 Nicholas R. LaBonte
Nicholas R. LaBonte Peng Zhao
Peng Zhao Keith Woeste
Keith Woeste- 1Department of Crop Sciences, University of Illinois Urbana-Champaign, Urbana, IL, United States
- 2Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, College of Life Sciences, Northwest University, Xi'an, China
- 3Hardwood Tree Improvement and Regeneration Center, Northern Research Station, USDA Forest Service, West Lafayette, IN, United States
Chestnuts (Castanea) are major nut crops in East Asia and southern Europe, and are unique among temperate nut crops in that the harvested seeds are starchy rather than oily. Chestnut species have been cultivated for three millennia or more in China, so it is likely that artificial selection has affected the genome of orchard-grown chestnuts. The genetics of Chinese chestnut (Castanea mollissima Blume) domestication are also of interest to breeders of hybrid American chestnut, especially if the low-growing, branching habit of Chinese chestnut, an impediment to American chestnut restoration, is partly the result of artificial selection. We resequenced genomes of wild and orchard-derived Chinese chestnuts and identified selective sweeps based on pooled whole-genome SNP datasets. We present candidate gene loci for chestnut domestication and discuss the potential phenotypic effects of candidate loci, some of which may be useful genes for chestnut improvement in Asia and North America. Selective sweeps included predicted genes potentially related to flower phenology and development, fruit maturation, and secondary metabolism, and included some genes homologous to domestication candidates in other woody plants.
Introduction
Traits relevant to plant domestication show genomic evidence of selection in diverse crop species, including grains (Cockram et al., 2007), legumes (Kaga et al., 2008; Lam et al., 2010; Li et al., 2013; Schmutz et al., 2014), annual fruit-bearing crops such as tomato and squash (Lefebvre et al., 1998; Frary et al., 2000; Ronen et al., 2000; Vrebalov et al., 2002; Rao and Paran, 2003; Guo et al., 2013), and woody perennial fruit crops (Cao et al., 2014; Khan et al., 2014). Some traits, such as flowering time and plant architecture, show genomic evidence of selection in many crops (Mao et al., 2000; Clark et al., 2006; Paran and van der Knaap, 2007; Tan et al., 2008; Zhou et al., 2009; Li et al., 2013). Other traits, such as fruit quality (Qi et al., 2013; Khan et al., 2014; Qin et al., 2014) or seed size (Shomura et al., 2008; Wang et al., 2008), are associated with genomic evidence of selection only in specific types of crops. Signatures of selection in the genomes of woody perennial crops may be obscured by longer generation times and more widespread self-incompatibility (Cornille et al., 2012) than annual crop plants. Nevertheless, parts of the genomes of grape (Zhou et al., 2017), peach (Cao et al., 2014; Akagi et al., 2016), and apple (Khan et al., 2014; Duan et al., 2017) have been identified as candidates for selection during domestication.
Chestnut (primarily Castanea mollissima) was first deliberately cultivated as a food plant in China at least 2000 years before present (ybp) (Rutter et al., 1991; Wang, 2004), likely more recently than the domestication of apples (4,000 ybp: Cornille et al., 2012) or of peach and almond (5,000 ybp: Velasco et al., 2016). It is possible that humans began artificially selecting chestnuts earlier than 2,000 ybp: an increase in chestnut pollen, at the expense of conifers, is noted in the archaeological record of northwest China around 4,600 ybp, which coincides with the appearance of grain cultivation (Li et al., 2007). Today, chestnut is an economically valuable crop and China is the world's largest producer (Metaxas, 2013). Chestnut orchards in China include both seedling trees and grafted cultivars, mostly of C. mollissima, with some regional use of C. henryi, C. crenata, or interspecific hybrids (Wang, 2004). The timing of flower development, pollination, and fertilization of ovules is crucial for optimizing chestnut yield (Shi and Stoesser, 2005); self-pollination does not normally occur (Pereira-Lorenzo et al., 2016). Characteristics currently under selection in improvement programs for Chinese orchard chestnuts include attractive (shiny) appearance of nuts, early maturation and bearing, stable yield, high sugar content, pest and disease resistance, and adaptation to orchard environments that are hotter and drier than the mountains where most wild C. mollissima occur (Zhang et al., 2010). Shorter catkins are also desired (Huang et al., 2009), as are large seeds (~20 g) (Xu et al., 2010), especially for commercial paste-production cultivars, and a pellicle that is easy to peel (Takada et al., 2012). Post-harvest diseases that destroy chestnuts in storage are a major concern (Ma et al., 2000). A study in Japanese chestnut (Nishio et al., 2017) recently revealed quantitative trait loci associated with a set of traits including harvest date, nut weight, and pericarp splitting. Broad-sense heritability estimates for these traits ranged from 0.40 (nut weight) to 0.91 (harvest date) (Nishio et al., 2014).
Traits possibly under selection during chestnut domestication include the traits currently targeted for improvement, as well as others, including plant architecture. A small, branchy tree is more manageable in an orchard setting than a very tall one, especially in locales where chestnuts are picked by hand after climbing the tree (Rutter et al., 1991). Chinese chestnut in general has a shorter stature and less-pronounced apical dominance than the non-domesticated American chestnut (Clapper, 1954) which is a major consideration in the backcross blight resistance breeding program being carried out by the American Chestnut Foundation (Burnham et al., 1986). In forest settings, C. mollissima grow to 20–25 m in height (Fei et al., 2012), so the short stature of orchard trees may be, at least in part, an artificially selected trait. Chestnuts are highly perishable (Rutter et al., 1991) so genes related to pericarp thickness and wax coatings on the pericarp may be important if they confer improved storage qualities. Fruit quality genes, while they may not affect the flavor of the chestnut, could be under selection for human aesthetic preferences; Clapper (1954) noted variation in the color of Chinese chestnuts that was not seen in American chestnuts. Finally, although preference for large seeds varies across China (Wang, 2004; Yang et al., 2015), seed size is a likely cause for artificial selection in Chinese chestnut, especially for “processing” varieties intended for the industrial production of paste and flour (e.g., Xu et al., 2010).
In addition to differentiation between cultivated and wild Chinese chestnut, there is likely to be differential selection among regional subpopulations of wild trees: Chinese chestnut occupies a larger range than any other Asian or American species of Castanea (Fei et al., 2012). The natural selective pressure on Chinese chestnut populations is likely to vary considerably between its temperate, high-altitude habitat in the Qin Mountains (northwest China) and the subtropical provinces of Yunnan and Guizhou. Considerable rangewide genetic variation, at the whole-genome scale, has been identified in forest tree genomes, including poplar (Slavov et al., 2012) and whitebark pine (Syring et al., 2016). Genetic diversity of wild Chinese chestnut has been analyzed with varying results; southwest (Zhang and Liu, 1998) and northwest China (Shaanxi Province; Cheng et al., 2012) have been proposed as centers of genetic diversity for the species. Given its wide distribution, the census population size of Chinese chestnut is probably similar to the estimated 3-4 billion American chestnuts (Castanea dentata) that grew in eastern North America prior to the introduction of chestnut blight disease (Hebard, 2012); given its outcrossing habit, this likely corresponds to a very high effective population size in wild Chinese chestnut. While genetic diversity is higher in wild trees, it appears that a high level of genetic diversity has been maintained in orchard (domesticated) Chinese chestnuts (Pereira-Lorenzo et al., 2016), although the genetic diversity of new cultivars may be lower than traditional orchard trees (Ovesna et al., 2004).
Signatures of selection due to domestication are generally identified as regions of the genome where, using statistics related to nucleotide diversity and heterozygosity (Tajima's D, pi, FST), reduction of allelic diversity in domesticated lineages, vs. wild lineages, is determined to be signficant (Teshima et al., 2006; Purugganan and Fuller, 2009); these regions may be called “selective sweeps.” Dozens or hundreds of relatively small genomic intervals may show evidence of a selective sweep in a domesticated plant genome. Given the large number of statistical tests, the likelihood that sweeps will be observed by chance alone (false positives) is high (Thornton and Jensen, 2007), although statistical methods for ameliorating this problem are available(Burger et al., 2008). Genes identified in domestication regions, if subsequent investigation confirms their predicted function and phenotypic effects, could be important for further improvement of Chinese and other chestnut species for orchard production.
We investigated the following questions:
1) Is genetic diversity on the genomic scale lower in orchard-derived Chinese chestnut than it is in wild Chinese chestnut?
2) What regions of the genome show evidence of selective sweeps in the genome of domesticated Chinese chestnut, and are these regions syntenic with regions under selection in other woody plants?
3) Do northern (Shaanxi Province) and southern (Yunnan and Guizhou) gene pools of wild Chinese chestnut present different signatures of selection?
To answer these questions, we utilized whole-genome resequencing with a pool-seq approach. Because we investigated genetic differentiation among different groups (pools) of trees rather than individual trees, it was feasible to estimate allele frequencies and genetic statistics (pi and Tajima's D) from pools of samples rather than individual genome sequences (Lynch et al., 2014) Pool-seq may reduce the precision of allele frequency estimates, but becausethere was no individual phenotype information (e.g. disease resistance, seed size) available for most of our samples, the potential gains from sequencing individuals was limited. Because the sequencing cost per individual was less, more individuals (a larger sample of the total genetic variation among wild and orchard trees) could be used to estimate population genetics statistics (Schlöetterer et al., 2014; Chen et al., 2016). We validated candidate loci for selection under domestication, identified by the pool-seq analysis, by analyzing nucleotide diversity statistics and heterozygosity of the same genomic regions in an independent sample of high-coverage genome sequences of 17 orchard-derived Chinese chestnut accessions.
Materials and Methods
DNA Samples
Leaf samples were collected in China during 2015, rapidly dried using desiccant beads, and mailed to Purdue University for DNA isolation in 2016 following applicable regulations for the importation of plant DNA samples. Trees classified as wild were sampled from natural montane forests where it is relatively unlikely that groves of chestnut represent escapes from cultivation (Figure 1). Orchard trees were sampled from orchard settings in northeast China where most commercial growing takes place (Table 1). The United States sample of orchard-derived Chinese chestnut was grown at Empire Chestnut Company, Carrollton, OH, from Beijing-area source material. DNA from US samples was isolated from dormant twigs. For leaf and twig samples, tissue (about 16 cm2 of leaf or a 6 cm section of twig with buds) was ground to a fine powder in liquid nitrogen using a mortar and pestle, then added to a tube of heated (55°C) CTAB extraction buffer and incubated for 4–6 h. Following incubation, DNA isolation was performed in 15 mL conical tubes using a phenol-chloroform extraction protocol, and DNA was precipitated in 0.2 M sodium chloride and isopropanol. After pelleting and resuspension of DNA in TE buffer, samples were cleaned using OneStep PCR Inhibitor Removal kits (Zymo Research, Irvine, CA, USA). Samples were quantified and quality assessed using a NanoDrop 8000 (Thermo-Fisher Scientific, Waltham, MA, USA) prior to pooling. Samples were pooled by source location at equimolar concentrations at a final volume of 200 uL and submitted for sequencing.
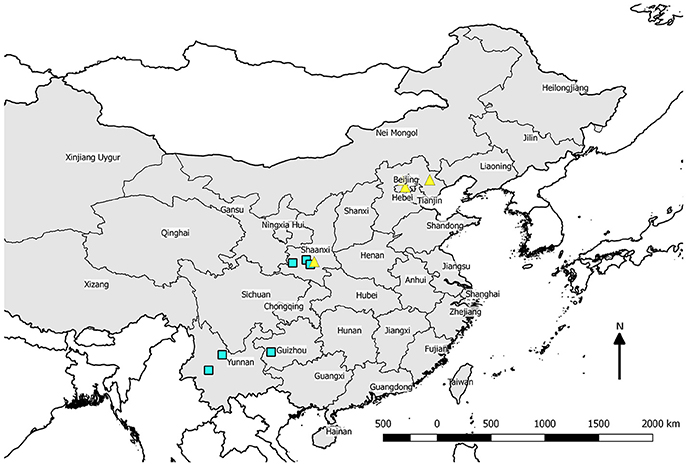
Figure 1. Map of the People's Republic of China showing locations from which wild trees were sampled (open squares) and the location of orchards sampled (filled triangles).
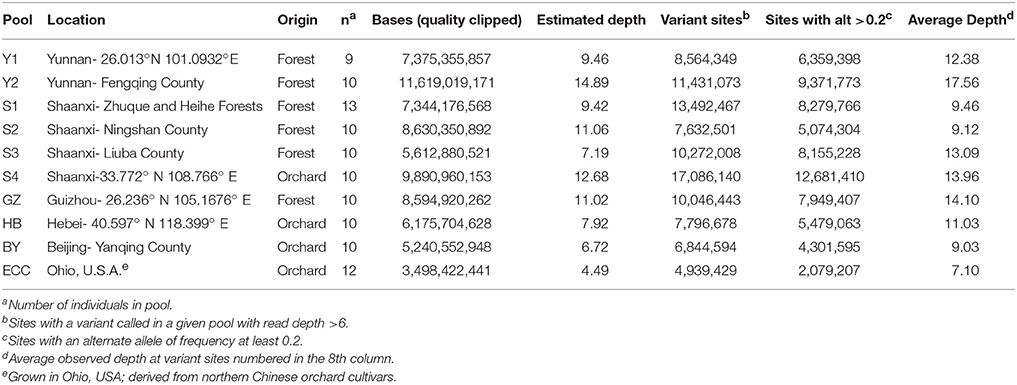
Table 1. Castanea mollissima DNA sample pools, with individuals (n) per sample site.
DNA Sequencing and Assembly
Sequencing of 100 bp paired-end reads was carried out with an Illumina HiSeq 2500 (Illumina Inc., San Diego, CA, USA) at the Purdue Genomics Core Facility. Six genomic DNA pools (about 10 individuals each; Table 1) were sequenced per lane, with the goal of obtaining ~10x coverage per pool. Low-quality reads were filtered prior to assembly using Trimmomatic version 0.32 (Bolger et al., 2014).
Chloroplasts were sequenced by assembling short reads to the complete Chinese chestnut chloroplast reference sequence (Jansen et al., 2011). The 1.0 version of the Linkage Group A (LGA) pseudochromosome assembly and beta versions of the LGB-LGL assemblies (12 total) were obtained from Dr. John Carlson of Penn State University (Staton et al., 2014). Short reads were assembled to reference sequences using BWA, duplicates were flagged and alignment files sorted using Picard Tools, and SNPs were called using the HaplotypeCaller tool from the Genome Analysis ToolKit (GATK), with a polyploid value equal to the number of individuals in the pool. The Samtools mpileup tool was used to generate pileup-formatted SNP files for the orchard and wild sets of sample pools.
Identification of Regions Under Selection in the Genome
Tajima's D and pi were calculated from mpileup files of orchard and wild assemblies using PoPoolation 2.0 (Kofler et al., 2011) over 10 kb windows for the entire genome. The difference in Tajima's D between orchard and wild pools was calculated and statistical significance tested using a permutation test encoded in a Perl script. Permutations were performed by assigning observed Tajima's D values within the orchard and wild pools of samples to a random base-pair interval of the genome and re-calculating the difference in Tajima's D between pools over the shuffled intervals. A p-value was assigned to each interval based on how many times a difference larger than the difference at that interval was observed in 1,000 shuffled genomes. Candidate loci for selection in orchard trees were intervals where the permuted p-value was less than 0.01. To reduce the false positive rate, we only considered for further analysis intervals where multiple consecutive 10 kb intervals showed significantly different (p < 0.01) values for Tajima's D and pi in orchard vs. wild trees, and/or a p-value less than 0.001. In addition, local false discovery rates for all 10 kb intervals were calculated using the qvalue package (Storey, 2002) in the R computing environment.
A second method for identifying regions in the genome under selection identified predicted gene intervals where the percent of SNPs that had one allele fixed was higher in one sample than in the other. The frequency of the major allele at SNP loci was averaged over all SNPs in a given predicted gene, and then the average major allele frequency was calculated for 10-gene intervals across the genome. Loci potentially under selection in orchard trees were identified based on the empirical distribution of the difference in the allele-frequency statistic over all predicted genes that had alignments to the UniProt database. A predicted gene was determined as potentially under selection if the difference in average major allele frequency between wild and orchard samples was greater than two standard deviations above the mean difference for all predicted genes in the genome. This method was used to identify genes under selection in orchard vs. wild trees, and also to identify loci with varying allele frequency among regional subpopulations of wild trees: northern (Shaanxi) vs. southern (Yunnan + Guizhou).
Gene Prediction and Filtering
De novo gene prediction was carried out using AUGUSTUS (Stanke et al., 2006) with Arabidopsis thaliana as the training protein set and default settings. To assign a putative function to predicted genes, the predicted gene file (.gff) was converted to fasta (.fa) format and aligned to the UniProt protein database using the blastp function of the DIAMOND sequence aligner (Buchfink et al., 2015) using default settings. The top hit annotation on the UniProt website was used to assign a putative function to each gene.
To provide a measure of validation to this predicted gene set, publicly available cDNA contig files for American chestnut, Chinese chestnut, European chestnut, and Japanese chestnut were downloaded from http://www.hardwoodgenomics.org/transcriptomes. These were each aligned using the blastx function of DIAMOND, using default settings, to a database created using the predicted Chinese chestnut protein set output by AUGUSTUS. Transcripts were matched to the protein that provided the top hit from the predicted protein set; a predicted protein was only counted as having transcript support if it was the best alignment for at least one cDNA contig. This was carried out using a custom Perl script.
Identification of Chloroplast Haplotypes
Chloroplast reads from whole-genome sequence data were assembled to the reference Chinese chestnut chloroplast genome using BWA and Picard Tools and SNPs were called using GATK with ploidy set equal to 10. A custom Perl script was developed that tallied the number of SNPs with a given alternate allele frequency (between 10 and 100%) in each pool as an approximation of the haplotype structure of the genome pools. For example, if a chloroplast haplotype with about 300 SNP variants vs. the reference was found in 30% of the samples from a pool, we expected to find about 300 SNP sites with 30% alternate allele frequency in that pool. Alternate chloroplast haplotypes were identified by peaks on a histogram of SNPs in allele frequency bins for each sample; the frequency of a haplotype was estimated by the bin where a “peak” occurred, and the haplotype identity estimated by the number of SNPs in an allele frequency bin (Figure S1). SNPs were compared with individual chloroplast sequences from Chinese chestnuts to determine whether haplotypes matched either of the two previously identified haplotypes.
Validation of Regions Under Selection
Whole-genome sequences of individual chestnuts were used to provide validation of regions under selection identified using pooled sequences. Tajima's D, nucleotide diversity, heterozygosity, and pi were calculated (VCFTools) using SNPs within exons of predicted genes for 18 Chinese chestnuts of southern Chinese and Korean provenance, as well as 2 American chestnuts, which represent non-domesticated trees. A negative value of Tajima's D, low values for pi, and proportion of heterozygous loci for a given predicted gene among individual orchard-derived Chinese chestnuts, were interpreted as support for a gene's selection during domestication. Synteny with other domesticated woody plants (peach, apple, and grapevine) was analyzed by aligning predicted proteins from domestication-related selective sweeps in peach (Cao et al., 2014), apple (Duan et al., 2017), and grape (Zhou et al., 2017) to predicted proteins from chestnut sweep regions. We considered there to be evidence of syntenic domestication regions if multiple chestnut proteins from a given regions were the best alignments for multiple proteins from a domestication region in another woody domestic plant. Correlation between the location of putative domestication selective sweeps and chestnut agronomic QTL was identified by aligning microsatellite and SNP markers from a QTL mapping experiment (Nishio et al., 2017) to the whole genome and calculating the distance (bp) between QTL-delimiting markers and putative domestication sweeps.
Results
Genome Sequencing and Assembly
Average estimated genome coverage for the pools sequenced was close to 1x per individual tree in a pool for most of the sequenced pools (Table 1) and was greater than 7x for all but two of the pools sequenced. The number of polymorphisms with alternate allele frequencies >0.2, which are less likely to result from sequencing errors, was highest in the Shaanxi orchard sample and lowest in the Beijing-derived orchard sample from Ohio (Table 2). The genomes of most of the orchard samples had fewer polymorphisms than wild trees.
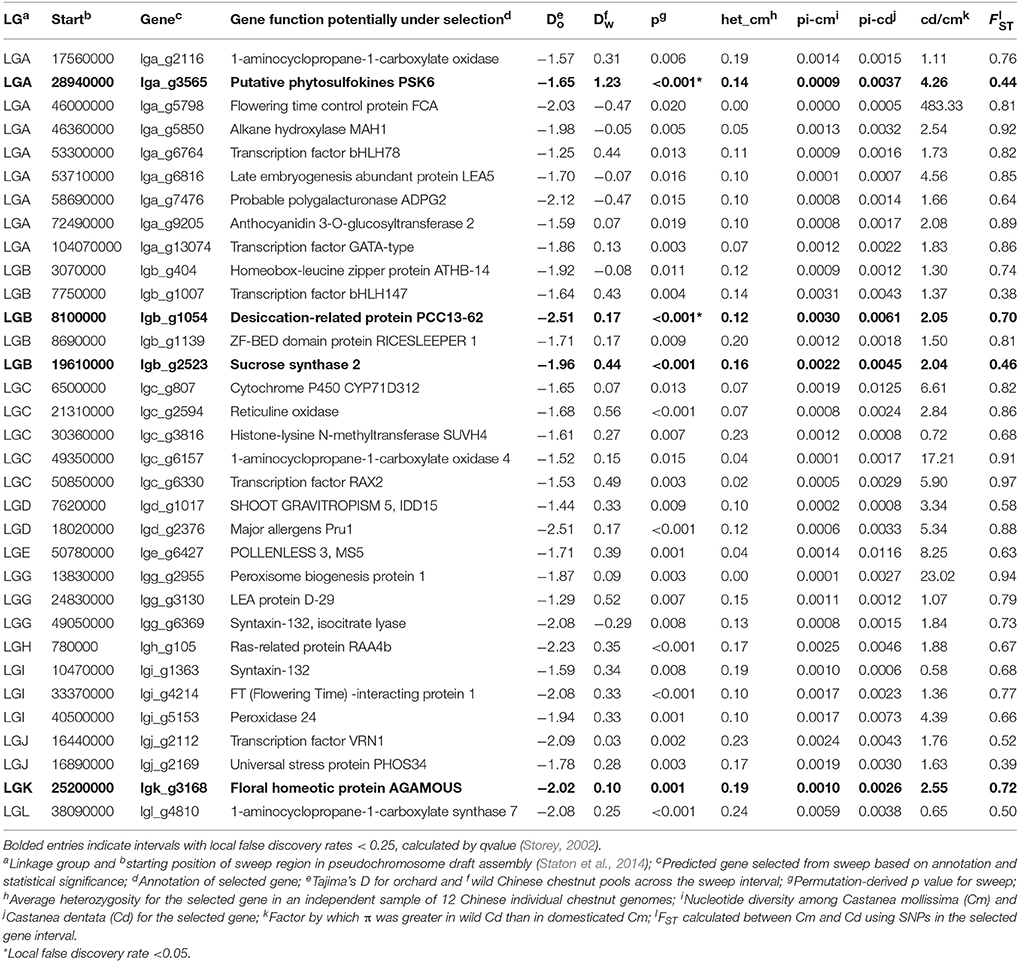
Table 2. Notable regions under selection during chestnut domestication with statistical support and functional annotations.
Regions Under Selection
Tajima's D, used as a measure of selection pressure, was on average lower in orchard pools (−0.64) than in wild (−0.50). Using the Tajima's D and pi outlier method, >100 intervals were significantly different between wild and orchard trees, as determined by permutation tests with a significance cutoff of p < 0.01 for a given 10,000 base-pair interval (Table S1); several intervals with large differences in Tajima's D were chosen for further annotation (Table 1). The major allele frequency across predicted gene sequences was slightly higher for orchard chestnuts (0.693) than for wild chestnuts (0.685). Using the allele frequency method to identify regions under selection, the standard deviation of the difference in major allele frequency between orchard and wild pools was used to identify outliers (cutoff: >3 standard deviations greater than mean difference for orchard vs. wild and >2 sd for regional differences), which led to the identification of approximately 25 candidate loci for domestication and 15 for regional genetic differences (Tables 3, 5, Table S3). The identified candidate loci contained predicted flowering-time genes, genes involved in the synthesis of ethylene, genes influencing male fertility, cell wall structure, secondary metabolites, and disease resistance (Tables S2, S4). Candidate loci under selection showed lower-than average heterozygosity and nucleotide diversity in Chinese chestnut and, in many cases, greater nucleotide diversity in American chestnut than Chinese chestnut (Table S5). Several predicted proteins in putative selective sweeps of chestnut were likely homologs of predicted proteins in selective sweep regions of peach, apple, and grapevine (Tables S2, S4, S6); in total, 11 of the identified sweep regions in chestnut showed evidence of synteny with domestication candidate regions with at least one other woody plant.
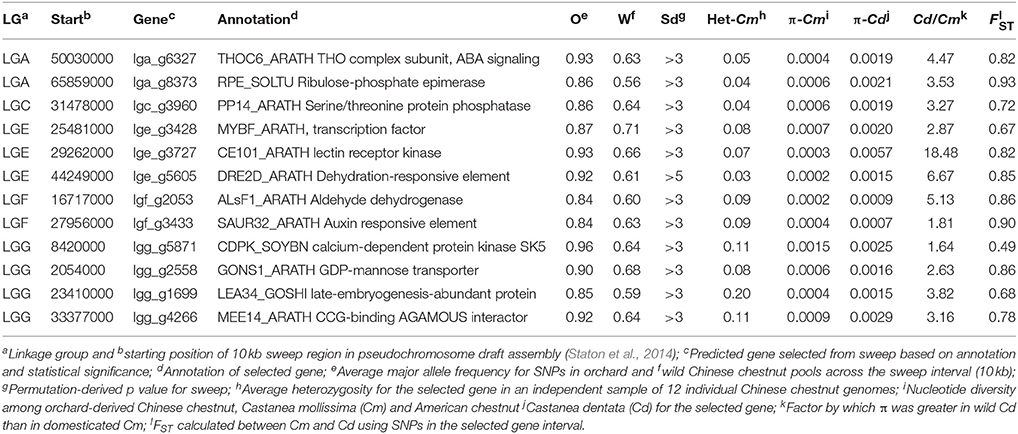
Table 3. Additional regions under selection due to domestication and regional climatic variation identified by allelic fixation at SNPs.
Chloroplast Haplotypes
The reference chloroplast haplotype was found at its highest frequency in one Yunnan sample (100%) and the Guizhou sample (~60%), and at its lowest frequencies in the Hebei and ECC orchard samples (~10%) (Figure S2). One alternate haplotype was present in the Guizhou (~40%), Hebei (~90%), ECC (90%), Beijing (~20%), and Shaanxi-3 (~90%) pooled samples (Figures S2, S3). This haplotype, which had about 260 SNP polymorphisms different from the reference, was found to be the same as the (non-reference) C. mollissima chloroplast of “Clapper” (LaBonte et al., in preparation). Other polymorphic sites did not correspond to the “Clapper” haplotype, so additional haplotypes must have been present in some of the sampled populations. A highly divergent (1000+ SNPs different from reference) haplotype appears to be present at relatively low frequency in the Shaanxi-1, Shaanxi-4, and Yunnan-2 samples (Figure S3), and an additional haplotype with low divergence from the reference, about 75 SNPs, appears to be present in the Shaanxi-1 sample (Figure S2).
Discussion
Chloroplast Assemblies and Genetic Diversity
Genotyping of pooled chloroplasts indicated the presence of several haplotypes not identified in a previous survey of Castanea mollissima chloroplast genome assemblies (LaBonte et al., in preparation). Other than the reference haplotype, the most common and widely-distributed haplotype was variant at ~250 sites and was most abundant in northern Chinese orchard samples, but is not particularly common in American orchard germplasm. The reference haplotype was most abundant in Southern Chinese wild samples; its abundance in the US population of Chinese chestnut supports a southern origin for most US chestnut germplasm. The Shaanxi orchard chestnut sample's chloroplast genotype profile resembled the wild Shaanxi-1 chloroplast profile more than it did the other orchard samples, which indicated that admixture between local wild populations and orchard trees is probably extensive in cultivated Chinese chestnut. The chloroplast haplotype shared by “Clapper” and two of the orchard pools (Hebei and ECC) was also found at high frequency in the Shaanxi-3 wild sample. The diversity of chloroplast haplotypes evident in the three wild Shaanxi samples supports earlier findings that the Qinling (=Dabashan) range in Shaanxi province represents a center of genetic diversity for C. mollissima (Cheng et al., 2012; Liu et al., 2013a). More sampling of whole chloroplast genomes is needed determine the true number of unique haplotypes, especially in the Shaanxi and Yunnan chestnut populations, where the strongest evidence for diversity was observed.
Previous studies of genetic diversity in wild and orchard Chinese chestnuts found relatively high genetic diversity maintained in orchard trees (Pereira-Lorenzo et al., 2016). It appears to be the case that, like other perennial woody food plants (Cornille et al., 2012) the overall reduction in genetic diversity in chestnut due to domestication has been limited. Despite this, the number of 50–100 kb regions in the genome where orchard trees had low genetic diversity relative to wild trees was about 10 times larger than the number of regions where orchard trees had higher nucleotide diversity than wild trees. It is possible that lower genome coverage in orchard samples led to underestimates of heterozygosity. The same minimum coverage filter (8x) was implemented for the SNP sets from orchard and wild pools during data analysis, however, to minimize bias due to lower coverage of orchard tree genomes. Using individual whole-genome SNP data from 17 orchard-grown Chinese chestnuts and two American chestnuts, we were able to identify several loci that showed strong evidence of low genetic diversity both in orchard pools and in orchard chestnuts relative to the non-domesticated American chestnut. These predicted genes (Tables 2, 3; also highlighted in Tables S2, S4) we consider the best candidates for chestnut domestication. Several putative chestnut sweeps (on LGA, LGC, LGD, LGI, and LGL) contained multiple predicted genes with >60% amino acid identity to predicted genes from sweeps in apple (Duan et al., 2017), peach (Cao et al., 2014), and grape (Zhou et al., 2017) (Figure 2, Table 4, Table S6), indicating that some syntenic loci have likely been selected in multiple domesticated woody plants.
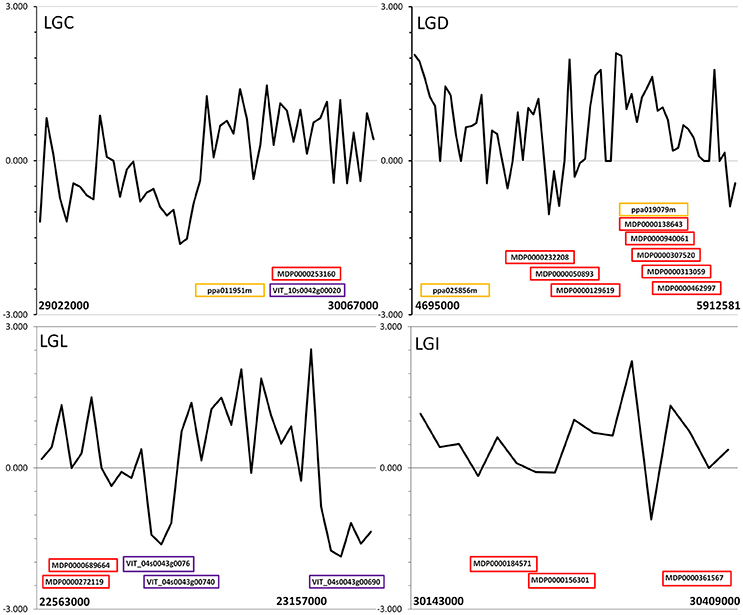
Figure 2. Tajima's D statistic in an independent sample of 8 orchard-derived chestnut whole-genome sequences, graphed over putative selective sweeps on LGC, LGD, LGL, and LGI of the Chinese chestnut genome identified using pooled whole-genome data. Approximate locations of predicted chestnut genes that were the best alignment for genes in domestication-associated selective sweeps of apple (red), grape (purple) and peach (orange) are labeled with the name of the aligned apple, grape, or peach gene.
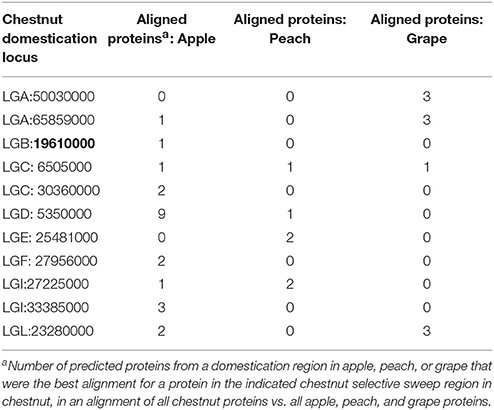
Table 4. Evidence of synteny between chestnut domestication candidate loci and domestication-associated chromosomal regions in other woody plant crops.
Functional Annotation of Regions Under Selection in Chestnut Domestication
Domestication candidate loci with the strongest statistical evidence, considering permutation tests, local false discovery rate calculations, and nucleotide diversity in independent whole-genome SNP datasets from orchard-derived Chinese chestnuts (Table 3) included several predicted genes with annotations that indicate a potential role in chestnut domestication. One locus on LGA included a predicted gene similar to a putative phytosulfokines 6 protein from Arabidopsis, which is a growth regulator active during embryogenesis (Matsubayashi et al., 2006). Additional highly significant loci included a dessication-related protein and a sucrose-synthase (Angeles-Nunez and Tiessen, 2010) like protein on LGB; the latter protein is highly similar (90.9% peptide identity) to a domestication candidate (MDP0000859573) on chromosome 13 of apple (Duan et al., 2017).
Several additional loci contained predicted gene annotations pointing to potential roles in chestnut domestication. One, also on LGA, was similar to anthocyanidin 3-O-glucosyltransferase 2 (LGA) of wine grapes (Vitis vinifera), which is responsible for the synthesis of red wine pigments (Ford et al., 1998). The existence of Chinese chestnut cultivars with enhanced red coloration in their leaves and twigs (Junhao et al., 2000) indicates that increased anthocyanin production was selected for during domestication.
Genes that regulate flower development and timing are among the most frequently identified in selective sweeps related to plant domestication (e.g., Kaga et al., 2008; Schmutz et al., 2014). Predicted genes similar to known flowering-time regulatory genes were found at several putative selective sweep loci. Putative domestication sweep regions included predicted genes similar to FLOWERING LOCUS C (FLC), a MADS-box protein that functions as major floral development repressor (Choi et al., 2009); FTIP1 of Arabidopsis, which exports the essential flowering control protein FLOWERING TIME (FT) into phloem sieve elements (Liu et al., 2012), POLLENLESS, a male fertility locus (Glover et al., 1998), AGAMOUS, which controls organ identity in developing flowers (Drews et al., 1991), and SUVH4 which suppresses a transcriptional regulator (Jackson et al., 2002) involved in female floral development (Sakai et al., 1995). The FLOWERING LOCUS C homolog showed a particularly strong signature of selection in the 17 whole-genome sequences we obtained from orchard-derived Chinese chestnuts (Tables S2, S4, S6) vs. non-domesticated American chestnut. The POLLENLESS_like gene is intriguing because a short-catkin mutation of Chinese chestnut has previously been identified (Feng et al., 2011), and some Castanea sativa cultivars with exceptionally large nuts (“marron” types) actually produce astaminate catkins that are sterile (Pereira-Lorenzo et al., 2006, 2016).
A number of the predicted genes in the regions with signatures of selection in orchard trees were similar to genes in model plants that are involved in the regulation of plant development and cell wall modification: a shoot gravitropism regulator (SGR5 or IDD15) of Arabidopsis, which regulates branch orientation (Cui et al., 2013) and starch levels (Tanimoto et al., 2008), a cell-number regulation enzyme of maize (LGC) that affects plant organ size and is homologous to a major fruit weight QTL gene in tomato (Guo et al., 2010), Arabidopsis RABA4B, a Golgi-network trafficking regulatory protein that may involved in the secretion of cell wall components (Preuss et al., 2004), and a polygalacturonase similar to ADPG2 in Arabidopsis, which is involved in pod shattering (González-Carranza et al., 2007; Ogawa et al., 2009) Modification of cell walls is a major part of fruit ripening, which is why polygalacturonases, cellulases, and other cell-wall enzymes have been discovered in selective sweeps in the genomes of domesticated tomato and pepper (Paran and van der Knaap, 2007). The IDD15-like locus may correspond to a Japanese chestnut nut weight QTL (Nishio et al., 2017), and the RABA4-like and polygalacturonase-containing loci correspond closely to QTL identified for harvest time in Japanese chestnut (Nishio et al., 2017).
Management of environmental stresses—heat and drought tolerance, as well as insect pests and fungal diseases—is currently a goal of chestnut breeding programs in China (Gaoping et al., 2001). It is likely that stress tolerance has been under selection throughout Chinese chestnut's history of cultivation. Management of disease and environmental stress was the inferred role of several predicted genes within the putative domestication intervals: one similar to the ethylene-responsive transcription factor ERF3; late-embryogenesis-abundant (LEA) proteins from orange (Citrus aurantium var. chinensis) and cotton (Gossypium hirsutum), which are believed to have a role in desiccation tolerance of seeds and vegetative tissues (Battaglia et al., 2008); homeobox-leucine zipper transcription factor proteins ATHB-6, involved in water deficit responses (Söederman et al., 1999); and a predicted peroxidase similar to a protein in Arabidopsis which is upregulated in response to cold (Fowler and Thomashow, 2002).
Phytohorome metabolism, and transcription factors that regulate plant development, are commonly associated with domestication-related selective sweeps, such as the bHLH and MYB-family transcription factors identified in domestication sweep regions of the genomes of peach (Cao et al., 2014) and apple (Khan et al., 2014; Duan et al., 2017), as well as other plants (e.g., Schmutz et al., 2014). Several MYB- and bHLH-type transcription factors were found in regions that showed evidence of strong selection in the genomes of orchard chestnuts. One basic helix-loop-helix (bHLH)—type transcription factor in a sweep region may be a homolog to the Arabidopsis bHLH78 transcription factor, which promotes the expression of the Flowering Time gene and therefore is involved in the initiation of flowering (Liu et al., 2013b). One putative selective sweep (LGD) containing a predicted MYB-type transcription factor that corresponded to a QTL (Nishio et al., 2017) for bur number/tree in Japanese chestnut. Two individual selective sweeps on different linkage groups (LGA, LGC) contained predicted genes that were similar to 1-aminocyclopropane-1-carboxylate oxidase genes from Arabidopsis and a third (LGL) contained one that was similar to 1-aminocyclopropane-1-carboxylate synthase. The products of these genes together regulate the production and degradation of the plant hormone ethylene (Yamagami et al., 2003; Qin et al., 2007). It is not clear, however, whether these ethylene-related genes influence nut ripening, stress response, or other processes.
Most loci with regional differences in allele frequency were closer to fixation in the southern samples of wild trees (Yunnan and Guizhou) than in the northern sample (Shaanxi), with the exception of one interval on LGE that contained a predicted gene similar to cinnamoyl alcohol dehydrogenase from Eucalyptus botryoides, and another on LGH that was similar to a senescence-associated protein from Arabidopsis (Table 5). The locus on LGE is intriguing because it may correspond to a QTL for resistance to Phytophthora cinammomi resistance in hybrids of Chinese and American chestnut (Olukolu et al., 2012). It is possible that more alleles for this gene are present in southern Chinese populations of chestnut to combat variable races of P. cinnammomi, which thrive in warm climates. Several other genes in regions with differentiated allele frequencies among regional subpopulations included several lignin-synthesis genes, and a DRE1B-type gene, all of which are probably involved in cold-tolerance. Interestingly, one predicted gene that had decreased allele frequency in southern China was similar to a transcription factor in Arabidopsis that controls trichome density (Schnellmann et al., 2002). Increased trichome density could be favorable in warmer climates where water loss is more severe during hot weather.
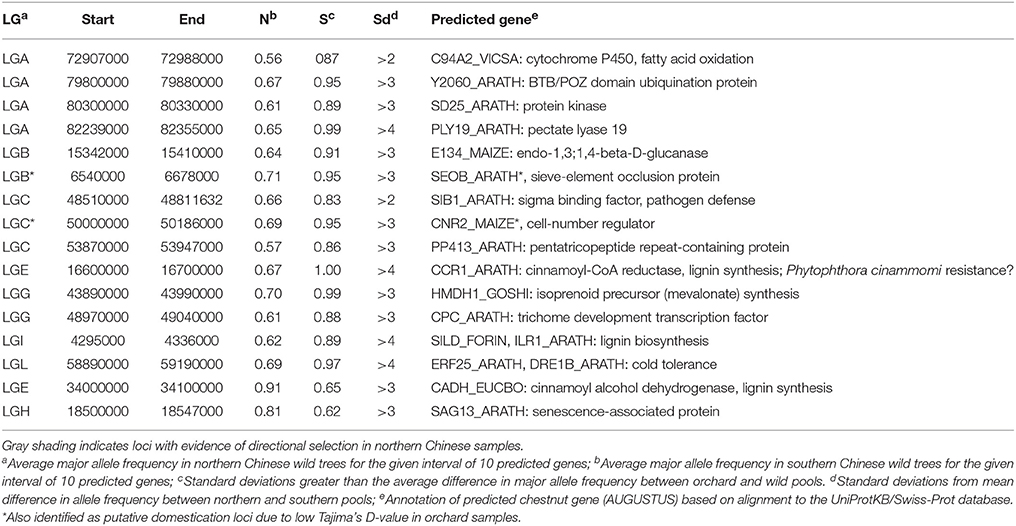
Table 5. Putative loci differentially selected among northern and southern samples of wild Chinese chestnut, identified by comparing allele frequencies among pools of chestnut, with annotations based on the best UniProt alignments of predicted genes.
Conclusions
Our study provides a first glimpse into the complex pathways of selection by which humans transformed a forest tree into a reliable food crop, but also has practical importance for chestnut improvement. For breeders who are interested in improving Chinese chestnut for increased nut production or nut size, genes that were selected during domestication to promote heavier fruiting, such as the male-sterility genes identified here, could be a pathway to trees with shorter catkins and more female flowers. Many of the genes potentially involved in cuticular wax synthesis, stress tolerance, and synthesis of secondary compounds could be used for improving storage quality and pest resistance of chestnuts. For breeders who are interested in transferring disease resistance from Chinese chestnut into other species, genes involved in orchard-type crown architecture might be desirable or undesirable, depending on the phenotypic goals of the program. Conversely, some of the genes identified in these sweep regions may be desirable for improving the resistance of other chestnut species to pests like Asian gall wasp and Phytophthora root rot. More research is needed to determine the actual phenotypic effects of the gene loci identified here, but our results provide a glimpse of selective pressure on the chestnut genome during the tree's domestication, and a rough sketch of a map for future genomics-assisted chestnut improvement.
Data Statement
All sequence data associated with this project is stored in a sequence read archive (SRA) on the GenBank website with accession number (PENDING). Custom Perl scripts (e.g., the permutation test) used in this research are available upon request from the corresponding author.
Author Contributions
NL carried out DNA extraction, sequencing, and analysis as part of his doctoral research. PZ supervised collection of Chinese chestnut samples from wild and orchard populations. As NRL's doctoral advisor KW provided guidance for the research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Special thanks are due to Aziz Ebrahimi for helping with DNA extractions, (Morgan's students) for collecting Chinese chestnut leaf samples, and Greg Miller of the Empire Chestnut Company for providing orchard chestnut samples and helpful comments on Chinese chestnut orchard culture. This work was funded by a Frederick M. Van Eck scholarship in the Forestry and Natural Resources department at Purdue University. Thanks also to the Purdue Genomics Core Facility for their role in preparing and sequencing libraries. Mention of a trademark, proprietary product, or vendor does not constitute a guarantee or warranty of the product by the U.S. Department of Agriculture and does not imply its approval to the exclusion of other products or vendors that also may be suitable.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00810/full#supplementary-material
References
Akagi, T., Hanada, T., Yaegaki, H., Gradziel, T. M., and Tao, R. (2016). Genome-wide view of genetic diversity reveals paths of selection and cultivar differentiation in peach domestication. DNA Res. 23, 271–282. doi: 10.1093/dnares/dsw014
Angeles-Nunez, J. G., and Tiessen, A. (2010). Arabidopsis sucrose sythase 2 and 3 modulate metabolic homeostasis and direct carbon towards starch synthesis in developing seeds. Planta 232, 701–718. doi: 10.1007/s00425-010-1207-9
Battaglia, M., Olvera-Carrillo, Y., Garciarubio, A., Campos, F., and Covarrubias, A. A. (2008). The enigmatic LEA proteins and other hydrophobins. Plant Physiol. 148, 6–24. doi: 10.1104/pp.108.120725
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina Sequence Data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buchfink, B., Xie, C., and Huson, D. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Burger, J. C., Chapman, M. A., and Burke, J. M. (2008). Molecular insights into the evolution of crop plants. Am. J. Bot. 95, 113–122. doi: 10.3732/ajb.95.2.113
Burnham, C. R., Rutter, P. A., and French, D. W. (1986). Breeding Blight-Resistant Chestnuts. Plant Breed. Rev. 4, 347–397. doi: 10.1002/9781118061015.ch11
Cao, K., Zheng, Z., Wang, L., Liu, X., Zhu, G., Fang, W., et al. (2014). Comparative population genomics reveals the domestication history of the peach, Prunus persica, and human influences on perennial fruit crops. Genome Biol. 15:415. doi: 10.1186/s13059-014-0415-1
Chen, J., Kaellman, T., Ma, X.-F., Zaina, G., Morgante, M., and Lascoux, M. (2016). Identifying genetic signatures of natural selection using pooled population sequencing in Picea abies. G3 Genes Genomes Genet. 6, 1979–1989. doi: 10.1534/g3.116.028753
Cheng, L.-L., Feng, H.-D., Rao, Q., Wu, W., Zhou, M., Hu, G.-L., et al. (2012). Diversity of wild Chinese chestnut chloroplast DNA SSRs in Shiyan. J Fruit Sci 3, 382–386. doi: 10.17660/ActaHortic.2010.866.29
Choi, J., Hyun, Y., Kang, M. J., In Yun, H., Yun, J. Y., Lister, C., et al. (2009). Resetting and regulation of Flowering Locus C expression during Arabidopsis reproductive development. Plant J. 57, 918–931. doi: 10.1111/j.1365-313X.2008.03776.x
Clapper, R. B. (1954). Chestnut breeding, techniques and results. I. Breeding material and pollination techniques. J. Hered. 45, 106–114.
Clark, R. M., Nussbaum-Wagler, T., Quijada, P., and Doebley, J. (2006). A distant upstream enhancer at the maize domestication gene tb1 has pleiotropic effects on plant and inflorescence architecture. Nat. Genet. 38, 594–597. doi: 10.1038/ng1784
Cockram, J., Jones, H., Leigh, F. J., O'Sullivan, D., Powell, W., Laurie, D. A., et al. (2007). Control of flowering time in temperate cereals: genes, domestication, and sustainable productivity. J. Exp. Bot. 58, 1231–1244. doi: 10.1093/jxb/erm042
Cornille, A., Gladieux, P., Smulders, M. J., Roldán-Ruiz, I., Laurens, F., Le Cam, B., et al. (2012). New insight into the history of domesticated apple: secondary contribution of the European wild apple to the genome of cultivated varieties. PLoS Genet. 8:e1002703. doi: 10.1371/journal.pgen.1002703
Cui, D., Zhao, J., Jing, Y., Fan, M., Liu, J., Xin, W., et al. (2013). The Arabidopsis IDD14, IDD15, and IDD16 cooperatively regulate lateral organ morphogenesis and gravitropism by promoting auxin biosynthesis and transport. PLoS Genet 9:E1003759. doi: 10.1371/journal.pgen.1003759
Drews, G. N., Bowman, J. L., and Meyerowitz, E. M. (1991). Negative regulation of the Arabidopsis homeotic gene AGAMOUS by the APETALA2 product. Cell 65, 991–1002.
Duan, N., Bai, Y., and Chen, X. (2017). Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nat. Commun. 8:249. doi: 10.1038/s41467-017-00336-7
Fei, S., Liang, L., Paillet, F. L., Steiner, K. C., Fang, J., Shen, Z., et al. (2012). Modeling chestnut biogeography for American chestnut restoration. Div. Distrib. 18, 754–768. doi: 10.1111/j.1472-4642.2012.00886.x
Feng, Y.-Q., Shen, Y.-Y., Qin, L., Cao, Q.-Q., and Han, Z.-H. (2011). Short catkin1, a novel mutant of Castanea mollissima, is associated with programmed cell death during chestnut staminate flower differentation. Sci. Horticult. 130, 431–435. doi: 10.1016/j.scienta.2011.07.014
Ford, C. M., Boss, P. K., and Hoj, P. B. (1998). Cloning and characterization of Vitis vinifera UDP-glucose:flavonoid 3-O-glucosyltransferase, a homologue of the enzyme encoded by the maize Bronze-1 locus that may primarily serve to glucosylate anthocyanidins in vivo. J. Biol. Chem. 273, 9224–9233.
Fowler, S., and Thomashow, M. F. (2002). Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell 14, 1675–1690. doi: 10.1105/tpc.003483
Frary, A., Nesbitt, T. C., Grandillo, S., van der Knaap, E., Cong, B., Liu, J., et al. (2000). fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289, 85–88. doi: 10.1126/science.289.5476.85
Gaoping, W., Qing, Y., Kai, Z., and Ciesla, W. M. (2001). Factors affecting production of Chinese chestnut in Xinxian County, Henan Province, China. Forest. Chronicle 77:839. doi: 10.5558/tfc77839-5
Glover, J., Grelon, M., Craig, S., Chaudhury, A., and Dennis, E. (1998). Cloning and characterization of MS5 from Arabidopsis: a gene critical in male meiosis. Plant J. 15, 345–356. doi: 10.1046/j.1365-313X.1998.00216.x
González-Carranza, Z. H., Elliott, K. A., and Roberts, J. A. (2007). Expression of polygalacturonases and evidence to support their role during cell separation processes in Arabidopsis thaliana. J. Exp. Bot. 58, 3719–3730. doi: 10.1093/jxb/erm222
Guo, M., Rupe, M. A., Dieter, J. A., Zou, J., Spielbauer, D., Duncan, K. E., et al. (2010). Cell Number Regulator1 affects plant and organ size in maize: implications for crop yield enhancement and heterosis. Plant Cell 22, 1057–1073. doi: 10.1105/tpc.109.073676
Guo, S., Zhang, J., Sun, H., Salse, J., Lucas, W. J., Zhang, H., et al. (2013). The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 45, 51–58. doi: 10.1038/ng.2470
Hebard, F. V. (2012). “The American Chestnut Foundation breeding program,” in Proceedings of the Fourth International Workshop on the Genetics of Host-Parasite Interactions in Forestry, R. A. Sniezko, A.Yanchuk, D., J. Kliejunas, T., K. Palmieri, M., J.Alexander, M., and S.Frankel, J. (tech. coords.) (Albany, CA: USDA For. Serv., Gen. Tech. Rep. PSW-GTR-240, Pacific Southwest Research Station), 221–234.
Huang, W. G., Zhou, Z. J., Cheng, L. L., Chen, S. F., and He, X. S. (2009). A new variety of Chinese chestnut 'Heishanzhai 7'. Sci. Silvae Sin. 45:177. doi: 10.11707/j.1001-7488.20090632
Jackson, J. P., Lindroth, A. M., Cao, X., and Jacobsen, S. E. (2002). Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 416, 556–560. doi: 10.1038/nature731
Jansen, R. K., Saski, C., Lee, S. B., Hansen, A. K., and Daniell, J. (2011). Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 28, 835–847. doi: 10.1093/molbev/msq261
Junhao, D., Mingqing, Z., Zongwen, L., and Zhongfu, H. (2000). Breeding research of Lantian bright red Chinese chestnut. J. Northw. Forest. Coll. 1:4.
Kaga, A., Isemura, T., Tomooka, N., and Vaughan, D. A. (2008). The genetics of domestication of the azuki bean (Vigna angularis) Genetics 178, 1013–1036. doi: 10.1534/genetics.107.078451
Khan, M. A., Olsen, K. M., Sover, V., Kushad, M. M., and Korban, S. S. (2014). Fruit quality traits have played critical roles in domestication of the apple. Plant Genome 7, 1–18. doi: 10.3835/plantgenome2014.04.0018
Kofler, R., Pandey, P. V., and Schlötterer, C. (2011). PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinfomatics 27, 3435–3436. doi: 10.1093/bioinformatics/btr589
Lam, H. M., Xu, X., Liu, X., Chen, W., Yang, G., Wong, F. L, et al. (2010). Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053–1059. doi: 10.1038/ng.715
Lefebvre, V., Kunst, M., Camara, B., and Palloix, A. (1998). The capsanthin-capsorubin synthase gene: a candidate gene for the y locus controlling red fruit color in pepper. Plant Mol. Biol. 36, 785–789. doi: 10.1023/A:1005966313415
Li, X., Dodson, J., Zhou, X., Zhang, H., and Masutomoto, R. (2007). Early cultivated wheat and the broadening of agriculture in Neolithic China. Holocene 17:555. doi: 10.1177/0959683607078978
Li, Y., Zhao, S. C., Ma, J. X., Li, D., Yan, L., Li, J., et al. (2013). Molecular footprints of domestication and improvement in soybean revealed by whole genome re-sequencing. BMC Genomics 14:579. doi: 10.1186/1471-2164-14-579
Liu, L., Liu, C., Hou, X., Xi, W., Shen, L., Tao, Z., et al. (2012). FTIP1 is an essential regulator required for florigen transport. PLoS Biol. 10:E1001313. doi: 10.1371/journal.pbio.1001313
Liu, W., Kang, M., Tian, H., and Huang, H. (2013a). A range wide geographic pattern of genetic diversity and population structure of Castanea mollissima populations inferred from nuclear and chloroplast microsatellites. Tree Genet. Genomes 9, 975–987. doi: 10.1007/s11295-013-0610-3
Liu, Y., Li, X., Li, K., Liu, H., and Lin, C. (2013b). Multiple bHLH proteins form heterodimers to mediate CRY2-dependent regulation of flowering time in Arabidopsis. PLoS Genet 9:E1003861. doi: 10.1371/journal.pgen.1003861
Lynch, M., Bost, D., Wilson, S., Maruki, T., and Harrison, S. (2014). Population-genetic inference from pooled-sequencing data. Genome Bio. Evol. 6, 1210–1218. doi: 10.1093/gbe/evu085
Ma, G. S., Guo, H., and Jian, C. (2000). Study on the pattern of diseases in Chinese chestnuts in storage. Plant Protect. 26, 29–31.
Mao, L., Begum, D., Chuang, H. W., Budiman, M. A., Szymkowiak, E. J., Irish, E. E., et al. (2000). Jointless is a MADS-box gene controlling tomato flower abscission zone development. Nature 406, 910–913. doi: 10.1038/35022611
Matsubayashi, Y., Ogawa, M., Kihara, H., Niwa, M., and Sakagami, Y. (2006). Disruption and overexpression of Arabidopsis phytosulfokine receptor gene affects cellular longevity and potential for growth. Plant Physiol. 142, 45–53. doi: 10.1104/pp.106.081109
Metaxas, A. (2013). Chestnut (Castanea spp.) Cultivar Evaluation for Commercial Chestnut Production in M.S.E.S Thesis, University of Tennessee at Chattanooga, Hamilton County, TN.
Nishio, S., Terakami, S., Matsumoto, T., Yamamoto, T., Takada, N., Kato, H., et al. (2017). Identification of QTLs for agronomic traits in the Japanese chestnut (Castanea crenata) breeding. Hortic. J. 87, 43–54. doi: 10.2503/hortj.OKD-093
Nishio, S., Yamada, M., Takada, N., Kato, H., Onoue, N., Sawamura, Y., et al. (2014). Environmental variance and broad-sense heritability of nut traits in Japanese chestnut breeding. Hortscience 49, 696–700.
Ogawa, M., Kay, P., Wilson, S., and Swain, S. M. (2009). ARABIDOPSIS DEHISCENCE ZONE POLYGALACTURONASE1 (ADPG1), ADPG2, and QUARTET2 are polygalacturonases required for cell separation during reproductive development in Arabidopsis. Plant Cell 21, 216–233. doi: 10.1105/tpc.108.063768
Olukolu, B. A., Nelson, C. D., and Abbott, A. G. (2012). “Mapping resistance to Phytophthora cinnamomi in chestnut (Castanea sp.),” in Proceedings of the Fourth International Workshop on the Genetics of Host-Parasite Interactions in Forestry: Disease and Insect Resistance in Forest Trees. Gen. Tech. Rep. PSW-GTR-240, eds R. A. Sniezko, Yanchuk, A. D., Kliejunas, J. T., et al. (Albany, CA: Pacific Southwest Research Station, Forest Service, U.S. Department of Agriculture), 177.
Ovesna, J., Kucera, L., Jiang, L. J., and Vagnerova, D. (2004). Characterisation of Chinese elite cultivars and genetic resources of chestnut by AFLP. Biol. Plant. 49, 125–127. doi: 10.1007/s10535-005-5127-7
Paran, I., and van der Knaap, E. (2007). Genetic and molecular regulation of fruit and plant domestication traits in tomato and pepper. J. Exp. Bot. 58, 3841–3852. doi: 10.1093/jxb/erm257
Pereira-Lorenzo, S., Lourenço Costa, R., and Anagnostakis, S. (2016). “Chapter: 15: Polyploidy and Hybridization for Crop Improvement,” in Interspecific Hybridization of Chestnut, A. S. Mason (Boca Raton, FL: CRC Press), 379–408.
Pereira-Lorenzo, S., Ramos-Cabrer, A. M., Ciordia-Ara, M., and Rios-Mesa, D. (2006). Chemical composition of chestnut cultivars from Spain. Sci. Hortic. 9, 134–142. doi: 10.1016/j.scienta.2005.08.008
Preuss, M. L., Serna, J., Falbel, T. G., Bednarek, S. Y., and Nielsen, E. (2004). The Arabidopsis Rab GTPase RABA4b localizes to the tips of growing root hair cells. Plant Cell 16, 1589–1603. doi: 10.1105/tpc.021634
Purugganan, M. D., and Fuller, D. Q. (2009). The nature of selection during plant domestication. Nature 457:843–848. doi: 10.1038/nature07895
Qi, J., Liu, X., Shen, D., Miao, H., Xie, B., Li, X., et al. (2013). A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat. Genet. 45, 1510–1515. doi: 10.1038/ng.2801
Qin, C., Yu, C., Shen, Y., Fang, X., Chen, L., Min, J., et al. (2014). Whole-genome sequencing of cultivated and wild pepper provides insights into Capsicum domestication and specialization. Proc. Nat. Acad. Sci. U.S.A. 111, 5135–5140. doi: 10.1073/pnas.1400975111
Qin, Y.-M., Hu, C.-Y., Pang, Y., Kastaniotis, A. J., Hiltunen, J. K., and Zhu, Y.-X. (2007). Saturated very-long-chain fatty acids promote cotton fiber and Arabidopsis cell elongation by activating ethylene biosynthesis. Plant Cell 19, 3692–3704. doi: 10.1105/tpc.107.054437
Rao, G. U., and Paran, I. (2003). Polygalacturonase: a candidate gene for the soft flesh and deciduous fruit mutation in Capsicum. Plant Mol. Biol. 51, 135–141. doi: 10.1023/A:1020771906524
Ronen, G., Carmel-Goren, L., Zamir, D., and Hirschberg, J. (2000). An alternative pathway to beta-carotene formation in plant chloroplasts discovered by map-based cloning of beta and old-gold color mutations in tomato. Proc. Natl. Acad. Sci. U.S.A. 97, 11102–11107. doi: 10.1073/pnas.190177497
Rutter, P. A., Miller, G., and Payne, J. A. (1991). Chestnuts (Castanea). Acta Hortic. 290, 761–790. doi: 10.17660/ActaHortic.1991.290.17
Sakai, H., Medrano, L. J., and Meyerowitz, E. M. (1995). Role of SUPERMAN in maintaining Arabidopsis floral whorl boundaries. Nature 378, 199–203.
Schlöetterer, C., Tobler, R., Kofler, R., and Nolte, V. (2014). Sequencing pools of individuals- mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 15, 749–763. doi: 10.1038/nrg3803
Schmutz, J., McClean, P., Mamdia, S., Wu, G. A., Cannon, S. B., Grimwood, J., et al. (2014). A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 46, 707–713. doi: 10.1038/ng.3008
Schnellmann, S., Schnittger, A., Kirik, V., Wada, T., Okada, K., Beermann, A., et al. (2002). TRIPTYCHON and CAPRICE mediate lateral inhibition during trichome and rooth hair patterning in Arabidopsis. EMBO J. 21, 5036–5046. doi: 10.1093/emboj/cdf524
Shi, Z., and Stoesser, R. (2005). Reproductive biology of Chinese chestnut (Castanea mollissima Blume). Europ. J. Hort. Sci. 70, 96–103. Available online at: http://www.jstor.org/stable/24126314
Shomura, A., Izawa, T., Ebana, K., Ebitani, T., Kanegae, H., Konishi, S., et al. (2008). Deletion in a gene associated with grain size increased yields during rice domestication. Nat. Genet. 40, 1023–1029. doi: 10.1038/ng.169
Slavov, G. T., DiFazio, S. P., Martin, J., Schackwitz, W., Muchero, W., Rodgers-Melnick, E., et al. (2012). Genome resequencing reveals multiscale geographic structure and extensive linkage disequilibrium in the forest tree Populus trichocarpa. New Phytol. 196, 713–725. doi: 10.1111/j.1469-8137.2012.04258.x
Söederman, E., Hjellströem, M., Fahleson, J., and Engstroem, P. (1999). The HD_zip gene ATHB6 in Arabidopsis is expressed in developing leaves, roots, and carpels and up-regulated by water deficit conditions. Plant Mol. Biol. 40, 1073–1083. doi: 10.1023/A:1006267013170
Stanke, M., Schöeffmann, O., Morgenstern, B., and Waack, S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformat. 7:62. doi: 10.1186/1471-2105-7-62
Staton, M. E., Addo-Quaye, C., Cannon, N., Tomsho, L. P., Drautz, D., Wagner, T. K., et al. (2014). The Chinese chestnut (Castanea mollissima) Genome Version 1.1. Available online at: https://www.hardwoodgenomics.org/organism/Castanea/mollissima (Accessed August 2, 2016).
Syring, J. V., Tennessen, J. A., Jennings, T. N., Wegrzyn, J., Scelfo-Dalbey, C., and Cronn, R. (2016). Targeted capture sequencing in whitebark pine reveals range-wide demographic and adaptive patterns despite challenges of a large, repetitive genome. Front. Plant. Sci. 7:484. doi: 10.3389/fpls.2016.00484
Takada, N., Nishio, S., Yamada, M., Sawamura, Y., Sato, A., Hirabayashi, T., et al. (2012). Inheritance of the easy-peeling pellicle trait of Japanese chestnut cultivar Porotan. HortScience 47, 845–847.
Tan, L., Li, X., Liu, F., Sun, X., Li, C., Zhu, Z., et al. (2008). Control of a key transition from prostrate to erect growth in rice domestication. Nat. Genet. 40, 1360–1364. doi: 10.1038/ng.197
Tanimoto, M., Tremblay, R., and Colasanti, J. (2008). Altered gravitropic response, amyloplast sedimentation, and circumnutation in the Arabidopsis shoot gravitropism 5 mutant are associated with reduced starch levels. Plant Mol. Biol. 67, 57–59. doi: 10.1007/s11103-008-9301-0
Teshima, K. M., Coop, G., and Przeworski, M. (2006). How reliable are empirical genomic scans for selective sweeps? Genome Res. 16, 702–712. doi: 10.1101/gr.5105206
Thornton, K. R., and Jensen, J. D. (2007). Controlling the false-positive rate in multilocus genome scans for selection. Genetics 175, 737–750. doi: 10.1534/genetics.106.064642
Velasco, D., Hough, J., Aradhya, M., and Ross-Ibarra, J. (2016). Evolutionary genomics of peach and almond domestication. G3 (Bethesda) 6, 3985–3993. doi: 10.1534/g3.116.032672
Vrebalov, J., Ruezinsky, D., Padmanabhan, V., White, R., Medrano, D., Drake, R., et al. (2002). A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (rin) locus. Science 196, 343–346. doi: 10.1126/science.1068181
Wang, C. (2004). Ban Li (Chestnut). Journal of the American Chestnut Foundation 38(1), 17 pp. Translated from: Anonymous, 1979. Chestnut, Science Publishing House, Beijing Institute of Botanical Research, Jiangsu.
Wang, E., Wang, J., Zhu, X., Hao, W., Wang, L., Li, Q., et al. (2008). Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 40, 1370–1374. doi: 10.1038/ng.220
Xu, Y. H., Jiang, Y. C., Wang, Z. J., Fang, B., Wang, Q. F., Zhang, L. T., et al. (2010). Breeding of a new processing Chinese chestnut cultivar Jinliwang. J. Fruit Sci. 27, 156–157.
Yamagami, T., Tsuchisaka, A., Yamada, K., Haddon, W. F., Harden, L. A., and Theologis, A. (2003). Biochemical diversity among the 1-amino-cyclopropane-carboxylate synthase isozymes encoded by the Arabidopsis gene family. J. Biol. Chem. 278, 49102–49112. doi: 10.1074/jbc.M308297200
Yang, F., Liu, Q., Pan, S., Xu, C., Youling, L., and Xiong, L. (2015). Chemical composition and quality traits of Chinese chestnuts (Castanea mollissima) produced in different ecological regions. Food BioSci. 11, 33–42. doi: 10.1016/j.fbio.2015.04.004
Zhang, H., and Liu, L. (1998). The genetic diversity of Castanea mollissima and the effect of artificial selection. Acta Botanica Yunnanica 20, 81–88.
Zhang, Y. L., Shao, Z. X., Yang, W. M., Ning, D. L., and Du, C. H. (2010). Selection of a new Chinese chestnut cultivar Yunxia. J. Fruit Sci. 27, 475–476.
Zhou, Y., Massonnet, M., Sanjak, J. S., Cantu, D., and Gaut, B. S. (2017). Evolutionary genomics of grape (Vitis vinifera ssp. vinifera) domestication. Proc. Natl. Acad. Sci. U.S.A. 114, 11715–11720. doi: 10.1073/pnas.1709257114
Keywords: chestnut, Fagaceae, crop domestication, Illumina sequencing, nut tree, pool-seq, selective sweep, woody perennial
Citation: LaBonte NR, Zhao P and Woeste K (2018) Signatures of Selection in the Genomes of Chinese Chestnut (Castanea mollissima Blume): The Roots of Nut Tree Domestication. Front. Plant Sci. 9:810. doi: 10.3389/fpls.2018.00810
Received: 28 February 2018; Accepted: 25 May 2018;
Published: 25 June 2018.
Edited by:
S. Hong Lee, University of South Australia, AustraliaReviewed by:
Guo-Bo Chen, Zhejiang Provincial People's Hospital, ChinaChaeyoung Lee, Soongsil University, South Korea
Copyright © 2018 LaBonte, Zhao and Woeste. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicholas R. LaBonte, bnJsYWJvbnRlQGdtYWlsLmNvbQ==