 Bartosz Sekula
Bartosz Sekula Milosz Ruszkowski
Milosz Ruszkowski Zbigniew Dauter
Zbigniew Dauter- Synchrotron Radiation Research Section, Macromolecular Crystallography Laboratory, National Cancer Institute, Argonne, IL, United States
Phosphoserine aminotransferase (PSAT) is a pyridoxal 5′-phosphate (PLP)-dependent enzyme that catalyzes the conversion of 3-phosphohydroxypyruvate (3-PHP) to 3-phosphoserine (PSer) in an L-glutamate (Glu)-linked reversible transamination reaction. This process proceeds through a bimolecular ping–pong mechanism and in plants takes place in plastids. It is a part of the phosphorylated pathway of serine biosynthesis, one of three routes recognized in plant organisms that yield serine. In this three-step biotransformation, 3-phosphoglycerate (3-PGA) delivered from plastidial glycolysis and Calvin cycle is oxidized by 3-PGA dehydrogenase. Then, 3-PHP is subjected to transamination with Glu to yield PSer and α-ketoglutarate (AKG). In the last step of the pathway, serine is produced by the action of phosphoserine phosphatase. Here we present the structural characterization of PSAT isoform 1 from Arabidopsis thaliana (AtPSAT1), a dimeric S-shaped protein that truncated of its 71-residue-long chloroplast-targeting signal peptide. Three crystal structures of AtPSAT1 captured at different stages of the reaction: (i) internal aldimine state with PLP covalently bound to the catalytic K265, (ii) holoenzyme in complex with pyridoxamine-5′-phosphate (PMP) after transfer of the amino group from glutamate and (iii) the geminal diamine intermediate state wherein the cofactor is covalently bound to both, K265 and PSer. These snapshots over the course of the reaction present detailed architecture of AtPSAT1 and allow for the comparison of this plant enzyme with other PSATs. Conformational changes of the protein during the catalytic event concern (i) the neighborhood of K265 when the amino group is transferred to the cofactor to form PMP and (ii) movement of the gate-keeping loop (residues 391–401) upon binding of 3-PHP and PSer. The latter conformational change of the loop may likely be one of key elements that regulate catalytic activity of PSATs.
Introduction
Serine is one of the endogenous proteinogenic amino acids which also acts as the precursor of glycine, tryptophan, and cysteine (Ireland and Hiltz, 1995). Moreover, it is an important intermediate in the biosynthesis of phospholipids, porphyrins, and nucleobases (Munoz-Bertomeu et al., 2013). Ser is also the major source of one-carbon units, essential for the methylation of nucleic acids and proteins (Ho and Saito, 2001).
Plant organisms have developed three independent routes for Ser biosynthesis (Supplementary Figure S1): two non-photorespiratory pathways, (i) phosphorylated and (ii) glycerate pathways, and also (iii) glycolate pathway that is linked with photorespiration (Ros et al., 2014). Both non-photorespiratory routes start with 3-PGA. In the phosphorylated pathway, which operates in plastids, 3-PGA is delivered from plastidial glycolysis and Calvin cycle. In contrast, the glycerate cycle is fed with 3-PGA from cytosolic glycolysis and enzymes of this pathway are localized in either cytosol or peroxisomes. Although both routes start with the same substrate, their organization is very different. The first reaction of phosphorylated pathway is NAD+-dependent oxidation of 3-PGA to 3-PHP by 3-PGA dehydrogenase. In the following step, PSer aminotransferase catalyzes transfer of amino group from Glu to 3-PHP to yield PSer and AKG. The last step involves cleavage of the PSer phosphate group by PSer phosphatase. In the glycerate pathway, the order of reactions is somewhat shuffled, with 3-PGA dephosphorylation followed by oxidation and finally aminotransfer. The glycolate route takes place in mitochondria, mostly within photosynthetic cells, whose activity is strongly connected with the day-night cycle (Cascales-Minana et al., 2013) and is regulated by the circadian clock (McClung et al., 2000). It starts with the biotransformation of two Gly molecules. The first molecule is utilized by GCS (Bauwe, 2017) which transforms THF to 5,10-CH2-THF. The reaction of 5,10-CH2-THF with the other Gly molecule, catalyzed by SHMT, yields Ser. In mitochondrion, direction of this thermodynamically unfavorable reaction toward Ser synthesis is attained through a high activity of the GCS, which delivers 5,10-CH2-THF for the SHMT reaction (Rebeille et al., 1994). However, plants have several isoforms of SHMT, localized in mitochondria, chloroplasts, nuclei, and cytoplasm (Ruszkowski et al., 2018). SHMTs outside the mitochondrial matrix preferentially catalyze a reversed Ser-to-Gly conversion to generate one-carbon units.
Plants have developed the alternate routes for Ser production probably because there was a necessity to bypass Ser biosynthesis through the glycolate pathway and make it independent of daylight to supply Ser equally to all tissues (Ros et al., 2014). Out of the two non-respiratory pathways, more Ser is produced through the phosphorylated pathway, which is important during the night and in non-photosynthetic tissues (Ros et al., 2014). Phosphorylated pathway activity regulates the glycolytic flux, affects the Krebs cycle and tryptophan biosynthesis rate (Munoz-Bertomeu et al., 2013). It is important for the plant development, especially at its early stages with respect to root formation (Benstein et al., 2013). Under environmental stress (high salinity, flooding or low temperature), the activity of phosphorylated pathway is alleviated which suggests that supplying with Ser is important for non-photosynthetic cells under harsh conditions (Ho and Saito, 2001). The phosphorylated pathway also affects other metabolic pathways like glycolysis, the tricarboxylic acid cycle, and amino acid biosynthesis (Toujani et al., 2013). Moreover, the phosphorylated pathway is induced upon infection of plants by pathogens (Benstein et al., 2013).
Phosphoserine aminotransferases belong to the class IV of ATs with the α-type fold (Liepman and Olsen, 2004). The α family of ATs predominantly catalyzes reactions at substrate Cα, that is the carbon atom of the substrate which is imine-bonded to PLP during the catalysis (Liepman and Olsen, 2004). PSATs are PLP-dependent enzymes that catalyze the second step of the phosphorylated pathway – reversible conversion of 3-PHP to PSer in a Glu-linked reaction (Basurko et al., 1999). In general, this class of enzymes is characterized by the presence of two domains with mixed α/β fold. Also, the conserved catalytic lysine, which directly follows a hydrophobic β-strand, is localized closer to the C-terminus than the Gly-rich region (Grishin et al., 1995). Another common feature of PSATs is an aspartate residue which hydrogen bonds the pyridoxal ring N1 atom and precedes the Schiff base lysine by 20–50 amino acids.
The transamination reaction catalyzed by PSAT consists of two reversible half-reactions (Figure 1; Eliot and Kirsch, 2004). At the beginning of the catalytic event, the internal aldimine is protonated at Nζ of the catalytic lysine residue by the hydroxyl (O3) group of PLP which allows for the alignment of the imine and pyridine ring in a plane. In the first step, the amine nitrogen atom of Glu performs a nucleophilic attack on the iminium carbon of the internal aldimine and, through PLP-Glu geminal diamine intermediate, substitutes lysine Nζ with the creation of PLP-Glu external aldimine. Because PLP acts as an “electron sink,” it withdraws electrons from the intermediate and triggers the rearrangement of the double bond to form a ketimine. Afterwards, hydrolysis of the imine bond releases the newly created AKG. Now, the enzyme holding PMP is ready to accommodate 3-PHP, which then forms a covalent bond with PMP. The subsequent release of a water molecule produces another ketimine, and afterwards PLP-PSer external aldimine. Then, Nζ of the catalytic lysine performs a nucleophilic attack on the C4′ of PLP to form PLP-PSer geminal diamine intermediate. The reaction ends when the initial internal aldimine state is restored and PSer is released from the active site.
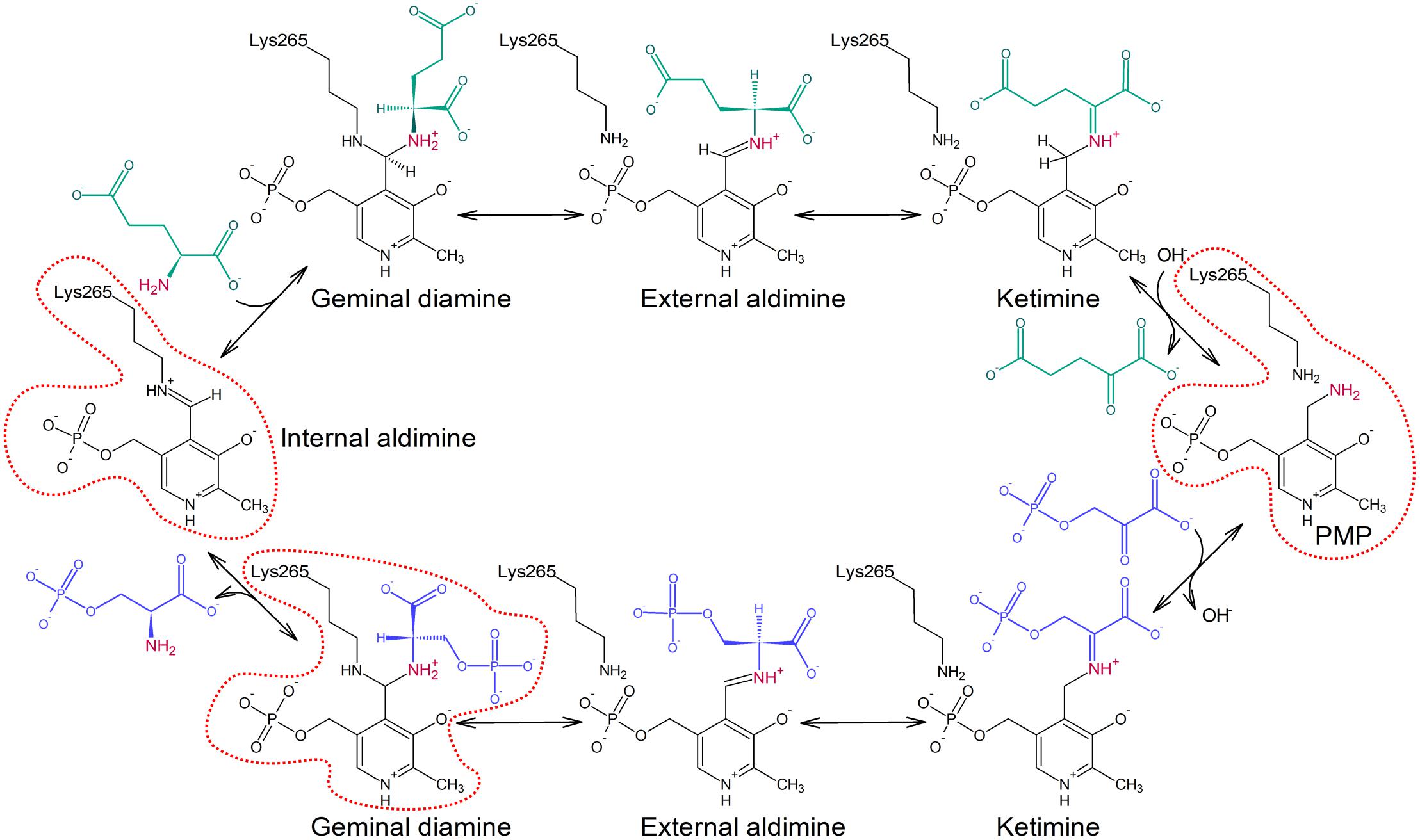
FIGURE 1. Schematic representation of the transamination reaction catalyzed by PSAT isoform 1 from Arabidopsis thaliana (AtPSAT1). Reaction states presented in the crystallographic snapshots in this work are contoured with red dotted line.
In this work, we describe a structure of the isoform 1 of chloroplastic PSAT from A. thaliana (At), which is further referred to in the manuscript as AtPSAT1. The gene coding for AtPSAT1 is localized in the lower arm of chromosome 4 and encodes the protein with an N-terminal plastidial transit peptide (Ho and Saito, 2001). AtPSAT1 mRNA is expressed in all tissues with the highest expression level observed in the light-grown roots and shoots, with significantly lower rates of the dark-treated individuals (Ho et al., 1998). AtPSAT1 is accumulated in the stele, especially in the cells close to the xylem in leaf, stem, and root sections. The full-length recombinant protein was reported previously to be inactive and insoluble, however, a variant truncated of approximately sixty N-terminal residues was active, with the KM-values for Glu and 3-PHP of 70 μM and 5 mM, respectively (Ho et al., 1998). The authors reported that cysteine at high concentration presented some inhibitory properties toward AtPSAT1, but no inhibition was observed by 5–50 mM serine, threonine, valine, glycine, tryptophan, or O-acetyl-L-serine. Our crystal structures capture the enzyme with cleaved signal sequence in three different states, as: (i) internal aldimine with PLP covalently bound to the catalytic K265, (ii) the holoenzyme in complex with PMP after the transfer of the amino group from Glu, and (iii) the PLP-PSer geminal diamine intermediate wherein the cofactor is covalently bound to both K265 and to PSer. We discuss the structural adaptability of AtPSAT1 over the course of the catalytic event and we also compare AtPSAT1 structure with other ATs.
Materials and Methods
Cloning, Overexpression, and Purification of AtPSAT1
AtPSAT1 was cloned and purified using a modified protocol recently applied for the production of Medicago truncatula N-carbamoylputrescine amidohydrolase (Sekula et al., 2016) and thermospermine synthase (Sekula and Dauter, 2018). Isolation of total RNA from leaves of A. thaliana was performed with RNeasy Plant Mini Kit (Qiagen). SuperScript II reverse transcriptase (Life Technologies) and oligo dT (15 and 18) primers were used for the preparation of the complementary DNA (cDNA). The following primers, forward: TACTTCCAATCCAATGCCCGTGTCTTCAACTTCGCGGCG and reverse: TTATCCACTTCCAATGTTACTAAGCATGCTTAGCCTGGAAATCTTTC, and cDNA as a template were used in polymerase chain reaction to obtain the AtPSAT1 open reading frame with the encoded protein starting from the codon number 72. The incorporation of AtPSAT1 gene into pMCSG68 vector (Midwest Center for Structural Genomics) was performed according to the ligase-independent cloning protocol (Kim et al., 2011). The vector introduces N-terminal His6-tag followed by the Tobacco Etch Virus (TEV) protease cleavage site to the cloned protein. In the next step, the BL21 Gold Escherichia coli competent cells (Agilent Technologies) were transformed with the vector containing AtPSAT1 gene. The cells were precultured at 30°C in LB medium with ampicillin (150 μg/ml) overnight. Next, 1.5% v/v of the culture was used as inoculum of the fresh LB medium with ampicillin. It was cultured at 37°C until OD600 reached the value of 1.0. In the next step, the culture was cooled to 10°C for 2 h and then the protein production was induced with 0.5 mM of isopropyl-β-D-thiogalactopyranoside. At this point K2HPO4 (40 mM) was added. The culture continued for 16 h at 18°C. Before pelleting the cells at 3,500 × g for 30 min, the culture was cooled to 4°C. The cell pellets were resuspended in 35 ml of the binding buffer (50 mM HEPES pH 7.4; 500 mM NaCl; 20 mM imidazole; 1 mM tris(2-carboxyethyl)phosphine, TCEP) and frozen at -80°C. Thawed cells were disrupted by sonication in an ice/water bath for 4 min (bursts of 4 s with 26 s intervals). Then, they were pelleted by centrifugation at 25,000 ×g for 30 min at 4°C.
The first step of AtPSAT1 purification was performed using a column packed with 5 ml of HisTrap HP resin (GE Healthcare) connected to VacMan (Promega). The supernatant was applied to the column and washed five times with 40 ml of the binding buffer. The protein elution was performed with 20 ml of elution buffer (50 mM HEPES pH 7.4; 500 mM NaCl; 400 mM imidazole; 1 mM TCEP). His6-tagged TEV protease (final concentration of 0.1 mg/ml) was used to cleave the His6-tag from AtPSAT1. This step was simultaneous to the overnight dialysis at 4°C against the dialysis buffer (50 mM HEPES pH 7.4; 500 mM NaCl; 1 mM TCEP). After dialysis, the sample was applied onto HisTrap HP resin to remove the cleaved His6-tag and TEV protease. The final step of the purification of AtPSAT1 was size exclusion chromatography on HiLoad Superdex 200 16/60 column (GE Healthcare) connected to the AKTA FPLC system (Amersham Biosciences). The running buffer was 25 mM HEPES pH 7.4, 100 mM KCl, 50 mM NaCl, and 1 mM TCEP.
Crystallization and Data Collection
AtPSAT1 was concentrated with Amicon concentrators (Millipore) to the final concentration of approximately 15 mg/ml, determined by the absorbance measurement at 280 nm, with the extinction coefficient of 34380. Full saturation of the cofactor was obtained by addition of 1 mM PLP (final concentration) to the concentrated protein sample. AtPSAT1 crystallized in the 77th condition of the Index Screen (Hampton Research), which contains 0.2 M lithium sulfate, 25% PEG 3350, and 0.1 M Tris at pH 8.5. Significantly better-diffracting crystals were obtained by streak seeding of the drops which were set up with the PEG concentration lowered to 17%. The protein was crystallized by sitting drop method. Such conditions were used for the crystallization of AtPSAT1-PLP and AtPSAT1-PMP complexes, but the AtPSAT1-PMP complex was obtained by addition of 10 mM Glu to the protein sample before crystallization setup. The AtPSAT1-PSer complex was cocrystallized with 50 mM PSer in 19% PEG 3350 and 0.1 M Tris at pH 8.5, without lithium sulfate. Crystals were transferred to the mixture of the well solution with 2-methyl-2,4-pentanediol at the volume ratio 2:1 to provide a sufficient cryoprotection before freezing in liquid nitrogen. In case of crystals of AtPSAT1-PMP and AtPSAT1-PSer, 10 mM Glu or 50 mM PSer were included in the cryoprotectant solution, respectively.
The diffraction data were collected at SER-CAT 22-ID and SBC 19-ID beamlines at the Advanced Photon Source (APS), Argonne National Laboratory, United States. The diffraction data were processed with XDS (Kabsch, 2010); for details see Table 1.
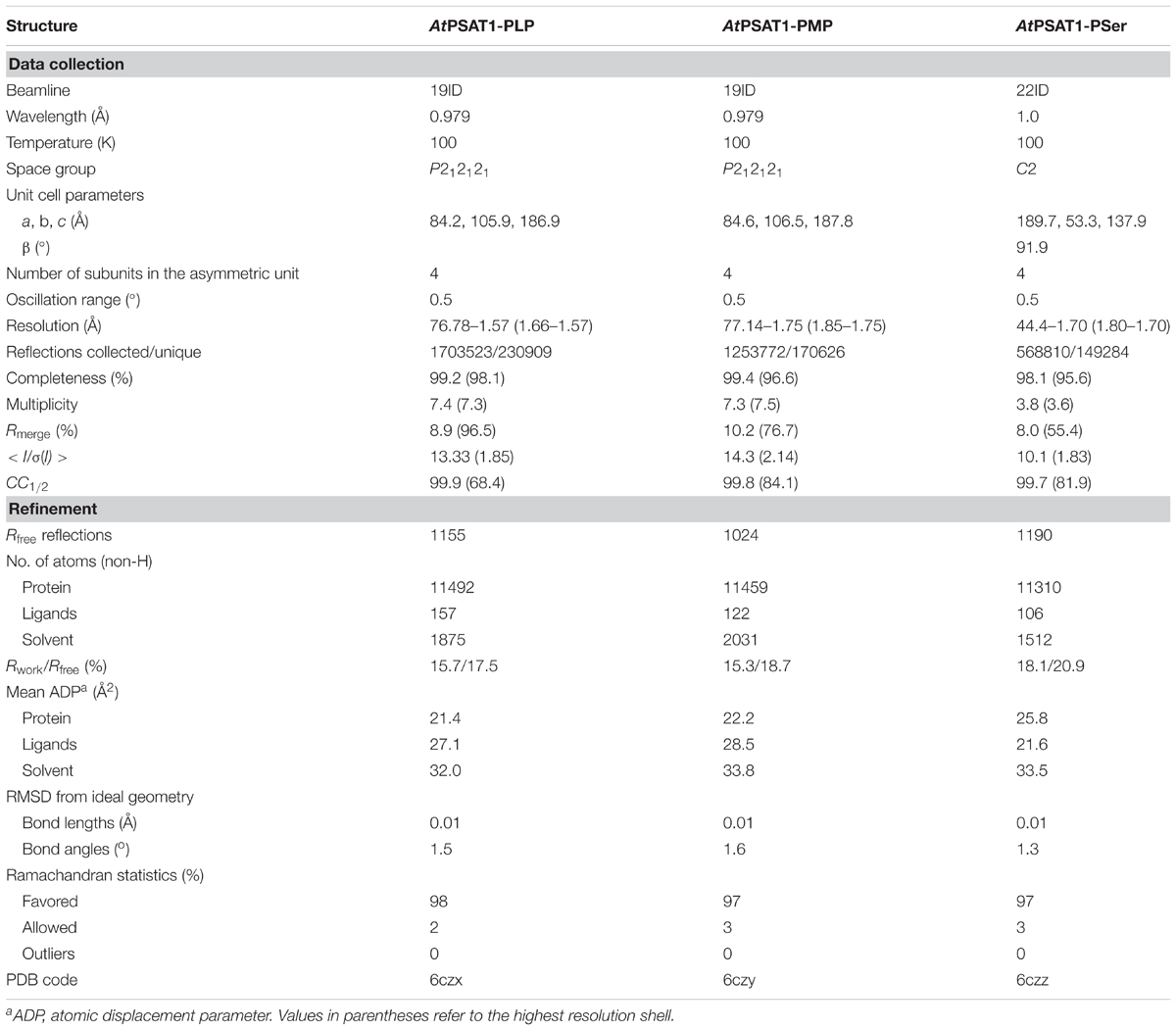
TABLE 1. Data-collection and refinement statistics.
Structure Determination and Refinement
The structure of AtPSAT1 was solved by molecular replacement in Phaser (McCoy et al., 2007); the structure of Pseudomonas aeruginosa PSAT (PaPSAT, PDB ID: 4xk1) was used as a search model. The initial model was rebuilt in ARP/wARP (Langer et al., 2008). Then, the structure was subjected to manual and automatic refinement with Coot (Emsley et al., 2010), Refmac (Murshudov et al., 2011), and Phenix (Adams et al., 2010). At the later stages of the structure refinement, TLS parameters (Winn et al., 2003) were applied. The refined structure of AtPSAT1 was used as the model for determination of the structures of AtPSAT1 with ligands. The stereochemical restraints for ligands were generated in Sketcher from CCP4 (Winn et al., 2011) and Elbow (Moriarty et al., 2009). The quality of refined structures was controlled by Rwork, Rfree factors (Brunger, 1992) and geometric parameters. The evaluation of the final structures was performed in PROCHECK (Laskowski et al., 1993) and MolProbity (Chen et al., 2010). The final refinement statistics are given in Table 1.
Other Software Used
Molecular illustrations were created with UCSF Chimera (Pettersen et al., 2004). Ramachandran plot was calculated in Rampage (Lovell et al., 2003). Secondary structure was recognized with ProMotif (Hutchinson and Thornton, 1996) within PDBsum server (de Beer et al., 2014). Sequence alignment was performed in CLUSTAL W (Thompson et al., 1994), and edited in BioEdit (Hall, 1999). Sequence conservation was calculated in ConSurf (Ashkenazy et al., 2016) based on the sequence alignment performed in Muscle (Edgar, 2004) under the MEGA7 suite (Kumar et al., 2016) with sequences classified to the eukaryotic PSAT family (IPR022278) by InterPro (Finn et al., 2017). Surface electrostatic potential was calculated in PDB2PQR and APBS (Baker et al., 2001; Dolinsky et al., 2004).
Results and Discussion
AtPSAT1 With the Fold of α-Type Aminotransferases
The subunit of AtPSAT1 has the length of 430 residues, including the predicted N-terminal transit peptide. According to the previous reports, transit peptide with the length of 60- (Ho et al., 1998) or 76-residues (Liepman and Olsen, 2004) was predicted. The previous study (Ho et al., 1998) also confirmed that the full-length protein was insoluble and inactive, however, the construct truncated of about 60-residues regained its catalytic activity. An additional analysis in the ChloroP 1.1. server (Emanuelsson et al., 1999) indicated an 86-residues-long signal sequence, however, based on the sequence homology with different PSATs (Supplementary Figure S2) and the analysis of available structural data of PSATs from other kingdoms of life, the designed construct was truncated before residue R72. Thus, the expressed protein (residues 72–430) has the length of 359 residues, plus the N-terminal SNA tag (the remaining sequence after the TEV protease cleavage). Truncation of the 71-residue peptide seems to be adequate since the electron density maps for the N-terminal part of AtPSAT1 structure is clearly defined (except for the SNA tag). On the other hand, a longer transit peptide is very doubtful – the polypeptide chain in the structure of AtPSAT1 starts with a 12-residue long bent coil with a short fragment (residues 74–75) in β-strand conformation and reaches the PLP binding site around G79. Thus, a more pronounced truncation of the N-termini would most likely affect the active site of the enzyme.
The protein mass calculated from the retention volume from size exclusion chromatography (∼80 kDa) and the crystal structure analyzed with PISA server (Krissinel, 2015) confirm the assembly of AtPSAT1 to be homodimeric (Figure 2). Therefore, the asymmetric unit with four subunits represents two functional AtPSAT1 dimers. The subunit of AtPSAT1 consists of two domains with mixed α/β topology (Figure 3A). This is a typical fold of class IV of the α-type ATs with a much larger N-terminal catalytic domain and a smaller C-terminal domain. Hydrophobic core of the N-terminal domain is constituted by a seven-stranded β-sheet with one anti-parallel strand (↑β2-↓β10-↑β9-↑β8-↑β5-↑β3-↑β4). The sheet is covered from the inner side of the AtPSAT1 dimer by helices α4, α5, η9, α10, α11, and by long loops from the outer side. An integral part of this domain is also a helical bundle (α1, η2, α3, α12) which builds a substantial part of the dimer interface. The small C-terminal domain comprises residues 326–430 which form a three-stranded anti-parallel β-sheet (↑β11-↓β12-↑β14) and a bundle of three helices (α13, η15/α15, α16). The symmetrical S-shaped dimer of AtPSAT1 (Figure 2) is formed through an extensive interface area (Figure 3B) which is over 2300 Å2, or about 14% of the subunit surface. The dimer interface is created by residues from the N-terminal coil, α1, η2, the loop between α11 and α12, the loop after η9, η10, the loop between β9 and β10, and the helices α4 and α5. The small domain does not directly take part in formation of the AtPSAT1 dimer, however, through a small β-sheet (↑β1-↑β13) it stabilizes the N-terminal coil. The dimer interface is rather flat with no significant cavities. Conservation of the residues at the interface is generally high among eukaryotic PSATs (Figure 3B). However, the highest conservation is, of course, observed around the PLP binding site, thus around the catalytic venue.
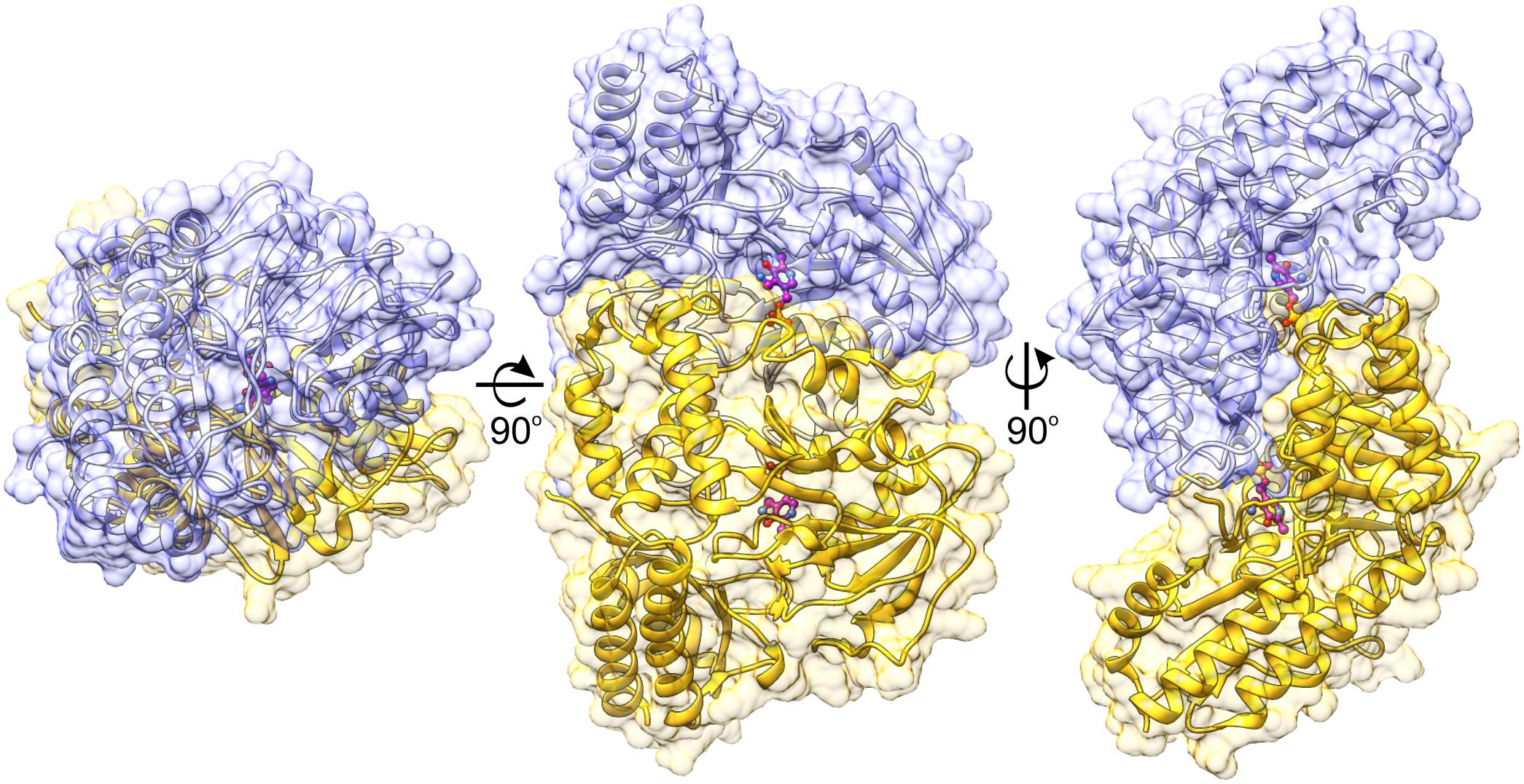
FIGURE 2. Biological assembly of AtPSAT1 dimer. Chains A and B are colored in yellow and light blue; the bound cofactor (PMP) is shown in balls-and-sticks representation (magenta). Coloring is used consequently throughout the manuscript unless stated otherwise.
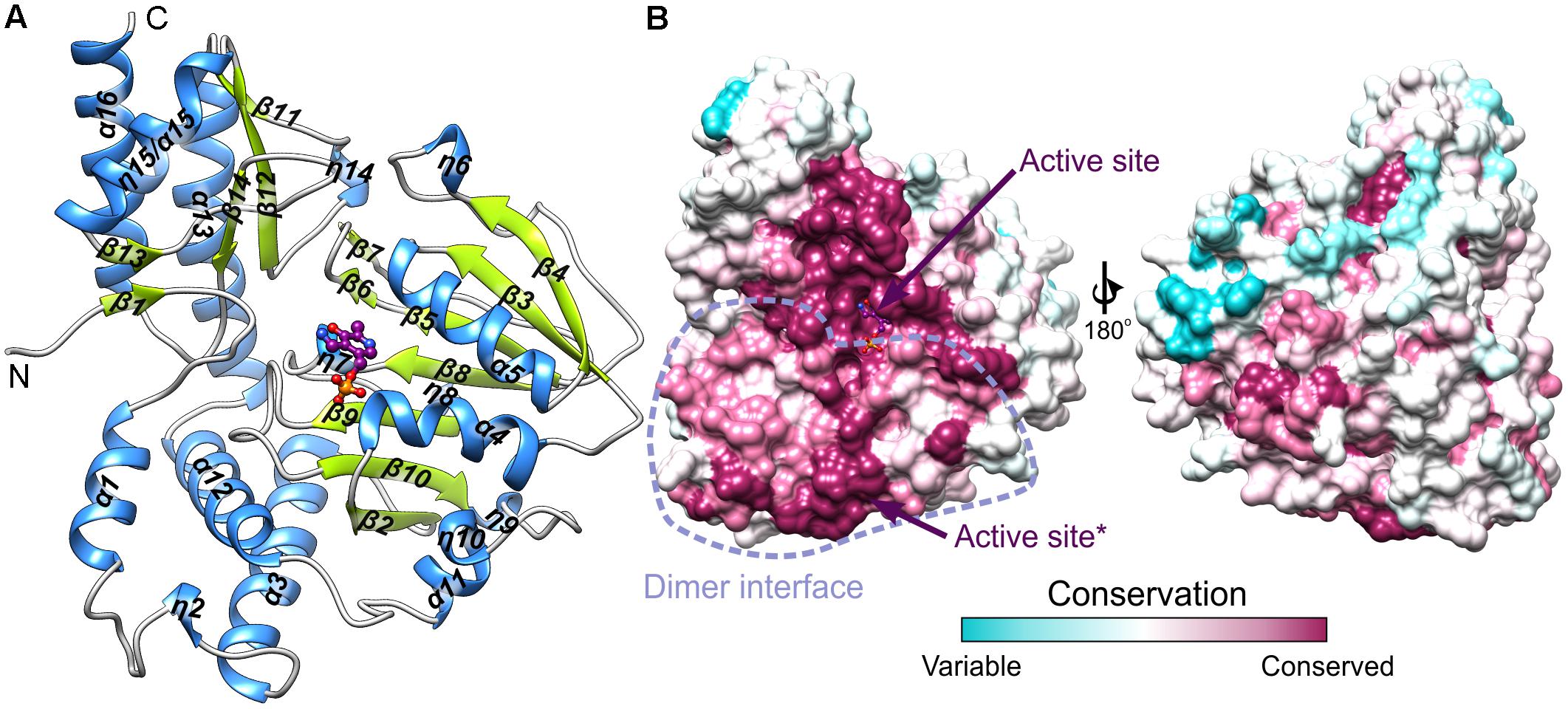
FIGURE 3. The subunit of AtPSAT1. (A) Arrangement of the secondary structure elements of AtPSAT1, helices (blue) and sheets (lime green); cofactor (PMP) is shown in balls-and-sticks representation (magenta). (B) Sequence conservation of the eukaryotic PSATs mapped the surface of AtPSAT1 monomer with marked location of the active site and schematic boundaries of dimer interface; asterisk indicates active site of the other subunit of the dimer.
The Dali server (Holm and Rosenstrom, 2010) search through the Protein Data Bank (PDB) (Berman et al., 2000) shows a few protein structures with a significant sequence identity (Supplementary Figure S2), which are structurally very similar to AtPSAT1: PaPSAT (PDB ID: 4xk1, r.m.s.d. = 1.0 Å, Z = 53.8), Yersinia pestis PSAT (YpPSAT, PDB ID: 3qbo, r.m.s.d. = 1.3 Å, Z = 51.9), Homo sapiens PSAT (HsPSAT, PDB ID: 3e77, r.m.s.d. = 1.2 Å, Z = 51.2), E. coli PSAT (EcPSAT, PDB ID: 1bjo, r.m.s.d. = 1.3 Å, Z = 52.0) (Hester et al., 1999), Bacillus alcalophilus PSAT (BaPSAT, PDB ID: 4azj, r.m.s.d. = 1.1 Å, Z = 54.1) (Battula et al., 2013), Salmonella enterica (SePSAT, PDB ID: 3qm2, r.m.s.d. = 1.4 Å, Z = 48.0). It is not surprising that the overall active site architecture is similar among PSATs of different species since they catalyze the same reaction. In all PSATs PLP is π-stacked with Trp (W171 in AtPSAT1) that has been shown by mutagenesis to be necessary for the full PSAT activity (Mishra et al., 2012). However, a detailed look into other PSATs shows that the enzymes are not identical. For example, in the structure of HsPSAT (PDB ID: 3e77), the N-terminal coil at first look seems to be placed significantly further from the active site, leaving about 6–7 Å free space in the neighborhood of the catalytic Lys. Longer coil in AtPSAT1 has in this region P80 in cis conformation; the structure of HsPSAT lacks it. In fact, the expressed HsPSAT started with L17, thus at the end of the N-terminal coil. Residues 8–16 visible in the structure came from the expression tag. Sequence alignment (Supplementary Figure S2) and structural comparison of different PSATs allow to predict similar conformation of this coil to AtPSAT1. Another distinguishing feature of HsPSAT, is C80 corresponding to T146 of AtPSAT1 which interacts with the phosphate group of PLP. In other PSATs there is Ser (in BaPSAT and PaPSAT) or Arg (EcPSAT, SePSAT, and YpPSAT). Long sidechain of this Arg residue in EcPSAT, SePSAT, and YpPSAT increases the positive charge around the substrate binding site and may directly take part in stabilization of the bound ligands, but it is unlikely to H-bond the phosphate of PLP. These three proteins also present significantly different conformation of the loop between α3 and β2, although this feature concerns region rather far away from the catalytic site. However, the D100A mutation of HsPSAT (corresponding to D164 of AtPSAT1), which is very close to this loop, causes significant solubility decrease and activity loss (Hart et al., 2007). Noteworthy, this mutation in HsPSAT is one of the causes of the rare disorder named PSAT deficiency (Hart et al., 2007).
Among the plant ATs with structures deposited in the PDB there are two, although more divergent from AtPSAT1: A. thaliana tryptophan aminotransferase (AtTrpAT, PDB ID: 3bwn, r.m.s.d. = 3.4 Å, 14% sequence identity) (Tao et al., 2008) and Hordeum vulgare alanine aminotransferase (HvAlaAT, PDB ID: 3tcm, r.m.s.d. = 3.3 Å, 13% identity) (Duff et al., 2012). The structural comparison of AtPSAT1 with other structures of plant ATs shows that these proteins share the S-like shape of the dimer (Figure 4), although some, like HvAlaAT, are bulkier, as if the “font” was changed. This is caused by the bigger C-terminal domains and additional helices in the PLP-binding domain, which in HvAlaAT form a β-sandwich fold, not observed in PSATs. PSATs have fewer helical fragments in their catalytic domains than the standard of eight to nine helices in other ATs (John, 1995). ATs are most of the times dimers, however, there are examples of ATs which are active as tetramers, like the unusual Archean aspartate AT (Tanaka et al., 1992), or monomers, as for instance AlaAT from Trypanosoma cruzi (de Morais et al., 2016). The distinctive feature of PSATs, shared between species, is a rather open and vast, positively charged region, localized not only around the PLP-binding site, but also around the entire catalytic site (Figure 4). It is easily explainable by the necessity of PSATs to attract phosphorylated, thus negatively charged substrate, 3-PHP. In other plant ATs (AtTrpAT and HvAlaAT), which do not utilize phosphorylated substrates, this positively charged patch covers a much smaller area, only directly around the PLP-binding sites in a deep tunnel (AtTrpAT) or is even completely covered by residues from the mostly helical extended N-terminus (HvAlaAT, Figure 4).
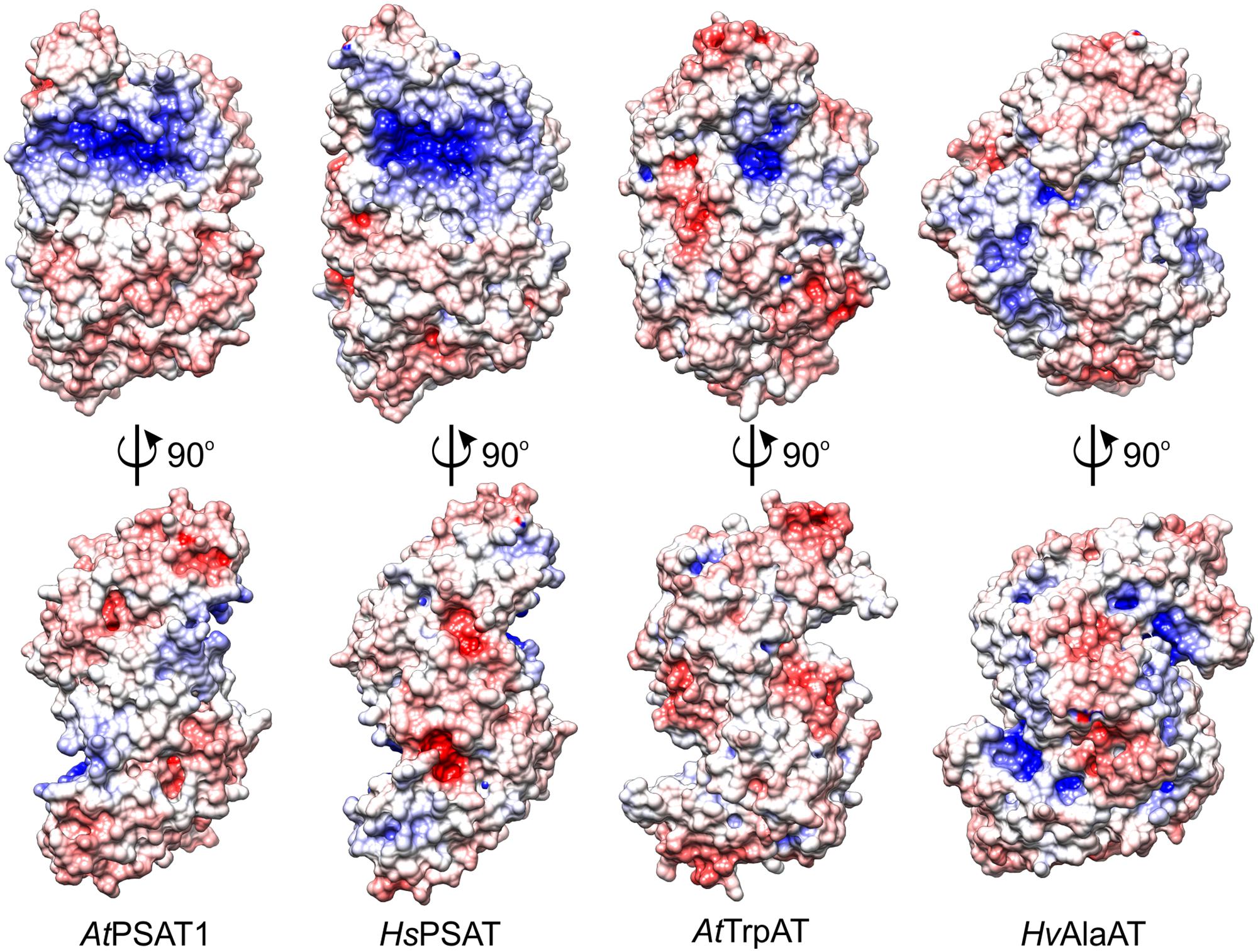
FIGURE 4. Charge distribution on the surface of selected aminotransferases (ATs): A. thaliana phosphoserine aminotransferase (AtPSAT1, this work), Homo sapiens PSAT (HsPSAT, PDB ID: 3e77), A. thaliana tryptophan aminotransferase (AtTrpAT, PDB ID: 3bwn), Hordeum vulgare alanine aminotransferase (HvAlaAT, PDB ID: 3tcm).
PLP Bound in the Active Site Forms Internal Aldimine With K265
The positive electrostatic potential around the catalytic venue is achieved by the contribution of K174, H396, R397, R403, and R110∗, H111∗ (asterisks indicate residues from the other subunit within the dimer), and secures the binding of both, PLP and the negatively charged substrates. The two active sites in the dimer are distant from each other by about 30 Å and are placed in the cavities on the opposite sides of the dimer (Figure 2). The PLP prosthetic group creates a Schiff base (internal aldimine) between C4′ and Nζ of K265 (Figures 5A,B), the residue placed in the coil between β9 and β10. Most of the residues interacting with PLP are placed on the C-terminal sides of the β-strands of the large domain core. PLP is bound inside the active pocket with its pyridoxal ring π-stacked with W171 on the re-face of the cofactor (Figure 5C). The protonated N1 of the pyridoxal ring is H-bonded to the carboxylate of D241. Another hydrogen bond is created between the hydroxyl group of PLP and T221. Phosphate of PLP creates a much more pronounced hydrogen bonding network with the protein. It includes direct H-bonds with backbone amides of A145 and T146, which, due to the placement at the N-terminal part of the α4 helix, possess an increased positive charge. Hydrogen bonds of PLP phosphate group also involve interactions with the side chain amide of Q264 and the hydroxyl group of T146. The phosphate also forms contacts with residues of the AtPSAT1 dimer mate, particularly hydrogen bonds with: the side chain amide of N306∗, the main chain amide of T307∗, and the side chain hydroxyl group of T307∗. Moreover, the phosphate of PLP creates water-mediated hydrogen bonds with the hydroxyl group of S243, the backbone amide of G262, the carbonyl oxygen of T307∗ and N𝜀 of Q142∗. Altogether, before the catalytic event, the PLP prosthetic group in AtPSAT1 structure presents a very conserved conformation in the internal aldimine state similar to that observed in other ATs, where C4′ atom of PLP is exposed to the entrance of the pocket.
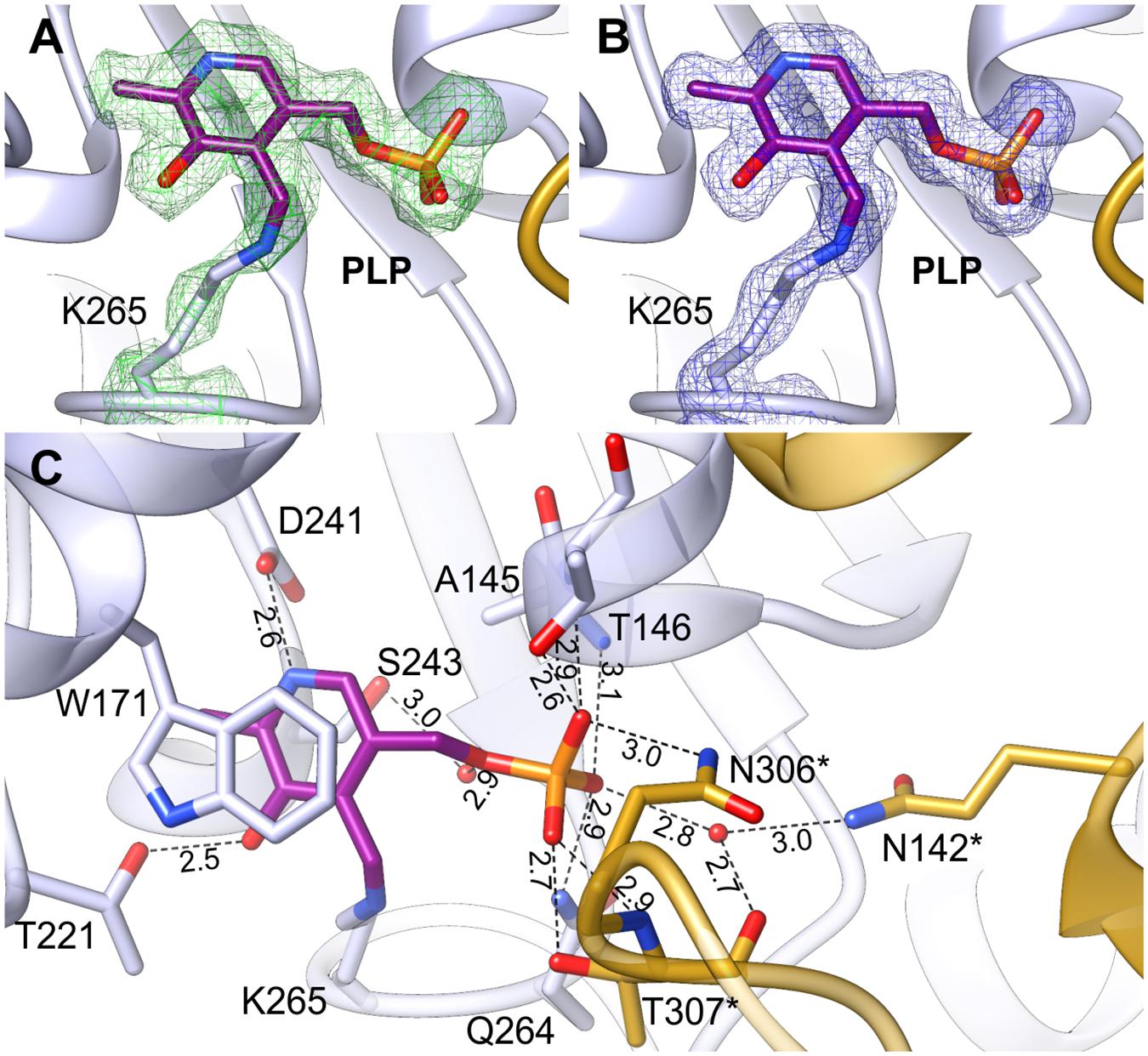
FIGURE 5. Internal aldimine in the AtPSAT1-pyridoxal 5′-phosphate (PLP) structure. (A) OMIT Fo–Fc electron density map for K265 and PLP contoured at 3σ; (B) 2Fo–Fc map electron density map for K265 and PLP contoured at 1σ. (C) A detailed PLP binding mode; asterisks indicate residues from the other subunit.
Complex With PMP Shows the Enzyme Primed for a Covalent Binding of 3-PHP
The AtPSAT1-PMP complex was obtained by crystallization of AtPSAT1-PLP with Glu. Neither Glu nor AKG (after the transfer of the amino group to PLP) were present in the crystal structure. However, electron density maps (Figures 6A,B) revealed the cofactor with the amino group already transferred from Glu and no longer covalently bound to K265, clearly showing that the first half of transamination reaction had taken place. The overall AtPSAT1-PMP structure is very similar to AtPSAT1-PLP (r.m.s.d. = 0.4 Å). Position of PMP is almost identical as PLP, thus its interactions with the protein are preserved (Figure 6C), excluding of course the broken covalent bond with K265. Position of the amine transferred to C4′ is almost in plane with the PMP pyridoxal ring, where it creates an intramolecular H-bond with the hydroxyl group of PMP. Although there is no movement of the prosthetic group observed during the transfer of the amino group from Glu and transition from the internal aldimine state to form PMP, the protein modifies conformation in the neighborhood of K265. The change concerns the main chain of Q264 and residues K265 and N266 (Figure 6D). The side chain of K265 is more bent with the Nζ amine moved by about 2 Å from its position in AtPSAT1-PLP structure. The Cα position of K265 and N265 is shifted away from the PMP by about 0.7 Å. This conformational change influences the position of the carbonyl oxygen of Q264 (which is rotated toward PMP by over 30°) and the conformation of N266 side chain as well. Such adaptation of the protein allows to achieve the position of the cofactor amino group exposed slightly toward the entrance of the active site. This way, PMP is ready for a nucleophilic attack on the Cα of 3-PHP bound in the active site in the next stage of the transamination reaction.
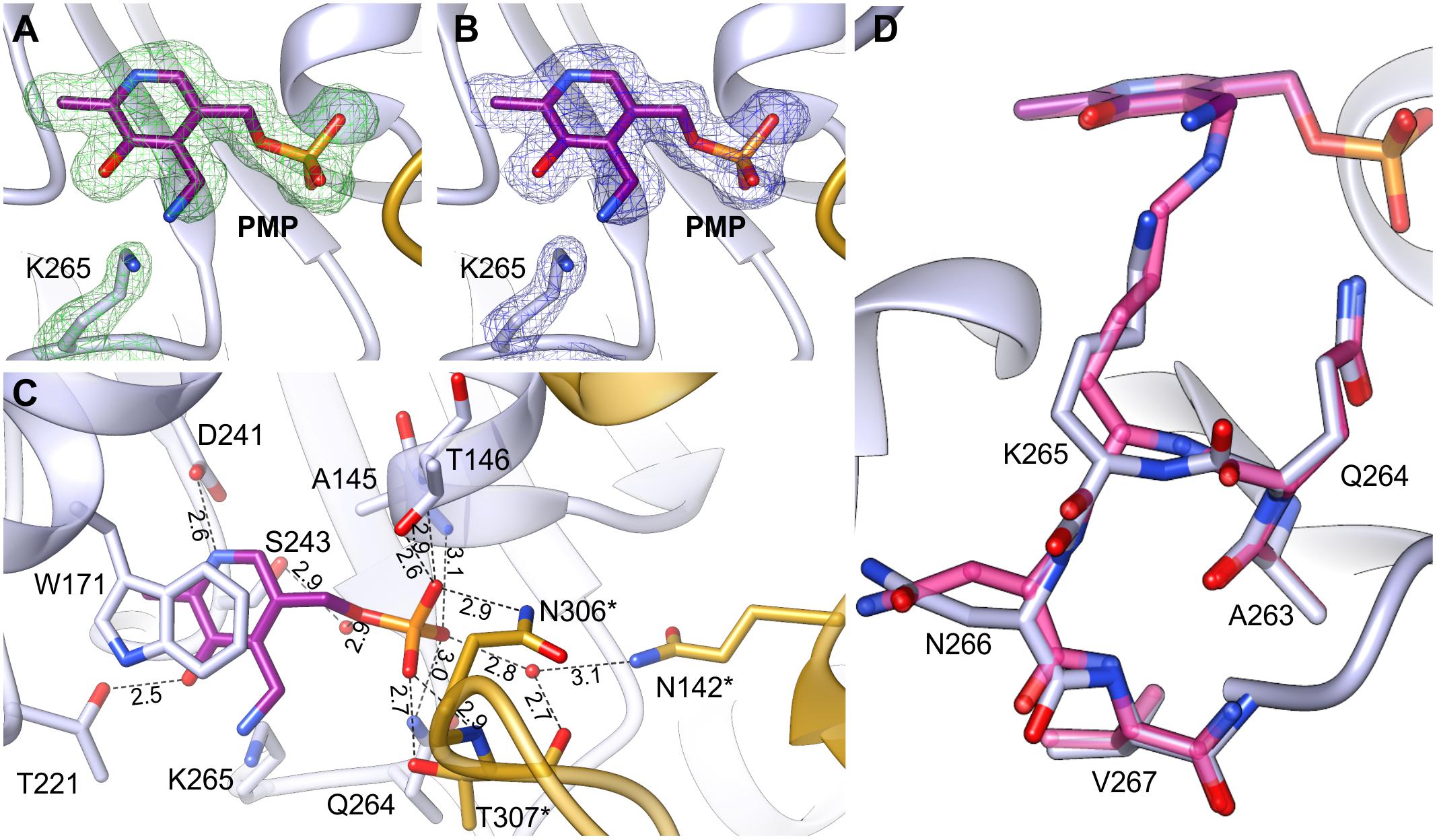
FIGURE 6. AtPSAT1-PMP complex. (A) OMIT Fo–Fc electron density map for K265 and PMP contoured at 3σ; (B) 2Fo–Fc map electron density map for K265 and PMP contoured at 1σ. (C) A detailed PMP binding mode; asterisks indicate residues from the other subunit. (D) Comparison of the conformation of AtPSAT1-PMP (light blue) with AtPSAT1-PLP internal aldimine (pink).
AtPSAT1-PSer Structure Brings Insights Into the Unexpected Geminal Diamine Intermediate State
Initial unsuccessful attempts to determine the structure of AtPSAT1-PSer complex resulted in a very “blurry” difference electron density maps around the catalytic site. Finally, crystallization of AtPSAT1 with 50 mM PSer and without sulfate ions in the crystallization conditions succeeded and the structure has shown somewhat unexpected results. The electron density maps (Figure 7A and Supplementary Figure S3) revealed not only the position of PSer in the active site itself, but they also indicated the presence of two covalent bonds: between the nitrogen atom of PSer and C4′ of PLP, and between C4′ of PLP and Nζ atom of K265. Thus, the structure does not show neither the last step of the transamination reaction, in the internal aldimine state (with PSer non-covalently bound just before its release from the active site) nor the PLP-PSer external aldimine. The reaction snapshot in the AtPSAT1-PSer structure actually shows one of the reaction intermediates – the PLP-PSer geminal diamine (Figure 1).
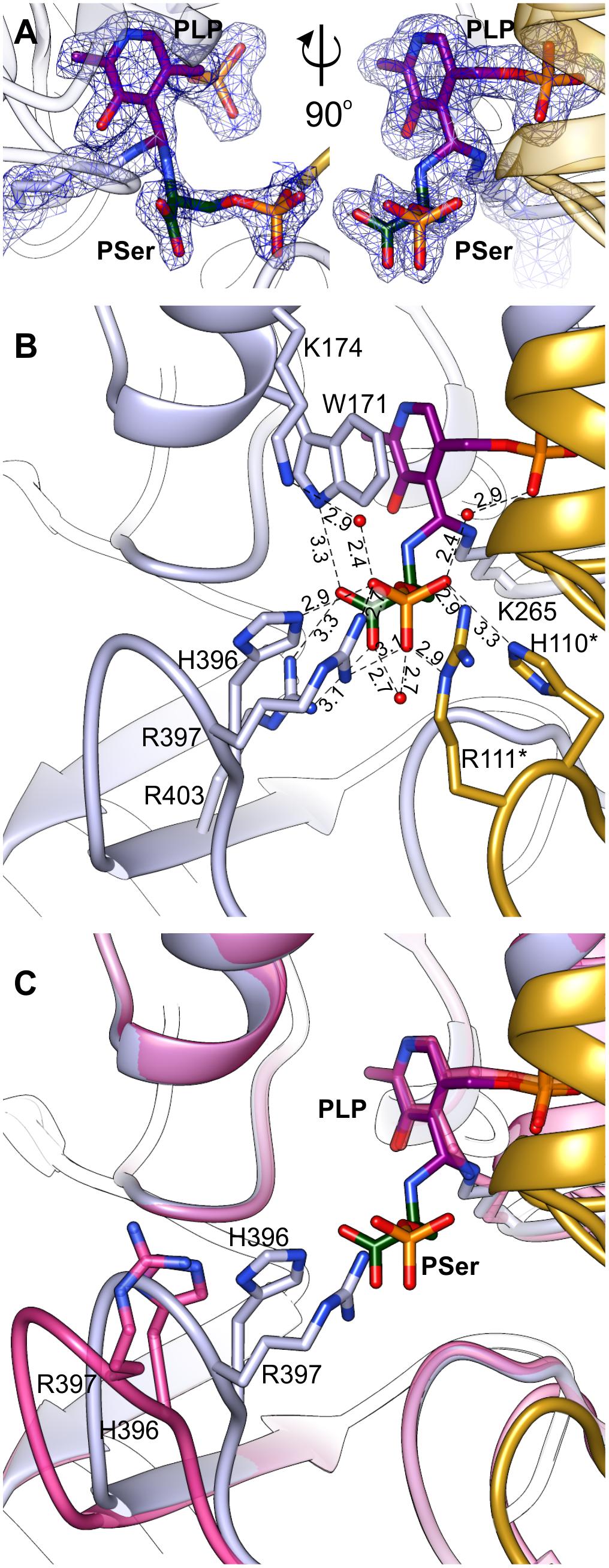
FIGURE 7. Geminal diamine in AtPSAT1-PSer structure. (A) 2Fo–Fc map electron density map for K265, PLP, and PSer contoured at 1σ. (B) A detailed PSer binding mode; asterisks indicate residues from the other subunit. (C) Comparison of the 391–401 loop conformation in AtPSAT1-PSer structure (light blue) and AtPSAT1-PLP structure (pink).
In this complex, the prosthetic group, similarly to the other structures (AtPSAT1-PLP and AtPSAT1-PMP) preserves interactions with the protein, with a maintained position of the phosphate group. The pyridoxal ring presents a subtle in-plane rotation (by about 8°). As a result of creation of two covalent bonds, the C4′ atom of PLP presents sp3 hybridization. In the PLP-PSer geminal diamine state, K265 and its neighborhood present almost identical conformation as in the internal aldimine state (the AtPSAT1-PLP structure). The bound PSer creates several hydrogen bonds in the active site, which include: a salt bridge between PSer carboxylate and guanidine group of R403 placed in the β14 strand, an H-bond between one of the oxygen atoms from the carboxyl group of PSer and N𝜀 of the indole ring of W171 from the α5 helix. Oxygen atoms from the phosphate group of PSer create several hydrogen bonds, either directly with protein side chains or through water molecules (Figure 7B). These include direct hydrogen bonds with H396, R397 (loop between β13 and β14), and with R111∗, H110∗ (loop between η2 and α3). The water-mediated hydrogen bonds include interaction with K174, H-bond with the carboxyl group of PSer, and interaction with oxygen from PLP phosphate group.
The superposition of AtPSAT1-PLP and AtPSAT1-PSer shows a pronounced movement of the loop 391–401 toward the catalytic site associated with PSer binding (Figure 7C). This clearly shows that during the binding of phosphorylated ligands (3-PHP or PSer) this gate-keeping loop transits from open to close conformation in order to stabilize the ligand during the amino group transfer, in a very similar way to the structure of BaPSAT (PDB ID: 4azj), where authors captured PLP-PSer external aldimine state (Battula et al., 2013). Therefore, it is very likely that sulfate ions, which competed with PSer to interact with H396, R397, R111∗, and H110∗, prevented from stable PSer binding in the initial crystallization attempts of AtPSAT1-PSer complex.
Previous report shows that the transamination reaction catalyzed by PSAT is able to proceed in both directions (Basurko et al., 1999) and the donor of the amino group can be Glu or PSer. Of course, kinetics and thermodynamics of the reversed reaction may be completely different than physiological transamination with Glu and 3-PHP in the direction of PSer synthesis. We observe PLP-PSer geminal diamine in our structure, which suggests a relative stability of this intermediate state. It is, therefore, very likely that the direction of Ser production is controlled by the activity of PSer phosphatase, catalyzing the final and irreversible step of Ser synthesis (Wang et al., 2002).
Binding the Substrates Requires a Conformational Change of AtPSAT1
It has been proposed that ATs reach their full catalytic activity only in the closed conformation (John, 1995). Taking this into account, a requirement for the catalytic event to occur is that the loop 391–401 of AtPSAT1 has to adopt an optimal conformation, with H396 and R397 poised closer to the PLP binding site to direct the substrates in the appropriate position (Figure 7C). This suggests that open-to-close transition of the 391–401 loop most likely guides the substrates toward the catalytic site. It is worth noting that the electron density maps for this region in the AtPSAT1-PSer structure clearly show the position for residues within this loop. In case of the other two structures, the electron density maps are blurrier and with no meaningful signal for the side chain of R397, which indicates a flexibility of this region. Since binding of PSer apparently stabilizes conformation of the gate-keeping loop, it is very likely that a similar stabilization secures 3-PHP as well. Such a bracing would therefore promote the catalytic event by locking the substrate in an optimal position.
Conclusion
The presented in this work crystal structures of AtPSAT1 bring detailed insights into the conversion of 3-PHP to PSer. Therefore, they expand the knowledge about the phosphorylated pathway of serine metabolism in plants. AtPSAT1 is a dimeric PLP-dependent AT that, similarly to other α-type ATs, binds the prosthetic group in the cavity within the large domain. The three crystal structures of AtPSAT1 present snapshots along the catalytic event showing PLP internal aldimine state, complex with PMP after the first half-reaction and PSer-PLP geminal diamine intermediate state. The structures give detailed information about the reaction catalyzed by AtPSAT1, revealing structural changes in the neighborhood of the catalytic K265 during the transition from the internal aldimine to the PMP complex. Moreover, the structure of AtPSAT1-PSer shows the closed conformation of the gate-keeping loop between β13 and β14 (residues 391–401), required for the stabilization of AtPSAT1-PSer geminal diamine and most likely for the catalysis as well. The transition from open to close conformation of the gate-keeping loop probably improves loading of 3-PHP and discarding PSer after the catalysis. AtPSAT1-PSer crystal structure additionally shows that the reverse reaction follows only to the PLP-PSer geminal diamine intermediate state, at least in the crystalline state.
Accession Numbers
Coordinates and structure factors of the related structures were deposited in the Protein Data Bank: 6czx (AtPSAT1-PLP), 6czy (AtPSAT1-PMP), 6czz (AtPSAT1-PSer).
Author Contributions
BS and MR planned and performed the experiments, analyzed the results, and wrote the manuscript. ZD analyzed the results and supervised the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported in part by the Intramural Research Program of the NCI, Center for Cancer Research. Diffraction data were collected at the Advanced Photon Source (APS), Argonne National Laboratory (ANL) at the SER-CAT beamline 22-ID, supported by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences under Contract W-31-109-Eng-38, and 19-ID beamline of the Structural Biology Center (operated by UChicago Argonne, LLC, for the DOE, Office of Biological and Environmental Research under contract DE-AC02-06CH11357).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00876/full#supplementary-material
Abbreviations
3-PGA, 3-phosphoglycerate; 3-PHP, 3-phosphohydroxypyruvate; 5,10-CH2-THF, N5,N10-methylene-tetrahydrofolate; AKG, α-ketoglutarate; AT, aminotransferase; GCS, glycine cleavage system; PLP, pyridoxal 5′-phosphate; PMP, pyridoxamine-5′-phosphate; PSAT, phosphoserine aminotransferase; PSer, 3-phosphoserine; SHMT, serine hydroxymethyltransferase; THF, tetrahydrofolate.
References
Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221. doi: 10.1107/S0907444909052925
Ashkenazy, H., Abadi, S., Martz, E., Chay, O., Mayrose, I., Pupko, T., et al. (2016). ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350. doi: 10.1093/nar/gkw408
Baker, N. A., Sept, D., Joseph, S., Holst, M. J., and McCammon, J. A. (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041. doi: 10.1073/pnas.181342398
Basurko, M. J., Marche, M., Darriet, M., and Cassaigne, A. (1999). Phosphoserine aminotransferase, the second step-catalyzing enzyme for serine biosynthesis. IUBMB Life 48, 525–529. doi: 10.1080/713803557
Battula, P., Dubnovitsky, A. P., and Papageorgiou, A. C. (2013). Structural basis of L-phosphoserine binding to Bacillus alcalophilus phosphoserine aminotransferase. Acta Crystallogr. D 69, 804–811. doi: 10.1107/s0907444913002096
Bauwe, H. (2017). Measurement of enzyme activities. Methods Mol. Biol. 1653, 31–50. doi: 10.1007/978-1-4939-7225-8_3
Benstein, R. M., Ludewig, K., Wulfert, S., Wittek, S., Gigolashvili, T., Frerigmann, H., et al. (2013). Arabidopsis phosphoglycerate dehydrogenase1 of the phosphoserine pathway is essential for development and required for ammonium assimilation and tryptophan biosynthesis. Plant Cell 25, 5011–5029. doi: 10.1105/tpc.113.118992
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nucleic Acids Res. 28, 235–242. doi: 10.1093/nar/28.1.235
Brunger, A. T. (1992). Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475. doi: 10.1038/355472a0
Cascales-Minana, B., Munoz-Bertomeu, J., Flores-Tornero, M., Anoman, A. D., Pertusa, J., Alaiz, M., et al. (2013). The phosphorylated pathway of serine biosynthesis is essential both for male gametophyte and embryo development and for root growth in Arabidopsis. Plant Cell 25, 2084–2101. doi: 10.1105/tpc.113.112359
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., et al. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21. doi: 10.1107/S0907444909042073
de Beer, T. A., Berka, K., Thornton, J. M., and Laskowski, R. A. (2014). PDBsum additions. Nucleic Acids Res. 42, D292–D296. doi: 10.1093/nar/gkt940
de Morais, S. B., de Arruda Campos Brasil de Souza, T., Weiler, A. V., and Murakami, M. T. (2016). Biophysical characterization of alanine aminotransferase from Trypanosoma cruzi. Protein Pept. Lett. 23, 1118–1122. doi: 10.2174/0929866523666161025120511
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A., and Baker, N. A. (2004). PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667. doi: 10.1093/nar/gkh381
Duff, S. M., Rydel, T. J., McClerren, A. L., Zhang, W., Li, J. Y., Sturman, E. J., et al. (2012). The enzymology of alanine aminotransferase (AlaAT) isoforms from Hordeum vulgare and other organisms, and the HvAlaAT crystal structure. Arch. Biochem. Biophys. 528, 90–101. doi: 10.1016/j.abb.2012.06.006
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eliot, A. C., and Kirsch, J. F. (2004). Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 73, 383–415. doi: 10.1146/annurev.biochem.73.011303.074021
Emanuelsson, O., Nielsen, H., and von Heijne, G. (1999). ChloroP, a neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci. 8, 978–984. doi: 10.1110/ps.8.5.978
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010). Features and development of Coot. Acta Crystallogr. D 66, 486–501. doi: 10.1107/S0907444910007493
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199. doi: 10.1093/nar/gkw1107
Grishin, N. V., Phillips, M. A., and Goldsmith, E. J. (1995). Modeling of the spatial structure of eukaryotic ornithine decarboxylases. Protein Sci. 4, 1291–1304. doi: 10.1002/pro.5560040705
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Hart, C. E., Race, V., Achouri, Y., Wiame, E., Sharrard, M., Olpin, S. E., et al. (2007). Phosphoserine aminotransferase deficiency: a novel disorder of the serine biosynthesis pathway. Am. J. Hum. Genet. 80, 931–937. doi: 10.1086/517888
Hester, G., Stark, W., Moser, M., Kallen, J., Markovic-Housley, Z., and Jansonius, J. N. (1999). Crystal structure of phosphoserine aminotransferase from Escherichia coli at 2.3 A resolution: comparison of the unligated enzyme and a complex with alpha-methyl-l-glutamate. J. Mol. Biol. 286, 829–850. doi: 10.1006/jmbi.1998.2506
Ho, C. L., Noji, M., Saito, M., Yamazaki, M., and Saito, K. (1998). Molecular characterization of plastidic phosphoserine aminotransferase in serine biosynthesis from Arabidopsis. Plant J. 16, 443–452. doi: 10.1046/j.1365-313x.1998.00313.x
Ho, C. L., and Saito, K. (2001). Molecular biology of the plastidic phosphorylated serine biosynthetic pathway in Arabidopsis thaliana. Amino Acids 20, 243–259. doi: 10.1007/s007260170042
Holm, L., and Rosenstrom, P. (2010). Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549. doi: 10.1093/nar/gkq366
Hutchinson, E. G., and Thornton, J. M. (1996). PROMOTIF–a program to identify and analyze structural motifs in proteins. Protein Sci. 5, 212–220. doi: 10.1002/pro.5560050204
Ireland, R. J., and Hiltz, D. A. (1995). “Glycine and serine synthesis in non-photosynthetic tissues,” in Amino Acids and their Derivatives in Higher Plants, ed. R. M. Wallsgrove (Cambridge: Cambridge University Press), 111–118. doi: 10.1017/CBO9780511721809.009
John, R. A. (1995). Pyridoxal phosphate-dependent enzymes. Biochim. Biophys. Acta 1248, 81–96. doi: 10.1016/0167-4838(95)00025-P
Kim, Y., Babnigg, G., Jedrzejczak, R., Eschenfeldt, W. H., Li, H., Maltseva, N., et al. (2011). High-throughput protein purification and quality assessment for crystallization. Methods 55, 12–28. doi: 10.1016/j.ymeth.2011.07.010
Krissinel, E. (2015). Stock-based detection of protein oligomeric states in jsPISA. Nucleic Acids Res. 43, W314–W319. doi: 10.1093/nar/gkv314
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Langer, G., Cohen, S. X., Lamzin, V. S., and Perrakis, A. (2008). Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179. doi: 10.1038/nprot.2008.91
Laskowski, R. A., Macarthur, M. W., Moss, D. S., and Thornton, J. M. (1993). Procheck - a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. doi: 10.1107/S0021889892009944
Liepman, A. H., and Olsen, L. J. (2004). Genomic analysis of aminotransferases in Arabidopsis thaliana. Crit. Rev. Plant Sci. 23, 73–89. doi: 10.1080/07352680490273419
Lovell, S. C., Davis, I. W., Arendall, W. B. III, de Bakker, P. I., Word, J. M., Prisant, M. G., et al. (2003). Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50, 437–450. doi: 10.1002/prot.10286
McClung, C. R., Hsu, M., Painter, J. E., Gagne, J. M., Karlsberg, S. D., and Salome, P. A. (2000). Integrated temporal regulation of the photorespiratory pathway. Circadian regulation of two Arabidopsis genes encoding serine hydroxymethyltransferase. Plant Physiol. 123, 381–392. doi: 10.1104/pp.123.1.381
McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. doi: 10.1107/s0021889807021206
Mishra, V., Kumar, A., Ali, V., Nozaki, T., Zhang, K. Y. J., and Bhakuni, V. (2012). Role of conserved active site tryptophan-101 in functional activity and stability of phosphoserine aminotransferase from an enteric human parasite. Amino Acids 43, 483–491. doi: 10.1007/s00726-011-1105-x
Moriarty, N. W., Grosse-Kunstleve, R. W., and Adams, P. D. (2009). Electronic ligand builder and optimization workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D 65, 1074–1080. doi: 10.1107/S0907444909029436
Munoz-Bertomeu, J., Anoman, A., Flores-Tornero, M., Toujani, W., Rosa-Tellez, S., Fernie, A. R., et al. (2013). The essential role of the phosphorylated pathway of serine biosynthesis in Arabidopsis. Plant Signal. Behav. 8:e27104. doi: 10.4161/psb.27104
Murshudov, G. N., Skubak, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., et al. (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 67, 355–367. doi: 10.1107/S0907444911001314
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Rebeille, F., Neuburger, M., and Douce, R. (1994). Interaction between glycine decarboxylase, serine hydroxymethyltransferase and tetrahydrofolate polyglutamates in pea leaf mitochondria. Biochem. J. 302, 223–228. doi: 10.1042/bj3020223
Ros, R., Munoz-Bertomeu, J., and Krueger, S. (2014). Serine in plants: biosynthesis, metabolism, and functions. Trends Plant Sci. 19, 564–569. doi: 10.1016/j.tplants.2014.06.003
Ruszkowski, M., Sekula, B., Ruszkowska, A., and Dauter, Z. (2018). Chloroplastic serine hydroxymethyltransferase from Medicago truncatula: a structural characterization. Front. Plant Sci. 9:584. doi: 10.3389/fpls.2018.00584
Sekula, B., and Dauter, Z. (2018). Crystal structure of thermospermine synthase from Medicago truncatula and substrate discriminatory features of plant aminopropyltransferases. Biochem. J. 475, 787–802. doi: 10.1042/bcj20170900
Sekula, B., Ruszkowski, M., Malinska, M., and Dauter, Z. (2016). Structural investigations of N-carbamoylputrescine amidohydrolase from Medicago truncatula: insights into the ultimate step of putrescine biosynthesis in plants. Front. Plant Sci. 7:350. doi: 10.3389/fpls.2016.00350
Tanaka, T., Yamamoto, S., Taniguchi, M., Hayashi, H., Kuramitsu, S., Kagamiyama, H., et al. (1992). Further studies on aspartate aminotransferase of thermophilic methanogens by analysis of general properties, bound cofactors, and subunit structures. J. Biochem. 112, 811–815. doi: 10.1093/oxfordjournals.jbchem.a123981
Tao, Y., Ferrer, J. L., Ljung, K., Pojer, F., Hong, F., Long, J. A., et al. (2008). Rapid synthesis of auxin via a new tryptophan-dependent pathway is required for shade avoidance in plants. Cell 133, 164–176. doi: 10.1016/j.cell.2008.01.049
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Toujani, W., Muñoz-Bertomeu, J., Flores-Tornero, M., Rosa-Téllez, S., Anoman, A. D., Alseekh, S., et al. (2013). Functional characterization of the plastidial 3-phosphoglycerate dehydrogenase family in Arabidopsis. Plant Physiol. 163, 1164–1178. doi: 10.1104/pp.113.226720
Wang, W., Cho, H. S., Kim, R., Jancarik, J., Yokota, H., Nguyen, H. H., et al. (2002). Structural characterization of the reaction pathway in phosphoserine phosphatase: crystallographic “snapshots” of intermediate states. J. Mol. Biol. 319, 421–431. doi: 10.1016/s0022-2836(02)00324-8
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242. doi: 10.1107/S0907444910045749
Keywords: serine metabolism, PSAT, PLP, pyridoxal 5′-phosphate, transaminase, geminal diamine
Citation: Sekula B, Ruszkowski M and Dauter Z (2018) Structural Analysis of Phosphoserine Aminotransferase (Isoform 1) From Arabidopsis thaliana– the Enzyme Involved in the Phosphorylated Pathway of Serine Biosynthesis. Front. Plant Sci. 9:876. doi: 10.3389/fpls.2018.00876
Received: 10 April 2018; Accepted: 05 June 2018;
Published: 06 July 2018.
Edited by:
Roc Ros, Universitat de València, SpainReviewed by:
Peng Zhang, Shanghai Institutes for Biological Sciences (CAS), ChinaTakahiro Mori, The University of Tokyo, Japan
John F. Honek, University of Waterloo, Canada
Copyright © 2018 Sekula, Ruszkowski and Dauter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bartosz Sekula, YmFydG9zei5zZWt1bGFAbmloLmdvdg==; c2VrdWxhLmJhcnRvc3pAZ21haWwuY29t