 Gaurav Sablok1,2
Gaurav Sablok1,2 Ali Amiryousefi
Ali Amiryousefi Xiaolan He
Xiaolan He Jaakko Hyvönen
Jaakko Hyvönen Péter Poczai
Péter Poczai- 1Finnish Museum of Natural History (Botany Unit), University of Helsinki, Helsinki, Finland
- 2Organismal Evolution and Biology, Faculty of Biology and Environmental Sciences, Viikki Plant Science Centre, University of Helsinki, Helsinki, Finland
We report the first plastome sequence of giant ragweed (Ambrosia trifida); with this new genome information, we assessed the phylogeny of Asteraceae and the transcriptional profiling against glyphosate resistance in giant ragweed. Assembly and genic features show a normal angiosperm quadripartite plastome structure with no signatures of deviation in gene directionality. Comparative analysis revealed large inversions across the plastome of giant ragweed and the previously sequenced members of the plant family. Asteraceae plastid genomes contain two inversions of 22.8 and 3.3 kb; the former is located between trnS-GCU and trnG-UCC genes, and the latter between trnE-UUC and trnT-GGU genes. The plastid genome sequences of A. trifida and the related species, Ambrosia artemisiifolia, are identical in gene content and arrangement, but they differ in length. The phylogeny is well-resolved and congruent with previous hypotheses about the phylogenetic relationship of Asteraceae. Transcriptomic analysis revealed divergence in the relative expressions at the exonic and intronic levels, providing hints toward the ecological adaptation of the genus. Giant ragweed shows various levels of glyphosate resistance, with introns displaying higher expression patterns at resistant time points after the assumed herbicide treatment.
Introduction
Plant invasions are accelerating on a global scale and cannot be fully understood without analyzing the genetic background of the source and invading populations. Biological invasions may threaten both global and local biodiversity, ecosystem functions, agriculture, and public health (Vitousek et al., 1997). Human-induced global climate change might further complicate the effects of the invasions. In Europe, as in many other regions of the world, the number of invasive plant species has increased considerably in the past 200 years due to increased trade, tourism, and disturbance (Pyšek et al., 2009). Europe suffers from invasive species in many ways, and a crude estimate of monetary impact (diminished yield and control measures) suggests that these additional costs due to invasive species exceed €12 billion annually (Kettunen et al., 2009), although this may be an underestimate (Vilá et al., 2010).
The genus Ambrosia L. (ragweeds) of the Asteraceae (tribe Heliantheae, subtribe Ambrosiinae) includes 40–50 species (Payne, 1964; Martin et al., 2018). Most are dioecious desert shrubs, both annual and perennial, while some species are weedy pioneers that have become established as exotics outside of their original range (Martin et al., 2018). Based on their current diversity and distribution, Ambrosia most likely evolved in the SW USA and adjacent Mexico, then subsequently radiated to many areas of North America (Payne, 1964). Giant ragweed (Ambrosia trifida L.) has raised awareness as an invasive plant in Europe — together with its relatives, common (Ambrosia artemisiifolia L.) and perennial ragweed (A. psilostachya DC.), it represents an example of aggressive invasion all over the continent. Ragweeds are among the most economically destructive weeds, as they interfere with the growth and establishment of various crops (Cowborough et al., 2003; Kong et al., 2007). A. trifida was primarily a weed of floodplains and ditch banks, but in the past decades it has expanded its native range in North America, causing considerable economic loss in the Corn Belt (Regnier et al., 2016). It can dominate in common croplands due to its rapid growth and large leaf area. Notably, its ability to adjust its resource utilization responses and extend its germination period has allowed it to tolerate changing environments, adding to its success as an invasive species (Abul-fatih and Bazzaz, 1979). Ragweeds also produce large amounts of pollen that causes allergenic rhinitis, presenting a burden to public health (Kazinczi et al., 2008). Currently half of all hay fever cases in North America are caused by ragweeds (Taramarcaz et al., 2005), making it one of the most powerful aeroallergens. In fact, it has been estimated that areas in Europe affected by severe ragweed allergy are likely to increase substantially by the year 2100 (Rasmussen et al., 2017). Pollen production of these plants is enhanced by special anemophily-specialized floral organs, including the pistillodium (thought to be universal within Ambrosia), which is a vestigial pistil in staminate flowers that forces pollen away from the corolla (Payne, 1963; Martin et al., 2009; Martin et al., 2010).
Giant ragweed is often controlled with the broad-spectrum herbicide glyphosate [N-(phosphonomethyl) glycine], which has become a dominant herbicide worldwide since its introduction in 1974. Although glyphosate belongs to the herbicide group with the greatest increase in resistance cases, it is still the most widely used non-selective systemic herbicide worldwide (Bracamonte et al., 2018), despite its debated environmental effects (Vandenberg et al., 2017). Recently, it was shown that glyphosate affects the gut microbiome of honeybees, inducing susceptibility to infections and death from pathogenic bacteria (Motta et al., 2018). The continuous application of glyphosate, along with reduced use of other weed management practices has also caused many weeds, including A. trifida, to become glyphosate-resistant (Ganie et al., 2017). Since the introduction of transgenic glyphosate-resistant (GR) crops in 1996, 19 weeds have evolved resistance to glyphosate; about half evolved in GR crops (Heap, 2018). In 2016, the area of transgenic crops reached 185.1 million ha globally, and approximately 80% were planted solely with GR crops (International Service for the Acquisition of Agri-biotech Applications [ISAAA], 2016). This means that growers continually applied glyphosate alone over these vast areas to control genetically variable and prolific weeds. The high initial efficacy of glyphosate often leads to a decline in the use of other herbicide options and less investment by industry to discover new herbicide active ingredients (Green and Owen, 2011). No matter how effective the herbicide is, weed management programs cannot rely heavily on only one tactic otherwise weeds will ultimately adapt and survive in large numbers (Green and Owen, 2011).
Our knowledge about glyphosate resistance is still limited (Duke and Powles, 2008; Sammons and Gaines, 2014), and several mechanisms have been reported in various weeds (Norsworthy et al., 2011). A specific type of resistance, called target-site resistance, may occur when single or several mutations take place in the conserved region of the EPSPS gene and/or its duplications (Yu et al., 2015; Patterson et al., 2018; Sammons et al., 2018). This gene codes the 5-enolpyruvylshikimate-3-phosphate synthase (EPSPS) enzyme that is inactivated by glyphosate, thus disrupting aromatic amino acid synthesis in plants. Such target-site resistance appears rapidly in weed populations. However, the evaluation of several glyphosate-resistant A. trifida accessions revealed no changes in the EPSPS gene sequence and copy number. Rather, a form of non-target-site resistance (NTSR) known as metabolic resistance has been suggested (Moretti et al., 2018; Van Horn et al., 2018). This type of metabolic resistance involves more changes than just the target site alone. Plants with NTSR rapidly metabolize or break down the herbicide before it is able to cause toxic effects (Sammons and Gaines, 2014), thus representing a major issue for the chemical control of weeds. NTSR is hard to manage because weeds have unpredictable resistance to herbicides with different chemical structures and/or target proteins (Délye, 2013). Furthermore, recent studies have demonstrated that weeds with NTSR can transmit cross-resistance to other herbicides with different modes of action, even to those not yet marketed (Petit et al., 2010). This is because NTSR is much more of a general adaptive response to herbicides (Yuan et al., 2007). According to the current view, herbicide application is a stress to the plant, which triggers response pathways in weeds irrespective of their sensitivity to the herbicide (Délye, 2013). These stress–response pathways are pre-existent in plant species, coded by multiple genes and alleles. The selection of these genes starts with the uneven application of the herbicide in the field. For example, some individuals may survive herbicide spraying because they receive less of the chemical, hence their genes are carried to future generations, providing resistance to some degree. Further NTSR evolution likely requires several generations of sexual recombination within a population until individuals accumulate enough alleles to possess a resistance level that would enable survival of the full dose of herbicide (Délye et al., 2013). Recurrent selection experiments have convincingly provided support for this theory (Neve and Powels, 2005; Busi et al., 2012). Studies of the genetic basis of NTSR are sparse, and only the key role of glutathione transferase genes have been investigated (Cummins et al., 2013). Research addressing NTSR in weeds has long been hampered by the absence of ‘omics’ resources for the vast majority of weed species. The increasing accessibility of genomics and transcriptomics supported by next-generation sequencing (NGS) technologies should rapidly enable the identification of genes governing NTSR (Délye et al., 2013). For such purposes, transcriptomes are already available for various weeds (Peng et al., 2010; Riggins et al., 2010; Zhang et al., 2012) including A. artemisiifolia (Taller et al., 2016b; Virág et al., 2016).
Genetic research of ragweeds has mostly been focused on A. artemisiifolia, with the use of microsatellite markers to determine the origins of invading populations (Genton et al., 2005a,b; Gaudeul et al., 2011). Population genetics of A. artemisiifolia has also been investigated (Cseh and Taller, 2008; Chun et al., 2010; Gladieux et al., 2011; Mátyás et al., 2012; Martin et al., 2016; von Boheemen et al., 2017), and genomic tools are under development for the analysis of historical specimens (Sánchez Barreiro et al., 2017) and fresh plant material (Cseh et al., 2009; Cseh, 2010; Mátyás et al., 2011; Taller et al., 2016a; Virág et al., 2016; Nagy et al., 2017). The history of common ragweed distribution was revealed based on comprehensive studies of herbarium specimens (Chauvel et al., 2006; Csontos et al., 2010; Martin et al., 2014). However, the genetics of other ragweed species in Europe have been less studied. As such, the relationships of ragweeds are poorly understood and thus a taxonomic update of Payne (1964), with newly described species and a solid phylogenetic framework of the genus is necessary. Toward this goal, herbarium specimens accumulated over the past 100 years that are readily available for sampling would be valuable material to assess ragweed diversity, and to perform evolutionary studies. Accordingly, a recent study by Martin et al. (2018) demonstrated the utility of herbarium specimens in phylogenetic analyses.
Until recently, the use of herbarium collections for obtaining molecular-level data has been difficult due to the generally poor condition of their DNA (Besnard et al., 2014). However, advances in NGS technologies and subsequent development of techniques for extracting historical DNA (Gutaker et al., 2017; Shepherd, 2017) have recently made herbarium specimens an attractive option. In fact, most NGS methods are designed for using short fragmented DNA molecules (100–400 bp) as templates, which suits herbarium specimens (Staats et al., 2013). Gutaker and Burbano (2017) demonstrated that NGS is better suited than PCR-based approaches for ancient DNA sequencing, because adapter ligation allows interrogation of the molecule ends so that longer molecules can be obtained for sequencing. As such, this approach has enabled access to genetic information on type or other important historical specimens, which are crucial for resolving taxonomic uncertainties. Moreover, NGS is both feasible and cost-effective (Bakker et al., 2015; Bakker, 2017). Staats et al. (2011) showed that it is also possible to use genome skimming to analyze DNA that is otherwise too degraded for PCR-based approaches, thus offering the possibility to include rare and extinct species from natural history collections in phylogenetic analyses. Several phylogenomic studies have explored the potential of herbarium specimens of different ages (Staats et al., 2013; Besnard et al., 2014; Aubriot et al., 2018; McManus et al., 2018; Zeng et al., 2018). For example, the entire nuclear genome of a 43-year-old Arabidopsis thaliana (L.) Heynh. (Brassicaceae) herbarium specimen with high and uniform sequence coverage has been reported using a reference-based assembly approach (Staats et al., 2013). NGS techniques can also be used to further acquire mitochondrial genomic data, which provides the possibility to address questions related to hybridization and introgression (e.g., Rydin et al., 2017; Vargas et al., 2017), as well as assess the phylogenetic congruence between topologies based on mitochondrial regions and plastomes (Van de Paer et al., 2018). For example, the evolutionary history of an extinct plant lineage Hesperelaea A. Gray, known from an 1875 collection from Guadalupe Island, has been revealed based on both the plastome (Zedane et al., 2016) and mitogenome (Van de Paer et al., 2016).
Herbarium specimens also offer a unique opportunity to study the plastid genome compartments preserved in specimens. Plastomes of angiosperms are small (typically approximately 120–190 kb in size) and have a highly conserved quadripartite structure containing two inverted repeats (IRa and IRb), which separate the large and small single-copy regions (LSC and SSC). The plastid genome includes 110 to 130 genes that participate primarily in photosynthesis, transcription, and translation (Palmer, 1985; Olmstead and Palmer, 1994; Daniell et al., 2016). Their conserved gene content, order, and organization make plastid genomes fairly well-suited for studies about gene loss, structural rearrangement, pseudogenes, or additional mutation events that might be characteristic of some lineages (Poczai and Hyvönen, 2013, 2017; Beck and Semple, 2015; Wicke and Schneeweiss, 2015). Herbarium specimens appear to yield enough reads for effective plastome assembly, similar to fresh specimens (Bakker et al., 2015). This is because plant cell walls are composed of cellulose microfibrils (Cosgrove, 1999) that provide additional protection for preservation of the genomic DNA (Bakker, 2017). The total assembly length of plastid genomes is comparable between fresh material and herbarium specimens. Those from herbarium specimens simply need slightly more editing and “scaffolding” because on average they yield shorter contigs (Bakker et al., 2015; Bakker, 2017; Twyford and Ness, 2017).
In the current study, we sought to develop further resources to facilitate genomic research of ragweeds. By using a herbarium specimen, we aimed to demonstrate the utility of such collections for weed genomics. We present the complete chloroplast genome sequence of giant ragweed (A. trifida) using high-throughput sequencing, and report the assembly, annotation, gene expression, and unique structure characterization of its plastome. Comparison of the gene order and inverted repeat (IR) length across Asteraceae is also presented, as well as transcriptional profiling against glyphosate resistance.
Materials and Methods
DNA Isolation and Plastome Sequencing
Giant ragweed leaves (0.5 g) were obtained from a herbarium specimen of the Finnish Museum of Natural History (H), University of Helsinki, Finland (H1645542; Mansfield, OH, United States, 1886). Leaf samples were rinsed with deionized water and 70% ethanol, and total genomic DNA was isolated using the NucleoSpin Plant II kit (Macherey-Nagel, Düren, Germany). All work was carried out in a dedicated laboratory with UV sterilized equipment; blank samples were processed during DNA extraction. DNA concentration was measured with a Qubit fluorometer (Invitrogen) and verified on an 0.8% agarose gel. A paired-end genomic library was constructed using the Nextera DNA library preparation kit (Illumina, San Diego, CA, United States). Fragment analysis was conducted with an Agilent Technologies 2100 Bioanalyzer using a DNA 1000 chip. Sequencing was performed on an Illumina MiSeq platform from both ends with 150 bp read length.
Genome Assembly and Annotation
To obtain high-quality clean data, raw reads were first filtered by removing low-quality reads with a sliding window quality cutoff of Q20 using Trimmomatic (Bolger et al., 2014). Plastid reads were obtained by reference mapping using A. artemisiifolia L. (MG019037; NC035875) plastid genome (Amiryousefi et al., 2017; Nagy et al., 2017) with Geneious R10 (Kearse et al., 2012). Clean reads were used for de novo assembly, which was performed with three different algorithms of the programs Velvet v1.2.10 (Zerbino and Birney, 2008), Geneious assembler, and NOVOPlasty (Dierckxsens et al., 2017). General genome structure and ambiguous nucleotide positions were evaluated by an additional assessment. Sequence inconsistencies were checked by mapping reads to the plastome as described in Wysocki et al. (2015). The final de novo assembly was used to map the reads and calculate the mean coverage of the plastid genome using Geneious R10. Sanger-based gap closure and IR junction verification was performed following Poczai and Hyvönen (2017). Annotation was performed using a two-step procedure. First, we transferred all annotations in Geneious from the reference A. artemisiifolia to the A. trifida genome using a similarity threshold of 80%. The genome sequence was also annotated using GeSeq (Tillich et al., 2017), since this program performed better than other algorithms in a comparative study (Amiryousefi et al., 2018b). In a second step, we inspected, compared, and curated all annotations manually. This included extracting all coding regions per genome, confirming start/stop codons and features for each gene, and aligning extracted regions across all plastid genomes to confirm approximate gene lengths based on their conserved gene order. Final curated annotations were transferred to the complete plastid genome sequence of A. trifida, which was deposited in GenBank (NC036810). The resulting genome map was drawn with OGDraw v.1.2 (Lohse et al., 2007).
Comparative Plastome Analysis of Sequenced Asteraceae With Ambrosia trifida
To highlight the significance of the sequenced A. trifida plastome compared to the previously available plastomes of Asteraceae, we first investigated the repeat proportion of the genome using MISA (Thiel et al., 2003; Beier et al., 2017) and REPUTER (Kurtz et al., 2001) to identify the stretches of the perfect, compound, and long-forward repeats. MISA was used to analyze the perfect microsatellites [often abbreviated as simple sequence repeats (SSRs)] with a defined length of n = 10 in mononucleotide repeats, n = 6 in dinucleotide repeats, and n = 3 in tri-, tetra-, penta-, and hexa-nucleotide repeats. For the compound repeats, two defined SSRs should be interrupted by 100 bp. For comparative analysis, a repeat profile was mined across the 10 species to observe any divergence in the occurrence of the repeat motifs. For the identification of the forward repeats, REPUTER was used with a defined length of 30 bp and a hamming distance of 3. For the identification of microstructural events, pairwise alignments of the Asteraceae plastomes with A. trifida were performed using LASTZ (Harris, 2007) and MuMMER version 3.1 (Kurtz et al., 2004). The show-snps feature was used to evaluate the identification of plastomic variations. Once microstructural events were identified, they were further plotted using Circos (Krzywinski et al., 2009) and ggbio (Yin et al., 2012).
Comparative Phylogenomics
We based our sampling on the results of Panéro and Funk (2002), updated in Panéro et al. (2014), considering the availability of currently deposited plastid genomes in the Organelle Genome Resources of NCBI (Wolfsberg et al., 2001) accessed on 12.12.2017. Of the currently accepted 13 subfamilies of Asteraceae, only three (Carduoideae [8], Cichorioideae [8], and Asteroideae [128]) had complete plastid genome sequences deposited with a prominent bias in the genomes of Asteroideae. Genomes within subfamilies were chosen to represent each (super)tribe from the available sequences. For Carduoideae, we included one species from all accessible genera that were sequenced and available at the time we conducted the phylogenetic analysis. In Cichorioideae, only two — Lactuca L. and Taraxacum F. H. Wigg — were available. From Asteroideae, we aimed to include at least one species from each of the 21 tribes. We also included genome sequences of Carum carvi L. and Foeniculum vulgare Mill. of Apiaceae from the campanulid clade (Angiosperm Phylogeny Group, 2016) as outgroups. Accession numbers are provided in Supplementary Table S1. For phylogenetic analysis, we used a matrix of 50 protein-coding genes representing 43 species, with a total concatenated matrix alignment of 31,356 bp. We estimated congruence between different sources of information by comparing the whole genome alignment matrix, coding region matrix, and non-coding region matrix (intergenic regions). We observed ambiguity in the repeat expansion, which might affect the gene composition. Since large microstructural variations such as single nucleotide polymorphism (SNPs), and insertions/deletions (INDELs) were observed, phylogenetic analyses were limited to the coding regions (Curci et al., 2015).
From all the sequenced Asteraceae and A. trifida, coding alignments were constructed for the following fifty protein-coding genes: cemA, infA, matK, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK, petA, petG, petL, psaA, psaB, psaC, psaI, psaJ, psbA, psbB, psbC, psbD, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, rbcL, rpl14, rpl20, rpl22, rpl32, rpl33, rpl36, rpoA, rpoB, rps2, rps3, rps4, rps8, rps11, rps14, rps18, and ycf4, using MACSE (Ranwez et al., 2011), which uses a frameshift alignment algorithm for aligning coding sequences. Following the MACSE alignments, each frameshift was masked and the subsequent alignment was trimmed using trimAL (Capella-Gutiérrez et al., 2009). Finally, prior to the construction of the super-matrix, terminal stop codons were identified and subsequently removed from the trimmed alignments. Concatenation of the phylogenetic matrix was performed using SequenceMatrix version 1.8 (Vaidya et al., 2011). The concatenated sequence matrix was analyzed by maximum likelihood (ML) and parsimony methods. ML analyses were performed with IQ-TREE (Nguyen et al., 2015). The best fitting model (GY+F+I+G4) was determined by ModelFinder (Kalyaanamoorthy et al., 2017) as implemented in IQ-TREE according to the Akaike information criterion (AIC), and Bayesian information criterion (BIC). To assess branch support, all IQ-TREE analyses used the ultrafast bootstrap approximation (UFBoot; Hoang et al., 2018) with 1,000 replicates and the SH-like approximate likelihood ratio test (SH-aLRT) also with 1,000 bootstrap replicates.
Parsimony analyses were performed using nona (Goloboff, 1994) within winclada (Nixon, 2002) shell. Prior to analyses, the command “mop uninformative characters” was used to exclude parsimony uninformative characters. This resulted in a matrix of 3,769 characters. Two separate searches were performed (using processor time as a seed to randomize the order of the terminals) with the following settings: hold 30,000 (holding defined maximum number of trees), 100 replications (search performed with multiple tree-bisection-reconnection algorithm mult∗max∗), hold/3 (keeping three starting trees for each replication). In addition, we also performed larger analyses by keeping 20 starting trees for each replication (hold/20) with 1,000 replications. To assess whether the longer genes have any compositional heterogeneity, we created a partition-specific matrix of the long genes ndhD, ndhH, psaA, psaB, psbA, psbB, psbC, and rbcL and evaluated the skewness and the compositional heterogeneity across the combined and partition-specific variations.
To reveal the placement of the genes near the inverted repeat junction sites of A. trifida, an IR plot of this species and nine other Asteraceae were obtained with IRscope (Amiryousefi et al., 2018a).
Transcriptome Profiling
Despite their small size, plastids represent a classical example of miniature genomes containing mono- and polycistronic transcripts. With the advent of plastome sequencing, there has been considerable interest in understanding the transcriptional activity of plastid genes, as well as other events related to RNA editing and post-transcriptional splicing. To understand the transcriptional divergence associated with glyphosate resistance, which is defined as the amount of the transcriptional plasticity between the sensitive and resistant treatment in the giant ragweed plastome, we mapped the RNA-seq reads previously deposited to the currently sequenced A. trifida plastid genome using ChloroSeq (Castandet et al., 2016), with the defined exonic and intronic localization in the annotated GFF3 (NCBI SRA: PRJNA267208; Supplementary Table S2). For mapping the RNA-seq reads, tophat2 (Kim et al., 2013) was used with -g 2 and -no-novel-junctions functions to minimize the identification of the novel splice sites. IR repeat diversity, mapping read estimation, and genome coverage was subsequently calculated using SAMtools version 1.15 (Li et al., 2009) and bedtools version 2.25 (Quinlan and Hall, 2010). Expression values in terms of the RPKM values were mapped to the genome features and were visualized using Circos (Krzywinski et al., 2009) and ggbio (Yin et al., 2012).
Results and Discussion
Genome Assembly and Plastome Features
Plastid DNA sequencing generated 145,207 paired-end reads, with an average fragment length of 267 bp. De novo assemblies of reads resulted a total of 38 contigs with an N50 of 13,231 bp. Ten contigs were alignable and covered 100% of the A. artemisiifolia reference genome. Mapping of the reads to the de novo assembled plastid genome resulted in a 180× mean coverage. The chloroplast genome of A. trifida was 152,040 bp and showed a quadripartite structure of long (83,966 bp) and small (17,894 bp) single-copy regions, separated by two inverted repeat regions of 25,090 bp (Figure 1). As in other species of Asteraceae, the A. trifida chloroplast genome contains 80 protein-coding, 28 tRNA, and four rRNA genes comprising a total of 112 unique genes (Supplementary Tables S3, S4). The distribution of the genes also exhibited similarity with other Asteraceae and angiosperms, with 13 genes found in the SSC, 19 genes in the IR, and 80 genes in the LSC. The overall GC content of the chloroplast genome was 37.2%. Only 21% of the whole genome is non-coding. There were 18 intron-containing genes in the giant ragweed plastome (Supplementary Table S4). From these, 16 (10 protein-coding and six tRNA) genes had a single intron, and two (ycf3, clpP) had two introns. 12 (eight protein-coding and four tRNA) genes are located in the LSC, one (protein-coding) gene in the SSC, and five (three protein-coding and two tRNA) genes in the IR region. The largest intron (2,565 bp) was located in the trnK-UUU gene, including the highly diverse matK gene. The trnK intron is of interest because it represents an unusual form of a group II intron derived from a mobile group of mitochondrial-like intron ORFs (Hausner et al., 2006). The rps12 gene was trans-spliced with the 5′ end exon located in the LSC region and the two remaining exons found in the IR regions. We also observed three cases of overlapping genes, namely psbD/psbC, atpE/atpB, and rps3/rpl22. In the ndhD and psbL genes, we observed that ACG is used as an alternative start codon instead of the common AUG in A. trifida and in most species of Asteraceae. It has been shown that this exceptional ACG start codon is RNA edited in all Solanaceae (Amiryousefi et al., 2018b) except Datura stramonium L., while the start codon of psbL is unedited. In Asteraceae, the canonical AUG form is found for both psbL and ndhD in Ageratina adenophora (Spreng.) King & H. Rob., Pericallis hybrida (Willd.) R. Nordenstam, Silybum marianum (L.) Gaertn. However, only ndhD possesses the AUG start codon in Centaurea diffusa Lam., Chrysanthemum indicum L., Jacobaea vulgaris Gaertn., Mikania micrantha Kunth and Saussurea involucrata Matsum. & Koidz.
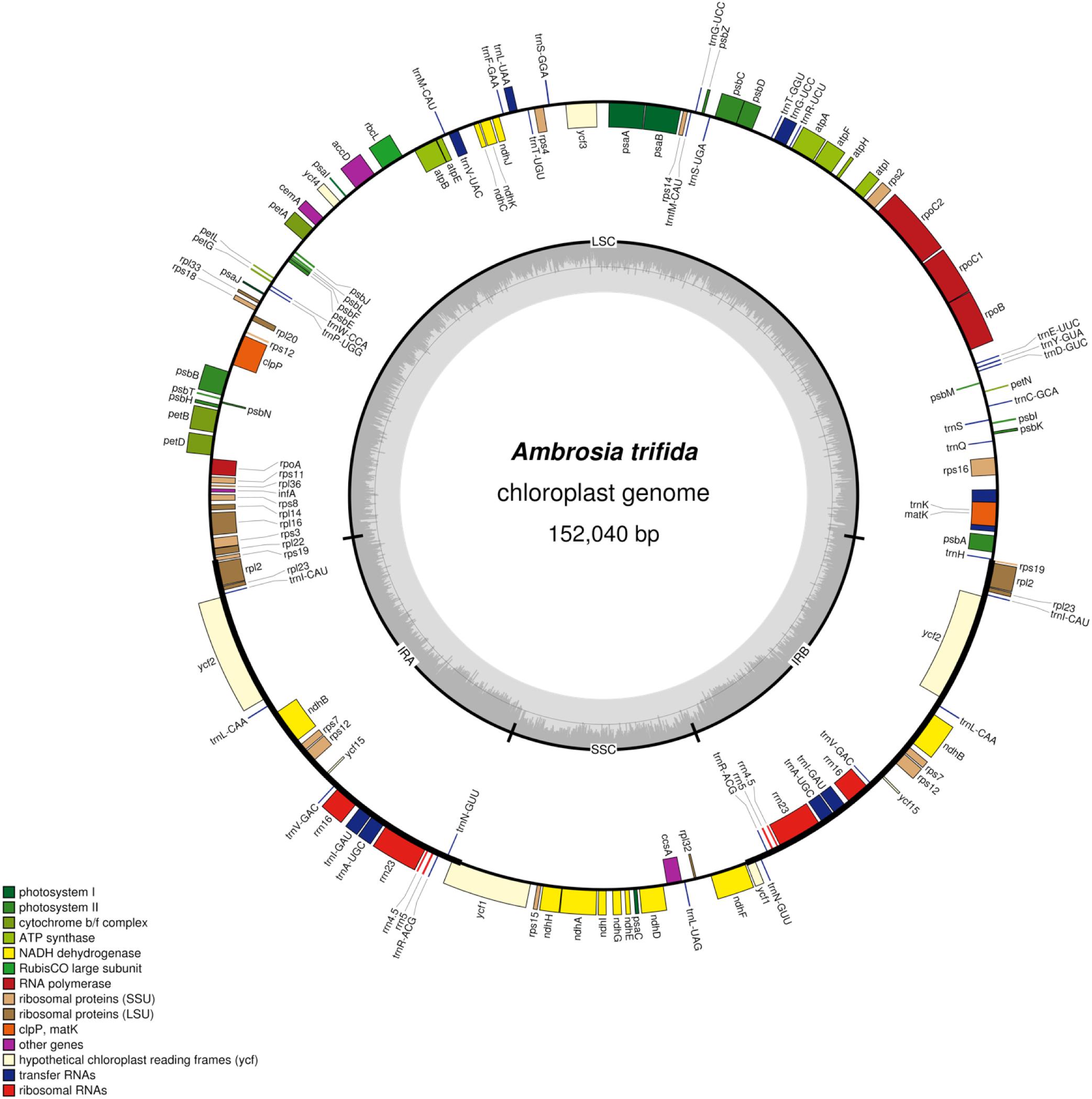
Figure 1. Plastome map of Ambrosia trifida. Gene features and distribution of the direction of genes with respect to their localization. Genes on the inner side of the outer circle are transcribed counterclockwise while those on the outer side are transcribed clockwise. Genes belonging to different functional groups are color-coded; the GC and AT content of the genome are plotted on the inner circle as dark and light gray, respectively. Inverted repeats (IR); large single copy (LSC) regions; small single copy (SSC) regions.
Genomic Repeats and Rearrangements
Microsatellites (or SSRs) are valuable molecular markers of high-degree variations within the same species. These markers have been previously used in population genetics and polymorphism investigation of ragweeds (Genton et al., 2005a,b; Gaudeul et al., 2011). We analyzed the distribution of SSRs according to the defined length criteria in the giant ragweed plastome. A total of 100 SSRs were observed, with eight present in compound form. To understand whether their distribution varies among Asteraceae species, we further compared 10 previously sequenced plastomes (Table 1). The most abundant motifs of the SSRs were poly-A/T stretches characteristic of angiosperm plastid genomes. These results are consistent with previous findings that the SRs are generally composed of short poly-A or poly-T repeats and rarely contain tandem G or C repeats h. In addition to mononucleotide stretches, we observed tetra- (AGAT/ATCT) and hexa- (AAGGAT/ATCCTT) nucleotide repeats, which could be of specific interest for future cpSSR marker development in cross-species amplifications or population genetic studies of ragweeds. We further identified 25 larger repeats (>30 bp) using the defined parameters in REPUTER (Kurtz et al., 2001). Most of these long repeats were present in the intergenic spacers (Table 2). Among these repeats, a large portion were found within ycf genes, which have been shown to have high divergence rates in most embryophyte lineages and have undergone pseudogenization (Michelangeli et al., 2003). The nucleotide sequence similarity among embryophyte ycf2 is extraordinarily low compared to other plastid-encoded genes: it is less than 50% across bryophytes, ferns, and seed plants (Wicke et al., 2011). This divergence is not surprising since ycf genes have experienced many insertions/deletions. For example, these deletions account for the reduction in the chloroplast genome size among members of the Graminid clade (Poczai and Hyvönen, 2017).
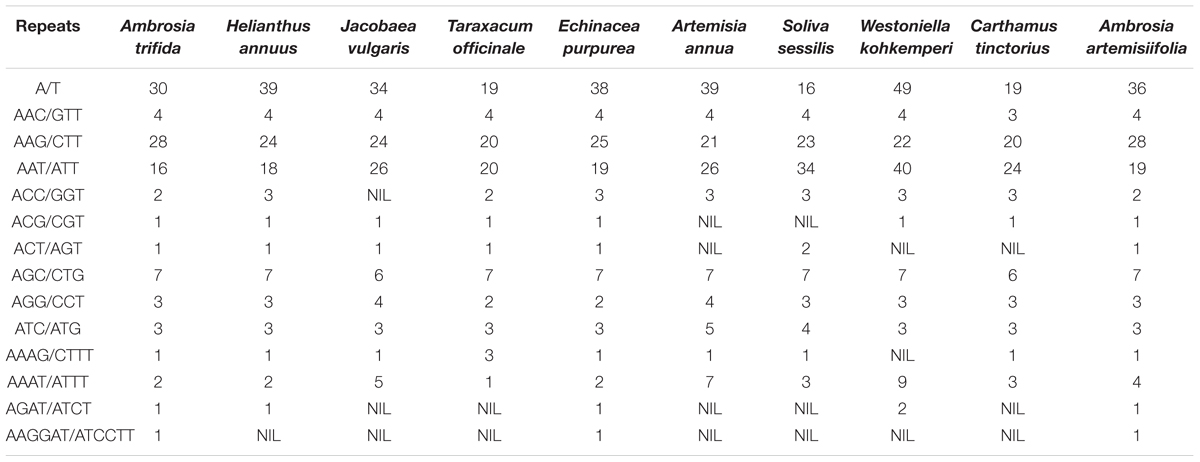
Table 1. Distribution and summary of the shared SSRs across the Asteraceae plastomes.

Table 2. Summary of the identified forward repeat stretched across the Ambrosia trifida plastome.
Asteraceae plastid genomes contain two inversions of 22.8 and 3.3 kb as compared with the outgroup terminals Carum carvi and Foeniculum vulgare of Apiaceae. The larger inversion is located between the trnS-GCU and trnG-UCC genes, and the smaller between the trnE-UUC and trnT-GGU genes. In addition to these large inversions, another smaller inversion of 3.3 kb is located within the larger inversion, between the trnC-GCA and rpoB genes. These rearrangements are assumed to have originated in the late Eocene (36–42 My BP) and are commonly reported for Asteraceae (Jansen and Palmer, 1987; Kim et al., 2005). They are absent in the species of the basal subfamily Barnadesioideae as assessed by restriction endonuclease digestions (Jansen and Palmer, 1987). This interesting finding should be confirmed by future research, since complete plastid genome data is currently missing for this group. To further understand the level of the microstructural events, we performed a pairwise comparison of the Asteraceae plastomes with the sequenced A. trifida, revealing a high number of substitutions compared to insertions and deletions. Interestingly, we observed a large number of inversions compared with Parthenium argentatum A. Gray and Ageratina adenophora (Spreng.) King & H. RobAs in other angiosperms, the coding regions of Asteraceae are more conservative than the non-coding regions; rpoC1 is the most divergent of all the genes. The invasive weed Ageratina adenophora rpoC1 contains two introns, while only one intron is found in the plastid genomes of other Asteraceae (Nie et al., 2012). Our LASTZ dot-plot comparison with the complete plastome of Saussurea involucrata Matsum. & Koidz. showed rearrangement patterns (Figure 2 and Supplementary Figure S1) caused by a large shift (approximately 93 kb) in residue numbering that caused problems during de-circularization of the genome. The published genome of this species (Xie et al., 2015) also contains numerous annotation errors with several key genes unannotated (for example, rpl16 exon 2, rps12, and truncated rps19). We found that SSC regions showed signatures of inversions (Figure 2), which is consistent with previous reports (Liu et al., 2013; Walker et al., 2014; Gruenstaeudl et al., 2017). These major spots of inversions have also been reported previously in the plastomes of several plant lineages (Schwarz et al., 2015; Hsu et al., 2016; Graham et al., 2017; Sinn et al., 2018), It is important to note that the SSC region occurs in two inversion isomers that exist in equimolar proportions in the same individual, which are identical in sequence but different in orientation (Palmer, 1983). Presently, whole plastid genome sequences are uploaded in GenBank without preference for the orientation of the SSC region, which appears as a genome structural inversion but is truly just chloroplast heteroplasmy (Walker et al., 2015). Many plastid genome studies use reference-guided mapping, where the orientation of the SSC is copied along novel genomes. For example, SSC regions occur in inverted orientations in Solanaceae compared to Asteraceae (Salih et al., 2017). Another good example in Asteraceae is the plastid genome of Lactuca sativa L., which has been published twice independently (NC007578 and DQ383816). Besides minor polymorphisms among these genomes, the major difference is the orientation of the SSC, which appears to be inverted. Due to the rapid increase in amount of de novo assembled plastid genomes that are often deposited independently and parallel to each other, this phenomenon should not be overlooked, and SSC orientations should not be regarded as diversity hot-spots.
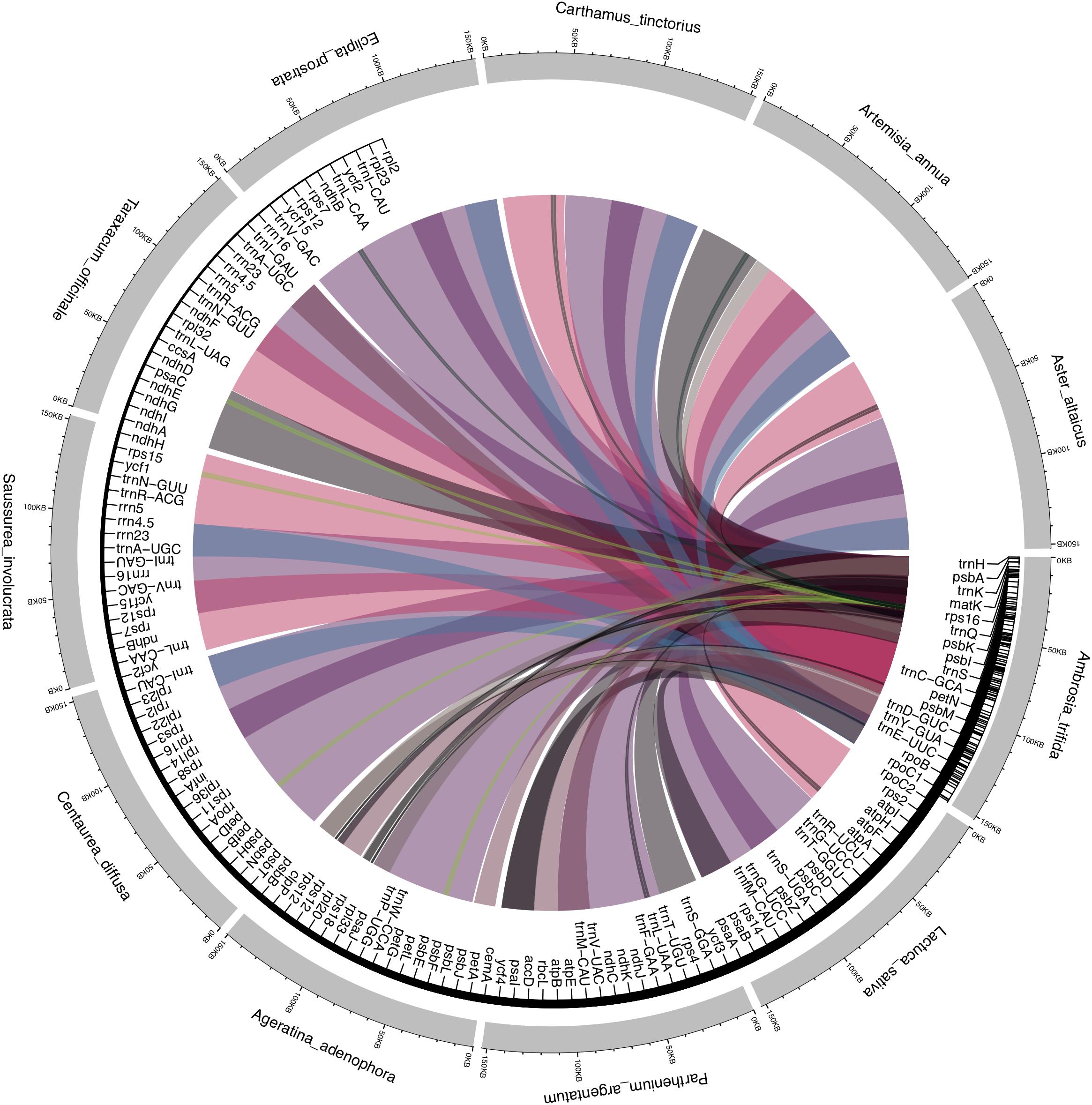
Figure 2. Segmental synteny and conservation of Ambrosia trifida across Asteraceae plastomes. LASTz alignments were filtered to keep only colored tracks showing > 70% alignment coverage, with the colored ribbons are species-specific. The inner tract displays the corresponding gene annotations of A. trifida, with the corresponding ribbons showing the inversions and segmental rearrangement across the gene regions.
Inverted Repeat (IR) Diversity
Most of the length variation in angiosperms is attributed to expansion or contraction of the IR regions. It is rare that this variation is due to gene losses, which could vary within a single genus (Sloan et al., 2014). To obtain comprehensive insight into the IR regions of the Asteraceae, a survey of over 40 species was performed using a recently published tool IRscope (Amiryousefi et al., 2018a). This revealed the placement of rps19 in the JLB (LSC/IRb) in almost all cases (Figure 3 and Supplementary Figure S2). The inversion of the SSC region, and hence its reversed gene annotation, is a main distinguishing factor in the visual representation of the species. The fixation of the extended ycf1 in the JSB (IRb/SSC) was confirmed for the majority of the species (approximately 80%). For the same set of species, we observed a pseudo ycf1 gene often tangental to the JSA (SSC/IRa) on the IRa side and the ndhF gene near the JSA on the SSC side. In some species, the SSC occurred in the reverse orientation and with ycf1 extending on the JSA site and a pseudo ycf1 and ndhF plotted on the JSB. The JLA has the rps19 and trnH genes in its vicinity, with psbA further in the LSC region in almost all cases. The pseudo rps19 gene was unannotated in the original files of the first four species in Figure 3. Hence, we obtained the corresponding annotations using GeSeq and inverted the SSC region with IRscope for the first five species to improve visual comparison. Unlike the others, the new annotation of A. artemisiifolia confirmed the existence of the rps19 and its corresponding pseudo fragment on the LSC adjacent to IRb and IRa, respectively (Figure 3). Non-identical IR features were observed for A. artemisiifolia (NC035875), where the IRb contains a single C base insertion near rpl2 (84,242 bp). The trnH gene was also assigned to the opposite DNA strand, which we have manually corrected for Figure 3. After these adjustments, we obtained a corrected size of 25,086 bp for the IRs (cf. Nagy et al., 2017).

Figure 3. Inverted repeat plot of Ambrosia trifida. rps19 gene on the LSC/IRb regions with corresponding 184/95 bp. The positive strand synteny of the rpl2, rps19, and rpl22 on the IRb, JLB, and LSC, respectively, is also confirmed. In accordance with the other selected nine Asteraceae, the JSB of A. trifida is hosting the extended functional version of the ycf1 gene, while the pseudo version of this gene beside ndhF is placed near the JSA site. Another copy of the rps19 and trnH, along rpl2 are gathered near the JLA.
The conservation of the length of the IRs, SSC, and LSC is another interesting point. These regions were between 24 to 26, 18 to 19, and 82 to 84 kb, respectively. The maximum length of plastid genomes is 153,014 bp for Conyza bonariensis (L.) Cronquist, while the shortest length of 149, 510 bp was found in Aster spathulifolius Maxim. As in other Asteraceae, the inverted repeat plot of the A. trifida showed the rps19 in the LSC/IRb regions with a corresponding 184/95 bp. The synteny of the rpl2, rps19, and rpl22 (for IRb, JLB, and LSC, respectively) of the positive strand was also confirmed. As in the majority of other angiosperms, the JSB of A. trifida has the extended functional version of the ycf1 gene while the pseudo version of this gene beside ndhF is placed near the JSA site. Another copy of the genes rps19, trnH and rpl2 can be found near the JLA.
Comparative Phylogenomics
Asteraceae is one of the largest families of flowering plants, with approximately 1,500 genera and 23,000 species (Kumar et al., 2009). While several studies have been conducted to resolve the phylogeny (Denda et al., 1999; Panéro and Funk, 2002, 2008; Panéro et al., 2014), many questions remain open. Plastome sequences can now be easily acquired for phylogenomic analyses at relatively low costs, thus providing rich sources of phylogenetic information. To assess the phylogenetic position of A. trifida relative to the previously sequenced Asteraceae species, we performed maximum likelihood (Figure 4) and parsimony analysis. The latter resulted in six equally parsimonious trees with length of 7,297 steps, with consistency index (Kluge and Farris, 1969) CI 0.65 and retention index (Farris, 1989) RI 0.83 (Supplementary Figure S3). The tree is congruent with that obtained by using maximum likelihood as an optimality criterion, and no bias in compositional heterogeneity was detected in the sequences (Supplementary Figure S4).
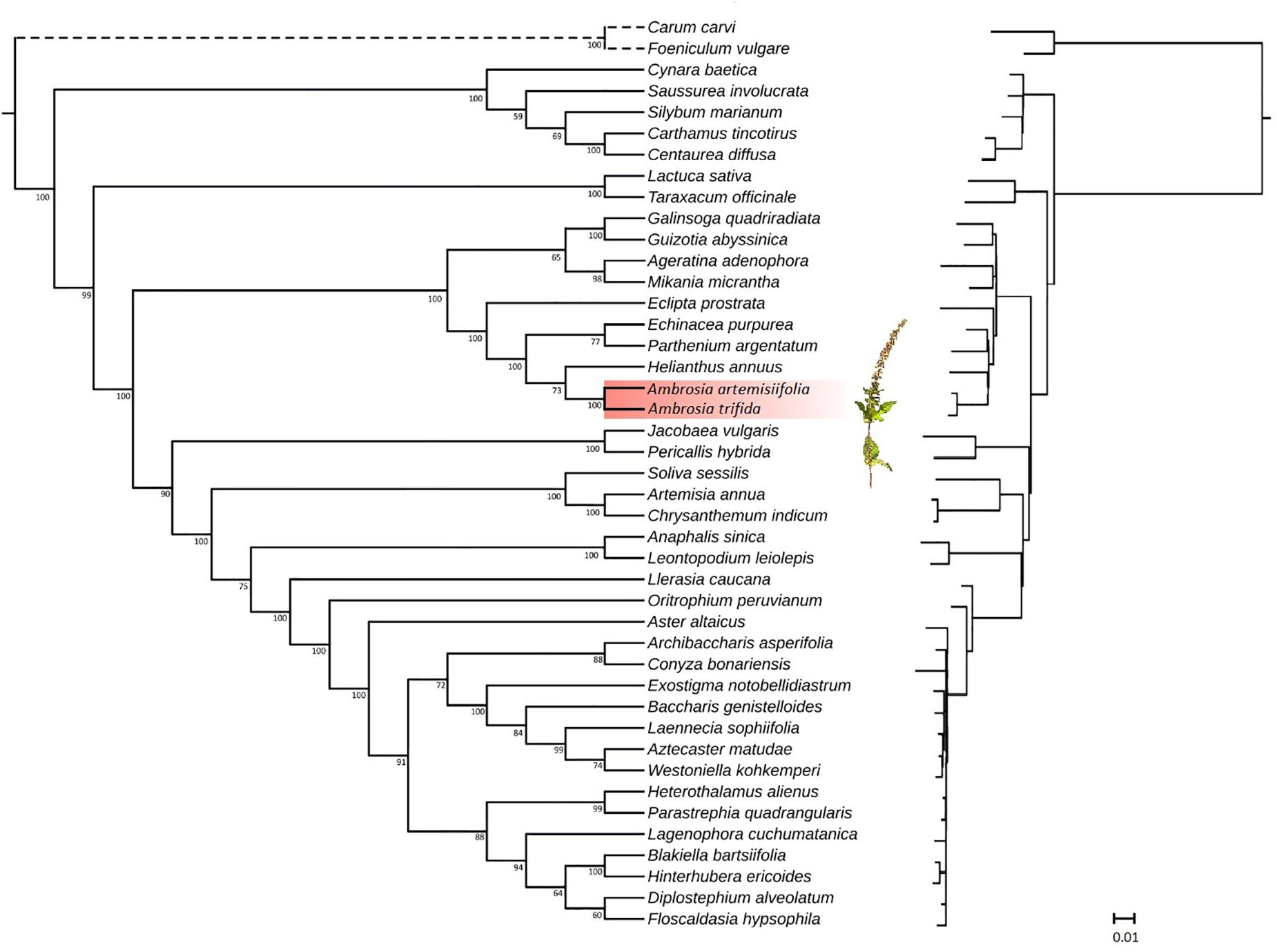
Figure 4. Phylogenetic tree (left in cladogram style, right with branch lengths) inferred from plastid genome data. Maximum likelihood phylogeny illustrating the phylogenetic relationships of Asteraceae based on fifty protein-coding genes. Species of the genus Amrbosia are highlighted in red. Branch lengths are proportional to the number of substitutions, while numbers represent bootstrap support values for each node. The given scale represents substitutions per site.
Historically, Asteraceae were divided into two large subfamilies (Asteroideae and Cichorioideae) and 13 tribes (Bentham, 1873). Major changes in classification have been made during the past decade, resulting in a better phylogenetic framework. Based on the analyses of chloroplast DNA markers (Panéro and Funk, 2002, 2008; Panéro et al., 2014), 13 major clades (subfamilies) were identified in Asteraceae. From these subfamilies, our study included representatives of Carduoideae, Cichorioideae and Asteroideae with complete plastid genome sequences available from public databases. The Asteroideae subfamily was sister to Cichorioideae, including Lactuca sativa L. and Taraxacum officinale (L.) Weber ex F. H. Wigg of the tribe Cichorieae. Taxa within the subfamily Carduoideae were represented by members of the tribe Cynareae, and formed two clades. The first was composed of Carthamus tinctorius L. and Centaurea diffusa Lam. The second included Cynara baetica (Spreng.) Pau and Silybum marianum (L.) Gaertn. as sisters, while Saussurea involucrata Matsum. & Koidz. were resolved in a basal position. This phylogeny obtained for the Cynareae is consistent with the results of a more detailed molecular analysis (Barres et al., 2013) including larger number of terminals. As expected, our tree resolved three supertribes Asteroideae, Helianthidae, and Senecionodae within the subfamily Asteroideae. Jacobea vulgaris Gaertn., together with Pericallis hybrida (Willd.) R. Nordenstam, grouped in the supertribe Senecionodae. In the Asterodae, all three sampled tribes of Anthemidae, Gnaphalieae, and Astereae were distinct with high support values compared to previous plastid studies where tribes of Asteroideae were not resolved (Curci et al., 2015). Helianthus annuus L, A. artemisiifolia, and A. trifida grouped in the Heliantheae alliance indicating a group with high support values within the supertribe Helianthodae, and separate of Millerideae (Galinsoga quadriradiata Ruiz & Pav., Guizotia abyssinica) and Eupatorieae (Ageratina adenophora [Spreng.] King & H. Rob.; Mikania micrantha Kunth). Deeper level relationships within the alliance Heliantheae were unresolved. Whole chloroplast genome sequences were unavailable for the cockleburs (Xanthium L.) thus we were unable to provide support for the previously reported sister genus relationship to Ambrosia (Miao et al., 1995). Both genera are monoecious with pistilate florets surrounded by woody involucres, a morphological trait that has been assumed to indicate their close relationship (Peterson and Payne, 1973).
Comparative Transcriptional Profiles Against Glyphosate Resistance
As a first contribution toward deciphering the complete genetic information of giant ragweed, we determined the complete sequence of its plastome. Since assembling plastid genomes from herbarium specimens is possible, the complete sequences can be used in further applications. The analysis presented here is an example of the use of herbarium genomics in other fields like weed research where, besides the biology of ragweeds, their control with herbicides is a major area of focus for research. Herbicides have a pivotal role in controlling weeds and sustaining food security by restricting weeds while being as harmless as possible for crops (Duhoux et al., 2017). Reaching this specific goal with various mechanisms of action, herbicides induce changes in gene expression to alter or terminate plant physiological pathways. Knowledge about the regulation of plastid gene expression in response to herbicide treatments is central to our understanding of photosynthesis and other plastid-localized metabolic pathways associated with herbicide resistance. To date, the molecular basis of herbicide resistance has been largely investigated by single-gene sequencing to identify single-point mutations in the target site of the herbicide. More recently, second-generation sequencing technologies have also enabled transcriptomic approaches (e.g., RNA-seq) to identify candidate genes underlying more complex NTSR mechanisms, such as herbicide metabolism and translocation (Ravet et al., 2018). Weed genomics offers the promise to go beyond transcriptomics and provide further novel insights into the biological processes such as NTSR.
Despite its small size, the transcriptional apparatus of plastids is relatively divergent at the expression level compared to the nuclear genome, which is the first line in transcriptional-mediated responses pertaining to abiotic and biotic stress including herbicide resistance. These transcriptional events, as defined by the variations observed in the transcriptional expression levels, play an important role in understanding plastid metabolism at the cellular and energetic levels by altering the transport of solutes across the membrane, or by regulating their sequestration through membrane trafficking (Sammons and Gaines, 2014). One such example is glyphosate, which is sequestered by a transport mechanism and inhibits the shikimate pathway in the chloroplast (Sammons and Gaines, 2014). Plastid gene expression and its regulation has been intensely studied in chloroplasts (see the review of Leister et al., 2017). By contrast, knowledge of gene expression under herbicide treatment in plastids is still very limited. Since a large fraction of plastid protein-coding genes is involved in photosynthesis, it is generally believed that plastid gene expression is affected in plant tissues treated with herbicides. Until now, limited knowledge has been accumulated to understand the transcriptional events related to glyphosate resistance, although it has been used globally to control noxious weeds such as A. trifida (Sammons and Gaines, 2014). As previously mentioned, since glyphosate sequestration and harvesting through the chloroplast mediated shikimate pathway play an important role in herbicide resistance, we compared the transcriptional events in the plastid genome of A. trifida. For this, we used RNA-seq reads previously deposited in the Sequence Read Archive (Supplementary Table S2) gathered from total plant cell transcriptomes, which capture both primary and processed mRNA sequences of the plastome. Therefore, we first isolated the plastid genome data from the total transcriptomes of glyphosate resistant and sensitive A. trifida plants sequenced across four different time points – 0, 3, 8, and 12 h after spraying with the herbicide. In particular, we observed that exonic divergence (change in the patterns of the expression levels of exons across the sensitive and the resistant time points) to be more pronounced compared to the intronic divergence (change in the patterns of the expression levels of introns across the sensitive and the resistant time points; Figure 5 and Supplementary Table S5). Considering the divergence of the glyphosate uptake, a higher expression of plastid genes was found in resistant as compared to the sensitive conditions. As a first line of defense against any abiotic stress, the primary focus of the gene expression alternation at the nuclear level is mainly pertaining to the genes involved in photosynthetic responses, heat shock or those involved in the membrane transport. We observed a higher expression of the tRNA genes across all of the time points, which is conserved across all of the embryophytes and suggests that the higher expression of the relative tRNAs is required for rapid transcription activity, which might be required to circumvent the herbicide-mediated changes in the membrane transport of the generated ATPs across the NADPH complex. Additionally, it might indicate that the expression of the SIG2, responsible for the transcription of tRNA genes, is pre-dominant as compared to the SIG6, which is mostly associated with the photosynthetic genes (Figure 6).
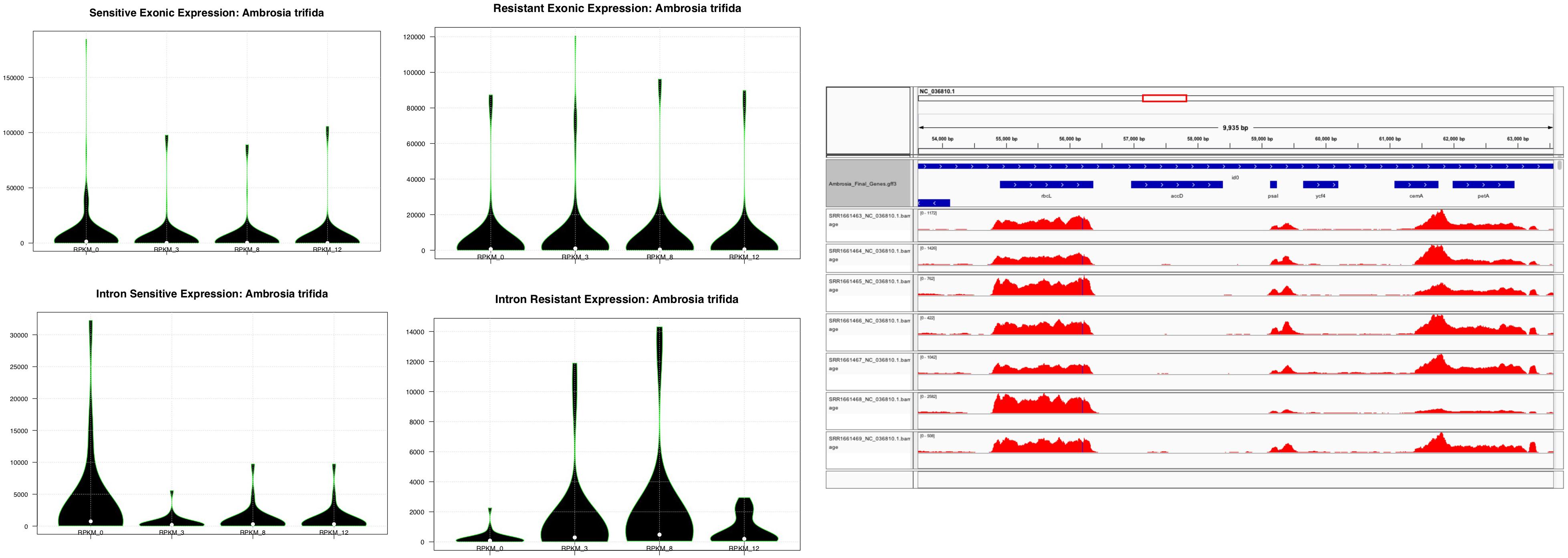
Figure 5. Violin plots showing the exonic and intronic RPKM distribution across the sensitive and resistant glyphosate ragweeds. Time points indicate sample collection: 0, 3, 8, and 12 h after herbicide application.
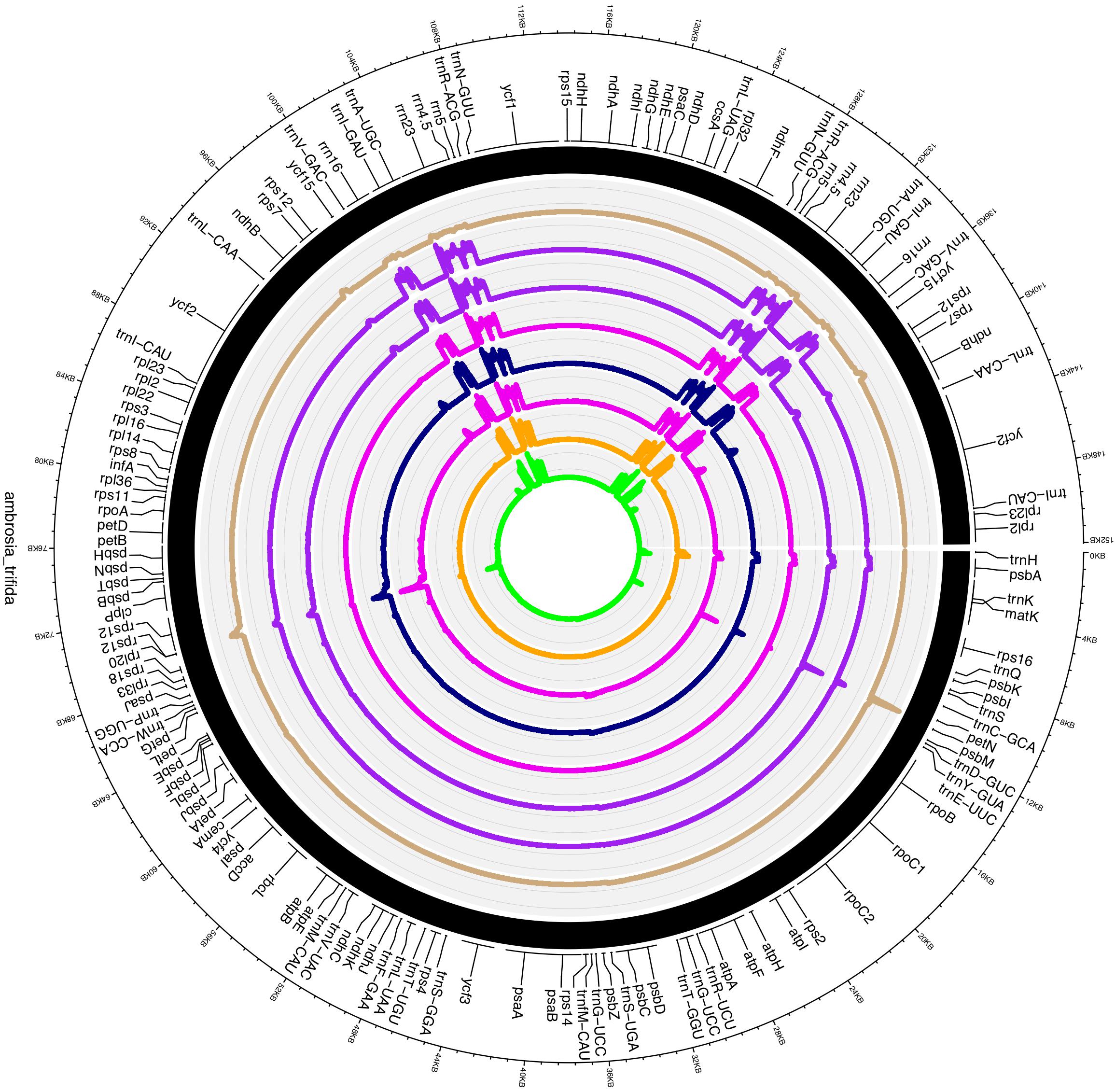
Figure 6. Circos plot visualization of the transcriptional mapping of Ambrosia trifida. Single-base resolution of the mapped RNA-seq reads across the A. trifida plastome with represented gene annotations. Track ribbons order according to the following SRA accession number: SRR1661420, SRR1661463, SRR1661464, SRR1661465, SRR1661466, SRR1661467, SRR1661468 and SRR1661469.
Furthermore, A. trifida had relatively low expression levels, which might hint toward the uptake of the glyphosate and the rate of disruption of the photosynthetic apparatus and the metabolic pathways associated with plastid genes. It has been widely demonstrated that glyphosate also affects growth by contaminating the environment and accumulating in plant organs, and can be a major limiting factor for agricultural productivity (Fartyal et al., 2018). According to our observations, plastid intronic expression is higher in glyphosate-resistant giant ragweed at all sequenced time points compared to the sensitive time points after the herbicide treatment (Supplementary Table S5). The expression of introns associated with ycf3 increased during the 8 and 12 h time points in sensitive A. trifida, which might present a defense mechanism to combat glyphosate (Figure 6). Relative expression divergence among the sensitive and resistant time points for the photosystem I (PSI) assembly protein ycf3 revealed an up-regulation in the resistant phenotype compared to the sensitive phenotype. This clearly reflects the role of glyphosate as an interfering herbicide in plant growth through the inhibition of the photosynthetic complex (Fartyal et al., 2018). Higher expression of ribosomal-associated genes has also been observed in sensitive time points compared to the glyphosate resistant time points, which might reflect the higher activity rates of the translation and ribosomal machinery required for the efficient translation of plastid genes during glyphosate resistance. We also observed down-regulation of atpF, which is an H+ATPase (CF0 subunit of the CF0CF1) and a critical component for energy production also associated with the down-regulation of ndhA and ndhB. Members of the NADH-like dehydrogenases play an important role in the PSI cyclic electron flow (Suorsa et al., 2009). Overall, these findings suggest that glyphosate has immediate effects on the photo-accumulation events and thus alters the transport and the energy production in plastid genes. Similar disruptions were reported in mitochondrial ATP influx-outflux during glyphosate resistance (Gomes et al., 2017).
Conclusion
The inclusion of historical museum specimens in phylogenomic analyses of biodiversity provides new possibilities for both fundamental and applied plant biology research. Our study is a good example of how herbarium specimens can be used to investigate phylogeny and genomic patterns of herbicide resistance. However, it should be kept in mind that museomics are limited by the amount of plant material, and disruptive sampling should be cautiously carried out with such specimens. Herbarium genomics of weeds could also improve our understanding of resistance to herbicides. As demonstrated in our study, plastid genomes reconstructed from herbarium specimens, coupled with transcriptomic resources can also be used to investigate herbicide resistance with potential for further applications in weed management.
Author Contributions
PP conceived the study, performed the sampling, laboratory experiments, genome assembly, annotation, and data curation, and prepared the original draft of the manuscript. GS conceived the methodologies and performed the further analyses. JH and AA performed parsimony and inverted repeat analyses, respectively. JH, XH, and PP carried out the project administration and provided resources for the work. PP and JH supervised the work. GS, AA, and PP performed the validation and visualization of the work. All authors reviewed and edited the manuscript.
Funding
This study was funded by the Systematics Research Fund (United Kingdom), the Finnish Museum of Natural History Luomus Trigger Funds (PP), and the Academy of Finland (Grant No. 295595 to JH and XH).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the staff and colleagues of the Viikki Biocenter who kindly contributed reagents and materials for our study. We thank CSC – IT Center for Sciences, Espoo (Finland) for providing computational servers for the data analysis. We also thank Derek Ho and Jacquelin DeFaveri for critical reading of the manuscript and valuable comments on an earlier version.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00218/full#supplementary-material
FIGURE S1 | LASTZ dot-plot comparison among Asteraceae complete plastid genome sequences.
FIGURE S2 | Inverted repeat plot of 30 Asteraceae chloroplast genomes.
FIGURE S3 | Phylogenetic tree of Asteraceae based on the analysis of fifty protein coding genes with parsimony used as an optimality criterion.
FIGURE S4 | Evaluation of the skewness and compositional heterogeneity across the combined and partition-specific variations of the long genes ndhD, ndhH, psaA, psaB, psbA, psbB, psbC, and rbcL.
TABLE S1 | List of the sequenced plastomes used for the phylogenomic analysis.
TABLE S2 | Summary of the aligned transcriptomics reads.
TABLE S3 | List of genes in the chloroplast genome of Ambrosia trifida.
TABLE S4 | Genes having an intron in the Ambrosia trifida plastid genome, and the length of the exons and introns.
TABLE S5 | Intronic reads per kb per million mapped (RPKM) across the herbicide treatments in the plastid genome of giant ragweed (Ambrosia trifida).
References
Abul-fatih, H. A., and Bazzaz, F. A. (1979). Biology of Ambrosia trifida L. 2. Germination, emergence, growth and survival. New Phytol. 83, 817–827. doi: 10.1111/j.1469-8137.1979.tb02313.x
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2017). The plastid genome sequence of the invasive plant common ragweed (Ambrosia artemisiifolia, Asteraceae). Mitochondrial DNA B 2, 753–754. doi: 10.1080/23802359.2017.1390423
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2018a). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2018b). The chloroplast genome sequence of bittersweet (Solanum dulcamara): plastid genome structure evolution in Solanaceae. PLoS One 13:e0196069. doi: 10.1371/journal.pone.0196069
Angiosperm Phylogeny Group (2016). An updated of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1016/j.jep.2015.05.035
Aubriot, X., Knapp, S., Syfert, M. M., Poczai, P., and Buerki, S. (2018). Shedding new light on the origin and spread of the brinjal eggplant (Solanum melongena L.) and its wild relatives. Am. J. Bot. 105, 1175–1187. doi: 10.1002/ajb2.1133
Bakker, F. T. (2017). Herbarium genomics: skimming and plastomics from archival specimens. Webbia 72, 35–45. doi: 10.1080/00837792.2017.1313383
Bakker, F. T., Lei, D., Yu, J., Mohammadin, S., Wei, Z., van de Kerke, S., et al. (2015). Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an iterative organelle genome assembly pipeline. Biol. J. Linn. Soc. 117, 33–43. doi: 10.1111/bij.12642
Barres, L., Sanmartín, I., Anderson, C. L., Susanna, A., Buerki, S., Galbany-Casals, M., et al. (2013). Reconstructing the evolution and biogeographic history of tribe Cardueae (Compositae). Am. J. Bot. 100, 867–882. doi: 10.3732/ajb.1200058
Beck, J. B., and Semple, J. C. (2015). Next-generation sampling: pairing genomics with herbarium specimens provides species-level signal in Solidago (Asteraceae). Appl. Plant Sci. 3:1500014. doi: 10.3732/apps.1500014
Beier, S., Thiel, T., Münch, T., Scholz, U., and Mascher, M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Bentham, G. (1873). “Compositae,” in Genera Plantarum, Vol. 2, eds G. Bentham and J. D. Hooker (London: Reeve), 163–533.
Besnard, G., Christin, P.-A., Malé, P.-J. G., Lhuillier, E., Lauzeral, C., Coissac, E., et al. (2014). From museums to genomics: old herbarium specimens shed light on C3 to C4 transition. J. Exp. Bot. 65, 6711–6721. doi: 10.1093/jxb/eru395
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bracamonte, E., da Silveira, H. M., Alcántara-de la Cruz, R., Domínguez-Valenzuela, J. A., Cruz-Hipolito, H. E., and De Prado, R. (2018). From tolerance to resistance: mechanisms governing the differential response to glyphosate in Chloris barbata. Pest Manag. Sci. 74, 1118–1124. doi: 10.1002/ps.4874
Busi, R., Gaines, T. A., Walsh, M. J., and Powels, S. B. (2012). Understanding the potential for resistance evolution to the new pyroxasulfone: field selection at high doses versus recurrent selection at low doses. Weed Res. 52, 489–499. doi: 10.1111/j.1365-3180.2012.00948.x
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Castandet, B., Hotto, A. M., Strickler, S. R., and Stern, D. B. (2016). ChloroSeq, an optimized chloroplast RNA-Seq bioinformatic pipeline, reveals remodeling of the organellar transcriptome under heat stress. G3. 6, 2817–2827. doi: 10.1534/g3.116.030783
Chauvel, B., Dessaint, F., Cardinal-Legrand, C., and Bretagnolle, F. (2006). The historical spread of Ambrosia artemisiifolia L. in France from herbarium records. J. Biogeogr. 33, 665–673. doi: 10.1111/j.1365-2699.2005.01401.x
Chun, Y. J., Fumanal, B., Laitung, B., and Bretagnolle, F. (2010). Gene flow and population admixture as the primary post-invasion process in common ragweed (Ambrosia artemisiifolia) populations in France. New Phytol. 185, 1100–1107. doi: 10.1111/j.1469-8137.2009.03129.x
Cosgrove, D. J. (1999). Enzymes and other agents that enhance cell wall extensibility. Ann. Rev. Plant Phys. Plant. Mol. Biol. 50, 391–417. doi: 10.1146/annurev.arplant.50.1.391
Cowborough, M. J., Brown, R. B., and Tardif, F. J. (2003). Impact of common ragweed (Ambrosia artemisiifolia) aggregation on economic thresholds in soybean. Weed Sci. 51, 947–954. doi: 10.1614/02-036
Cseh, A. (2010). The Molecular Genetic Analysis of the Most Important Weed Biological Characteristics and Health Effects of Common Ragweed. Ph.D. thesis, University of Pannonia, Hungary.
Cseh, A., Cernák, I., and Taller, J. (2009). Molecular characterization of atrazine resistance in common ragweed (Ambrosia artemisiifolia L.). J. Appl. Genet. 50, 321–327. doi: 10.1007/BF03195690
Cseh, A., and Taller, J. (2008). Genetic diversity of ragweed (Ambrosia artemisiifolia L.) a comparison of maternally inherited cpDNA and mtDNA. J. Plant Dis. Prot. 21, 389–394.
Csontos, P., Vitalos, M., Barina, Z., and Kiss, L. (2010). Early distribution and spread of Ambrosia artemisiifolia in Central and Eastern Europe. Bot. Helv. 120, 75–78. doi: 10.1007/s00035-010-0072-2
Cummins, I., Wortley, D. J., Sabbadin, F., He, Z., Coxon, C. R., Straker, H. E., et al. (2013). Key role for glutathione transferase in multiple-herbicide resistance in grass weeds. Proc. Natl. Acad. Sci. U.S.A. 110, 5812–5817. doi: 10.1073/pnas.1221179110
Curci, P. L., De Paola, D., Danzi, D., Vendramin, G. G., and Sonnante, G. (2015). Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other Asteraceae. PLoS One 10:e0120589. doi: 10.1371/journal.pone.0120589
Daniell, H., Lin, C.-S., Yu, M., and Chang, W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Délye, C. (2013). Unravelling the genetic bases of non-target-site based resistance (NTSR) to herbicides: a major challenge for weed science in the forthcoming decade. Pest Manag. Sci. 69, 176–187. doi: 10.1002/ps.3318
Délye, C., Jasieniuk, M., and Le Corre, V. (2013). Deciphering the evolution of herbicide resistance in weeds. Trend. Genet. 29, 649–658. doi: 10.1016/j.tig.2013.06.001
Denda, T., Watanabe, K., Kosuge, K., Yahara, T., and Ito, M. (1999). Molecular phylogeny of Brachycome (Asteraceae). Plant Syst. Evol. 217, 299–311. doi: 10.1007/BF00984372
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucl. Acid Res. 45:e18. doi: 10.1093/nar/gkw955
Duhoux, A., Pernin, F., Desserre, D., and Délye, C. (2017). Herbicide safeners decrease sensitivity to herbicides inhibiting acetolactate-synthase and likely activate non-target-site-based resistance pathways in the major grass weed Lolium sp. (Rye-grass). Front. Plant Sci. 8:1310. doi: 10.3389/fpls.2017.01310
Duke, S. O., and Powles, S. B. (2008). Glyphosate: a once-in-a-century herbicide. Pest Manag. Sci. 64, 319–325. doi: 10.1002/ps.1518
Farris, J. S. (1989). The retention index and homoplasy excess. Syst. Zool. 38, 406–407. doi: 10.1016/j.jtbi.2014.10.033
Fartyal, D., Agarwal, A., James, D., Borphukan, B., Ram, B., Sheri, V., et al. (2018). Co-expression of P173S mutant rice EPSPS and igrA genes results in higher glyphosate tolerance in transgenic rice. Front. Plant Sci. 9:144. doi: 10.3389/fpls.2018.00144
Ganie, Z. A., Lindquist, J. L., Jugulam, M., Kruger, G. R., Marx, D. B., and Jhala, A. J. (2017). An integrated approach to control glyphosate-resistant Ambrosia trifida with tillage and herbicides in glyphosate-resistant maize. Weed Res. 57, 112–122. doi: 10.1111/wre.12244
Gaudeul, M., Giraud, T., Kiss, L., and Shykoff, J. A. (2011). Nuclear and chloroplast microsatellites show multiple introductions in the worldwide invasion history of common ragweed, Ambrosia aretemisiifolia. PLoS One 6:e17658. doi: 10.1371/journal.pone.0017658
Genton, B. J., Jonot, O., Thévenet, D., Fournier, E., Blatrix, R., Vautrin, D., et al. (2005a). Isolation of five polymorphic microsatellite loci in the invasive weed Ambrosia artemisiifolia (Asteraceae) using an enrichment protocol. Mol. Ecol. Res. 5, 381–383. doi: 10.1111/j.1471-8286.2005.00934.x
Genton, B. J., Shykoff, J. A., and Giraud, T. (2005b). High genetic diversity in French invasive populations of common ragweed, Ambrosia artemisiifolia, as a result of multiple sources of introduction. Mol. Ecol. 14, 4275–4285.
Gladieux, P., Giraud, T., Kiss, L., Genton, B. J., Jonot, O., and Shykoff, J. A. (2011). Distinct invasion sources of common ragweed (Ambrosia artemisiifolia) in Eastern and Western Europe. Biol. Inv. 13, 933–944. doi: 10.1007/s10530-010-9880-y
Goloboff, P. A. (1994). NONA. Version 2.0. Tucumán, AR: Computer software distributed by the author.
Gomes, M. P., da Silva Cruz, F. V., Bicalho, E. M., Borges, F. V., Fonseca, M. B., Juneau, P., et al. (2017). Effects of glyphosate acid and the glyphosate-commercial formulation (Roundup) on Dimorphandra wilsonii seed germination: interference of seed respiratory metabolism. Environ. Pollut. 220, 452–459. doi: 10.1016/j.envpol.2016.09.087
Graham, S. W., Lam, V. K. Y., and Merckx, V. S. F. T. (2017). Plastomes on the edge: the evolutionary breakdown of mycoheterotroph plastid genomes. New Phytol. 214, 48–55. doi: 10.1111/nph.14398
Green, J. M., and Owen, M. D. K. (2011). Herbicide-resistant crops: utilities and limitations for herbicide-resistant weed management. J. Agric. Food Chem. 59, 5819–5829. doi: 10.1021/jf101286h
Gruenstaeudl, M., Nauheimer, L., and Borsch, T. (2017). Plastid genome structure and phylogenomics of Nymphaeales: conserved gene order and new insights into relationships. Plant Syst. Evol. 303, 1251–1270. doi: 10.1007/s00606-017-1436-5
Gutaker, R. M., and Burbano, H. A. (2017). Reinforcing plant evolutionary genomics using ancient DNA. Curr. Opin. Plant Biol. 36, 38–45. doi: 10.1016/j.pbi.2017.01.002
Gutaker, R. M., Reiter, E., Furtwängler, A., Schuenemann, V. J., and Burbano, H. A. (2017). Extraction of ultrashort DNA molecules from herbarium specimens. Biotechniques 62, 76–79. doi: 10.2144/000114517
Harris, R. S. (2007). Improved Pairwise Alignment of Genomic DNA. Ph.D. thesis, Pennsylvania State University, University Park, PA.
Hausner, G., Olson, R., Simon, D., Johnosn, I., Sanders, E. R., Karol, K. G., et al. (2006). Origin and evolution of the chloroplast trnK (matK) intron: a model for evolution of group II intron RNA strucutres. Mol. Biol. Evol. 23, 380–391. doi: 10.1093/molbev/msj047
Heap, I. (2018). The International Survey of Herbicide Resistant Weeds. Available at: http://www.weedscience.org/Summary/home.aspx [accessed March 7, 2018].
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Hsu, C. Y., Wu, C. S., and Chaw, S. M. (2016). Birth of four chimeric plastid gene clusters in Japanese umbrella pine. Genome Biol. Evol. 8, 1776–1784. doi: 10.1093/gbe/evw109
International Service for the Acquisition of Agri-biotech Applications [ISAAA] (2016). Global status of commercialized biotech/GM crops. ISAAA Breif No. 52. Ithaca, NY: ISAAA.
Jansen, R. K., and Palmer, J. D. (1987). A chloroplast DNA inversion marks an ancient evolutionary split in the sunflower family (Asteraceae). Proc. Natl. Acad. Sci. U.S.A. 84, 5818–5822. doi: 10.1073/pnas.84.16.5818
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kazinczi, G., Beres, I., Pathy, Z., and Novak, R. (2008). Common ragweed (Ambrosia artemisiifolia L.): a review with special regards to the results in Hungary: II. Importance and harmful effect, allergy, habitat, allelopathy and beneficial characteristics. Herbologia 9, 93–118.
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kettunen, M., Genovesi, P., Gollasch, S., Pagad, S., Starfinger, U., ten Brink, P., et al. (2009). Technical support to EU strategy on invasive alien species (IAS) - Assessment of the impacts of IAS in Europe and the EU. Brussels: Institute for European Environmental Policy.
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Kim, K.-L., Choi, K.-S., and Jansen, R. K. (2005). Two chloroplast DNA inversions originated simultaneously during the early evolution of the sunflower family (Asteraceae). Mol. Biol. Evol. 22, 1783–1792. doi: 10.1093/molbev/msi174
Kluge, A. G., and Farris, J. S. (1969). Quantitative phyletics and the evolution of Anurans. Syst. Zool. 18, 1–32. doi: 10.2307/2412407
Kong, C.-H., Wang, P., and Xu, X.-X. (2007). Allelopathic interference of Ambrosia trifida with wheat (Triticum aestivum). Agr. Ecosyst. Environ. 119, 416–420. doi: 10.1016/j.agee.2006.07.014
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kuang, D. Y., Wu, H., Wang, Y. L., Gao, L. M., Zhang, S. Z., and Lu, L. (2011). Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome 54, 663–673. doi: 10.1139/G11-026
Kumar, S., Hahn, F. H., McMahan, C. M., Cornish, K., and Whalen, M. C. (2009). Comparative analysis of the complete sequence of the plastid genome of Parthenium argentatum and identification of DNA barcodes to differentiate Parthenium species and lines. BMC Plant Biol. 9:131. doi: 10.1186/1471-2229-9-131
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., Giegerich, R., et al. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Leister, D., Wang, L., and Kleine, T. (2017). Organellar gene expression and acclimation of plants to environmental stress. Front. Plant. Sci. 8:387. doi: 10.3389/fpls.2017.00387
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liu, Y., Huo, N., Dong, L., Wang, Y., Zhang, S., Young, H. A., et al. (2013). Complete chloroplast genome sequences of Mongolia medicine Artemisia frigida and phylogenetic relationships with other plants. PLoS One 8:e57533. doi: 10.1371/journal.pone.0057533
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW) – a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Martin, M. D., Chamecki, M., and Brush, G. S. (2010). Anthesis synchronization and floral morphology determine diurnal patterns of ragweed pollen dispersal. Agric. For. Meteorol. 150, 1307–1317. doi: 10.1016/j.agrformet.2010.06.001
Martin, M. D., Chamecki, M., Brush, G. S., Meneveau, C., and Parlange, M. B. (2009). Pollen clumping and wind dispersal in an invasive angiosperm. Am. J. Bot. 96, 1703–1711. doi: 10.3732/ajb.0800407
Martin, M. D., Olsen, M. T., Samaniego, J. A., Zimmer, E. A., and Gilbert, M. T. P. (2016). The population genomic basis of geographic differentiation in North American common ragweed (Ambrosia artemisiifolia L.). Ecol. Evol. 6, 3760–3771. doi: 10.1002/ece3.2143
Martin, M. D., Quiroz-Claros, E., Brush, G. S., and Zimmer, E. A. (2018). Herbarium collection-based phylogenetics of the ragweeds (Ambrosia. Asteraceae). Mol. Phylogenet. Evol. 120, 335–341. doi: 10.1016/j.ympev.2017.12.023
Martin, M. D., Zimmer, E. A., Olsen, M. T., Foote, A. D., Gilbert, M. T. P., and Brush, G. S. (2014). Herbarium specimens reveal a historical shift in phylogeographic structure of common ragweed during native range disturbance. Mol. Ecol. 23, 1701–1716. doi: 10.1111/mec.12675
Mátyás, K. K., Taller, J., Cseh, A., Poczai, P., and Cernák, I. (2011). Development of a simple PCR-based assay for the identification of triazine resistance in the noxious plant common ragweed (Ambrosia artemisiifolia) and its applicability in higher plants. Biotechnol. Lett. 33, 2509–2515. doi: 10.1007/s10529-011-0714-5
Mátyás, K. K., Vignesh, M., and Taller, J. (2012). Population genetic analysis of common ragweed (Ambrosia artemisiifolia L.) in the Carpathian-basin. Magy. Gyomkut. Tech. 13, 21–36.
McManus, H. A., Fučíková, K., Lewis, P. O., Lewis, L. A., and Karol, K. G. (2018). Organellar phylogenomics inform systematics in the green algal family Hydrodictyaceae (Chlorophyceae) and provide clues to the complex evolutionary history of plastid genomes in the green algal tree of life. Am. J. Bot. 105, 315–329. doi: 10.1002/ajb2.1066
Miao, B., Turner, B., Simpson, B., and Mabry, T. J. (1995). Chloroplast DNA study of the genera Ambrosia s.l. and Hymenoclea (Asteraceae): systematic implications. Plant Syst. Evol. 194, 241–255. doi: 10.1007/BF00982858
Michelangeli, F. A., Davis, J. I, and Stevenson, D. W. (2003). Phylogenetic relationships among Poaceae and related families as inferred from morphology, inversions in the plastid genome, and sequence data from the mitochondrial and plastid genomes. Am. J. Bot. 90, 93–106. doi: 10.3732/ajb.90.1.93
Moretti, M. L., Van Horn, C. R., Robertson, R., Segobye, K., Weller, S. C., Young, B. G., et al. (2018). Glyphosate resistance in Ambrosia trifida: Part 2. Rapid response physiology and non-target-site resistance. Pest Manag. Sci. 74, 1079–1088. doi: 10.1002/ps.4569
Motta, E. V. S., Raymann, K., and Moran, N. A. (2018). Glyphosate perturbs the gut microbiota of honey bees. Proc. Natl. Acad. Sci. U.S.A. 115, 10305–10310. doi: 10.1073/pnas.1803880115
Nagy, E., Hegedûs, G., Taller, J., Kutasy, B., and Virág, E. (2017). Illumina sequencing of the chloroplast genome of common ragweed (Ambrosia artemisiifolia L.). Data Brief 15, 606–611. doi: 10.1016/j.dib.2017.10.009
Neve, P., and Powels, S. (2005). Recurrent selection with reduced herbicide rates results in the rapid evolution of herbicide resistance in Lolium rigidum. Theor. Appl. Genet. 110, 1154–1166. doi: 10.1007/s00122-005-1947-2
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nie, X., Lv, S., Zhang, Y., Du, X., Wang, L., Biradar, S. S., et al. (2012). Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS One 7:e36869. doi: 10.1371/journal.pone.0036869
Nixon, K. C. (2002). WinClada ver. 1.00. 08. Ithaca, NY: Published by the Author. Available at: http://www.diversityoflife.org/winclada/
Norsworthy, J. K., Riar, D., Jha, P., and Scott, R. C. (2011). Confirmation, control, and physiology of glyphosate-resistant giant ragweed (Ambrosia trifida) in Arkansas. Weed Technol. 25, 430–435. doi: 10.1614/WT-D-10-00155.1
Olmstead, R. G., and Palmer, J. D. (1994). Chloroplast DNA systematic: a review of methods and data analysis. Am. J. Bot. 81, 1205–1224. doi: 10.1002/j.1537-2197.1994.tb15615.x
Palmer, J. D. (1983). Chloroplast DNA exists in two orientations. Nature 301, 92–93. doi: 10.1038/301092a0
Palmer, J. D. (1985). Comparative organization of chloroplast genomes. Ann. Rev. Genet. 19, 325–354. doi: 10.1146/annurev.ge.19.120185.001545
Panéro, J. L., Freire, S. E., Ariza Espinar, L., Crozier, B. S., Barboza, G. E., and Cantero, J. J. (2014). Resolution of deep nodes yields an improved backbone phylogeny and a new basal lineage to study early evolution of Asteraceae. Mol. Phylogenet. Evol. 80, 43–53. doi: 10.1016/j.ympev.2014.07.012
Panéro, J. L., and Funk, V. A. (2002). Toward a phylogenetic subfamilial classification for the Compositae (Asteraceae). Proc. Biol. Soc. Wash. 115, 909–922.
Panéro, J. L., and Funk, V. A. (2008). The value of sampling anomalous taxa in phylogenetic studies: major clades of the Asteraceae revealed. Mol. Phylogenet. Evol. 47, 757–782. doi: 10.1016/j.ympev.2008.02.011
Patterson, E. L., Pettinga, D. J., Ravet, K., Neve, P., and Gaines, T. A. (2018). Glyphosate resistance and EPSPS gene duplication: convergent evolution in multiple plant species. J. Hered. 109, 117–125. doi: 10.1093/jhered/esx087
Payne, W. W. (1963). The morphology of the inflorescence of ragweeds (Ambrosia-Franseria). Am. J. Bot. 50, 872–880. doi: 10.1002/j.1537-2197.1963.tb06566.x
Payne, W. W. (1964). A re-evaluation of the genus Ambrosia (Compositae). J. Arnold Arboretum 45, 401–430.
Peng, Y., Abercrombie, L. L. G., Yuan, J. S., Riggins, C. W., Sammons, R. D., Tranel, P. J., et al. (2010). Characterization of the horseweed (Conyza canadensis) transcriptome using GS-FLX 454 pyrosequencing and its application for expression analysis of candidate non-target herbicide resistance genes. Pest. Manag. Sci. 66, 1053–1062. doi: 10.1002/ps.2004
Peterson, K., and Payne, W. W. (1973). The genus Hymenoclea (Compositae: Ambrosieae). Brittonia 25, 243–256. doi: 10.2307/2805586
Petit, C., Bay, G., Pernin, F., and Délye, C. (2010). Prevalence of cross- or multiple resistance to the acetyl-coenzyme A carboxylase inhibitors fenoxaprop, clodinafop and pinoxaden in black-grass (Alopecurus myosuroides Huds.) in France. Pest. Manag. Sci. 66, 168–177. doi: 10.1002/ps.1851
Poczai, P., and Hyvönen, J. (2013). Discovery of novel plastid phenylalanine (trnF) pseudogenes defines a distinctive clade in Solanaceae. Springerplus 2:459. doi: 10.1186/2193-1801-2-459
Poczai, P., and Hyvönen, J. (2017). The complete chloroplast genome sequence of the CAM epiphyte Spanish moss (Tillandsia usneoides, Bromeliaceae) and its comparative analysis. PLoS One 12:e0187199. doi: 10.1371/journal.pone.0187199
Pyšek, P., Jarošík, V., Pergl, J., Randall, R., Chytrý, M., Kühn, I., et al. (2009). The global invasion success of Central European plants is related to distribution characterisitcs in their native range and species traits. Divers. Distrib. 15, 891–903. doi: 10.1111/j.1472-4642.2009.00602.x
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Ranwez, V., Harispe, S., Delsuc, F., and Douzery, E. J. P. (2011). MACSE: multiple alignment of coding SEquences Accounting for frameshifts and stop codons. PLoS One 6:e22594. doi: 10.1371/journal.pone.0022594
Rasmussen, K., Thyrring, J., Muscarella, R., and Borchsenius, F. (2017). Climate-change-induced range shifts of three allergenic ragweeds (Ambrosia L.) in Europe and their potential impact on human health. PeerJ 5:e3104. doi: 10.7717/peerj.3104
Ravet, K., Patterson, E. L., Krähmer, H., Hamouzová, K., Fan, L., Jasieniuk, M., et al. (2018). The power and potential of genomics in weed biology and management. Pest. Manag. Sci. 74, 2216-2225. doi: 10.1002/ps.5048
Regnier, E. E., Harrison, S. K., Loux, M. M., Holloman, C., Venkatesh, R., Diekmann, F., et al. (2016). Certified crop advisors’ perceptions of giant ragweed (Ambrosia trifida) distribution, herbicide resistance, and management in the Corn Belt. Weed Sci. 64, 361–377. doi: 10.1614/WS-D-15-00116.1
Riggins, C. W., Peng, Y., and Stewart, C. N. Jr. (2010). Characterization of de novo transcriptome for waterhemp (Amaranthus tuberculatus) using GS-FLX 454 pyrosequencing and its application for studies of herbicide target-site genes. Pest Manag. Sci. 66, 1042–1052. doi: 10.1002/ps.2006
Rydin, C., Wikström, N., and Bremer, B. (2017). Conflicting results from mitochondrial genomic data challenge current views of Rubiaceae phylogeny. Am. J. Bot. 104, 1522–1532. doi: 10.3732/ajb.1700255
Salih, M. R. H., Majeský, L’, Schwarzacher, T., Gornall, R., and Heslop-Harrison, P. (2017). Complete chloroplast genomes from apomictic Taraxacum (Asteraceae): identity and variation between three microspecies. PLoS One 12:e0168008. doi: 10.1371/journal.pone.0168008
Sammons, R. D., and Gaines, T. A. (2014). Glyphosate resistance: state of knowledge. Pest Manag. Sci. 70, 1367–1377. doi: 10.1002/ps.3743
Sammons, R. D., You, J., Qi, Y., Flasinski, S., Kavanaugh, C., Washam, J., et al. (2018). Evaluation of glyphosate resistance in Arabidopsis thaliana expressing an altered target site EPSPS. Pest Manag. Sci. 74, 1174–1183. doi: 10.1002/ps.4654
Sánchez Barreiro, F., Vieira, F. G., Martin, M. D., Haile, J., Gilbert, M. T. P., and Wales, N. (2017). Characterizing restriction enzyme-associated loci in historic ragweed (Ambrosia artemisiifolia) voucher specimens using custom-designed RNA probes. Mol. Ecol. Res. 17, 209–220. doi: 10.1111/1755-0998.12610
Schwarz, E. N., Ruhlman, T. A., Sabir, J. S. M., Hajrah, N. H., Alharbi, N. S., Al-Malki, A. L., et al. (2015). Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. J. Syst. Evol. 53, 458–468. doi: 10.1111/jse.12179
Shepherd, L. D. (2017). A non-destructive DNA sampling technique for herbarium specimens. PLoS One 12:e0183555. doi: 10.1371/journal.pone.0183555
Sinn, B. T., Sedmak, D. D., Kelly, L. M., and Freudenstein, J. V. (2018). Total duplication of the small single copy region in the angiosperm plastome: rearrangement and inverted repeat instability in Asarum. Am. J. Bot. 105, 71–84. doi: 10.1002/ajb2.1001
Sloan, D. B., Triant, D. A., Forrester, N. J., Bergner, L. M., Wu, M., and Taylor, D. R. (2014). A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol. Phylogenet. Evol. 72, 82–89. doi: 10.1016/j.ympev.2013.12.004
Staats, M., Cuenca, A., Richardson, J. E., Vrielink-van Ginkel, R., Petersen, G., Seberg, O., et al. (2011). DNA damage in plant herbarium tissue. PLoS One 6:e28448. doi: 10.1371/journal.pone.0028448
Staats, M., Erkens, R. H. J., van de Vossenberg, B., Wieringa, J. J., Kraaijeveld, K., Stielow, B., et al. (2013). Genomic treasure troves: complete genome sequencing of herbarium and insect museum specimens. PLoS One 8:e6918. doi: 10.1371/journal.pone.0069189
Suorsa, M., Sirpiö, S., and Aro, E.-M. (2009). Towards characterization of the chloroplast NAD(P)H dehydrogenase complex. Mol. Plant 2, 1127–1140. doi: 10.1093/mp/ssp052
Taller, J., Decsi, K., Farkas, E., Nagy, E., Mátyás, K. K., Kolics, B., et al. (2016a). De novo transcriptome sequencing based identification of Amb a 3-like pollen allergen in common ragweed (Ambrosia artemisiifolia). J. Bot. Sci. 5, 12–16.
Taller, J., Nagy, E., Decsi, K., Kutasy, B., Mátyás, K., Farkas, E., et al. (2016b). Utilization of transcriptomics in weed research. A case study on common ragweed (Ambrosia artemisiifolia L.) the most widespread weed in Hungary. Magy. Gyomkut. Technol. 17, 5–15.
Taramarcaz, P., Lambelet, C., Clot, B., Kleimer, C., and Hauser, C. (2005). Ragweed (Ambrosia) progression and its health risks: will Switzerland resist this invasion? Swiss Med. Wkly. 135, 538–548.
Thiel, T., Michalek, W., Varshney, R. K., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucl. Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Twyford, A. D., and Ness, R. W. (2017). Strategies for complete plastid genome sequencing. Mol. Ecol. Res. 17, 858–868. doi: 10.1111/1755-0998.12626
Vaidya, G., Lohman, D. J., and Meier, R. (2011). SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. doi: 10.1111/j.1096-0031.2010.00329.x
Van de Paer, C., Bouchez, O., and Besnard, G. (2018). Prospects on the evolutionary mitogenomics of plants: a case study on the olive family (Oleaceae). Mol. Ecol. Res. 18, 407–423. doi: 10.1111/1755-0998.12742
Van de Paer, C., Hong-Wa, C., Jeziorski, C., and Besnard, G. (2016). Mitogenomics of Hesperelaea, an extinct genus of Oleaceae. Gene 594, 197–202. doi: 10.1016/j.gene.2016.09.007
Van Horn, C. P., Moretti, M. L., Robertson, R. R., Segobye, K., Weller, S. C., Young, B. G., et al. (2018). Glyphosate resistance in Ambrosia trifida: part 1. Novel rapid cell death response to glyphosate. Pest Manag. Sci. 74, 1071–1078. doi: 10.1002/ps.4567
Vandenberg, L. N., Blumberg, B., Antoniou, M. N., Benbrook, C. M., Carroll, L., Colborn, T., et al. (2017). Is it time to reassess current safety standards for glyphosate-based herbicides? J. Epidemiol. Comm. Health 71, 613–618. doi: 10.1136/jech-2016-208463
Vargas, O. M., Ortiz, E. M., and Simpson, B. B. (2017). Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high-Andean diversification (Asteraceae: Astereae: Diplostephium). New Phytol. 214, 1736–1750. doi: 10.1111/nph.14530
Vilá, M., Basnou, C., Pyšek, P., Josefsson, M., Genovesi, P., Gollasch, S., et al. (2010). How well do we understand the impacts of alien species on ecosystem services? A pan-European, cross-taxa assessment. Front. Ecol. Environ. 8, 135–144. doi: 10.1890/080083
Virág, E., Hegedûs, G., Barta, E., Nagy, E., Mátyás, K., Kolics, B., et al. (2016). Illumina sequencing of common (short) ragweed (Ambrosia artemisiifolia L.) reproductive organs and leaves. Front. Plant. Sci. 7:1506. doi: 10.3389/fpls.2016.01506
Vitousek, P. M., D’Antonio, C. M., Loope, L. L., Rejmanek, M., and Westbrooks, R. (1997). Introduced species: a significant component of human-caused global change. New. Zeal. J. Ecol. 21, 1–16. doi: 10.1007/s00114-014-1153-7
von Boheemen, L. A., Lombaert, E., Nurkowski, K. A., Gauffre, B., Rieseberg, L. H., and Hodgins, K. A. (2017). Multiple introductions, admixture and bridgehead invasion characterize the introduction history of Ambrosia artemisiifolia in Europe and Australia. Mol. Ecol. 26, 5421–5434. doi: 10.1111/mec.14293
Walker, J. F., Jansen, R. K., Zanis, M. J., and Emery, N. C. (2015). Sources of inversion variation in the small single copy (SSC) region of chloroplast genomes. Am. J. Bot. 102, 1751–1752. doi: 10.3732/ajb.1500299
Walker, J. F., Zanis, M. J., and Emery, N. C. (2014). Comparative analysis of complete chloroplast genome sequence and inversion variation in Lasthenia burkei (Madieae, Asteraceae). Am. J. Bot. 101, 722–729. doi: 10.3732/ajb.1400049
Wicke, S., and Schneeweiss, G. M. (2015). “Next-generation organellar genomics: potentials and pitfalls of high-throughput technologies for molecular evolutionary studies and plant systematics,” in Nextgeneration Sequencing in Plant Systematics, eds E. Hörandl and M. S. Appelhans (Bratislava: International Association for Plant Taxonomy (IAPT)), 9–50.
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant. Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wolfsberg, T. G., Schafer, S. S., Tatusov, R. L., and Tatusov, T. A. (2001). Organelle genome resouces at NCBI. Tends Biochem. Sci. 26, 199–203. doi: 10.1016/S0968-0004(00)01773-4
Wysocki, W. P., Clark, L. G., Attigala, L., Ruiz-Sanchez, E., and Duvall, M. R. (2015). Evolution of the bamboos (Bambusoideae; Poaceae): a full plastome phylogenomic analysis. BMC Evol. Biol. 15:50. doi: 10.1186/s12862-015-0321-5
Xie, Q., Shen, K.-N., Hao, X., Nam, P. N., Hieu, B. T. N., Chen, C.-H., et al. (2015). The complete chloroplast genome of Tianshan snow lotus (Saussurea involucrata), a famous traditional Chinese medicinal plant of the family Asteraceae. Mitochondrial DNA B Resour. 28, 294–295. doi: 10.3109/19401736.2015.1118086
Yin, T., Cook, D., and Lawrence, M. (2012). ggbio: an R package for extending the grammar of graphics for genomic data. Genome Biol. 13:R77. doi: 10.1186/gb-2012-13-8-r77
Yu, Q., Jalaludin, A., Han, H., Chen, M., Sammons, R. D., and Powels, S. B. (2015). Evolution of a double amino acid substitution in the 5-enolpyruvylshikimate-3-phosphate synthases in Eleusine indica conferring high-level glyphosate resistance. Plant Physiol. 167, 1440–1447. doi: 10.1104/pp.15.00146
Yuan, J. S., Tranel, P. J., and Stewart, C. N. Jr. (2007). Non-target-site herbicide resistance: a family business. Trends Plant Sci. 12, 6–13. doi: 10.1016/j.tplants.2006.11.001
Zedane, L., Hong-Wa, C., Murienne, J., Jeziorski, C., Baldwin, B. G., and Besnard, G. (2016). Museuomics illuminate the history of an extinct, paleoendemic plant lineage (Hesperelaea, Oleaceae) known from an 1875 collection from Guadalupe Island, Mexico. Biol. J. Linn. Soc. 117, 44–57. doi: 10.1111/bij.12509
Zeng, C.-X., Hollingsworth, P. M., Yang, J., He, Z.-S., Zhang, Z.-R., Li, D.-Z., et al. (2018). Genome skimming herbarium specimens for DNA barcoding and phylogenomics. Plant Methods 14:43. doi: 10.1186/s13007-018-0300-0
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using Bruijin graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Keywords: Ambrosia trifida, chloroplast genome, genome skimming, glyphosate, herbicide resistance, invasive plants, noxious weeds, ragweed
Citation: Sablok G, Amiryousefi A, He X, Hyvönen J and Poczai P (2019) Sequencing the Plastid Genome of Giant Ragweed (Ambrosia trifida, Asteraceae) From a Herbarium Specimen. Front. Plant Sci. 10:218. doi: 10.3389/fpls.2019.00218
Received: 08 June 2018; Accepted: 08 February 2019;
Published: 28 February 2019.
Edited by:
Freek T. Bakker, Wageningen University & Research, NetherlandsReviewed by:
Denis Baurain, University of Liège, BelgiumJinming Chen, Wuhan Botanical Garden, Chinese Academy of Sciences, China
Copyright © 2019 Sablok, Amiryousefi, He, Hyvönen and Poczai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Péter Poczai, cGV0ZXIucG9jemFpQGhlbHNpbmtpLmZp