 Sagar Banerjee1,2†
Sagar Banerjee1,2† Sarvajeet S. Gill2
Sarvajeet S. Gill2 Bharat H. Gawade3
Bharat H. Gawade3 Pradeep K. Jain4
Pradeep K. Jain4 Kuppuswamy Subramaniam5
Kuppuswamy Subramaniam5 Anil Sirohi1*
Anil Sirohi1*- 1Division of Nematology, ICAR-Indian Agricultural Research Institute, New Delhi, India
- 2Stress Physiology and Molecular Biology Lab, Centre for Biotechnology, Maharshi Dayanand University, Rohtak, India
- 3Division of Plant Quarantine, ICAR-National Bureau of Plant Genetic Resources, New Delhi, India
- 4ICAR-National Research Centre on Plant Biotechnology, New Delhi, India
- 5Department of Biotechnology, Indian Institute of Technology, Chennai, India
Root-knot nematodes have emerged as devastating parasites causing substantial losses to agricultural economy worldwide. Tomato is the most favored host for major species of root-knot nematodes. Control strategies like use of nematicides have proved to be harmful to the environment. Other control methods like development of resistant cultivars and crop rotation have serious limitations. This study deals with the application of host generated RNA interference toward development of resistance against root-knot nematode Meloidogyne incognita in tomato. Two cuticle collagen genes viz. Mi-col-1 and Lemmi-5 involved in the synthesis and maintenance of the cuticle in M. incognita were targeted through host generated RNA interference. Expression of both Mi-col-1 and Lemmi-5 was found to be higher in adult females followed by egg masses and J2s. Tomato var. Pusa Ruby was transformed with the RNAi constructs of these genes to develop transgenic lines expressing the target dsRNAs. 30.80–35.00% reduction in the number of adult females, 50.06–65.73% reduction in the number of egg mass per plant and 76.47–82.59% reduction in the number of eggs per egg mass were observed for the T1 events expressing Mi-col-1 dsRNA. Similarly, 34.14–38.54% reduction in the number of adult females, 62.34–66.71% reduction in number of egg mass per plant and 67.13–79.76% reduction in the number of eggs per egg mass were observed for the T1 generation expressing Lemmi-5 dsRNA. The multiplication factor of M. incognita reduced significantly in both the cases and the structure of adult females isolated from transgenic plants were heavily distorted. This study demonstrates the role of the cuticle collagen genes Mi-col-1 and Lemmi-5 in the structure and development of M. incognita cuticle inside the host and reinforces the potential of host generated RNA interference for management of plant parasitic nematodes (PPNs).
Introduction
Plant parasitic nematodes (PPNs) pose a major threat to world agriculture causing an estimated economic loss of around US $173 billion annually (Elling, 2013). Root knot nematodes (RKNs) are one of the most devastating PPNs with a wide host range of more than 3000 plant species. Meloidogyne incognita is the most predominant species of the root-knot nematodes and possibly the most damaging PPN all across the world (Trudgill and Blok, 2001). Jain et al. (2007), have attributed 27.21% losses annually in India to M. incognita in the solanacious crop tomato. M. incognita is an apomictic (mitotic parthenogenetic) and biotrophic sedentary endoparasite. Host plant roots are penetrated by infective second stage juveniles (J2s) which migrate further until they reach vascular tissues. Here, they induce the formation of multinucleate giant cells (GCs) by injecting esophageal gland secretions into root cells which become their feeding sites. The developing nematodes receive food from the feeding cells and molt thrice to form third stage juveniles (J3s), fourth stage juveniles (J4s) and adult females. Meanwhile, the cells around the developing nematode become enlarged and proliferated leading to the formation of galls (Jones and Northcote, 1972). The formation of galls interferes with upward translocation of nutrients and water in the affected roots leading to reduction in crop yield (Moens et al., 2009).
Over the years, different approaches like use of chemical nematicides, development of resistant cultivars and cultural practices have been employed for management of these devastating parasites. Chemical nematicides have been most effective as control measures for RKNs, however, use of chemical nematicides leads to serious health and environmental issues such as contamination of ground water, environmental persistence, mammalian toxicity etc. Therefore, many countries have imposed bans on major nematicides. Resistance breeding continues to be a strategy of nematode control against Meloidogyne spp., Globodera rostochiensis and Heterodera glycines on tomato, potato and soybean respectively (Atkinson et al., 2003); however, its success depends on the host gene pool and availability of resistant genes. This scenario indicates that the measures for the control of RKNs are currently limited. Therefore, novel approaches need to be applied for the control of RKNs. RNA interference (RNAi) is an effective tool that can be utilized to engineer resistance against RKNs. In this approach, dsRNA precursors corresponding to the target gene are processed by RNase III domain containing specialized enzymes called Dicer leading to the generation of small RNA duplexes called short interfering RNAs (siRNAs). These RNA duplexes are then incorporated into the RNA-induced silencing complex (RISC). RISC loaded with siRNA is guided to the target RNA by the core catalytic component of RISC which consists of argonoute family protein, which induces the silencing of the target gene in a sequence specific manner.
RNAi in PPNs was first demonstrated by Urwin et al. (2002) by soaking J2s of H. glycines and G. palida in dsRNA solutions. This method was later used in several studies in different PPNs (Bakhetia et al., 2005; Rosso et al., 2005; Huang et al., 2006; Dubreuil et al., 2007; Kimber et al., 2007; Shingles et al., 2007; Gleason et al., 2008; Park et al., 2008) for functional analysis of potentially lethal target genes and paved way for host generated RNAi which is a suitable approach for management of PPNs owing to their obligate parasitic nature. The target genes for host generated RNAi are selected on the basis of their involvement in the various stages of nematode development, parasitism, reproduction etc. Host generated RNAi was first demonstrated by Yadav et al. (2006) in tobacco against M. incognita by targeting two housekeeping genes, integrase and splicing factor. Since then numerous studies have reported host generated RNAi against RKNs targeting various housekeeping genes, parasitism genes and genes associated with nematode development (Huang et al., 2006; Fairbairn et al., 2007; Niu et al., 2012, 2016; Antonino de Souza Júnior et al., 2013; Papolu et al., 2013; Xue et al., 2013; Dinh et al., 2014a,b; Dutta et al., 2015; Lourenço-Tessutti et al., 2015; Zhuo et al., 2016; Kumar et al., 2017) with varying degree of success toward achieving resistance in Arabidopsis thaliana, tobacco, tomato and potato.
Cuticle collagens are the key components of RKN cuticle which maintains the shape of the nematode, protects it from external environment and plays important role in its interaction with the host and soil environment (Davies and Curtis, 2011). Cuticle collagen genes have been well explored in the model free living nematode Caenorhabditis elegans (Johnstone, 2000), however, these are still understudied in PPNs. M. incognita molts multiple times throughout its life cycle and needs to form new cuticle during each molting process. Cuticle collagen genes of PPNs have not been targeted till date for host generated RNAi. However, they can be effective targets for silencing owing to their involvement in the development of the nematodes during their parasitic life cycle. In this study, we have targeted two cuticle collagen genes of M. incognita; Mi-col-1 and Lemmi-5 for host generated RNAi in tomato to assess its effect on the development and parasitism of the nematode.
Materials and Methods
Nematode Culture and Its Maintenance
Meloidogyne incognita Chitwood race I was maintained on young tomato plants (Solanum lycopersicum L. cv. Pusa Ruby) inoculated with 2 J2s/gram of soil. Infected plants were uprooted 30 days post inoculations and egg masses were handpicked after washing the roots with double distilled water. The egg masses were kept in a cavity block and surface sterilized with 0.1% HgCl2 for 1 min. The surface sterilizing agent was then removed by washing the egg masses thrice with double distilled water. The egg masses were allowed to hatch at 26–28°C through a wire gauge covered with double layered tissue paper into a petri plate filled with double distilled water (Hooper, 1986). Freshly hatched J2s were used for further experiments. Adult females were also isolated from the roots of the infected tomato plants 30 days post inoculation under the microscope using a needle.
Target Genes for Silencing and Their in Silico Characterization
Cuticle collagen genes, Mi-col-1 and Lemmi-5 were selected for silencing through host delivered RNAi to demonstrate the effect of downregulation of these structural genes in the parasitic life cycle of M. incognita. The nucleotide sequences of both the genes and their corresponding amino acid sequences were retrieved from NCBI database. Domains were predicted from the amino acid sequences of these genes using SMART (Letunic et al., 2015). A phylogenetic tree was constructed using the amino acid sequences of the cuticle collagens of the plant parasitic and free living nematodes available in the database using MEGA6 (Tamura et al., 2013). TMHMM 2.0 server was used for prediction of transmembrane domains.
Cloning and Sequencing of Mi-col-1 and Lemmi-5
Total RNA was extracted from adult females of M. incognita using TRIzol reagent (Invitrogen) following manufacturer's protocol followed by DNase I (Thermo Fischer Scientific) treatment. The total RNA was quantified using Nanodrop spectrophotometer (Thermo Fischer Scientific) and first strand cDNA was synthesized with 1 μg of the total RNA using PrimeScript™ first strand cDNA synthesis kit (Takara). 416 and 799 bp long fragments of Lemmi-5 and Mi-col-1, respectively were amplified using gene specific primers (Table 1). The amplified products were run on 1% agarose gel and subsequently eluted by using GeneJET gel extraction kit (Thermo Fischer Scientific). The eluted products were cloned into pGEMT easy vector (Promega) following manufacturer's protocol. The ligated products were transformed into E. coli DH5α and positive colonies were screened through blue white selection and colony PCR. Plasmids were isolated from PCR positive colonies using GeneJET plasmid miniprep kit (Thermo Fischer Scientific) following manufacturer's protocol and confirmed for the presence of the inserts by restriction digestion by Eco RI. The inserts in the plasmid were custom sequenced (ABI SOLiD sequencing system) to reassert their identity.
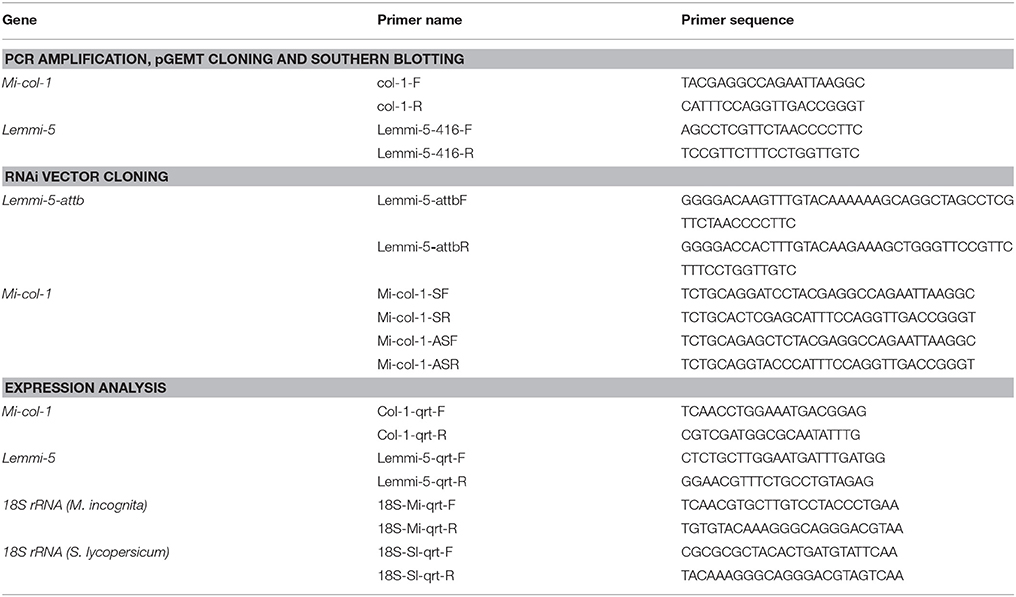
Table 1. List of primers used for PCR amplification, pGEMT cloning and southern blotting, RNAi vector cloning and expression analysis.
Differential Expression of Mi-col-1 and Lemmi-5 at Different Life Stages of M. incognita
Expression of Mi-col-1 and Lemmi-5 was quantified at different life stages of M. incognita through quantitative real time PCR (qRT-PCR) using cDNAs from J2s, egg masses and adult females. Total RNA was isolated and from these developmental stages, quantified and first stranded cDNA was synthesized as described above. A constitutively expressed gene, 18s rRNA was used as an internal control to normalize the gene expression levels. qRT-PCR was performed in a CFX96 real time system (Biorad) using 2X brilliant III SYBR Green q-PCR master mix (Agilent). 20 μL reaction mixture for each sample consisted of 10 μL of 2X SYBR green qPCR master mix, 500 nM of each gene specific primer (Table 1) and 100 ng of first strand cDNA. Two biological and three technical replicates were taken for each sample. The amplification reactions included initial denaturation of 95°C for 5 min, followed by 35 cycles of 95°C for 30 s and 60°C for 1 min. A melt curve analysis at 60–95°C was performed after 35 cycles for assessment of the amplification specificity. Fold change in gene expression was quantified by 2−ΔΔCT method using mean Ct values for each amplification (Livak and Schmittgen, 2001).
Development of RNAi Constructs for Mi-col-1 and Lemmi-5
Partial sequences of Mi-col-1 (799 bp) and Lemmi-5 (416 bp) were amplified from their respective pGEMT clones. Lemmi-5 fragment was further cloned into pDONR 221 entry vector using attB1 and attB2 sites at the upstream and downstream of the gene sequence, respectively. Primer details are given in Table 1. pHELLSGATE12 [Obtained from Dr. Tushar Kanti Dutta, Division of Nematology, Indian Agricultural Research Institute, New Delhi] was used as a destination binary vector for development of the Lemmi-5 RNAi construct through recombination based gateway cloning technology mediated by LR recombination reaction catalyzed by LR clonase enzyme (Invitrogen). Mi-col-1 RNAi construct was developed using conventional cloning method with the binary vector pBC-6 (Yadav et al., 2006). The sense strand for Mi-col-1 was cloned with BamHI and XhoI while the antisense strand was cloned with KpnI and SacI restriction enzymes. The recombinant clones for both the genes were transformed to E. coli DH5α cells. Presence of the inserts was confirmed by colony PCR and restriction digestion. Binary plasmids containing the RNAi constructs of Lemmi-5 and Mi-col-1 were independently transformed into Agrobacterium tumefaciens EHA 105 using freeze and thaw method (Jyothishwaran et al., 2007). Selection of positive clones was done through colony PCR and restriction digestion. The positive clones were maintained in yeast extract peptone medium supplemented with selection antibiotic rifampicin (25 μg mL−1). In addition, spectinomycin (100 μg mL−1) was used for the selection of pHELLSGATE12 clones while kanamycin (100 μg mL−1) was used for the selection of pBC-6 clones.
Mobilization of the RNAi Constructs in Tomato through Agrobacterium Mediated Transformation
Tomato (cv. Pusa Ruby) seeds were treated with tween 20 for 15 min followed by 3–4 washes with double distilled water. The seeds were treated 70% ethanol for 1 min and washed with autoclaved double distilled water for 3–4 times. These seeds were further sterilized with 4% NaOCl for 8–10 min followed by 4–5 washes with autoclaved double distilled water. Sterile seeds were germinated on half strength Murashige and Skoog (MS) agar medium (pH 5.8). Cotyledonary leaves taken from 12 to 15 days old tomato seedlings were pre-cultured in Petri dishes containing pre-cultivation media (MS + 0.5 mg L−1 IAA + 1 mg L−1 zeatin) with their abaxial surface in contact with the medium at 25°C for 2 days. Healthy explants after pre-culturing were infected with A. tumefaciens EHA105 harboring the RNAi constructs for 15 min with gentle shaking. These explants were blotted on sterile filter paper and co-cultured on the same pre-cultivation medium for 48 h at 28°C with 20–25 explants in each 9 cm Petri plate. After 48 h of co-cultivation under dark conditions, the co-cultivated explants were washed with 250 mg/L cefotaxime for 30 min and blotted on sterile filter paper and transferred to selection medium (MS + 0.5 mg L−1 IAA + 1 mg L−1 zeatin + 100 mg L−1 kanamycin + 250 mg L−1cefotaxime). The selection plates were incubated at 24°C with 16/8 h of light/dark photoperiod. Healthy calluses were cut and transferred to shooting medium (MS + 0.5 mg L−1 IAA + 2 mg L−1 zeatin + 100 mg L−1 kanamycin + 250 mg L−1 cefotaxime) for shoot induction and elongation. After every 15 days, the healthy explants were sub-cultured into fresh shooting medium for shoot induction and elongation. The regenerated shoots were excised from the callus and transferred on to the rooting medium (MS + 0.5 mg L−1 IAA + 100 mg L−1 kanamycin + 250 mg L−1cefotaxime). After 20–25 days, tomato plantlets with well-developed shoots and roots were transferred to 10 cm diameter pots containing 50% soil rite mixed with autoclaved soil for hardening. After hardening, the plants were transferred to growth chambers and maintained under the controlled condition at 25 ± 2°C with a photoperiod of 16/8 h (light/dark) at National Phytotron Facility, ICAR-IARI, New Delhi.
Molecular Confirmation of T0 Transgenic Events by PCR
Healthy leaves from T0 events were used for genomic DNA isolation from cetyltrimethyl-ammonium bromide (CTAB) method (Murray and Thompson, 1980). Approximately one gram of fresh, green and healthy leaf tissues were ground to fine powder in pre-chilled mortar and pestle using liquid nitrogen. 15 mL of pre-warmed (65°C) CTAB-DNA extraction buffer (1 M Tris-HCl; 0.5 M EDTA pH 8.0; 4 M NaCl and 0.1 M β-mercaptoethanol) was added to each powdered leaf sample and each sample was transferred to autoclaved Oakridge centrifuge tubes. These tubes containing the samples were incubated at 65°C in the water bath for an hour with intermittent mixing. 15 mL of phenol: chloroform: isoamyl alcohol (25:24:1) was added to each sample and mixed gently by inverting the tubes followed by centrifugation at 20,000 × g for 15 min at room temperature. The upper aqueous layer from the tubes were carefully pipetted out and transferred into new Oakridge tubes. 12 mL of chloroform: isoamyl alcohol (24:1) mixture was added to each tube containing the aqueous layer obtained from previous step followed by centrifugation at 20,000 × g for 15 min at room temperature. The upper aqueous layer from the tubes were carefully pipetted out and transferred into new Oakridge tubes and 0.6 volume of chilled isopropanol was added to each tube and stored overnight at −20°C for precipitation. After overnight incubation, the tubes were centrifuged at 10,000 × g for 10 min at 4°C. The supernatant from each tube was discarded and pellets were washed with 2 mL of 70% ethanol by centrifugation at 10,000 × g for 10 min at 4°C. The supernatant from each tube was discarded completely and the pellets were allowed to air-dry. Each pellet sample was dissolved in 200 μl of nuclease free water. For purification of DNA, 1 μl of RNase A (10 mg/ml stock) was added to each tube and incubated at 37°C for 2 h in a dry bath. Equal volumes of phenol: chloroform: isoamyl alcohol in the ratio of 25:24:1 was added to each tube, mixed by inversion and centrifuged at 10,000 × g for 5 min at room temperature. The upper aqueous phase was collected in fresh microcentrifuge tubes, 2 volumes of chilled isopropanol was added to each tube, mixed properly by inverting and incubated overnight at −20°C. Next day, the tubes were subjected to centrifugation at 10,000 × g for 5 min at 4°C and each pellet was washed with 2 ml of 70% ethanol, air dried and dissolved in 100 μl of nuclease free water. Purity of the DNA was checked on agarose gel (0.8%) and quantification was done using nanodrop spectrophotometer (Thermo Fischer Scientific).
The presence of the transgenes in the putative T0 plants was confirmed through PCR using gene specific primers (Table 1). Each PCR mixture (25 μl) contained 100 ng of first strand cDNA, 1 × Taq buffer, 10 mmol/L dNTP, 20 μmol/L of each primer, 3.5 mmol/L MgCl2 and 1.5 U Taq DNA polymerase (Fermentas). The PCR products were separated on 1.2% agarose gel through gel electrophoresis.
Development of T0 Clones and T1 Transgenic Events
Clones of T0 tomato plants were developed through hydroponics. Growing branches from T0 plants were cut appropriately and put on conical flasks filled with water at the National Phytotron Facility. T0 clones with well-developed roots were transferred to 10 cm pots after the development of healthy roots. Fruits were harvested from T0 tomato plants, seeds were extracted, germinated and the developing seedlings were transferred to 10 cm pots.
Molecular Analysis of T1 Transgenic Tomato Events
DNA Isolation from T1 Transgenic Tomato Events and PCR
Genomic DNA was isolated and purified as described previously from putative T1 tomato plants. The presence of the transgenes in the putative T1 plants was confirmed through PCR using gene specific primers for Mi-col-1 and Lemmi-5 as described previously.
Southern Blot to Confirm the Transgene Integrity
Twenty microgram of genomic DNA isolated from T1 tomato transgenic plants were used for southern blotting through non-radioactive biotin labeling method. The genomic DNA of each transgenic event and control was digested with 50 units of BamHI and HindIII restriction enzymes in the recommended 1X NEB buffer (New England Biolabs) separately. The 50 μL reaction mixtures were incubated at 37°C overnight. The digested DNA was separated on 0.8% agarose gel. The gel was pre-run at 30 V for 10 min followed by loading of sample in the gel. The voltage applied across the gel was 90 V for 4–5 h. The gel was stained with ethidium bromide (30 μL/300 mL) for 15 min and visualized under UV light. The gel was then soaked in gel tray containing 250 mL of de-purination solution for 15 min. Subsequently, the gel was washed with single distilled water three times and soaked in 250 mL of denaturation solution with gentle shaking for 45 min on rocker platform followed by four times washing with sterile distilled water and again soaked in 250 mL sterile neutralization buffer with gentle shaking for 15 min on a rocker platform.
The assembly for dry capillary blotting was set up. Whatman paper (No. 3) wick was kept on platform in such a way that the ends dipped well in the 10X SSC. 10X SSC was poured on the wick and bubbles were removed by rolling a clean glass rod over it. The gel was carefully kept inverted on the wick. A positively charged nylon membrane (Axiva) was cut to the same size as the gel, soaked in 10X SSC for 10 min and placed over the gel. Three pieces of Whatman paper (No. 3) of the same size as the gel were soaked in 10X SSC and placed over the membrane. A clean glass rod was rolled over to remove the excess of SSC buffer and air bubbles which may interfere with the transfer process. On this, 5–7 cm thick stack of dry blotting sheets (of same size as the gel) were placed, followed by a glass-plate and a weight of 500 g. The transfer was allowed to continue for 18 h, after which the assembly was dismantled and membrane was washed with 2X SSC buffer. The DNA transferred onto the membrane was cross-linked by exposing the membrane to UV rays (120 mJ) for 90 s and the blot was stored at 4°C for further use. The probes were prepared using biotin decalabel DNA labeling kit (Thermo Fischer Scientific) according to manufacturer's protocol. The membranes were treated with the prehybridization solution for 3–4 h with shaking at 42°C followed by subsequent treatment with hybridization solution overnight at 42°C. The presence of the transgenes was detected using biotin chromogenic detection kit using manufacturer's protocol (Thermo Fischer Scientific).
Quantitative Expression of Mi-col-1 and Lemmi-5 dsRNAs in Transgenic Lines
To quantify the expression of Mi-col-1 and Lemmi-5 in the transgenic tomato events, leaves were cut from the PCR positive T1 plants and total RNA was isolated, quantified and first strand cDNA were synthesized. For analyzing the expression of the target genes, qRT-PCR was performed as described above. 18sRNA gene (S. lycopersicum) was used as an internal control. The primer details are mentioned in Table 1.
Nematode Infection Assays for Transgenic Plants Expressing Mi-col-1 and Lemmi-5 dsRNAs
T1 transgenic plants were grown in 10 cm pots in a growth chamber at 24°C with 16/8 h light/dark photoperiod. 20 days old T1 and untransformed control plants were inoculated with approximately 1000 freshly hatched J2s and were grown at National Phytotron Facility, New Delhi at 24°C with 16/8 h light/dark photoperiod for 35 days. After 35 days, the plants were uprooted, the roots were washed, and total number of egg masses, eggs per egg mass, galls and adult females were counted for each individual plant. The size and shape of the adult females were observed under the microscope (Eclipse 80i and stereomicroscope SMZ1000, Tokyo, Japan) and images were captured using Nikon DS-Fi2 camera attached to the microscope. The area and diameter of the females isolated from wild type and transgenic plants were measured using the microscope software. Nematode multiplication factor [(number of egg masses × number of eggs per egg mass) ÷ nematode inoculum level] was also calculated for each plant to assess the effect of host generated RNAi on reproductive potential of M. incognita. Sets of data obtained for T1 transgenic plants expressing Mi-col-1 and Lemmi-5 dsRNAs were compared with that of wild type untransformed plants. Six replicates per treatment were used for this study and the data sets were subjected to one-way analysis of variance (ANOVA). The means were reported as significant or non-significant according to the Tukey's test (P < 0.05) using SPSS statistics software (Version-2.0, IBM, Chicago, USA).
Target Gene Expression Analysis in Adult Females Extracted from Transgenic Plants
Adult females were extracted from untransformed wild type and T1 transgenic plants expressing Mi-col-1 and Lemmi-5 dsRNAs 35 days post inoculation. Total RNA was isolated from these adult females and first strand cDNA was synthesized as described above. Transcript abundance of Mi-col-1 and Lemmi-5 was quantified using real time PCR as described above. M. incognita 18s rRNA was used as internal control gene. Primer details are given in Table 1. Two biological and three technical replicates were used for this study.
Results
In Silico Analysis of Mi-col-1 and Lemmi-5
Nucleotide sequences of Mi-col-1 (Accession no. U40766) and Lemmi-5 (Accession no. AF006727) were retrieved from NCBI Genbank. The nucleotide sequences of these two genes showed no significant similarities. Further, the conserved cystein patterns of the deduced amino acid sequences revealed that Mi-col-1 belongs to group 3 of nematode cuticle collagens, while Lemmi-5 belongs to group 1a according to the classification proposed by Johnstone (2000) (Figure S1). Primary structural properties of the deduced amino acid sequences are listed in Table S1. SMART domain analysis revealed the presence of nematode cuticle collagen N terminal domains in Mi-col-1 (between amino acids 12–64), as well as Lemmi-5 (between amino acids 5–58). In Mi-col-1, two pfam collagen domain between amino acids 160–219 and 220–219 were also predicted (Figure S2). Presence of a transmembrane domain was predicted for both the sequences (Figure S3), however, amino acid composition in the domain varied for Mi-col-1 and Lemmi-5. Phylogenetic tree based on predicted amino acid sequences showed two clusters. Lemmi-5 formed a separate sub-cluster within cluster I, while Mi-col-1 was placed in cluster II and showed close relationship with Mi-col-2 and Mj-col-3 (Figure 1).
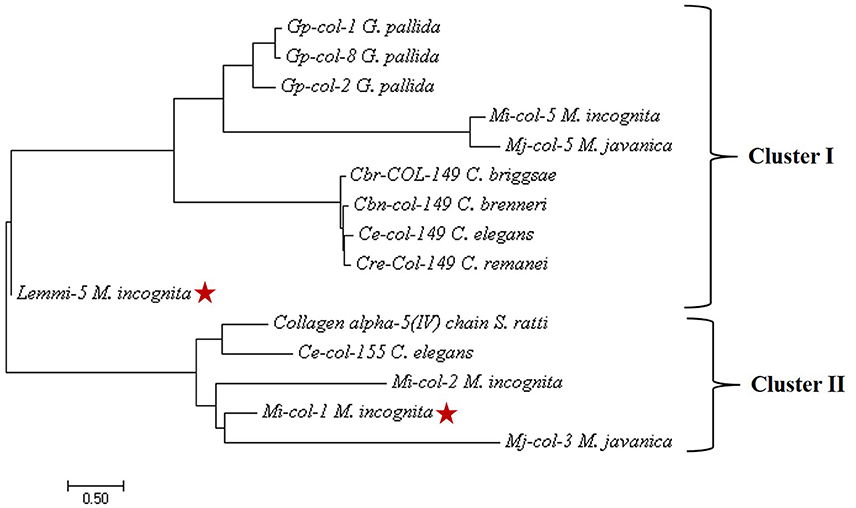
Figure 1. Phylogenetic tree constructed by maximum likelihood method using MEGA 7 showing evolutionary relationship of Mi-col-1 and Lemmi-5 with cuticle collagens of other plant parasitic, animal parasitic and free living nematodes.
Differential Expression of Mi-col-1 and Lemmi-5 at Different Life Stages of M. incognita
Relative expression of Mi-col-1 and Lemmi-5 in egg masses, J2s and adult female stages of M. incognita was quantified through qRT-PCR. Expression of both the genes were found to be maximum in adult females followed by egg masses and J2s. Using expression levels in pre parasitic J2s as reference, 11.8 folds higher expression of Mi-col-1 was observed in adult females, while around 7 folds higher expression of Mi-col-1 was found in egg masses. Similarly, expression of Lemmi-5 was around 22.5 and 11.3 folds higher in adult females and egg masses, respectively compared to its expression in pre parasitic J2s (Figure 2).
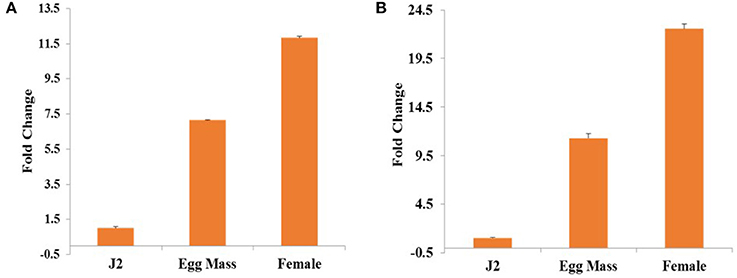
Figure 2. Differential expression of (A) Mi-col-1 and (B) Lemmi-5 in different life stages of M. incognita through qRT-PCR. 18s rRNA (M. incognita) gene has been used as an internal control. Each bar represents the mean ± SE of n = 3.
Transformation of Tomato Plants and Their Molecular Analysis for T-DNA Integration
dsRNA constructs of Mi-col-1 and Lemmi-5 were transformed into tomato plants through A. tumifaciens mediated transformation to generate T0 population (Figure 3). Preliminary screening of T0 events was carried out by PCR with target gene specific primers. Amplification of 799 bp gene specific DNA of Mi-col-1 was observed in all the nine T0 events of Mi-col-1. Similarly, 416 bp long fragment was amplified from all the eight putative events of Lemmi-5 (Figure 4).

Figure 3. Different stages in the development in transgenic tomato plants carrying Mi-col-1 and Lemmi-5 dsRNAs independently. (A) Tomato leaf explants in the precultivation medium, (B) Explants in the selection medium, (C) Development of callus in the selection medium, (D) Development of shoots in the shooting medium, (E) Development of roots in the rooting medium, (F) Putative transgenics in the pots for hardening, (G) Further growth and hardening of putative transgenic plants in the National Phytotron Facility, (H) Development of fruits in the putative transgenic plants.
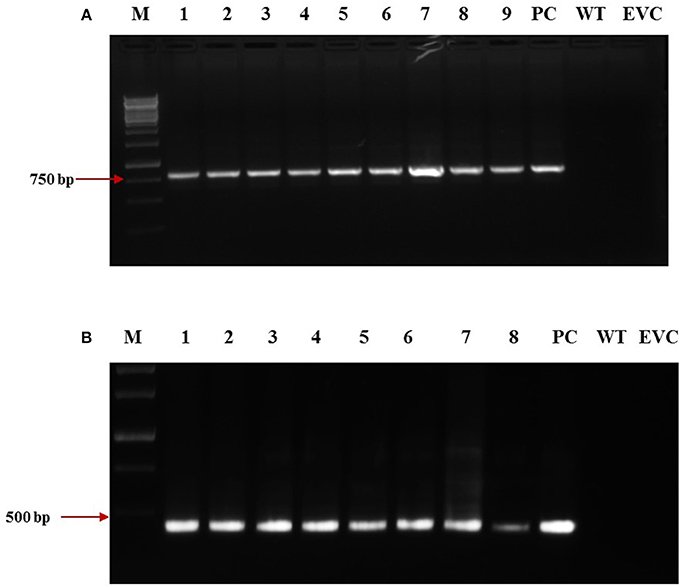
Figure 4. Molecular confirmation of T0 transgenic events for the presence of the transgene through PCR using gene specific primers. (A) Amplification of Mi-col-1 insert from the DNA isolated from putatively transgenic events of Mi-col-1. Lane M: 1 kb ladder, lanes 1–9: T0 transgenic events, lane PC: positive control (pGEMT clone of Mi-col-1), lane WT, Wild type; lane EVC, Empty vector control; (B) Amplification of Lemmi-5 insert from the DNA isolated from putatively transgenic events of Lemmi-5. Lane M: 1 kb ladder, lanes 1–8: T0 transgenic events, lane PC, positive control (pGEMT clone of Lemmi-5); lane WT, Wild type; lane EVC, Empty vector control.
Molecular Analysis of T1 Transgenic Plants through PCR and Southern Blotting
T1 plants were generated from the seeds obtained from T0 plants grown in the NPF, ICAR-IARI, New Delhi. A total of 7 T1 events were generated for Mi-col-1, while 6 events were generated for Lemmi-5. Preliminary screening of T1 plants was carried out by PCR with target gene specific primers. Genomic DNA was isolated from leaf tissues of putative T1 plants of Mi-col-1 and Lemmi-5. Presence of transgene was detected in all the T1 events for Mi-col-1 and Lemmi-5 RNAi transgenic lines. The PCR confirmed T1 events for Mi-col-1 and Lemmi-5 were further subjected to southern blotting for T-DNA integration and copy number analysis. Out of the seven Mi-col-1 transgenic events, C1.4, had two insertions, while all other events had single insertions. All the six transgenic events for Lemmi-5 had single insertions. Hybridization bands were not detected in wild type untransformed and empty vector controls (Figure 5).
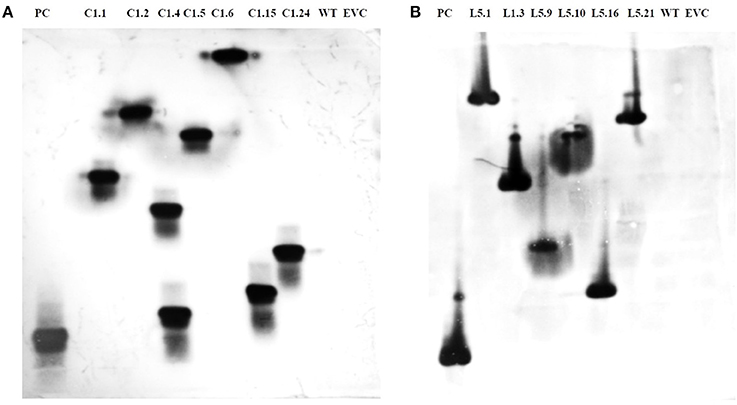
Figure 5. Southern blot depicting T-DNA integration in T1 plants harboring dsRNA of (A) Mi-col-1 and (B) Lemmi-5. Mi-col-1events are C1.1, C1.2, C1.4, C1.5, C1.6, C1.15 and C1.24; Lemmi-5 events are L1.1, L1.3, L1.9, L1.10, L1.16, and L1.21; PC denotes positive control probe of respective genes; WT denotes untransformed wild type tomato “Pusa Ruby,” EVC denotes empty vector controls for the respective gene constructs.
Quantitative Expression of Mi-col-1 and Lemmi-5 in T1 Transgenic Events
dsRNA transcript accumulation and expression was validated by qRT-PCR analysis of the selected transgenic events of Mi-col-1 and Lemmi-5. An increased transcript expression was observed in all the events for Mi-col-1 and Lemmi-5 as compared to the wild type. However, variation in the expression was observed among the respective events. Event C1.4 showed highest expression level of Mi-col-1 dsRNA among all the events in terms of average ΔCT values, while Lemmi-5 dsRNA expression was found to be highest in the event L5.16 (Figure 6). These results further insured the expression of the desired dsRNAs in the transgenic events of Mi-col-1 and Lemmi-5.
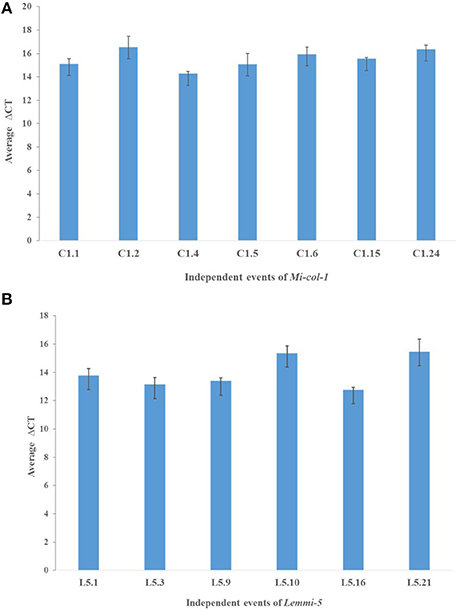
Figure 6. Expression analysis of (A) Mi-col-1 and (B) Lemmi-5 dsRNAs in their respective transgenic tomato events using qRT-PCR. 18s rRNA (S. lycopersicum) gene has been used as an internal control. Each bar represents the mean ± SE of n = 3.
Effect of Host Delivered RNAi on M. incognita Infection on T1 Tomato Lines
In order to study the effect of host delivered RNAi of Mi-col-1 and Lemmi-5 on M. incognita infection in tomato, T1 plants confirmed for the presence and expression of Mi-col-1 and Lemmi-5 were inoculated with 1,000 freshly hatched J2s/pot. T1 plants of seven independent events were evaluated in case of Mi-col-1, while six independent events were evaluated for Lemmi-5 transgenic plants 35 days post infection. Nematode infection was scored and assessed by means of number of galls, females, egg masses, eggs per egg masses and size of adult females in T1 plants in comparison with wild type plants (Figures 7, 8). Reduction in the number of adult females was observed in T1 plants as compared to the wild type and the percentage of reduction was 30.80–35.00% for Mi-col-1 transgenic lines and 34.15–38.54% for Lemmi-5 transgenic lines. Furthermore, the size of the adult females was heavily distorted and deformed in both Mi-col-1 and Lemmi-5 transgenic lines (Figure 9). Mean area of females isolated from wild type plants was calculated as 86,580.10 μm2. Whereas, the mean area of females isolated from transgenic plants expressing Mi-col-1 and Lemmi-5 dsRNA were calculated as 50,854.10 and 37,681.40 μm2, respectively. Similarly, mean diameter of females isolated from wild type plants was calculated as 360.10 μm; whereas, the mean diameter of females isolated from transgenic plants expressing Mi-col-1 and Lemmi-5 dsRNA were calculated as 231.00 and 199.70 μm, respectively. Statistical analysis revealed significant reduction in the area and diameter of the females isolated from transgenic plants expressing Mi-col-1 and Lemmi-5 as compared to those isolated from wild type plants (Table 2). The fecundity of the nematodes also got hampered as indicated by the reduction in number of egg masses/plant in the range of 50.06–65.73% and 57.30–66.44% in Mi-col-1 and Lemmi-5 RNAi transgenic lines, respectively. The number of eggs/egg masses also reduced significantly in the range of 76.07–82.59% and 67.13–79.56% in Mi-col-1 and Lemmi-5 RNAi lines, respectively. Nematode multiplication factor (MF) determines successful establishment of the concerned nematode in the host plants by representing parasitic and reproductive fitness of the nematode. The multiplication factor of the nematodes in the transgenic events expressing Mi-col-1 was calculated to be 1.51–2.17 among the events as compared to a multiplication factor of 20.39 in the wild type untransformed plants. Similarly, in transgenic events expressing Lemmi-5 dsRNA, the multiplication factor ranged from 1.48 to 2.49 as compared to a multiplication factor of 20.35 in the wild type untransformed plants. However, No significant reduction in number of galls was observed in the transgenic events as compared to the wild type.
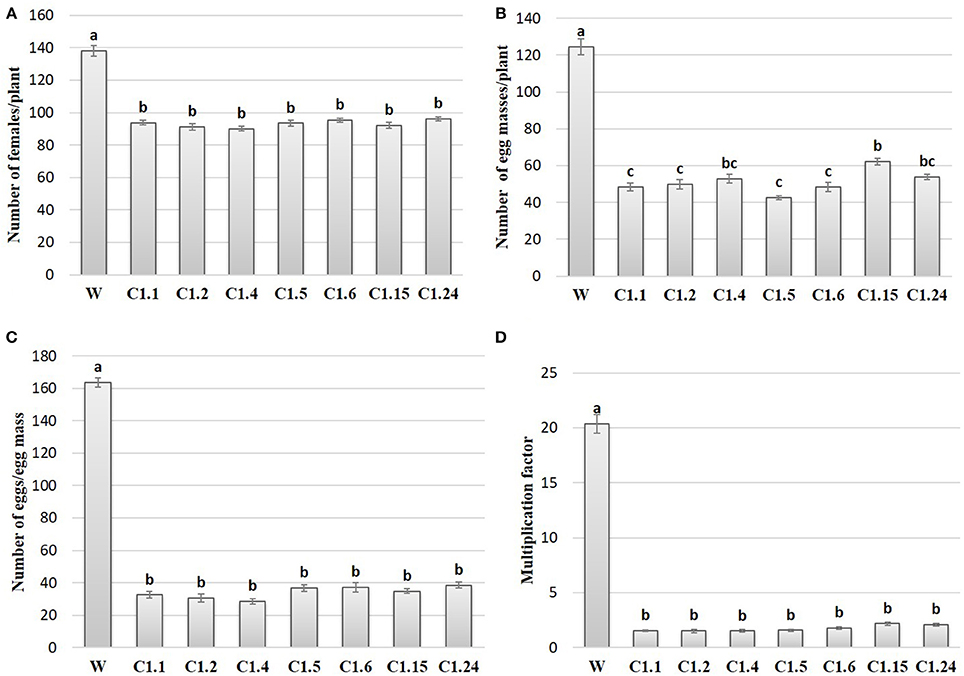
Figure 7. Effect of host generated RNAi of Mi-col-1 on (A) relative number of females/plant; (B) egg masses/plant; (C) eggs/egg mass; and (D) multiplication factor in transgenic tomato events (C1.1, C1.2, C1.4, C1.5, C1.6, C1.15, and C1.24) and wild type (W) tomato plants after 35 days of inoculation. Each bar represents mean ± standard error of six different plants of each event. Bars with different letters indicate statistically significant difference between the treatments (P < 0.05) according to Tukey's test (Tukey's HSD).
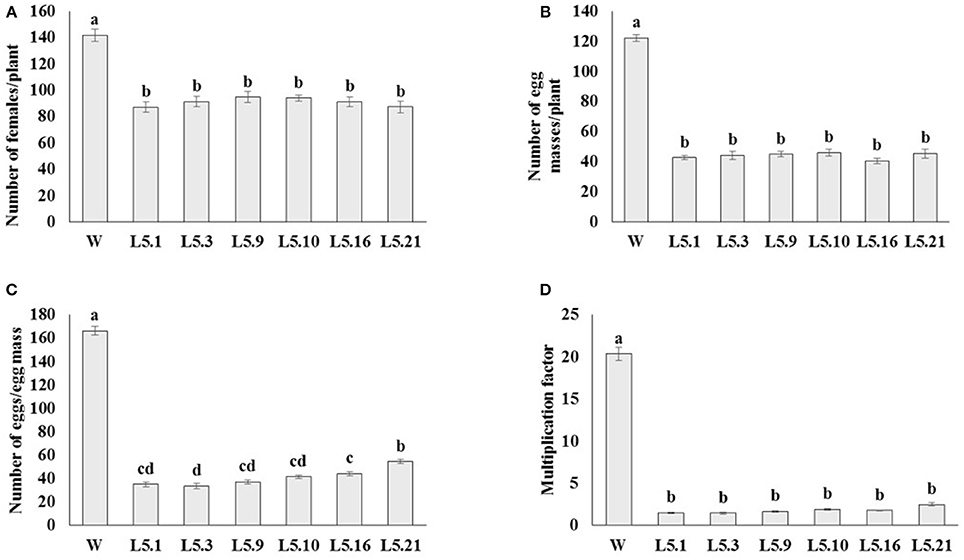
Figure 8. Effect of host generated RNAi of Lemmi-5 on (A) relative number of females/plant, (B) egg masses/plant, (C) eggs/egg mass, and (D) multiplication factor in transgenic tomato events (L5.1, L5.3, L5.9, L5.10, L5.16, and L5.21) and wildtype (W) tomato plants after 35 days of inoculation. Each bar represents mean ± standard error of six different plants of each event. Bars with different letters indicate statistically significant difference between the treatments (P < 0.05) according to Tukey's test (Tukey's HSD).
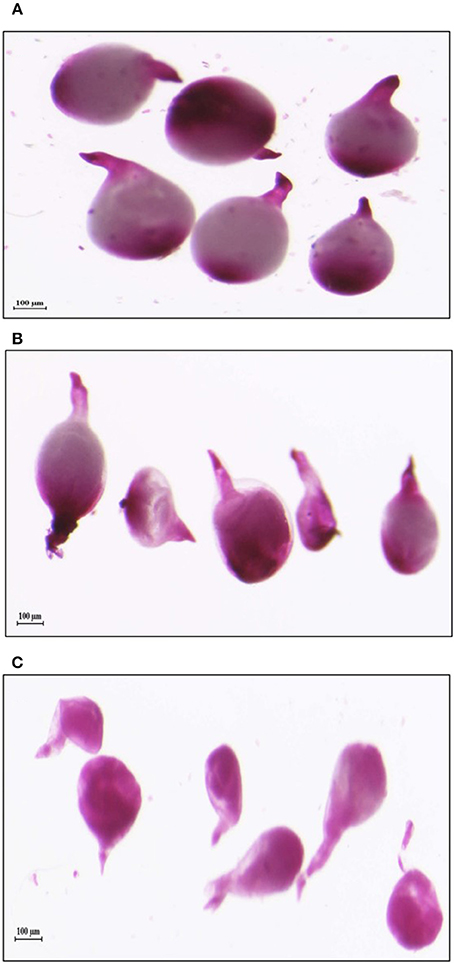
Figure 9. Deformation in the shape of mature females isolated from tomato plants expressing dsRNAs of (B) Mi-col-1 and (C) Lemmi-5 as compared to the healthy females isolated from (A) Wild type, 35 days post inoculation.

Table 2. Area and diameter of Meloidogyne incognita females isolated from wild type and transgenic RNAi lines.
qRT-PCR analysis of Mi-col-1 and Lemmi-5 gene expression in the females isolated from the roots of transgenic tomato events expressing the respective dsRNAs revealed significant down regulation of the target genes further indicating successful host delivered RNAi in transgenic plants. Expression levels of Mi-col-1 was reduced in the range of 67.69–91.30% in the females isolated from tomato lines expressing Mi-col-1 dsRNA. Similarly, the reduction in the expression levels of Lemmi-5 was found to be in the range of 79.55–96.12% in the females extracted from the tomato transgenic lines expressing Lemmi-5 dsRNA (Figure 10).
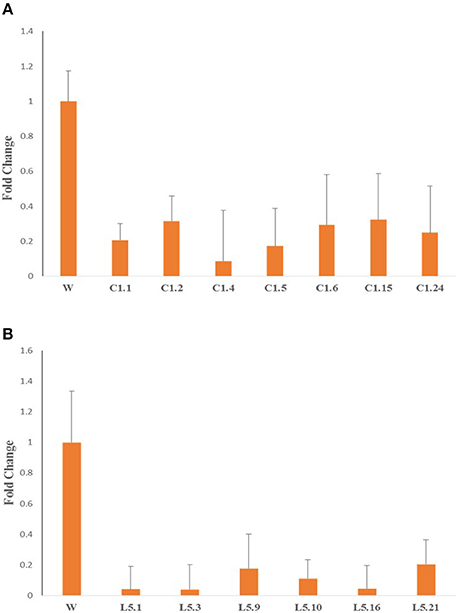
Figure 10. Expression of target genes (A) Mi-col-1, and (B) Lemmi-5 in adult females isolated from respective transgenic tomato lines 35 days post inoculation.
Discussion
The cuticle of RKNs like M. incognita performs multiple functions like protection from external environment, interaction with soil and host environment, movement and locomotion and defines the shape and development of the nematode during its pre-parasitic and parasitic life cycle. About 80% of the nematode cuticle consists of collagens (Kingston, 1991). Although more than 150 cuticle collagen genes have been characterized in C. elegans, only a few genes have been identified in PPNs. In M. incognita; Mi-col-1, Mi-col-2, Lemmi-5, and Mi-col-5 have been characterized and are reported to have differential expression at different stages of its life cycle (Van Der Eycken et al., 1994; Ray and Hussey, 1995; Wang et al., 1998; Banerjee et al., 2017a). Mutations in the cuticle collagen genes in C. elegans have resulted in morphological defects like dumpy, roller and blister phenotypes leading to larval and embryonic death (Johnstone, 2000; Page and Johnstone, 2007). RNAi of four cuticle collagen genes known as dumpy genes (Bx-dpy-2, 4, 10, and 11) resulted in morphological aberrations like small dumpy body size and reduced body length in the pinewood nematode, Bursaphelenchus xylophilus (Wang et al., 2016). The authors demonstrated that the nematodes fed on filamentous fungus Fusarium oxysporum transformed with the target dsRNA constructs brought about RNAi silencing of the target dumpy genes in B. xylophilus. However, our study is the first report on the host generated RNAi of cuticle collagen genes in PPNs. Multiple molting of M. incognita during its life cycle makes the cuticle collagen genes potential targets for silencing to understand the effect and lethality of individual genes and possible chances of its effect on the overall development and parasitism of M. incognita.
The target genes Mi-col-1 and Lemmi-5 did not show similarities at the nucleotide and amino acid level. The conserved pattern of cystein residues showed that they belong to different groups of cuticle collagen genes. Phylogenetic analysis based on amino acid sequences also placed them in different clusters. However, pattern of differential expression of these genes at different life stages of the life cuticle of M. incognita is similar. In agreement with the earlier reports (Ray and Hussey, 1995; Wang et al., 1998), both the genes showed maximum expression in adult females followed by egg masses and J2s. However, relative folds of expression in adult females and egg masses compared to J2s were higher in case of Lemmi-5. Hence, despite their structural differences, both the genes appear to be involved in the cuticle development, maintenance and thickening of adult females and eggs. Unlike Mi-col-1 and Lemmi-5, a recently characterized cuticle collagen gene Mi-col-5 was reported to express more in egg masses followed by adult females and J2s (Banerjee et al., 2017a). Therefore, like C. elegans, the cuticle collagen formation seems to be governed by different collagen genes at different stages of the life cycle of PPNs like M. incognita.
Transgenic tomato plants independently expressing Mi-col-1 and Lemmi-5 dsRNAs were developed through Agrobacterium mediated transformation. The integration and inheritance of the transgenes were demonstrated by PCR and southern blotting. The expression of the dsRNAs in the respective transgenic lines analyzed through qRT-PCR also indicated stable transformation of the T-DNA harboring the dsRNA constructs of the target genes. Transgenic tomato lines were inoculated with M. incognita J2s to evaluate the effect of silencing of the target cuticle collagen genes through host delivered RNAi. Significant reduction in number of adult females, number of egg masses per plant and number of eggs per egg masses was observed. This indicates that both Mi-col-1 and Lemmi-5 are involved in the development and maintenance of the adult female cuticle which also seems to have affected the fecundity of nematodes and their ability to lay eggs as reflected by the steep reduction in the number of eggs and eggs per egg masses in the independent tomato lines expressing Mi-col-1 and Lemmi-5 dsRNA. Reduction in the nematode multiplication factor was also significant indicating successful repression of the nematode establishment and reproduction in the transgenic lines. Microscopic observation also revealed clear deformations in the structure of adult females feeding on these transgenic tomato lines compared to those feeding on wild type plants. The deformed structure of the adult females might have affected their reproductive potential which can be correlated to the higher expression of both the genes at the adult female stage of M. incognita. This could best be answered by studying the cuticle ultrastructure of these females in comparison to normally developed nematode females, but it is beyond the scope of this research work. However, it can be thought that because of improper structural development of the cuticle, the nematode female may experience non-conducive effect of the host environment, thereby affecting the female nematode's fecundity. Permeability of host molecules because of improper development of cuticle of the adult female might also have affected their reproductive potential. However, the knockdown of Mi-col-1 and Lemmi-5 was not able to bring about any significant reduction in gall development which indicates that these genes might not be involved in the early stages of nematode development and cuticle formation. In tomato, host generated RNAi of Mi-Rpn7 gene of M. incognita also showed reduction in number of egg mass per gram of root tissue and number of eggs per gram of root tissue without any significant reduction in the number of galls (Niu et al., 2012). Similarly, gall formation was not affected by host delivered RNAi of isocitrate lyase (ICL) of M. incognita in tobacco, while substantial reduction in egg oviposition was reported in transgenic lines compared to wild type (Lourenço-Tessutti et al., 2015).
Expression of Mi-col-1 and Lemmi-5 dsRNAs in respective transgenic T1 plants was confirmed through real time PCR. This approach was used in earlier studies to analyze the target transcript abundance in the T1 plants (Papolu et al., 2013; Dutta et al., 2015). The average ΔCT values reflect the expression of target dsRNAs in the transgenic plants. Furthermore, qRT-PCR analysis of the target genes was carried out from cDNA of the adult females developed on transgenic tomato plants independently expressing Mi-col-1 and Lemmi-5 dsRNAs to analyse the long term effect and heritable nature of host generated RNAi. Significant reduction in the expression of the target genes was observed in adult females extracted from the transgenic plants. This indicates successful silencing of the target genes, Mi-col-1 and Lemmi-5 through uptake of dsRNAs/siRNAs via host generated RNAi in M. incognita and also suggests transmittance of the RNAi effect along subsequent moltings of M. incognita to the adult female stage. This systemic and heritable nature of the RNAi in PPNs was also reported in earlier studies (Fairbairn et al., 2007; Papolu et al., 2013; Dutta et al., 2015; Zhuo et al., 2016). However, it is not clear whether M. incognita directly ingested the dsRNAs from the host and generated the siRNAs through their own machinery or they ingested the plant processed siRNAs to bring about silencing of the target genes. Both the mechanisms are possible since RKNs are reported to ingest relatively large biomolecules (Urwin et al., 1997; Dutta et al., 2015).
Host generated RNAi of Mi-col-1 and Lemmi-5 has significantly hampered structure and reproduction in M. incognita. However, complete resistance against M. incognita could not be obtained. This could be due to the fact that cuticle collagen genes belong to multigene family, therefore possibility of compensatory functions of similar genes could not be denied. However, the reduction in the nematode numbers and nematode multiplication factor is comparable to the previously reported studies on host delivered RNAi to combat PPNs (reviewed by Banerjee et al., 2017b). Stacking of multiple genes for developing RNAi constructs for targeting more than one system of the nematode may be an efficient approach to achieve better degree of resistance in future studies. Application of conventional breeding for crossing two RNAi lines expressing different target leading to combined expression of both the dsRNAs also has additive effect on reduction in nematode numbers and development (Charlton et al., 2010). Gene pyramiding of PPN genes and other pathogens like virus and bacteria can also provide broad spectrum resistance against different pathogens (Walawage et al., 2013). RNAi based transgenics provide a relatively more biosafe option for developing genetically modified crops as functional proteins are not produced from the dsRNAs, which minimizes non-target effects. Moreover, humans commonly consume plants infected with virus, which produce molecules quite similar to dsRNAs or siRNAs. Therefore, these are not alien entities to human body and cells. Identification of suitable lethal target genes can be facilitated by data mining from the whole genome sequences of the key PPNs for engineering better resistance. Use of tissue specific and nematode induced promoters may address the biosafety concerns better by limiting the dsRNA expression in response to specific nematodes and in specific plant tissues.
Author Contributions
AS, SG, PJ, KS, and SB conceived and designed the experiments. SB performed the experiments. SB and BG analyzed the data. SB wrote the manuscript. AS, KS, PJ, and SG critically revised the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors gratefully acknowledge the financial support from Indian Council of Agricultural Research (ICAR) through National Agricultural Innovative Project (NAIP/C4/C1092) and National Fund for Basic Strategic and Frontier Application Research in Agriculture (NFBSFARA/RNA-3022/2012-13).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2017.02266/full#supplementary-material
References
Antonino de Souza Júnior, J. D., Ramos Coelho, R., Tristan Lourenço, I., da Rocha Fragoso, R., Barbosa Viana, A. A., Lima Pepino de Macedo, L., et al. (2013). Knocking- down Meloidogyne incognita proteases by plant-delivered dsRNA has negative pleiotropic effect on nematode vigor. PLoS ONE 8:e85364. doi: 10.1371/journal.pone.0085364
Atkinson, H. J., Urwin, P. E., and Mcpherson, M. J. (2003). Engineering plants for nematode resistance. Annu. Rev. Phytopathol. 41, 615–639. doi: 10.1146/annurev.phyto.41.052002.095737
Bakhetia, M., Charlton, W., Atkinson, H. J., and McPherson, M. J. (2005). RNA interference of dual oxidase in the plant nematode Meloidogyne incognita. Mol. Plant-Microbe Interact. 18, 1099–1106. doi: 10.1094/MPMI-18-1099
Banerjee, S., Gill, S. S., Jain, P. K., and Sirohi, A. (2017a). Isolation, cloning and characterization of a cuticle collagen gene, Mi-col-5 in Meloidogyne incognita. 3 Biotech. 7, 64. doi: 10.1007/s13205-017-0665-1
Banerjee, S., Banerjee, A., Gill, S. S., Gupta, O. P., Dahuja, A., Jain, P. K., et al. (2017b). RNA interference: a novel source of resistance to combat plant parasitic nematodes. Front. Plant Sci. 8:834. doi: 10.3389/fpls.2017.00834
Charlton, W. L., Harel, H. Y. M., Bakhetia, M., Hibbard, J. K., Atkinson, H. J., and Mcpherson, M. J. (2010). Additive effects of plant expressed double-stranded RNAs on root-knot nematode development. Int. J. Parasitol. 40, 855–864. doi: 10.1016/j.ijpara.2010.01.003
Davies, K. G., and Curtis, R. S. G. (2011). Cuticle surface coat of plant-parasitic nematodes. Ann. Rev. Phytopathol. 49, 135–156. doi: 10.1146/annurev-phyto-121310-111406
Dinh, P. T. Y., Brown, C. R., and Elling, A. A. (2014a). RNA interference of effector gene Mc16D10L confers resistance against Meloidogyne chitwoodi in Arabidopsis and Potato. Phytopathology 104, 1098–1106. doi: 10.1094/PHYTO-03-14-0063-R
Dinh, P. T. Y., Zhang, L., Brown, C. R., and Elling, A. A. (2014b). Plant-mediated RNA interference of effector gene Mc16D10L confers resistance against Meloidogyne chitwoodi in diverse genetic backgrounds of potato and reduces pathogenicity of nematode offspring. Nematology 16, 669–682. doi: 10.1163/15685411-00002796
Dubreuil, G., Magliano, M., Deleury, E., Abad, P., and Rosso, M. N. (2007). Transcriptome analysis of root knot nematode functions induced in early stage of parasitism. New Phytol. 176, 426–436. doi: 10.1111/j.1469-8137.2007.02181.x
Dutta, T. K., Papolu, P. K., Banakar, P., Choudhary, D., Sirohi, A., and Rao, U. (2015). Tomato transgenic plants expressing hairpin construct of a nematode protease gene conferred enhanced resistance to root-knot nematodes. Front. Microbiol. 6:260. doi: 10.3389/fmicb.2015.00260
Elling, A. A. (2013). Major emerging problems with minor Meloidogyne species. Phytopathology 103, 1092–1102. doi: 10.1094/PHYTO-01-13-0019-RVW
Fairbairn, D. J., Cavallaro, A. S., Bernard, M., Mahalinga-Iyer, J., Graham, M. W., and Botella, J. R. (2007). Host-delivered RNAi: an effective strategy to silence genes in plant parasitic nematodes. Planta 226, 1525–1533. doi: 10.1007/s00425-007-0588-x
Gleason, C. A., Liu, Q. L., and Williamson, V. M. (2008). Silencing a candidate nematode effector gene corresponding to the tomato resistance gene Mi-1 leads to acquisition of virulence. Mol. Plant Microbe Interact. 21, 576–585. doi: 10.1094/MPMI-21-5-0576
Hooper, D. J. (1986). “Extraction of free-living stages from soil,” in Laboratory methods for work with plant and soil nematodes, ed J. F. Southey (London: Ministry of agriculture, fisheries and food), 5–30.
Huang, G., Allen, R., Davis, E. L., Baum, T. J., and Hussey, R. S. (2006). Engineering broad root-knot resistance in transgenic plants by RNAi silencing of a conserved and essential root-knot nematode parasitism gene. Proc. Natl. Acad. Sci. U.S.A. 103, 14302–14306. doi: 10.1073/pnas.0604698103
Jain, R. K., Mathur, K. N., and Singh, R. V. (2007). Estimation of losses due to plantparasitic nematodes on different crops in India. Ind. J. Nematol. 37, 219–220. doi: 10.1016/j.proenv.2015.07.270
Johnstone, I. L. (2000). Cuticle collagen genes expression in Caenorhabditis elegans. Trends Genet. 16, 21–27. doi: 10.1016/S0168-9525(99)01857-0
Jones, M. G. K., and Northcote, D. H. (1972). Multinucleate transfer cells induced in coleus roots by the root-knot nematode, Meloidogyne arenaria. Protoplasma 75, 381–395. doi: 10.1007/BF01282117
Jyothishwaran, G., Kotresha, D., Selvaraj, T., Srideshikan, S. M., Rajvanshi, P. K., and Jayabaskaran, C. (2007). A modified freeze-thaw method for efficient transformation of Agrobacterium tumefaciens. Curr. Sci. 6, 770–772. Available online at: http://www.jstor.org/stable/24099118
Kimber, M. J., McKinney, S., McMaster, S., Day, T. A., Flemming, C. C., and Maule, A. G. (2007). Flp gene disruption in a parasitic nematode reveals motor disfunction and unusual neuronal sensitivity to RNA interference. FASEB J. 21, 1233–1243. doi: 10.1096/fj.06-7343com
Kingston, I. B. (1991). Nematode collagen genes. Parasitol. Today 7, 11–15. doi: 10.1016/0169-4758(91)90077-2
Kumar, A., Kakrana, A., Sirohi, A., Subramaniam, K., Srinivasan, R., Abdin, M. Z., et al. (2017). Host-delivered RNAi-mediated root-knot nematode resistance in Arabidopsis by targeting splicing factor and integrase genes. J. Gen. Plant Pathol. 83, 91–97. doi: 10.1007/s10327-017-0701-3
Letunic, I., Doerks, T., and Bork, P. (2015). SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 43, D257–D260. doi: 10.1093/nar/gku949
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lourenço-Tessutti, I. T., Souza Junior, J. D. A., Martins-de-Sa, D., Viana, A. A. B., Carneiro, R. M. D. G., Togawa, R. C., et al. (2015). Knock-down of heat-shock protein 90 and isocitrate lyase gene expression reduced root-knot nematode reproduction. Phytopathology 105, 628–637. doi: 10.1094/PHYTO-09-14-0237-R
Moens, M., Perry, R. N., and Starr, J. L. (2009). “Meloidogyne spp.-a diverse group of novel and important plant parasite,” in Root-Knot Nematodes, eds R. N. Perry, M. Moens and J. L. Starr (Wallingford; Oxfordshire: CAB International), 1–17.
Murray, M. G., and Thompson, W. F. (1980). Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4326. doi: 10.1093/nar/8.19.4321
Niu, J. H., Jian, H., Xu, J., Chen, C., and Guo, Q. (2012). RNAi silencing of the Meloidogyne incognita Rpn7 gene reduces nematode parasitic success. Eur. J. Plant Pathol. 134, 131–144. doi: 10.1007/s10658-012-9971-y
Niu, J., Liu, P., Liu, Q., Chen, C., Guo, Q., Yin, J., et al. (2016). Msp40 effector of root-knot nematode manipulates plant immunity to facilitate parasitism. Sci. Rep. 6:19443. doi: 10.1038/srep19443
Page, A. P., and Johnstone, I. L. (2007). “The cuticle,” in WormBook. The C. elegans Research Community (Pasadena, CA: WormBook).
Papolu, P. K., Gantasala, N. P., Kamaraju, D., Banakar, P., Sreevathsa, R., and Rao, U. (2013). Utility of host delivered RNAi of two FMRF amide like peptides, flp-14 and flp-18, for the management of root knot nematode, Meloidogyne incognita. PLoS ONE 8:e80603. doi: 10.1371/journal.pone.0080603
Park, J. E., Lee, K. Y., Lee, S. J., Oh, W. S., Jeong, P. Y., Woo, T., et al. (2008). The efficiency of RNA interference in Bursaphelenchus xylophilus. Mol. Cells 26, 81–86.
Ray, C., and Hussey, R. S. (1995). Evidence for proteolytic processing of a cuticle collagen in a plant-parasitic nematode. Mol. Biochem. Parasit. 72, 243–246. doi: 10.1016/0166-6851(95)00082-C
Rosso, M. N., Dubrana, M. P., Cimbolini, N., Jaubert, S., and Abad, P. (2005). Application of RNA interference to root-knot nematode genes encoding esophageal gland proteins. Mol. Plant Microbe Interact. 18, 615–620. doi: 10.1094/MPMI-18-0615
Shingles, J., Lilley, C. J., Atkinson, H. J., and Urwin, P. E. (2007). Meloidogyne incognita: molecular and biochemical characterization of a cathepsin L cysteine proteinase and the effect on parasitism following RNAi. Exp. Parasitol. 115, 114–120. doi: 10.1016/j.exppara.2006.07.008
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Trudgill, D. L., and Blok, V. C. (2001). Apomictic, polyphagous root-knot nematodes: exceptionally successful and damaging biotrophic root pathogens. Annu. Rev. Phytopathol. 39, 53–77. doi: 10.1146/annurev.phyto.39.1.53
Urwin, P. E., Lilley, C. J., and Atkinson, H. J. (2002). Ingestion of double-stranded RNA by pre-parasitic juvenile cyst nematodes leads to RNA interference. Mol. Plant Microbe Interact. 15, 747–752. doi: 10.1094/MPMI.2002.15.8.747
Urwin, P. E., Moller, S. G., Lilley, C. J., McPherson, M. J., and Atkinson, H. J. (1997). Continual green fluorescent protein monitoring of cauliflower mosaic virus 35S promoter activity in nematode-induced feeding cells in Arabidopsis thaliana. Mol. Plant Microbe Interact. 1, 394–400. doi: 10.1094/MPMI.1997.10.3.394
Van Der Eycken, W., Dealmeidaengler, J., Vanmontagu, M., and Gheysen, G. (1994). Identification and analysis of a cuticular collagen coding gene from the plant-parasitic nematode Meloidogyne incognita. Gene 151, 237–242. doi: 10.1016/0378-1119(94)90663-7
Walawage, S. L., Britton, M. T., Leslie, C. A., Uratsu, S. L., Li, Y., and Dandekar, M. (2013). Stacking resistance to crown gall and nematodes in walnut rootstocks. BMC Genomics 14:668. doi: 10.1186/1471-2164-14-668
Wang, M., Wang, D., Zhang, X., Wang, X., Liu, W., Hou, X., et al. (2016). Double-stranded RNA-mediated interference of dumpy genes in Bursaphelenchus xylophilus by feeding on filamentous fungal transformants. Int. J. Parasitol. 46, 351–360. doi: 10.1016/j.ijpara.2016.01.008
Wang, T., Deom, C. M., and Hussey, R. S. (1998). Identification of a Meloidogyne incognita cuticle collagen gene and characterization of the developmental expression of three collagen genes in parasitic stages. Mol. Biochem. Parasit. 93, 131–134. doi: 10.1016/S0166-6851(98)00018-8
Xue, B., Hamamouch, N., Li, C., Huang, G., Hussey, R. S., Baum, T. J., et al. (2013). The 8D05 parasitism gene of Meloidogyne incognita is required for successful infection of host roots. Phytopathology 103, 175–181. doi: 10.1094/PHYTO-07-12-0173-R
Yadav, B. C., Veluthambi, K., and Subramaniam, K. (2006). Host generated double stranded RNA induces RNAi in plant parasitic nematodes and protects the host from infection. Mol. Biochem. Parasitol. 148, 219–222. doi: 10.1016/j.molbiopara.2006.03.013
Keywords: RNAi, dsRNA, siRNA, plant parasitic nematodes, Mi-col-1, Lemmi-5, cuticle collagens
Citation: Banerjee S, Gill SS, Gawade BH, Jain PK, Subramaniam K and Sirohi A (2018) Host Delivered RNAi of Two Cuticle Collagen Genes, Mi-col-1 and Lemmi-5 Hampers Structure and Fecundity in Meloidogyne incognita. Front. Plant Sci. 8:2266. doi: 10.3389/fpls.2017.02266
Received: 10 October 2017; Accepted: 27 December 2017;
Published: 22 January 2018.
Edited by:
Marcelo Menossi Menossi, Universidade Estadual de Campinas, BrazilReviewed by:
Paola Leonetti, Consiglio Nazionale delle Ricerche (CNR), ItalyLei Zhang, Washington State University, United States
Copyright © 2018 Banerjee, Gill, Gawade, Jain, Subramaniam and Sirohi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anil Sirohi, YW5pbHNpcm9oaUB5YWhvby5jb20=
†Present Address: Sagar Banerjee, Centre for Biological Engineering, Shobhit University, Gangoh, India