Abstract
Plants are immobile and often face stressful environmental conditions, prompting the evolution of genes regulating environmental responses. Such evolution is achieved largely through gene duplication and subsequent divergence. One of the most important gene families involved in regulating plant environmental responses and development is the AP2/ERF superfamily; however, the evolutionary history of these genes is unclear across angiosperms and in major angiosperm families adapted to various ecological niches. Specifically, the impact on gene copy number of whole-genome duplication events occurring around the time of the origins of several plant families is unknown. Here, we present the first angiosperm-wide comparative study of AP2/ERF genes, identifying 75 Angiosperm OrthoGroups (AOGs), each derived from an ancestral angiosperm gene copy. Among these AOGs, 21 retain duplicates with increased copy number in many angiosperm lineages, while the remaining 54 AOGs tend to maintain low copy number. Further analyses of multiple species in the Brassicaceae family indicated that family-specific duplicates experienced differential selective pressures in coding regions, with some paralogs showing signs of positive selection. Further, cis regulatory elements also exhibit extensive divergence between duplicates in Arabidopsis. Moreover, comparison of expression levels suggested that AP2/ERF genes with frequently retained duplicates are enriched for broad expression patterns, offering increased opportunities for functional diversification via changes in expression patterns, and providing a mechanism for repeated duplicate retention in some AOGs. Our results represent the most comprehensive evolutionary history of the AP2/ERF gene family, and support the hypothesis that AP2/ERF genes with broader expression patterns are more likely to be retained as duplicates than those with narrower expression profiles, which could lead to a higher chance of duplicate gene subfunctionalization. The greater tendency of some AOGs to retain duplicates, allowing expression and functional divergence, may facilitate the evolution of complex signaling networks in response to new environmental conditions.
Introduction
Plants face numerous environmental stresses, including salinity, temperature extremes, toxicity, pathogen attack, and wounding. Such environmental conditions constrain plant development and productivity, thereby affecting their geographical distribution and limiting their economic value and ecological impact (Zou et al., 2009). How plants cope with abiotic stress conditions is a particularly intriguing topic, given the extraordinary diversity of habitats and ecological niches that plants have colonized, spanning extremely arid, hot, and cold environments on land, as well as marine, brackish, and freshwater aquatic habitats. Colonization of these habitats would have imposed novel challenges and requirements for genes responsive to abiotic stressors. That is, diverse ecological niches present varying evolutionary pressures on plant genes, particularly those responsive to the stresses. To survive and reproduce in various ecological niches, plants have evolved multiple adaptive traits governed by complex signaling and regulatory systems, with downstream networks regulated by transcription factors (TFs) (Licausi et al., 2013; Phukan et al., 2017). For example, the AP2/ERF family of plant TFs has important roles in regulating responses to environmental stressors, growth and development, and metabolism (Licausi et al., 2013; Phukan et al., 2017; Supplementary Table S1).
Plant AP2/ERF genes are members of a superfamily comprising several families, namely the AP2, DREB/ERF, RAV, and Soloist families, as defined previously (Sakuma et al., 2002; Kim et al., 2006; Nakano et al., 2006; Zumajo-Cardona and Pabón-Mora, 2016). The DREB/ERF family can be further subdivided into the DREB and ERF subfamilies, each of which has several subgroups, referred to as Groups I–IV and Groups V–X, respectively (Nakano et al., 2006). For simplicity, hereafter we will use DREB or ERF “family” for the respective groups collectively and “subfamily” to refer to these groups individually. Proteins in the AP2 family contain two AP2 domains, whereas members of the other families have only one AP2 domain (Nakano et al., 2006). Many AP2/ERF family members have known functions in plant development and stress responses (Licausi et al., 2013; Phukan et al., 2017). AP2 family genes also control floral organ identity (AP2), and embryo development and flowering time (TOE1) (Licausi et al., 2013; Zhang et al., 2015; Phukan et al., 2017). DREB genes are crucial for responses to abiotic stressors, including drought (DREB2) (Krishnaswamy et al., 2011), high salt (ESE) (Zhang et al., 2011), low temperature (DREB1/CBFs) (Jia et al., 2016; Zhou et al., 2017), wounding (WIND) (Iwase et al., 2016), and floral organ senescence (FUF1) (Chen et al., 2015). ERF members participate in pathogen responses (Licausi et al., 2013; Phukan et al., 2017), water availability (SNORKEL1 and SNORKEL2) (Hattori et al., 2009), root and shoot growth (TINY) (Wilson, 1996; Wei et al., 2005), low oxygen responses (RAP2.12, RAP2.2, AtRAP2.3, AtHRE1, AtHRE2) (Licausi et al., 2010, 2011; Weits et al., 2014; Bui et al., 2015; Kosmacz et al., 2015), and floral meristem identity (PUCHI, DRN, DRNL) (Chandler and Werr, 2017). RAV family members have roles in regulating hormone and stress responses, independent of ABA (Fu et al., 2014).
In plants, gene duplication is widespread and often results from whole-genome duplication (WGD) (Jiao et al., 2011, 2012; Vanneste et al., 2014; Huang et al., 2016b; Xiang et al., 2016; Ren et al., 2018), providing raw genetic materials for functional innovations, including regulation of development and physiology and contributing to the success of plants under different environmental conditions (Li et al., 2016; Panchy et al., 2016). Duplication of the AP2/ERF genes has been investigated in phylogenetic studies with limited sampling of angiosperms, mostly restricted to core eudicot and monocot models, or single species (Song et al., 2013; Cui et al., 2016; Dossa et al., 2016; Zeng et al., 2016; Liu and Zhang, 2017; Li et al., 2018). Some studies have focused on a specific family or subclade of genes [i.e., the AP2 family (Rashotte and Goertzen, 2010; Zumajo-Cardona and Pabón-Mora, 2016)]. Thus, the patterns of AP2/ERF gene retention following WGD across angiosperms remain unclear, as do the phylogenetic relationships among AP2/ERF genes of multiple members of the same plant family. Specifically, it is not clear when and where AP2/ERF duplicates were retained in angiosperm history, or whether the same duplicate(s) generated in an ancestor tend to be retained in multiple species adapted to different ecological niches, or retained in some species but lost in others. Further, how duplication and divergence of AP2/ERF genes contribute to the evolution of stress response pathways is also not well understood. The patterns of retention and loss of AP2/ERF duplicates have the potential to provide insights into the evolution of regulatory genes for plant development and environmental responses more generally (LehtiShiu et al., 2015).
It has been proposed that speciation in plants involves rapid evolution of stress response genes (Zou et al., 2009). AP2/ERF duplicates are associated with environmental adaptation, including habitat colonization. For example, natural variation in the DREB family members, C-repeat-binding factor (CBF) genes, among populations of the model plant, Arabidopsis thaliana, along the Yangtze River in China, is the molecular basis of divergence in freezing tolerance (Kang et al., 2013); however, it is unclear whether AP2/ERF genes are associated with adaptation in other plants. Relatives of A. thaliana in the Brassicaceae, Eutrema salsugineum (Yang et al., 2013) and Schrenkiella parvula (Dassanayake et al., 2011), are tolerant of alkaline soils, but it is unknown whether this is associated with lineage-specific expansions of AP2/ERF genes. The species Medicago truncatula, Ananas comosus, Zea mays, Sorghum bicolor, and Setaria italica are drought-tolerant. This property and the phylogenetic placements among angiosperms provide great opportunities for investigation of the evolution of AP2/ERF genes from an ecological perspective. Among angiosperms with sequenced genomes, three, the sacred lotus (Nelumbo nucifera) (Ming et al., 2013), duckweed (Spirodela polyrhiza) (Wang et al., 2014), and seagrass (Zostera marina) (Olsen et al., 2016), are aquatic, and have independently colonized their habitats. As AP2/ERFs are involved in plant responses to water availability, examination of the evolutionary patterns of AP2/ERF genes in these parallel lineages would be a worthwhile exercise.
To better understand the diversity and evolution of the AP2/ERF superfamily and to obtain clues about their functional evolution and their potential contributions to the radiations of angiosperms, and different angiosperm groups in various habitats, we performed comprehensive analyses of AP2/ERF genes in 37 species, covering the major lineages of angiosperms. First, we detailed the initial history of AP2/ERF gene duplication in representative major angiosperm groups, identifying multiple duplications in the core eudicots, Brassicaceae, Fabaceae, Asteraceae, Commelinids, Poales, and Poaceae, and differential expansion of the AP2/ERF family. Next, we identified preferential retention of recent AP2/ERF gene duplicates that had undergone previous duplication and/or exhibited broad expression. Then, we examined patterns of sequence divergence and evidence of positive selection, and found that disordered protein regions evolved more rapidly than those that were ordered. Finally, we discuss experimental support from the literature for a strong correlation between duplicate retention, resulting in expanded AP2/ERF Angiosperm OrthoGroups (AOGs) and broad expression patterns, allowing subfunctionalization in expression patterns. This study provides rich information supporting hypotheses on how AP2/ERF genes have expanded and evolved during angiosperm history through adaptation to different ecological niches.
Materials and Methods
Data Collection
The genome sequences of 38 species were downloaded from public databases, 32 species (including the outgoup, Selaginella moellendorffii) from Phytozome v12.0 (Goodstein et al., 2012)1; three species, Aethionema arabicum, Leavenworthia alabamica, and Sisymbrium irio (Rosenbloom et al., 2015) using the University of California at Santa Cruz Genome Browser2; S. parvula3 (Dassanayake et al., 2011); N. nucifera from LOTUS-DB4 (Ming et al., 2013); and Helianthus annuus from the Genome Database5 (Badouin et al., 2017). Species information is detailed in Supplementary Table S2.
Sequence Retrieval and Identification
For this study, AP2/ERF genes were identified using HMMER v3.16 (Finn et al., 2011) using the AP2 entry [A profile hidden Markov model (HMM) from the seed alignment] (Pfam accession number PF00847) of Pfam-A, downloaded from Pfam v27.07 (Finn et al., 2016), and named with a four-letter species designations, including the first two letters of the genus and the species (Supplementary Table S3). All retrieved protein sequences containing the AP2 domain were verified by a batch search of the NCBI Conserved Domain Database8 (Marchler-Bauer et al., 2015), with a threshold of e-value < 10-5; sequences lacking the AP2 domain were discarded before further analyses.
Phylogenetic Analysis
We employed OrthoFinder (Emms and Kelly, 2015) to infer AOGs, each with one copy in the most recent common ancestor (MRCA) of angiosperms, for AP2/ERF proteins. Next, protein sequences from each AOG were aligned using Multiple Sequence Alignment (MSA) in Fast Fourier Transform (MAFFT) v7.273 (Katoh and Standley, 2013), and regions of low quality were automatically removed using trimAl v1.4 (Capella-Gutiérrez et al., 2009) with default settings. Then, a preliminary approximate maximum-likelihood (ML) phylogenetic tree was generated based on the alignments, using FastTree v2.1.7 (Price et al., 2009). If a tree indicated a duplication event at a node before the MRCA of angiosperms, we split it into two subtrees to ensure that each subtree represented a single ancestral angiosperm gene and its descendants. A second round of MSA was performed for each AOG separately. To improve the alignment of relatively variable N-terminal and C-terminal regions, the “–localpair” and “–maxiterrate 1000” commands were used. As AP2/ERF sequences often lacked sufficient phylogenetic information to yield well-supported gene tree topologies, thereby affecting subsequent analyses, we incorporated a gene tree improvement procedure using TreeFix (Wu et al., 2013), a gene tree–species tree reconciliation algorithm that performs topological changes to gene trees and searches for alternative topologies that minimize the duplication/loss cost, while having a likelihood not statistically significantly worse than that of the ML topology. Briefly, ML trees were first constructed for the AOGs using IQ-TREE (Nguyen et al., 2015), based on trimmed protein sequence alignments previously obtained using the amino acid substitution (LG+F+G) model (Le and Gascuel, 2008). The trees were rooted arbitrarily, as rooting is needed for TreeFix, whereas the specific position of the root is not important. Subsequently, using a reference species tree (Zeng et al., 2014, 2017; Huang et al., 2016a), a file was generated that mapped leaves of the gene tree onto those of the species tree. In addition, codon-based CDS (DNA sequence) alignments, generated from the amino acid sequence alignments, were back translated using TreeBeST v1.9.2 (Vilella et al., 2009) and used for analysis with TreeFix, under the default reconciliation model (duplication/loss cost), with rooted gene trees to generate reconstructed gene trees, increasing the number of iterations to 1000. Furthermore, branch lengths and SH-aLRT support for the reconstructed gene trees generated by TreeFix (Wu et al., 2013) were computed using IQ-TREE v.1.5.5, with the best-fit codon model, as determined using ModelFinder (Kalyaanamoorthy et al., 2017).
Evolutionary Analysis of Brassicaceae Genes
Paralogous pairs of Brassicaceae-specific duplicates and their orthologs in Theobroma cacao and Carica papaya were aligned using PRANK v140603 (Veidenberg et al., 2016) and back translated using TreeBeST v1.9.2 (Vilella et al., 2009). Gene trees reconciled with the species tree were generated using the same pipeline described in the section “Phylogenetic analysis.” To detect sequence differences, as evidence for relaxed selection or positive selection, the gene tree and codon-based alignment were imported into RELAX9 (Wertheim et al., 2015), to detect whether selective strength was relaxed in post-duplication branches relative to their pre-duplication branches, and codeml implemented in the program to estimate non-synonymous (Ka) to synonymous (Ks) substitutions rates (ω = Ka/Ks) values and test for positive selection using the PAML 4.8 package (Yang, 2007).
To detect different selective pressures that affect specific lineages, we compared the two-ratios model (Model 2), which allows a different ω ratio for foreground lineages, with the one-ratio model, which assumes that the ω ratio is constrained along all branches in the phylogeny (one-ratio Model 0) (Yang, 1998; Yang et al., 1998). The positive selection that affected some sites in the post-duplication branches was detected by comparing the branch-site model (Model A), assuming one class of sites in the foreground lineage, ω > 1, with null model A (Zhang et al., 2005), where ω was fixed and ≤1 (no positive selection). To detect differences (or divergence) in selective constraints among entire clades that had evolved following duplication events, we compared the site-specific discrete Model 1a, which assumes two classes of sites with different ω ratios, to clade Model C (Bielawski and Yang, 2003), which allows selective pressure at one class of sites (foreground clade) to differ from the rest of the phylogeny. The likelihood ratio test statistic (similar to a chi-square distribution) was used to compare nested likelihood models. These test statistics are calculated as 2 × abs (lnl1 – lnl0) (i.e., twice the difference between the lnl value under each model), with critical values of 2.71 at 5% and 5.41 at 1%. Pairs of orthologous codon-based CD alignments were obtained using the same procedure described in the section “Phylogenetic analysis.” Ka, Ks, and ω were estimated with default parameters using PAML (Yang, 2007).
Analysis of Sequence Difference as Evidence for Functional Divergence
Following duplication, the sequence difference between paralogs can suggest functional divergence. To detect such sequence divergence, we used the program DIVERGE v3.0 (Gu et al., 2013) to analyze sequence differences in Brassicaceae-specific duplicates. In this program, two types of sequence differences are defined as evidence for functional divergence: Type-I functional divergence is defined as amino acid patterns that are highly conserved in one duplicate among various species, but highly variable in the other duplicate among the same set of species; Type-II functional divergence describes amino acid patterns that differ between paralogs in terms of biochemical properties, but are highly conserved among the set of species under consideration. These types of sequence divergence between paralogs were tested using ML estimates of θ and standard error (SE). Z-values were calculated by dividing θ by the SE, and P-values were calculated using a two-tailed Z-score test (normal distribution test). The effective number of sites exhibiting such functional divergence was also counted using DIVERGE (Gu et al., 2013).
Amino Acid Substitution Rates and Sequence-Based Predictions
Per site amino acid substitution rates were estimated in Rate4Site v3.2 using an empirical Bayesian method (Mayrose et al., 2004). The default JTT amino acids substitution model was used as the model of evolution, with 16 categories of gamma distribution. Structural disorder was predicted using DISOPRED3 (Jones and Cozzetto, 2015). The amino acid substitution rates and confidence scores for each sequence were plotted using the ggplot program in the R package. The borders of the AP2 domain were predicted using the SMART Web server (Schultz et al., 1998).
Cis-Elements and Expression Pattern Analyses
Cis-elements in A. thaliana AP2/ERF gene promoters were obtained from AGRIS (Yilmaz et al., 2011) and A. thaliana AP2/ERF gene expression data were obtained from different datasets: the At-TAX tilling array dataset (Laubinger et al., 2008), AtGenExpress (Schmid et al., 2005), and Expression Atlas (Petryszak et al., 2016). AP2/ERF gene expression data from Glycine max and Oryza sativa were obtained from Severin et al. (2010) and Davidson et al. (2012), respectively. Tissue specificity was measured using the index, τ (Yanai et al., 2005).
Results
Phylogenetic Classification and Expansion of AP2/ERF Genes
To systematically analyze genes of the AP2/ERF superfamily, we selected 37 angiosperm species (26 eudicots, 10 monocots, 1 basal angiosperm) with genome sequences to represent the major angiosperm lineages. The numbers of AP2/ERF genes varied greatly (Figure 1A), from 76 (basal angiosperm Amborella trichopoda; Amborella hereafter), to 79–268 (monocots), and 93–348 (eudicots), with 348 in G. max (Fabaceae, eudicot). Species with large numbers (≥190) of AP2/ERF genes were associated with proposed recent WGD events [i.e., Brassica rapa (286), Populus trichocarpa (209), G. max (348), and Z. mays (194)], suggesting that WGD events likely increase AP2/ERF gene numbers at the family or genus levels. In contrast, aquatic plants, such as seagrass (Z. marina) (105) and duckweed (S. polyrhiza) (79), have relatively few AP2/ERF genes, despite having putative historical WGD events, presumably because they do not need the function of AP2/ERF genes for drought response in their aquatic habitats. AP2 protein sequences from 37 angiosperms were classified into 75 AOGs (see the section “Materials and Methods”), each likely derived from a single ancestral angiosperm copy and usually supported by genes from Amborella, a sister of other extant angiosperms. The AOGs were classified into subfamilies according to AP2/ERF genes from A. thaliana or O. sativa in each AOG. The Soloist subfamily showed least variation, with one copy in most species and two in some species with recent WGD. Other subfamilies experienced expansion from smaller numbers of ancestral angiosperm genes during angiosperm history (Figure 1A).
FIGURE 1

Copy number variation AP2/ERF genes in each species. (A) A species phylogeny and number of AP2/ERF genes in each subfamily. The species tree in the study was inferred from based on the phylogeny of conserved nuclear genes (Zeng et al., 2014, 2017; Huang et al., 2016a). Genome duplication and triplication events were according to the literature (Jiao et al., 2011, 2012; Vanneste et al., 2014; Huang et al., 2016b; Xiang et al., 2016; Ren et al., 2018), and are depicted by stars. The copy number of each subfamily was indicated. Only sequences that can be aligned and clearly classified are included. (B) Copy number variation in each AOGs inferred from gene trees. A heat map showing clustering according to copy number. Rows represent species and columns represent the AOGs.
Next, we examined differences in duplicate retention among AOGs by constructing a copy number profile matrix (Figure 1B), which provided the number of genes in each AOG for a species. Furthermore, we clustered AOGs based their copy number profiles, using Ward’s hierarchical clustering method, and found that AOGs can be clustered into two groups (Figure 1B): Group 1 contains 21/75 (28%) AOGs covering 87/146 (59.59%) Arabidopsis AP2/ERF genes, and 81/159 (50.9%) rice AP2/ERF genes; while Group 2 contains 54 AOGs covering 59/146 (40.4%) Arabidopsis genes and 78/159 (49.1%) rice AP2/ERF genes. AOGs in Group 1 have multiple (generally ≥ 3) copies in most species; however, usually only one in Amborella, which lacks evidence of WGD events after divergence from other angiosperms (Amborella Genome Project, 2013). In contrast, AOGs in Group 2 usually have one or a few (generally ≤ 3) genes in a species, and they are even absent in some species. Thus Group 1 is referred to hereafter as the multi-copy group, and Group 2 as the low-copy group. Group 2 can be further divided into three subgroups: Group 2a (generally contains 0–3 genes in the majority of angiosperms), Group 2b (generally contains 0 or 1 copy in the majority of angiosperms, except in some species with recent WGD events), and Group 2c (generally contains 0 or 1 copy in eudicots, but two copies in monocots). Within Group 2, some species show increased copy number, indicating that low-copy AOGs likely experienced lineage-specific duplications, whereas others exhibit a strong tendency to return to single-copy after WGD events. Ten AOGs (four in the ERF V family) in low-copy Group 2b and three in low-copy Group 2c were not recovered from any Brassicaceae genomes we analyzed, supporting their losses in the ancestor of Brassicaceae (Figure 1B). There were also 4 and 14 losses in the MRCA of Fabaceae and Poaceae, respectively.
Repeated Retention of AP2/ERF Genes in the Multi-copy Group in Multiple Angiosperm Lineages
To further understand the duplication histories of the different AOGs, we investigated the duplication patterns of AP2/ERF genes in each AOG (Figure 2) for lineages with known WGD events using gene tree reconciliation (see the section “Materials and Methods”). The results (Figure 3 and Supplementary Figures S1–S13) showed that AOGs in the multi-copy group have retained duplicates corresponding to both WGD events shared by multiple families [γ shared by core eudicots, the Commelinids-specific WGD (τ), or the Poales-specific WGD (σ)] and family-level WGD events [i.e., Brassicaceae WGD (α/β), Poaceae WGD (ρ), Fabaceae WGD, and Asteraceae WGD] (Figure 3). However, duplicates generated by the core eudicot-specific γ were not equally retained in the three eudicot families, Brassicaceae, Fabaceae, and Asteraceae, following their family-level WGD events. For example, three duplicates were generated by γ in DREBIb-AOG3: AtWIND1/AtWIND2, AtWIND3, and AtWIND4/AtERF056/AtERF060 [Figure 3(I) and Supplementary Figure S2D]. Among these, only AtWIND1/AtWIND2 retained duplicates from family-specific WGD in Brassicaceae, Fabaceae, and Asteraceae. AtWIND3 was not duplicated in Brassicaceae and Asteraceae, and was lost in the Fabaceae. The AtWIND4/AtERF056/AtERF060 genes were duplicated in the Brassicaceae, corresponding to two duplicates in the Fabaceae, but without a counterpart in the Asteraceae. In another example, two duplicates were generated by the core eudicot-specific γ WGD in DREBIIIc-AOG2: AtCBFs and AtDDFs. These were followed by additional duplicates from family-specific WGD events in the Brassicaceae and Poaceae; however, only AtCBFs, and not AtDDFs, were subject to additional duplication during family-specific WGD within the Asteraceae [Figure 3(III) and Supplementary Figure S4F]. In addition, almost all species had several copies of AtCBF homologs, generated by species or lineage-specific duplications, whereas this was not the case for AtDDFs genes. For example, in M. truncatula there are 13 members in the AtCBF clade, while there are only 4 in the AtDDF clade (Supplementary Figure S4F). In contrast, AOGs in Group 2 tend to have retained a few duplicates from WGD events, either shared by multiple families or specific to a family, thus retaining their low-copy status, even after lineage-specific losses (Supplementary Figures S1–S13). For example, no duplicates from γ were retained and there was Commelinids–Asparagales-specific WGD (τ) in AP2-AOG1 (Supplementary Figure S1A). Nevertheless, some duplicates were generated from family-specific WGD in the Brassicaceae (AtAP2/AtTOE3), Fabaceae, and Asteraceae, and order-specific WGD in Poales. Two duplicates were generated by γ in VIIIb-AOG3: the AtLEP and AtERF087 lineages (Supplementary Figure S9G); however, no duplicates were retained from subsequent family-specific WGD events. In IIIc-AOG1, genes were lost in the Brassicaceae, whereas three duplicates were retained in the Fabaceae (Supplementary Figure S4E). In IVb-AOG2, three duplicates related to AtABI4 were found in the Poaceae; however, the copy number was unchanged in the Brassicaceae, Fabaceae, and Asteraceae (Supplementary Figure S5C). The VI-AOG1 gene was lost in the Brassicaceae, whereas two duplicates were retained in the Fabaceae and Asteraceae (Supplementary Figure S7A). In Xa-AOG3, two duplicates were retained from the τ WGD, and duplicates were again retained from the Poaceae-specific WGD (ρ) (Supplementary Figure S11C), whereas no duplicates were generated by WGD events in eudicots. These and other results indicate that low-copy AOGs experienced limited and differential expansion in different angiosperm lineages.
FIGURE 2
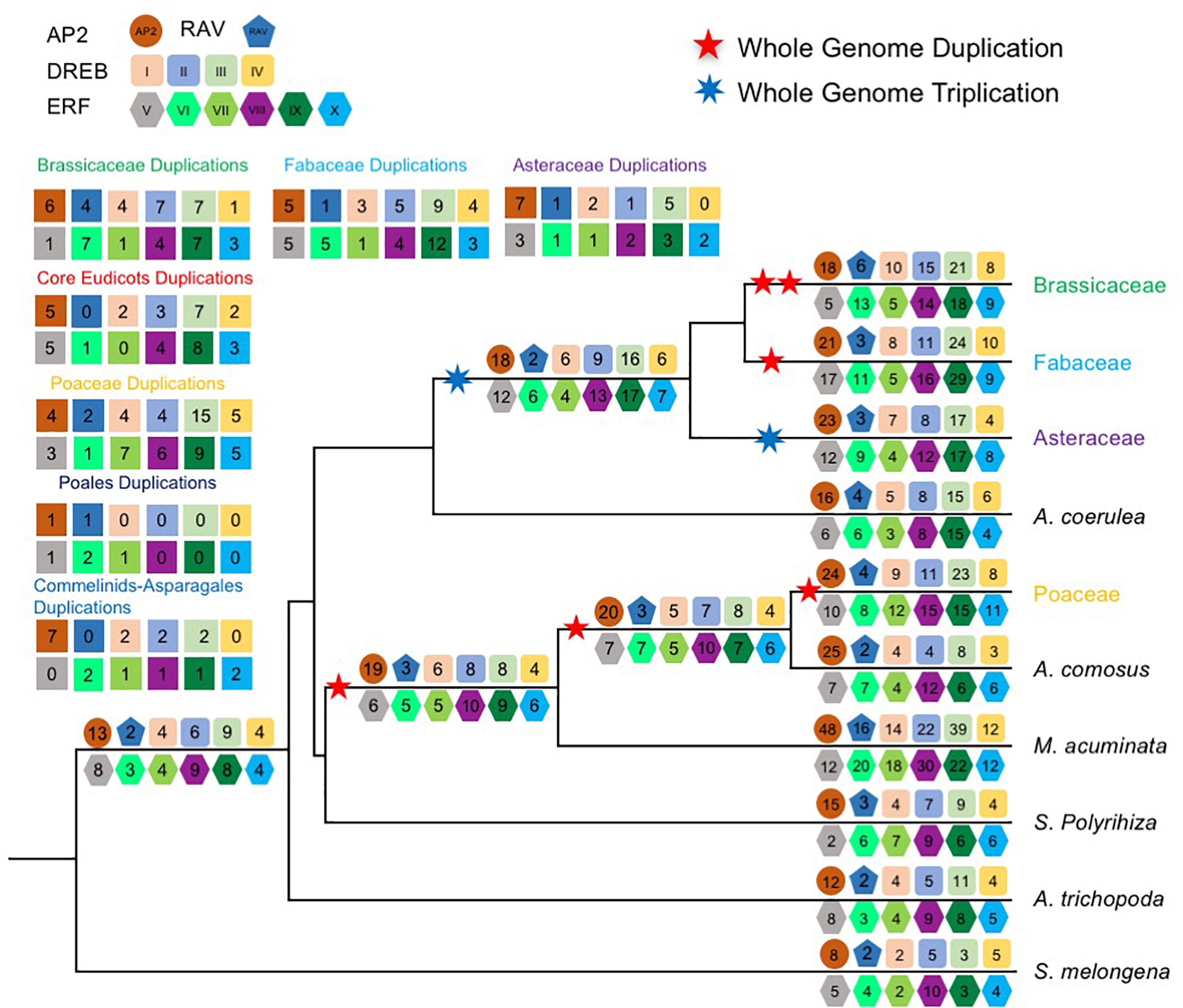
Duplication events inferred from gene trees. Schematic representation of the minimum inferred AP2/ERF protein complement and gene duplication events in the last common ancestor of each major plant group. A numbered geometry represents the copy number in each AP2/ERF subfamily. The numbers in the top and left geometries represent the minimum inferred gene duplication events in the last common ancestor of each major angiosperm groups. Duplication events were inferred from gene trees and species tree.
FIGURE 3
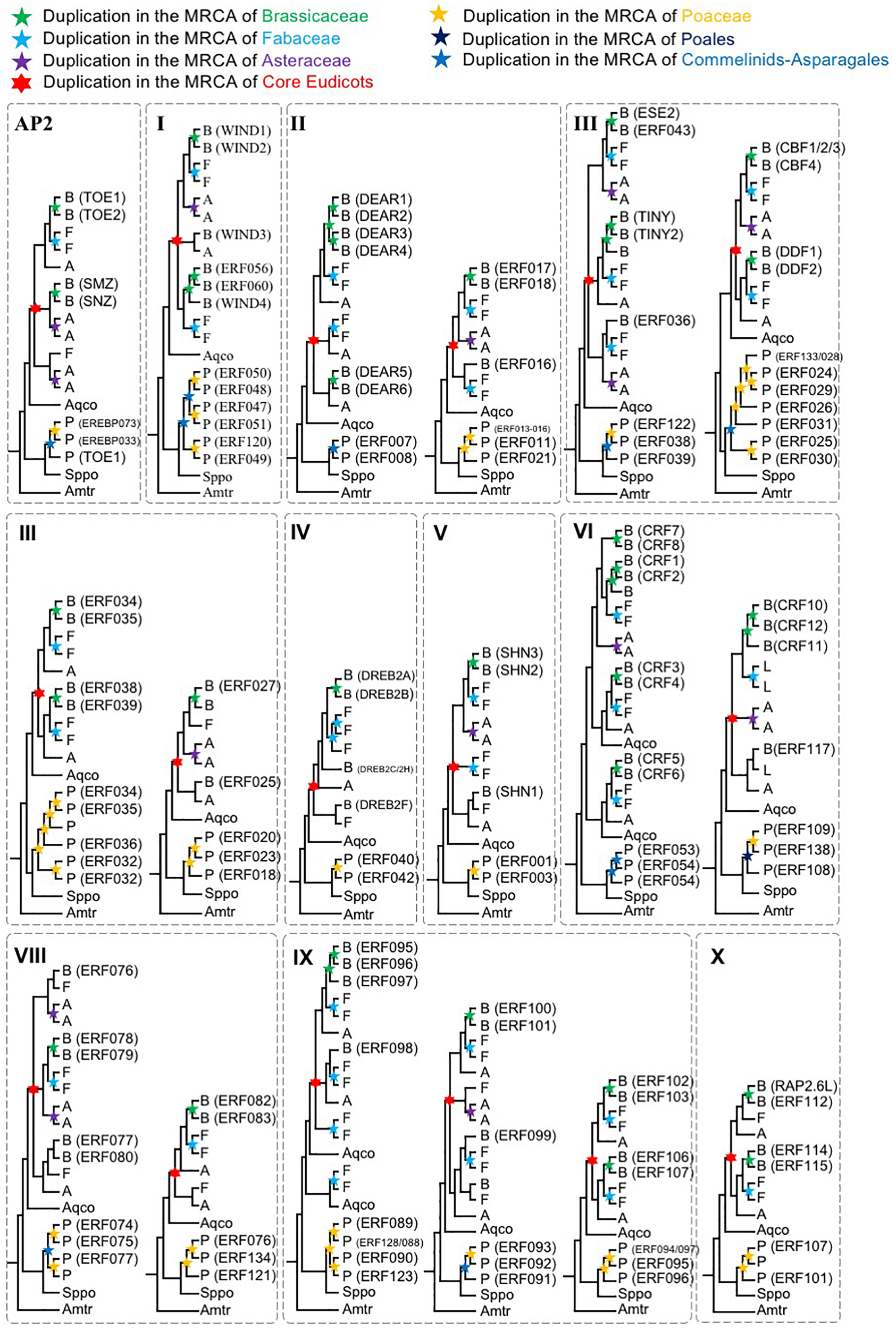
Schematic phylogenetic diagrams of AP2/ERF genes (simplified phylogenetic trees). The topologies are shown for each AOG generated by combining sequence data and species tree information. See the section “Materials and Methods” and (Supplementary Figures S1–S13) for more complete information on taxon sampling and support values for all nodes. Duplication events of Brassicaceae, Fabaceae, Asteraceae, Core Eudicots, Poaceae, Commelinids, Poales, are indicated by green, light blue, purple, red, orange, blue, and deep blue stars, respectively. Genes from Aquilegia coerulea, Spirodela polyrhiza, and Amborella trichopoda were shown as Aqco, Sppo, and Amtr, respectively, and are not necessarily single copy. The abbreviations used are as follows: B, Brassicaceae; F, Fabaceae; A, Asteraceae.
Additional Gene Clade-Specific Expansions of AP2/ERF Genes
In addition to the duplications described above, we observed that AP2/ERF genes exhibit a high incidence of gene clade-specific expansions in different angiosperms. For example, in the clades IIIe-AOG2 (Supplementary Figure S4I) and VIIa-AOG3 (Supplementary Figure S8C), seagrass genes have undergone expansion to a degree not observed in the freshwater duckweed (S. polyrhiza), suggesting that the increased gene copies may be important for adaptation to the marine environment. In the sacred lotus, several specific duplications occurred in clades IIId-AOG1 (Supplementary Figure S4G), IVa-AOG1 (Supplementary Figure S5A), and IXc-AOG2 (Supplementary Figure S10F). IIId-AOG1 also contained three lineage-specific duplicates in seagrass and two specific duplicates in duckweed (Supplementary Figure S4G). In IVa-AOG1, nine lineage-specific duplicates were found in lettuce (Lactuca sativa) (Supplementary Figure S5A), while in VIIIa-AOG1, one gene clade generated by the γ WGD has undergone specific expansions in M. truncatula (six genes); however, their close homologs have been lost in the Brassicaceae (Supplementary Figure S9A). These gains may have contributed to the adaption of corresponding angiosperm lineages to their habitats.
Duplicates Evolved Under Purifying Selection No Less Than Single Copy Genes
Previous genome-wide studies found that the retention of genes is skewed toward duplicates that have minimal phenotypic impact (O’Toole et al., 2017), and some analyses also indicated that conserved proteins tend to exhibit higher gene duplicability (Davis and Petrov, 2004; Liang and Li, 2007). To test these two possibilities for AP2/ERF genes, we measured the ratio of non-synonymous (Ka) to synonymous (Ks) substitutions rates (ω = Ka/Ks), as a proxy for the evolutionary rate, using codon-based models in PAML (Yang, 2007). We measured ω values between one-to-one orthologous pairs from two representative species in each of the three angiosperm families. We found that the ω-values had broad distributions for genes in both the multi-copy and low-copy groups, with median ω-values slightly lower in the multi-copy, compared with the low-copy, groups, except for one Fabaceae orthologous pair (Phaseolus vulgaris versus Vigna unguiculata), perhaps because the phylogenetic relationship of these legume species is too close (Figure 4). The differences in ω-values between multi-copy and low-copy group genes were significant in Poaceae (between S. bicolor and S. italic; and between S. bicolor and O. sativa, P = 0.020 and P = 0.004, respectively; Wilcoxon rank-sum test) (Figure 4). The low ω-values detected suggest both groups have experienced purifying selection during the history of these families, and further imply that genes in the multi-copy group have generally not been under relaxed selection during the evolutionary histories of these families, but may even be under higher evolutionary constraint.
FIGURE 4
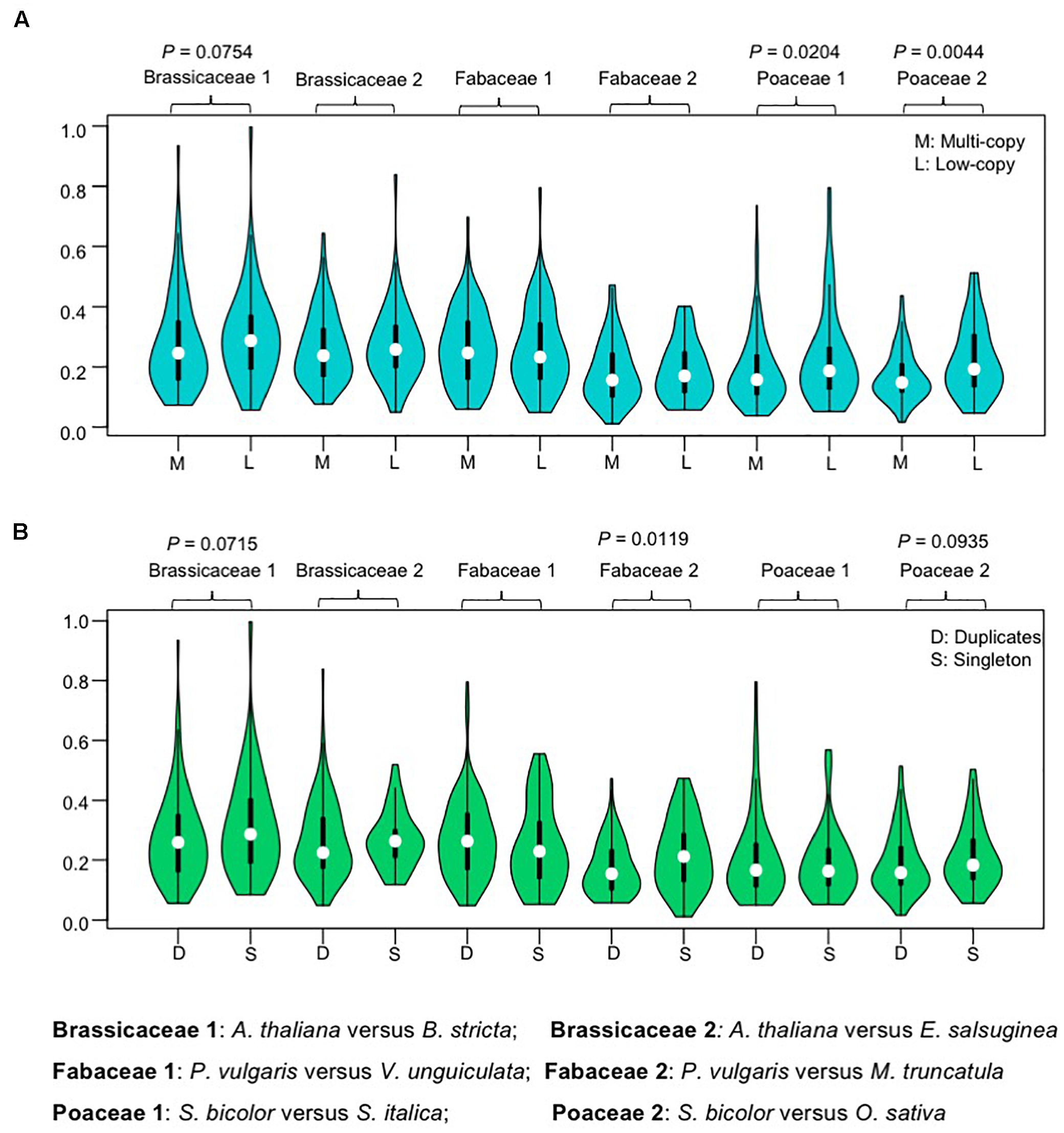
Violin plots showing ω (Ka/Ks) ratio for one-to-one orthologous pairs. (A) Violin plots showing ω (Ka/Ks) values for multi-copy group and low copy group gene sets. The data are separated into multi-copy group and low copy group according to the copy number profile matrix. (B) Violin plots showing ω (Ka/Ks) values for recent duplicates and singleton sets. The data are separated into recent duplicates (family-level WGDs) and singletons according to previous described gene trees. All evolutionary rates were calculated by comparison of orthologous pairs from selected species. Values were compared using the Wilcoxon rank sum test and P-values less than 0.1 are shown above each pair of Violin plots.
The effect of recent gene duplication on evolutionary constraint was also assessed by sequence comparisons between orthologs in groups that retained duplicates from recent WGD (duplicates) and those that did not (singletons). Duplicates of Brassicaceae, Fabaceae, and Poaceae AP2/ERF genes were identified from gene trees. The median -values of recent duplicates were slightly lower than those for singletons, except in one Fabaceae pair 1, where there was a statistically significant difference in the -values of orthologous pairs between the groups with duplicates or singletons in the Fabaceae 2 (P. vulgaris versus M. truncatula) (Figure 4B). These results further support the idea that AP2/ERF duplicates did not experience relaxed selection relative to singletons after the speciation event, and may have experienced higher evolutionary constraints.
To test whether the observed higher evolutionary constraints in duplicated genes reflect post-duplicate neofunctionalization or persistence of an ancestral state, we sought to estimate the evolutionary rates of genes prior to, and independent of, duplication events. We chose to examine two species, L. alabamica (Haudry et al., 2013) and B. rapa (Wang et al., 2011), both Brassicaceae, which have undergone recent lineage-specific WGD events. In contrast, A. thaliana, Boechera stricta, and Eutrema salsuginea exhibited no evidence of WGD after divergence from other species in the same family. As evolutionary rates for a particular gene are highly correlated within the same family, we reasoned that the evolutionary rates of genes in these outgroups can serve as proxies for the rate of the ancestral gene in the common ancestor of Arabidopsis and B. rapa in the Brassicaceae. We then partitioned every pair of family-specific duplicates (corresponding to Brassicaceae WGD events) into two categories with greater and fewer gene copies relative to one another. We found that gene clades from the outgroup contained more retained duplicates, and had evolved significantly more slowly, than the sister clade, supporting the model of preferential retention of slower evolving AP2/ERF genes (Supplementary Figure S14). As AP2/ERF gene copy numbers in G. max and Z. mays were the same in most family-specific (Fabaceae or Poaceae) duplicate pairs, we could not conduct this analysis for these two families.
AP2/ERF Clades With Duplicates From WGD Events Are Enriched for Broad Gene Expression Patterns
Highly and broadly expressed genes tend to evolve slowly (Liao et al., 2007). To test whether these trends held true for different clades within the AP2/ERF superfamily, and to investigate a possible relationship between duplicate retention and expression pattern, we compared gene expression profiles among AP2/ERF gene duplication categories for species with expression data. The results showed that multi-copy genes were expressed more broadly (higher expression breadth) than low-copy genes in A. thaliana, G. max, and O. sativa (Figure 5A–D). Further, we compared the expression patterns of relatively recent duplicates from family-specific WGD events with those of single-copy genes, with similar results (Figure 5A–D). We also tested whether the observed high expression breadth in duplicated genes reflected post-duplicate neofunctionalization or persistence of an ancestral state, based on the suggestion that the expression of orthologous genes is generally conserved within the same family. We found that gene clades containing more retained duplicates had lower tissue specificity than sister clades in the outgroup (Figure 5E,F), supporting the model of preferential retention of genes with lower tissue specificity.
FIGURE 5
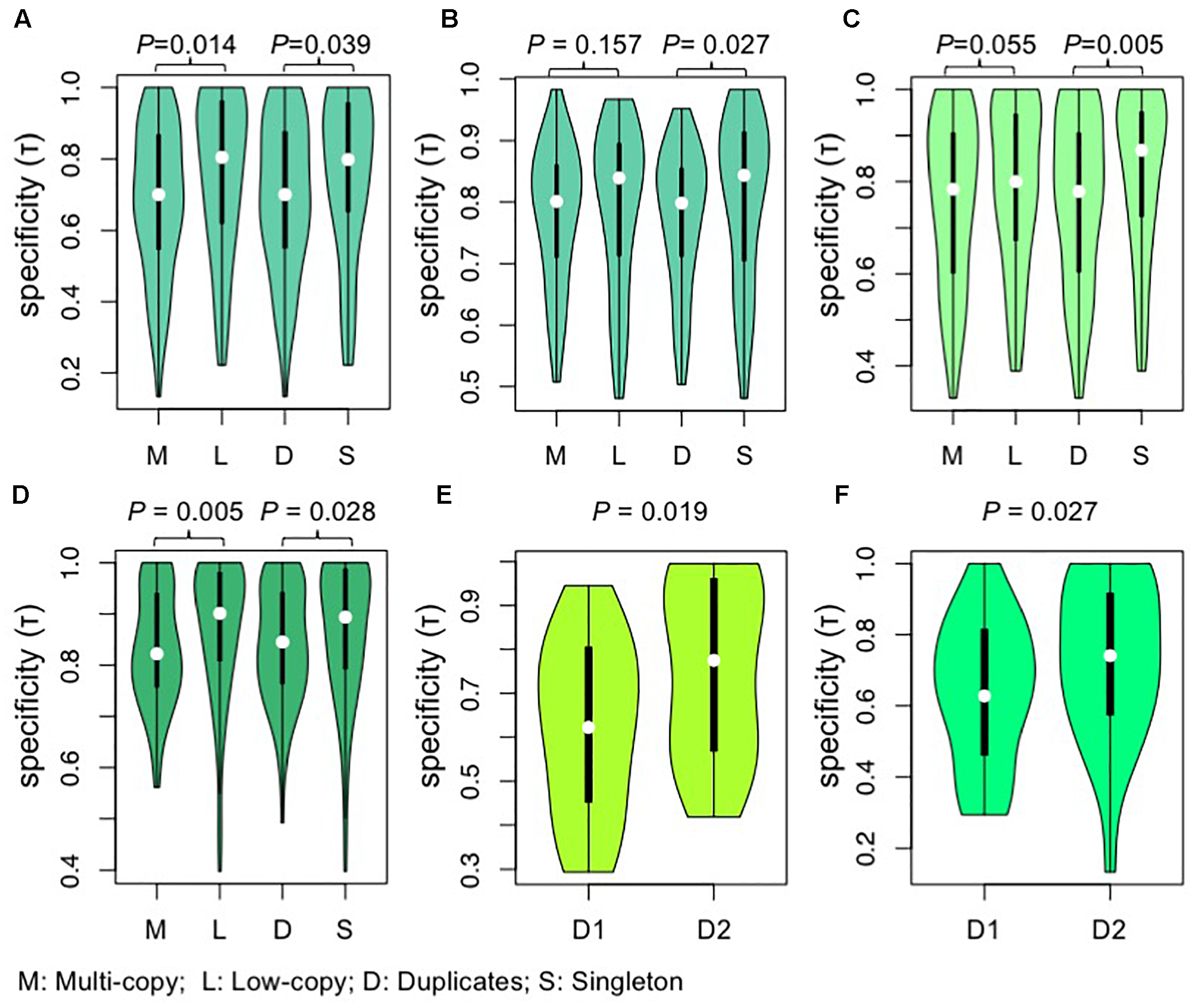
Violin plots showing the comparisons of tissue specificity (τ) among different groups of AP2/ERF genes. (A,B) The comparisons of tissue specificity (τ) among different groups of genes from A. thaliana. (A) Gene expression data of roots, leaves, flowers, and siliques were obtained by high-throughput sequencing of A. thaliana from expression atlas (Petryszak et al., 2016). (B) Gene expression data of 79 samples were obtained from AtGenExpress expression atlas (Schmid et al., 2005). (C) The comparisons of tissue specificity (τ) among different groups of genes from G. max. Gene expression data of young leaf, flower, pods, seeds, roots, and nodules were obtained by high-throughput sequencing of G. max. Gene expression data were download from RNA-Seq atlas of G. max (Severin et al., 2010). (D) The comparisons of tissue specificity (τ) among different groups of genes from O. Sativa. Gene expression data of 10 tissues (seedling four-leaf stage, leaves-20 days, shoots, pre-emergence inflorescence, emerging inflorescence, anther, pistil, seed-5 DAP, seed-10 DAP, endosperm-25 DAP) were download from Rice Genome Annotation Project (Davidson et al., 2012). Values were compared using the Wilcoxon rank-sum test and P-values are shown above each pair of Violin plots. (E, F) Tissue specificity (τ) differences between two paralogous clades in Brassicaceae. D that each duplicate is responsive to. The duplicate pairs generated near the origin of Brassicaceae are separated into D1 and D2 according to their copy number of AP2/REF genes from Leavenworthia alabamica(E) or B. rapa(F), with the D1 having more gene copy in L. alabamica(E) or B. rapa(F). Values were compared using the Wilcoxon rank-signed tests and P-values are shown above each pair of Violin plots. M, multi-copy genes; L, low-copy genes; D, duplicates; S, singleton; DAP, days after pollination.
Divergence in Cis-Elements, Partitioning, and Expression Patterns Between Paralogs in Brassicaceae
Generally, duplicated genes are retained after functional differentiation (Zou et al., 2009), and some analyses indicate that dosage balance sensitivity is a general characteristic of repeatedly retained genes (Gout and Lynch, 2015; Xie et al., 2016; Tasdighian et al., 2017). To test these possibilities, we analyzed the divergence of 96 duplicates (44 paralogous pairs/trios) generated by the WGD that occurred near the origin of the Brassicaceae. Mutations in cis-regulatory regions have been hypothesized to serve as the mechanistic basis for functional differentiation (Zou et al., 2009); therefore, we investigated whether there were changes in the cis-element content in the promoter regions of duplicate AP2/ERF genes in A. thaliana. Specifically, we asked whether there were duplicate pairs with asymmetric partitioning of cis-elements. First, we examined the pattern of partitioning of cis-elements among duplicates and found that it tended to be asymmetric (Supplementary Figure S15). The number of intersections between cis-elements of duplicates was small, suggesting that the cis-elements are retained in both duplicates, possibly causing the duplicates to function in ways complementary to one another. To further test for divergence in expression, we examined expression datasets (Laubinger et al., 2008) and found that paralogs mainly exhibited three differential gene expression patterns (Supplementary Figure S16): (1) one copy predominates in all tissues (TOE1 versus TOE2, ERF017 versus ERF018, DEAR1 versus DEAR2, ERF042 versus ERF043, and ERF114 versus ERF115); (2) one copy had more expression in some tissues (ERF034 versus ERF035, TINY versus TINY2, and TEM1 versus TEM2); and different paralogs had high expression levels in different tissues (AIL6 versus AIL7, ERF110 versus ERF108, and ERF112 versus ERF113).
Sequence Divergence of AP2/ERF Duplicates and Evidence of Positive Selection
Duplicated genes often evolve more rapidly than the ancestral gene before duplication, in a time-dependent process (Pegueroles et al., 2013), presumably due to relaxed selection for the duplicates, compared with the single-copy ancestor. To tested whether post-duplication AP2/ERF genes experienced relaxation of selective constraints relative to their pre-duplication ancestral genes, we used the RELAX program to analyze codon-based alignments of Brassicaceae-specific duplicates and their orthologs in T. cacao and C. papaya and the corresponding phylogenetic tree. Given two categories of branches within a phylogeny, a set of test branches and a set of “reference” branches, RELAX tests whether the strength of selection has been relaxed or intensified in test branches compared with “reference” branches (Wertheim et al., 2015). To compare the strength of selection of different branches before and after duplication, we focused on genes duplicated before the divergence of the extant Brassicaceae species included in this study. We divided the branches in gene trees into three types (Figure 6): Pre-Duplication Branches (PreDB, before the α/β WGD events shared by Brassicaceae taxa), Post-Duplication Branches (PostDB, after the α/β WGD events, but before the divergence of extant Brassicaceae taxa), and Post-Speciation Branches (PostSB, since the first divergence of Brassicaceae lineages). In the AP2/ERF trees, there were 44 pairs/trios (PreDBs) in the Brassicaceae with 96 PostDBs. Compared with the PreDBs, only four PostDBs underwent relaxed selection (Supplementary Tables S4, S5), suggesting that most duplicates have been under selective constraint to a similar extent as their ancestors.
FIGURE 6
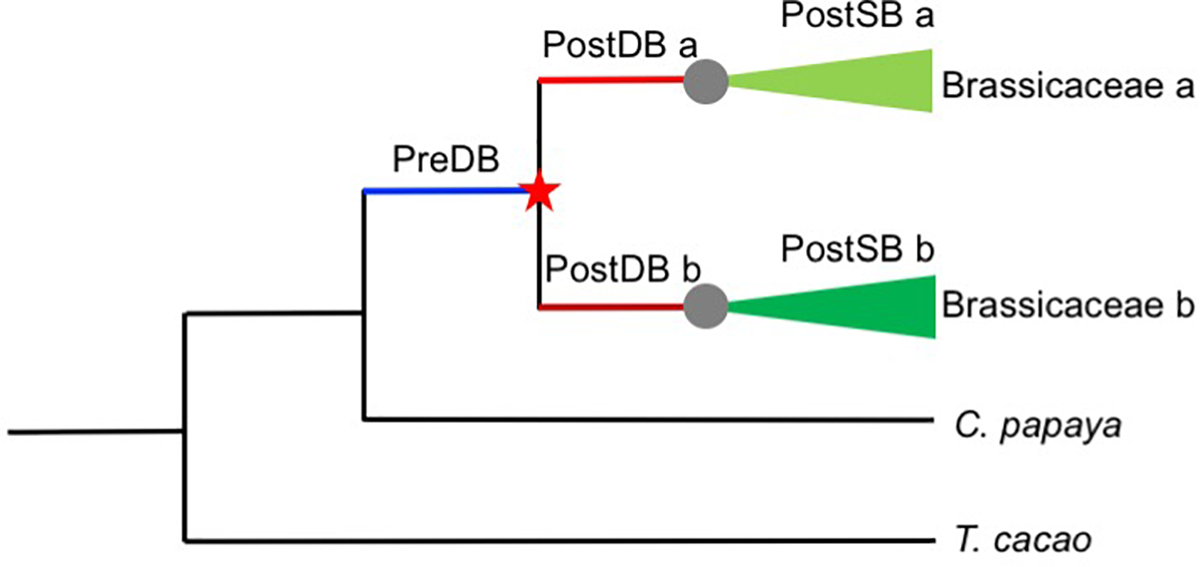
Schematic representation of the AP2/ERF gene duplication data sets employed in this study. Red star indicates a duplication event and gray circles speciation event. Pre-duplication branch (PreDB), Post-duplication branch (PostDB), and Post-speciation (PostSB).
To further examine sequence divergence as evidence for positive selection and functional divergence of duplicates from the α/β WGD events in Brassicaceae, we determined the ω ratio [Ka/Ks, using codon-based models in PAML (Yang, 2007) for 96 PostDBs, as an indication of selective pressure. The mean ω, estimated using the one-ratio model (Model 0), ranged from 0.056 to 0.219 for these duplicates, suggesting that they were under purifying selection during the history of the Brassicales. In addition, analyses using the two-ratio branch model showed that only 17/96 (17.71%) PostDBs differed significantly from the background ω value (Supplementary Table S6, yellow background; P < 0.05), indicating that the majority of PostDBs did not experience a shift in selective pressure, was consistent with the results of analyses using RELAX (see above). Among these 17 branches, 7 were in the AP2 subfamily (12 PostDBs in total), 9 genes were in the DREB/ERF subfamilies (78 PostDBs in total), and 1 gene was in the RAV subfamily (6 PostDBs in total). To examine whether PostDBs were under positive selection before the radiation of the Brassicaceae taxa, we used branch-site models (Yang et al., 2005; Zhang et al., 2005). Of the 96 PostDBs we tested, signals of positive selection were detected in 20 (20.83%) branches from 14 (31.82%) pairs/trios (Table 1 and Supplementary Figure S17, green background; P < 0.05), indicating that functional divergence of PostDBs was, at least in part, related to positive selection and the potential for adaptive evolution in these genes along the Brassicaceae. Among the 20 branches, 4 branches were in the AP2 subfamily, 15 in the DREB/ERF subfamilies, and 1 in the RAV subfamily.
Table 1
| Foreground branch | Model | In L | 2Δl | Site class | Positive selection |
|---|---|---|---|---|---|
| PLT2 | Model A null | 9245.76 | 14.59∗∗ | ω0 = 0.041 ω1 = 1.000 ω2 = 998.999 | 412S∗∗ 498D∗∗ |
| Model A | 9238.47 | ||||
| PLT1 | Model A null | 9245.06 | 9.37∗∗ | ω0 = 0.041 ω1 = 1.000 ω2 = 998.999 | 539T∗∗ |
| Model A | 9240.38 | ||||
| AIL6 | Model A null | 7797.22 | 2.89∗ | ω0 = 0.062 ω1 = 1.000 ω2 = 9.034 | 346N#∗ |
| Model A | 7795.78 | ||||
| TOE1 | Model A null | 4054.23 | 3.59∗ | ω0 = 0.099 ω1 = 1.000 ω2 = 9.123 | 106S∗ |
| Model A | 4052.44 | ||||
| WIND1 | Model A null | 3882.48 | 11.29∗∗ | ω0 = 0.055 ω1 = 1.000 ω2 = 999.000 | 294A∗∗ |
| Model A | 3876.83 | ||||
| ERF053 | Model A null | 3883.26 | 6.07∗∗ | ω0 = 0.099 ω1 = 1.000 ω2 = 12.640 | 270S∗ 282A∗ |
| Model A | 3880.22 | ||||
| ERF017 | Model A null | 3064.50 | 10.10∗∗ | ω0 = 0.057 ω1 = 1.000 ω2 = 999.000 | 79N∗ |
| Model A | 3059.45 | ||||
| ERF014 | Model A null | 3526.90 | 36.35∗∗ | ω0 = 0.056 ω1 = 1.000 ω2 = 1.284 | 31R 32M∗ 109N∗∗ 115S∗ 125A∗∗ |
| Model A | 3508.72 | ||||
| ERF043 | Model A null | 3955.99 | 8.69∗∗ | ω0 = 0.046 ω1 = 1.000 ω2 = 999.000 | 55M#∗∗ 93R∗∗ |
| Model A | 3951.65 | ||||
| CBF2 | Model A null | 4800.62 | 3.83∗ | ω0 = 0.032 ω1 = 1.000 ω2 = 999.000 | 37P |
| Model A | 4798.71 | ||||
| RAP2.2 | Model A null | 4219.29 | 7.69∗∗ | ω0 = 0.078 ω1 = 1.000 ω2 = 999.000 | 152E#∗ |
| Model A | 4215.45 | ||||
| RAP2.10 | Model A null | 2956.35 | 6.48∗∗ | ω0 = 0.112 ω1 = 1.000 ω2 = 998.998 | 180K∗ 181V∗ |
| Model A | 2953.11 | ||||
| ESR1 | Model A null | 4953.85 | 3.01∗ | ω0 = 0.035 ω1 = 1.000 ω2 = 998.993 | 103F 232K∗ |
| Model A | 4952.34 | ||||
| ESR2 | Model A null | 4955.52 | 2.82∗ | ω0 = 0.039 ω1 = 1.000 ω2 = 998.932 | 6M 197I |
| Model A | 4954.11 | ||||
| ERF015 | Model A null | 5763.05 | 2.78∗ | ω0 = 0.065 ω1 = 1.000 ω2 = 9.358 | 25N 133T# |
| Model A | 5761.66 | ||||
| ORA59 | Model A null | 5762.71 | 3.36∗ | ω0 = 0.066 ω1 = 1.000 ω2 = 999.000 | 129L# |
| Model A | 5761.03 | ||||
| ERF100 | Model A null | 3890.53 | 7.91∗∗ | ω0 = 0.051 ω1 = 1.000 ω2 = 999.000 | 104E∗∗ |
| Model A | 3886.57 | ||||
| ERF101 | Model A null | 3892.97 | 16.45∗∗ | ω0 = 0.057 ω1 = 1.000 ω2 = 999.000 | 82D |
| Model A | 3884.75 | ||||
| RAP2.6L | Model A null | 3654.68 | 2.91∗ | ω0 = 0.051 ω1 = 1.000 ω2 = 999.000 | 192Q |
| Model A | 3653.22 | ||||
| RAV1 | Model A null | 10,643.41 | 3.55∗∗ | ω0 = 0.059 ω1 = 1.000 ω2 = 34.664 | 275S∗∗ |
| Model A | 10,641.65 | ||||
List of detected positive selection on the post-duplication branches of AP2/ERF family in Brassicaceae using branch-site model.
Model A null (ω2 is fixed as 1) vs. Model A; degree of freedom (df) = 1 for all the comparisons; In L: the log-likelihood difference between the two models; 2Δl: twice the log-likelihood difference between the two models. The amino acid sequence of A. thaliana was used as the reference sequence, and positive selected sites were identified with posterior probability. For positive selected sites, posterior probabilities (PP) > 90% (>95% indicated with asterisks, >99% indicated with double asterisks) are shown. The arrangement of positive selected sites in A. thaliana AP2 domains are indicated with pound sign.
In addition, paralogs might experience different selective pressures, allowing differential sequence changes, and potentially resulting in functional divergence. To detect such differences between paralogous clades, as evidence for differential functional constraints and selective pressure, we examined paralogous clades for differences in sequence divergence using Clade Model C (Bielawski and Yang, 2003) implemented in PAML (Yang, 2007). This model allows the selective constraint on a proportion of sites to vary among three partitions (0 < ω0 < 1,ω1 = 1,ω2 and 3) for different clades of the phylogeny. Through comparison to a null model that allows two partitions (0 < ω0 < 1,ω1 = 1), Clade Model C tests for a long-term shift in the intensity of selection (i.e., divergent selective pressures). Our results showed that the functional constraints (as evidence for shift in selective pressure) were not similar between paralogous clades in 43 of 44 pairs/trios (97.73%; P < 0.05) (Supplementary Table S6), suggesting that natural selection, likely mediated via ecological niches, has had a substantial influence on AP2/ERF duplicate evolution in the Brassicaceae. To identify amino acid sites likely responsible for functional changes between two paralogs, we used the DIVERGE program (Gu et al., 2013), which is based on functional constraint patterns in MSAs and corresponding gene trees. Significant support for Type-I functional divergence (see the section “Materials and Methods”) was identified between paralog clades in 44/58 pairs (75.86%) (Supplementary Tables S7, S8). Hence, sequence divergence results from both Clade Model C and DIVERGE analyses strongly support that a majority of AP2/ERF paralogous pairs generated in the early Brassicaceae have subsequently experienced differential selective pressures and undergone functional divergence. This is likely due to the long-term selection pressures acting on Brassicaceae species.
Evolutionary Rates and Structural Disorder Across Brassicaceae Paralogs
To examine whether different regions/sites of AP2 proteins experienced more rapid or slower transition than other regions/sites, evolutionary rates for amino acid sequence substitutions were calculated based on a MSA of Brassicaceae-specific paralogous pairs and their orthologs in T. cacao and C. papaya, with the corresponding gene trees, using Rate4Site v3.2 (Mayrose et al., 2004). The results showed that amino acids located in the AP2 domain substituted more slowly than those in regions flanking this domain (Supplementary Figure S17). In addition, there were also some other slowly transitioning sites, such as those C-terminal of the AP2 domain in subfamilies II and III, suggesting that they are functionally important and experienced purifying selection. Rapidly transitioning sites were usually only conserved on the level of pairs of paralogs, rather than the subfamily level, suggesting different functional constraints in different paralogs.
In addition, we investigated the propensity for structural disorder and potential protein binding sites in the amino acid sequences of AP2/ERF duplicates retained in Arabidopsis using DISOPRED3 (Jones and Cozzetto, 2015). Disordered regions were predicted in each AP2/ERF protein and their locations in pairs of paralogs were generally conserved. The presence of disordered regions in each AP2/ERF protein suggests the importance of conformational flexibility for their functions (Supplementary Figure S18). The regions C-terminal of the AP2 domains in members of the DREB II and III subfamilies are predicted to be ordered. In addition, members of the DREB III subfamily also possess ordered regions near the C terminus (Supplementary Figure S18). In the ERF VI subfamily, the N-terminal region flanking the AP2 domain is ordered, with an adjacent disordered region. Also, the linker regions between two AP2 domains are generally ordered in members of the AP2 subfamily. These profiles indicate that the positions of disordered regions are similar among different subfamilies, suggesting that these regions are prone to be disordered, or that these disordered regions are functionally important. We then compared the evolutionary rates of the predicted disordered and ordered regions, and found that disordered regions have higher evolutionary rates than those of ordered regions (P < 2.2e-16, Wilcoxon Rank-Sum test) (Supplementary Figure S19).
Inferred Ancestral Expression Supports Subfunctionalization of Widely Expressed AP2/ERF Genes With Repeated Retention Patterns
Several duplicate AP2/ERF genes show differential expression patterns, providing an opportunity to test the idea that widely expressed AP2/ERF genes with strong, repeated retention patterns experience subfunctionalization following duplication. In one case, three Arabidopsis AP2/ERF genes in the same clade differed in spatial and temporal expression patterns, with one encoding SHINE1/WAX INDUCER1 (SHN1/WIN1), a regulator of cuticular wax synthesis and flower development (Aharoni, 2004). Reconstruction of the ancestral expression states allows inference of gain and loss of expression among gene duplicates (Zou et al., 2009). Therefore, we inferred the expression patterns of the ancestral SHINE clade genes in root, stem, leaf, flower, and fruit, as described below. In the SHINE clade, Va-AOG2, three copies were generated by the core eudicot-specific WGD (γ): the AtSHN1 clade, the AtSHN2/AtSHN3 clade, and another clade lacking genes in A. thaliana (Figure 7A and Supplementary Figure S6B). In the AtSHN1 clade, AtSHN1 expression was detected in flower, fruit, and root tissues, but not in stem and leaf (Aharoni, 2004); GmSHN1 and GmSHN2 expression were detected only in flower, and GmSHN8 expression was detected in leaf and fruit (Xu et al., 2016). In the AtSHN2/AtSHN3 clade, AtSHN2 expression was detected in flower and fruit; AtSHN3 was most broadly expressed, with transcription in all plant organs (Aharoni, 2004); GmSHN9 was expressed in adult stem and leaf; GmSHN5 in adult stem, flower, and fruit; and GmSHN10 in adult stem, leaf, and fruit (Xu et al., 2016). In the clade without A. thaliana genes, GmSHN3 and GmSHN4 were detected in all plant organs, with GmSHN6 and GmSHN7 in root, stem, and flower (Xu et al., 2016). AtSHN3 and GmSHN3/GmSHN4 were detected in all plant organs; however, they are in different clades. According to Occam’s Razor and the rule, “degenerative mutations are much more frequent than beneficial mutations” (Force et al., 1999), we speculate that the single ancestral gene in the MRCA of extant angiosperms of SHINEs should be expressed in all plant organs (Figure 7A). Expression of some genes in the SHINE clade was lost or significantly reduced in some tissues. We speculate that the ancestral gene of the AtSHN2/AtSHN3 clade was expressed in all plant organs and that the ancestral gene of the AtSHN1 clade was expressed in root, leaf, flower, and fruit. The tissue specificity of the ancestral gene of the AtSHN2/AtSHN3 clade was lower than that of the ancestral gene of the AtSHN1 clade. We observed more retention after WGD events in the AtSHN2/AtSHN3 clade than the AtSHN1 clade in different plant groups, confirming the model, “preferential retention of AP2/ERF genes with wide expression.”
FIGURE 7
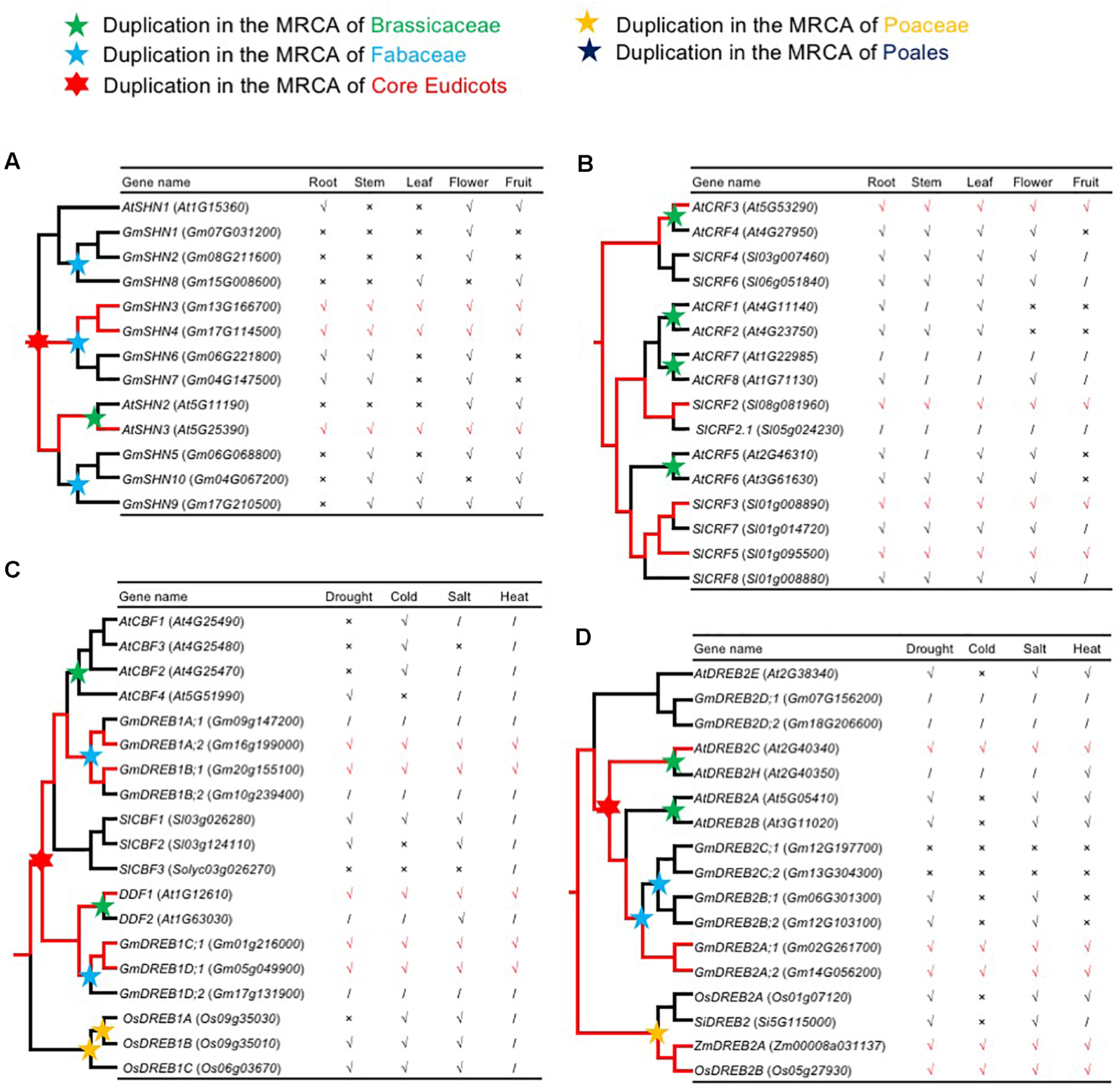
The reconstruction of ancestral expression state. (A,B) The ancestral expression profiles of SHNs and CRFs in roots, stems, leafs, flowers, and fruits were inferred. Tick, Cross, and Slash indicate that gene expression is detected expression, not detected expression, and none data, respectively. Putative ancestral expression state is highlighted in red. (A) The reconstruction of ancestral expression of SHNs. Expression data of A. thaliana SHNs were obtained from previous studies (Aharoni, 2004). Expression data of G. max SHNs were obtained from previous studies (Xu et al., 2016). (B) The reconstruction of ancestral expression of CRFs. Expression data of CRFs from A. thaliana (Zwack et al., 2012; Jeon et al., 2016) and S. lycopersicum (Shi et al., 2012; Gupta and Rashotte, 2014; Shi et al., 2014) were obtained from previous studies. (C,D) The ancestral expression profiles of DREB1s and DREB2s in response to cold, drought, salt, and heat stress were inferred. Tick, Cross, and Slash indicate that gene expression is up-regulated and not changed and none data, respectively. Putative ancestral stress responses are highlighted in red. (C) The reconstruction of ancestral stress responses of DREB1s. Expression data of DREB1s from A. thaliana (Medina et al., 1999; Kang et al., 2011) and G. max (Kidokoro et al., 2015) were obtained from previous studies. (D) The reconstruction of ancestral stress responses of DREB2s. Expression data of DREB2s from A. thaliana (Krishnaswamy et al., 2011), S. italica (Lata et al., 2011), O. sativa (Matsukura et al., 2010), and Z. mays (Qin et al., 2007) were obtained from previous studies.
Cytokinin response factor genes (CRFs) encode TFs belonging to a small family within the ERF VI subfamily [Figure 3(VI) and Supplementary Figure S7B]. Recent studies have revealed the biological functions of some Arabidopsis CRFs, providing insight into the role of these plant TFs in integrating environmental and hormonal signals for plant adaptation. There are three clades in ERF VI-AOG2, each of which had at least one gene with expression detected in all organs (AtCRF3, SlCRF2, SlCRF3, and SlCRF5) (Shi et al., 2012; Zwack et al., 2012; Gupta and Rashotte, 2014; Shi et al., 2014; Jeon et al., 2016) (Figure 7B), indicating that their single ancestral gene, in the MRCA of extant angiosperms, was also expressed in all plant organs, and that other genes originated from this single ancestral gene may have lost, or significantly reduced, their expression in some tissues.
Some AP2/ERF genes are responsive to various environmental stressors, while others are only responsive to a few environmental stressors. To test the hypothesis that some AP2/ERF genes lost responsiveness, or became less responsive to stressors, we inferred the stress response patterns of ancestral AP2/ERF genes. CBF/DREB1 is reported to be associated with regulation of gene expression under stress conditions. DDF1 and DDF2 are induced by salt and confer tolerance to salt stress when ectopically expressed (Magome et al., 2004). These two clades were generated by core eudicot-specific WGD (γ) in DREB IIIc-AOG2 [Figure 3(III) and Supplementary Figure S4F], and each clade has genes responsive to drought, salt, cold, and heat (GmDREB1A;2 and GmDREB1B;1 in the CBF clade; AtDDF1, GmDREB1C;1, and GmDREB1D;1 in the DDF clade) (Medina et al., 1999; Magome et al., 2004; Kang et al., 2011; Kidokoro et al., 2015). These results indicate that the single ancestral gene of IIIc-AOG2 was likely drought, salt, cold, and heat responsive, and that some genes have retained more ancestral stress responses, while others have lost, or significantly reduced, these responses (Figure 7C). AtCBF1-3 genes are induced under cold stress, while AtCBF4 gene expression is up-regulated by drought stress, but not by low temperature. Overexpression of AtCBF4 in transgenic Arabidopsis plants results in activation of downstream genes involved in cold acclimation and drought adaptation. Consequently, the transgenic plants are more tolerant to freezing and drought stress (Novillo et al., 2004). Given the physiological similarity between freezing and drought stress, and the sequence and structural similarity of the AtCBF/DREB1 and AtCBF4 proteins, we propose that a plant responsive to cold and drought evolved with a common CBF-like TF, first through gene duplication and then via the evolution of promoter and coding sequences. Some DREB2 genes in DREB IVa-AOG1 [Figure 3(IV) and Supplementary Figure S4F] are also responsive to drought, salt, cold, and heat (Qin et al., 2007; Matsukura et al., 2010; Krishnaswamy et al., 2011; Lata et al., 2011), which also indicates that the single ancestral gene of DREB IVa-AOG1 was broadly responsive to stress (Figure 7D), further implying that broadly responsive AP2/ERF genes are more likely to be retained after WGD events.
Discussion
Highly Similar Patterns of AP2/ERF Duplicate Retention Across all Angiosperms
Gene duplication is a major source of evolutionary novelty and plays a key role in speciation (Ren et al., 2018). Here, we found that the MRCA of extant angiosperms possessed at least 75 AP2/ERF genes, indicating numerous duplication events and substantial preexisting functional diversity before angiosperm radiation. Further analyses of the duplicate retention patterns of AP2/ERF genes in 37 angiosperm genomes indicated an important contribution of WGD to the increase in AP2/ERF numbers. Although most duplicates originating from WGD were subsequently lost (Li et al., 2016; Ren et al., 2018), a substantial fraction of AP2/ERF duplicates was retained after WGD events of different ages in the MRCA of core eudicots, Brassicaceae, Fabaceae, Asteraceae, Commelinids, Poales, and Poaceae. The AP2/ERF duplicate retention patterns following these WGD events were highly similar across angiosperm genomes, with over 72% (54/75) of AOGs maintaining a low-copy status, whereas a smaller set (28%, 21/75) retained duplicates from multiple lineage-specific WGD events. Relatively few gene families have expanded greatly during evolution (i.e., there is “dominance by a selected few”) (Karev et al., 2002, 2003, 2004; Novozhilov et al., 2006), with the size distribution of gene families in many genomes being similar to a generalized Pareto distribution. Our results, indicating that only 21 (28%) among 75 AOGs have expanded, suggesting that “dominance by a selected few” may also hold true at the level of OrthoGroups within a gene family.
An inspection of the annotated functions of AP2/ERF genes shows that stress-related AP2/ERF genes are enriched in the multi-copy group in Arabidopsis. Among 87 Arabidopsis AP2/ERF genes in the multi-copy group, 38 (43.68%) are related to stress responses, including AtWINDs (Iwase et al., 2016), AtDREB2s (Chen et al., 2012), AtCBFs (Kang et al., 2013; Haake et al., 2016), AtSHN1 (Aharoni, 2004), AtCRFs (Jeon et al., 2016; Kim, 2016), AtDDFs (Kang et al., 2011), AtHREs (Licausi et al., 2010), and AtRAP2.6L (Krishnaswamy et al., 2011) (Figure 3 and Supplementary Table S1). Indeed, every annotated AOG in the DREB/ERF family multi-copy group had at least one stress response-related gene. Among 57 Arabidopsis AP2/ERF genes in the low-copy group, only 12 (20.34%) are related to stress responses, with 18 (30.51%) associated with development, including WRI1 (fatty acid biosynthesis) (To et al., 2012), FUF1 (flower senescence) (Chen et al., 2015), TINY (shoot development) (Wilson, 1996; Wei et al., 2005), DRN and DRNL (morphogenesis), PUCHI (morphogenesis) (Chandler and Werr, 2017), and genes in the AP2 family (Licausi et al., 2013). Thus, in addition to previous findings of preferential retention of duplicates involved in signaling and transcriptional regulation (Li et al., 2016; Ren et al., 2018), our results suggest further differential retention of duplicates in the same TF family, with preference for OrthoGroups involved in environmental responses, possibly contributing to adaption to highly variable environmental conditions, consistent with morphological and ecological evolutionary findings in angiosperms (Zeng et al., 2014).
Lineage-Specific Expansions and Their Role in Adaptation to Aquatic Environments
Tolerance to submergence and low oxygen availability (hypoxia) are influenced by different members of subfamily VII in Arabidopsis [AtRAP2.12; AtRAP2.2 (Licausi et al., 2011; Paul et al., 2016); AtRAP2.3 (Paul et al., 2016); AtHRE1 and AtHRE2 (Licausi et al., 2010)] and rice [(OsSUB1s (Xu et al., 2006); OsSK1 and OsSK2 (Hattori et al., 2009)]. The copy number of the VII subfamily was almost unchanged from the MRCA of angiosperms (4) to the MRCA of each major plant group [(Brassicaceae (5), Fabaceae (5), Asteraceae (4), core eudicots (4), Commelinids (5), and Poales (5)], but it was significantly expanded in Seagrasses (17), and slightly expanded in duckweed (7) and sacred lotus (7). Specifically, AtRAP2.12, AtRAP2.2, and AtRAP2.3 are important for hypoxia survival (Paul et al., 2016), and their orthologs are also duplicated in seagrasses, duckweed, and sacred lotus (Supplementary Figures S8A,D). There are three AtRAP2.3 orthologs in sacred lotus. Similarly, rice SUB1A, SUB1B, SUB1C (Xu et al., 2006), SK1, and SK2, are orthologs of AtHRE1, and contribute to adaptation to deep water (Hattori et al., 2009) and improved water use efficiency, as do paralogs in VIIa-AOG3 from Poaceae-specific and rice-specific duplications (Supplementary Figure S8C). The orthologs of AtHRE1 in seagrasses (eight duplicates) also expanded independently, while those in duckweed and sacred lotus did not (Supplementary Figure S8C), suggesting different pathways during these parallel, but independent, colonization events. AtHRE2 has five orthologs in seagrass, three in duckweed, and two in sacred lotus (Supplementary Figure S8B). The rice ERF-VII proteins contain an N-terminal sequence, the MC motif, that is important for oxygen-dependent degradation of these proteins, rendering them more stable under hypoxia and causing increased transcriptional activation of genes induced by low-oxygen (Licausi et al., 2011). Six seagrass homologs of OsSUB1A-1 and one duckweed homolog (Nenu.14844) of AtRAP2.3 lack the MC motif, likely making their stable insensitivity to oxygen constitutively active. In addition, several ERF-VII TFs in seagrass and duckweed have altered amino acids in the MC motif (Supplementary Figure S20). These changes in crucial regulators of low-oxygen responses may have had important functions in adaptation to persistent hypoxic environments.
Preferential Retention of Widely Expressed AP2/ERF Genes Following Subfunctionalization
Our data reveal that the multi copy AP2/ERF genes, which exhibit repeated duplicate retention patterns in angiosperms (i.e., the AP2/ERF genes that show the strongest patterns of preferential retention after WGD are repeatedly retained in the genomes of angiosperms), did not evolve more rapidly, and some even evolved more slowly, on average than low-copy genes. Slower rates of protein evolution correlate with higher levels, and greater breadth, of expression, as well as a larger number of protein interactions (Liao et al., 2007; Yang and Gaut, 2011). Hence, we compared the expression patterns of the two groups of genes and found that, across the angiosperms as a whole, widely expressed AP2/ERF genes were more likely to be retained than those with narrower expression profiles, consistent with the preferential subfunctionalization of slow-evolving genes (Sémon and Wolfe, 2008). These findings suggest that, consistent with previous findings, the evolutionary trajectory of AP2/ERF duplicates may possess two stages: post-duplication functional divergence, followed by a generally slow evolutionary rate, owing to the higher level of functional constraints after functional divergence (Davis and Petrov, 2004; Pegueroles et al., 2013). Next, we asked a different type of question: namely, why are some types of gene are more likely to be duplicated than others? Several models of duplicate gene preservation have explained why slowly evolving genes may have an increased likelihood of being preserved. Some models predict that repeatedly retained genes will be retained as duplicates following WGD events, simply because they are beneficial for gene dosage (Gout and Lynch, 2015; Xie et al., 2016; Tasdighian et al., 2017). Other models predict the preferential preservation of genes expressed in numerous tissues (Force et al., 1999), or of genes that encode multidomain proteins (Liang and Li, 2007). As degenerative mutations are much more frequent than beneficial alterations, it follows that subfunctionalization may be a much more common mechanism of duplicate-gene preservation than neofunctionalization (Force et al., 1999); our results, and several examples of AP2/ERF genes, support the latter model. These observations lead us to propose that widely expressed genes are more easily subfunctionalized, and therefore more easily retained, after WGD events. Our model of gene evolution after WGD is illustrated in Figure 8.
FIGURE 8
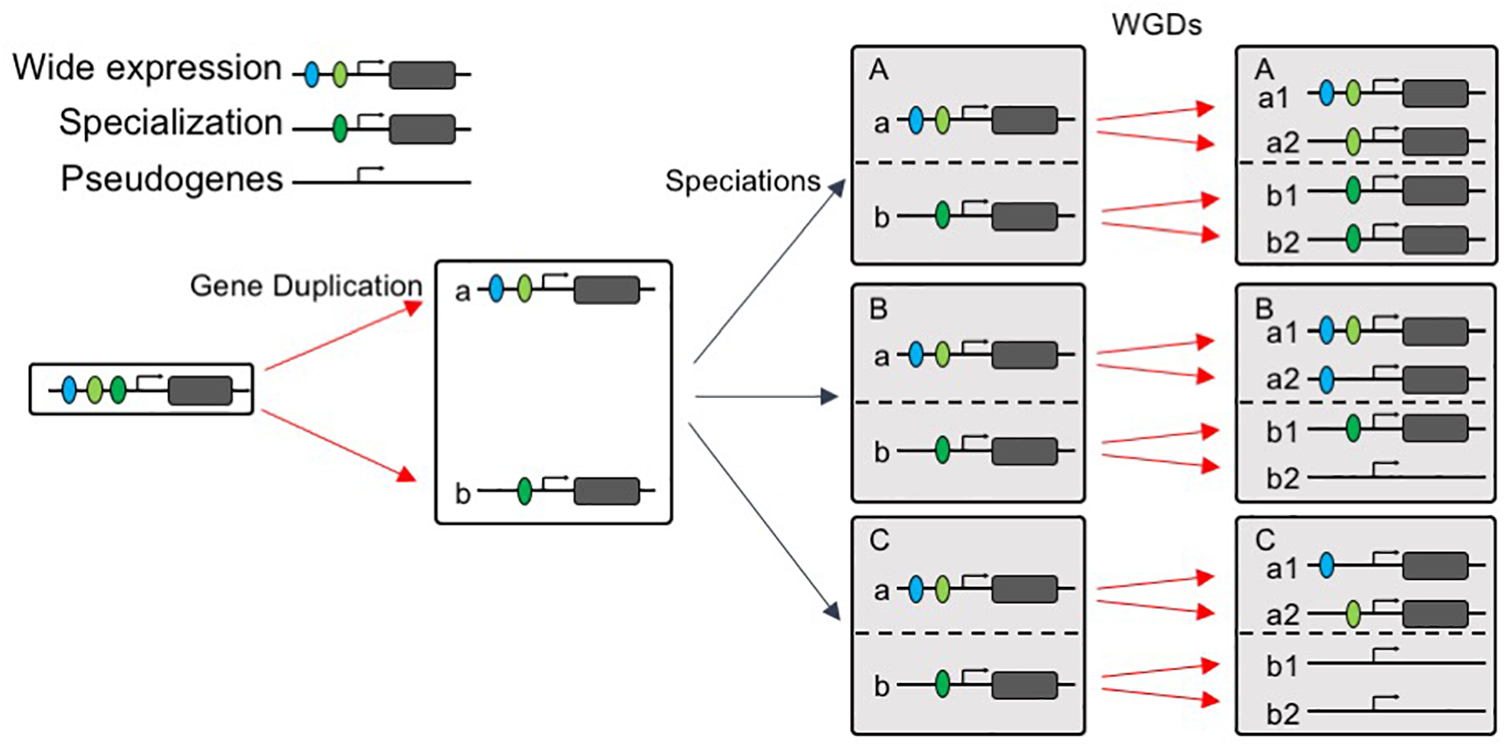
“Wide expression-high duplicability” model. A widely expressed gene were subfunctionalized into gene a and gene b after duplication, with gene a expressed more widely than gene b. After speciations, species A, B, and C all retained gene a and gene b, when species A, B, and C experienced WGD, wide expressed gene a tends to be retained in all species and one or both copies subfunctionalized, but gene b with narrower expression tends to be retained in few species.
Paralogs exhibit differential rates of evolution and differential shifts in expression patterns (Wagner and Conant, 2003). In our model, pairs of diverging sequences also accumulate differences in gene duplicability, but to a greater extent in genes with narrower expression than those with wide expression patterns. When another WGD event occurs, the widely expressed AP2/ERF genes are more likely to be retained again and subfunctionalized into various expression patterns in different species. The fact that early WGD gene duplicates are expressed more specifically than those from recent WGD events (Yang and Gaut, 2011) may also be the result of preferential retention of the more widely expressed paralog. In contrast, the AP2/ERF genes that are already specialized only tended to be duplicated in specific lineages, possibly allowing further adaptation to specialized niches.
Specialization of AP2/ERF Genes With Broad Abiotic Responses Can Enhance Resistance and Minimize Growth Inhibition
AP2/ERF genes are important for stress responses and development. For example, AtCRFs (1–8) are paralogs from a single ancestral angiosperm gene, with important roles in multiple aspects of plant defense, growth, and development (Kim, 2016). Genes in the SHINE clade activate wax biosynthesis, confer drought tolerance when overexpressed in Arabidopsis (Aharoni, 2004), and influence the surface pattern of Arabidopsis flower organs (Shi et al., 2011). Also, some AP2/ERF TFs can function as a nexus between stress and development. For example, RAP2.4 acts at, or downstream of, the convergence point of light and ethylene signaling pathways, to coordinately regulate multiple developmental processes and stress responses (Lin et al., 2008). These functions of AP2/ERF genes in growth and defense may originate from common ancestral functions.
Plants must both grow and defend themselves to survive and reproduce, with trade-offs between growth and defense (Huot et al., 2014). While the deployment of defense mechanisms is imperative for plant survival, defense activation generally comes at the expense of plant growth. Therefore, evolutionarily, plants have had to develop efficient strategies to balance growth and defense. Overexpression of some AP2/ERF genes in transgenic Arabidopsis plants often causes dwarfed phenotypes, because they induce strong expression of target genes under unstressed conditions (Supplementary Table S1) (Licausi et al., 2013). Some AP2/ERF genes, such as AtDDF1, AtDREB2C, ZmDREB2A, and OsDREB2B, are broadly up-regulated by cold, drought, and heat, which may improve plant adaptability to adversity; however, plants may invest in an abiotic defense against one stress factor, but not require defenses against other stressors. Some AP2/ERF genes are only up-regulated by single abiotic stressors. For example, AtCBF1–3 are induced under cold stress (Novillo et al., 2004; Kang et al., 2013), while AtCBF4 gene expression is up-regulated by drought stress (Finn et al., 2016). Overexpression of AtCBF4 results in constitutive expression of both cold- and drought-inducible AtCBF1–3 target genes, and the transgenic plants are more tolerant to freezing and drought stress (Haake et al., 2016); these phenotypes have also been observed for the constitutive overexpression of AtCBF1–3 (Novillo et al., 2004; Kang et al., 2013; Zhou et al., 2017), indicating that cold and drought resistance may have been the ancestral function of the MRCA of AtCBF1–4. Therefore, we propose that the response of the plant to cold and drought has evolved from a common CBF-like TF and that, after duplication, AtCBF1–3 and AtCBF4 subfunctionalized to become cold- and drought-inducible, respectively. AtCBF1, 2, and 3 also exhibit different patterns of induction in response to low temperatures (Novillo et al., 2004), which may facilitate more precise regulation of cold responses. There are also some specific abiotic-responsive and broadly abiotic-responsive DREB2 genes in DREB IVa-AOG1 (Figure 8; Supplementary Figure S4F). For example, both AtDREB2A and AtDREB2C can be up-regulated by heat stress (Chen et al., 2012); however, AtDREB2C also has an important role in promoting oxidative stress tolerance in Arabidopsis (Hwang et al., 2012). Hence, we speculate that other specific abiotic responsive AP2/ERF genes may also have evolved from broadly abiotic responsive AP2/ERF genes by subfunctionalization. Subfunctionalization of broadly abiotic responsive genes can simultaneously result in enhanced resistance to specific abiotic stress and minimize the inhibition of plant growth. Considering the diversity of ecological niches colonized by angiosperms, it is conceivable that having multiple AP2/ERF paralogs does not merely increase the expression of AP2/ERF genes, but also leads to the expression of different specialized forms of the AP2/ERF TFs, facilitating adaption to new environmental conditions.
Divergence of AP2/ERF Duplicates Facilitates More Sophisticated Regulation of Signaling Networks and Diversification
Some AP2/ERF duplicates generated by WGD in early core eudicots (γ) are conserved and diversified in their biological functions, such as CRFs (Supplementary Figure S7B), which regulate various aspects of plant growth and development, and mediate interactions between hormonal signaling and the environmental stress response (Supplementary Table S1) (Kim, 2016). In contrast, some AP2/ERF duplicates, corresponding to WGD in early core eudicots (γ) are already involved in different pathways, suggesting functional specialization or neofunctionalization. For example, the two clades AtEBE/AtERF115 and AtRAP2.6L/AtERF112 arose due to WGD in early core eudicots (γ) (Supplementary Figure S11B). ERF115 controls root quiescent center cell division and stem cell replenishment (Heyman et al., 2013). EBE influences shoot architecture in Arabidopsis (Mehrnia et al., 2013), and RAP2.6L functions in graft healing and delayed waterlogging-induced premature senescence (Matsuoka et al., 2018). The spatiotemporal expression of PUCHI, DRN, and DRNL is largely discrete in the inflorescence meristem and early floral meristem; however, they contribute interdependently to cell fate decisions (Chandler and Werr, 2017). The Clade Model C and DIVERGE analyses implemented in this study identified substantial divergent selection in Brassicaceae AP2/ERF duplicates, showing that retained AP2/ERF duplicates likely underwent functional divergence, potentially in adapting to various ecological niches (Supplementary Tables S6, S7). Some Brassicaceae-specific AP2/ERF duplicates have reported differences in expression, protein–protein interaction, and binding preferences (Supplementary Table S9).
Transcription factors are intricately regulated by conformational changes involving allosterism (Phukan et al., 2017). Disordered peptides in proteins can provide conformational flexibility, and such flexible disordered peptides are strongly associated with signaling and regulation (Wright and Dyson, 2014). Some functions of disordered regions have been experimentally verified. Both the N- and C-terminal regions of AP2/ERFs often act as transactivation or repression domains, which regulate expression of downstream genes (Phukan et al., 2017). For example, deletion of the C-terminal intrinsically disordered region of WRI1 affects its stability (Ma et al., 2015). Also, the disordered C-terminal region of TOE1 (294–449) is necessary and sufficient for interaction with the flowering time protein, CO (Zhang et al., 2015). The CRF domain from the N-terminal region of CRFs alone is sufficient for homo- and hetero-dimerization of CRF proteins and for interaction with the histidine-phosphotransfer proteins, AHP1–AHP5 (Cutcliffe et al., 2011). Increased purifying selection means greater functional constraint and a lower evolutionary rate, thus changes in evolutionary rate can be interpreted as “changes in functional constraints,” which is an indication of functional divergence (Gu et al., 2013). We found that disordered regions have higher evolutionary rates than ordered regions, suggesting that duplicates may diverge in these regions. Thus, mutations could contribute to structural flexibility, conferring a selective advantage in novel regulatory mechanisms, such as interactions and post-translational modifications. For example, TOE1 can interact with the related CO, COL1, COL2, COL3, and COL5 proteins; however, TOE2 can only interact with COL1 and COL5 (Zhang et al., 2015). In addition, among the proteins CRF1–CRF6, only CRF2 and CRF3 do not interact with AHP2 (Cutcliffe et al., 2011). Additional studies in the near future will likely further improve the understanding of the function and evolution of AP2/ERF genes and their impact on development and environmental responses.
Conclusion
Our study reveals the evolutionary landscape of the AP2/ERF family in angiosperms. We found that AP2/ERF protein sequences can be classified into 75 AOGs. For most AOGs, we observed a highly consistent pattern of gene duplicability within the same AOG across species. Specifically, one group (multi-copy) comprising 21 of 75 AOGs has experienced significant expansion in multiple angiosperm lineages, except the basal angiosperms and basal eudicots. In contrast, a second group (low-copy), comprising the other 54 AOGs, tends to have maintained a low-copy status, with some having only a few lineage-specific duplicates and others with lineage-specific losses. We also found that multi-copy genes did not evolve more rapidly than low-copy genes, and that multi-copy genes are expressed with lower specificity than low-copy genes. Moreover, in gene groups with two successive duplications, between two older AP2/ERF duplicates, the one with relatively broad expression and slower evolutionary rates was more likely to retain both younger duplicates. Duplicates of widely expressed and slower evolving AP2/ERF genes are retained repeatedly and relatively often, and continue to experience functional constraint. One possible explanation is that the widely expressed AP2/ERF genes may have a greater chance of subfunctionalization (e.g., tissue specialization, specific responses) following duplication, and the newly subfunctionalized genes can facilitate more precise regulation of signaling networks or adaption to new environmental conditions. AP2/ERF genes that were already specialized (narrower expression) tended to be duplicated in specific lineages, possibly allowing further adaptation to special niches, or lost in some lineages, which changed their ecological niches.
Statements
Author contributions
HM and JL designed the study. LW carried out evolutionary analysis. LW and JL drafted the manuscript and HM revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by National Natural Science Foundation of China (31370346 to JL and 91531301 and 31670209 to HM) and State Key Laboratory of Genetic Engineering.
Acknowledgments
We are grateful to Drs. Yaqiong Wang, Ren Ren, Chien-Hsun Huang, Caifei Zhang, Chunce Guo for assistance and discussion.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00196/full#supplementary-material
Figure S1Phylogenetic trees of the AP2 subfamily from representative Angiosperms. SH-aLRT supports (approximate likelihood-ratio test and relies on a nonparametric, Shimodaira–Hasegawa–like procedure) above 50% are labeled on internal nodes. The MRCA (circles) and gene duplication events (stars) of each major plant group (Brassicaceae, Fabaceae, Asteraceae, core eudicots, Poaceae, Poales, Commelinids, Monocots, and Angiosperms) are indicated on internal nodes in different colors (green, light blue, purple, red, orange, deep blue, blue, deep red, and black). The numbers in the top left textbox represent the minimum inferred AP2/ERF protein complement and gene duplication events in the last common ancestor of each major plant group.
Figure S2Phylogenetic trees of the DREB I subfamily from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S3Phylogenetic trees of the DREB II subfamily from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S4Phylogenetic trees of DREB III subfamily from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S5Phylogenetic trees of DREB IV subfamily from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S6Phylogenetic trees of ERF V subfamily from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S7Phylogenetic trees of ERF VI subfamily from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S8Phylogenetic trees of ERF VII subfamily from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S9Phylogenetic trees of ERF VIII subfamily from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S10Phylogenetic trees of ERF IX subfamily from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S11Phylogenetic trees of ERF X subfamily from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S12Phylogenetic trees of RAV subfamily from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S13A phylogenetic tree of Soloist from representative Angiosperms. SH-aLRT supports above 50% are labeled on internal nodes. The labeling is the same as in Supplementary Figure S1.
Figure S14Boxplots showing Ka, Ks, and ω (Ka/Ks) values for each duplicated clades generated near the origin of Brassicaceae. The duplicates pairs generated near the origin of Brassicaceae are separated into D1 and D2 according to their copy number of AP2/REF genes from L. alabamica(A–E) or B. rapa(F–J), with the D1 having more gene copy in L. alabamica(A–E) or B. rapa(F–J). All Ka, Ks, and ω (Ka/Ks) values were calculated by comparison of orthologous pairs from selected species. Values in A–C and F–H were calculated by comparison of orthologous pairs between A. thaliana and B. stricta sequence. Values in D–F and I–K were calculated by comparison of orthologous pairs between A. thaliana and E. salsuginea sequence. Values were compared using the Wilcoxon rank signed rank tests and P-values are shown above each pair of boxplots.
Figure S15Cis-regulatory element partitioning among duplicates. (A) Subfunction measure (Sub) and asymmetry measure (Asy) definition. Asy was calculated based on the number of cis-regulatory element. Extreme asymmetry was defined based on Asy = 1. Sub was calculated based on the intersection and union of cis-regulatory element of duplicate pairs. Extreme subfunctionalization was defined based on Sub = 0. The cis-regulatory element of each genes is de-duplicated, before the Sub was calculated. (B) Relative frequencies of the number of duplicate pairs with various degrees of asymmetric partitioning. (C) Relative frequencies of the number of duplicate pairs with various degrees of subfunctionalized partitioning. Majority of cis-regulatory element of extant genes was assumed to be ancestral, new cis-regulatory element was assumed to be very little.
Figure S16Gene expression data from At-TAX tilling array in 11 tissues of Arabidopsis AP2/ERF genes. (1) Roots, (2) seedlings, (3) expanding leaves, (4) senescing leaves, (5) stem, (6) vegetative shoot meristem, (7) inflorescence shoot meristem, (8) whole inflorescences, (9) whole inflorescences (clv3-7), (10) flowers, and (11) fruits. Brassicaceae-specific AP2/ERF paralogs were indicated by green left brackets. AOGs duplicates corresponding core eudicots WGD (γ) and Brassicaceae WGD (α/β) were indicated by red brackets. Values to the left of brackets are Pearson’s correlation coefficients for correlated expression between Brassicaceae-specific AP2/ERF paralogs.
Figure S17Normalized evolutionary rates per site mapped onto the amino acid sequence. All evolutionary rates were normalized with a mean of zero and standard deviation of one. The confidence interval for the rate estimates was show as error bar. The AP2 domain were colored as cornsilk, the B3 domain were colored as light yellow. Coiled coil region was colored as green. Positive selected sites were indicated by red point; Type I functional divergence amino acid sites that are highly conserved were indicated by yellow point; Type I functional divergence amino acid sites that are variable were indicated by skyblue point. Blue braces represent subfamilies; red brace represent Brassicaceae-specific paralogous pairs.
Figure S18The region besides AP2 domain is intrinsically disordered. Disorder states are indicated as blue lines; protein-binding disordered residues are indicated as orange lines. Blue braces represent subfamilies; red brace represents Brassicaceae-specific paralogous pairs. The AP2 domain were colored as cornsilk, the B3 domain were colored as lightyellow.
Figure S19Violin plots of evolutionary rates per site. The median number of evolutionary rates are shown by the red dot. Values were compared using the Wilcoxon rank-sum test and P-values are shown above the Violin plots.
Figure S20MAFFT alignment of ERF VII proteins from representative species with a N-terminal MC-motif (Met–Cys–Gly–Gly–Ala–Ile/Leu/Val, MCGGAI/L/V).
Table S1Functionally characterized divergences of AP2/ERF gene from Arabidopsis.
Table S2Taxa included in this study and data collection.
Table S3A list of AP2/RF genes included in this study.
Table S4Test for shifts in selective pressures Using RELAX (Wertheim et al., 2015). The parameter k is the estimated selection intensity; values under and above 1 indicate relaxed and intensified selection in relative Branch.
Table S5RELAX model fits. ω distributions under the Null model and the Alternative model.
Table S6Estimates of parameters used in codon-substitution evolutionary models for AP2/ERF paralogs in Brassicaceae.
Table S7Analysis of FD by DIVERGE.
Table S8Sites with Type I functional divergence (FD-I) in AP2/ERF functional diversification.
Table S9Functionally characterized divergences of AP2/ERF genes from A. thaliana.
Footnotes
1.^https://phytozome.jgi.doe.gov/pz/portal.html
5.^https://www.sunflowergenome.org
References
1
AharoniA. (2004). The SHINE clade of AP2 domain transcription factors activates wax biosynthesis, alters cuticle properties, and confers drought tolerance when overexpressed in Arabidopsis.Plant Cell162463–2480. 10.1105/tpc.104.022897
2
Amborella Genome Project (2013). The Amborella genome and the evolution of flowering plants.Science342:1241089. 10.1126/science.1241089
3
BadouinH.GouzyJ.GrassaC. J.MuratF.StatonS. E.CottretL.et al (2017). The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution.Nature546148–152. 10.1038/nature22380
4
BielawskiJ. P.YangZ. (2003). “Maximum likelihood methods for detecting adaptive evolution after gene duplication,” in Genome Evolution, edsMeyerA.Van de PeerY. (Dordrecht: Springer), 201–212.
5
BuiL. T.GiuntoliB.KosmaczM.ParlantiS.LicausiF. (2015). Constitutively expressed ERF-VII transcription factors redundantly activate the core anaerobic response in Arabidopsis thaliana.Plant Sci.23637–43. 10.1016/j.plantsci.2015.03.008
6
Capella-GutiérrezS.Silla-MartínezJ. M.GabaldónT. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses.Bioinformatics251972–1973. 10.1093/bioinformatics/btp348
7
ChandlerJ. W.WerrW. (2017). DORNRÖSCHEN, DORNRÖSCHEN-LIKE, and PUCHI redundantly control floral meristem identity and organ initiation in Arabidopsis.J. Exp. Bot.683457–3472. 10.1093/jxb/erx208
8
ChenH.JeJ.SongC.HwangJ. E.LimC. O. (2012). A proximal promoter region of Arabidopsis DREB2C confers tissue-specific expression under heat stress.J. Integr. Plant Biol.54640–651. 10.1111/j.1744-7909.2012.01137.x
9
ChenW. H.LiP. F.ChenM. K.LeeY. I.YangC. H. (2015). FOREVER YOUNG FLOWER negatively regulates ethylene response DNA-binding factors by activating an ethylene-responsive factor to control Arabidopsis floral organ senescence and abscission.Plant Physiol.1681666–1683. 10.1104/pp.15.00433
10
CuiL.FengK.WangM.WangM.DengP.SongW.et al (2016). Genome-wide identification, phylogeny and expression analysis of AP2/ERF transcription factors family in Brachypodium distachyon.BMC Genomics17:636. 10.1186/s12864-016-2968-8
11
CutcliffeJ. W.HellmannE.HeylA.RashotteA. M. (2011). CRFs form protein-protein interactions with each other and with members of the cytokinin signalling pathway in Arabidopsis via the CRF domain.J. Exp. Bot.624995–5002. 10.1093/jxb/err199
12
DassanayakeM.OhD. H.HaasJ. S.HernandezA.HongH.AliS.et al (2011). The genome of the extremophile crucifer Thellungiella parvula.Nat. Genet.43913–918. 10.1038/ng.889
13
DavidsonR. M.GowdaM.MogheG.LinH. N.VaillancourtB.ShiuS. H.et al (2012). Comparative transcriptomics of three Poaceae species reveals patterns of gene expression evolution.Plant J.71492–502. 10.1111/j.1365-313X.2012.05005.x
14
DavisJ. C.PetrovD. A. (2004). Preferential duplication of conserved proteins in eukaryotic genomes.PLoS Biol.2:E55. 10.1371/journal.pbio.0020055
15
DossaK.WeiX.LiD.FoncekaD.ZhangY.WangL.et al (2016). Insight into the AP2/ERF transcription factor superfamily in sesame and expression profiling of DREB subfamily under drought stress.BMC Plant Biol.16:171. 10.1186/s12870-016-0859-4
16
EmmsD. M.KellyS. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy.Genome Biol.16:157. 10.1186/s13059-015-0721-2
17
FinnR. D.ClementsJ.EddyS. R. (2011). HMMER web server: interactive sequence similarity searching.Nucleic Acids Res.3929–37. 10.1093/nar/gkr367
18
FinnR. D.CoggillP.EberhardtR. Y.EddyS. R.MistryJ.MitchellA. L.et al (2016). The Pfam protein families database: towards a more sustainable future.Nucleic Acids Res.44D279–D285. 10.1093/nar/gkv1344
19
ForceA.LynchM.PickettF. B.AmoresA.YanY. L.PostlethwaitJ. (1999). Preservation of duplicate genes by complementary, degenerative mutations.Genetics1511531–1545.
20
FuM.KangH. K.SonS. H.KimS. K.NamK. H. (2014). A subset of arabidopsis RAV transcription factors modulates drought and salt stress responses independent of ABA.Plant Cell Physiol.551892–1904. 10.1093/pcp/pcu118
21
GoodsteinD. M.ShuS.HowsonR.NeupaneR.HayesR. D.FazoJ.et al (2012). Phytozome: a comparative platform for green plant genomics.Nucleic Acids Res.401178–1186. 10.1093/nar/gkr944
22
GoutJ. F.LynchM. (2015). Maintenance and loss of duplicated genes by dosage subfunctionalization.Mol. Biol. Evol.322141–2148. 10.1093/molbev/msv095
23
GuX.ZouY.SuZ.HuangW.ZhouZ.ArendseeZ.et al (2013). An update of DIVERGE software for functional divergence analysis of protein family.Mol. Biol. Evol.301713–1719. 10.1093/molbev/mst069
24
GuptaS.RashotteA. M. (2014). Expression patterns and regulation of SlCRF3 and SlCRF5 in response to cytokinin and abiotic stresses in tomato (Solanum lycopersicum).J. Plant Physiol.171349–358. 10.1016/j.jplph.2013.09.003
25
HaakeV.CookD.RiechmannL.PinedaO.ThomashowM. F.ZhangJ. Z.et al (2016). Transcription factor CBF4 is a regulator of drought adaptation in Arabidopsis.Plant Physiol.130639–648. 10.1104/pp.006478
26
HattoriY.NagaiK.FurukawaS.SongX. J.KawanoR.SakakibaraH.et al (2009). The ethylene response factors SNORKEL1 and SNORKEL2 allow rice to adapt to deep water.Nature4601026–1030. 10.1038/nature08258
27
HaudryA.PlattsA. E.VelloE.HoenD. R.LeclercqM.WilliamsonR. J.et al (2013). An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions.Nat. Genet.45891–898. 10.1038/ng.2684
28
HeymanJ.CoolsT.VandenbusscheF.HeyndrickxK. S.Van LeeneJ.VercauterenI.et al (2013). ERF115 controls root quiescent center cell division and stem cell replenishment.Science342860–863. 10.1126/science.1240667
29
HuangC. H.SunR.HuY.ZengL.ZhangN.CaiL.et al (2016a). Resolution of brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution.Mol. Biol. Evol.33394–412. 10.1093/molbev/msv226
30
HuangC. H.ZhangC.LiuM.HuY.GaoT.QiJ.et al (2016b). Multiple polyploidization events across asteraceae with two nested events in the early history revealed by nuclear phylogenomics.Mol. Biol. Evol.332820–2835. 10.1093/molbev/msw157
31
HuotB.YaoJ.MontgomeryB. L.HeS. Y. (2014). Growth-defense tradeoffs in plants: a balancing act to optimize fitness.Mol. Plant71267–1287. 10.1093/mp/ssu049
32
HwangJ. E.LimC. J.ChenH.JeJ.SongC.LimC. O. (2012). Overexpression of Arabidopsis Dehydration-responsive element-binding protein 2C confers tolerance to oxidative stress.Mol. Cells33135–140. 10.1007/s10059-012-2188-2
33
IwaseA.HarashimaH.IkeuchiM.RymenB.OhnumaM.KomakiS.et al (2016). WIND1 promotes shoot regeneration through transcriptional activation of ENHANCER OF SHOOT REGENERATION1 in Arabidopsis.Plant Cell2954–69. 10.1105/tpc.16.00623
34
JeonJ.ChoC.LeeM. R.Van BinhN.KimJ. (2016). CYTOKININ RESPONSE FACTOR2(CRF2) and CRF3 regulate lateral root development in response to cold stress in Arabidopsis.Plant Cell281828–1843. 10.1105/tpc.15.00909
35
JiaY.DingY.ShiY.ZhangX.GongZ.YangS. (2016). The cbfs triple mutants reveal the essential functions of CBFs in cold acclimation and allow the definition of CBF regulons in Arabidopsis.New Phytol.212345–353. 10.1111/nph.14088
36
JiaoY.Leebens-MackJ.AyyampalayamS.BowersJ. E.McKainM. R.McNealJ.et al (2012). A genome triplication associated with early diversification of the core eudicots.Genome Biol.13:R3. 10.1186/gb-2012-13-1-r3
37
JiaoY.WickettN. J.AyyampalayamS.ChanderbaliA. S.LandherrL.RalphP. E.et al (2011). Ancestral polyploidy in seed plants and angiosperms.Nature47397–100. 10.1038/nature09916
38
JonesD. T.CozzettoD. (2015). DISOPRED3: precise disordered region predictions with annotated protein-binding activity.Bioinformatics31857–863. 10.1093/bioinformatics/btu744
39
KalyaanamoorthyS.MinhB. Q.WongT. K. F.von HaeselerA.JermiinL. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates.Nat. Methods14587–589. 10.1038/nmeth.4285
40
KangH. G.KimJ.KimB.JeongH.ChoiS. H.KimE. K.et al (2011). Overexpression of FTL1/DDF1, an AP2 transcription factor, enhances tolerance to cold, drought, and heat stresses in Arabidopsis thaliana.Plant Sci.180634–641. 10.1016/j.plantsci.2011.01.002
41
KangJ.ZhangH.SunT.ShiY.WangJ.ZhangB.et al (2013). Natural variation of C-repeat-binding factor (CBFs) genes is a major cause of divergence in freezing tolerance among a group of Arabidopsis thaliana populations along the Yangtze River in China.New Phytol.1991069–1080. 10.1111/nph.12335
42
KarevG. P.WolfY. I.BerezovskayaF. S.KooninE. V. (2004). Gene family evolution: an in-depth theoretical and simulation analysis of non-linear birth-death-innovation models.BMC Evol. Biol.4:32. 10.1186/1471-2148-4-32
43
KarevG. P.WolfY. I.KooninE. V. (2003). Simple stochastic birth and death models of genome evolution: was there enough time for us to evolve?Bioinformatics191889–1900. 10.1093/bioinformatics/btg351
44
KarevG. P.WolfY. I.RzhetskyA. Y.BerezovskayaF. S.KooninE. V. (2002). Birth and death of protein domains: a simple model of evolution explains power law behavior.BMC Evol. Biol.2:18. 10.1186/1471-2148-2-18
45
KatohK.StandleyD. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability.Mol. Biol. Evol.30772–780. 10.1093/molbev/mst010
46
KidokoroS.WatanabeK.OhoriT.MoriwakiT.MaruyamaK.MizoiJ.et al (2015). Soybean DREB1/CBF-type transcription factors function in heat and drought as well as cold stress-responsive gene expression.Plant J.81505–518. 10.1111/tpj.12746
47
KimJ. (2016). CYTOKININ RESPONSE FACTORs gating environmental signals and hormones.Trends Plant Sci.21993–996. 10.1016/j.tplants.2016.10.004
48
KimS.SoltisP. S.WallK.SoltisD. E. (2006). Phylogeny and domain evolution in the APETALA2-like gene family.Mol. Biol. Evol.23107–120. 10.1093/molbev/msj014
49
KosmaczM.ParlantiS.SchwarzländerM.KraglerF.LicausiF.Van DongenJ. T. (2015). The stability and nuclear localization of the transcription factor RAP2.12 are dynamically regulated by oxygen concentration.Plant Cell Environ.381094–1103. 10.1111/pce.12493
50
KrishnaswamyS.VermaS.RahmanM. H.KavN. N. V. (2011). Functional characterization of four APETALA2-family genes (RAP2.6, RAP2.6L, DREB19 and DREB26) in Arabidopsis.Plant Mol. Biol.75107–127. 10.1007/s11103-010-9711-7
51
LataC.BhuttyS.BahadurR. P.MajeeM.PrasadM. (2011). Association of an SNP in a novel DREB2-like gene SiDREB2 with stress tolerance in foxtail millet [Setaria italica (L.)].J. Exp. Bot.623387–3401. 10.1093/jxb/err016
52
LaubingerS.ZellerG.HenzS. R.SachsenbergT.WidmerC. K.NaouarN.et al (2008). At-TAX: a whole genome tiling array resource for developmental expression analysis and transcript identification in Arabidopsis thaliana.Genome Biol.9:R112. 10.1186/gb-2008-9-7-r112
53
LeS. Q.GascuelO. (2008). An improved general amino acid replacement matrix.Mol. Biol. Evol.251307–1320. 10.1093/molbev/msn067
54
LehtiShiuM. D.UygunS.MogheG. D.PanchyN.FangL.HufnagelD. E.et al (2015). Molecular evidence for functional divergence and decay of a transcription factor derived from whole-genome duplication in Arabidopsis thaliana.Plant Physiol.1681717–1734. 10.1104/pp.15.00689
55
LiX. L.TaoS.WeiS. W.MingM. L.HuangX. S.ZhangS. L.et al (2018). The mining and evolutionary investigation of AP2/ERF genes in pear (Pyrus).BMC Plant Biol.18:14. 10.1186/s12870-018-1265-x
56
LiZ.DefoortJ.TasdighianS.MaereS.Van de PeerY.De SmetR. (2016). Gene duplicability of core genes is highly consistent across all angiosperms.Plant Cell28326–344. 10.1105/tpc.15.00877
57
LiangH.LiW. H. (2007). Gene essentiality, gene duplicability and protein connectivity in human and mouse.Trends Genet.23375–378. 10.1016/j.tig.2007.04.005
58
LiaoB. Y.ScottN. M.ZhangJ. Z. (2007). Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins.Mol. Biol. Evol.241091–1091. 10.1093/molbev/ms1076
59
LicausiF.KosmaczM.WeitsD. A.GiuntoliB.GiorgiF. M.VoesenekL. A. C. J.et al (2011). Oxygen sensing in plants is mediated by an N-end rule pathway for protein destabilization.Nature479419–422. 10.1038/nature10536
60
LicausiF.Ohme-TakagiM.PerataP. (2013). APETALA2/ETHYLENE Responsive Factor (AP2/ERF) transcription factors: mediators of stress responses and developmental programs.New Phytol.199639–649. 10.1111/nph.12291
61
LicausiF.Van DongenJ. T.GiuntoliB.NoviG.SantanielloA.GeigenbergerP.et al (2010). HRE1 and HRE2, two hypoxia-inducible ethylene response factors, affect anaerobic responses in Arabidopsis thaliana.Plant J.62302–315. 10.1111/j.1365-313X.2010.04149.x
62
LinR. C.ParkH. J.WangH. Y. (2008). Role of Arabidopsis RAP2.4 in regulating lightand ethylene-mediated developmental processes and drought stress tolerance.Mol. Plant142–57. 10.1093/mp/ssm004
63
LiuC.ZhangT. (2017). Expansion and stress responses of the AP2/EREBP superfamily in cotton.BMC Genomics18:118. 10.1186/s12864-017-3517-9
64
MaW.KongQ.GrixM.MantylaJ. J.YangY.BenningC.et al (2015). Deletion of a C-terminal intrinsically disordered region of WRINKLED1 affects its stability and enhances oil accumulation in Arabidopsis.Plant J.83864–874. 10.1111/tpj.12933
65
MagomeH.YamaguchiS.HanadaA.KamiyaY.OdaK. (2004). Dwarf and delayed-flowering 1, a novel Arabidopsis mutant deficient in gibberellin biosynthesis because of overexpression of a putative AP2 transcription factor.Plant J.37720–729. 10.1111/j.1365-313X.2003.01998.x
66
Marchler-BauerA.DerbyshireM. K.GonzalesN. R.LuS.ChitsazF.GeerL. Y.et al (2015). CDD: NCBI’s conserved domain database.Nucleic Acids Res.43D222–D226. 10.1093/nar/gku1221
67
MatsukuraS.MizoiJ.YoshidaT.TodakaD.ItoY.MaruyamaK.et al (2010). Comprehensive analysis of rice DREB2-type genes that encode transcription factors involved in the expression of abiotic stress-responsive genes.Mol. Genet. Genomics283185–196. 10.1007/s00438-009-0506-y
68
MatsuokaK.YanagiR.YumotoE.YokotaT.YamaneH.SatohS.et al (2018). RAP2.6L and jasmonic acid–responsive genes are expressed upon Arabidopsis hypocotyl grafting but are not needed for cell proliferation related to healing.Plant Mol. Biol.96531–542. 10.1007/s11103-018-0702-4
69
MayroseI.GraurD.Ben-TalN.PupkoT. (2004). Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior.Mol. Biol. Evol.211781–1791. 10.1093/molbev/msh194
70
MedinaJ.BarguesM.TerolJ.Perez-AlonsoM.SalinasJ. (1999). The Arabidopsis CBF gene family is composed of three genes encoding AP2 domain-containing proteins whose expression is regulated by low temperature but not by abscisic acid or dehydration.Plant Physiol.119463–469. 10.1104/Pp.119.2.463
71
MehrniaM.BalazadehS.ZanorM. I.Mueller-RoeberB. (2013). EBE, an AP2/ERF transcription factor highly expressed in proliferating cells, affects shoot architecture in Arabidopsis.Plant Physiol.162842–857. 10.1104/pp.113.214049
72
MingR.VanBurenR.LiuY.YangM.HanY.LiL. T.et al (2013). Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.).Genome Biol.14:R41. 10.1186/gb-2013-14-5-r41
73
NakanoT.SuzukiK.FujimuraT.ShinshiH. (2006). Genome-wide analysis of the ERF gene family.Plant Physiol.140411–432. 10.1104/pp.105.073783.currently
74
NguyenL. T.SchmidtH. A.Von HaeselerA.MinhB. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies.Mol. Biol. Evol.32268–274. 10.1093/molbev/msu300
75
NovilloF.AlonsoJ. M.EckerJ. R.SalinasJ. (2004). CBF2/DREB1C is a negative regulator of CBF1/DREB1B and CBF3/DREB1A expression and plays a central role in, stress tolerance in Arabidopsis.Proc. Natl. Acad. Sci. U.S.A.1013985–3990. 10.1073/pnas.0303029101
76
NovozhilovA. S.KarevG. P.KooninE. V. (2006). Biological applications of the theory of birth-and-death processes.Brief. Bioinform.770–85. 10.1093/bib/bbk006
77
OlsenJ. L.RouzéP.VerhelstB.LinY. C.BayerT.CollenJ.et al (2016). The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea.Nature530331–335. 10.1038/nature16548
78
O’TooleÁN.HurstL. D.McLysaghtA. (2017). Faster evolving primate genes are more likely to duplicate.Mol. Biol. Evol.35107–118. 10.1093/molbev/msx270
79
PanchyN.Lehti-ShiuM. D.ShiuS. H. (2016). Evolution of gene duplication in plants.Plant Physiol.1712294–2316. 10.1104/pp.16.00523
80
PaulM. V.IyerS.AmerhauserC.LehmannM.van DongenJ. T.GeigenbergerP. (2016). Oxygen sensing via the ethylene response transcription factor RAP2.12 affects plant metabolism and performance under both normoxia and hypoxia.Plant Physiol.172141–153. 10.1104/pp.16.00460
81
PeguerolesC.LaurieS.AlbàM. M. (2013). Accelerated evolution after gene duplication: a time-dependent process affecting just one copy.Mol. Biol. Evol.301830–1842. 10.1093/molbev/mst083
82
PetryszakR.KeaysM.TangY. A.FonsecaN. A.BarreraE.BurdettT.et al (2016). Expression Atlas update - an integrated database of gene and protein expression in humans, animals and plants.Nucleic Acids Res.44D746–D752. 10.1093/nar/gkv1045
83
PhukanU. J.JeenaG. S.TripathiV.ShuklaR. K. (2017). Regulation of Apetala2/Ethylene response factors in plants.Front. Plant Sci.8:150. 10.3389/fpls.2017.00150
84
PriceM. N.DehalP. S.ArkinA. P. (2009). Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix.Mol. Biol. Evol.261641–1650. 10.1093/molbev/msp077
85
QinF.KakimotoM.SakumaY.MaruyamaK.OsakabeY.TranL. S. P.et al (2007). Regulation and functional analysis of ZmDREB2A in response to drought and heat stresses in Zea mays L.Plant J.5054–69. 10.1111/j.1365-313X.2007.03034.x
86
RashotteA. M.GoertzenL. R. (2010). The CRF domain defines Cytokinin Response Factor proteins in plants.BMC Plant Biol.10:74. 10.1186/1471-2229-10-74
87
RenR.WangH.GuoC.ZhangN.ZengL.ChenY.et al (2018). Wide-spread whole genome duplications contribute to genome complexity and species diversity in angiosperms.Mol. Plant11414–428. 10.1016/j.molp.2018.01.002
88
RosenbloomK. R.ArmstrongJ.BarberG. P.CasperJ.ClawsonH.DiekhansM.et al (2015). The UCSC genome browser database: 2015 update.Nucleic Acids Res.43D670–D681. 10.1093/nar/gku1177
89
SakumaY.LiuQ.DubouzetJ. G.AbeH.ShinozakiK.Yamaguchi-ShinozakiK. (2002). DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression.Biochem. Biophys. Res. Commun.290998–1009. 10.1006/bbrc.2001.6299
90
SchmidM.DavisonT. S.HenzS. R.PapeU. J.DemarM.VingronM.et al (2005). A gene expression map of Arabidopsis thaliana development.Nat. Genet.37501–506. 10.1038/ng1543
91
SchultzJ.MilpetzF.BorkP.PontingC. P. (1998). SMART, a simple modular architecture research tool: identification of signaling domains.Proc. Natl. Acad. Sci. U.S.A.955857–5864. 10.1073/pnas.95.11.5857
92
SémonM.WolfeK. H. (2008). Preferential subfunctionalization of slow-evolving genes after allopolyploidization in Xenopus laevis.Proc. Natl. Acad. Sci. U.S.A.1058333–8338. 10.1073/pnas.0708705105
93
SeverinA. J.WoodyJ. L.BolonY. T.JosephB.DiersB. W.FarmerA. D.et al (2010). RNA-Seq Atlas of Glycine max: a guide to the soybean transcriptome.BMC Plant Biol.10:160. 10.1186/1471-2229-10-160
94
ShiJ. X.MalitskyS.de OliveiraS.BraniganC.FrankeR. B.SchreiberL.et al (2011). SHINE transcription factors act redundantly to pattern the archetypal surface of Arabidopsis flower organs.PLoS Genet.7:e1001388. 10.1371/journal.pgen.1001388
95
ShiX. L.GuptaS.RashotteA. M. (2012). Solanum lycopersicum cytokinin response factor (SlCRF) genes: characterization of CRF domain-containing ERF genes in tomato.J. Exp. Bot.63973–982. 10.1093/jxb/err325
96
ShiX. L.GuptaS.RashotteA. M. (2014). Characterization of two tomato AP2/ERF genes, SlCRF1 and SlCRF2 in hormone and stress responses.Plant Cell Rep.3335–45. 10.1007/s00299-013-1510-6
97
SongX.LiY.HouX. (2013). Genome-wide analysis of the AP2/ERF transcription factor superfamily in Chinese cabbage (Brassica rapa ssp. pekinensis).BMC Genomics14:573. 10.1186/1471-2164-14-573
98
TasdighianS.Van BelM.LiZ.Van de PeerY.Carretero-PauletL.MaereS. (2017). Reciprocally retained genes in the angiosperm lineage show the hallmarks of dosage balance sensitivity.Plant Cell292766–2785. 10.1105/tpc.17.00313
99
ToA.JoubesJ.BartholeG.LecureuilA.ScagnelliA.JasinskiS.et al (2012). WRINKLED transcription factors orchestrate tissue-specific regulation of fatty acid biosynthesis in Arabidopsis.Plant Cell245007–5023. 10.1105/tpc.112.106120
100
VannesteK.BaeleG.MaereS.PeerY. V. D. (2014). Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous - Paleogene boundary.Genome Res.321334–1347. 10.1101/gr.168997.113
101
VeidenbergA.MedlarA.LöytynojaA. (2016). Wasabi: an integrated platform for evolutionary sequence analysis and data visualization.Mol. Biol. Evol.331126–1130. 10.1093/molbev/msv333
102
VilellaA. J.SeverinJ.Ureta-VidalA.HengL.DurbinR.BirneyE. (2009). EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates.Genome Res.19327–335. 10.1101/gr.073585.107
103
WagnerA.ConantG. C. (2003). Asymmetric sequence divergence of duplicate genes.Genome Res.132052–2058. 10.1101/gr.1252603
104
WangW.HabererG.GundlachH.GläßerC.NussbaumerT.LuoM. C.et al (2014). The Spirodela polyrhiza genome reveals insights into its neotenous reduction fast growth and aquatic lifestyle.Nat. Commun.5:3311. 10.1038/ncomms4311
105
WangX.WangH.WangJ.SunR.WuJ.LiuS.et al (2011). The genome of the mesopolyploid crop species Brassica rapa.Nat. Genet.431035–1040. 10.1038/ng.919
106
WeiG.PanY.LeiJ.ZhuY.-X. (2005). Molecular cloning, phylogenetic analysis, expressional profiling and in vitro studies of TINY2 from Arabidopsis thaliana.J. Biochem. Mol. Biol.38440–446. 10.5483/BMBRep.2005.38.4.440
107
WeitsD. A.GiuntoliB.KosmaczM.ParlantiS.HubbertenH. M.RieglerH.et al (2014). Plant cysteine oxidases control the oxygen-dependent branch of the N-end-rule pathway.Nat. Commun.5:3425. 10.1038/ncomms4425
108
WertheimJ. O.MurrellB.SmithM. D.PondS. L. K.SchefflerK. (2015). RELAX: detecting relaxed selection in a phylogenetic framework.Mol. Biol. Evol.32820–832. 10.1093/molbev/msu400
109
WilsonK. (1996). A dissociation insertion causes a semidominant mutation that increases expression of TINY, an Arabidopsis gene related to APETALA2.Plant Cell8659–671. 10.1105/tpc.8.4.659
110
WrightP. E.DysonH. J. (2014). Intrinsically disordered proteins in cellular signalling and regulation.Nat. Rev. Mol. Cell Biol.1618–29. 10.1038/nrm3920
111
WuY. C.RasmussenM. D.BansalM. S.KellisM. (2013). TreeFix: statistically informed gene tree error correction using species trees.Syst. Biol.62110–120. 10.5061/dryad.44cb5
112
XiangY.HuangC. H.HuY.WenJ.LiS.YiT.et al (2016). Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication.Mol. Biol. Evol.34262–281. 10.1093/molbev/msw242
113
XieT.YangQ. Y.WangX. T.McLysaghtA.ZhangH. Y. (2016). Spatial colocalization of human ohnolog pairs acts to maintain dosage-balance.Mol. Biol. Evol.332368–2375. 10.1093/molbev/msw108
114
XuK.XuX.FukaoT.CanlasP.Maghirang-RodriguezR.HeuerS.et al (2006). Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice.Nature442705–708. 10.1038/nature04920
115
XuY. Y.WuH. Y.ZhaoM. M.WuW.XuY. N.GuD. (2016). Overexpression of the transcription factors GmSHN1 and GmSHN9 differentially regulates wax and cutin biosynthesis, alters cuticle properties, and changes leaf phenotypes in Arabidopsis.Int. J. Mol. Sci.17E587. 10.3390/ijms17040587
116
YanaiI.BenjaminH.ShmoishM.Chalifa-CaspiV.ShklarM.OphirR.et al (2005). Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification.Bioinformatics21650–659. 10.1093/bioinformatics/bti042
117
YangL.GautB. S. (2011). Factors that contribute to variation in evolutionary rate among arabidopsis genes.Mol. Biol. Evol.282359–2369. 10.1093/molbev/msr058
118
YangR.JarvisD. E.ChenH.BeilsteinM. A.GrimwoodJ.JenkinsJ.et al (2013). The reference genome of the palophytic plant Eutrema salsugineum.Front. Plant Sci.4:46. 10.3389/fpls.2013.00046
119
YangZ. (1998). Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution.Mol. Biol. Evol.15568–573. 10.1093/oxfordjournals.molbev.a025957
120
YangZ. (2007). PAML 4: phylogenetic analysis by maximum likelihood.Mol. Biol. Evol.241586–1591. 10.1093/molbev/msm088
121
YangZ.NielsenR.HasegawaM. (1998). Models of amino acid substitution and applications to mitochondrial protein evolution.Mol. Biol. Evol.151600–1611. 10.1093/oxfordjournals.molbev.a025888
122
YangZ.WongW. S. W.NielsenR. (2005). Bayes empirical Bayes inference of amino acid sites under positive selection.Mol. Biol. Evol.221107–1118. 10.1093/molbev/msi097
123
YilmazA.Mejia-GuerraM. K.KurzK.LiangX. Y.WelchL.GrotewoldE. (2011). AGRIS: the Arabidopsis gene regulatory information server, an update.Nucleic Acids Res.39D1118–D1122. 10.1093/nar/gkq1120
124
ZengL.YinY.YouC.PanQ.XuD.JinT.et al (2016). Evolution and protein interactions of AP2 proteins in Brassicaceae: evidence linking development and environmental responses.J. Integr. Plant Biol.58549–563. 10.1111/jipb.12439
125
ZengL.ZhangN.ZhangQ.EndressP. K.HuangJ.MaH. (2017). Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets.New Phytol.2141338–1354. 10.1111/nph.14503
126
ZengL.ZhangQ.SunR.KongH.ZhangN.MaH. (2014). Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times.Nat. Commun.5:4956. 10.1038/ncomms5956
127
ZhangB.WangL.ZengL.ZhangC.MaH. (2015). Arabidopsis TOE proteins convey a photoperiodic signal to antagonize CONSTANS and regulate flowering time.Genes Dev.29975–987. 10.1101/gad.251520.114
128
ZhangJ.NielsenR.YangZ. (2005). Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level.Mol. Biol. Evol.222472–2479. 10.1093/molbev/msi237
129
ZhangL.LiZ.QuanR.LiG.WangR.HuangR. (2011). An AP2 domain-containing gene, ESE1, targeted by the ethylene signaling component EIN3 is important for the salt response in Arabidopsis.Plant Physiol.157854–865. 10.1104/pp.111.179028
130
ZhouM.ChenH.WeiD.MaH.LinJ. (2017). Arabidopsis CBF3 and DELLAs positively regulate each other in response to low temperature.Sci. Rep.7:39819. 10.1038/srep39819
131
ZouC.LehtiShiuM. D.ThomashowM.ShiuS. H. (2009). Evolution of stress-regulated gene expression in duplicate genes of Arabidopsis thalianae.PLoS Genet.5:e1000581. 10.1371/journal.pgen.1000581
132
Zumajo-CardonaC.Pabón-MoraN. (2016). Evolution of the APETALA2 gene lineage in seed plants.Mol. Biol. Evol.331818–1832. 10.1093/molbev/msw059
133
ZwackP. J.ShiX. L.RobinsonB. R.GuptaS.ComptonM. A.GerkenD. M.et al (2012). Vascular expression and C-Terminal sequence divergence of cytokinin response factors in flowering plants.Plant Cell Physiol.531683–1695. 10.1093/pcp/pcs110
Summary
Keywords
AP2/ERF genes, gene duplication, preferential retention, functional divergence, stress response, angiosperm evolution, ecological niche
Citation
Wang L, Ma H and Lin J (2019) Angiosperm-Wide and Family-Level Analyses of AP2/ERF Genes Reveal Differential Retention and Sequence Divergence After Whole-Genome Duplication. Front. Plant Sci. 10:196. doi: 10.3389/fpls.2019.00196
Received
05 November 2018
Accepted
05 February 2019
Published
26 February 2019
Volume
10 - 2019
Edited by
Michael Eric Schranz, Wageningen University & Research, Netherlands
Reviewed by
Liangsheng Zhang, Fujian Agriculture and Forestry University, China; Kui Lin, Beijing Normal University, China
Updates
Copyright
© 2019 Wang, Ma and Lin.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Ma, hongma@fudan.edu.cn; hxm16@psu.edu Juan Lin, linjuan@fudan.edu.cn
This article was submitted to Plant Systematics and Evolution, a section of the journal Frontiers in Plant Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.