 Dinusha C. Maheepala1
Dinusha C. Maheepala1 Christopher A. Emerling2†Alex Rajewski1Jenna Macon1Maya Strahl3†
Christopher A. Emerling2†Alex Rajewski1Jenna Macon1Maya Strahl3† Natalia Pabón-Mora4
Natalia Pabón-Mora4 Amy Litt1*
Amy Litt1*- 1Department of Botany and Plant Sciences, University of California, Riverside, Riverside, CA, United States
- 2Institut des Sciences de l’Évolution de Montpellier, Université de Montpellier, Centre National de la Recherche Scientifique, Institut de Recherche pour le Développement, École Pratique des Hautes Études, Montpellier, France
- 3The New York Botanical Garden, Bronx, NY, United States
- 4Instituto de Biología, Universidad de Antioquia, Medellín, Colombia
Ecologically and economically important fleshy edible fruits have evolved from dry fruit numerous times during angiosperm diversification. However, the molecular mechanisms that underlie these shifts are unknown. In the Solanaceae there has been a major shift to fleshy fruits in the subfamily Solanoideae. Evidence suggests that an ortholog of FRUITFULL (FUL), a transcription factor that regulates cell proliferation and limits the dehiscence zone in the silique of Arabidopsis, plays a similar role in dry-fruited Solanaceae. However, studies have shown that FUL orthologs have taken on new functions in fleshy fruit development, including regulating elements of tomato ripening such as pigment accumulation. FUL belongs to the core eudicot euFUL clade of the angiosperm AP1/FUL gene lineage. The euFUL genes fall into two paralogous clades, euFULI and euFULII. While most core eudicots have one gene in each clade, Solanaceae have two: FUL1 and FUL2 in the former, and MBP10 and MBP20 in the latter. We characterized the evolution of the euFUL genes to identify changes that might be correlated with the origin of fleshy fruit in Solanaceae. Our analyses revealed that the Solanaceae FUL1 and FUL2 clades probably originated through an early whole genome multiplication event. By contrast, the data suggest that the MBP10 and MBP20 clades are the result of a later tandem duplication event. MBP10 is expressed at weak to moderate levels, and its atypical short first intron lacks putative transcription factor binding sites, indicating possible pseudogenization. Consistent with this, our analyses show that MBP10 is evolving at a faster rate compared to MBP20. Our analyses found that Solanaceae euFUL gene duplications, evolutionary rates, and changes in protein residues and expression patterns are not correlated with the shift in fruit type. This suggests deeper analyses are needed to identify the mechanism underlying the change in FUL ortholog function.
Introduction
Fleshy fruits are agriculturally and economically important plant organs that have evolved from dry fruits many times during angiosperm evolution. However, the genetic changes that are required for this shift to occur are as yet unknown (Bolmgren and Eriksson, 2010). In the agriculturally, pharmacologically, and horticulturally important plant family Solanaceae (nightshades), there was a shift to fleshy fruit in the subfamily Solanoideae from plesiomorphic dry fruit (Figure 1) (Knapp, 2002). In the family two independent transitions to fleshy fruits have also occurred in the genera Duboisia (subfamily Anthocercideae) and Cestrum (subfamily Cestroideae), as well as a reversal to dry fruit in the genus Datura (subfamily Solanoideae) (Knapp, 2002).
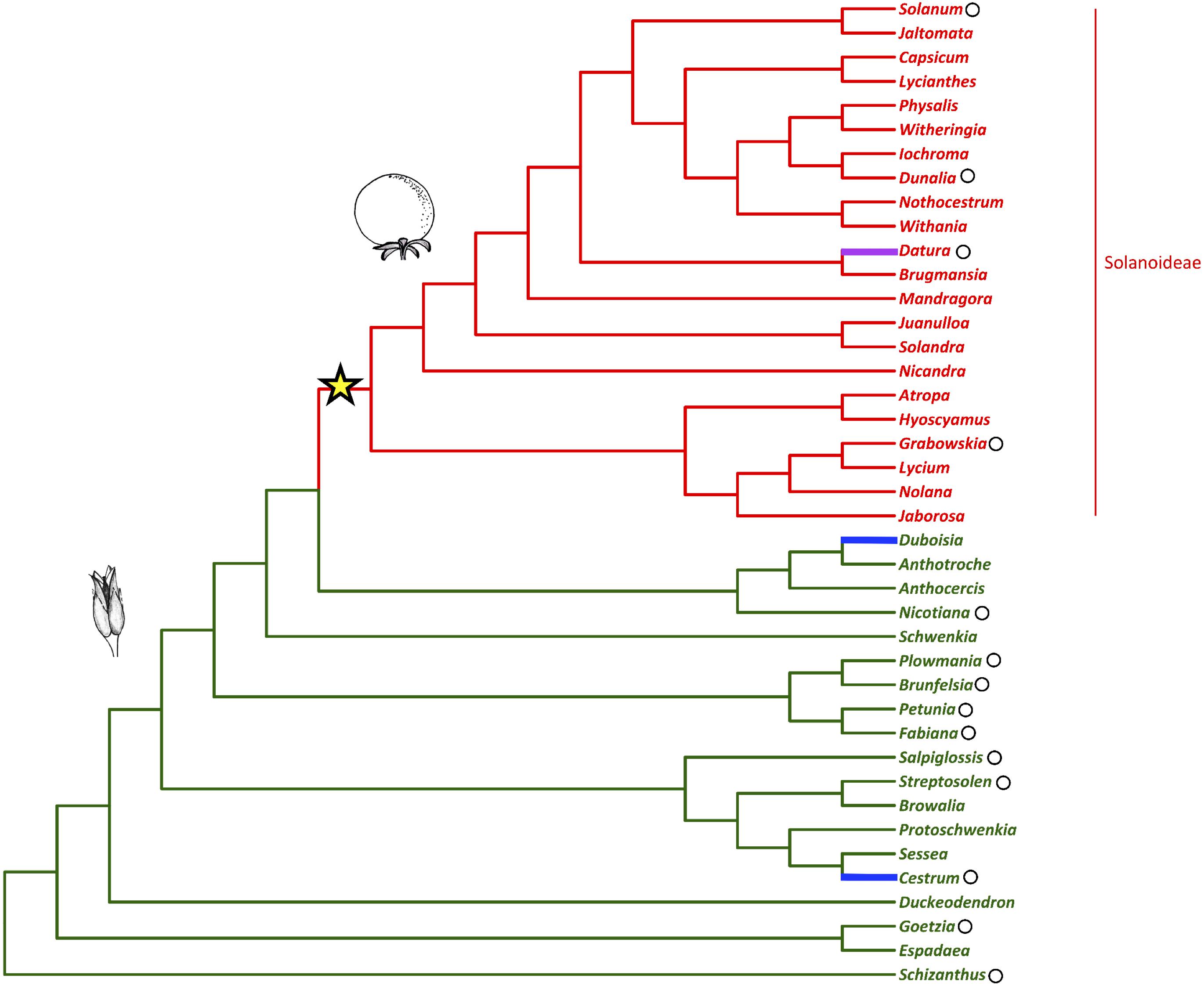
Figure 1. Solanaceae phylogeny with fruit type (dry vs. fleshy) mapped (adapted from Knapp, 2002; Olmstead et al., 2008). The shift to fleshy fruit in the sub-family Solanoideae is indicated with the star. The capsule represents the ancestral fruit-type while the berry represents the generic fruit-type following this shift. The reversal to dry fruit and the independent evolutionary origins of fleshy fruit are highlighted in magenta and blue, respectively. Black circles mark the genera referred to in the text.
Evidence from tomato (Solanum lycopersicum, subfamily Solanoideae) indicates that FRUITFULL (FUL) transcription factors (TFs) have novel functions in fleshy fruit development compared to Arabidopsis (Brassicaceae) and Nicotiana (Solanaceae, subfamily Nicotianoideae) (Gu et al., 1998; Smykal et al., 2007; Bemer et al., 2012; Shima et al., 2013, 2014; Wang et al., 2014). FUL is a MADS-box TF that plays pleiotropic roles in both reproductive and vegetative development in the model plant Arabidopsis thaliana (Spence et al., 1996; Gu et al., 1998; Liljegren et al., 2000; Rajani and Sundaresan, 2001; Melzer et al., 2008). FUL controls cell proliferation in the fruit valves and spatially limits the formation of the dehiscence zone in the dry silique of A. thaliana, enabling the mature fruits to dehisce (Spence et al., 1996; Gu et al., 1998; Liljegren et al., 2000, 2004; Rajani and Sundaresan, 2001). Overexpression of a Nicotiana tabacum FUL ortholog in woodland tobacco (Nicotiana sylvestris) resulted in indehiscent fruits with reduced lignification at the dehiscence zones, suggesting a role similar to that observed in silique development in A. thaliana (Smykal et al., 2007). Several groups have examined the function of euFUL genes, the core-eudicot clade to which FUL belongs, in tomato (Bemer et al., 2012; Shima et al., 2014; Wang et al., 2014). All studies showed defects in fruit pigmentation during ripening when FUL ortholog expression was downregulated, and some studies also suggested roles in ethylene production and pericarp and cuticle thickness (Bemer et al., 2012; Shima et al., 2014; Wang et al., 2014). These data indicate that euFUL genes are controlling different processes in dry and fleshy fruits in the Solanaceae.
Early in the diversification of core-eudicots, there was a duplication in the euFUL gene clade, which resulted in the euFULI and euFULII clades (Litt and Irish, 2003; Shan et al., 2007). The A. thaliana FUL gene belongs to the euFULI clade while its paralog, AGL79 which plays a role in lateral root development, branching, leaf morphology, and transition to flowering, belongs to the euFULII clade (Gao et al., 2018). The euFULI clade has duplicated in Solanaceae resulting in two subclades, designated here as FUL1 and FUL2; likewise the euFULII clade has two Solanaceae-specific subclades, here designated MBP10 and MBP20 (Hileman et al., 2006; Bemer et al., 2012; The Tomato Genome Consortium, 2012). We studied the evolution of euFUL genes in Solanaceae to characterize patterns of selection, duplication, and sequence evolution to identify changes that might be correlated with the shift to fleshy fruit. We tested the following hypotheses: (1) following the duplication of euFUL genes, there was a relaxation of selection in some or all of the resulting clades that resulted in sequence diversification; (2) changes in amino acid sequences are correlated with the origin of fleshy fruit. Although we found several sites showing changes in amino acid residues that might have resulted in changes in protein function, none of these were associated with the evolution of fleshy fruit. Consistent with our hypothesis, we found that the FUL1 and MBP10 genes are evolving at significantly faster rates in comparison to FUL2 and MBP20. In combination with the relatively weak expression of MBP10 and loss of potential regulatory elements, our data suggest that the MBP10 lineage may be undergoing pseudogenization.
Materials and Methods
Plant Material for Sequencing
Sources of plant and tissue material for sequencing are listed in Table S1 in Supplementary Data Sheet S1. Plants were grown in temperature controlled glasshouses at University of California, Riverside (UCR), The New York Botanical Garden, NY (NYBG), and The University of Antioquia, Colombia (UdeA) or collected from the grounds at UCR and the Universidad de Antioquia or the field at Parque Arvi, Vereda Santa Elena, El Tambo, Colombia.
For ease of reference and to simplify language, throughout the paper, members of Solanoideae, including the dry-fruited Datura, will be referred to as “fleshy-fruited species” (rather than “fleshy-fruited species and Datura”). Likewise non-Solanoideae, including the fleshy-fruited Cestrum and Duboisia, will be referred to as “dry-fruited species” (rather than “dry-fruited species and Cestrum and Duboisia”).
RNA Isolation, cDNA Synthesis/Library Preparation, and Sequencing
RNA was extracted from fruit, floral/inflorescence or leaf tissue using RNeasy Plant Mini Kits (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol. For Grabowskia glauca, Dunalia spinosa, Fabiana viscosa, and Salpiglossis sinuata RNA extractions, lysis buffer RLC was used instead of RLT and 2.5% (w/v) polyvinylpyrrolidone (PVP) was added. The RLT buffer was used for extracting RNA from all other species. RNA quality was checked using a BioSpectrometer Basic (Eppendorf, Hamburg, Germany) and stored at −80°C. cDNA was synthesized using SuperScript III Reverse Transcriptase (Thermo Fisher, San Diego, CA, United States) according to the manufacturer’s protocol and the product was checked by amplifying ACTIN. Clade-specific degenerate primers were designed to target specific euFUL gene homologs based on conserved regions in Solanaceae euFUL gene alignments (Supplementary Table S5). PCR was run for two initial cycles with an annealing temperature between 40 and 45°C followed by 30 cycles at 55°C annealing temperature. The PCR products were visualized on a 1% agarose gel. If multiple amplicon band sizes were present, the annealing temperature of the first two cycles was increased until only one product size was achieved.
PCR products were purified using QIAquick PCR Purification Kit (QIAGEN) according to the manufacturer’s protocol. The purified product was then cloned using TOPO TA Cloning Kit (Life Technologies, Carlsbad, CA, United States) according to the manufacturer’s protocol, and the ligated plasmids were transformed into chemically competent TOP10 strain of Escherichia coli. Transformants were plated on LB plates with kanamycin selection (50 μg/mL) coated with 40 μL of 25 mg/mL X-Gal and IPTG, and incubated at 37°C overnight. Individual positive (white) colonies were used as templates in amplification with M13F and M13R primers (Life Technologies, Carlsbad, CA, United States) to identify those colonies with inserts of the expected size between 500 bp and 1 kb. These were grown overnight in 5 mL liquid LB medium supplemented with kanamycin (50 μg/mL) in an incubator-shaker at 250 RPM and 37°C. Plasmids were extracted from the liquid cultures using Plasmid Miniprep Kit (QIAGEN) according to the manufacturer’s protocol, and sequenced using M13 reverse primer at the Institute for Integrative Genome Biology (IIGB) at UCR or Eton Bioscience, Inc. (San Diego, CA, United States).
For library preparation, RNA quality was checked using a Bioanalyzer (Agilent, Santa Clara, CA, United States). RNAseq library preparation was done according to the manufacturer’s Poly(A) mRNA Magnetic Isolation Module protocol for NEBNext Ultra Directional RNA library Prep Kit for Illumina (New England BioLabs, Ipswich, MA, United States). Cestrum diurnum, C. nocturnum, and Schizanthus grahamii libraries were sequenced on an Illumina NextSeq v2 platform with high-output runs of 75 bp paired-end reads while Dunalia spinosa, Fabiana viscosa, Grabowskia glauca, and Salpiglossis sinuata libraries were sequenced on an Illumina NextSeq v2 platform with high-output runs of both 75 bp paired-end reads and 150 bp single-end reads at IIGB, UCR. Nicotiana obtusifolia libraries were generated at NYBG and sequenced at the Beijing Genomics Institute (Shenzhen, China), and Brunfelsia australis and Streptosolen jamesonii libraries (Ortiz-Ramírez et al., 2018) were generated at UdeA and sequenced at Macrogen (Korea). All resulting euFUL sequences from both degenerate primer PCRs and transcriptomes are listed in Table S2 in Supplementary Data Sheet S1. Individual sequences from PCR-based methods have been deposited in the GenBank (for accession numbers, see Table S1 in Supplementary Data Sheet S1) and transcriptome data for N. obtusifolia, C. diurnum, C. nocturnum, D. spinosa, F. viscosa, G. glauca, and S. grahamii have been deposited on the SolGenomics network1.
Mining euFUL Sequences From de novo Transcriptome Assembly and Databases
For transcriptome assembly, raw paired-end reads and single-end reads from Illumina sequencing were first quality trimmed using Trimmomatic v0.36 (Bolger et al., 2014) or TrimGalore (Krueger, 2017) and de novo assembled on the UCR High Performance Computing Cluster (HPCC) using the default settings of Trinity v2.4.0 (Grabherr et al., 2011). Dunalia spinosa, Fabiana viscosa, Grabowskia glauca, and Salpiglossis sinuata libraries were assembled by combining both 75 bp paired-end and 150 bp single-end reads. Each assembled transcriptome was then used to create a custom Basic Local Alignment Tool (BLAST) (Altschul et al., 1990) database. The BLAST database for each species was queried on the HPCC with both blastn and tblastx using all available sequences in our euFUL sequence file using a UNIX command line that sequentially matched each sequence in our query file against the database (BLAST® Command Line Applications User Manual, 2008). BLAST analyses were also conducted on the NCBI2 (NCBI Resource Coordinators, 2017) and oneKP3 (Matasci et al., 2014) databases using A. thaliana FUL and various Solanaceae FUL homologs as query. Matching output sequences (Table S1 in Supplementary Data Sheet S1) from both transcriptomes assemblies and database mining were further confirmed by compiling a gene tree as described below. We confirmed the accuracy of our sequences using gene specific primers and Sanger sequencing. Unless specified otherwise, all sequences referred to in this manuscript are the full or partial mRNA sequences.
Gene-Tree Generation
The Multiple Sequence Comparison by Log-Expectation (MUSCLE) (Edgar, 2004) tool was used to align euFUL sequences (Supplementary Data Sheet S2). The appropriate model for tree building, GTR+G, was determined with jModelTest 2.0 (Darriba et al., 2012). Ten independent maximum likelihood (ML) analyses starting with random trees were performed using GARLI v2.1 (Genetic Algorithm for Rapid Likelihood Inference) (Bazinet et al., 2014). euFUL genes from Convolvulaceae (Convolvulus, Cuscuta and Ipomoea species), which were retrieved from the oneKP database4, were designated as the outgroup in each analysis, which meant these sequences were automatically excluded from the ingroup clades. Each ML run was set to terminate when there was no significantly better scoring topology for 20,000 consecutive generations. The ten resulting trees were checked for agreement by calculating the pairwise Robinson–Foulds distance using ‘ape’ and ‘phangorn’ packages on R (Robinson and Foulds, 1981; Paradis et al., 2004; Schliep, 2010; R Core Team, 2018). The tree with the largest ML value was chosen as the starting tree in a bootstrap analysis involving 1,000 replicates. The results of the replicates were summarized and bootstrap values were calculated using SumTrees tool of DendroPy package on Python ver. 2.7 (Python Language Reference, 2010; Sukumaran and Holder, 2010) or Geneious 10.2 (Darling et al., 2010; Kearse et al., 2012).
Any sequences that did not group with any of the subclades were aligned with the paralogs to investigate whether these may have been splice isoforms. Any such isoform was expected to have large insertions/deletions at splice junctions. None were noted.
Selection Pressure Analysis
The CODEML program within the Phylogenetic Analysis by Maximum Likelihood (PAML) (Yang, 1997) v 1.35 software package was run on the HPCC at UCR to analyze the selection pressure acting on euFUL genes. These analyses were performed to test if different gene lineages as well as sub-groups within those lineages were evolving at significantly different rates. Further scenarios were considered in which each gene, the transition branches from dry to fleshy fruit trait, or specific sites in the sequences were tested for significantly different rates of evolution. Model 0 (M0) was used to estimate a single evolutionary rate for all genes when the clades being analyzed encompassed the entire dataset. Model 2 (M2) was used when two groups encompassing the entire data set have different rates or when two groups that are being compared do not encompass the entire data set. In the latter case, the two clades being compared were grouped together to obtain a single evolutionary rate in comparison to the rate for the remaining data (background). This single rate for the two clades grouped together was then compared to the rates for each clade separately to determine if the separate rates were significantly different from the combined rate. The test statistic, 2ΔL (twice the difference of the resulting log-likelihood values), and the degrees of freedom (df), were then used in chi-squared tests to check for statistical significance. In any comparison where the P-value was less than 0.05, the second hypothesis was considered to have the better fit than the first, implying there is statistical power to support that the gene clades are evolving at different rates. Since Solanaceae has a well-supported phylogeny (Olmstead et al., 2008; Särkinen et al., 2013), for PAML analyses, the branches of the gene-tree described above were adjusted to match the phylogenetic relationships of the species included in the analysis. In the euFUL gene groups that are evolving faster, sites undergoing positive selection were analyzed using mixed effects model of evolution (MEME)6 (Murrell et al., 2012).
The gene alignments for the euFUL subclades that are evolving at statistically significantly faster than the other subclades were translated using AliView (Larsson, 2014). In these protein alignments, the sites that changed from hydrophilic to hydrophobic or vice versa were identified manually. Those changes that might have been functionally deleterious versus those that might have been neutral were identified using the PROVEAN Protein tool7 (Choi, 2012; Choi et al., 2012; Choi and Chan, 2015).
MADS (M), intervening/interacting (I) and keratin-like (K) domains of the proteins were identified using a published MADS-box protein model (Kaufmann et al., 2005).
The structure of M, I, and K domains of tomato FUL1 and MBP10 were predicted using PHYRE2 server8 (Kelley et al., 2015).
MBP10/MBP20 Synteny and Intron Analyses
One-million-base-pair regions surrounding tomato MBP10 and MBP20 were analyzed for synteny using the progressive Mauve alignment tool on Geneious 10.29 (Darling et al., 2010; Kearse et al., 2012).
Putative TF binding site searches for MBP10 and MBP20 first introns were done using PROMO 3.010 at a maximum matrix dissimilarity rate of zero (Messeguer et al., 2002; Farré et al., 2003).
Solanaceae euFUL Expression Analysis
The expression patterns of euFUL genes were analyzed using RT-PCR data for Solanum pimpinellifolium organs, and transcriptome data from this study for five stages of fruit development in S. pimpinellifolium and tomato following stages identified by Gillaspy et al. (1993) and Tanksley (2004). Additional expression data were obtained from the eFP browser11 for tomato, S. pimpinellifolium, potato (S. tuberosum) (Massa et al., 2011; Potato Genome Sequencing Consortium et al., 2011; The Tomato Genome Consortium, 2012) and from the Gene Expression Atlas12 for Nicotiana benthamiana (Nakasugi et al., 2014), and other publications (Hileman et al., 2006; Burko et al., 2013).
The TF binding sites for the 2 kb and 5 kb regions upstream of the euFUL gene transcription start sites of tomato (GCF_000188115.4) (The Tomato Genome Consortium, 2012), potato (GCF_000226075.1) (Potato Genome Sequencing Consortium et al., 2011) and N. sylvestris (GCA_000393655.1) (Sierro et al., 2013) were predicted using PlantPAN 2.013 (Chang et al., 2008). Due to the limitations of available contig length, the longest promoter region used for N. sylvestris MBP10 was 3.3 kb.
Results
Solanaceae Have Four Clades of euFUL Genes
Our analysis consisted of 106 sequences from 45 species in 26 genera obtained from direct amplification, transcriptomes, and online genomic databases (Table S1 in Supplementary Data Sheet S1). Of these, 64 sequences belonged to species from the Solanoideae, characterized by the derived fleshy fruit, whereas the other 42 sequences were from species with the ancestral dry-fruit trait. We designated euFUL genes from Convolvulaceae, the sister-group of Solanaceae, as the outgroup (Stefanoviæ et al., 2003). For many species in the analysis, we have an incomplete set of paralogs; however, we had substantial and diverse representation from across the phylogeny, which allows us to test hypotheses regarding the evolution of this gene lineage in Solanaceae.
We used maximum likelihood methods (Garli v2.1) (Bazinet et al., 2014) to reconstruct the relationships of Solanaceae euFUL genes (Figure 2). The resulting tree shows two major lineages of euFUL genes, with 80% and 100% bootstrap support, respectively, that correspond to the previously identified core eudicot euFULI and euFULII lineages (Litt and Irish, 2003; Shan et al., 2007). A Solanaceae whole-genome triplication has been proposed (The Tomato Genome Consortium, 2012; Albert and Chang, 2014; Vanneste et al., 2014; Bombarely et al., 2016), which would suggest that all Solanaceae should have three euFULI and three euFULII genes. However, others have suggested a duplication (Blanc and Wolfe, 2004; Schlueter et al., 2004; Song et al., 2012). Our data and other studies, as well as searches of the tomato genome have shown that tomato has four euFUL genes: two euFULI and two euFULII (Hileman et al., 2006; Bemer et al., 2012; The Tomato Genome Consortium, 2012) instead of the six predicted by a triplication. Additional genome sequencing (e.g., potato, Capsicum annuum) (Potato Genome Sequencing Consortium et al., 2011; Hulse-Kemp et al., 2018), transcriptome sequencing, and PCR-based analyses (this study) have also found two euFULI and two euFULII genes. This suggests the loss of one paralog from each of the euFULI and euFULII clades following a whole-genome triplication (The Tomato Genome Consortium, 2012; Albert and Chang, 2014; Vanneste et al., 2014; Bombarely et al., 2016) or, alternatively one or more duplication events (Blanc and Wolfe, 2004; Schlueter et al., 2004; Song et al., 2012).
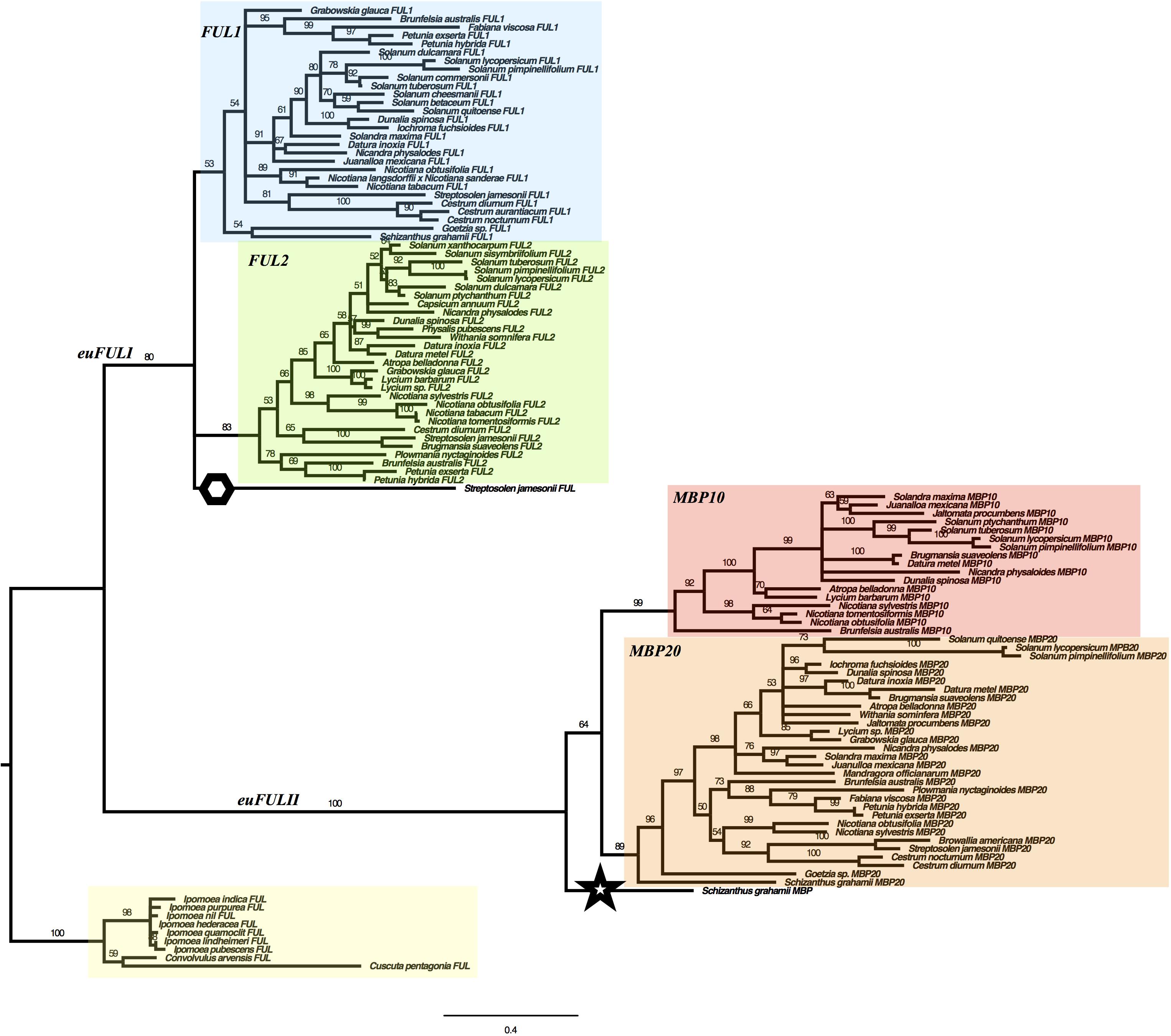
Figure 2. Solanaceae euFUL Maximum Likelihood gene tree. FUL1, FUL2, MBP10, and MBP20 clades are colored in blue, green, red, and orange, respectively. A hexagon is placed next to the Streptosolen gene that is sister to FUL1 and FUL2 clades, and a star is placed next to the Schizanthus gene that is sister to the euFULII clade. The Convolvulaceae outgroup is highlighted in yellow. The numbers on the branches indicate the bootstrap support.
For the purposes of this paper, we will refer to the euFULI and euFULII subclades by the name currently used for the tomato gene in each subclade (Hileman et al., 2006; Bemer et al., 2012). Thus, the two euFULI subclades will be referred to as the FUL1 and FUL2 clades, and the euFULII subclades will be referred to as the MPB10 and MBP20 subclades (Figure 2). In our gene tree, while the FUL2, MBP10, and MBP20 clades had high bootstrap support of 83, 99 and 89%, respectively, the FUL1 clade had only 53% support (Figure 2). A single gene from Streptosolen grouped sister to the FUL1 and FUL2 clades, while a gene from Schizanthus, one of the earliest diverging genera (Olmstead et al., 2008; Särkinen et al., 2013), grouped as sister to the euFULII clade. To confirm the above were not artifacts, we re-assembled the Streptosolen transcriptome while searching for reads supporting the gene contig, and amplified the Schizanthus sequence using gene-specific primers.
The presence of both FUL1 and FUL2 genes in species from across the phylogeny is consistent with the event that produced these two clades being part of a family-wide, whole-genome duplication or triplication (Blanc and Wolfe, 2004; Schlueter et al., 2004; Song et al., 2012; The Tomato Genome Consortium, 2012; Albert and Chang, 2014; Vanneste et al., 2014; Bombarely et al., 2016). However, we did not find a FUL2 ortholog in Schizanthus, using transcriptome data, or Goetzia, using PCR. These two genera are among the earliest diverging in the family (Olmstead et al., 2008; Särkinen et al., 2013), and are the earliest that we sampled. This raises the possibility that the FUL1/FUL2 clades resulted from a duplication that occurred following the diversification of Schizanthus and Goetzia. In addition, although we obtained MBP10 sequences from Nicotiana and most of the genera that diversified subsequently (Figure 1 and Figure S2 in Supplementary Data Sheet S1), we did not find members of the MBP10 clade in genera that diverged prior to Brunfelsia. This suggests that the MBP10 and MBP20 subclades were produced by a duplication that occurred later in Solanaceae diversification, after the euFULI duplication and any proposed family-wide whole-genome events.
The euFULII Clades Are the Result of a Tandem Gene Duplication
To investigate the nature of the MBP10/MBP20 duplication, we mapped the location of the four euFUL paralogs to the genome of cultivated tomato. FUL1 and FUL2 are located on chromosomes 6 and 3, respectively, consistent with their origin from a whole genome multiplication. By contrast, MBP10 and MBP20 are both located on chromosome 2, about 14.3 million base pairs apart (Figure 3). The location of both euFULII genes on the same chromosome, and the presence of only one ortholog in early diverging species, support the hypothesis that these paralogs may be the result of a tandem gene duplication. Moreover, comparing a 1-million-base-pair region surrounding both MBP10 and MBP20 shows synteny, further supporting a tandem duplication (Figure 3). Annotations indicate that these syntenic zones contain 17 homologous regions. The regions that show homology are located on the opposite sides of MBP10 and MBP20, suggesting an inversion of the tandemly duplicated region.
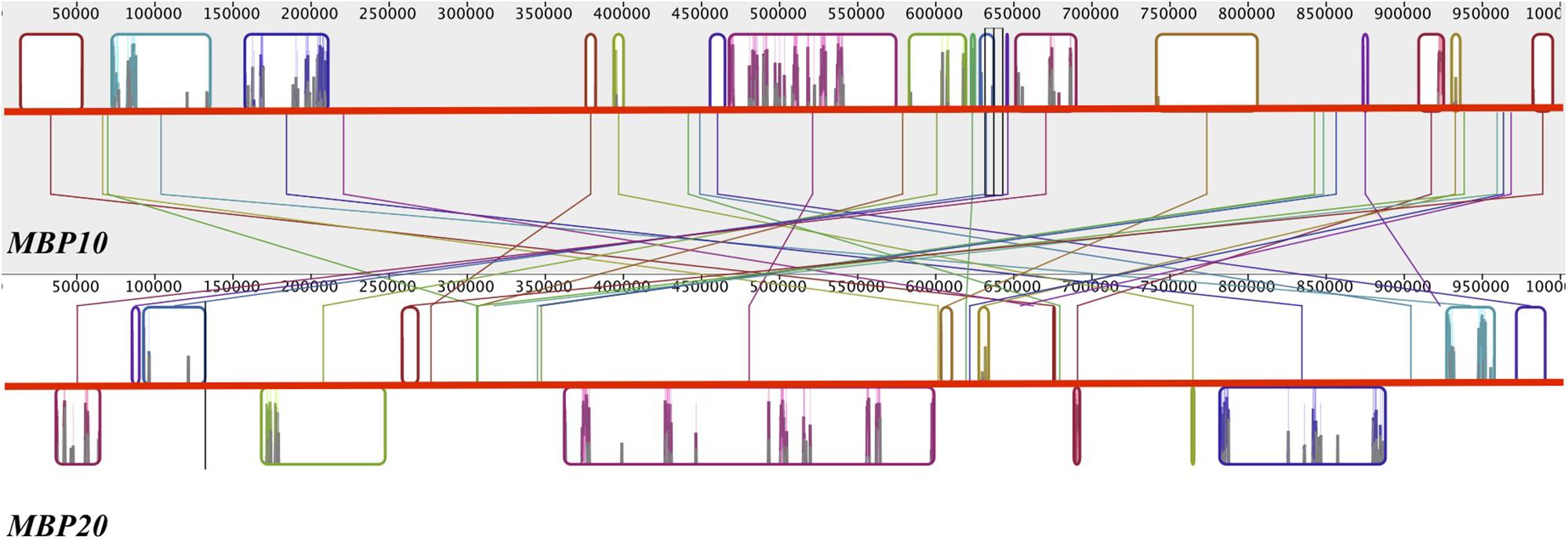
Figure 3. Reverse synteny of the regions surrounding MBP10 and MBP20 on tomato chromosome 2. The gray block at the top contains the 1 Mbp region surrounding MBP10 and the white block at the bottom contains the 1 Mbp region surrounding MBP20. A colored box in one block is homologous to a box with the identical color in the other block. MBP10 and MBP20 genomic sequences are in the center homologous region of the respective block. In MBP20, the boxed regions below the red horizontal line are in reverse orientation to the corresponding homologous regions in MBP10.
Although we recovered an MBP10-clade member in Brunfelsia australis using transcriptome analysis, we were unable to amplify this gene from leaf or floral tissue of Fabiana or Plowmania, genera that are most closely related to Brunfelsia (Figure 1 and Figure S2 in Supplementary Data Sheet S1). In addition, Petunia is also a member of the clade that includes Brunfelsia, and searches of the published Petunia genomes (Bombarely et al., 2016) also failed to turn up an MBP10-clade member. However, the Brunfelsia sequence in our analysis, obtained from transcriptome data, falls in the expected place in the phylogeny, and we confirmed the presence of MBP10 transcript in Brunfelsia floribunda floral RNA. This suggests that the MBP10/MBP20 duplication occurred before the divergence of the Brunfelsia/Fabiana/Petunia/Plowmania clade but the MBP10 paralog was lost in Fabiana, Petunia and Plowmania.
MBP10 Has a Short First Intron With No TF Binding Sites
A long first intron ranging from 1 to 10 kb, with multiple potential TF binding sites, is a general feature of FUL homologs (Table 2) (Takumi et al., 2011). By contrast, MBP10 has a short first intron of about 80 bp in both cultivated tomato and its closest wild relative, S. pimpinellifolium, and about 110 bp in Nicotiana obtusifolia (Table 2). The expression of most euFUL genes is strong across nearly all vegetative and reproductive organs (Ferrándiz et al., 2000; Shchennikova et al., 2004; Kim et al., 2005; Hileman et al., 2006; Bemer et al., 2012; Pabón-Mora et al., 2012, 2013; Scorza et al., 2017); however, diverse analyses using both quantitative and non-quantitative methods indicate that MBP10 expression is relatively weak in tomato, S. pimpinellifolium, and N. obtusifolia in most organs (Massa et al., 2011; Potato Genome Sequencing Consortium et al., 2011; The Tomato Genome Consortium, 2012; Nakasugi et al., 2014), however, some studies have suggested moderate expression in leaves (Figure S1 in Supplementary Data Sheet S1 and unpublished data). To determine if the short first intron lacks putative TF binding sites, we searched the first intron of MBP10 and MBP20 in tomato (Promo v3.0) (Messeguer et al., 2002; Farré et al., 2003). We found that the first intron of MBP10 contains no putative TF binding sites, while that of MBP20 contains 88 putative TF binding sites for eight different TFs. These TFs belong to five main families (Figure S3 in Supplementary Data Sheet S1): MYB (MYB2, C1), HSF (HSF1), Dof (Dof1, MNB1a, PBF), WRKY (SPF1) and MADS-box (SQUA). A similar situation was observed for Nicotiana obtusifolia, which had 133 putative binding sites in the first intron of MBP20 for a similar array of TFs, while MBP10 had only four such sites. In addition, we searched the first intron of AGL79, the euFULII paralog of FUL in A. thaliana, and found 49 putative binding sites, also for similar TFs and TF families. This suggests a loss of regulatory motifs in MBP10.
FUL1 and MBP10 Genes Are Evolving at a Faster Rate Than FUL2 and MBP20
Using the Solanaceae euFUL sequence data (Table S1 in Supplementary Data Sheet S1), we conducted selection pressure analyses (PAML v1.3) (Yang, 1997) to investigate if there was a shift in evolutionary rate following the FUL1/FUL2 or MBP10/MBP20 duplication. Selection pressure (ω) acting upon different euFUL gene subclades was calculated as the ratio of the rate of non-synonymous substitutions to the rate of synonymous substitutions (dN/dS) (Yang, 1997; Yang and Nielsen, 2000). An ω value of less than 1 means the coding regions are under purifying selection and that protein function is conserved. By contrast, an ω of more than 1 means that the coding regions are under diversifying selection (Yang and Nielsen, 2000). This is interpreted as allowing potential divergence in protein function (Torgerson et al., 2002; Almeida and Desalle, 2009). The nucleotide alignments we used in these analyses excluded the C-termini for all sequences except for those in the FUL2 clade, due to the high variability of this region, which prevents reliable alignment.
Our results indicate that all Solanaceae euFUL gene clades are undergoing purifying selection (ω ≤ 0.20; Table 1 and Table S3 in Supplementary Data Sheet S1), suggesting conservation of function. The two main lineages, euFULI (ω = 0.13) and euFULII (ω = 0.16) are evolving at statistically indistinguishable rates. However, within the euFULI clade, genes of the FUL1 clade are evolving at a significantly higher rate (ω = 0.17) compared to those of the FUL2 clade (ω = 0.11). Within the euFULII clade, MBP10 genes are also evolving at a significantly higher rate (ω = 0.19) compared to MBP20 (ω = 0.15). Comparing each clade against all other clades showed that FUL2 ortholog sequences are the most conserved while MBP10 ortholog sequences have the weakest purifying selection rates, followed by FUL1, implying the possibility of diversifying functions in the latter two subclades (Table 1 and Table S3 in Supplementary Data Sheet S1). None of the gene groups showed a change in evolutionary rates in comparisons between dry- and fleshy-fruited species (Table S3 in Supplementary Data Sheet S1).
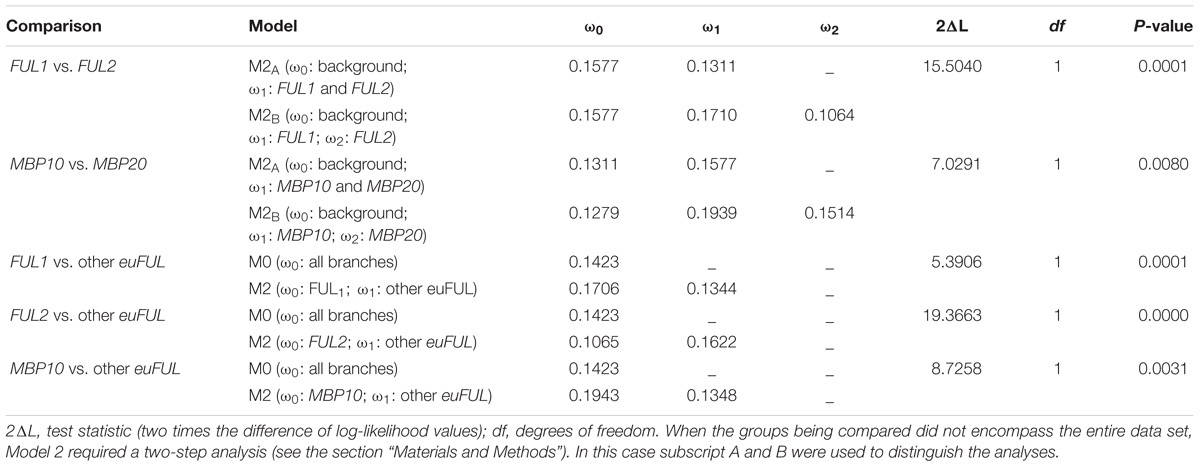
Table 1. Evolutionary rates of euFUL gene clades that are evolving at statistically different rates.
Rapidly Evolving Sites Are in Regions Responsible for Protein Complex Formation
We further analyzed the sequences to identify changes at individual amino acid sites, specifically those that involved a change between polar/charged and non-polar, that might have resulted in a change in protein conformation and function and that were correlated with the change from dry to fleshy fruit. The euFUL genes belong to the Type II MADS-domain containing proteins, which are characterized by a MADS (M) domain, which functions in DNA binding and DNA-protein dimer specificity, an intervening/interacting (I) domain that also has a role in dimer specificity, a keratin-like (K) domain important for protein–protein interactions, and a C-terminal (C) domain, implicated in protein-multimerization, transcription activation, and additional functions (Cho et al., 1999; Heijmans et al., 2012). The C-termini were excluded from this analysis. We selected comparisons in which our results showed two gene groups evolving at significantly different rates (e.g., FUL1 vs. FUL2; Table 1 and Table S3 in Supplementary Data Sheet S1). In the faster evolving group, we searched for sites in the M, I, and K regions that are undergoing diversifying selection (>1) using mixed effects model of evolution (MEME) (see footnote 6)14 (Murrell et al., 2012). The results (Figure S4 in Supplementary Data Sheet S1) suggest that sites undergoing diversifying selection are located mainly between amino acids 90 and 180 (out of ∼210 amino acids in the protein). This region corresponds to the K domain (∼90 to ∼180 amino acids) (Kaufmann et al., 2005). In comparison, the M (∼1 to ∼60 amino acids) and the I domains (∼60 to ∼90 amino acids) had relatively few sites undergoing diversifying selection. Since these TFs function in complexes with other MADS-domain proteins as well as other proteins, novel interactions made possible by amino acid changes in this region might lead to changes in transcriptional activity.
The K domain had 14 sites undergoing diversifying selection in the FUL1 proteins and four of those showed a change in polarity (Figure S4 in Supplementary Data Sheet S1). Of those four, a site that corresponds to the 153rd residue in the tomato protein had negatively charged glutamate (E) in most of the non-Solanoideae (mainly dry-fruited) species (11 out of 15 sequences) while all Solanoideae (mainly fleshy-fruited) species had a non-polar residue: valine (V; 13 species) or methionine (M; 1 species) (Figure S5 in Supplementary Data Sheet S1). This change was due to a single nucleotide change from an A to T in the former and G to A in the latter. All other changes in FUL1 proteins that result in a change in charge appeared to be reversible, and none were correlated with the phylogeny nor with phenotypic changes. We used the PROVEAN tool on all four K-domain sites that showed a change in charge to predict whether these transitions were likely to be deleterious or neutral (Choi, 2012; Choi et al., 2012; Choi and Chan, 2015). Two of these sites, one with a histidine (H) to glutamine/asparagine (Q/N) shift at the 95th residue, and one with a lysine (K) to glutamine/threonine (Q/T) shift at the 157th residue (Figure S5 in Supplementary Data Sheet S1), were predicted to be functionally deleterious while the other two sites, including the 153rd residue with E to V change, were predicted to be neutral. There were five rapidly changing sites in the M domain and six sites undergoing positive selection in the I domain of FUL1. None of the sites in the M domain showed a change in polarity. Only one site in the I domain showed a change in polarity, but this site was predicted to be neutral functionally. MBP10 proteins had 20 sites undergoing diversifying selection in the K domain, only 1 such site in the M domain and 3 in the I domain (Figures S4, S5 in Supplementary Data Sheet S1). Of these, only three sites in the I domain showed a change in charge, all of which were also predicted not to have a negative effect on function.
Solanaceae euFULI and euFULII Homologs May Have Experienced Distinct Mechanisms of Cis-Regulatory Evolution
We compared euFUL expression data for the cultivated and wild tomato species, potato and Nicotiana benthamiana to identify any patterns that might be the result of changes in the regulatory regions following the duplications of these genes. Not all data from online sources were comparable across species, as different studies included different organs and developmental stages in their analyses, limiting cross-species comparisons. The analysis shows similar spatial expression patterns for FUL1 and FUL2 (Figure 4 and Figure S1 in Supplementary Data Sheet S1). These two paralogs are broadly expressed in leaves, flowers and fruits of tomato, potato, and tobacco. Although the eFP browser data (Figure 4) shows no expression for FUL1 and FUL2 in tomato leaves, our RT-PCR data (Figure S1 in Supplementary Data Sheet S1) and previous publications (Hileman et al., 2006; Burko et al., 2013) show expression of all four euFUL homologs in these organs. Both euFULI genes are expressed relatively weakly in the roots of tomato, potato, and tobacco (Massa et al., 2011; Potato Genome Sequencing Consortium et al., 2011; The Tomato Genome Consortium, 2012; Nakasugi et al., 2014) (Figure 4 and Figure S1 in Supplementary Data Sheet S1). Although spatial domains of expression are similar for the euFULI genes, they differ in temporal expression over the course of fruit developmental stages in tomato. Although both FUL1 and FUL2 are expressed in the fruits of all species, in tomato FUL2 is highly expressed during the early stages of fruit development and then tapers off, whereas FUL1 expression increases with time (Figure 4 and Figure S1 in Supplementary Data Sheet S1).

Figure 4. The euFUL expression profiles in Solanum lycopersicum, S. pimpinellifolium, S. tuberosum, from eFP browser (http://bar.utoronto.ca), and Nicotiana benthamiana, from the Gene Expression Atlas (http://benthgenome.qut.edu.au) data.
In comparison to the euFULI genes, the two euFULII paralogs show more striking differences in spatial expression at the organ level (Figure 4 and Figure S1 in Supplementary Data Sheet S1), and also between species. In all species for which expression is reported, MBP10, alone among the euFUL genes in Solanaceae, is not expressed in fruits, or is expressed at barely detectable levels. In tomato, MBP20 is expressed strongly in roots while MBP10 is not. By contrast, in potato tubers, MBP10 expression is high and MBP20 is not expressed (Figure 4). The online sources and our RT-PCR data also show subtle intra-specific differences in expression between MBP10 and MBP20 in flowers (Figure 4 and Figure S1 in Supplementary Data Sheet S1). In addition, our RT-PCR data show that MBP10 is expressed relatively weakly in petals and stamens in tomato while MBP20 is expressed throughout the flower (Figure S1 in Supplementary Data Sheet S1). However, these differences seem to be a matter of expression intensity in comparison to the more striking contrasts seen in roots, tubers, and fruits.
The types of differences in expression between FUL1 and FUL2 versus MBP10 and MBP20 might be due to differences in the regulatory environment as a result of the different ways in which these duplicates arose. A tandem duplication and inversion may have disrupted regulatory regions in ways that would not be associated with a whole genome duplication or triplication (Tanimoto et al., 1999; Kmita et al., 2000; Vogel et al., 2009; Lupiáñez et al., 2015; Puig et al., 2015). To investigate this, we searched for putative TF binding sites in the promoter regions (2 and 5 kb upstream from the transcription start site) of euFUL genes in tomato, potato, and woodland tobacco to compare the differences between the pairs of paralogs (Table S4 in Supplementary Data Sheet S1). Woodland tobacco was used rather than N. benthamiana since relatively longer promoter sequence lengths for euFUL genes were available for this genome assembly (Sierro et al., 2013). Despite this, the maximum available promoter length for NsMBP10 was about 3.3 kb. We found that the differences in types and numbers of predicted TF binding sites between FUL1 and FUL2 were comparable to the differences between MBP10 and MBP20 (Table S4 in Supplementary Data Sheet S1). Nonetheless we did find some differences that may underlie observed differences in expression between paralogs. Some of these differences were presence/absence of binding sites for a particular TF, and some were in the number and distribution of sites. Putative binding sites for AUXIN RESPONSE FACTORS (ARF) were absent from the tomato FUL2 promoter while they were present in the promoters of all other euFUL genes in all species examined. Only FUL2 in tomato, FUL1 in potato, and MBP10 in woodland tobacco contained binding sites for STOREKEEPER (STK). ETHYLENE INSENSITIVE 3 (EIN3) has three sites in tomato FUL1 and five in tomato FUL2, but the distribution of the sites differs. In FUL1, there are no sites within 2 kb of the coding sequence, and three within 5 kb, whereas in FUL2 there is one site in the 2 kb region and four in the full 5 kb region. In woodland tobacco, there are three EIN3 sites in FUL1, all of which are within the 2 kb region, and only one in FUL2, which is located between 2 and 5 kb. These types of differences may underlie observed differences in expression.
Discussion
Solanaceae euFUL Gene Tree Shows the History of Duplications in This Lineage
In Solanaceae, there has been a major shift to fleshy fruit in the Solanoideae (Knapp, 2002). However, we do not know the molecular basis of this economically and ecologically important evolutionary event. FUL negatively regulates lignification in the dehiscence zone in the dry silique of A. thaliana, and functions in cauline leaf development, the transition to flowering and determinacy (Spence et al., 1996; Gu et al., 1998; Liljegren et al., 2000, 2004; Rajani and Sundaresan, 2001; Melzer et al., 2008). Studies of FUL ortholog function across the angiosperms have shown that it is labile, and orthologs have acquired diverse roles over evolutionary time. VEGETATIVE 1 (VEG1), an ortholog of FUL in pea (Pisum sativum), is involved in secondary inflorescence meristem identity (Berbel et al., 2012). AGAMOUS-like 79 (AGL79), the A. thaliana euFULII paralog of FUL, is mainly expressed in the root and has functions in lateral root development and may also play a role function in leaf shape, leaf number, branching, and time to flowering (Gao et al., 2018). However, the overexpression of an AGL79 ortholog from snapdragon (Antirrhinum majus) in A. thaliana resulted in indehiscent siliques, suggesting a role more similar to A. thaliana FUL (Müller et al., 2001). Evidence suggests that in tomato, one of the AGL79 orthologs, MBP20, plays a role in leaf development (Burko et al., 2013). VERNALIZATION 1 (VRN1) genes, which are FUL-like orthologs in grass species such as wheat (Triticum spp.) and barley (Hordeum vulgare), function in the vernalization response (Preston and Kellogg, 2008). Evidence to date, therefore, suggests that euFUL function is labile, and has changed substantially in different plant lineages during the course of angiosperm evolution. Thus it is not surprising to find a change in function of euFUL orthologs in Solanaceae.
There is evidence to suggest that Solanaceae euFUL orthologs play a role similar to that of A. thaliana FUL in the development of dry dehiscent fruits (Smykal et al., 2007). However, studies suggest that in the fleshy fruit of Solanoideae, FUL orthologs play roles in pigmentation as well as ethylene response, cell wall modification, glutamic acid degradation, volatile production, and pericarp and cuticle thickness (Bemer et al., 2012; Shima et al., 2014; Wang et al., 2014). To determine if we could identify changes in euFUL sequences or selection that might shed light on this change in function, we analyzed euFUL gene evolution in Solanaceae.
We performed a maximum likelihood phylogenetic analysis (Garli v2.1) (Bazinet et al., 2014) on a data set that consisted of 106 Solanaceae members of the euFUL gene lineage (Litt and Irish, 2003; Shan et al., 2007), which we obtained through amplification and sequencing (37 sequences), generating transcriptome sequence data (29 sequences), or mining databases (40 sequences). As outgroup we used 10 euFUL genes from Convolvulaceae, the sister family to Solanaceae (Figure 2 and Supplementary Table S1) (Stefanoviæ et al., 2003). The resulting tree shows the two major clades of core-eudicot euFUL genes, the euFULI and euFULII lineages (Litt and Irish, 2003; Shan et al., 2007). Within each of these clades there is evidence of a Solanaceae-specific duplication, resulting in two subclades in each lineage. Within each subclade, the order of branches correlates well with the topology of the Solanaceae phylogeny (Olmstead et al., 2008; Särkinen et al., 2013); discrepancies at the genus level are likely due to the short length of some sequences and sequence divergence in some taxa. Each of the subclades includes orthologs from both fleshy- and dry-fruited species, indicating that the subclade duplications preceded the origin of fleshy fruit.
Although duplications in these genes are common (Litt and Irish, 2003; Preston and Kellogg, 2007; Pabón-Mora et al., 2013), we did not find significant evidence of taxon-specific duplications. We did, however, find two genes that did not fall into a specific subclade. A third Streptosolen gene grouped sister to the rest of the euFULI clade (76% identity among the three Streptosolen genes), potentially the result of a taxon-specific duplication followed by sequence divergence. In addition, a Schizanthus gene grouped sister to the euFULII clade (77% pairwise identity with Schizanthus MBP20). This may also be a divergent genus-specific paralog, but since Schizanthus is one of the earliest diverging genera (Olmstead et al., 2008; Särkinen et al., 2013), it is also possible this gene might be a remaining paralog from the reported whole genome duplication/triplication that occurred early in Solanaceae diversification (Blanc and Wolfe, 2004; Schlueter et al., 2004; Song et al., 2012; The Tomato Genome Consortium, 2012; Albert and Chang, 2014; Bombarely et al., 2016). Examination of sequences showed that these are not likely to be splice isoforms. We also found potential evidence of loss – not every Solanaceae species we studied had a copy of each euFUL gene. We did not, for example, find FUL2 genes in Iochroma, Fabiana, Solandra, Juanulloa, Schizanthus, or Goetzia, even though these all had genes in the FUL1 clade (see Table S1 in Supplementary Data Sheet S1 for a complete list). However, although this may represent paralog loss, it is possible we did not recover all gene copies due to PCR primer mismatches, low expression levels, or the absence of transcript in the sampled tissue.
In addition to the major shift to fleshy fruit in the Solanoideae subfamily, fleshy fruits have independently evolved in Cestrum and Duboisia, and there has also been a reversal to a dry fruit in Datura (Knapp, 2002). Our analysis does not include genes from Duboisia, but the euFUL genes from Cestrum and Datura grouped in positions in the tree that were expected based on their phylogenetic position, and did not show any notable differences in sequence from the euFUL genes of their close relatives.
euFULI and euFULII Clade Duplicates Have Experienced Different Levels of Purifying Selection
We compared dN/dS ratios between and among Solanaceae euFULI and euFULII lineages, as well as between sequences before and after the transition to fleshy fruit, to investigate if any changes in selection might be correlated with sequence diversification. All ω values from our analyses are closer to 0 than to 1 (Table 1 and Table S3 in Supplementary Data Sheet S1), which indicates that all euFUL gene clades are under strong purifying selection (Yang, 1997; Yang and Nielsen, 2000; Torgerson et al., 2002; Almeida and Desalle, 2009). Studies suggest that this is the norm for most protein coding genes, and that under such stringent evolutionary constraints, slight differences in evolutionary rates may result in functional diversification (Yu et al., 2017). Our data show a weakening of purifying selection in FUL1 genes relative to FUL2 genes (ω = 0.17 vs. 0.11, p < 0.0005) and in MBP10 genes relative to MBP20 genes (ω = 0.19 vs. 0.15, p < 0.01). Immediately after the euFULI duplication, the FUL1 and FUL2 lineage genes would have been fully redundant, which might have allowed the reduction in purifying selection on the FUL1 genes resulting in potential functional divergence. Similarly, the duplication that resulted in the two euFULII gene clades would have resulted in redundancy in the MBP10 and MBP20 lineages, possibly allowing the more rapid diversification of MBP10 genes.
Although studies indicate that the euFULI genes of tomato have novel functions compared to those in dry fruit (Gu et al., 1998; Smykal et al., 2007; Bemer et al., 2012; Shima et al., 2014; Wang et al., 2014), it remains unclear whether the new functions are the result of changes in coding sequences, regulatory regions, or downstream gene targets. Our analysis shows that euFUL genes in both dry- and fleshy-fruited species are evolving at similar rates (Table S3 in Supplementary Data Sheet S1). This suggests conservation of the coding sequences in both fleshy- and dry-fruited species despite the central roles in the development of these distinct fruit morphologies.
Sixty-four of the sequences in our analysis were from fleshy-fruited species whereas only 42 were from dry-fruited species. Although, we had broad representation across the dry grade, it is possible with additional representation from dry fruited species, more evolutionary patterns would be revealed (Anisimova et al., 2001; Domazet-Loso, 2003; Nielsen et al., 2005).
FUL1 and MBP10 Proteins Show Amino Acid Changes in Conserved Functional Domains
An analysis of selection across an entire sequence may indicate different types of selection for the whole gene, but this overlooks the fact that key residues may be undergoing rapid evolution that may result in functional changes (Ota and Nei, 1994; Nei et al., 1997; Yang and Bielawski, 2000; Piontkivska et al., 2002; Martinez-Castilla and Alvarez-Buylla, 2003; Jeffares et al., 2006). Other empirical studies have further described functional changes due to a change in a single amino acid residue (Ingram, 1957; Hanzawa et al., 2005; Hichri et al., 2011; Zhao et al., 2012; Fourquin et al., 2013; Dai et al., 2016; Sakuma et al., 2017) specifically associated with changes in polarity (Schröfelbauer et al., 2004; Hoekstra et al., 2006) or conformation (Aseev et al., 2012). Studies in A. thaliana, show that a single amino acid mutation in GLABRA1 (GL1) results in the inhibition of trichome formation (Dai et al., 2016) and a change of a single residue is sufficient to convert the function of TERMINAL FLOWER 1 (TFL1), which inhibits flower formation, to that of the closely related FLOWERING LOCUS T (FT), which promotes flowering (Hanzawa et al., 2005). Three-dimensional modeling has also shown that a single amino acid change in a highly conserved domains may lead to changes in protein–protein interactions (Teng et al., 2009; David et al., 2012; Li et al., 2014). We searched for individual sites in the predicted amino acid sequences that showed evidence of positive selection within the gene groups that, although under purifying selection, were found to have statistically significantly accelerated evolutionary rates (i.e., the FUL1 and MBP10 clades) to determine if any amino acid changes at these sites had the potential to result in a change in protein function.
Our findings show that more residues are rapidly changing in the K domain compared to the M and I domains (Figure S4 in Supplementary Data Sheet S1). The K domain is predicted to have an α-helix structure that facilitates protein–protein interactions (Figure S5 in Supplementary Data Sheet S1) (Yang et al., 2003a,b; Kaufmann et al., 2005; Immink et al., 2010). The α-helix structure depends on conserved hydrophobic residues spaced through the domain (Eisenberg et al., 1982). Therefore, changes to protein residues that alter charge and/or conformation in this region can lead to changes in such interactions. Most of the rapidly evolving sites did not show an amino acid change specifically associated with the shift to fleshy fruit, but rather showed changes and reversals over the course of gene evolution. Interestingly, in the FUL1 proteins, we found one site in the K domain, corresponding to the 153rd residue in the tomato protein (Slugina et al., 2018), at which 11 out of 15 sequences from dry-fruited species have a negatively charged glutamate (E) residue. In comparison, 100% of the fleshy clade contains a non-polar residue: valine (V) (13 species) or methionine (1 species). However, since the remaining four FUL1 sequences from dry-fruited species have non-polar glutamine (Q) or V at this site, the change from charged to non-polar is not associated with the shift to fleshy fruit. In addition, a PROVEAN analysis predicted the changes at this site to be neutral with regards to function.
Two other sites in the FUL1 K domain show changes that are predicted to have functionally deleterious consequences according to our PROVEAN analysis (Choi, 2012; Choi et al., 2012; Choi and Chan, 2015). These include a charged histidine (H) to a non-polar glutamine/asparagine (Q/N) transition at the 95th residue and a charged lysine (K) to non-polar glutamine/threonine (Q/T) transition at the 157th residue (Figure S5 in Supplementary Data Sheet S1). Polar residues are important for protein–protein interactions of the K domain α-helix (Sheinerman et al., 2000; Curran and Engelman, 2003; Ma et al., 2003; Zhou et al., 2018) and changes might disrupt interactions with other proteins (Liu et al., 2014). However, since these changes are not correlated with the fruit type, it seems unlikely that any alteration to protein function affects fruit morphology. It is also plausible that any negative effect at these sites is masked by the FUL2 paralog, which is likely to be functionally redundant (Bemer et al., 2012; Wang et al., 2014). This is consistent with FUL1 evolving relatively faster (Table 1 and Table S3 in Supplementary Data Sheet S1), thus enabling divergence compared to FUL2, which appears to be more highly functionally conserved based on stricter sequence conservation.
None of the sites undergoing positive change in the K domain of MBP10 showed a change in charge, suggesting these changes are not likely to affect protein function. We also observed residues in the M domain that are under diversifying selection in both the FUL1 and MBP10 clades. These residues are located not in the α-helix region that directly binds to DNA, but in the β-sheet region of the MADS domain (Figure S4 in Supplementary Data Sheet S1) (Immink et al., 2010). β-sheets are important for protein arrangement in three dimensional space. Therefore, any changes in this region might change protein conformation, influencing DNA binding of the α-helix as well as the ability of the euFUL proteins to form higher order complexes (Pellegrini et al., 1995). However, these shifts were reversible, with no phylogenetic pattern or change in charge, and there was no correlation with the fruit type. Therefore it is unlikely that these shifts have significant functional impact.
A previous report that investigated the evolution of MADS-box genes in A. thaliana also found rapidly evolving sites in the M and K domains of Type II MADS-box proteins, which might have been involved in the functional diversification of this group, but did not report changes in the I domain (Martinez-Castilla and Alvarez-Buylla, 2003). Residues in this domain that are directly involved in forming an α-helix structure are expected to be highly conserved, whereas the remaining residues may not be under such constraints (Yang et al., 2003a,b; Kaufmann et al., 2005). We found residues in the conserved region of the I domain that are undergoing diversifying selection in both FUL1 and MBP10 clades. Of these, one site in FUL1 and three sites in MBP10 had undergone changes in charge but none were predicted to negatively affect the function (Figures S4, S5 in Supplementary Data Sheet S1). In addition, as with the sites in the M and K domains, none of these was correlated with the Solanaceae phylogeny or changes in fruit morphology. It has been reported that higher rates of substitution in lineages that show weakened purifying selection or even diversifying selection may be occurring at residues of minimal functional importance (Jacobsen et al., 2016). This might explain the apparent ease of reversibility and lack of phylogenetic signal among the rapidly changing sites we observed.
The MBP10 and MBP20 Clades Are the Result of a Tandem Duplication Event
The FUL1 and FUL2 genes of tomato are located on different chromosomes (6 and 3, respectively), which is consistent with the proposed Solanaceae whole genome duplication (Blanc and Wolfe, 2004; Schlueter et al., 2004; Song et al., 2012) or triplication (The Tomato Genome Consortium, 2012; Albert and Chang, 2014; Vanneste et al., 2014; Bombarely et al., 2016) followed by loss of one paralog. The lack of a FUL2 ortholog in our dataset from Goetzia or Schizanthus (Figure 2), the two earliest diverging genera that we included in our analyses, raises the possibility that the FUL1/FUL2 clades originated via a duplication that occurred after the diversification of these genera, and not as a result of a whole genome event that preceded the diversification of the family. Whole genome sequences from multiple early diverging lineages will be needed to determine the timing and nature of these early events.
We did not recover an MBP10 ortholog from any of the genera that diverged prior to Brunfelsia (Figure 2 and Figure S2 in Supplementary Data Sheet S1). Our investigation revealed that in tomato, both MBP10 and MBP20 are located on chromosome 2, about 14.3 million base pairs apart. The 1 million base-pair region surrounding each gene shows synteny, but the order of the homologous regions is reversed (Figure 3). Together, this suggests that the MBP10/MBP20 clades are the result of a tandem duplication accompanied by an inversion (Purugganan et al., 1995; Vision et al., 2000; Achaz et al., 2001; Prince and Pickett, 2002). Supporting this, a previous report that investigated genomic duplication events in tomato also found evidence for large-scale intra-chromosomal duplications in chromosome 2 (Song et al., 2012). Although the authors suggest this event was concurrent with a whole genome duplication at the origin of the family, they give a large window, 36–82 million years ago (MYA), for the timing of this event. The stem age of the family is predicted to be approximately 49 MYA (Särkinen et al., 2013), indicating that this duplication might have happened later in Solanaceae diversification. Our data suggest that this duplication event is independent of the reported whole genome events, occurring prior to the diversification of the Brunfelsia clade but after the event that produced the FUL1 and FUL2 clades (Figure 2 and Figure S2 in Supplementary Data Sheet S1).
The expected topology for the euFULII clade, based on a duplication prior to the divergence of the Brunfelsia clade, would be a paraphyletic grade of pre-duplication euFULII genes, from species that diversified prior to Brunfelsia, and nested MBP10 and MBP20 clades that would include post-duplication genes from all species that diversified subsequent to the duplication. However, in our tree, the pre-duplication genes do not form such a basal grade (Figure 2). Rather, they form a clade with the post-duplication MBP20 genes. The results of our PAML analyses indicate that the MBP20-clade genes show less sequence divergence than MBP10 genes; this higher degree of similarity among pre-duplication sequences and post-duplication MBP20 genes may underlie their grouping into one clade (Pegueroles et al., 2013).
Our results indicate that the euFULII duplication occurred prior to the origin of the clade containing Brunfelsia. We would therefore expect to find both an MBP10 and an MBP20 in all species of that clade. However, we did not find an MBP10 ortholog in members of this clade other than Brunfelsia. MBP10 appears to have been lost from the genome of Petunia, based on analyses of multiple fully sequenced genomes (Bombarely et al., 2016), and potentially from Plowmania and Fabiana. We were able to recover MBP10 orthologs from Nicotiana and most other later-diverging genera. However, our analysis includes fewer species from the dry grade of the Solanaceae phylogeny than the fleshy-fruited Solanoideae clade (17 out of 45) and even fewer species that diverged prior to Brunfelsia (7). In the MBP10 clade in particular, our analysis includes 13 orthologs from species in the fleshy-fruited clade but just four from the dry-fruited species, and our analysis only includes sequence data from four genera that diverged prior to the origin of the Brunfelsia clade (Streptosolen, Cestrum, Goetzia, Schizanthus) (Figures 1, 2). Thus there may be genera that originated prior to Brunfelsia that contain MBP10 that our sampling did not include. Floral and fruit transcriptomes, which provided MBP10 orthologs from later diverging species, yielded no MBP10 sequences from Cestrum and Schizanthus; nonetheless, whole genome sequences of early diverging species are needed to determine the timing of the MBP10/MBP20 duplication.
euFULII Expression Divergence May Be Associated With Cis-Regulatory Re-coupling
Our analysis of Solanaceae euFUL homologs show that FUL1 and FUL2 are broadly expressed in leaves, flowers, and fruit (Figure 4 and Figure S1 in Supplementary Data Sheet S1). This overall similarity in expression may indicate a conservation of cis-regulatory elements in gene copies following duplication (Haberer et al., 2004). Supporting this, our investigation into the number of putative TF binding sites in the promoter region of euFULI homologs did not reveal statistically significant differences (Table S4 in Supplementary Data Sheet S1). In tomato fruit development, FUL1 expression increases with time, whereas FUL2 expression reaches a maximum at early stages and then decreases over later stages (Figure 4 and Figure S1 in Supplementary Data Sheet S1). This variation in expression associated with the developmental stages might be due to changes in cis-elements as a result of the accumulation of random mutations over time (Force et al., 1999; Haberer et al., 2004).
Our analysis did find differences in the number and location of predicted binding sites for specific TFs or families, for instance for ARF, STK, and EIN3 TFs, which may account for the types of differences in expression seen between euFUL paralogs. The 5 kb region upstream of the FUL1 transcription start site in tomato contains three putative ARF binding sites but the corresponding region of FUL2 in tomato contains no such motifs (Table S4 in Supplementary Data Sheet S1). ARF TFs, important in tomato fruit development, are activated in response to auxin and may upregulate or repress downstream genes (de Jong et al., 2010, 2015; Liu et al., 2018); the absence of binding sites from the FUL2 promoter is the type of factor that might underlie differences in expression observed between FUL1 and FUL2. Predicted STK binding sites are only found in the promoters of potato FUL1, tomato FUL2 and woodland tobacco MBP10. STK and STK-like proteins appear to function in storage protein synthesis, glucose reception, and vegetative and reproductive development (Zourelidou et al., 2002; Curaba et al., 2003; Bömer et al., 2011; Chung et al., 2016; Nietzsche et al., 2017). Meanwhile, the 2 kb upstream region of FUL2 contains a putative site for EIN3. This protein is involved in the development of tomato in response to ripening-associated ethylene production (Tieman et al., 2001). No such motifs are found in the corresponding region of FUL1. In contrast, the 2–5 kb region in FUL2 contains four putative sites for EIN3 while the corresponding region in FUL1 contains three such sites (Table S4 in Supplementary Data Sheet S1). Such variation in number and location of TF binding sites has been shown to be associated with the temporal differences in gene expression (Lebrecht et al., 2005; Liu et al., 2006; Giorgetti et al., 2010; Guertin and Lis, 2010; White et al., 2013; Ezer et al., 2014; Levo et al., 2015; Payne and Wagner, 2015).
Whereas the euFULI members largely overlap in spatial expression with some variation associated with developmental stages, the euFULII homologs show less consistent spatial expression patterns. Only MBP20 is expressed in tomato roots and potato fruit while only MBP10 is expressed in potato tubers (Figure 4). However, these “on” or “off” expression patterns cannot be explained by the presence or absence of any putative TF binding sites (Table S4 in Supplementary Data Sheet S1). These two paralogs, which appear to be the result of a tandem duplication and inversion, are located approximately 14.3 Mbp apart (Figure 3) on chromosome 2. Although gene clusters resulting from tandem duplications are often coexpressed, this is not the case when there are large physical distances between the genes (Lercher et al., 2003). An investigation into the expression of human transgenes in mice also found changes in expression as a consequence of an inversion, possibly through disrupting enhancer activity or changes to chromatin structure (Tanimoto et al., 1999; Vogel et al., 2009; Puig et al., 2015). Chromosomal rearrangements such as inversions may also result in novel connections between coding regions and other promoters or long distance regulatory motifs while disrupting the original regulatory mechanisms (Kmita et al., 2000; Lupiáñez et al., 2015). This sort of re-coupling of one of the two paralogs might lead to the types of contrasting expression patterns observed for MBP10 and MBP20. However, the expression patterns are not consistent across species (Figure 4 and Figure S1 in Supplementary Data Sheet S1) and this might be due to additional changes following the inversion (Cosner et al., 1997; Lupski, 1998; Haberle et al., 2008; Chiang et al., 2012). An in-depth analysis of the entire loci and their genomic environment for all paralogs in multiple species would be necessary to determine if the tandem duplication and inversion are associated with changes in proximity to heterochromatin, additional rearrangements, or other phenomena that might have altered gene expression.
MBP10 Shows Signs of Pseudogenization
The first intron of some MADS-box genes contains cis-elements important for the regulation of expression (Gazzani et al., 2003; Michaels et al., 2003; Schauer et al., 2009). Studies have found that deletions in the first intron of a FUL-like gene in Aegilops tauschii alters expression and results in the loss of the vernalization requirement (Fu et al., 2005; Takumi et al., 2011). Consistent with this, the first introns of angiosperm euFUL orthologs are generally in the range of 1–10 kb (Table 2) (Takumi et al., 2011). In contrast, tomato MBP10 has a short first intron of 80 bp. We compared the putative TF binding sites in the first introns of MBP10 and MBP20 in tomato to characterize potential loss of such sites, which might suggest reduced gene regulation. The first intron of MBP10 is predicted to have no TF binding sites, while the first intron of MBP20 is predicted to contain 88 TF binding sites (Figure S3 in Supplementary Data Sheet S1). These included binding sites for MYB, HSF, Dof, WRKY, and MADS-box TFs. Specific TFs predicted to bind to these sites include MYB2 and C1 (MYB), which play roles in anthocyanin accumulation and lignin biosynthesis, PBF (Dof), which plays a role in endosperm storage protein accumulation, and SPF1 (WRKY), thought to function in fruit ripening (Bovy et al., 2002; Fei et al., 2004; Hwang et al., 2004; Lee et al., 2010; Xu et al., 2014; Jun et al., 2015). A similar pattern was found in analysis of the first intron of MBP10 in Nicotiana obtusifolia, which is 110 bp (Table 2). This analysis found three putative TF binding sites for MYB2 and one for PBF. By contrast, the first intron of N. obtusifolia MBP20 is predicted to have 133 TF binding sites and include a repertoire similar to those found for tomato MBP20. To determine whether the difference in TF binding site number between the paralogs represented a gain of sites in the MBP20 genes or a loss in the MBP10 genes, we also searched for TF binding sites in the first intron of AGL79, the single euFULII ortholog in A. thaliana (Gao et al., 2018). We found that it contains 49 predicted TF binding sites for five different TFs in four families: MYB (MYB2, GAMYB), HSF (HSF1), WRKY (SPF1), and GT-box (GT-1). Although this number is substantially smaller than the number of sites predicted in the first introns of the Solanaceae MBP20 genes, the results suggest that there has been a loss of TF binding sites in MBP10.

Table 2. Approximate lengths of the first introns of several FUL homologs.
Core-eudicot euFUL and basal-eudicot FUL-like genes frequently have broad expression patterns and are generally expressed in fruit (Ferrándiz et al., 2000; Shchennikova et al., 2004; Kim et al., 2005; Hileman et al., 2006; Bemer et al., 2012; Pabón-Mora et al., 2012, 2013; Scorza et al., 2017). Therefore, the absence or extremely weak expression of MBP10 in fruits of all species, and its weak expression in most organs of tomato and potato is notable (Figure 4 and Figure S1 in Supplementary Data Sheet S1). This relatively weak expression may at least in part be due to the loss of TF binding sites in the first intron and suggests a potentially reduced role in regulating fruit-related developmental processes. Importantly, the loss of putative TF binding sites and low expression, combined with the faster evolutionary rate, suggest MBP10 might be in the process of becoming a pseudogene. Further support for this hypothesis comes from an examination of the MBP10 sequences, which suggests that at least two of the sequences in our study (from N. sylvestris and Dunalia spinosa) show a frameshift that would result in an premature stop codon.
Conclusion
Our results suggest that there was a weakening in purifying selection following the euFUL gene duplications in Solanaceae, resulting in coding sequence diversification in FUL1 and MBP10 clades relative to FUL2 and MBP20. Expression of the euFULI genes is broad, while the euFULII genes have contrasting patterns at the organ level, potentially resulting from cis-regulatory changes associated with the inversion event. We also found evidence to suggest that the MBP10 clade is becoming a pseudogene. Although at least some clades of Solanaceae euFUL genes took on new functions associated with the development of fleshy fruit we did not find any amino acid shifts that were correlated with the change in fruit type. It is also possible that the novel functions are a consequence of downstream changes, perhaps as the result of changes in binding partners or targets. Therefore, the mechanism underlying the shift in euFUL function from dry to fleshy fruit in Solanaceae awaits additional analyses.
Author Contributions
AL designed and supervised research, and assisted in writing the paper. DM contributed to the design of the study, generated Cestrum diurnum, C. nocturnum, and Schizanthus grahamii transcriptome libraries, retrieved sequences from PCR-based methods and database mining, analyzed the data, and wrote the paper. CE assisted with PAML analysis, contributed suggestions for analyses, and made suggestions on the paper. AR generated Dunalia spinosa, Fabiana viscosa, Grabowskia glauca, and Salpiglossis sinuata transcriptome libraries, contributed suggestions for analyses, contributed in recording the associated protocols, and commented on the paper. JM retrieved sequences from PCR-based methods. MS generated the Nicotiana obtusifolia transcriptome libraries and additional sequences using PCR-based methods. NP-M generated Brunfelsia australis and Streptosolen jamesonii transcriptome libraries, contributed suggestions for analyses, and made suggestions on this paper.
Funding
This research was funded by the National Science Foundation (IOS 1456109). DM was partly supported by UCR Graduate Assistance in Areas of National Need (GAANN) and Graduate Research Mentoring Program (GRMP) fellowships. NP-M acknowledges grants by COLCIENCIAS (111565842812), the Convocatoria Programaticas 2017–2018, and the Estrategia de Sostenibilidad 2018–2019, in the Universidad de Antioquia given to the group Evo-Devo en Plantas.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful for the help with transcriptome library compilation and analyses by Glenn Hicks, John Weger, Holly Eckelhoefer, and Clay Clark at the UCR IIGB Core Facility, the advice on the gene tree generation by John Heraty, feedback by Elizabeth McCarthy, Jacob Landis, and Patricia Springer and Jaimie Van Norman lab members, and greenhouse and lab support by Arman Baghaei, Alan Le, and Victor Herrera.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00043/full#supplementary-material
Footnotes
- ^ ftp://ftp.solgenomics.net/manuscripts/Litt_2018
- ^ https://www.ncbi.nlm.nih.gov/blast
- ^ https://db.cngb.org/blast4onekp/
- ^ https://sites.google.com/a/ualberta.ca/onekp
- ^ http://abacus.gene.ucl.ac.uk/software/paml.html
- ^ http://datamonkey.org/meme
- ^ http://provean.jcvi.org
- ^ http://www.sbg.bio.ic.ac.uk/~phyre2
- ^ https://www.geneious.com
- ^ http://alggen.lsi.upc.es/recerca/frame-recerca.html
- ^ http://bar.utoronto.ca
- ^ http://benthgenome.qut.edu.au/
- ^ http://plantpan2.itps.ncku.edu.tw
- ^ http://datamonkey.org/meme
References
Achaz, G., Netter, P., and Coissac, E. (2001). Study of intrachromosomal duplications among the eukaryote genomes. Mol. Biol. Evol. 18, 2280–2288. doi: 10.1093/oxfordjournals.molbev.a003774
Albert, V. A., and Chang, T.-H. (2014). Evolution of a hot genome. Proc. Natl. Acad. Sci. U.S.A. 111, 5069–5070. doi: 10.1073/pnas.1402378111
Almeida, F. C., and Desalle, R. (2009). Orthology, function and evolution of accessory gland proteins in the Drosophila repleta group. Genetics 181, 235–245. doi: 10.1534/genetics.108.096263
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anisimova, M., Bielawski, J. P., and Yang, Z. (2001). Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol. Biol. Evol. 18, 1585–1592. doi: 10.1093/oxfordjournals.molbev.a003945
Aseev, L. V., Chugunov, A. O., Efremov, R. G., and Boni, I. V. (2012). A single missense mutation in a coiled-coil domain of Escherichia coli ribosomal protein S2 confers a thermosensitive phenotype that can be suppressed by ribosomal protein S1. J. Bacteriol. 195, 95–104. doi: 10.1128/JB.01305-12
Bazinet, A. L., Zwickl, D. J., and Cummings, M. P. (2014). A gateway for phylogenetic analysis powered by grid computing featuring GARLI 2.0. Syst. Biol. 63, 812–818. doi: 10.1093/sysbio/syu031
Bemer, M., Karlova, R., Ballester, A. R., Tikunov, Y. M., Bovy, A. G., Wolters-Arts, M., et al. (2012). The tomato FRUITFULL homologs TDR4/FUL1 and MBP7/FUL2 regulate ethylene-independent aspects of fruit ripening. Plant Cell 24, 4437–4451. doi: 10.1105/tpc.112.103283
Berbel, A., Ferrándiz, C., Hecht, V., Dalmais, M., Lund, O. S., Sussmilch, F. C., et al. (2012). VEGETATIVE1 is essential for development of the compound inflorescence in pea. Nat. Commun. 3:797. doi: 10.1038/ncomms1801
Blanc, G., and Wolfe, K. H. (2004). Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16, 1667–1678. doi: 10.1105/tpc.021345
BLAST® Command Line Applications User Manual (2008). BLAST® Command Line Applications User Manual. Bethesda, MD: National Center for Biotechnology Information.
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolmgren, K., and Eriksson, O. (2010). Seed mass and the evolution of fleshy fruits in angiosperms. Oikos 119, 707–718. doi: 10.1111/j.1600-0706.2009.17944.x
Bombarely, A., Moser, M., Amrad, A., Bao, M., Bapaume, L., Barry, C. S., et al. (2016). Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants 2:16074. doi: 10.1038/nplants.2016.74
Bömer, M., Uhrig, J., Jach, G., and Müller, K. (2011). Increased vegetative development and sturdiness of storekeeper-transgenic tobacco. Open Life Sci. 6, 342–351. doi: 10.2478/s11535-011-0009-9
Bovy, A., de Vos, R., Kemper, M., Schijlen, E., Almenar Pertejo, M., Muir, S., et al. (2002). High-flavonol tomatoes resulting from the heterologous expression of the maize transcription factor genes LC and C1. Plant Cell 14, 2509–2526. doi: 10.1105/tpc.004218
Burko, Y., Shleizer-Burko, S., Yanai, O., Shwartz, I., Zelnik, I. D., Jacob-Hirsch, J., et al. (2013). A role for APETALA1/fruitfull transcription factors in tomato leaf development. Plant Cell 25, 2070–2083. doi: 10.1105/tpc.113.113035
Chang, W.-C., Lee, T.-Y., Huang, H.-D., Huang, H.-Y., and Pan, R.-L. (2008). PlantPAN: plant promoter analysis navigator, for identifying combinatorial cis-regulatory elements with distance constraint in plant gene groups. BMC Genomics 9:561. doi: 10.1186/1471-2164-9-561
Chiang, C., Jacobsen, J. C., Ernst, C., Hanscom, C., Heilbut, A., Blumenthal, I., et al. (2012). Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat. Genet. 44, 390–397, S1. doi: 10.1038/ng.2202
Cho, S., Jang, S., Chae, S., Chung, K. M., Moon, Y. H., An, G., et al. (1999). Analysis of the C-terminal region of Arabidopsis thaliana APETALA1 as a transcription activation domain. Plant Mol. Biol. 40, 419–429. doi: 10.1023/A:100627312
Choi, Y. (2012). “A fast computation of pairwise sequence alignment scores between a protein and a set of single-locus variants of another protein,” in Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine-BCB, Orlando, FL. doi: 10.1145/2382936.2382989
Choi, Y., and Chan, A. P. (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747. doi: 10.1093/bioinformatics/btv195
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS One 7:e46688. doi: 10.1371/journal.pone.0046688
Chung, M.-S., Lee, S., Min, J.-H., Huang, P., Ju, H.-W., and Kim, C. S. (2016). Regulation of Arabidopsis thaliana plasma membrane glucose-responsive regulator (AtPGR) expression by A. thaliana storekeeper-like transcription factor, AtSTKL, modulates glucose response in Arabidopsis. Plant Physiol. Biochem. 104, 155–164. doi: 10.1016/j.plaphy.2016.03.029
Cosner, M. E., Jansen, R. K., Palmer, J. D., and Downie, S. R. (1997). The highly rearranged chloroplast genome of Trachelium caeruleum (Campanulaceae): multiple inversions, inverted repeat expansion and contraction, transposition, insertions/deletions, and several repeat families. Curr. Genet. 31, 419–429. doi: 10.1007/s002940050225
Curaba, J., Herzog, M., and Vachon, G. (2003). GeBP, the first member of a new gene family in Arabidopsis, encodes a nuclear protein with DNA-binding activity and is regulated by KNAT1. Plant J. 33, 305–317. doi: 10.1046/j.1365-313X.2003.01622.x
Curran, A. R., and Engelman, D. M. (2003). Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr. Opin. Struct. Biol. 13, 412–417. doi: 10.1016/S0959-440X(03)00102-7
Dai, X., Zhou, L., Zhang, W., Cai, L., Guo, H., Tian, H., et al. (2016). A single amino acid substitution in the R3 domain of GLABRA1 leads to inhibition of trichome formation in Arabidopsis without affecting its interaction with GLABRA3. Plant Cell Environ. 39, 897–907. doi: 10.1111/pce.12695
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
David, A., Razali, R., Wass, M. N., and Sternberg, M. J. E. (2012). Protein-protein interaction sites are hot spots for disease-associated nonsynonymous SNPs. Hum. Mutat. 33, 359–363. doi: 10.1002/humu.21656
de Jong, M., Wolters-Arts, M., García-Martínez, J. L., Mariani, C., and Vriezen, W. H. (2010). The Solanum lycopersicum AUXIN RESPONSE FACTOR 7 (SlARF7) mediates cross-talk between auxin and gibberellin signalling during tomato fruit set and development. J. Exp. Bot. 62, 617–626. doi: 10.1093/jxb/erq293
de Jong, M., Wolters-Arts, M., Schimmel, B. C. J., Stultiens, C. L. M., de Groot, P. F. M., Powers, S. J., et al. (2015). Solanum lycopersicum AUXIN RESPONSE FACTOR 9 regulates cell division activity during early tomato fruit development. J. Exp. Bot. 66, 3405–3416. doi: 10.1093/jxb/erv152
Domazet-Loso, T. (2003). An evolutionary analysis of orphan genes in Drosophila. Genome Res. 13, 2213–2219. doi: 10.1101/gr.1311003
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Eisenberg, D., Weiss, R. M., and Terwilliger, T. C. (1982). The helical hydrophobic moment: a measure of the amphiphilicity of a helix. Nature 299, 371–374. doi: 10.1038/299371a0
Ezer, D., Zabet, N. R., and Adryan, B. (2014). Homotypic clusters of transcription factor binding sites: a model system for understanding the physical mechanics of gene expression. Comput. Struct. Biotechnol. J. 10, 63–69. doi: 10.1016/j.csbj.2014.07.005
Farré, D., Roset, R., Huerta, M., Adsuara, J. E., Roselló, L., Albà, M. M., et al. (2003). Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 31, 3651–3653. doi: 10.1093/nar/gkg605
Fei, Z., Tang, X., Alba, R. M., White, J. A., Ronning, C. M., Martin, G. B., et al. (2004). Comprehensive EST analysis of tomato and comparative genomics of fruit ripening. Plant J. 40, 47–59. doi: 10.1111/j.1365-313X.2004.02188.x
Ferrándiz, C., Gu, Q., Martienssen, R., and Yanofsky, M. F. (2000). Redundant regulation of meristem identity and plant architecture by FRUITFULL, APETALA1 and CAULIFLOWER. Development 127, 725–734.
Force, A., Lynch, M., Pickett, F. B., Amores, A., Yan, Y. L., and Postlethwait, J. (1999). Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531–1545.
Fourquin, C., del Cerro, C., Victoria, F. C., Vialette-Guiraud, A., de Oliveira, A. C., and Ferrándiz, C. (2013). A change in SHATTERPROOF protein lies at the origin of a fruit morphological novelty and a new strategy for seed dispersal in Medicago genus. Plant Physiol. 162, 907–917. doi: 10.1104/pp.113.217570
Fu, D., Szücs, P., Yan, L., Helguera, M., Skinner, J. S., von Zitzewitz, J., et al. (2005). Large deletions within the first intron in VRN-1 are associated with spring growth habit in barley and wheat. Mol. Genet. Genomics 274, 442–443. doi: 10.1007/s00438-005-0045-0
Gao, R., Wang, Y., Gruber, M. Y., and Hannoufa, A. (2018). miR156/SPL10 modulates lateral root development, branching and leaf morphology in Arabidopsis by silencing AGAMOUS-LIKE 79. Front. Plant Sci. 8:2226. doi: 10.3389/fpls.2017.02226
Gazzani, S., Gendall, A. R., Lister, C., and Dean, C. (2003). Analysis of the molecular basis of flowering time variation in Arabidopsis accessions. Plant Physiol. 132, 1107–1114. doi: 10.1104/pp.103.021212
Gillaspy, G., Ben-David, H., and Gruissem, W. (1993). Fruits: a developmental perspective. Plant Cell 5, 1439–1451. doi: 10.1105/tpc.5.10.1439
Giorgetti, L., Siggers, T., Tiana, G., Caprara, G., Notarbartolo, S., Corona, T., et al. (2010). Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol. Cell 37, 418–428. doi: 10.1016/j.molcel.2010.01.016
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gu, Q., Ferrándiz, C., Yanofsky, M. F., and Martienssen, R. (1998). The FRUITFULL MADS-box gene mediates cell differentiation during Arabidopsis fruit development. Development 125, 1509–1517.
Guertin, M. J., and Lis, J. T. (2010). Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet. 6:e1001114. doi: 10.1371/journal.pgen.1001114
Haberer, G., Hindemitt, T., Meyers, B. C., and Mayer, K. F. X. (2004). Transcriptional similarities, dissimilarities, and conservation of cis-elements in duplicated genes of Arabidopsis. Plant Physiol. 136, 3009–3022. doi: 10.1104/pp.104.046466
Haberle, R. C., Fourcade, H. M., Boore, J. L., and Jansen, R. K. (2008). Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA genes. J. Mol. Evol. 66, 350–361. doi: 10.1007/s00239-008-9086-4
Hanzawa, Y., Money, T., and Bradley, D. (2005). A single amino acid converts a repressor to an activator of flowering. Proc. Natl. Acad. Sci. U.S.A. 102, 7748–7753. doi: 10.1073/pnas.0500932102
Heijmans, K., Morel, P., and Vandenbussche, M. (2012). MADS-box genes and floral development: the dark side. J. Exp. Bot. 63, 5397–5404. doi: 10.1093/jxb/ers233
Hichri, I., Deluc, L., Barrieu, F., Bogs, J., Mahjoub, A., Regad, F., et al. (2011). A single amino acid change within the R2 domain of the VvMYB5b transcription factor modulates affinity for protein partners and target promoters selectivity. BMC Plant Biol. 11:117. doi: 10.1186/1471-2229-11-117
Hileman, L. C., Sundstrom, J. F., Litt, A., Chen, M., Shumba, T., and Irish, V. F. (2006). Molecular and phylogenetic analyses of the MADS-box gene family in tomato. Mol. Biol. Evol. 23, 2245–2258. doi: 10.1093/molbev/msl095
Hoekstra, H. E., Hirschmann, R. J., Bundey, R. A., Insel, P. A., and Crossland, J. P. (2006). A single amino acid mutation contributes to adaptive beach mouse color pattern. Science 313, 101–104. doi: 10.1126/science.1126121
Hulse-Kemp, A. M., Maheshwari, S., Stoffel, K., Hill, T. A., Jaffe, D., Williams, S. R., et al. (2018). Reference quality assembly of the 3.5-Gb genome of Capsicum annuum from a single linked-read library. Hortic. Res. 5:4. doi: 10.1038/s41438-017-0011-0
Hwang, Y.-S., Ciceri, P., Parsons, R. L., Moose, S. P., Schmidt, R. J., and Huang, N. (2004). The maize O2 and PBF proteins act additively to promote transcription from storage protein gene promoters in rice endosperm cells. Plant Cell Physiol. 45, 1509–1518. doi: 10.1093/pcp/pch173
Immink, R. G. H., Kaufmann, K., and Angenent, G. C. (2010). The “ABC” of MADS domain protein behaviour and interactions. Semin. Cell Dev. Biol. 21, 87–93. doi: 10.1016/j.semcdb.2009.10.004
Ingram, V. M. (1957). Gene mutations in human hæmoglobin: the chemical difference between normal and sickle cell hæmoglobin. Nature 180, 326–328. doi: 10.1038/180326a0
Jacobsen, M. W., da Fonseca, R. R., Bernatchez, L., and Hansen, M. M. (2016). Comparative analysis of complete mitochondrial genomes suggests that relaxed purifying selection is driving high nonsynonymous evolutionary rate of the NADH2 gene in whitefish (Coregonus ssp.). Mol. Phylogenet. Evol. 95, 161–170. doi: 10.1016/j.ympev.2015.11.008
Jeffares, D. C., Pain, A., Berry, A., Cox, A. V., Stalker, J., Ingle, C. E., et al. (2006). Genome variation and evolution of the malaria parasite Plasmodium falciparum. Nat. Genet. 39, 120–125. doi: 10.1038/ng1931
Jun, J. H., Liu, C., Xiao, X., and Dixon, R. A. (2015). The transcriptional repressor MYB2 regulates both spatial and temporal patterns of proanthocyandin and anthocyanin pigmentation in Medicago truncatula. Plant Cell 27, 2860–2879. doi: 10.1105/tpc.15.00476
Kaufmann, K., Melzer, R., and Theissen, G. (2005). MIKC-type MADS-domain proteins: structural modularity, protein interactions and network evolution in land plants. Gene 347, 183–198. doi: 10.1016/j.gene.2004.12.014
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kim, S., Koh, J., Yoo, M.-J., Kong, H., Hu, Y., Ma, H., et al. (2005). Expression of floral MADS-box genes in basal angiosperms: implications for the evolution of floral regulators. Plant J. 43, 724–744. doi: 10.1111/j.1365-313X.2005.02487.x
Kmita, M., Kondo, T., and Duboule, D. (2000). Targeted inversion of a polar silencer within the HoxD complex re-allocates domains of enhancer sharing. Nat. Genet. 26, 451–454. doi: 10.1038/82593
Knapp, S. (2002). Tobacco to tomatoes: a phylogenetic perspective on fruit diversity in the Solanaceae. J. Exp. Bot. 53, 2001–2022. doi: 10.1093/jxb/erf068
Krueger, F. (2017). Trim Galore!. Available at: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
Larsson, A. (2014). AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278. doi: 10.1093/bioinformatics/btu531
Lebrecht, D., Foehr, M., Smith, E., Lopes, F. J. P., Vanario-Alonso, C. E., Reinitz, J., et al. (2005). Bicoid cooperative DNA binding is critical for embryonic patterning in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 102, 13176–13181. doi: 10.1073/pnas.0506462102
Lee, S., Chung, E.-J., Joung, Y.-H., and Choi, D. (2010). Non-climacteric fruit ripening in pepper: increased transcription of EIL-like genes normally regulated by ethylene. Funct. Integr. Genomics 10, 135–146. doi: 10.1007/s10142-009-0136-9
Lercher, M. J., Blumenthal, T., and Hurst, L. D. (2003). Coexpression of neighboring genes in Caenorhabditis elegans is mostly due to operons and duplicate genes. Genome Res. 13, 238–243. doi: 10.1101/gr.553803
Levo, M., Zalckvar, E., Sharon, E., Dantas Machado, A. C., Kalma, Y., Lotam-Pompan, M., et al. (2015). Unraveling determinants of transcription factor binding outside the core binding site. Genome Res. 25, 1018–1029. doi: 10.1101/gr.185033.114
Li, M., Petukh, M., Alexov, E., and Panchenko, A. R. (2014). Predicting the impact of missense mutations on protein–protein binding affinity. J. Chem. Theory Comput. 10, 1770–1780. doi: 10.1021/ct401022c
Liljegren, S. J., Ditta, G. S., Eshed, Y., Savidge, B., Bowman, J. L., and Yanofsky, M. F. (2000). SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature 404, 766–770. doi: 10.1038/35008089
Liljegren, S. J., Roeder, A. H. K., Kempin, S. A., Gremski, K., Østergaard, L., Guimil, S., et al. (2004). Control of fruit patterning in Arabidopsis by INDEHISCENT. Cell 116, 843–853. doi: 10.1016/S0092-8674(04)00217-X
Litt, A., and Irish, V. F. (2003). Duplication and diversification in the APETALA1/FRUITFULL floral homeotic gene lineage: implications for the evolution of floral development. Genetics 165, 821–833.
Liu, M., Watson, L. T., and Zhang, L. (2014). Quantitative prediction of the effect of genetic variation using hidden Markov models. BMC Bioinformatics 15:5. doi: 10.1186/1471-2105-15-5
Liu, S., Zhang, Y., Feng, Q., Qin, L., Pan, C., Lamin-Samu, A. T., et al. (2018). Tomato AUXIN RESPONSE FACTOR 5 regulates fruit set and development via the mediation of auxin and gibberellin signaling. Sci. Rep. 8:2971. doi: 10.1038/s41598-018-21315-y
Liu, X., Lee, C.-K., Granek, J. A., Clarke, N. D., and Lieb, J. D. (2006). Whole-genome comparison of Leu3 binding in vitro and in vivo reveals the importance of nucleosome occupancy in target site selection. Genome Res. 16, 1517–1528. doi: 10.1101/gr.5655606
Lupiáñez, D. G., Kraft, K., Heinrich, V., Krawitz, P., Brancati, F., Klopocki, E., et al. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. doi: 10.1016/j.cell.2015.04.004
Lupski, J. R. (1998). Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 14, 417–422. doi: 10.1016/S0168-9525(98)01555-8
Ma, B., Elkayam, T., Wolfson, H., and Nussinov, R. (2003). Protein-protein interactions: structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl. Acad. Sci. U.S.A. 100, 5772–5777. doi: 10.1073/pnas.1030237100
Martinez-Castilla, L. P., and Alvarez-Buylla, E. R. (2003). Adaptive evolution in the Arabidopsis MADS-box gene family inferred from its complete resolved phylogeny. Proc. Natl. Acad. Sci. U.S.A. 100, 13407–13412. doi: 10.1073/pnas.1835864100
Massa, A. N., Childs, K. L., Lin, H., Bryan, G. J., Giuliano, G., and Buell, C. R. (2011). The transcriptome of the reference potato genome Solanum tuberosum Group Phureja clone DM1-3 516R44. PLoS One 6:e26801. doi: 10.1371/journal.pone.0026801
Matasci, N., Hung, L.-H., Yan, Z., Carpenter, E. J., Wickett, N. J., Mirarab, S., et al. (2014). Data access for the 1,000 plants (1KP) project. Gigascience 3:17. doi: 10.1186/2047-217X-3-17
Melzer, S., Lens, F., Gennen, J., Vanneste, S., Rohde, A., and Beeckman, T. (2008). Flowering-time genes modulate meristem determinacy and growth form in Arabidopsis thaliana. Nat. Genet. 40, 1489–1492. doi: 10.1038/ng.253
Messeguer, X., Escudero, R., Farré, D., Núñez, O., Martínez, J., and Albà, M. M. (2002). PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18, 333–334. doi: 10.1093/bioinformatics/18.2.333
Michaels, S. D., He, Y., Scortecci, K. C., and Amasino, R. M. (2003). Attenuation of FLOWERING LOCUS C activity as a mechanism for the evolution of summer-annual flowering behavior in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 100, 10102–10107. doi: 10.1073/pnas.1531467100
Müller, B. M., Saedler, H., and Zachgo, S. (2001). The MADS-box gene DEFH28 from Antirrhinum is involved in the regulation of floral meristem identity and fruit development. Plant J. 28, 169–179. doi: 10.1046/j.1365-313X.2001.01139.x
Murrell, B., Wertheim, J. O., Moola, S., Weighill, T., Scheffler, K., and Kosakovsky Pond, S. L. (2012). Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8:e1002764. doi: 10.1371/journal.pgen.1002764
Nakasugi, K., Crowhurst, R., Bally, J., and Waterhouse, P. (2014). Combining transcriptome assemblies from multiple de novo assemblers in the allo-tetraploid plant Nicotiana benthamiana. PLoS One 9:e91776. doi: 10.1371/journal.pone.0091776
NCBI Resource Coordinators (2017). Database resources of the national center for biotechnology information. Nucleic Acids Res. 45, D12–D17. doi: 10.1093/nar/gkw1071
Nei, M., Gu, X., and Sitnikova, T. (1997). Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc. Natl. Acad. Sci. U.S.A. 94, 7799–7806. doi: 10.1073/pnas.94.15.7799
Nielsen, R., Bustamante, C., Clark, A. G., Glanowski, S., Sackton, T. B., Hubisz, M. J., et al. (2005). A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 3:e170. doi: 10.1371/journal.pbio.0030170
Nietzsche, M., Guerra, T., Alseekh, S., Wiermer, M., Sonnewald, S., Fernie, A. R., et al. (2017). STOREKEEPER RELATED1/G-element binding protein (STKR1) interacts with protein kinase SnRK1. Plant Physiol. 176, 1773–1792. doi: 10.1104/pp.17.01461
Olmstead, R. G., Bohs, L., Migid, H. A., Santiago-Valentin, E., Garcia, V. F., and Collier, S. M. (2008). A molecular phylogeny of the Solanaceae. Taxon 57, 1159–1181.
Ortiz-Ramírez, C. I., Plata-Arboleda, S., and Pabón-Mora, N. (2018). Evolution of genes associated with gynoecium patterning and fruit development in Solanaceae. Ann. Bot. 121, 1211–1230. doi: 10.1093/aob/mcy007
Ota, T., and Nei, M. (1994). Divergent evolution and evolution by the birth-and-death process in the immunoglobulin VH gene family. Mol. Biol. Evol. 11, 469–482.
Pabón-Mora, N., Ambrose, B. A., and Litt, A. (2012). Poppy APETALA1/FRUITFULL orthologs control flowering time, branching, perianth identity, and fruit development. Plant Physiol. 158, 1685–1704. doi: 10.1104/pp.111.192104
Pabón-Mora, N., Sharma, B., Holappa, L. D., Kramer, E. M., and Litt, A. (2013). The Aquilegia FRUITFULL-like genes play key roles in leaf morphogenesis and inflorescence development. Plant J. 74, 197–212. doi: 10.1111/tpj.12113
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Payne, J. L., and Wagner, A. (2015). Mechanisms of mutational robustness in transcriptional regulation. Front. Genet. 6:322. doi: 10.3389/fgene.2015.00322
Pegueroles, C., Laurie, S., and Albà, M. M. (2013). Accelerated evolution after gene duplication: a time-dependent process affecting just one copy. Mol. Biol. Evol. 30, 1830–1842. doi: 10.1093/molbev/mst083
Pellegrini, L., Tan, S., and Richmond, T. J. (1995). Structure of serum response factor core bound to DNA. Nature 376, 490–498. doi: 10.1038/376490a0
Piontkivska, H., Rooney, A. P., and Nei, M. (2002). Purifying selection and birth-and-death evolution in the histone H4 gene family. Mol. Biol. Evol. 19, 689–697. doi: 10.1093/oxfordjournals.molbev.a004127
Potato Genome Sequencing Consortium, Xu, X., Pan, S., Cheng, S., Zhang, B., Mu, D., et al. (2011). Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195. doi: 10.1038/nature10158
Preston, J. C., and Kellogg, E. A. (2007). Conservation and divergence of APETALA1/FRUITFULL-like gene function in grasses: evidence from gene expression analyses. Plant J. 52, 69–81. doi: 10.1111/j.1365-313X.2007.03209.x
Preston, J. C., and Kellogg, E. A. (2008). Discrete developmental roles for temperate cereal grass VERNALIZATION1/FRUITFULL-like genes in flowering competency and the transition to flowering. Plant Physiol. 146, 265–276. doi: 10.1104/pp.107.109561
Prince, V. E., and Pickett, F. B. (2002). Splitting pairs: the diverging fates of duplicated genes. Nat. Rev. Genet. 3, 827–837. doi: 10.1038/nrg928
Puig, M., Casillas, S., Villatoro, S., and Cáceres, M. (2015). Human inversions and their functional consequences. Brief. Funct. Genomics 14, 369–379. doi: 10.1093/bfgp/elv020
Purugganan, M. D., Rounsley, S. D., Schmidt, R. J., and Yanofsky, M. F. (1995). Molecular evolution of flower development: diversification of the plant MADS-box regulatory gene family. Genetics 140, 345–356.
Python Language Reference (2010). Python Software Foundation. Available at: http://www.python.org
R Core Team (2018). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rajani, S., and Sundaresan, V. (2001). The Arabidopsis myc/bHLH gene ALCATRAZ enables cell separation in fruit dehiscence. Curr. Biol. 11, 1914–1922. doi: 10.1016/S0960-9822(01)00593-0
Robinson, D. F., and Foulds, L. R. (1981). Comparison of phylogenetic trees. Math. Biosci. 53, 131–147. doi: 10.1016/0025-5564(81)90043-2
Sakuma, S., Lundqvist, U., Kakei, Y., Thirulogachandar, V., Suzuki, T., Hori, K., et al. (2017). Extreme suppression of lateral floret development by a single amino acid change in the VRS1 transcription factor. Plant Physiol. 175, 1720–1731. doi: 10.1104/pp.17.01149
Särkinen, T., Bohs, L., Olmstead, R. G., and Knapp, S. (2013). A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): a dated 1000-tip tree. BMC Evol. Biol. 13:214. doi: 10.1186/1471-2148-13-214
Schauer, S. E., Schlüter, P. M., Baskar, R., Gheyselinck, J., Bolaños, A., Curtis, M. D., et al. (2009). Intronic regulatory elements determine the divergent expression patterns of AGAMOUS-LIKE6 subfamily members in Arabidopsis. Plant J. 59, 987–1000. doi: 10.1111/j.1365-313X.2009.03928.x
Schliep, K. P. (2010). phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593. doi: 10.1093/bioinformatics/btq706
Schlueter, J. A., Dixon, P., Granger, C., Grant, D., Clark, L., Doyle, J. J., et al. (2004). Mining EST databases to resolve evolutionary events in major crop species. Genome 47, 868–876. doi: 10.1139/g04-047
Schröfelbauer, B., Chen, D., and Landau, N. R. (2004). A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc. Natl. Acad. Sci. U.S.A. 101, 3927–3932. doi: 10.1073/pnas.0307132101
Scorza, L. C. T., Hernandes-Lopes, J., Melo-de-Pinna, G. F. A., and Dornelas, M. C. (2017). Expression patterns of Passiflora edulis APETALA1/FRUITFULL homologues shed light onto tendril and corona identities. Evodevo 8:3. doi: 10.1186/s13227-017-0066-x
Shan, H., Zhang, N., Liu, C., Xu, G., Zhang, J., Chen, Z., et al. (2007). Patterns of gene duplication and functional diversification during the evolution of the AP1/SQUA subfamily of plant MADS-box genes. Mol. Phylogenet. Evol. 44, 26–41. doi: 10.1016/j.ympev.2007.02.016
Shchennikova, A. V., Shulga, O. A., Immink, R., Skryabin, K. G., and Angenent, G. C. (2004). Identification and characterization of four chrysanthemum MADS-box genes, belonging to the APETALA1/FRUITFULL and SEPALLATA3 subfamilies. Plant Physiol. 134, 1632–1641. doi: 10.1104/pp.103.036665
Sheinerman, F. B., Norel, R., and Honig, B. (2000). Electrostatic aspects of protein–protein interactions. Curr. Opin. Struct. Biol. 10, 153–159. doi: 10.1016/S0959-440X(00)00065-8
Shima, Y., Fujisawa, M., Kitagawa, M., Nakano, T., Kimbara, J., Nakamura, N., et al. (2014). Tomato FRUITFULL homologs regulate fruit ripening via ethylene biosynthesis. Biosci. Biotechnol. Biochem. 78, 231–237. doi: 10.1080/09168451.2014.878221
Shima, Y., Kitagawa, M., Fujisawa, M., Nakano, T., Kato, H., Kimbara, J., et al. (2013). Tomato FRUITFULL homologues act in fruit ripening via forming MADS-box transcription factor complexes with RIN. Plant Mol. Biol. 82, 427–438. doi: 10.1007/s11103-013-0071-y
Sierro, N., Battey, J. N. D., Ouadi, S., Bovet, L., Goepfert, S., Bakaher, N., et al. (2013). Reference genomes and transcriptomes of Nicotiana sylvestris and Nicotiana tomentosiformis. Genome Biol. 14:R60. doi: 10.1186/gb-2013-14-6-r60
Slugina, M. A., Shchennikova, A. V., Pishnaya, O. N., and Kochieva, E. Z. (2018). Assessment of the fruit-ripening-related FUL2 gene diversity in morphophysiologically contrasted cultivated and wild tomato species. Mol. Breed. 38:82. doi: 10.1007/s11032-018-0842-x
Smykal, P., Gennen, J., De Bodt, S., Ranganath, V., and Melzer, S. (2007). Flowering of strict photoperiodic Nicotiana varieties in non-inductive conditions by transgenic approaches. Plant Mol. Biol. 65, 233–242. doi: 10.1007/s11103-007-9211-6
Song, C., Guo, J., Sun, W., and Wang, Y. (2012). Whole genome duplication of intra- and inter-chromosomes in the tomato genome. J. Genet. Genomics 39, 361–368. doi: 10.1016/j.jgg.2012.06.002
Spence, J., Vercher, Y., Gates, P., and Harris, N. (1996). “Pod shatter” in Arabidopsis thaliana, Brassica napus and B. juncea. J. Microsc. 181, 195–203. doi: 10.1046/j.1365-2818.1996.111391.x
Stefanović, S., Austin, D. F., and Olmstead, R. G. (2003). Classification of Convolvulaceae: a phylogenetic approach. Syst. Bot. 28, 791–806.
Sukumaran, J., and Holder, M. T. (2010). DendroPy: a Python library for phylogenetic computing. Bioinformatics 26, 1569–1571. doi: 10.1093/bioinformatics/btq228
Takumi, S., Koyama, K., Fujiwara, K., and Kobayashi, F. (2011). Identification of a large deletion in the first intron of the Vrn-D1 locus, associated with loss of vernalization requirement in wild wheat progenitor Aegilops tauschii Coss. Genes Genet. Syst. 86, 183–195. doi: 10.1266/ggs.86.183
Tanimoto, K., Liu, Q., Bungert, J., and Engel, J. D. (1999). Effects of altered gene order or orientation of the locus control region on human β-globin gene expression in mice. Nature 398, 344–348. doi: 10.1038/18698
Tanksley, S. D. (2004). The genetic, developmental, and molecular bases of fruit size and shape variation in tomato. Plant Cell 16(Suppl.), S181–S189. doi: 10.1105/tpc.018119
Teng, S., Madej, T., Panchenko, A., and Alexov, E. (2009). Modeling effects of human single nucleotide polymorphisms on protein-protein interactions. Biophys. J. 96, 2178–2188. doi: 10.1016/j.bpj.2008.12.3904
The Tomato Genome Consortium (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641. doi: 10.1038/nature11119
Tieman, D. M., Ciardi, J. A., Taylor, M. G., and Klee, H. J. (2001). Members of the tomato LeEIL (EIN3-like) gene family are functionally redundant and regulate ethylene responses throughout plant development. Plant J. 26, 47–58. doi: 10.1046/j.1365-313x.2001.01006.x
Torgerson, D. G., Kulathinal, R. J., and Singh, R. S. (2002). Mammalian sperm proteins are rapidly evolving: evidence of positive selection in functionally diverse genes. Mol. Biol. Evol. 19, 1973–1980. doi: 10.1093/oxfordjournals.molbev.a004021
Vanneste, K., Maere, S., and Van de Peer, Y. (2014). Tangled up in two: a burst of genome duplications at the end of the Cretaceous and the consequences for plant evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130353. doi: 10.1098/rstb.2013.0353
Vision, T. J., Brown, D. G., and Tanksley, S. D. (2000). The origins of genomic duplications in Arabidopsis. Science 290, 2114–2117. doi: 10.1126/science.290.5499.2114
Vogel, M. J., Pagie, L., Talhout, W., Nieuwland, M., Kerkhoven, R. M., and van Steensel, B. (2009). High-resolution mapping of heterochromatin redistribution in a Drosophila position-effect variegation model. Epigenetics Chromatin 2:1. doi: 10.1186/1756-8935-2-1
Wang, S., Lu, G., Hou, Z., Luo, Z., Wang, T., Li, H., et al. (2014). Members of the tomato FRUITFULL MADS-box family regulate style abscission and fruit ripening. J. Exp. Bot. 65, 3005–3014. doi: 10.1093/jxb/eru137
White, M. A., Myers, C. A., Corbo, J. C., and Cohen, B. A. (2013). Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc. Natl. Acad. Sci. U.S.A. 110, 11952–11957. doi: 10.1073/pnas.1307449110
Xu, Q., Yin, X.-R., Zeng, J.-K., Ge, H., Song, M., Xu, C.-J., et al. (2014). Activator- and repressor-type MYB transcription factors are involved in chilling injury induced flesh lignification in loquat via their interactions with the phenylpropanoid pathway. J. Exp. Bot. 65, 4349–4359. doi: 10.1093/jxb/eru208
Yang, Y., Fanning, L., and Jack, T. (2003a). The K domain mediates heterodimerization of the Arabidopsis floral organ identity proteins, APETALA3 and PISTILLATA. Plant J. 33, 47–59.
Yang, Y., Xiang, H., and Jack, T. (2003b). pistillata-5, an Arabidopsis B class mutant with strong defects in petal but not in stamen development. Plant J. 33, 177–188.
Yang, Z. (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556. doi: 10.1093/bioinformatics/13.5.555
Yang, Z., and Bielawski, J. P. (2000). Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15, 496–503. doi: 10.1016/S0169-5347(00)01994-7
Yang, Z., and Nielsen, R. (2000). Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43. doi: 10.1093/oxfordjournals.molbev.a026236
Yu, J., Wang, L., Guo, H., Liao, B., King, G., and Zhang, X. (2017). Genome evolutionary dynamics followed by diversifying selection explains the complexity of the Sesamum indicum genome. BMC Genomics 18:257. doi: 10.1186/s12864-017-3599-4
Zhao, H., Wang, X., Zhu, D., Cui, S., Li, X., Cao, Y., et al. (2012). A single amino acid substitution in IIIf subfamily of basic helix-loop-helix transcription factor AtMYC1 leads to trichome and root hair patterning defects by abolishing its interaction with partner proteins in Arabidopsis. J. Biol. Chem. 287, 14109–14121. doi: 10.1074/jbc.M111.280735
Zhou, P., Hou, S., Bai, Z., Li, Z., Wang, H., Chen, Z., et al. (2018). Disrupting the intramolecular interaction between proto-oncogene c-Src SH3 domain and its self-binding peptide PPII with rationally designed peptide ligands. Artif. Cells Nanomed. Biotechnol. 46, 1122–1131. doi: 10.1080/21691401.2017.1360327
Keywords: dry fruit, fleshy fruit, fruit development, fruit evolution, FRUITFULL, gene duplication, MADS-box transcription factors, Solanaceae
Citation: Maheepala DC, Emerling CA, Rajewski A, Macon J, Strahl M, Pabón-Mora N and Litt A (2019) Evolution and Diversification of FRUITFULL Genes in Solanaceae. Front. Plant Sci. 10:43. doi: 10.3389/fpls.2019.00043
Received: 29 September 2018; Accepted: 11 January 2019;
Published: 21 February 2019.
Edited by:
Jill Christine Preston, University of Vermont, United StatesReviewed by:
Renata Reinheimer, Instituto de Agrobiotecnología del Litoral (IAL), ArgentinaBharti Sharma, California State Polytechnic University, Pomona, United States
Copyright © 2019 Maheepala, Emerling, Rajewski, Macon, Strahl, Pabón-Mora and Litt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amy Litt, YW15LmxpdHRAdWNyLmVkdQ==
†Present address: Christopher A. Emerling, Whittier College, Whittier, CA, United States Maya Strahl, Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, United States