific DNA methylation analyses are divided into three types: (1) methods based on antibodies or specific proteins that exhibit affinity to methylcytosine; (2) methods that apply methyl-sensitive restriction enzymes, and (3) methods involving the treatment of DNA with sodium bisulfite, which converts unmethylated cytosine to uracil (Schmitz and Zhang, 2011; Ji et al., 2015). Randomly fragmented DNA that was subjected to immunoprecipitation (mCIP—methyl-CpG-immunoprecipitation) coupled with tiling arrays was used for the first genome-wide analysis of DNA methylation in A. thaliana (Zhang et al., 2006). This method allows for an enrichment of highly methylated regions; however, it does not identify the methylation status of particular cytosines. Moreover, it was shown that mCIP is biased toward heavily methylated regions, especially those containing CHG methylation (Lister et al., 2008). In 2008, the first complete A. thaliana methylomes were published and served as a reference for all plant species (Cokus et al., 2008; Lister et al., 2008). Analyses were based on large-scale bisulfite sequencing (methylC-seq), which is currently the most advanced, direct and specific approach. MethylC-seq allows for the identification of the level and pattern of methylation of specific cytosines within the whole genome. Nevertheless, methylC-seq is feasible only in model species that have small and simple genomes and low repetitive element content. This technique is also costly, especially for species with genomes that are significantly larger than Arabidopsis or rice. Moreover, because crop cereals have large, repetitive genomes with high levels of DNA methylation in all sequence contexts (CG, CHG, and CHH), data analysis is challenging. For species with large and complex genomes (typical for many crops), indirect methods to analyze DNA methylation are commonly used due to their simplicity and cost-effectiveness. Among the currently available techniques used to determine DNA methylation modulation, Methylation Sensitive Amplification Polymorphism (MSAP) is the most widely used. MSAP is a modification of the Amplified Fragment Length Polymorphism technique (AFLP; Vos et al., 1995) and utilizes cleavage with the methylation-sensitive restriction enzymes HpaII or MspI, followed by adapter ligation, amplification, and further gel-based visualization (Reyna-Lopez et al., 1997; Xiong et al., 1999). The cleavage capacities of HpaII and MspI are strongly affected by the methylation state of the external and internal cytosine residues within the recognized 5′-CCGG-3′ sequences. Thus, the methylation state can be determined for specific bands based on the ability of each enzyme to cleave the restriction site. MSAP-based analyses can be performed for a range of species regardless of their genome size and reference genome availability. The MSAP method was established in 1997 (Reyna-Lopez et al., 1997) and has been effective in many global analyses of DNA methylation in various plant species (Xiong et al., 1999; Peraza-Echeverria et al., 2001; Chakrabarty et al., 2003; Portis et al., 2004; Filek et al., 2006; Salmon et al., 2008; Tan, 2010; Wang et al., 2011; Guzy-Wrobelska et al., 2013; Marconi et al., 2013; Tang et al., 2014). This method is still widely used in model and non-model plants (A. thaliana—Li et al., 2015; Xu et al., 2015; Allium sativum—Gimenez et al., 2016; Brassica napus—Gautam et al., 2016; Gossypium hirsutum—Wang et al., 2016; Malus × Domestica—Kumar et al., 2016; O. sativa—Li et al., 2017; Vicia faba—Abid et al., 2017). Aside from its simplicity and usefulness, MSAP only provides a general overview of the methylation state and does not provide a specific sequence context.
In the present study, we aimed to develop a simple, high-throughput, and low-cost method for the direct identification of specific genomic sequences that undergo DNA methylation in plants with large genomes. We have designed and introduced a novel technique called Methylation Sensitive Amplification Polymorphism Sequencing (MSAP-Seq) for the analysis of DNA methylation patterns in Hordeum vulgare (Chwialkowska et al., 2016). This method is based on the conventional MSAP analysis, which was greatly improved by replacing the conventional separation of MSAP amplicons on polyacrylamide gels with direct high-throughput sequencing using Next Generation Sequencing (NGS) and automated data analysis. MSAP-Seq allows for the global and direct identification of a large set of sequences that undergo DNA methylation changes without laborious band excisions, reamplification, and subcloning, which are required for MSAP analysis. MSAP-Seq is a simple method that allows for the parallel identification of hundreds of thousands of sites at a low cost. The complexity of the assay is reduced by subsampling only the specific sites that are cut by restrictases and amplified with selective primers. In contrast to the expensive and complex MethylC-seq analysis, our method is well-suited for analyses with large sample sets. Because MSAP-Seq relies on methylation-sensitive restriction enzymes that recognize CCGG sites, only changes within these regions are identified. Therefore, other sequence contexts, which might also undergo important changes, are not detected. Consequently, the major limitation of MSAP-Seq is that it provides only a general overview of DNA methylation modulation within selected CG sites. We have optimized the MSAP-Seq method for the popular Illumina next generation sequencing platform. To complement the MSAP-Seq methodology, we developed the MSEQER software for automated MSAP-Seq data analysis (Korotko, 2017—personal communication). Reads are mapped to the reference genome to identify specific genomic sequences and their features. Changes in DNA methylation that are identified with MSAP-Seq are characterized qualitatively (as in conventional MSAP), based on the presence or absence of each amplicon, as well as quantitatively, by deep sequencing of the MSAP-Seq amplicons, which provides the fold change values of the abundance of normalized reads. MSAP-Seq can be used by researchers that utilize the traditional MSAP analysis. Our previous experiments with MSAP as well as MSAP-Seq revealed their comparability (Chwialkowska et al., 2016) and affirmed that MSAP-Seq can be reproducibly used in studies that are designed for traditional MSAP. MSAP-Seq can be easily applied to different plant species, even those with large and complex genomes, since (1) MSAP-Seq targets gene-rich regions and the most functionally important region of the methylome, due to restriction enzymes that recognize CCGG sequences, which are more frequent in genic than other genomic regions; (2) Application of selective primers enables the adjustment of the number of generated fragments and, consequently, for the selective analysis of a smaller portion of the methylome. This feature makes the method affordable and applicable for different experimental layouts in a wide variety of species, regardless of genome size. (3) The methodology is based on the widely used (especially in crops) MSAP technique, which has been successful in various model and non-model plants. Therefore, MSAP-Seq may prove valuable in other methylome analyses, such as estimating the influence of different factors on DNA methylation or methylome diversity analyses among different genotypes or populations.
Materials and methods
Plant material
We collected tissues from roots and second leaves of H. vulgare cv. “Karat” seedlings from three time points during water-deficiency treatment under strictly controlled conditions (described in detail in Chwialkowska et al., 2016). Plants were grown in a greenhouse (20/18°C; 16/8 h photoperiod) in pots filled with a mixture of clay and sand (7:2 ratio) with known physicochemical properties. The soil moisture was measured daily in each pot using a Time-domain Reflectometer EasyTest (Institute of Agrophysics, Polish Academy of Sciences, Poland) and water was applied to maintain a 12% volumetric water content (vwc) during the control phase (time point 1), which included the first 10 days after the pre-germinated seeds were sown in pots. On day 11, the soil moisture was decreased by withholding irrigation to attain 1.5–2% vwc (drought-stressed plants, time point 2). When the soil moisture of the water-stressed plants reached 3% vwc, plants were moved to a growth chamber with a temperature regime of 25 and 20°C during the day and night, respectively. Water deficiency treatment was maintained for 10 d. Afterwards, the plants were put back into the greenhouse and normal irrigation (12% vwc) was applied for 14 d (re-watering phase, time point 3). The detached tissue samples were immediately frozen in liquid nitrogen and stored at −80°C for DNA isolation.
DNA isolation
Genomic DNA was isolated using the micro C-TAB procedure from Doyle and Doyle (1987). The yield and purity of the DNA samples were determined using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, USA). The integrity of the gDNA was evaluated using agarose gel electrophoresis.
Identification of differentially methylated sites (DMS) using MSAP-Seq
Five hundred nanograms of genomic DNA was cut with 2.5 U of the frequent-cutting methylation sensitive restriction enzyme HpaII (New England Biolabs, Ipswich, USA) and 2.5 U of the rare-cutting EcoRI (New England Biolabs, Ipswich, USA) in a 20 μL reaction with 1x NEB1 buffer (New England Biolabs, Ipswich, USA) at 37°C for 6 h. Enzymes were inactivated at 80°C for 20 min. Next, 12 μL of ligation mixture containing 60 pmol of HpaII-related adapter, 6 pmol of EcoRI-related adapter (Table 1), 1x T4 ligase buffer (Thermo Scientific, Waltham, USA) and 1.2 U of T4 DNA ligase (Thermo Scientific, Waltham, USA) were added. This mixture was incubated at 37°C for 16 h. After that step, 5 μL of the ligation reaction was used for selective PCR amplification in a 51 μL reaction containing 75 ng of primers E-AC, 75 ng of primers H-TG (Table 1), 10 pmol of dNTPs, 5 U of Dynazyme II polymerase and 5 μL of dedicated 10x buffer (Thermo Scientific, Waltham, USA). PCR was performed under the following conditions: 30 s denaturation at 94°C, 40 s of annealing at 56°C and 50 s of extension at 72°C, for 30 cycles. Ten microliters of the amplification reaction was run on a 1.5% agarose gel. The remaining 40 μL of amplification reaction was purified using 1.8x Agencourt AMPure XP (Beckman Coulter, Brea, USA) and eluted into 50 μL 1x TE. To create tags, amplicons were fragmented with Bioruptor Plus (Diagenode, Liège, Belgium) using 10 cycles of 30 s ON followed by 30 s OFF under low power to obtain fragments with mean sizes of 300 bp. Fragmented tags were purified with 1.8x Agencourt AMPure XP (Beckman Coulter, Brea, USA) and eluted into 35 μL ddH2O.
Table 1
| Type | Name | Primer/oligo sequence |
|---|---|---|
| Adapter EcoRI | EcoRI_ A1 | CTCGTAGACTGCGTACC |
| EcoRI_ A2 | AATTGGTACGCAGTCTAC | |
| Adapter HpaII | HpaII_A1 | GACGATGAGTCTAGAA |
| HpaII_A2 | CGTTCTAGACTCATC | |
| PCR primer EcoRI-AC | E-AC | GACTGCGTACCAATTCAC |
| PCR primer HpaII-TG | H-TG | GATGAGTCTAGAACGGTG |
The sequences of the primers and adapters that were used for the MSAP-Seq analyses.
Sequencing libraries were subsequently prepared using the NEXTflex Rapid DNA-Seq Kit (BIOO Scientific, Austin, USA) according to the manufacturer's instructions with minor changes (option 1). Briefly, 32 μL of fragmented amplicons were added to 15 μL of end repair and adenylation buffer and 3 μL of end repair and adenylation enzyme mix. This mixture was incubated on a thermocycler under the following conditions: 22°C for 20 min, 72°C for 20 min and then 4°C on pause. Next, the barcoded adapters were ligated by adding 47.5 μL of ligase enzyme mix and 2.5 μL of undiluted DNA barcode adapter to 50 μL of end-repaired and adenylated DNA fragments. Reactions were incubated on a thermocycler for 15 min at 22°C. The ligation products were purified twice with Agencourt AMPure XP (Beckman Coulter, Brea, USA); first with 0.6x beads and an elution into 52 μL of resuspension buffer, and second using 50 μL of sample with 0.8x beads and an elution into 22 μL of resuspension buffer. Prepared fragments were subjected to PCR amplification in a total volume of 50 μL, including 5 μL of sample, 12 μL of dedicated PCR master mix and 2 μL of primer mix. PCR was performed in a thermocycler under the following conditions: initial denaturation at 98°C followed by 6 cycles of 98°C for 30 s, 65°C for 30 s and 72°C for 60 s, with a final elongation at 72°C for 4 min and then 4°C on pause. Finally, amplicons were purified with 0.8x Agencourt AMPure XP (Beckman Coulter, Brea, USA) and eluted into 21 μL of resuspension buffer. The quality of the prepared Illumina libraries was analyzed using Agilent Bioanalyzer and the Agilent High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, USA), and the quantities were estimated using a Qubit Fluorometer (Thermo Fisher Scientific, Waltham, USA). For cluster generation, the libraries were diluted to 15 pM and were sequenced using the Illumina HiSeq 1500 system (Illumina, San Diego, USA) with 24 barcoded samples per lane.
MSAP-Seq data analysis
Sequencing read processing and differential methylation analysis was performed using the automatic pipeline MSEQER (available at: http://mseqer.us.edu.pl). Reads were filtered based on the presence of the HpaII-related adapter. Only reads with the HpaII-related adapter were retained for further analysis. Next, reads were trimmed using BBDuk software, and those with a CGG sequence on the 3′ or 5′ in addition to a minimum length of 50 bp were retained. Reads were then mapped to the H. vulgare genome version ASM32608v1.31 using Bowtie2 (Langmead and Salzberg, 2012). Reads that mapped to the same CCGG location in the genome were counted and normalized using RPM (reads per million). Reads were annotated using information from the Ensembl Plants database (Yates et al., 2016) and classified into four categories: genes [classified from transcription start site (TSS) to the end, including exons and introns], putative promoters (1,000 bp upstream of the TSS), repeat regions and intergenic regions (without annotation in the genome used in the study). Then, reads were annotated functionally based on gene ontology (GO). Hierarchical clustering was performed to enable sample comparison. Differences in the levels of methylation among the samples were calculated as fold change values for normalized read counts. Statistical analysis was performed using DESeq2 software (Love et al., 2014). Data were gathered and further processed using Microsoft Excel 2016. The raw MSAP-Seq data were deposited into the SRA repository under the accession numbers PRJNA407808 and PRJNA407754, which correspond to datasets from case study 1 and 2, respectively.
Quantitative validation using single-locus DNA methylation assay—methylation sensitive restriction enzyme qPCR (MSRE-qPCR)
Equal amounts of genomic DNA (1.25 μg) were digested with 12.5 U of HpaII in 1x NEB1 buffer (New England Biolabs, Ipswich, USA) at 37°C overnight. The enzyme was inactivated at 80°C for 20 min. Mock (undigested) samples were treated simultaneously without the addition of restriction enzyme. All digests were performed in duplicate. After cleavage, samples were diluted by 5x and used as templates for qPCR amplification in a Roche LightCycler® 480 System (Roche, Basel, Switzerland) following the manufacturer's instructions with primers that flank the cut-sites (Supplementary Table S1). Amplification reactions were performed in duplicate, and their specificity was determined using a melting curve analysis. Raw data were processed using LinRegPCR (Ramakers et al., 2003). The relative methylation level was calculated using the formula FC = E∧ΔCp, where E is the mean amplification efficiency of a given gene and ΔCp corresponds to the difference between the Cp-values for a specific region, using the digested and undigested gDNA templates. Differences in relative methylation were analyzed using the Student's t-test at P ≤ 0.05 using STATISTICA 10 software (StatSoft).
Results and discussion
Technical overview of the MSAP-Seq method
MSAP is a common method used to evaluate DNA methylation in a wide range of plant species due to its simplicity, reliability and low cost. We greatly improved this method by applying the direct sequencing of amplicons using next-generation sequencing. As in typical MSAP, genomic DNA is first cut with the methylation-sensitive restriction enzyme HpaII and the rare-cutter EcoRI, which is not sensitive to DNA methylation (Figure 1). Next, specific adaptors that are complementary to the sticky ends generated by the restrictases are ligated and amplified with partially selective primers. The number and type of selective nucleotides can be easily adjusted to the species and application. With MSAP-Seq, amplicons are sequenced using large-scale NGS, such that the number of selective primers can be greatly reduced to obtain more sequences in the final analysis. We applied two selective nucleotides, equal to 16 primer combinations, with three selective nucleotides. Thus, only one PCR amplification step is necessary in MSAP-Seq. Amplicons were then subjected to fragmentation using sonication with pre- and post-fragmentation purification, to create short tags that can be sequenced using high-throughput sequencing. Rapid DNA library sequencing was performed using the following steps: end repair, adenylation, ligation of barcoded adapters, purification, PCR amplification and purification. Libraries were later evaluated based on product quantity and size distribution. Selected libraries were then subjected to cluster generation and high-throughput sequencing using the Illumina Hi-Seq platform. Using this platform, up to 24 differentially barcoded samples can be simultaneously sequenced, allowing MSAP-Seq to be a relatively low cost method. Data are obtained easily and can be processed with the user-friendly automatic pipeline MSEQER, which is software for MSAP-Seq that was developed by our group (Korotko, 2017—personal communication). MSEQER is available as a web-based service at www.mseqer.us.edu.pl. Single-end and paired-end reads can be analyzed in single or duplicate samples. Automatic analysis allows for the filtering of HpaII adapters, CGG sequence presence, adapter trimming, mapping to the reference genome, and includes genomic features and functional annotation. Read counts are then normalized and differential methylation analysis is performed among samples to obtain the FC (fold change) and p-values for statistical significance. The results are visualized in tables and graphs. A detailed and ready to use MSAP-Seq protocol is enclosed in Supplementary File 1. Additionally, MSAP-Seq is not restricted to species with sequenced reference genomes, as reads can be analyzed de novo without genome mapping for the quantitative comparison of tags among samples.
Figure 1
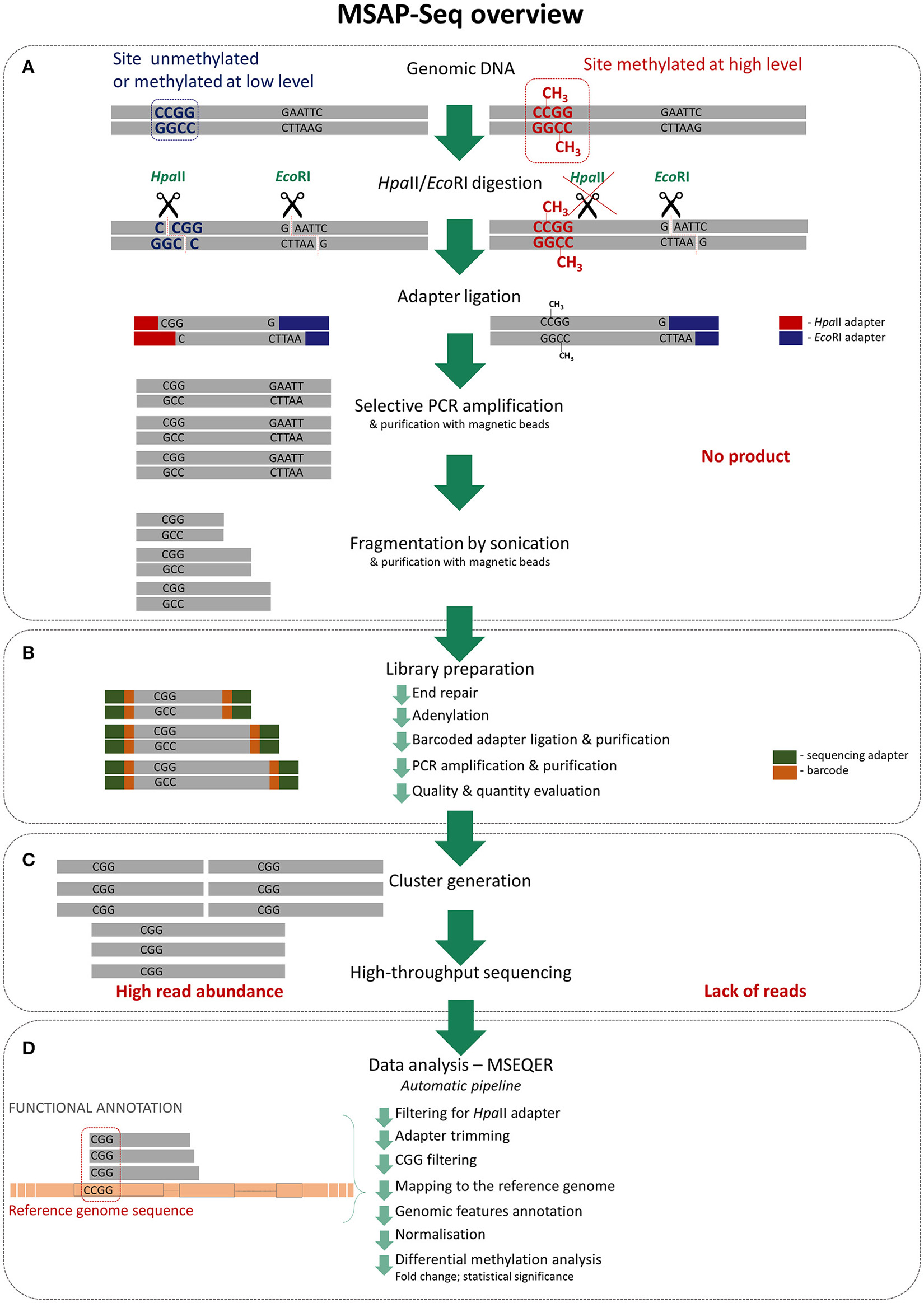
Detailed MSAP-Seq assay overview (A) Genomic DNA is cleaved using rare cutter (EcoRI) and methylation sensitive restriction enzyme (HpaII); only unmethylated recognition sites are digested by HpaII. Then adapters specific to sticky ends are ligated and obtained fragments are amplified in PCR using selective primers; only fragments generated from unmethylated regions are amplified as they contain ends complementary to adapters. Products are then purified and fragmented by sonication to create shorter tags. (B) Purified fragments are used for standard library preparation involving following steps: end repair, adenylation, barcoded adapters ligation and purification, PCR amplification and purification. Then libraries quality and quantity is estimated. (C) Prepared libraries are pooled and processed thru cluster generation and high-throughput sequencing. (D) Sequencing data are analyzed using dedicated automatic pipeline—MSEQER. Firstly, reads are filtered for presence of HpaII adapter and adapters are clipped. Then only reads containing CGG tags on the ends are mapped to the reference genome and functionally annotated. Obtained counts at each of the CCGG sites are normalized and differential methylation analysis among sets of samples is performed.
MSAP-Seq tag sequencing using the illumina platform
To validate this method and to evaluate the biological importance of MSAP-Seq data, we performed two studies that analyzed changes in DNA methylation using MSAP-Seq: (1) an analysis of changes in the barley methylome in leaves after water-deficiency stress and (2) a comparative analysis of DNA methylation in barley leaves and roots after water deficiency stress. All analyses were performed according to our previously described methods and the detailed MSAP-Seq protocol (Supplementary File 1). Both assays were performed in barley, which is representative of large genome crops and has a complex genome that is 5.3 Gbp (Ensembl Plants; Yates et al., 2016). We also found that the barley genome was densely methylated within CCGG sites (Chwialkowska et al., 2016).
Case study 1: analysis of changes in the barley methylome in leaves under water-deficiency stress
Our goal was to characterize the modulation of the methylome in barley leaves under conditions of water deficiency. We employed MSAP-Seq to identify changes in DNA methylation and compared three time points during drought stress: (1) control (normal watering); (2) drought (water deficiency treatment); (3) re-watering (after drought recovery with normal watering). In each time point, leaf material was harvested in triplicate, and MSAP-Seq analysis was performed as described.
We sequenced MSAP-Seq tags with the Illumina Hi-Seq 1500 system with 24 barcoded samples per lane. We obtained 10–20 mln paired-end reads per sample (Table 2). Reads were processed with the MSEQER pipeline. Five to ten million reads were filtered based on the presence of the HpaII adapter followed by a CGG sequence in addition to a read length greater or equal to 50 bp. Analyzed reads were between 50 and 91 bp with a modal value of 88 bp. Approximately 85% of reads were mapped to the barley reference genome. Not all reads could be mapped because the barley genome is incomplete and is still being assembled. On average, ~80,000 unique CCGG sites with a coverage minimum of 2 reads per sample were identified. Among them, 11,000 were gene-related sites, of which 80% were located within gene bodies and 20% within promoter regions. Clustering analysis of normalized reads counts showed that in each analyzed time point, biological replicates were similar and created one cluster (Figure 2). Separate clusters were formed for each time point. Thus, MSAP-Seq analysis was reproducible among biological replicates and allowed for the discrimination of different treatments.
Table 2
| Control A | Control B | Control C | Drought A | Drought B | Drought C | Re-watering A | Re-watering B | Re-watering C | Mean | |
|---|---|---|---|---|---|---|---|---|---|---|
| Initial number of reads [mln] | 14.16 | 19.88 | 9.57 | 16.45 | 14.17 | 15.18 | 16.62 | 10.10 | 13.66 | 14.42 |
| Number of reads after filtering* [mln] | 6.96 | 10.3 | 5.10 | 6.60 | 6.47 | 6.50 | 7.50 | 5.22 | 6.58 | 6.81 |
| Percentage of mapped reads [%] | 86.9 | 86.7 | 86.5 | 84.3 | 84.7 | 84.3 | 85.4 | 84.3 | 84.9 | 85.3 |
| Number of different sites | 65,980 | 79,929 | 52,164 | 91,374 | 86,706 | 89,271 | 85,101 | 74,211 | 84,738 | 78,830 |
| Number of different genes (gene bodies) | 7,437 | 8,351 | 6,333 | 9,403 | 9,311 | 9,470 | 9,028 | 8,202 | 8,900 | 8,493 |
| Number of different genes (promoters) | 1,830 | 2,140 | 1,910 | 2,418 | 2,430 | 2,417 | 2,372 | 2,083 | 2,344 | 2,216 |
The statistics of MSAP-Seq reads during different data processing steps during evaluation of changes in barley methylome in leaves under water-deficiency stress.
Filtering based on HpaII-related adapter presence followed by CGG sequence and 50 bp of minimal read length.
Figure 2
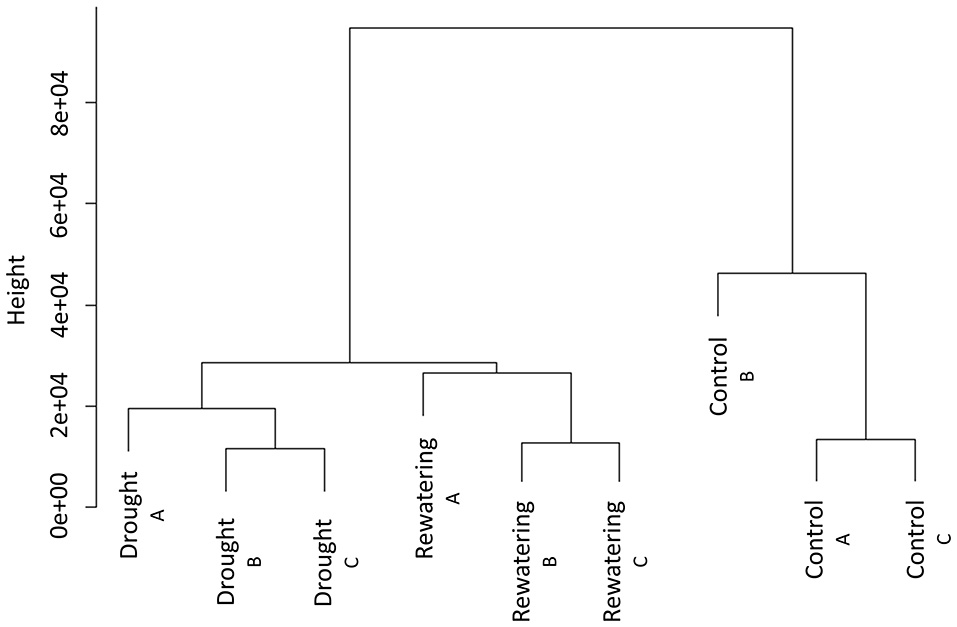
Hierarchical clustering of MSAP-Seq data of three samples (control, drought, and re-watering) performed in three independent biological replicates.
In total, approximately 190,000 different CCGG sites of known sequence and genomic location were identified and subjected to differential methylation analysis. This value is extremely high compared to traditional MSAP, where hundreds or thousands of amplicons are scored (Filek et al., 2006; Candaele et al., 2014; Chwialkowska et al., 2016). In MSAP, a limited quantity of differentially methylated bands are reamplified, subcloned and sequenced using the Sanger method, making this approach laborious and low-throughput. Importantly, we applied two selective nucleotides in the amplification step, equal to 16 primer combinations, compared to three selective nucleotides used in conventional MSAP. By adjusting the number of selective nucleotides in the amplification step, the number of amplicons is modified. Additionally, NGS-based sequencing with MSAP-Seq avoids the problem of homoplastic bands, which is common in gel-based MSAP studies (Vekemans et al., 2002).
When different genomic loci were accounted for, the majority of reads (62%) mapped to unannotated intergenic regions and 14% mapped to repetitive elements (Figure 3). However, because 84%of the barley genome are mobile elements and other repeat structures (International Barley Genome Sequencing Consortium, 2012), and detailed information of repetitive element annotation is not publicly available, we annotated only those present in the Ensembl Plants database. Thus, the vast majority of the unannotated regions also presumably consists of repetitive elements. Interestingly, 25% of the sites were gene related and over 20% were located within gene bodies. Because only 2% of the barley genome contains genes, we demonstrate that the MSAP-Seq method enriches for gene-containing regions. This is because MSAP-Seq utilizes the methylation-sensitive restriction enzyme HpaII, which recognizes the CCGG sequence that is frequently present in the GC-rich regions often found within genes (Glémin et al., 2014). In addition, we identified MSAP-Seq tags related to ~15,000 different genes. This is relatively high and represents ~60% of all barley genes. In conclusion, MSAP-Seq is an effective method for DNA methylation analysis for species with large genomes and low gene content.
Figure 3
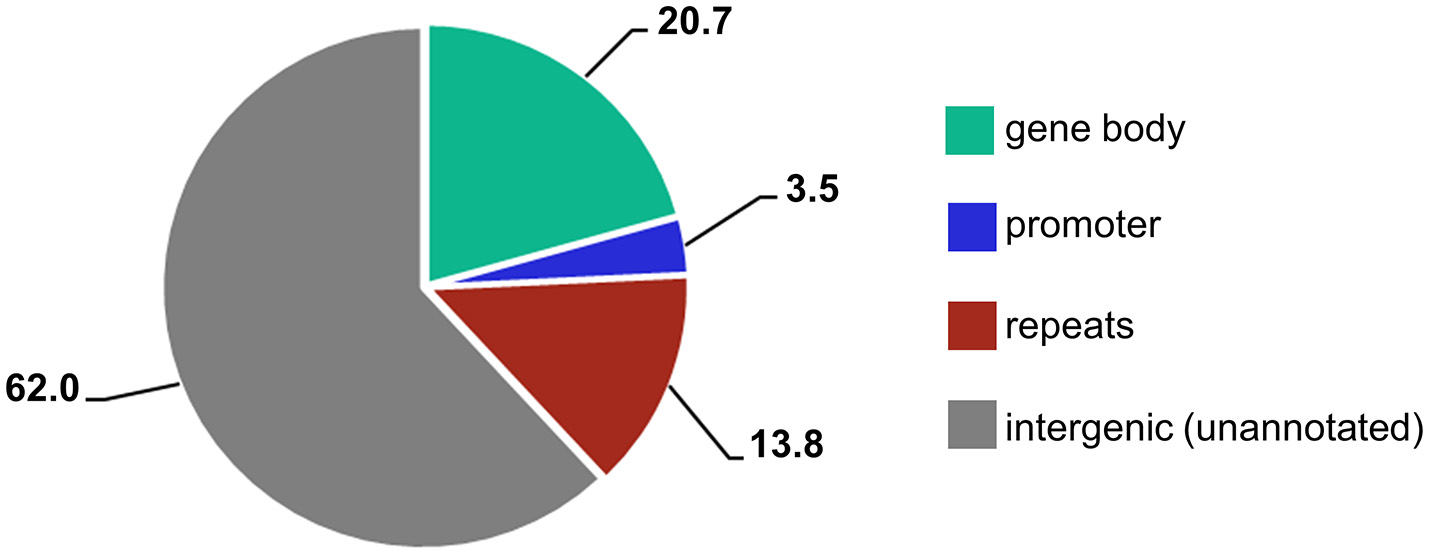
Percentages of different genic features identified within MSAP-Seq tags.
Differentially methylated sites (DMS) were determined from normalized reads counts and the FCs among samples and the statistical significance of those changes were evaluated. The methylation-sensitive restriction enzyme HpaII allowed for the distinction between two different methylation states within CCGG sequences: (1) unmethylated, which indicates that DNA is totally unmethylated or that methylation is present only on one strand (cleaved by HpaII), and (2) methylated, in which the cytosine is methylated on both strands (not cleaved by HpaII). The lower the read abundance is, the higher the DNA methylation level is, and conversely, the higher the read abundance is, the lower the DNA methylation level is. The normalized read counts were compared between the two datasets and changes were presented as FC-values. We identified 2,718 different CCGG sites that underwent DNA methylation changes under water-deficiency stress in barley leaves (P < 0.05). Fifty five percent of drought-related DMS were demethylation events and 45% were novel DNA methylations. Upon re-watering, 40% of demethylations and 50% of the new methylation events were reversible and reverted to their initial level, which was the same as the control. Within the drought-related DMS, we identified 219 new methylations and 288 demethylations that were located within genes. In both sets, 86% of the gene-related DMS were located within genes and 14% were within promoter regions. Interestingly, when the distribution of genomic features was accounted for, we found that within the drought-related DMS, genes were underrepresented and repetitive elements were overrepresented. This result suggests that changes in DNA methylation may target repetitive regions to regulate genome stability and to balance transposable element mobilization/inactivation under stress. Transposon insertions were found to be an important factor that affected adjacent gene expression under stress in maize, a plant with a highly repetitive genome (Makarevitch et al., 2015). Moreover, studies in A. thaliana revealed that DNA demethylases modulate the expression of stress-responsive genes by targeting changes in cytosine methylation within transposable elements (Le et al., 2014).
Case study 2: comparative analysis of DNA methylation changes in barley leaves and roots in response to water deficiency stress
Our previous study attempted to characterize and compare the barley methylome of leaves and roots under water-deficiency treatment, as well as during the subsequent re-watering phase, with an emphasis being placed on organ-specific changes in the DNA methylation pattern (Chwialkowska et al., 2016). This experiment involved methylome modulation analysis using MSAP-Seq in the leaves and roots under the same three time points: (1) control (normal watering); (2) drought (water deficiency treatment); and (3) re-watering (after drought recovery and normal watering). Samples at each time point were pooled from three independent plants. Each time point, as well as the leaves and root samples, were pooled and analyzed separately.
We performed MSAP-Seq using the Illumina Hi-Seq 1500 system with six barcoded MSAP-Seq samples (three time points for leaves and three time points for roots). We obtained 16–77 mln paired-end reads per sample (Table 3). Reads were processed using the automatic MSEQER pipeline. Three to nineteen million reads were filtered based on the presence of the HpaII-related adapter followed by a CGG sequence and a read length equal to or greater than 50 bp. Approximately 88% of filtered reads in the leaves and 49% in roots were mapped to the barley reference genome. On average, ~100,000 unique CCGG sites with a minimum coverage of 2 reads per sample were identified. Among the sites, more than 13,000 were gene-related sites, of which 80% were located within gene bodies and 20% within promoter regions.
Table 3
| LEAVES | ROOTS | |||||||
|---|---|---|---|---|---|---|---|---|
| Control | Drought | Re-watering | Mean | Control | Drought | Re-watering | Mean | |
| Initial number of reads [mln] | 77.52 | 72.00 | 56.46 | 68.66 | 31.18 | 37.13 | 16.05 | 28.12 |
| Number of reads after filtering* [mln] | 18.29 | 19.22 | 11.44 | 16.31 | 7.21 | 8.14 | 3.32 | 6.22 |
| Percentage of mapped reads [%] | 88.3 | 85.9 | 89.4 | 87.9 | 64.1 | 45.3 | 37.9 | 49.1 |
| Number of different sites | 117,259 | 122,631 | 95,172 | 111,687 | 107,044 | 111,879 | 57,749 | 92,224 |
| Number of different genes (gene bodies) | 10,961 | 11,248 | 10,336 | 10,848 | 11,819 | 11,905 | 7,728 | 10,484 |
| Number of different genes (promoters) | 3,067 | 3,247 | 2,805 | 3,039 | 3,107 | 3,050 | 1,785 | 2,647 |
The statistics of MSAP-Seq reads during different data processing steps during comparative evaluation of DNA methylation changes in barley leaf and root in response to water deficiency stress.
Filtering based on HpaII-related adapter presence followed by CGG sequence and 50 bp of minimal read length.
In total, ~250,000 different CCGG sites of known sequence and genomic location were identified and subjected to differential methylation analysis. When different genomic loci in the leaves and roots were accounted for, most reads (61%) mapped to unannotated intergenic regions and 13% mapped to repetitive elements. Similarly, 25% of the reads were located within genes and 21% were gene-body related, again demonstrating the enrichment for gene-containing regions. Interestingly, regarding both leaves and roots, we identified MSAP-Seq tags related to ~18,000 genes, including 75% of all barley genes.
Differentially methylated sites were determined from normalized read counts and the FCs among samples were obtained. Because samples were pooled from three plants without replicates, DMS with FCs greater than or equal to five were considered differentially methylated. MSAP-Seq analysis revealed 2,901 DMS in leaves and 6,098 in roots that exhibited changes under the water deficiency treatment. Among these sites, 496 DMS in leaves and 1,055 in roots were located within genes. In both leaves and roots, ~85% of the gene-related DMS were located within gene bodies and 15% were in promoter regions. Within the gene-related DMS in leaves, equal amounts of novel stress-induced methylation and demethylation events were observed. However, in roots, new methylation events dominated. Plant recovery after the re-watering phase led to the reversal of the major stress-induced demethylations within genes; however, this process was more efficient in leaves than in roots. In contrast, new methylation events were much more persistent in both organs. Repetitive elements preferentially underwent demethylation in leaves and novel methylation in roots. Interestingly, we found that genic regions were subjected to balanced methylome modulation rather than completely irreversible modification. This result suggests that methylation changes induced by water deficiency impacts the expression levels of stress-responsive genes. In contrast, most of the changes observed within the repetitive elements were of a stricter nature and almost all of them were irreversible methylations or completely reversible demethylations. Such tendency might allow for a reversal of most of the demethylations that could induce transposon mobilization and allow the maintenance of novel methylations that might silence them. We identified different biological processes within the subsets of the gene-related DMS in leaves compared to roots. Using Gene Ontology (GO) enrichment analysis, we determined the biological processes that were targeted by the epigenetic machinery under water-deficiency stress in both leaves and roots. For example, in the DMS of that were reversibly demethylated under conditions of drought, we observed an enrichment of metabolic processes such as xylogalacturonan and sphingolipid metabolism and raffinose biosynthesis. These processes are important in the adaptive response to water-deficiency stress (Ng et al., 2001; Liu et al., 2007; Le Gall et al., 2015). In roots, the reversibly demethylated DMS showed an enrichment of the hypersensitive response, carbohydrate metabolism, and the positive regulation of developmental processes. In conclusion, distinct biological processes were influenced by leaf- and root-specific methylation changes, which together contributed to complex stress-response networks. We offer a comprehensive catalog of the general properties of the barley leaf and root methylome, as well as its modulation under water-deficiency stress and after re-watering.
Quantitative validation of identified DMS
In Case Study 1, a random set of 15 DMSs were identified that exhibited novel methylations in genes under water deficiency stress in barley leaves (Chwialkowska et al., 2016) and were independently validated with a single-locus DNA methylation assay using Methylation Sensitive Restriction Enzyme qPCR (MSRE-qPCR). This method is based on the cleavage of genomic DNA with the HpaII enzyme followed by qPCR with primers flanking the cut-site. MSRE-qPCR was performed in two technical and three separate biological replicates that allowed for statistical analysis of the results. DNA methylation level changes exhibited similar tendencies and were comparable (Supplementary Table S2). This result demonstrates the reproducibility and reliability of detecting DMSs using MSAP-Seq.
Conclusions
We developed and validated the MSAP-Seq method, which provides sequence-based identification of changes in DNA methylation. MSAP-Seq can easily be applied to different plant species, including those with large, complex and highly repetitive genomes. MSAP-Seq is based on conventional MSAP analysis and relies on the differential cleavage of the methylation sensitive restrictase HpaII. This technique enables the fast, global, and reliable analysis of MSAP amplicons without laborious PAGE and sub-cloning assays. Thus, MSAP-Seq is as simple as the well-known and conventional MSAP but uses state-of-the-art NGS technology that enables the high-throughput, parallel and direct analysis of DNA methylation modulation in hundreds of thousands of sites. In contrast to traditional MSAP, it allows for the quantitative determination of DNA methylation changes as well as their direct localization. Additionally, the number of sequences obtained can be easily adjusted. NGS of up to 24 samples per Illumina Hi-Seq lane allows this analysis to be affordable, even for labs that conduct gel-based MSAP. One study can identify several thousand DMSs. MSAP-Seq is also well-suited for large analyses with large sets of samples. We have shown that the MSAP-Seq method enriches for gene-containing regions and that one analysis can cover 60–75% of all genes. MSAP-Seq is a method of choice for DNA methylation analysis in crop plants with large and complex genomes, due to its simplicity and low price.
Statements
Author contributions
KC and MK: Conceived and planned the assay; KC: Carried out all the experiments and data analyses under a supervision of MK; JK: Performed sequencing on Illumina platform; UK: Prepared pipeline for bioinformatics processing; KC: Wrote the manuscript under a supervision of MK and IS. All authors read and approved the final manuscript.
Funding
This work was financially supported by the Polish National Science Centre (NCN) grant no. 2014/13/N/NZ2/01153, the European Union project no. 289300 within The 7th Framework Programme, and the Ministry of Science and Higher Education grant 2486/7.PR/2012/2. KC and UK were granted a scholarship co-funded by the European Union within the framework of the European Social Fund “DoktoRIS Scholarship Program for Innovative Silesia.” KC was a beneficiary of the ETIUDA scholarship funded by the Polish National Science Centre (NCN) under grant no. 2016/20/T/NZ2/00577.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2017.02056/full#supplementary-material
References
1
AbidG.MingeotD.MuhovskiY.MergeaiG.AouidaM.AbdelkarimS.et al. (2017). Analysis of DNA methylation patterns associated with drought stress response in faba bean (Vicia faba L.) using methylation-sensitive amplification polymorphism (MSAP). Environ. Exp. Bot.142, 34–44. 10.1016/j.envexpbot.2017.08.004
2
BergerS. L.KouzaridesT.ShiekhattarR.ShilatifardA. (2009). An operational definition of epigenetics. Genes Dev.23, 781–783. 10.1101/gad.1787609
3
CandaeleJ.DemuynckK.MosotiD.BeemsterG. T.InzéD.NelissenH. (2014). Differential methylation during maize leaf growth targets developmentally regulated genes. Plant Physiol.164, 1350–1364. 10.1104/pp.113.233312
4
ChakrabartyD.YuK. W.PaekK. Y. (2003). Detection of DNA methylation changes during somatic embryogenesis of Siberian ginseng (Eleuterococcus senticosus). Plant Sci.165, 61–68. 10.1016/S0168-9452(03)00127-4
5
ChwialkowskaK.NowakowskaU.MroziewiczA.SzarejkoI.KwasniewskiM. (2016). Water-deficiency conditions differently modulate the methylome of roots and leaves in barley (Hordeum vulgare L.). J. Exp. Bot.67, 1109–1121. 10.1093/jxb/erv552
6
CokusS. J.FengS.ZhangX.ChenZ.MerrimanB.HaudenschildC. D.et al. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature452, 215–219. 10.1038/nature06745
7
DoyleJ. J.DoyleJ. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull.19, 11–15.
8
FengS.CokusS. J.ZhangX.ChenP. Y.BostickM.GollM. G.et al. (2010). Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. U.S.A.107, 8689–8694. 10.1073/pnas.1002720107
9
FilekM.JaniakA.SzarejkoI.GrabczynskaJ.MacháckováI.KrekuleJ. (2006). Does DNA methylation pattern mark generative development in winter rape?Z. Naturforsch. C.61, 387–396. 10.1515/znc-2006-5-615
10
GautamM.DangY.GeX.ShaoY.LiZ. (2016). Genetic and epigenetic changes in oilseed rape (Brassica napus L.) extracted from intergeneric allopolyploid and additions with Orychophragmus. Front. Plant Sci.7:438. 10.3389/fpls.2016.00438
11
GentJ. I.EllisN. A.GuoL.HarkessA. E.YaoY.ZhangX.et al. (2013). CHH islands: de novo DNA methylation in near-gene chromatin regulation in maize. Genome Res.23, 628–637. 10.1101/gr.146985.112
12
GimenezM. D.Yañez-SantosA. M.PazR. C.QuirogaM. P.MarfilC. F.ConciV. C.et al. (2016). Assessment of genetic and epigenetic changes in virus-free garlic (Allium sativum L.) plants obtained by meristem culture followed by in vitro propagation. Plant Cell Rep.35, 129–141. 10.1007/s00299-015-1874-x
13
GléminS.ClémentY.DavidJ.RessayreA. (2014). GC content evolution in coding regions of angiosperm genomes: a unifying hypothesis. Trends Genet.30, 263–270. 10.1016/j.tig.2014.05.002
14
Guzy-WrobelskaJ.KaliciakA.SzarejkoI.MachackovaI.KrekuleJ.BarciszewskaM. (2013). Vernalization and photoperiod-related changes in the DNA methylation state in winter and spring rapeseed. Acta Physiol. Plant.35, 817–827. 10.1007/s11738-012-1126-4
15
International Barley Genome Sequencing ConsortiumMayerK. F.WaughR.BrownJ. W.SchulmanA.LangridgeP.et al. (2012). A physical, genetic and functional sequence assembly of the barley genome. Nature491, 711–716. 10.1038/nature11543
16
JiL.NeumannD. A.SchmitzR. J. (2015). Crop epigenomics: identifying, unlocking, and harnessing cryptic variation in crop genomes. Mol. Plant8, 860–870. 10.1016/j.molp.2015.01.021
17
KapazoglouA.DrosouV.ArgiriouA.TsaftarisA. S. (2013). The study of a barley epigenetic regulator, HvDME, in seed development and under drought. BMC Plant Biol.13:172. 10.1186/1471-2229-13-172
18
KumarG.RattanU. K.SinghA. K. (2016). Chilling-mediated DNA methylation changes during dormancy and its release reveal the importance of epigenetic regulation during winter dormancy in apple (Malus x domestica Borkh.). PLoS ONE11:e0149934. 10.1371/journal.pone.0149934
19
LangmeadB.SalzbergS. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods9, 357–359. 10.1038/nmeth.1923
20
Le GallH.PhilippeF.DomonJ. M.GilletF.PellouxJ.RayonC. (2015). Cell wall metabolism in response to abiotic stress. Plants4, 112–166. 10.3390/plants4010112
21
LeT.-N.SchumannU.SmithN. A.TiwariS.Khang AuP. C.ZhuQ.-H.et al. (2014). DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol.15:458. 10.1186/s13059-014-0458-3
22
LiS.XiaQ.WangF.YuX.MaJ.KouH.et al. (2017). Laser irradiation-induced DNA methylation changes are heritable and accompanied with transpositional activation of mPing in rice. Front. Plant Sci.8:363. 10.3389/fpls.2017.00363
23
LiZ.LiuZ.ChenR.LiX.TaiP.GongZ.et al. (2015). DNA damage and genetic methylation changes caused by Cd in Arabidopsis thaliana seedlings. Environ. Toxicol. Chem.34, 2095–2103. 10.1002/etc.3033
24
ListerR.O'MalleyR. C.Tonti-FilippiniJ.GregoryB. D.BerryC. C.MillarA. H.et al. (2008). Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell133, 523–536. 10.1016/j.cell.2008.03.029
25
LiuH.DaiX.XuY.ChongK. (2007). Over-expression of OsUGE-1 altered raffinose level and tolerance to abiotic stress but not morphology in Arabidopsis. J. Plant Physiol.164, 1384–1390. 10.1016/j.jplph.2007.03.005
26
LoveM. I.HuberW.AndersS. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol.15, 1–21. 10.1186/s13059-014-0550-8
27
MakarevitchI.WatersA. J.WestP. T.StitzerM.HirschC. N.et al. (2015). Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet.11:e1004915. 10.1371/journal.pgen.1004915
28
MarconiG.PaceR.TrainiA.RaggiL.LuttsS.ChiusanoM.et al. (2013). Use of MSAP markers to analyse the effects of salt stress on DNA methylation in rapeseed (Brassica napus var. oleifera). PLoS ONE8:e75597. 10.1371/journal.pone.0075597
29
MeyerP. (2015). Epigenetic variation and environmental change. J. Exp. Bot.66, 3541–3548. 10.1093/jxb/eru502
30
NgC.CarrK.McAinshM.PowellB.HetheringtonA. (2001). Drought-induced guard cell signal transduction involves sphingosine-1-phosphate. Nature410, 596–599. 10.1038/35069092
31
Peraza-EcheverriaS.Herrera-ValenciaV. A.KayA. (2001). Detection of DNA methylation changes in micropropagated banana plants using methylation-sensitive amplification polymorphism (MSAP). Plant Sci.161, 359–367. 10.1016/S0168-9452(01)00421-6
32
PortisE.AcquadroA.CominoC.LanteriS. (2004). Analysis of DNA methylation during germination of pepper (Capsicum annuum L.) seeds using methylation-sensitive amplification polymorphism (MSAP). Plant Sci.166, 169–178. 10.1016/j.plantsci.2003.09.004
33
RamakersC.RuijterJ. M.DeprezR. H.MoormanA. F. (2003). Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett.339, 62–66. 10.1016/S0304-3940(02)01423-4
34
Reyna-LópezG. A.SimpsonJ.Ruiz-HerreraJ. (1997). Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol. Gen. Genet.253, 703–710. 10.1007/s004380050374
35
RichardsE. J.ElginS. C. (2002). Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell108, 489–500. 10.1016/S0092-8674(02)00644-X
36
SalmonA.ClotaultJ.JenczewskiE.ChableaV.Manzanares-DauleuxaM. J. (2008). Brassica oleracea displays a high level of DNA methylation polymorphism. Plant Sci.174, 61–70. 10.1016/j.plantsci.2007.09.012
37
SchmitzR. J.ZhangX. (2011). High-throughput approaches for plant epigenomic studies. Curr. Opin. Plant Biol.14, 130–136. 10.1016/j.pbi.2011.03.010
38
TanM. P. (2010). Analysis of DNA methylation of maize in response to osmotic and salt stress based on methylation-sensitive amplified polymorphism. Plant Physiol. Biochem.48, 21–26. 10.1016/j.plaphy.2009.10.005
39
TangX. M.TaoX.WangY.MaD. W.LiD.YangH.et al. (2014). Analysis of DNA methylation of perennial ryegrass under drought using the methylation-sensitive amplification polymorphism (MSAP) technique. Mol. Genet. Genomics289, 1075–1084. 10.1007/s00438-014-0869-6
40
VekemansX.BeauwensT.LemaireM.Roldán-RuizI. (2002). Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Mol. Ecol.11, 139–151. 10.1046/j.0962-1083.2001.01415.x
41
VosP.HogersR.BleekerM.ReijansM.van de LeeT.HornesM.et al. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res.23, 4407–4414. 10.1093/nar/23.21.4407
42
WangB.ZhangM.FuR.QianX.RongP.ZhangY.et al. (2016). Epigenetic mechanisms of salt tolerance and heterosis in Upland cotton (Gossypium hirsutum L.) revealed by methylation-sensitive amplified polymorphism analysis. Euphytica208, 477–491. 10.1007/s10681-015-1586-x
43
WangW. S.PanY. J.ZhaoX. Q.DwivediD.ZhuL. H.AliJ.et al. (2011). Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J. Exp. Bot.62, 1951–1960. 10.1093/jxb/erq391
44
XiongZ.XuC. G.Saghai MaroofM. A.ZhangQ. (1999). Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol. Gen. Genet.261, 439–446. 10.1007/s004380050986
45
XuW.WangT.XuS.XuS.WuL.WuY.et al. (2015). Radiation-induced epigenetic bystander effects demonstrated in Arabidopsis thaliana. Radiat. Res.183, 511–524. 10.1667/RR13909.1
46
YamauchiT.Johzuka-HisatomiY.TeradaR.NakamuraI.IidaS. (2014). The MET1b gene encoding a maintenance DNA methyltransferase is indispensable for normal development in rice. Plant Mol. Biol.85, 219–232. 10.1007/s11103-014-0178-9
47
YatesA.AkanniW.AmodeM. R.BarrellD.BillisK.Carvalho-SilvaD.et al. (2016). Ensembl 2016. Nucleic Acids Res.44, D710–D716. 10.1093/nar/gkv1157
48
ZhangX.YazakiJ.SundaresanA.CokusS.ChanS. W.ChenH.et al. (2006). Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell126, 1189–1201. 10.1016/j.cell.2006.08.003
Summary
Keywords
DNA methylation, MSAP, methylome analysis, new technique, next generation sequencing, large genomes
Citation
Chwialkowska K, Korotko U, Kosinska J, Szarejko I and Kwasniewski M (2017) Methylation Sensitive Amplification Polymorphism Sequencing (MSAP-Seq)—A Method for High-Throughput Analysis of Differentially Methylated CCGG Sites in Plants with Large Genomes. Front. Plant Sci. 8:2056. doi: 10.3389/fpls.2017.02056
Received
08 September 2017
Accepted
16 November 2017
Published
30 November 2017
Volume
8 - 2017
Edited by
Fabio Marroni, University of Udine, Italy
Reviewed by
Serena Varotto, Università degli Studi di Padova, Italy; Xue-liang Ren, Guizhou Academy of Tobacco Science, China
Updates
Copyright
© 2017 Chwialkowska, Korotko, Kosinska, Szarejko and Kwasniewski.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miroslaw Kwasniewski miroslaw.kwasniewski@umb.edu.pl
This article was submitted to Technical Advances in Plant Science, a section of the journal Frontiers in Plant Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.