 Xiang Li1
Xiang Li1 Lei Huang1
Lei Huang1 Jianhua Lu1Yihui Cheng1Qingbo You1Lijun Wang2Xuejiao Song3Xinan Zhou1
Jianhua Lu1Yihui Cheng1Qingbo You1Lijun Wang2Xuejiao Song3Xinan Zhou1 Yongqing Jiao1,4*
Yongqing Jiao1,4*- 1Key Laboratory of Biology and Genetic Improvement of Oil Crops, Ministry of Agriculture, Oil Crops Research Institute, Chinese Academy of Agricultural Sciences, Wuhan, China
- 2The College of Life Science, Yangtze University, Jingzhou, China
- 3State Key Laboratory of Crop Biology, College of Agronomy, Shandong Agricultural University, Tai’an, China
- 4Collaborative Innovation Center of Henan Grain Crops, College of Agronomy, Henan Agricultural University, Zhengzhou, China
Molecular breeding has become an important approach for crop improvement, and a prerequisite for molecular breeding is elucidation of the functions of genetic loci or genes. Soybean is one of the most important food and oil crops worldwide. However, due to the difficulty of genetic transformation in soybean, studies of its functional genomics lag far behind those of other crops such as rice, which severely impairs the progress of molecular improvement in soybean. Here, we describe an effective large-scale strategy to investigate the functions of soybean genes via overexpression of a full-length soybean cDNA library in Arabidopsis. The overexpression vector pJL12 was modified for use in the construction of a normalized full-length cDNA library. The constructed cDNA library showed good quality; repetitive clones represented approximately 4%, insertion fragments were approximately 2.2 kb, and the full-length rate was approximately 98%. This cDNA library was then overexpressed in Arabidopsis, and approximately 2000 transgenic lines were preliminarily obtained. Phenotypic analyses of the positive T1 transgenic plants showed that more than 5% of the T1 transgenic lines displayed abnormal developmental phenotypes, and approximately 1% of the transgenic lines exhibited potentially favorable traits. We randomly amplified 4 genes with obvious phenotypes (enlarged seeds, yellowish leaves, more branches, and dense siliques) and repeated the transgenic analyses in Arabidopsis. Subsequent phenotypic observation demonstrated that these phenotypes were indeed due to the overexpression of soybean genes. We believe our strategy represents an effective large-scale approach to investigate the functions of soybean genes and further reveal genes favorable for molecular improvement in soybean.
Introduction
Conventional crop breeding is challenging and often takes a great deal of time. Molecular breeding approaches, such as marker-assisted or transgenic breeding, have become an important approach for current crop improvement. The prerequisite for molecular breeding is elucidation of the functions of genetic loci or genes. Among the different crop species where molecular breeding has been successfully applied, rice is a good example. For seed-related traits, several loci affecting grain size have been successfully identified and elucidated, such as GW7 (Wang S. et al., 2015), GW2 (Song et al., 2007) and GL7 (Si et al., 2016). Further transgenic application of these genes greatly enhanced rice grain yield (Song et al., 2007; Wang Y. et al., 2015; Si et al., 2016). Plant architecture is another important factor affecting grain yield. Ideal Plant Architecture 1 (IPA1) is an important dominant gene that reduces tiller number and enlarges panicles (Jiao et al., 2010). Practical application of this gene could greatly enhance grain yield compared to that of the control.
Soybean (Glycine max (L.) Merr.) is one of the most important food and oil crops worldwide, providing abundant proteins, oil and nutrition sources for animal feed and the human diet (Lam et al., 2010; Korir et al., 2013). At present, over one-third of edible oils and two-thirds of protein meal in the world are derived from soybean (Sobhanian et al., 2010). Soybean researchers have made some progress in soybean functional genomics through either forward genetic or reverse genetic studies. For example, a series of soybean gene functions have been elucidated, including GmphyA1 and GmphyA2 (Liu et al., 2008), Arabidopsis thaliana EARLY FLOWERING 3 (ELF3) homologous J locus (Lu et al., 2017), GmGIP1 (Ma et al., 2017), GmAP1 (Chi et al., 2011), and GmZF351 (Li et al., 2017). However, due to the very large and complex genome, low transformation efficiency, and long transgenic process in soybean, studies of soybean functional genomics have lagged far behind those in other crops, such as rice (Oryza sativa). In addition, soybean is an ancient tetraploid plant with many redundant genes, which seriously influence the elucidation of gene function by means of studying loss-of-function soybean mutants. To date, many kinds of RNA and protein profiling in soybean have been conducted, and numerous regulated genes have been identified (Smith-Hammond et al., 2014; Lanubile et al., 2015; Yu et al., 2016). However, confirmation of the functions of these genes has been rare, let alone the application of these genes in molecular improvement. Therefore, a strategy is needed to conduct large-scale and efficient studies in soybean functional genomics and, especially, to mine a large number of candidate favorable genes for soybean genetic improvement.
Arabidopsis is a model plant for studies of dicotyledons and thus represents an important intermediate tool to confirm the functions of soybean genes (Liu et al., 2014; Wang F. et al., 2015; Wei et al., 2015; Pan et al., 2017). In this study, we overexpressed a full-length soybean cDNA library in Arabidopsis and investigated the functions of soybean genes by evaluating the phenotypes of transgenic Arabidopsis. Our strategy could provide an efficient and high-throughput approach to mining candidate favorable soybean genes that could facilitate soybean molecular improvement.
Materials and Methods
Plant Materials and Growth Conditions
The roots, stems, leaves and shoot apices of soybean cultivar Williams 82 were used as materials for RNA extraction. Arabidopsis ecotype Col-0 was used as transformation recipient. Arabidopsis Col-0 was planted in a greenhouse with a temperature of 23°C, humidity of approximately 75%, and a 16 h light and 8 h dark photoperiod.
Vector Modification and Construction of the Full-Length cDNA Library
For vector modification, we first analyzed the sequence of vector pJL12 and found an SfiI recognition site (downstream of the NOS terminator at position 3859). However, there was no SfiI site downstream of the 35S promoter. Thus, we needed to mutate the existing SfiI site (at position 3859) and add another downstream of the 35S promoter for the integration of cDNA inserts. Briefly, we artificially synthesized a 1397 nucleotide fragment from position 2563 to 3907 in pJL12, which contained BamHI and Sse232I adapters. This fragment was designed to mutate the SfiI recognition site at position 3896 by changing G to T and introduce a new SfiI site downstream of the 35S promoter, where the sequence ACTAGTTCTAGA was replaced by GGCCATTACGGCCAAGCTTGATATCGGCCGCCTCGGCC. Next, the pJL12 fragment from position 2563 (BamHI) to 3927 (Sse232I) was replaced by the newly synthesized fragment using BamHI and Sse232I digestion and T4 DNA ligase linkage.
For cDNA synthesis, RNA was extracted using TRIzol reagent (Invitrogen) and reverse-transcribed by a PrimeScript reagent kit (cat#RR047A, TaKaRa). Reverse transcription primers were a combination of 5SM4Z, 5SM4ZA, and 5SM4ZB in a ratio of 1:1:1 (5SM4Z: GCC ATT ACG GCC AAG TTA C XXXX X-3′, 5SM4ZA: GCC ATT ACG GCC AAG TTA C XXXX-3′, 5SM4ZB: GCC ATT ACG GCC AAG TTA C XXX-3′, X = modified G). Subsequently, normalization was performed according to Patanjali et al. (1991), Soares et al. (1994), Zhang et al. (2005), and Li et al. (2012). Later, the normalized cDNAs were integrated into the pJL12 vector using T4 DNA ligase (Thermo Scientific) and transformed into DH5α for propagation and storage.
Large-Scale Genetic Transformation of the cDNA Library Into Arabidopsis
The concentrations of the plasmids harboring the cDNA library were measured by a spectrophotometer (Nanodrop, Thermo fisher) and diluted to ensure values between 100 and 200 ng/μl. Then, 1 μl of each plasmid was transformed into Agrobacterium strain GV3101 by an electrotransformation apparatus (MicroPulser, Bio-Rad). After Agrobacterium colonies appeared, all colonies were scraped into 1 L LB medium (kanamycin 30 ng/μl, rifampicin 50 ng/μl) and propagated in an incubator. To avoid the possibility of growth priority in a prolonged culture period, the culture time for Agrobacterium propagation was limited to 4∼6 h. The transformation experiment was performed as described by Clough and Bent (1998), except that we used 500 μl Silwet L-77 per liter LB medium when performing the transformation, because Silwet L-77 could greatly improve the transformation rate (Martinez-Trujillo et al., 2004).
Phenotypic Evaluation and Genetic Analyses of Transgenic Arabidopsis
The transformed seeds overexpressing the full-length cDNA library were planted in a greenhouse. After screening with Basta, the surviving positive seedlings were compared to the seedlings expressing the empty vector at all developmental stages.
For the screening of homozygous transgenic lines, approximately 30 seeds derived from T2 plants were plated on Basta selective (1:1000 ratio dilution) plates, and the seeds of the plants whose seeds showed a 3:1 survival ratio were harvested. The phenotypes of the homozygous transgenic lines were further investigated without Basta selection.
For the A13, B12, C15 and D70 overexpression plasmids, the primers A13-f/A13-r, B12-f/B12-r, C15-f/C15-r and D70-f/D70-r were used to amplify each gene individually (Supplementary File 1). The PCR products were integrated into linearized pJL12 vector using the ClonExpress II One Step Cloning Kit (Vazyme, cat#c112) after digestion by the restriction enzyme BamHI. The correct constructs were retransformed into Col-0 to validate the observed phenotypes.
Chlorophyll Content Analysis
The total chlorophyll of the 4th and 5th rosette leaves was extracted as described in a previous report (Sakuraba et al., 2014b). Subsequently, the chlorophyll content was quantified according to another published report (Lichtenthaler, 1987).
Results
Construction of Soybean Full-Length cDNA Library
The vector pJL12, harboring the 35S promoter, is widely used in studies of functional genomics (Qiu et al., 2002, 2008a,b; Collins et al., 2003; Zou et al., 2011; Guo et al., 2014). To make this vector suitable for the construction of a full-length cDNA library, we first modified this vector. Because the cohesive ends produced by SfiI digestion were required, we first analyzed the sequence of the original pJL12 vector and found an SfiI restriction site in pJL12 (position 3859–3907, Figure 1A). However, downstream of the 35S promoter in the MCS position, there was no SfiI site. Therefore, we needed to mutate the existing SfiI restriction site (G to T transition) and add another in the MCS (Figure 1A). Considering the above, a nucleotide fragment with a length of 1397 containing the BamHI and Sse232I recognition sites was artificially synthesized (Supplementary File 2), in which one SfiI was mutated by transitioning G to T at position 3859–3907 and another SfiI was introduced downstream of the 35S promoter at position 2568. This fragment was later integrated into pJL12 by BamHI and Sse232I digestion. Thus, the modified pJL12 vector could allow the insertion of the cDNA library after digestion by the SfiI restriction enzyme and the overexpression of the library in plants through the 35S promoter.
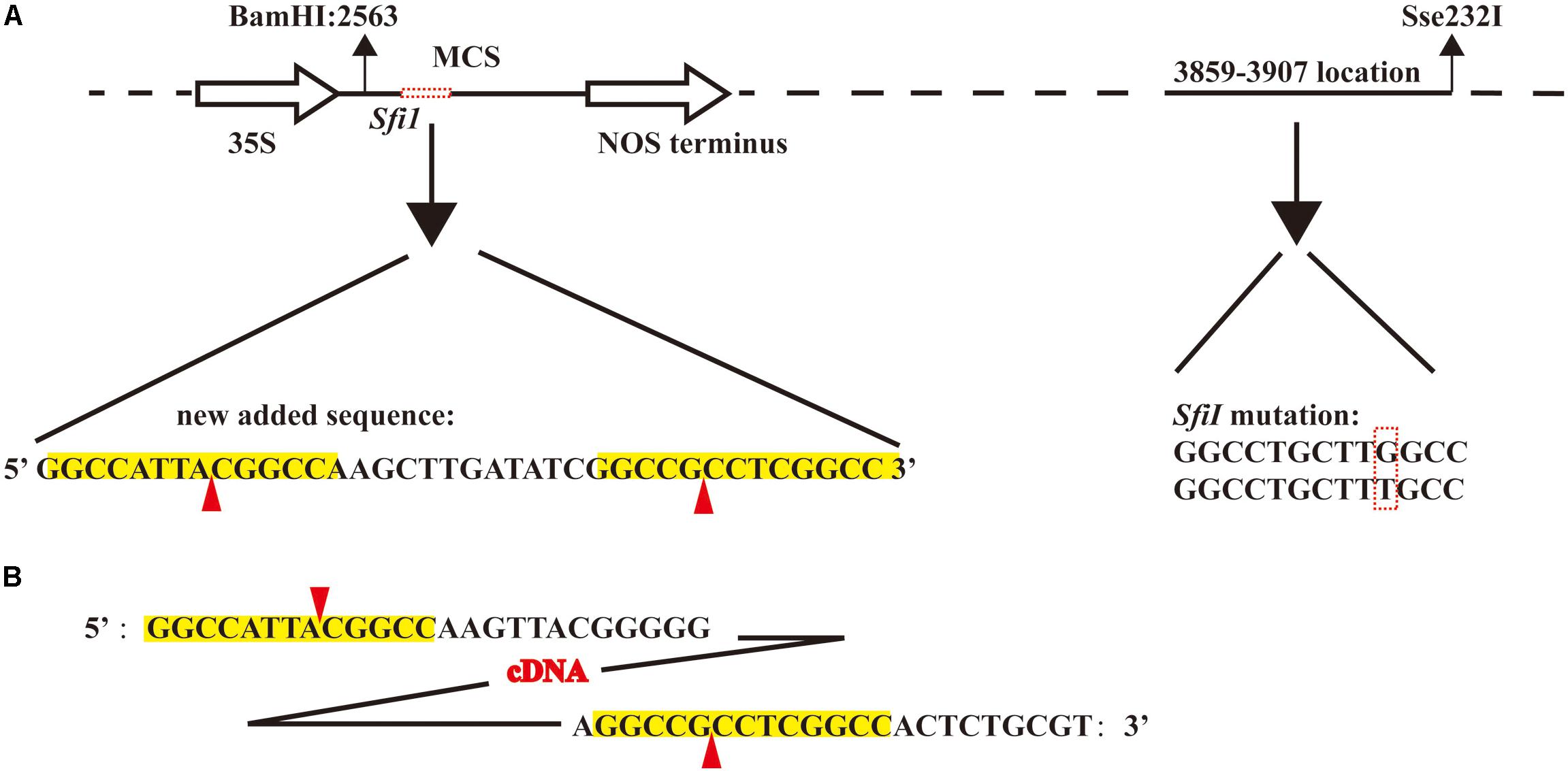
FIGURE 1. Construction of soybean full-length cDNA overexpression library. (A) Schematic of modified pJL12 vector. The solid and empty arrows represent the 35S promoter and NOS terminator, respectively. The black dashed line represents the pJL12 backbone sequence. The horizontal red dashed box indicates the artificially added sequence, which contains two SfiI recognition sequences, and the vertical red dashed box indicates the mutated SfiI base sequence. The red arrow indicates the SfiI cleavage site, and the yellow background represents the SfiI recognition site. (B) Reverse-transcribed and normalized cDNA sequence. The yellow background represents the SfiI recognition site, and the red arrow indicates the SfiI cleavage site.
We collected the roots, stems, leaves and shoot apices of Williams 82 plants for RNA extraction. Different adapters with SfiI cohesive ends at the 5′ and 3′ ends were used to perform reverse transcription (see section “Materials and methods”). Next, the cDNA was digested with SfiI and then separated by use of agarose gel electrophoresis. The approximately 2 kb fragments were recovered, normalized and ligated to the modified pJL12 vector (Figure 1B). In the end, we transformed the cDNA library into DH5α. In total, we obtained approximately 106 clones and retained all of the clones for propagation and storage (Table 1).
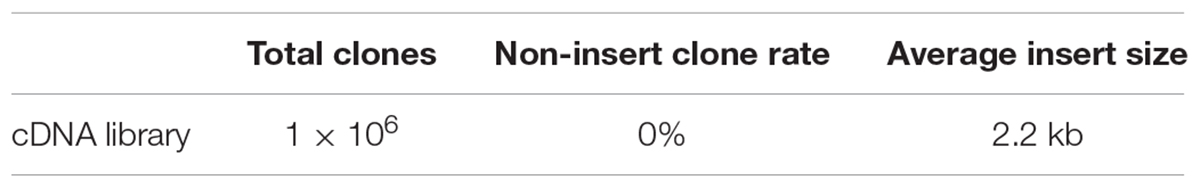
TABLE 1. Assessment summary of the constructed soybean full-length cDNA overexpression library.
Assessment of cDNA Library Quality
We randomly picked 96 colonies and used primers pJL12-f (in the 35S promoter) and pJL12-r (in the NOS terminator) to amplify the cDNA inserts. All 96 colonies were successfully amplified, which showed that each clone carried a cDNA insertion (Supplementary File 3). To evaluate the normalization rate of the constructed cDNA library, we sequenced the 96 PCR products. As shown in Figure 2A, non-repetitive cDNA sequences in the constructed cDNA library were approximately 96% and repetitive sequences were approximately 4%. To evaluate the genome coverage, we blasted the sequenced results against the Wm82.a2.v1 Transcript Sequences database1. Because soybean is a tetraploid plant, most of our sequencing data could match more than one gene locus. We retrieved gene matches with more than 95% identities for further analyses. All of the chromosomes had similar cDNA hit numbers (Figure 2B), which suggested that our constructed soybean full-length overexpressed cDNA library had good coverage. Later, we evaluated the full-length rate of our constructed cDNA library. Among the sequenced data, approximately 98% included full-length sequences, indicating that the majority of the inserts in the cDNA library were intact (Figure 2C). We also estimated the average insert size of our constructed cDNA library, which was approximately 2.2 kb (Table 1). Based on the above results, we concluded that the constructed full-length soybean cDNA library had high quality with high genome coverage, low repetitive frequency and high full-length ratio, and it could be used for further study.
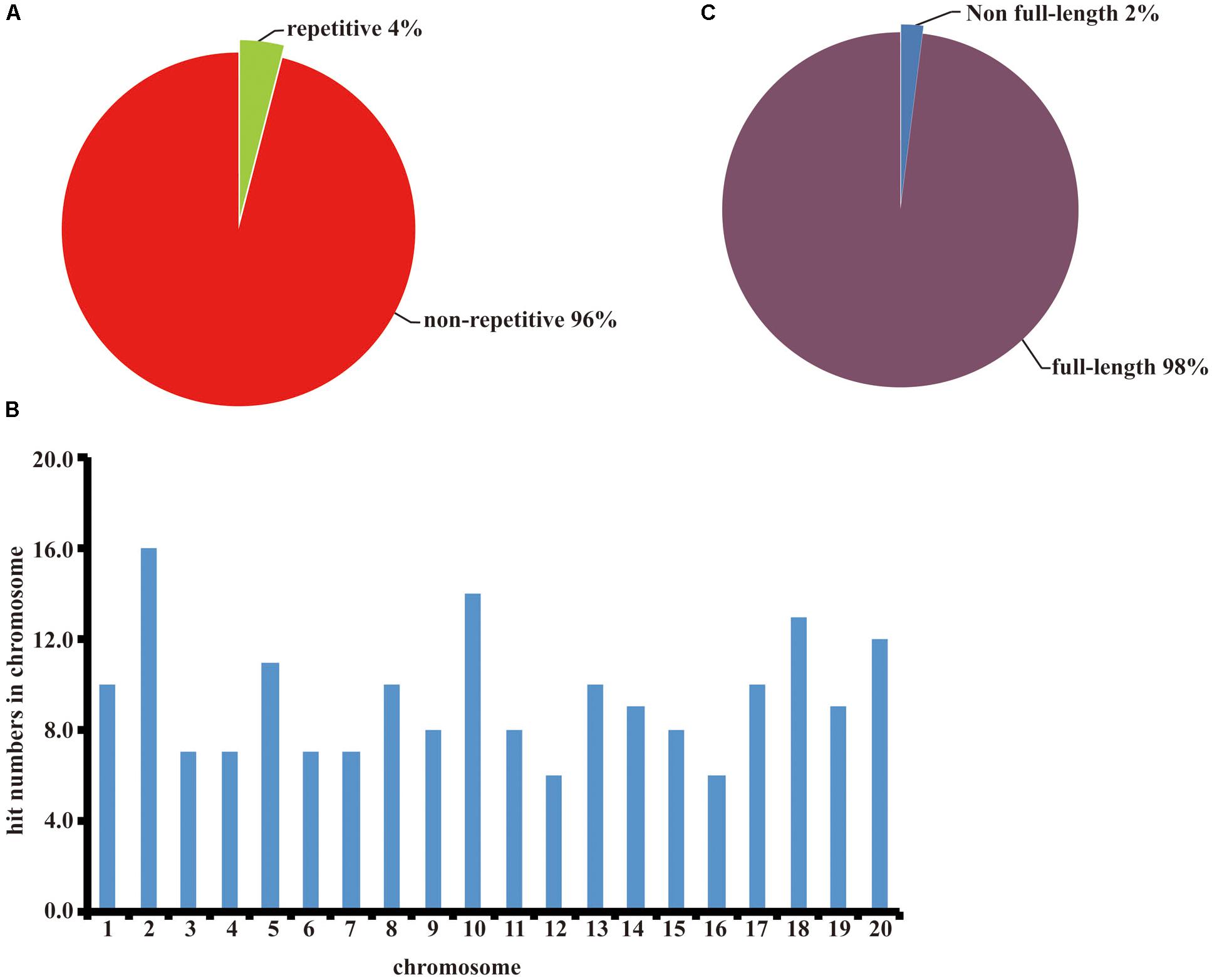
FIGURE 2. Quality assessment of the constructed soybean full-length cDNA overexpression library. (A) Repetitive frequency analysis of the constructed cDNA library. (B) Full-length proportion analysis of the constructed cDNA library. (C) Genome coverage analysis of the constructed cDNA library.
Transformation and Evaluation of Transgenic Arabidopsis
We extracted the plasmids from the cDNA library and transformed them into Agrobacterium cells. Next, the library was transformed into Arabidopsis through Agrobacterium tumefaciens–mediated transformation method with a slight modification (see section “Materials and methods”) (Clough and Bent, 1998). To obtain T1 positive transgenic lines overexpressing the soybean full-length cDNA library, T0 generation seeds were harvested and sown in a greenhouse, and Basta (1:1000) was sprayed for selection. In total, we obtained nearly 2000 T1 positive transgenic lines (Table 2). To observe phenotypes in the T1 transgenic lines, empty vector pJL12 (modified)–transformed lines were sown in parallel with positive cDNA-transformed T1 seedlings. Many of the T1 positive seedlings exhibited abnormal phenotypes (approximately 5%), including late flowering, yellowish color, loss of SAM (shoot apical meristem) activity, early senescence, more primary and secondary branches, curved leaves, smooth stems and early flowering (Supplementary File 4 and Table 2), whereas another 1% exhibited favorable traits, including large seeds, high silique density, strong drought resistance and vigorous growth appearance (Figure 3 and Table 2).
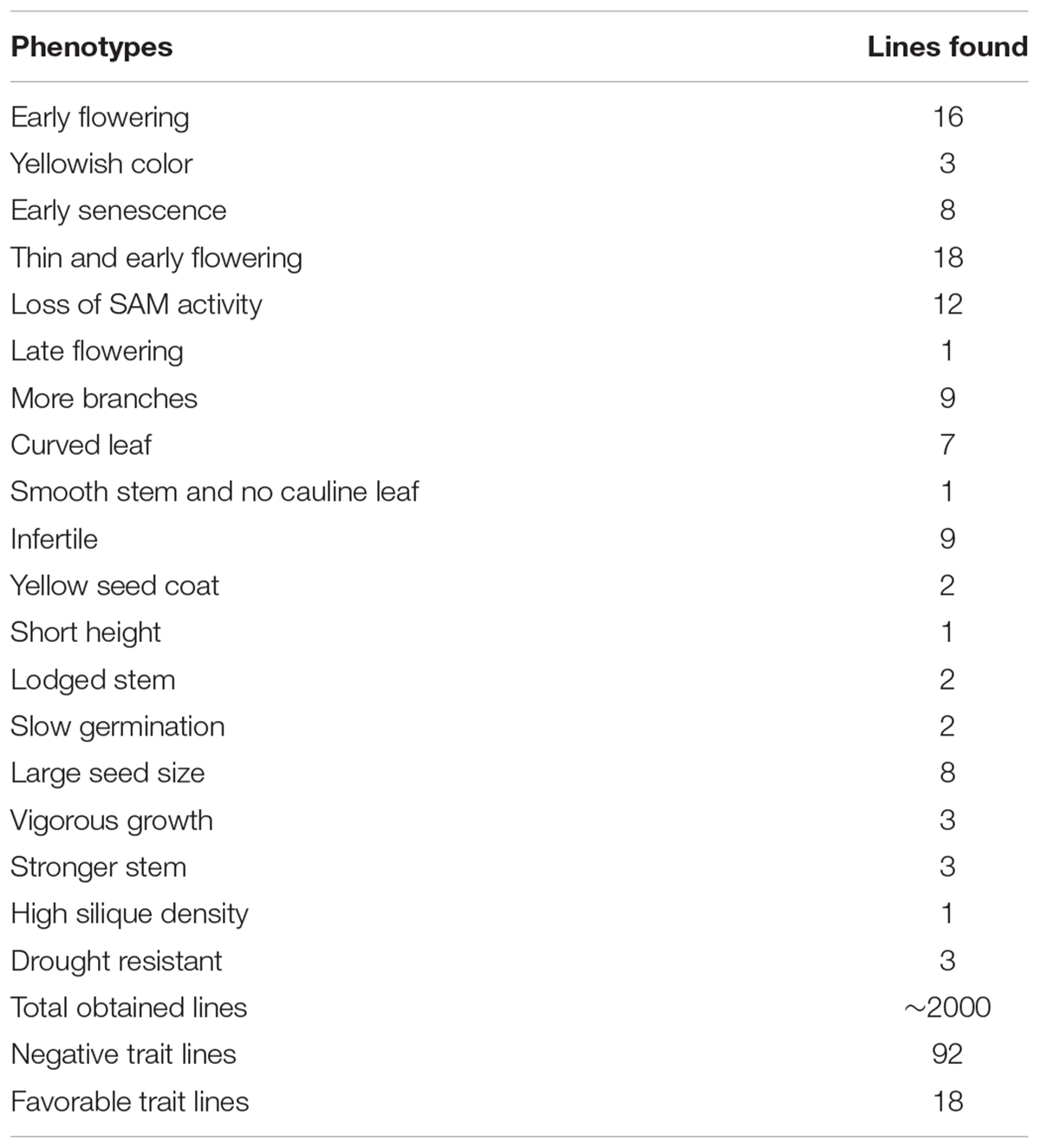
TABLE 2. Summary of phenotypes of T1 positive Arabidopsis lines transformed with the soybean full-length cDNA overexpression library.
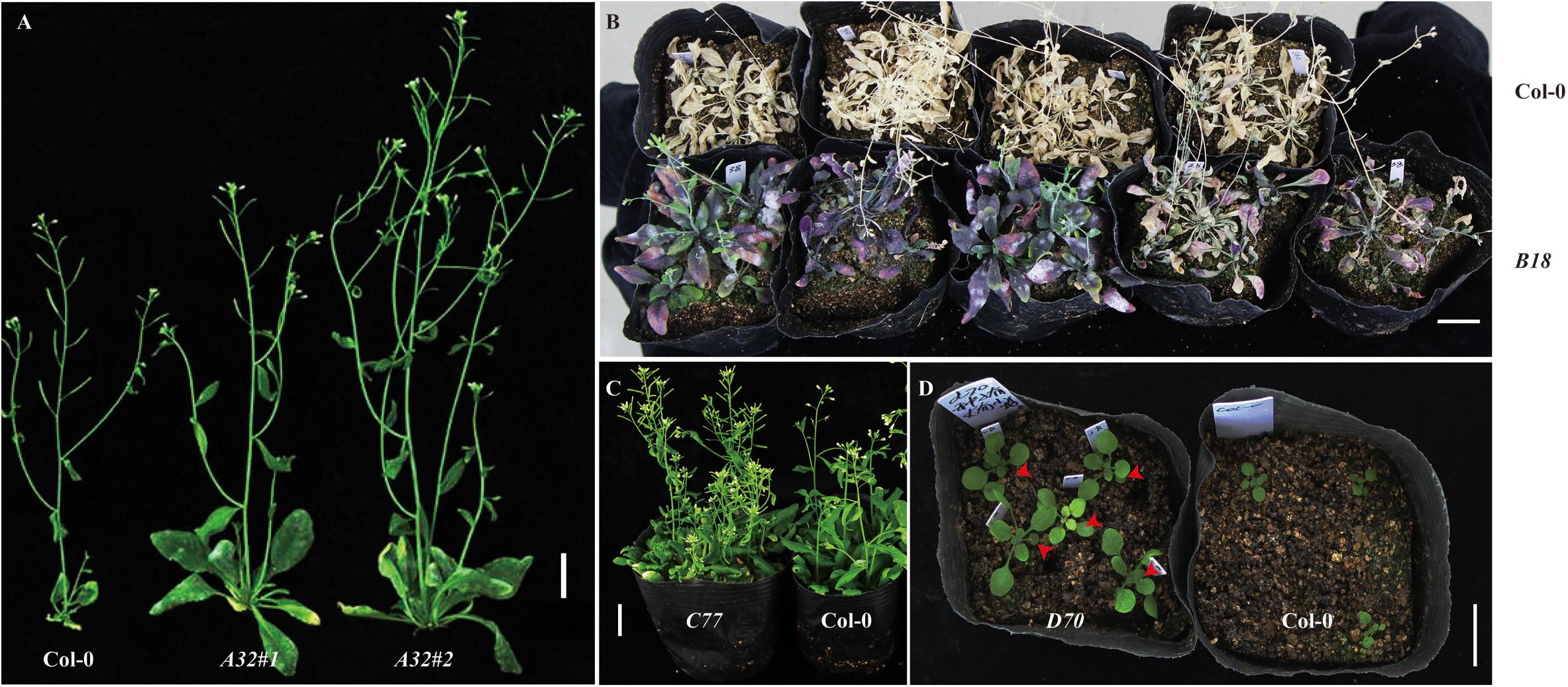
FIGURE 3. Homozygous T2 transformed Arabidopsis showed potential favorable traits. (A) A32 promotes plant growth. Two homozygous A32 lines showed more vigorous growth than that of Col-0. (B) B18 enhances drought resistance. Five homozygous B18 transgenic lines showed stronger drought resistance than that of Col-0. (C) C77 increases fruit density. Several homozygous C77 transgenic lines exhibited higher silique density than Col-0. (D) D70 exhibits enlarged seed size. Five homozygous D70 transgenic lines showed larger cotyledons than those of Col-0. The red arrows indicate enlarged cotyledons. Bar = 2 cm.
To inspect the distribution of cDNA inserts in the soybean genome, we extracted DNA from 96 randomly selected individual T1 seedlings and amplified their cDNA inserts using the primers pJL12-f and pJL12-r. Most of the 96 transgenic plants had successfully amplified products (Supplementary File 5A). Then, the amplified products were sequenced and blasted against the Wm82.a2.v1 Transcript Sequences database. The sequencing data showed that 97% of the cDNA inserts were non-repetitive, and all of the sequenced data were full-length (Supplementary File 5B). To examine the coverage of the cDNA inserts, we analyzed the distribution of sequencing data in the soybean genome. The results showed that all 20 soybean chromosomes had good match numbers (Supplementary File 5C).
Validation of Phenotypes of T1 Transgenic Arabidopsis by Retransformation of Corresponding Genes
To confirm whether the phenotypes of transgenic Arabidopsis were caused by the ectopic expression of soybean genes, we randomly selected 4 lines with obvious phenotypes for further study.
A13 homozygous transgenic plants displayed a yellowish phenotype (Figures 4A,B). The sequence analysis of the cDNA fragments showed that the A13 yellowish phenotype was caused by Glyma.06g119500, a homolog of Arabidopsis SGR1 (Shimoda et al., 2016). We reconstructed the overexpression vector for Glyma.06g119500 and transformed it into Arabidopsis. Phenotypic analysis showed that all homozygous transgenic lines exhibited a yellowing leaf phenotype (Figures 4A,B) consistent with the one from the transgenic line overexpressing the cDNA library.
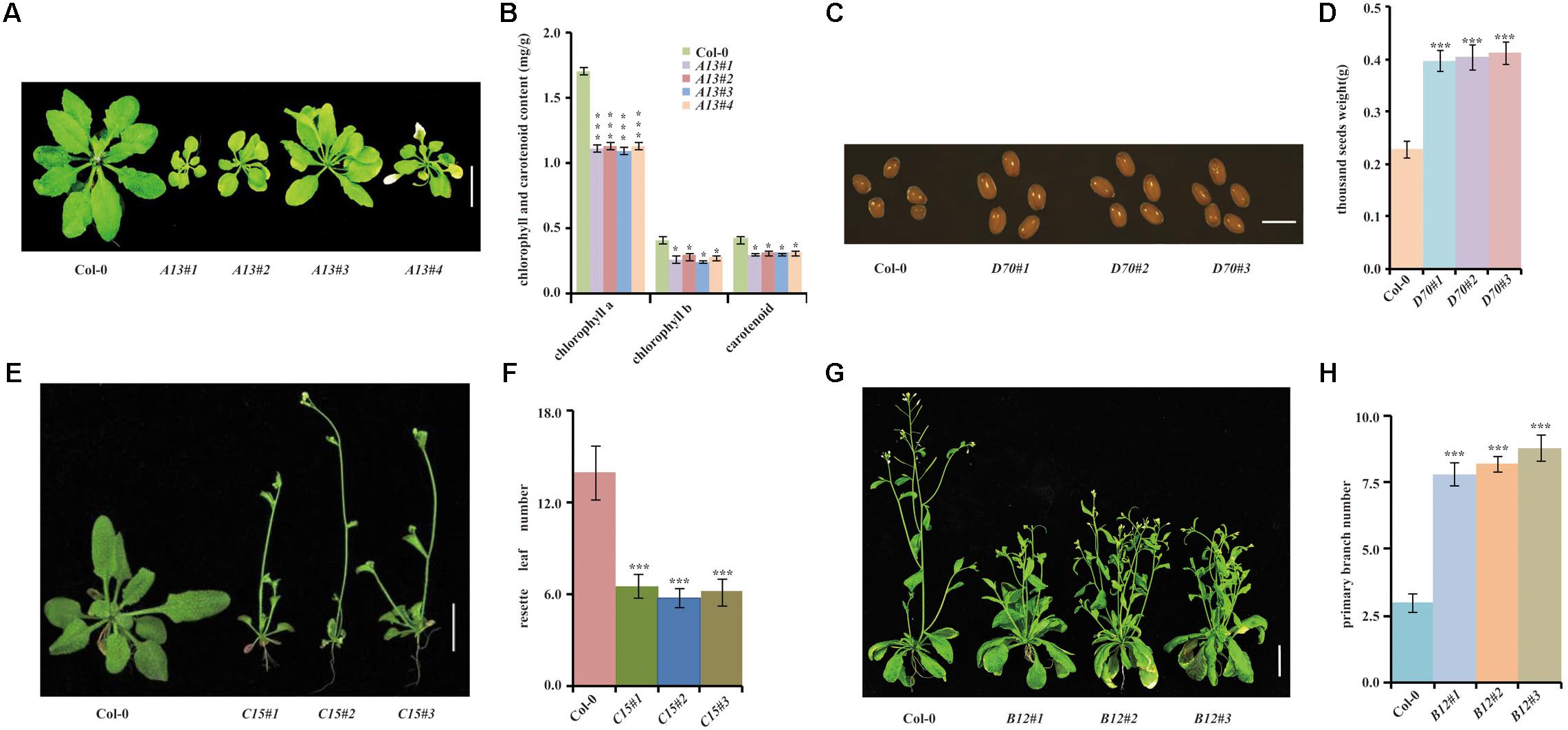
FIGURE 4. Phenotypic validation of 4 randomly selected transgenic lines by genetic retransformation. (A) Leaf phenotype of retransformed A13 homozygous lines and Col-0 control. (B) Chlorophyll and carotenoid contents of retransformed A13 homozygous lines and Col-0. (C) Photograph of seed size in retransformed D70 homozygous lines and Col-0 control. (D) Thousand seed weights of retransformed D70 homozygous lines and Col-0. (E) Phenotypes of retransformed C15 homozygous lines and Col-0 plants. (F) Rosette leaf numbers in retransformed C15 homozygous lines and Col-0 at bolting. (G) Branching phenotypes of retransformed B12 homozygous lines and Col-0 plants. (H) Primary branch statistical analysis in Col-0 and retransformed B12 homozygous lines. Bar = 2 cm, n ≥ 10. (∗∗∗p < 0.01, ∗p < 0.05, Student’s t-test).
B12 homozygous transgenic plants displayed a dwarfing and highly branched phenotype (Figures 4C,D). Sequence analysis showed that it might be caused by a predicted ANDROGEN INDUCED INHIBITOR OF PROLIFERATION (AS3) / PDS5-RELATED gene, Glyma.06g062700. To validate whether the B12 phenotype was caused by overexpression of Glyma.06g062700, we retransformed Glyma.06g062700 into Arabidopsis. Among 26 homozygous lines, 19 displayed obvious dwarfing and highly branched phenotypes (Figures 4C,D), suggesting that the B12 phenotype was due to overexpression of Glyma.06g062700.
C15 T1 transgenic plants were thin, with small stature and early flowering (Figures 4E,F). Sequence analysis revealed that the inserted gene was Glyma.11g199100, a nucleic acid binding protein with ATP-dependent RNA helicase activity. Retransformed Arabidopsis lines with overexpression of Glyma.11g199100 also showed the same thin, small and early-flowering phenotype (Figures 4E,F), demonstrating that the C15 phenotype was caused by overexpression of Glyma.11g199100.
The D70 T1 transgenic line displayed larger seeds (Figures 4G,H). Sequence analysis showed that the D70 insertion gene was Glyma.05g231900, which encoded an aldehyde dehydrogenase. To validate whether the large seeds in D70 were caused by Glyma.05g231900, we retransformed the overexpression vectors of Glyma.05g231900 into Arabidopsis. The results showed that 19 out of 24 homozygous lines showed larger seed sizes than those of the controls (Figures 4G,H), demonstrating that the aldehyde dehydrogenase Glyma.05g231900 exerts a specific influence on seed size.
Taking the above results together, we concluded that the phenotypes found in the T1 generation plants were most likely caused by the overexpression of the genes from the soybean full-length cDNA overexpression library.
Discussion
Soybean is an important crop that provides a well-balanced source of protein and oil. In addition, most of the components of soybean, such as α-linolenic acid and isoflavones, have beneficial health effects (Xia et al., 2013). However, studies of the functional genomics of soybean have lagged far behind those of other crops because of the relatively low efficiency and long transformation period and the relative complexity of the genome (Schmutz et al., 2010). Although some genes have been cloned (Lee et al., 2004; Gao et al., 2005; Gao and Bhattacharyya, 2008; Cook et al., 2012; Liu et al., 2012; Lu et al., 2017; Ma et al., 2017), the functions of many soybean genes remain unknown. As the demand for soybean is increasing, how to improve soybean yield and edible quality is an urgent challenge confronted by soybean breeders.
Rationality and Advantage of Our Strategy for Studies of Soybean Functional Genomics
Currently, when we mine favorable genes in soybean through the method of reverse genetics, we usually select dozens of candidate genes from transcriptome data or known homologous genes, and then investigate their functions one by one by use of Arabidopsis as the model plant. Subsequently, we confirm their functions in soybean. This process requires large amounts of time and labor, and the positive efficiency is relatively low, which severely influences the mining of favorable genes. On the other hand, soybean is an ancient tetraploid plant, and most of the soybean genes have duplicated copies, meaning that loss-of-function mutations in soybean genes usually have no apparent phenotypes. In addition, the successes of QTL cloning in soybean have been few to date, largely due to the complexity of the soybean genome (Kato et al., 2014; Lee et al., 2015). Due to the above, elucidation of the functions of soybean genes and, further, the mining of favorable genes have been much slower than those in other crops. Developing a large-scale and efficient strategy for studies of soybean functional genomics is an urgent need for soybean researchers.
Arabidopsis has diverse genetic resources and a large number of mutants created using various mutagenesis strategies2. Moreover, Arabidopsis has a small genome, short life cycle, and easy and effective transformation, making Arabidopsis a useful model plant (Koornneef and Meinke, 2010; Liepman et al., 2010). To date, many gene functions in Arabidopsis have been dissected and provided important references for other plant species3. Both soybean and Arabidopsis are dicotyledonous plants and share some similarities. These similarities provide a substantial potential for the use of Arabidopsis to carry out studies of soybean functional genomics. By using Arabidopsis as a model plant, the functions of many soybean genes have already been successfully elucidated (Li et al., 2016; Takeshima et al., 2016; Chu et al., 2017). For example, GmNAC20 and GmNAC11 are two NAC transcription factors in soybean. When GmNAC20 was overexpressed in Arabidopsis, it greatly enhanced salt and freezing tolerance, whereas overexpression of GmNAC11 improved only salt tolerance in transgenic Arabidopsis. The roles of these two NAC family transcription factors were also confirmed in soybean (Hao et al., 2011).
To mine gene functions at a larger scale, we constructed a full-length soybean cDNA library with a modified overexpression vector, pJL12, which contains a 35S promoter (Figure 1A). We assessed the resulting library and found that it had high quality (Figure 2). After transformation into Arabidopsis, we first obtained ∼2000 transgenic lines (Table 2), among which 5% of transgenic plants showed abnormal developmental phenotypes, whereas 1% of them showed potentially beneficial traits (Figure 3 and Table 2). Sequence analyses showed that all overexpressed soybean genes were different rather than redundant, suggesting the effectiveness of our strategy. We noticed that some soybean genes could be missed, including genes that exhibit phenotypic changes only in the loss-of-function condition and genes that have different functions in soybean from those in Arabidopsis. Moreover, some phenotypes exhibited by the transgenic Arabidopsis lines might be attributed to the insertion of vectors in the Arabidopsis genome rather than the overexpression of soybean genes. In addition, ectopic overexpression of a gene might not reflect its intrinsic function, which could be solved by studying the corresponding loss-of-function mutant or using the native promoter. Despite these imperfections, our results prove that this strategy is reasonable and advantageous for use in investigating the functions of soybean genes and further mining favorable soybean genes for future molecular improvements.
Dissection of Four Potentially Favorable Soybean Genes
In this study, we transformed the soybean full-length cDNA overexpression library into Arabidopsis. Many T1 generation positive seedlings exhibited various phenotypes (Table 2). The occurrences of phenotypes in these transgenic lines might be attributed to the overexpression of the soybean genes or to the destruction of Arabidopsis gene expression by T-DNA insertion in the Arabidopsis genome.
To confirm that the phenotypes occurring in the T1 positive seedlings were caused by cDNA ectopic overexpression, we randomly selected 4 lines for confirmation. Genetic retransformation proved that the overexpressed soybean cDNAs were responsible for each of the observed phenotypes. A13 encodes an Arabidopsis SGR1 homolog protein. In Arabidopsis, SGR1 interacts with Chl catabolic enzymes (CCE) to regulate chlorophyll metabolism, leading to a pale and yellowing leaf pattern during leaf senescence (Balazadeh, 2014; Sakuraba et al., 2014a,b, 2015; Bell et al., 2015). Furthermore, SGR1 regulates chlorophyll degradation in other plant species, such as rice (Park et al., 2007), pea (Pisum sativum) (Sato et al., 2007), tomato (Solanum lycopersicum), bell pepper (Capsicum annuum) (Barry et al., 2008), tall fescue (Festuca arundinacea) (Wei et al., 2011) and Medicago truncatula (Zhou et al., 2011). In this study, it also caused a yellowish appearance, indicating a conserved role of SGR1. B12 encodes an ANDROGEN INDUCED INHIBITOR OF PROLIFERATION (AS3) / PDS5-RELATED protein, which may inhibit cell proliferation, consistent with the dwarfing and highly branched phenotype of B12 (Yuan et al., 1993; Szelei et al., 1997). C15 encodes an ATP-dependent RNA helicase. RNA helicase plays diverse roles in plant growth and development (Linder and Owttrim, 2009), which may be a plausible reason for the small stature and early-flowering phenotype of C15. D70 encodes an aldehyde dehydrogenase (Brocker et al., 2013), which may affect the metabolism of energy in seeds. All of these genes are worthy of further study to elucidate their detailed functional mechanisms.
Conclusion
In this study, we used Arabidopsis as a recipient and developed a high-throughput strategy to investigate the functions of soybean genes by overexpressing a full-length soybean cDNA library in Arabidopsis. Many transgenic Arabidopsis lines exhibited abnormal phenotypes, among which some were found to contain potentially favorable genes for the molecular improvement of soybean. Although the functions of these genes must be further confirmed in soybean, we believe that this strategy may provide valuable sources of soybean genetic improvement.
Author Contributions
XL performed the majority of the experimental work. LH helped to construct the cDNA library. JL conducted part of the transformation work. YC constructed the overexpression vector. QY, LW, XS, and XZ gave valuable advice. XL and YJ wrote and revised the paper. YJ conceived the project and supervised the work.
Funding
This work was supported by the National Natural Science Foundation of China (31522042), the National Key R&D Program of China (2016YFD0100603 and 2016YFD0100504), the National Transgenic Project (2016ZX08004003), the Natural Science Foundation of Hubei (2016CFA049), and the Agricultural Science and Technology Innovation Program (Y2017JC14).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00631/full#supplementary-material
Footnotes
- ^ http://www.soybase.org/
- ^ http://signal.salk.edu/cgi-bin/tdnaexpress
- ^ http://www.arabidopsis.org/
References
Balazadeh, S. (2014). Stay-green not always stays green. Mol Plant 7, 1264–1266. doi: 10.1093/mp/ssu076
Barry, C. S., Mcquinn, R. P., Chung, M. Y., Besuden, A., and Giovannoni, J. J. (2008). Amino acid substitutions in homologs of the STAY-GREEN protein are responsible for the green-flesh and chlorophyll retainer mutations of tomato and pepper. Plant Physiol. 147, 179–187. doi: 10.1104/pp.108.118430
Bell, A., Moreau, C., Chinoy, C., Spanner, R., Dalmais, M., Le Signor, C., et al. (2015). SGRL can regulate chlorophyll metabolism and contributes to normal plant growth and development in Pisum sativum L. Plant Mol. Biol. 89, 539–558. doi: 10.1007/s11103-015-0372-4
Brocker, C., Vasiliou, M., Carpenter, S., Carpenter, C., Zhang, Y., Wang, X., et al. (2013). Aldehyde dehydrogenase (ALDH) superfamily in plants: gene nomenclature and comparative genomics. Planta 237, 189–210. doi: 10.1007/s00425-012-1749-0
Chi, Y., Huang, F., Liu, H., Yang, S., and Yu, D. (2011). An APETALA1-like gene of soybean regulates flowering time and specifies floral organs. J. Plant Physiol. 168, 2251–2259. doi: 10.1016/j.jplph.2011.08.007
Chu, S., Wang, J., Zhu, Y., Liu, S., Zhou, X., Zhang, H., et al. (2017). An R2R3-type MYB transcription factor, GmMYB29, regulates isoflavone biosynthesis in soybean. PLoS Genet 13:e1006770. doi: 10.1371/journal.pgen.1006770
Clough, S. J., and Bent, A. F. (1998). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743. doi: 10.1046/j.1365-313x.1998.00343.x
Collins, N. C., Thordal-Christensen, H., Lipka, V., Bau, S., Kombrink, E., Qiu, J. L., et al. (2003). SNARE-protein-mediated disease resistance at the plant cell wall. Nature 425, 973–977. doi: 10.1038/nature02076
Cook, D. E., Lee, T. G., Guo, X., Melito, S., Wang, K., Bayless, A. M., et al. (2012). Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science 338, 1206–1209. doi: 10.1126/science.1228746
Gao, H., and Bhattacharyya, M. K. (2008). The soybean-Phytophthora resistance locus Rps1-k encompasses coiled coil-nucleotide binding-leucine rich repeat-like genes and repetitive sequences. BMC Plant Biol 8:29. doi: 10.1186/1471-2229-8-29
Gao, H., Narayanan, N. N., Ellison, L., and Bhattacharyya, M. K. (2005). Two classes of highly similar coiled coil-nucleotide binding-leucine rich repeat genes isolated from the Rps1-k locus encode Phytophthora resistance in soybean. Mol. Plant Microbe Interact. 18, 1035–1045. doi: 10.1094/MPMI-18-1035
Guo, W., Zuo, Z., Cheng, X., Sun, J., Li, H., Li, L., et al. (2014). The chloride channel family gene CLCd negatively regulates pathogen-associated molecular pattern (PAMP)-triggered immunity in Arabidopsis. J. Exp. Bot. 65, 1205–1215. doi: 10.1093/jxb/ert484
Hao, Y. J., Wei, W., Song, Q. X., Chen, H. W., Zhang, Y. Q., Wang, F., et al. (2011). Soybean NAC transcription factors promote abiotic stress tolerance and lateral root formation in transgenic plants. Plant J. 68, 302–313. doi: 10.1111/j.1365-313X.2011.04687.x
Jiao, Y., Wang, Y., Xue, D., Wang, J., Yan, M., Liu, G., et al. (2010). Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 42, 541–544. doi: 10.1038/ng.591
Kato, S., Sayama, T., Fujii, K., Yumoto, S., Kono, Y., Hwang, T. Y., et al. (2014). A major and stable QTL associated with seed weight in soybean across multiple environments and genetic backgrounds. Theor. Appl. Genet. 127, 1365–1374. doi: 10.1007/s00122-014-2304-0
Koornneef, M., and Meinke, D. (2010). The development of Arabidopsis as a model plant. Plant J. 61, 909–921. doi: 10.1111/j.1365-313X.2009.04086.x
Korir, P. C., Zhang, J., Wu, K., Zhao, T., and Gai, J. (2013). Association mapping combined with linkage analysis for aluminum tolerance among soybean cultivars released in Yellow and Changjiang River Valleys in China. Theor. Appl. Genet. 126, 1659–1675. doi: 10.1007/s00122-013-2082-0
Lam, H. M., Xu, X., Liu, X., Chen, W., Yang, G., Wong, F. L., et al. (2010). Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053–1059. doi: 10.1038/ng.715
Lanubile, A., Muppirala, U. K., Severin, A. J., Marocco, A., and Munkvold, G. P. (2015). Transcriptome profiling of soybean (Glycine max) roots challenged with pathogenic and non-pathogenic isolates of Fusarium oxysporum. BMC Genomics 16:1089. doi: 10.1186/s12864-015-2318-2
Lee, G. J., Carter, TE Jr, Villagarcia, M. R., Li, Z., Zhou, X., Gibbs, M. O., et al. (2004). A major QTL conditioning salt tolerance in S-100 soybean and descendent cultivars. Theor. Appl. Genet. 109, 1610–1619. doi: 10.1007/s00122-004-1783-9
Lee, J. S., Yoo, M. H., Jung, J. K., Bilyeu, K. D., Lee, J. D., and Kang, S. (2015). Detection of novel QTLs for foxglove aphid resistance in soybean. Theor. Appl. Genet. 128, 1481–1488. doi: 10.1007/s00122-015-2519-8
Li, Q., Fang, C., Duan, Z., Liu, Y., Qin, H., Zhang, J., et al. (2016). Functional conservation and divergence of GmCHLI genes in polyploid soybean. Plant J. 88, 584–596. doi: 10.1111/tpj.13282
Li, Q. T., Lu, X., Song, Q. X., Chen, H. W., Wei, W., Tao, J. J., et al. (2017). Selection for a zinc-finger protein contributes to seed oil increase during soybean domestication. Plant Physiol. 173, 2208–2224. doi: 10.1104/pp.16.01610
Li, Z. C., An, L. H., Fu, Q., Liu, Y., Zhang, L., Chen, H., et al. (2012). Construction and characterization of a normalized cDNA library from the river snail Bellamya aeruginosa after exposure to copper. Ecotoxicology 21, 260–267. doi: 10.1007/s10646-011-0786-y
Lichtenthaler, H. K. (1987). Chlorophylls and carotenoids: pigments of photosynthetic biomembranes. Methods Enzymol. 148, 350–382. doi: 10.1016/0076-6879(87)48036-1
Liepman, A. H., Wightman, R., Geshi, N., Turner, S. R., and Scheller, H. V. (2010). Arabidopsis - a powerful model system for plant cell wall research. Plant J. 61, 1107–1121. doi: 10.1111/j.1365-313X.2010.04161.x
Linder, P., and Owttrim, G. W. (2009). Plant RNA helicases: linking aberrant and silencing RNA. Trends Plant Sci. 14, 344–352. doi: 10.1016/j.tplants.2009.03.007
Liu, B., Kanazawa, A., Matsumura, H., Takahashi, R., Harada, K., and Abe, J. (2008). Genetic redundancy in soybean photoresponses associated with duplication of the phytochrome A gene. Genetics 180, 995–1007. doi: 10.1534/genetics.108.092742
Liu, S., Kandoth, P. K., Warren, S. D., Yeckel, G., Heinz, R., Alden, J., et al. (2012). A soybean cyst nematode resistance gene points to a new mechanism of plant resistance to pathogens. Nature 492, 256–260. doi: 10.1038/nature11651
Liu, Y. F., Li, Q. T., Lu, X., Song, Q. X., Lam, S. M., Zhang, W. K., et al. (2014). Soybean GmMYB73 promotes lipid accumulation in transgenic plants. BMC Plant Biol. 14:73. doi: 10.1186/1471-2229-14-73
Lu, S., Zhao, X., Hu, Y., Liu, S., Nan, H., Li, X., et al. (2017). Natural variation at the soybean J locus improves adaptation to the tropics and enhances yield. Nat. Genet. 49, 773–779. doi: 10.1038/ng.3819
Ma, Z., Zhu, L., Song, T., Wang, Y., Zhang, Q., Xia, Y., et al. (2017). A paralogous decoy protects Phytophthora sojae apoplastic effector PsXEG1 from a host inhibitor. Science 355, 710–714. doi: 10.1126/science.aai7919
Martinez-Trujillo, M., Limones-Briones, V., Cabrera-Ponce, J. L., and Herrera-Estrella, L. (2004). Improving transformation efficiency of Arabidopsis thaliana by modifying the floral dip method. Plant Mol. Biol. Rep. 22, 63–70. doi: 10.1007/BF02773350
Pan, W. J., Tao, J. J., Cheng, T., Shen, M., Ma, J. B., Zhang, W. K., et al. (2017). Soybean NIMA-related kinase1 promotes plant growth and improves salt and cold tolerance. Plant Cell Physiol. 58, 1268–1278. doi: 10.1093/pcp/pcx060
Park, S. Y., Yu, J. W., Park, J. S., Li, J., Yoo, S. C., Lee, N. Y., et al. (2007). The senescence-induced stay green protein regulates chlorophyll degradation. Plant Cell 19, 1649–1664. doi: 10.1105/tpc.106.044891
Patanjali, S. R., Parimoo, S., and Weissman, S. M. (1991). Construction of a uniform-abundance (normalized) cDNA library. Proc. Natl. Acad. Sci. U.S.A. 88, 1943–1947. doi: 10.1073/pnas.88.5.1943
Qiu, J. L., Fiil, B. K., Petersen, K., Nielsen, H. B., Botanga, C. J., Thorgrimsen, S., et al. (2008a). Arabidopsis MAP kinase 4 regulates gene expression through transcription factor release in the nucleus. EMBO J. 27, 2214–2221. doi: 10.1038/emboj.2008.147
Qiu, J. L., Jilk, R., Marks, M. D., and Szymanski, D. B. (2002). The Arabidopsis SPIKE1 gene is required for normal cell shape control and tissue development. Plant Cell 14, 101–118. doi: 10.1105/tpc.010346
Qiu, J. L., Zhou, L., Yun, B. W., Nielsen, H. B., Fiil, B. K., Petersen, K., et al. (2008b). Arabidopsis mitogen-activated protein kinase kinases MKK1 and MKK2 have overlapping functions in defense signaling mediated by MEKK1, MPK4, and MKS1. Plant Physiol. 148, 212–222. doi: 10.1104/pp.108.120006
Sakuraba, Y., Kim, D., Kim, Y. S., Hortensteiner, S., and Paek, N. C. (2014a). Arabidopsis STAYGREEN-LIKE (SGRL) promotes abiotic stress-induced leaf yellowing during vegetative growth. FEBS Lett. 588, 3830–3837. doi: 10.1016/j.febslet.2014.09.018
Sakuraba, Y., Park, S. Y., Kim, Y. S., Wang, S. H., Yoo, S. C., Hortensteiner, S., et al. (2014b). Arabidopsis STAY-GREEN2 is a negative regulator of chlorophyll degradation during leaf senescence. Mol Plant 7, 1288–1302. doi: 10.1093/mp/ssu045
Sakuraba, Y., Park, S. Y., and Paek, N. C. (2015). The divergent roles of STAYGREEN (SGR) homologs in chlorophyll degradation. Mol. Cells 38, 390–395. doi: 10.14348/molcells.2015.0039
Sato, Y., Morita, R., Nishimura, M., Yamaguchi, H., and Kusaba, M. (2007). Mendel’s green cotyledon gene encodes a positive regulator of the chlorophyll-degrading pathway. Proc. Natl. Acad. Sci. U.S.A. 104, 14169–14174. doi: 10.1073/pnas.0705521104
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Shimoda, Y., Ito, H., and Tanaka, A. (2016). Arabidopsis STAY-GREEN, mendel’s green cotyledon gene, encodes magnesium-dechelatase. Plant Cell 28, 2147–2160. doi: 10.1105/tpc.16.00428
Si, L., Chen, J., Huang, X., Gong, H., Luo, J., Hou, Q., et al. (2016). OsSPL13 controls grain size in cultivated rice. Nat. Genet. 48, 447–456. doi: 10.1038/ng.3518
Smith-Hammond, C. L., Swatek, K. N., Johnston, M. L., Thelen, J. J., and Miernyk, J. A. (2014). Initial description of the developing soybean seed protein Lys-N(epsilon)-acetylome. J. Proteomics 96, 56–66. doi: 10.1016/j.jprot.2013.10.038
Soares, M. B., Bonaldo, M. F., Jelene, P., Su, L., Lawton, L., and Efstratiadis, A. (1994). Construction and characterization of a normalized cDNA library. Proc. Natl. Acad. Sci. U.S.A. 91, 9228–9232. doi: 10.1073/pnas.91.20.9228
Sobhanian, H., Razavizadeh, R., Nanjo, Y., Ehsanpour, A. A., Jazii, F. R., Motamed, N., et al. (2010). Proteome analysis of soybean leaves, hypocotyls and roots under salt stress. Proteome Sci. 8:19. doi: 10.1186/1477-5956-8-19
Song, X. J., Huang, W., Shi, M., Zhu, M. Z., and Lin, H. X. (2007). A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat. Genet. 39, 623–630. doi: 10.1038/ng2014
Szelei, J., Jimenez, J., Soto, A. M., Luizzi, M. F., and Sonnenschein, C. (1997). Androgen-induced inhibition of proliferation in human breast cancer MCF7 cells transfected with androgen receptor. Endocrinology 138, 1406–1412. doi: 10.1210/endo.138.4.5047
Takeshima, R., Hayashi, T., Zhu, J., Zhao, C., Xu, M., Yamaguchi, N., et al. (2016). A soybean quantitative trait locus that promotes flowering under long days is identified as FT5a, a FLOWERING LOCUS T ortholog. J. Exp. Bot. 67, 5247–5258. doi: 10.1093/jxb/erw283
Wang, F., Chen, H. W., Li, Q. T., Wei, W., Li, W., Zhang, W. K., et al. (2015). GmWRKY27 interacts with GmMYB174 to reduce expression of GmNAC29 for stress tolerance in soybean plants. Plant J. 83, 224–236. doi: 10.1111/tpj.12879
Wang, S., Li, S., Liu, Q., Wu, K., Zhang, J., Wang, Y., et al. (2015). The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat. Genet. 47, 949–954. doi: 10.1038/ng.3352
Wang, Y., Xiong, G., Hu, J., Jiang, L., Yu, H., Xu, J., et al. (2015). Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat. Genet. 47, 944–948. doi: 10.1038/ng.3346
Wei, Q., Guo, Y., and Kuai, B. (2011). Isolation and characterization of a chlorophyll degradation regulatory gene from tall fescue. Plant Cell Rep. 30, 1201–1207. doi: 10.1007/s00299-011-1028-8
Wei, W., Li, Q. T., Chu, Y. N., Reiter, R. J., Yu, X. M., Zhu, D. H., et al. (2015). Melatonin enhances plant growth and abiotic stress tolerance in soybean plants. J. Exp. Bot. 66, 695–707. doi: 10.1093/jxb/eru392
Xia, Z., Zhai, H., Lu, S., Wu, H., and Zhang, Y. (2013). Recent achievement in gene cloning and functional genomics in soybean. ScientificWorldJournal 2013:281367. doi: 10.1155/2013/281367
Yu, X., Yuan, F., Fu, X., and Zhu, D. (2016). Profiling and relationship of water-soluble sugar and protein compositions in soybean seeds. Food Chem. 196, 776–782. doi: 10.1016/j.foodchem.2015.09.092
Yuan, S., Trachtenberg, J., Mills, G. B., Brown, T. J., Xu, F., and Keating, A. (1993). Androgen-induced inhibition of cell proliferation in an androgen-insensitive prostate cancer cell line (PC-3) transfected with a human androgen receptor complementary DNA. Cancer Res. 53, 1304–1311.
Zhang, Z. X., Zhang, F. D., Tang, W. H., Pi, Y. J., and Zheng, Y. L. (2005). [Construction and characterization of normalized cDNA library of maize inbred Mo17 from multiple tissues and developmental stages]. Mol. Biol. 39, 198–206. doi: 10.1007/s11008-005-0026-8
Zhou, C., Han, L., Pislariu, C., Nakashima, J., Fu, C., Jiang, Q., et al. (2011). From model to crop: functional analysis of a STAY-GREEN gene in the model legume Medicago truncatula and effective use of the gene for alfalfa improvement. Plant Physiol. 157, 1483–1496. doi: 10.1104/pp.111.185140
Keywords: cDNA library, functional genomics, large-scale, gene mining, soybean
Citation: Li X, Huang L, Lu J, Cheng Y, You Q, Wang L, Song X, Zhou X and Jiao Y (2018) Large-Scale Investigation of Soybean Gene Functions by Overexpressing a Full-Length Soybean cDNA Library in Arabidopsis. Front. Plant Sci. 9:631. doi: 10.3389/fpls.2018.00631
Received: 09 January 2018; Accepted: 20 April 2018;
Published: 09 May 2018.
Edited by:
Paulo Arruda, Universidade Estadual de Campinas, BrazilReviewed by:
Zhixi Tian, Institute of Genetics and Developmental Biology (CAS), ChinaXianzhong Feng, Northeast Institute of Geography and Agroecology (CAS), China
Copyright © 2018 Li, Huang, Lu, Cheng, You, Wang, Song, Zhou and Jiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongqing Jiao, amlhb3lvbmdxaW5nQGNhYXMuY24=