 Sandra Odonkor1†
Sandra Odonkor1† Soyeon Choi2Debkanta Chakraborty3
Soyeon Choi2Debkanta Chakraborty3 Liliam Martinez-Bello1,4†
Liliam Martinez-Bello1,4† Xuewen Wang1,2,4Bochra A. Bahri1,4,5Maud I. Tenaillon6
Xuewen Wang1,2,4Bochra A. Bahri1,4,5Maud I. Tenaillon6 Olivier Panaud7
Olivier Panaud7 Katrien M. Devos1,4*
Katrien M. Devos1,4*- 1Institute of Plant Breeding, Genetics and Genomics, University of Georgia, Athens, GA, United States
- 2Department of Genetics, University of Georgia, Athens, GA, United States
- 3Institute of Bioinformatics, University of Georgia, Athens, GA, United States
- 4Department of Plant Biology, University of Georgia, Athens, GA, United States
- 5Laboratory of Bioagressors and Integrated Protection in Agriculture (LR14AGR02), The National Agronomic Institute of Tunisia, University of Carthage, Tunis, Tunisia
- 6UMR8120 Génétique Quantitative et Evolution Le Moulon, Institut National de la Recherche Agronomique, Université Paris-Sud, Centre National de la Recherche Scientifique, AgroParisTech, Université Paris-Saclay, Paris, France
- 7Laboratoire Génome et Développement des Plantes, UMR UPVD/CNRS, Université de Perpignan Via Domitia, Perpignan, France
Setaria (L.) P. Beauv is a genus of grasses that belongs to the Poaceae (grass) family, subfamily Panicoideae. Two members of the Setaria genus, Setaria italica (foxtail millet) and S. viridis (green foxtail), have been studied extensively over the past few years as model species for C4-photosynthesis and to facilitate genome studies in complex Panicoid bioenergy grasses. We exploited the available genetic and genomic resources for S. italica and its wild progenitor, S. viridis, to study the genetic basis of seed shattering. Reduced shattering is a key trait that underwent positive selection during domestication. Phenotyping of F2:3 and recombinant inbred line (RIL) populations generated from a cross between S. italica accession B100 and S. viridis accession A10 identified the presence of additive main effect quantitative trait loci (QTL) on chromosomes V and IX. As expected, enhanced seed shattering was contributed by the wild S. viridis. Comparative analyses pinpointed Sh1 and qSH1, two shattering genes previously identified in sorghum and rice, as potentially underlying the QTL on Setaria chromosomes IX and V, respectively. The Sh1 allele in S. italica was shown to carry a PIF/Harbinger MITE in exon 2, which gave rise to an alternatively spliced transcript that lacked exon 2. This MITE was universally present in S. italica accessions around the world and absent from the S. viridis germplasm tested, strongly suggesting a single origin of foxtail millet domestication. The qSH1 gene carried two MITEs in the 5′UTR. Presence of one or both MITEs was strongly associated with cultivated germplasm. If the MITE insertion(s) in qSH1 played a role in reducing shattering in S. italica accessions, selection for the variants likely occurred after the domestication of foxtail millet.
Introduction
Setaria italica, foxtail millet, and its wild ancestor Setaria viridis, green foxtail, are two of the ∼100 species that comprise the genus Setaria, tribe Paniceae, subtribe Cenchrinae, subfamily Panicoideae (Kellogg, 2017). The cultivated foxtail millet is grown both as a grain crop and for forage. The wild S. viridis is a weed found around the world in disturbed habitats. Cultivation of S. italica began approximately 5,900 years ago in the northern region of China, making it one of the oldest known domesticated crops (Barton et al., 2009). Domestication is the result of convergent selection for a limited number of traits aimed at making the crop better adapted to agricultural environments, higher yielding, and easier to harvest. The main traits favored in domesticated cereals are non-shattering seeds, larger grain size, increased grain number, larger panicle size, higher grain quality, and reduced sensitivity to photoperiod (Gepts, 2010).
Over the past decade, genes involved in a number of traits targeted by domestication have been characterized in cereal crops. One of the first genes isolated underlying a domestication quantitative trait locus was Teosinte-branched (Tb1) in maize, a transcription factor that controls axillary branch formation, sex expression, and inflorescence architecture (Doebley et al., 1997). Transition from the highly branched maize ancestor, teosinte, to the single-stalk phenotype of cultivated maize was caused by increased expression of Tb1, effected by the insertion of a transposable element 63 kb upstream of Tb1. Transcription factors have also been shown to be involved in the control of seed shattering in cereals (Doebley, 2006). SH4 and qSH1 are the two major disarticulation genes that underwent selection during rice domestication. SH4, located on rice chromosome 4, is a member of the trihelix family of transcription factors and carries a Myb3 DNA binding domain (Li et al., 2006). A single nucleotide polymorphism (SNP) in the Myb3 domain greatly reduced shattering. More recently, a frameshift mutation in SH4 was identified that completely eliminated shattering (Zhou et al., 2012). qSH1 on rice chromosome 1 encodes a BEL1-type transcription factor (Konishi et al., 2006). A SNP in the 5′ regulatory region eliminated qSH1 expression at the abscission layer leading to loss of shattering ability. In sorghum, selection for non-shattering phenotypes during domestication largely occurred at the Sh1 locus on chromosome 1 (Lin et al., 2012). At least three independent mutations have been identified that either inactivate the gene or reduce its expression significantly. Syntenic regions on rice chromosome 3, maize chromosomes 1 and 5, and Setaria chromosome IX have been shown to harbor disarticulation QTL, suggesting parallel selection at the Sh1 locus during the domestication of cereals (Lin et al., 2012). Free-threshing ability in wheat is conferred by mutations in the Q gene on wheat chromosome 5A, which encodes a transcription factor belonging to the APETALA2 family. Wild and cultivated alleles differ in their expression as well as by the presence of a non-synonymous SNP which causes reduced homodimer formation (Simons et al., 2006). Triticeae also display disarticulation of the rachis, which is controlled by two tightly linked genes on chromosome 3 (Komatsuda et al., 2007). Inactivation of one of the two genes is sufficient to yield a non-brittle rachis phenotype. One of the genes (BTR1) is predicted to encode a membrane-bound protein. The other gene (BTR2) likely encodes a soluble protein. BTR2 has some similarity to, but is not considered an ortholog of, the Arabidopsis thaliana INFLORESCENCE DEFICIENT IN ABSCISSION (Butenko et al., 2003).
Setaria has recently been brought to the spotlight as a model plant system to study agronomically relevant traits in C4-photosynthetic biofuel grasses and cereals (Doust et al., 2009; Li and Brutnell, 2011). S. italica and S. viridis have small diploid genomes (∼450 Mb), a short stature, rapid life cycle, and prolific seed production. Genetic and genomic resources include whole-genome sequence assemblies for both S. italica (Bennetzen et al., 2012; Zhang et al., 2012) and S. viridis (Setaria viridis v1.1, DOE-JGI1), a recombinant inbred line (RIL) population derived from a cross between B100 (S. italica) and A10 (S. viridis) (Bennetzen et al., 2012), high-density genetic maps (Bennetzen et al., 2012; Fang et al., 2016; Wang et al., 2017), a high-density haplotype map of genome variation (Jia et al., 2013) and transformation capability (Van Eck et al., 2017). QTL for seed shattering have previously been reported on Setaria chromosomes V and IX (Devos and Gale, 2000; Doust et al., 2014a). Here, we exploit the available genetic and genomic resources to further determine the genetic basis of shattering loss in foxtail millet.
Materials and Methods
Plant Material
Seed shattering was measured on F2:3 progeny (Devos et al., 1998; Wang et al., 1998) as well as on F10 recombinant inbred lines (RIL) (Bennetzen et al., 2012) from a cross between S. italica accession B100 and S. viridis accession A10. A total of 112 F2:3 families of 6–13 plants together with the parents were grown in a randomized block design with two replications during the period from May to September 1998 in the field at Orsay, France. Replicate 2 consisted of only 66 F2:3 families because of insufficient seed for some of the lines. For the RIL population, four plants per line for a total of 188 lines were grown in hill plots in a randomized block design with three replications at the University of Georgia (UGA) Plant Sciences Farm, Watkinsville, GA, during the period June to August, 2011. The S. italica accessions Yugu1 and B100 and the S. viridis accession A10 were part of the UGA Setaria collection. Their sources have been described previously (Wang et al., 1998; Bennetzen et al., 2012). S. italica accessions Ise-3 and Ise-5 were obtained from ICRISAT, Patancheru.
Genetic Maps and Markers
A linkage map for the F2 population had been developed by Wang and colleagues using restriction fragment length polymorphism (RFLP) markers (Devos et al., 1998; Wang et al., 1998). This map consisted of 257 loci spanning 1050 cM across the nine foxtail millet chromosomes. The genetic map generated in the RIL population and reported by Bennetzen et al. (2012) consisted of 990 single nucleotide polymorphism (SNP) markers and comprised a total map length of 1416 cM. The genotypic data used for the generation of both maps are attached as Supplementary Table S1.
Phenotypic Evaluation of Traits
Shattering ability, that is the ease with which spikelets are released from the pedicel, was scored subjectively from 0 (florets remaining intact as in the cultivated S. italica) to 3 (freely shattering as in the wild S. viridis) by clasping the panicle in a clenched fist. The phenotypic data used in the QTL mapping are provided in Supplementary Table S1.
QTL Mapping and Candidate Gene Identification
QTL mapping in both the F2:3 and RIL populations was done using composite interval mapping (CIM) in Windows QTL Cartographer 2.5 (Basten et al., 1995; Wang et al., 2012). We used model 6 with forward and backward regression, a 10 cM window size, and a mapping step size of 1 cM. The logarithm of odds (LOD) threshold that defined a significant QTL was determined based on 500 permutations and a significance level of P = 0.05. Analyses were done for each replicate separately, as well as across replicates. To find candidate genes underlying the QTL, QTL regions, delineated by the most distal flanking markers with LOD scores above the significance threshold, were located on the S. italica Yugu1 genome sequence assembly v2.22. Using comparative knowledge between Setaria and other grass species (Bennetzen et al., 2012; Devos et al., 2017), the orthologous regions in rice, sorghum, and maize were scanned for the presence of known shattering genes.
Comparative Analysis at the DNA Sequence Level of the S. italica and S. viridis Alleles at the Sh1 and qSH1 Loci
The sorghum Sh1 (Sobic.001G152901; Lin et al., 2012) and rice qSH1 (LOC_Os01g62920) coding sequences were used in BLASTN searches against the S. italica Yugu1 and S. viridis A10 whole genome sequence assemblies2 to identify the corresponding orthologs. For each gene, the genomic sequences of the S. italica and S. viridis alleles were aligned and compared. To determine whether the same structural variations were present in B100 as in Yugu1, we exploited the ∼43 Gb of sequence data that were available from a bulk of 48 B100 × A10 RIL lines (Bennetzen et al., 2012; SRX030717 and SRX030718). The RIL bulk sequencing data had been generated to identify the SNPs used in the construction of the RIL genetic map (Bennetzen et al., 2012). The Illumina reads (2 × 75 bp) were aligned against the Yugu1 genome assembly using Bowtie (Langmead and Salzberg, 2012) with default parameters. Alignments in the Sh1 and qSH1 regions were inspected visually using the Integrated genome viewer (IGV) v2.3 (Robinson et al., 2011).
The structural variants identified in Setaria in Sh1 and qSH1 were further validated by PCR-amplification using primer set SiSh1-91F (5′-GGATCATGCCTTGCACTCCT-3′) / SiSh1-91R (5′-CATGCATGCACATTTCGGCT-3′) for Sh1 and qSHIns1_F (5′-GCTTCCTGGAGTGGTCAAAC-3′) / qSHIns1_R (5′-GGACGTGGTAGAGCTGCTG-3′) for qSH1, and genomic DNA of S. italica accessions Yugu1 and B100, and S. viridis accession A10. PCR reactions consisted of 1.5 mM MgCl2, 200 nM dNTPs, 0.5 μM of forward and reverse primer and 4 U of GoTaq DNA polymerase (Promega) in 50 μl 1X GoTaq buffer (Promega) either with or without 5% DMSO. PCR conditions for Sh1 were initial denaturation at 95°C for 3 min, 40 cycles of denaturation at 95°C, annealing at 58°C for 40 s, ramping to 72°C at a rate of 1°C/s and extension at 72°C for 3 min, followed by a final extension of 5 min at 72°C after which reactions were held at 10°C. The same conditions were used for qSH1 except that reaction volumes were decreased to 10 or 20 μl, annealing was done at 60.9°C, and the number of cycles was reduced to 35. Amplicons were gel extracted using the ZymocleanTM Gel DNA Recovery Kit (The Epigenetics Company) and Sanger sequenced. Because of low amplification efficiency, Sh1 fragments amplified from B100 were reamplified following gel extraction using the same reaction conditions, gel extracted again, and then sequenced.
Assessing the Presence of the MITEs in Sh1 and qSH1 in S. italica and S. viridis Germplasm
We downloaded Illumina reads obtained from whole-genome resequencing of 200 S. italica accessions (Jia et al., 2013) and 100 S. viridis accessions3 and used sequences that spanned the boundary between genic and MITE sequence, as well as genic sequence that flanked the MITE insertion sites (S. viridis control) as queries in BLASTN analyses to test for the presence of the MITEs in the sequence data. The absence of hits when the S. viridis control was used as query combined with the presence of hits when the boundary region between genic sequence and MITE was used as query was taken as evidence for the presence of the MITE.
Transcript Analysis of Sh1 and qSH1 in S. italica and S. viridis
Total RNA was isolated from seedling leaves and from panicles 15 and 40 days after panicle emergence (15 DAPE) from A10 (S. viridis) and B100 (S. italica) using Trizol. Genomic DNA contamination was removed using the Turbo DNA-free kit (Invitrogen). cDNA was prepared using 500 ng of total RNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) according to the manufacturer’s instructions. To assess the effect of the MITE on Sh1 transcript processing in S. italica, 2 μg of cDNA was used as template in a PCR reaction (total volume 25 μl) consisting of 2 mM MgCl2, 200 nM dNTPs, 1 μM forward primer Sh1-09f-MITE (5′-GTGCTACGTACACTGCAACTT-3′; located in exon 1), 1 μM reverse primer Sh1-09r-MITE (5′-ATGATCCTGATCGCCTTTTG-3′; located in exon 3), and 0.025 U of GoTaq (Promega) in 25 μl of GoTaq Reaction Buffer (Promega). Amplicons were checked on a 1% agarose gel, purified from the gel using the GeneJET Gel Extraction Kit (Thermo Scientific), cloned into the pGEM-T Easy Vector (Promega) and Sanger sequenced. Semi-quantitative (semi-q) RT-PCR was conducted to determine transcript levels of qSH1 in A10 and B100 using primer sets qSH_expr1_F (Seq: 5′- TCCAACTGATGGTGACAAGC-3′) and qSH_expr1_R (Seq: 5′- CTTGTGGACCTGCCTCATCT-3′). Actin was used as control. Reaction conditions were the same as used for amplification of genomic DNA. The semi-qRT-PCR conditions for analyzing qSH1 expression levels were initial denaturation at 95°C for 5 min, 30 cycles of denaturation at 95°C for 45 s, annealing at 60°C for 45 s, and extension at 72°C for 1 min, followed by a final extension of 5 min at 72°C after which reactions were held at 10°C. A total of 5 μl reaction product was run on a 2.0% agarose gel and the intensity of the bands was measured using the software ImageJ (Abràmoff et al., 2004). Band intensity for qSH1 was normalized using the intensity of the actin amplicon. qSH1/Actin ratios of technical replicates were averaged for statistical analysis.
Results
Phenotypic Variation
Shattering in S. viridis acc. A10 was scored as 3 (highly shattering) and in S. italica acc. B100 as 0 (non-shattering). Some 39% of F2:3 families displayed little shattering (mean score ≤ 1.3) and 2% displayed high shattering (mean score ≥ 2.7). Similarly, in the RIL population, an average of 35% of RILs were low shattering and 5% were highly shattering.
QTL Detection
QTL for seed shattering were identified on chromosomes V, VI, and IX in the F2:3 population and on chromosomes V, VII, and IX in the RIL population (Table 1). For all QTL, the A10 allele led to increased shattering. The chromosome IX QTL in the F2:3 population was located between the rice cDNA sequences C0595 (Genbank accession number AU091626) and C1361 (C98269), which identify loci on the S. italica acc. Yugu1 genome sequence (Bennetzen et al., 2012) at positions 6.2 and 14.6 Mb, respectively. This QTL explained 30.6% of the variation in the across-replicate analysis. The main chromosome IX QTL in the RIL population was located in the region 6.2 Mb–11.9 Mb (Figure 1A) and explained 35.2% of the variation in the across-replicate analysis (Table 1). QTL on chromosome V were also identified in all replicates in both populations although the location of the QTL varied by replicate and population. The chromosome V QTL in the F2:3 population mapped to non-overlapping regions on the S. italica genome sequence (0.6–8.8 Mb and 29.5–to 43.7 Mb for replicate 1 and replicate 2, respectively). No QTL was identified on chromosome V when mean values across replicates 1 and 2 were used as trait data. In the RIL population, the chromosome V QTL was delineated to the region 35.8–36.9 Mb in replicate 1, 28.5–35.4 Mb in replicate 2, 32.3–35.4 Mb in replicate 3, and 28.1–36.9 Mb in the across-replicate analysis (Figure 1B). This QTL explained 12.5% of the variation in the across-replicate analysis (Table 1). The QTL on chromome VI was significant only in replicate 2 in the F2:3 population. Similarly, the QTL on chromosome VII was significant only in replicate 2 in the RIL population.
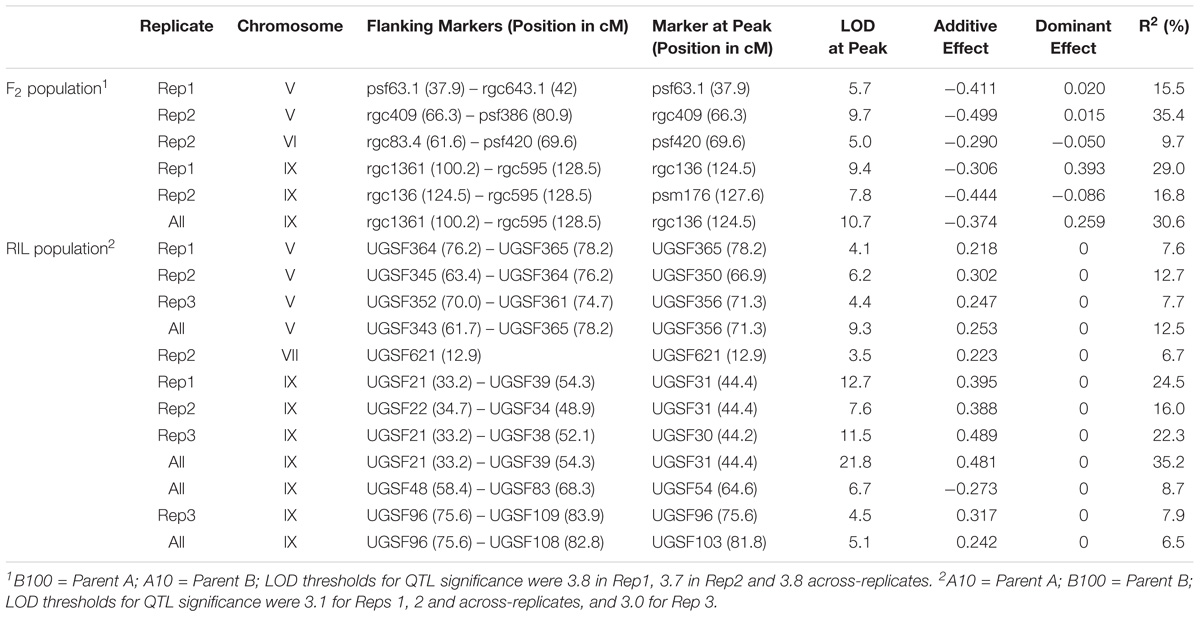
TABLE 1. QTL for seed shattering detected in the F2:3 and RIL populations.
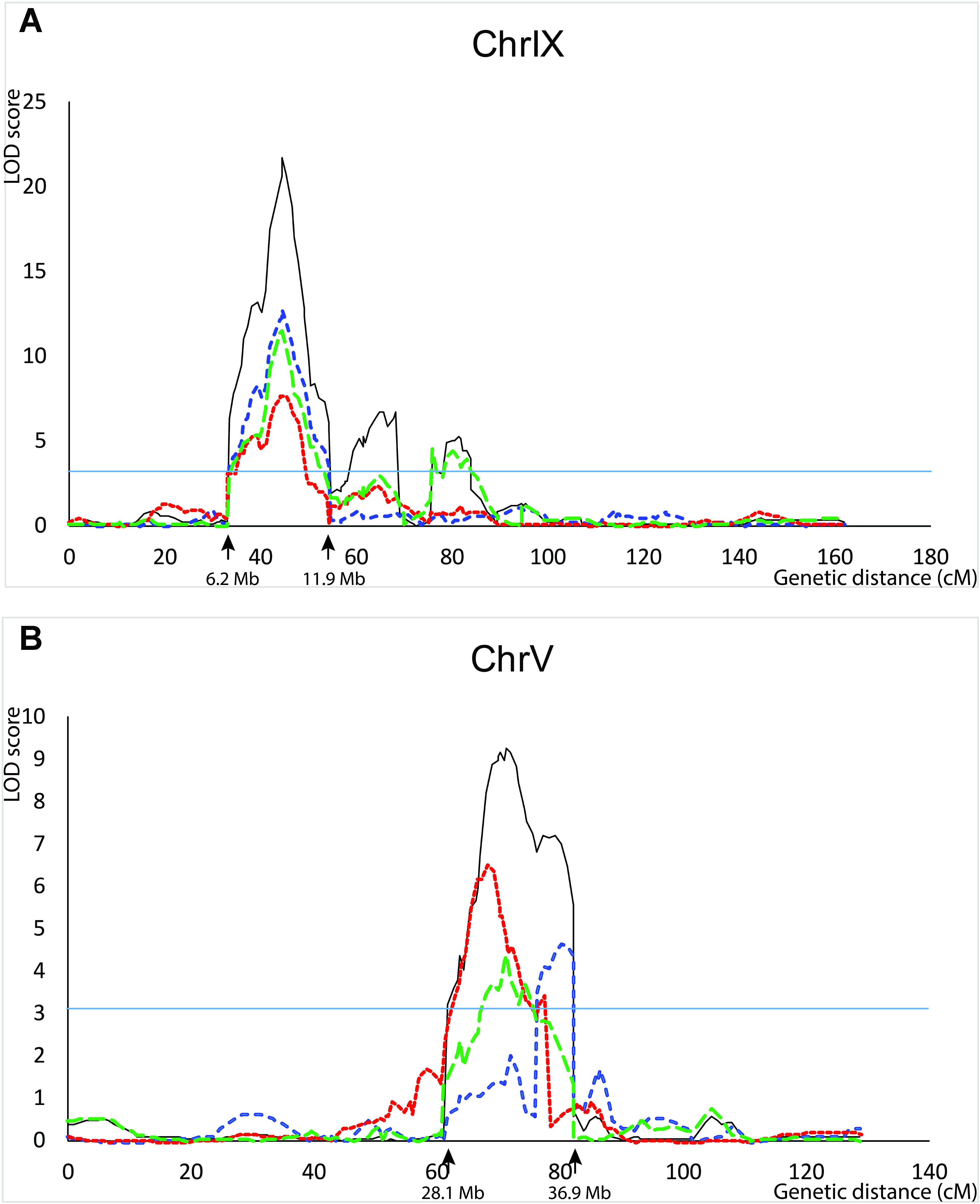
FIGURE 1. QTL identified in the B100 × A10 RIL population on chromosomes IX (A) and V (B). QTL in replicate 1 are in blue, in replicate 2 in red, in replicate 3 in green, and across-replicates in black. The horizontal line indicates the LOD threshold for significance at the 5% level.
Identification of Candidate Genes Underlying the Chromosome V and IX QTL
Several genes controlling seed shattering have been identified in cereals. Considering the known grass syntenic relationships (Gale and Devos, 1998; Devos, 2005), qSH1 (Konishi et al., 2006) and Sh1 (Lin et al., 2012) were of specific interest as candidate genes for the shattering QTL in our study. These genes had also been identified by Doust et al. (2014a,b) as candidates for shattering QTL they had mapped to Setaria chromosomes V and IX. qSH1 is a BEL1-type homeobox gene on rice chromosome 1 which is syntenic with Setaria chromosome V. Sh1 is a YABBY transcription factor on sorghum chromosome 1, which is syntenic with Setaria chromosome IX. The ortholog for Sh1 in S. italica accession Yugu1 (Seita.9G154300) was located at position 10.1 Mb (corresponding to ∼44.5 cM on the genetic map) and thus within the interval carrying the shattering QTL on chromosome IX (6.2–11.9 Mb; Figure 1A). Furthermore, the marker most highly associated with the shattering trait on chromosome IX in the RIL population, UGSF31, was also the marker most closely linked to the Sh1 gene. A comparison of the genomic and coding sequences of Seita.9G154300 and its ortholog in S. viridis acc. A10 (Sevir.9G153200) showed the presence of an 854 bp miniature inverted-repeat transposable element (MITE) in exon 2 in Seita.9G154300. The MITE belonged to the P-Instability Factor (PIF)/Harbinger family of DNA transposons. Scrutiny of this region in the alignment of Illumina reads generated from a pool of 48 B100 × A10 RILs showed the presence of reads that spanned the boundaries between Sh1 and MITE sequence, suggesting that the MITE was also present in S. italica accession B100. PCR amplification of the MITE-containing region in S. italica accessions Yugu1 and B100 using primers located in intron 1 and intron 2 yielded no product using standard PCR conditions, and a faint ∼1500 bp product when amplification was done in the presence of 5% DMSO. In contrast, both sets of reaction conditions yielded a strong ∼700 bp product in S. viridis accession A10. Sequencing of the B100 amplicon showed that this accession carried the same MITE that was present in S. italica accession Yugu1 (Supplementary Figure S1).
The Setaria orthologs for qSH1 were Seita.5G381300 in S. italica acc. Yugu1 and Sevir.5G386500 in S. viridis acc. A10. Seita.5G381300 maps to position 41.4 Mb (corresponding to ∼86 cM on the genetic map) in the S. italica Yugu1 genome assembly. Although none of the chromosome V QTL identified in our study spanned the qSH1 locus (Figure 1B), we nevertheless considered qSH1 as putatively underlying the shattering QTL on Setaria chromosome V because of its close proximity to the QTL. A comparison of Sevir.5G386500 and Seita.5G381300 showed that the two genes differed by a non-synonymous SNP in the coding region leading to a Q77 → H77 amino acid substitution, and by the presence of two MITEs belonging to the PIF/Harbinger family in the 5′UTR region. The insertion of both MITEs had been accompanied or followed by a rearrangement (Supplementary Figure S2). In case of the most 5′ located MITE (MITE 1), the deletion which spanned the 5′ boundary region of the qSH1 5′UTR and MITE was flanked by a short direct CGG repeat. This suggests that the deletion occurred by non-homologous end-joining during DNA break repair, possibly as part of the MITE insertion event. The insertion of the most 3′ located MITE (MITE 2) was accompanied/followed by both a deletion and an insertion (Supplementary Figure S2). Scrutiny of the alignment of the bulked B100 × A10 RIL reads against the Yugu1 genome assembly showed that the SNP and both MITEs were also present in S. italica accession B100. The presence of the two MITEs in the 5′UTR of B100 was confirmed by PCR and Sanger sequencing of the amplicons (Supplementary Figure S3). Semi-quantitative RT-PCR of qSH1 in leaves of B100 and A10 showed that transcript levels were significantly lower in B100 compared to A10 (P = 0.015) (Supplementary Table S2). qSH1 transcript levels in panicles were higher than in leaves (Supplementary Table S2), but not significantly different between A10 and B100 (P = 0.212).
Effect of the MITE on Transcript Processing of Sh1 in S. italica
Because the MITE in Yugu1 and B100 Sh1 was inserted close to the 3′ end of exon 2, we investigated whether its presence affected splicing. Sequencing of four clones from a single PCR reaction using S. viridis accession A10 cDNA as template with primer set sh1-09f-MITE/sh-09r-MITE, which was located in exon 1/exon 3 and hence amplified across the MITE insertion site in exon 2, indicated the presence of two potential 3′ splice sites for intron 2 that were separated by 9 bp (Figure 2 and Supplementary Figure S1). Reads corresponding to both splice products were also identified by BLASTN analysis against S. viridis A10 transcriptome data (NCBI SRA Experiment SRX875196), supporting the occurrence of alternative splicing. Sequencing of the same region (two clones from the same PCR reaction) in a S. italica B100 transcript also showed two alternative splice products that differed by nine basepairs. Furthermore, B100 transcripts lacked exon 2 (Figure 2 and Supplementary Figure S1). The presence of the MITE resulted in the simultaneous splicing of intron 1, exon 2 plus the MITE and intron 2. The transcript data obtained by RT-PCR were confirmed by alignment of available RNA-Seq data from S. italica Yugu1 (NCBI SRA Experiment SRX2832831) against Seita.9G154300. The alignments further suggested that the MITE presence led to an additional alternatively spliced product in which introns 1 and 2 were retained (Supplementary Figure S4). We were unable to confirm the formation of this putatively alternatively spliced product by RT-PCR with primers located in exon 2 and exon 3. This could be due to the difficulty in amplifying across the GC-rich MITE. The overall GC content of the MITE was 66%, but was as high as 90% in several 50 bp windows. Alternatively, the presence of introns 1 and 2 in the RNA-Seq data may have been caused by genomic DNA contamination. This, however, is unlikely, because no evidence was obtained of retention of introns 3, 4, and 5 (Supplementary Figure S4). Of the two alternative splice sites 9 bp apart between intron 2 and exon 3, use of only the most 5′ splice site was observed in the RNA-Seq reads.
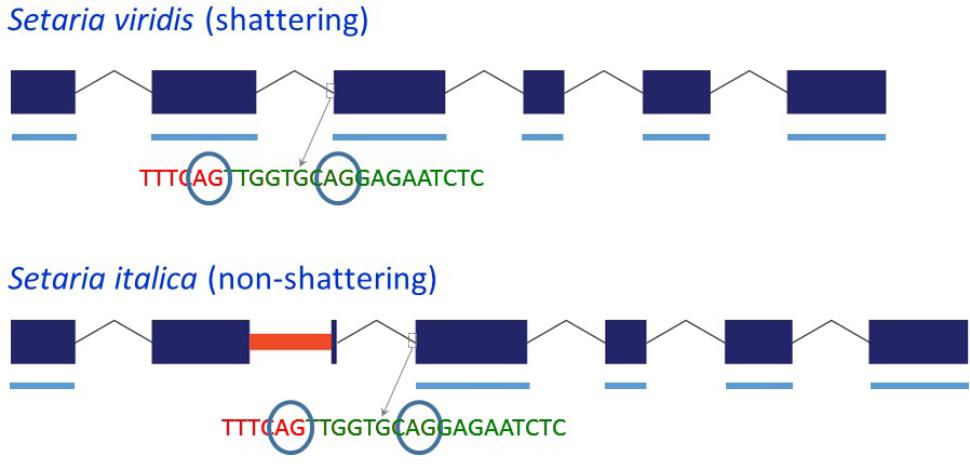
FIGURE 2. Schematic representation of the structure of the Sh1 gene and its transcript in S. viridis and S. italica. Blue boxes represent exons and thin lines represent introns. The red bar in exon 2 represents the MITE insertion. Thick blue lines underneath the exons represent exonic sequence present in the transcript. The circled “AG” indicates alternative splice sites.
Presence of the MITEs in S. italica and S. viridis Germplasm
To determine the prevalence of the MITEs in Sh1 and qSH1 in Setaria germplasm, regions of, on average 87 bp, that covered either the boundary between genic sequence and MITE sequence or were comprised of genic sequence flanking the MITEs, were used in BLASTN searches against resequencing data from 200 S. italica and 100 S. viridis accessions. One or more reads in 59 S. italica accessions had hits against the boundary region that covered the 3′ end of exon 2 and the 5′ end of the MITE in Sh1. A total of 95 S. italica accessions had hits using the last 40 bp of the MITE, the last 5 bp of exon 2 and the first 39 bp of intron 2 of Yugu1 Sh1 as query sequence. Thirty-five accessions had hits for both the 5′ and 3′ boundary regions, bringing the total number of S. italica accessions that carried a MITE to 119. None of the S. italica accessions had BLAST hits against the S. viridis control query sequence which covered the last 28 bp of exon 2 and the first 44 bp of intron 2 of A10 Sh1. In contrast, all 100 S. viridis accessions had BLAST hits using the MITE flanking sequence (S. viridis control) as query; none has BLAST hits using genic-MITE boundary regions as queries.
We found significant variation for the presence of the two MITEs in qSH1 in S. italica germplasm. Of the 122 S. italica accessions with one or more identified blast hits, 54% carried at least one of the two MITEs, 43% lacked one of the MITEs but the presence of the other MITE was unknown and 2.5% (3 accessions) lacked both MITEs (Supplementary Table S3). Both MITEs were absent from the 5′UTR in qSH1 in all S. viridis germplasm tested.
Discussion
Although shattering ability was evaluated in a simple manner by scoring seed loss on a 0–3 scale, QTL were consistently (i.e., across replicates and populations) identified on chromosomes V and IX. However, only the QTL on chromosome IX colocalized across populations and replicates. This suggests that the clenched-fist method was sufficiently accurate to identify shattering loci of large effect. Precise mapping of smaller-effect QTL, however, may require the use of a force gauge to more precisely measure the strength needed to disarticulate a spikelet. The presence of wild-type alleles at both the chromosomes V and IX loci was required for a high level of shattering, indicating that the chromosomes V and IX effects are additive. Although the two QTL combined explained less than 50% of the variation in shattering, this is likely an underestimation due to the imprecise phenotyping. Nevertheless, it is possible that smaller-effect QTL for shattering are present in the population which were not, or not consistently, detected. The QTL on chromosomes VI and VII which were identified in only one replicate in the F2:3 population and RIL population, respectively, could be such examples. Furthermore, additional QTL peaks were identified in some replicates on chromosome IX, and it is possible that they represent additional shattering genes that are present in the B100 × A10 population.
Although no visual abscission layer is present in Setaria (Hodge and Kellogg, 2016), our data show that the function of Sh1 in seed disarticulation is conserved with Sh1 function in other grass species despite differences in structure and/or location of the abscission zone (Doust et al., 2014b). The YABBY transcription factor (Sh1 gene) underlying the shattering QTL on chromosome IX has previously been identified as the main gene controlling disarticulation in domesticated sorghum (Lin et al., 2012). Sh1 also underlies shattering QTL on rice and maize, suggesting parallel selection in different grass crops on this gene during domestication (Lin et al., 2012). In S. italica, the insertion of a PIF/Harbinger MITE close to the 3′ end of exon 2 led to the formation of a transcript that lacked exon 2. Exon 2 comprises 123 basepairs. Deletion of 41 amino acids in the SH1 protein in S. italica will almost certainly reduce or eliminate its activity. The same is true for proteins translated from transcripts in which introns 1, 2 and the MITE are retained. Analysis of resequencing data of S. italica (Jia et al., 2013) showed that 119 of the 200 accessions analyzed carried the MITE in Sh1. Of the 119 accessions, 75 were from China, and the remainder originated from other countries in Asia, Europe, and Africa. For the remaining 81 accessions, we could not establish presence/absence of the MITE because of the overall low depth of the resequencing data (average and median sequencing depth: 0.94 and 1.04, respectively). Importantly, none of the S. italica accessions tested were positive for the S. viridis control query sequence, supporting further that all S. italica germplasm carried the MITE in Sh1. Two additional S. italica lines, Ise-3 and Ise-5, that originated from India were also shown to carry the MITE in Sh1 by PCR (Supplementary Figure S1).
Several hypotheses exist regarding the center of domestication of foxtail millet (reviewed by Diao and Jia, 2017). Some consider China the primary and possibly the only center of domestication for Setaria. Other studies advocate the existence of further independent centers of domestication in Europe and the region comprising central Asia, Pakistan, Afghanistan, and Northwest India. The presence of identical MITE insertions sites in Sh1 in all 119 S. italica accessions for which sequence reads were available in the target region, irrespective of their country of origin, and the absence of this MITE in all S. viridis lines analyzed, strongly suggest a single center of domestication for foxtail millet. This is supported by the fact that alleles at SNP positions extending to at least 100 kb on either side of Sh1 were close to fixation (99%) in our set of S. italica accessions (D. Chakraborty and K.M. Devos, unpublished data). The average frequency in this region of the S. italica SNP alleles in our set of S. viridis germplasm was 67%. The absence of the MITE in Sh1 in the S. viridis germplasm analyzed may be due to (1) a low frequency of the low-shattering Sh1 allele in wild populations, (2) insertion of the MITE early in the domestication process, or (3) a lack in our S. viridis collection of accessions from the center of domestication. Most of the resequencing data was for S. viridis accessions collected in the United States that belong to two major genetic subpopulations. Asian S. viridis germplasm largely forms a third subpopulation (Huang et al., 2014; Schröder et al., 2016)
It is unclear whether qSH1 is the gene that underlies the shattering QTL on Setaria chromosome V. The presence of MITEs near genes can affect transcript levels, and both positive and negative effects have been reported (Naito et al., 2009; Lu et al., 2012; Han et al., 2013; Mao et al., 2015). It is therefore conceivable that the presence of either of the two MITEs in the 5′UTR of qSH1 reduces expression of this transcription factor, leading to a strengthening of the abscission zone. In our analyses, transcript levels were lower in leaves of B100 than in A10, but no differences were observed in panicles. However, expression specifically in the abscission zone will need to be examined. In rice, qSH1 expression was observed in both non-shattering and shattering lines in the shoot apical meristem during floral transition and in the anther regions, but only in shattering lines at the position of the abscission layer (Konishi et al., 2006). Although the coding sequence of the qSH1 allele in S. italica acc. Yugu1 and B100 also differs from that in S. viridis accession A10 by the presence of a non-synonymous SNP, the lack of conservation of this region in different grass species (Supplementary Figure S5) suggests that this SNP is unlikely to affect qSH1 gene function. An analysis of the presence of the two MITEs in qSH1 across a worldwide collection of S. italica germplasm showed that 39% of Chinese germplasm (26 accessions) but only 7% (2) of accessions originating from countries other than China carried MITE 1. MITE 2 was predominantly present in S. italica worldwide. All S. viridis accessions tested lacked both MITEs. Genome-wide linkage disequilibrium in cultivated foxtail millet has been estimated at ∼100 kb (Jia et al., 2013), so our study could not differentiate between causal mutations and tightly linked variants. Of the three S. italica accessions that lacked both MITE 1 and MITE 2, two were from Hungary and one was from Turkey. The Turkish accession (Ci879) was listed as a “weedy foxtail” by Jia et al. (2013). Our study showed Ci879 to carry the MITE in Sh1 so this accession was presumably low shattering. More precise phenotyping is necessary to determine the degree of shattering in S. italica lines that differ in the presence of the MITEs in qSH1.
Conclusion
We identified Sh1 on Setaria chromosome IX as the main target for reduced shattering during the domestication of foxtail millet. The variant allele, which carries a PIF/Harbinger MITE, was present in S. italica lines from around the world and absent from all S. viridis accessions tested, supporting the hypothesis of a single center of domestication for foxtail millet. The S. italica accession B100 carries at least one other gene for reduced shattering located on chromosome V. Structural variation is present at the qSH1 locus between S. italica and S. viridis, and the presence of two MITEs in the 5′UTR of qSH1 is strongly associated with cultivated genotypes. Complementation tests or knock-out experiments are needed to unambiguously demonstrate that the MITE insertions represent the causal mutations to shattering rather than linked variants.
Author Contributions
SO participated in the experimental design and QTL analysis, conducted the comparative analysis, and drafted the manuscript. SC did the qSH1 transcript analysis, conducted the PCR to confirm the presence of the MITEs in Sh1 and qSH1, and analyzed the RNA-Seq reads in the Sh1 region. DC conducted the bioinformatic analysis of the presence of the MITEs in the S. viridis and S. italica germplasm. LM-B did the experimental Sh1 transcript analysis. MT and OP conducted the phenotyping of the F2 population. XW conducted the phenotyping of the RIL population. BB conducted the QTL analyses. KD designed the study, participated in all aspects of data analysis and data interpretation, and wrote the manuscript. All authors critically read and approved the manuscript.
Funding
SO was partially funded through a graduate fellowship from the Institute of Plant Breeding, Genetics and Genomics. KD acknowledges funding from the National Institute of Food and Agriculture Plant Feedstock Genomics for Bioenergy Program (grant #2008-35504-04851).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00918/full#supplementary-material
FIGURE S1 | Multi-sequence alignment of the Sh1 alleles and transcripts in S. viridis accession A10 and S. italica accessions Yugu1, B100, Ise-3 and Ise-5. Genomic and cDNA sequences are indicated with “genomic” and “cDNA,” respectively, in the sequence name. Intron sequences have a white background, exon sequences have brown (A), yellow (C), green (T), and purple (G) backgrounds, and MITE sequences have green (A), orange (C), blue (T), and red (G) backgrounds.
FIGURE S2 | Insertion of (A) MITE 1 and (B) MITE 2 in the 5′UTR of qSH1 was accompanied or followed by rearrangements. The MITE sequence is delineated by “|”. Deletions are in red, insertions in green. [ ] indicates SSR units. Microhomology flanking the deletion is highlighted in yellow.
FIGURE S3 | Multi-sequence alignment of the qSH1 alleles in S. viridis accession A10, and S. italica accessions B100 and Yugu1. MITE sequences have green (A), orange (C), blue (T), and red (G) backgrounds. The “ATG” start codon has a white background.
FIGURE S4 | Alignment of RNA-Seq reads against Sh1 genomic sequence. (A) A10 reads (SRX875196) against Sevir.9G153200 (only the read coverage is shown) and (B) Yugu1 reads (SRX2832831) against Seita.9G154300 (both read coverage and individual read alignments are shown). Exon locations are indicated with red boxes. The MITE location in Seita9G.154300 is shown in blue.
FIGURE S5 | Multi-sequence alignment at the protein level across different grass species of the qSH1 region carrying the non-synonymous SNP that differentiates S. italica from S. viridis (position 31 in this alignment).
TABLE S1 | Genotypic and phenotypic data for the B100 × A10 F2:3 and RIL populations.
TABLE S2 | Semi-quantitative RT-PCR of qSH1 in leaves and panicles of A10 and B100.
TABLE S3 | Presence/absence of MITEs 1 and 2 in qSH1 in S. italica germplasm.
Footnotes
References
Abràmoff, M. D., Magalhães, P. J., and Ram, S. J. (2004). Image processing with ImageJ. Biophotonics Int. 11, 36–42.
Barton, L., Newsome, S. D., Chen, F. H., Wang, H., Guilderson, T. P., and Bettinger, R. L. (2009). Agricultural origins and the isotopic identity of domestication in northern China. Proc. Natl. Acad. Sci. U.S.A. 106, 5523–5528. doi: 10.1073/pnas.0809960106
Basten, C. J., Weir, B. S., and Zeng, Z. B. (1995). QTL Cartographer: A Reference Manual and Tutorial for QTL Mapping. Raleigh, NC: North Carolina State University.
Bennetzen, J. L., Schmutz, J., Wang, H., Percifield, R., Hawkins, J., Pontaroli, A. C., et al. (2012). Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 30, 555–561. doi: 10.1038/nbt.2196
Butenko, M. A., Patterson, S. E., Grini, P. E., Stenvik, G.-E., Amundsen, S. S., Mandal, A., et al. (2003). INFLORESCENCE DEFICIENT IN ABSCISSION controls floral organ abscission in Arabidopsis and identifies a novel family of putative ligands in plants. Plant Cell 15, 2296–2307. doi: 10.1105/tpc.014365
Devos, K. M. (2005). Updating the ‘Crop Circle’. Curr. Opin. Plant Biol. 8, 155–162. doi: 10.1016/j.pbi.2005.01.005
Devos, K. M., and Gale, M. D. (2000). Genome relationships: the grass model in current research. Plant Cell 12, 637–646. doi: 10.1105/tpc.12.5.637
Devos, K. M., Wang, Z. M., Beales, J., Sasaki, T., and Gale, M. D. (1998). Comparative genetic maps of foxtail millet (Setaria italica) and rice (Oryza sativa). Theor. Appl. Genet. 96, 63–68. doi: 10.1111/tpj.12842
Devos, K. M., Wu, X., and Qi, P. (2017). “Genome structure and comparative genomics,” in Genetics and Genomics of Setaria – Plant Genetics and Genomics: Crops and Models, eds A. Doust and X. Diao (Cham: Springer), 135–147.
Diao, X., and Jia, G.(eds) (2017). “Origins and domestication of foxtail millet,” in Genetics and Genomics of Setaria – Plant Genetics and Genomics: Crops and Models, eds A. Doust and X. Diao (Cham: Springer), 61–72.
Doebley, J. (2006). Unfallen grains: How ancient farmers turned weeds into crops. Science 312, 1318–1319. doi: 10.1126/science.1128836
Doebley, J., Stec, A., and Hubbard, L. (1997). The evolution of apical dominance in maize. Nature 386, 485–488. doi: 10.1038/386485a0
Doust, A. N., Kellogg, E. A., Devos, K. M., and Bennetzen, J. L. (2009). Foxtail millet: a sequence-driven grass model system. Plant Physiol. 149, 137–141. doi: 10.1104/pp.108.129627
Doust, A. N., Lukens, L., Olsen, K. M., Mauro-Herrera, M., Meyer, A., and Rogers, K. (2014a). Beyond the single gene: How epistasis and gene-by-environment effects influence crop domestication. Proc. Natl. Acad. Sci. U.S.A. 111, 6178–6183. doi: 10.1073/pnas.1308940110
Doust, A. N., Mauro-Herrera, M., Francis, A. D., and Shand, L. C. (2014b). Morphological diversity and genetic regulation of inflorescence abscission zones in grasses. Am. J. Bot. 101, 1759–1769. doi: 10.3732/ajb.1400186
Fang, X., Dong, K., Wang, X., Liu, T., He, J., Ren, R., et al. (2016). A high density genetic map and QTL for agronomic and yield traits in Foxtail millet [Setaria italica (L.) P. Beauv.]. BMC Genomics 17:336. doi: 10.1186/s12864-016-2628-z
Gale, M. D., and Devos, K. M. (1998). Comparative genetics in the grasses. Proc. Natl. Acad. Sci. U.S.A. 95, 1971–1974. doi: 10.1073/pnas.95.5.1971
Gepts, P. (2010). Crop domestication as a long-term selection experiment. Plant Breed. Rev. 24, 1–44.
Han, Y., Qin, S., and Wessler, S. R. (2013). Comparison of class 2 transposable elements at superfamily resolution reveals conserved and distinct features in cereal grass genomes. BMC Genomics 14:71. doi: 10.1186/1471-2164-14-71
Hodge, J. G., and Kellogg, E. A. (2016). Abscission zone development in Setaria viridis and its domesticated relative, Setaria italica. Am. J. Bot. 103, 998–1005. doi: 10.3732/ajb.1500499
Huang, P., Feldman, M., Schroder, S., Bahri, B. A., Diao, X., Zhi, H., et al. (2014). Population genetics of Setaria viridis, a new model system. Mol. Ecol. 20, 4912–4925. doi: 10.1111/mec.12907
Jia, G., Huang, X., Zhi, H., Zhao, Y., Zhao, Q., Li, W., et al. (2013). A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet (Setaria italica). Nat. Genet. 45, 957–961. doi: 10.1038/ng.2673
Kellogg, E. A. (2017). “Evolution of Setaria,” in Genetics and Genomics of Setaria – Plant Genetics and Genomics: Crops and Models, eds A. Doust and X. Diao (Cham: Springer), 3–27. doi: 10.1007/978-3-319-45105-31
Komatsuda, T., Pourkheirandish, M., He, C. F., Azhaguvel, P., Kanamori, H., Perovic, D., et al. (2007). Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc. Natl. Acad. Sci. U.S.A. 104, 1424–1429. doi: 10.1073/pnas.0608580104
Konishi, S., Izawa, T., Lin, S. Y., Ebana, K., Fukuta, Y., Sasaki, T., et al. (2006). An SNP caused loss of seed shattering during rice domestication. Science 312, 1392–1396. doi: 10.1126/science.1126410
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, C., Zhou, A., and Sang, T. (2006). Rice domestication by reducing shattering. Science 311, 1936–1939. doi: 10.1126/science.1123604
Li, P., and Brutnell, T. (2011). Setaria viridis and Setaria italica, model genetic systems for the Panicoid grasses. J. Exp. Bot. 62, 3031–3037. doi: 10.1093/jxb/err096
Lin, Z., Li, X., Shannon, L. M., Yeh, C.-T., Wang, M. L., Bai, G., et al. (2012). Parallel domestication of the Shattering1 genes in cereals. Nat. Genet. 44, 720–724. doi: 10.1038/ng.2281
Lu, C., Chen, J., Zhang, Y., Hu, Q., Su, W., and Kuang, H. (2012). Miniature Inverted–Repeat Transposable Elements (MITEs) have been accumulated through amplification bursts and play important roles in gene expression and species diversity in Oryza sativa. Mol. Biol. Evol. 29, 1005–1017. doi: 10.1093/molbev/msr282
Mao, H., Wang, H., Liu, S., Li, Z., Yang, X., Yan, J., et al. (2015). A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat. Commun. 6:8326. doi: 10.1038/ncomms9326
Naito, K., Zhang, F., Tsukiyama, T., Saito, H., Hancock, C. N., Richardson, A. O., et al. (2009). Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 461, 1130–1134. doi: 10.1038/nature08479
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Schröder, S., Bahri, B. A., Eudy, D. M., Layton, D. J., Kellogg, E. A., and Devos, K. M. (2016). Genetic diversity and origin of North American green foxtail [Setaria viridis (L.) Beauv.] accessions. Genet. Resour. Crop Evol. 64, 367–378. doi: 10.1007/s10722-016-0363-6
Simons, K. J., Fellers, J. P., Trick, H. N., Zhang, Z. C., Tai, Y. S., Gill, B. S., et al. (2006). Molecular characterization of the major wheat domestication gene Q. Genetics 172, 547–555. doi: 10.1534/genetics.105.044727
Van Eck, J., Swartwood, K., Pidgeon, K., and Maxson-Stein, K. (2017). “Agrobacterium tumefaciens-mediated transformation of Setaria viridis,” in Genetics and Genomics of Setaria – Plant Genetics and Genomics: Crops and Models, eds A. Doust and X. Diao (Cham: Springer), 343–356.
Wang, J., Wang, Z., Du, X., Yang, H., Han, F., Han, Y., et al. (2017). A high-density genetic map and QTL analysis of agronomic traits in foxtail millet [Setaria italica (L.) P. Beauv.] using RAD-seq. PLoS One 12:e0179717. doi: 10.1371/journal.pone.0179717
Wang, S., Basten, C. J., and Zeng, Z.-B. (2012). Windows QTL Cartographer 2.5. Raleigh, NC: North Carolina State University.
Wang, Z. M., Devos, K. M., Liu, C. J., Wang, R. Q., and Gale, M. D. (1998). Construction of RFLP-based maps of foxtail millet, Setaria italica (L.) P.Beauv. Theor. Appl. Genet. 96, 31–36. doi: 10.1007/s001220050705
Zhang, G., Liu, X., Quan, Z., Cheng, S., Xu, X., Pan, S., et al. (2012). Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 30, 549–554. doi: 10.1038/nbt.2195
Keywords: domestication, miniature inverted-repeat transposable element (MITE), QTL analysis, seed shattering, Setaria italica, Setaria viridis, transcription factors
Citation: Odonkor S, Choi S, Chakraborty D, Martinez-Bello L, Wang X, Bahri BA, Tenaillon MI, Panaud O and Devos KM (2018) QTL Mapping Combined With Comparative Analyses Identified Candidate Genes for Reduced Shattering in Setaria italica. Front. Plant Sci. 9:918. doi: 10.3389/fpls.2018.00918
Received: 23 February 2018; Accepted: 11 June 2018;
Published: 19 July 2018.
Edited by:
Andrew Doust, Oklahoma State University, United StatesReviewed by:
Mingsheng Chen, Institute of Genetics and Developmental Biology (CAS), ChinaXinguang Zhu, University of Chinese Academy of Sciences (UCAS), China
Manoj Prasad, National Institute of Plant Genome Research (NIPGR), India
Copyright © 2018 Odonkor, Choi, Chakraborty, Martinez-Bello, Wang, Bahri, Tenaillon, Panaud and Devos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katrien M. Devos, a2Rldm9zQHVnYS5lZHU=
†Present address: Sandra Odonkor, West African Centre for Crop Improvement, University of Ghana, Legon, Ghana; Liliam Martinez-Bello, Horticultural Sciences Department, University of Florida, Gainesville, FL, United States