 Cristiane Santos1,2
Cristiane Santos1,2 Fábio C. S. Nogueira3
Fábio C. S. Nogueira3 Gilberto B. Domont3
Gilberto B. Domont3 Wagner Fontes4
Wagner Fontes4 Guilherme S. Prado1
Guilherme S. Prado1 Peyman Habibi1,5
Peyman Habibi1,5 Vanessa O. Santos1
Vanessa O. Santos1 Osmundo B. Oliveira-Neto1,6
Osmundo B. Oliveira-Neto1,6 Maria Fatima Grossi-de-Sá1,7
Maria Fatima Grossi-de-Sá1,7 Jesus V. Jorrín-Novo8
Jesus V. Jorrín-Novo8 Octavio L. Franco2,7,9*
Octavio L. Franco2,7,9* Angela Mehta1*
Angela Mehta1*- 1Embrapa Recursos Genéticos e Biotecnologia, Brasília, Brazil
- 2Departamento de Biologia, Universidade Federal de Juiz de Fora, Juiz de Fora, Brazil
- 3Proteomics Unit, Chemistry Institute, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 4Departamento de Biologia Celular, Universidade de Brasília, Brasília, Brazil
- 5Department of Bioprocess Engineering and Biotechnology, Universidade Federal do Paraná, Curitiba, Brazil
- 6Departamento de Bioquímica e Biologia Molecular, Escola de Medicina, Faculdades Integradas da União Educacional do Planalto Central, Brasília, Brazil
- 7Centro de Analises Proteomicas e Bioquimica, Pós-Graduação em Ciências Genômicas e Biotecnologia, Universidade Católica de Brasília, Brasília, Brazil
- 8Department of Biochemistry and Molecular Biology, Universidad de Córdoba, Córdoba, Spain
- 9S-Inova Biotech, Pós-Graduação em Biotecnologia, Universidade Católica Dom Bosco, Campo Grande, Brazil
Black rot is a severe disease caused by the bacterium Xanthomonas campestris pv. campestris (Xcc), which can lead to substantial losses in cruciferous vegetable production worldwide. Although the use of resistant cultivars is the main strategy to control this disease, there are limited sources of resistance. In this study, we used the LC-MS/MS technique to analyze young cabbage leaves and chloroplast-enriched samples at 24 h after infection by Xcc, using both susceptible (Veloce) and resistant (Astrus) cultivars. A comparison between susceptible Xcc-inoculated plants and the control condition, as well as between resistant Xcc-inoculated plants with the control was performed and more than 300 differentially abundant proteins were identified in each comparison. The chloroplast enriched samples contributed with the identification of 600 additional protein species in the resistant interaction and 900 in the susceptible one, which were not detected in total leaf sample. We further determined the expression levels for 30 genes encoding the identified differential proteins by qRT-PCR. CHI-B4 like gene, encoding an endochitinase showing a high increased abundance in resistant Xcc-inoculated leaves, was selected for functional validation by overexpression in Arabidopsis thaliana. Compared to the wild type (Col-0), transgenic plants were highly resistant to Xcc indicating that CHI-B4 like gene could be an interesting candidate to be used in genetic breeding programs aiming at black rot resistance.
Introduction
Black rot, caused by the bacterium X. campestris pv. campestris (Xcc), is one of the most severe diseases that affects cruciferous crops. The use of resistant cultivars is the most efficient strategy to control black rot and therefore, resistance genes have been studied in Brassica genomes including the genome A (B. rapa), genome BC (B. carinata, originated from B. nigra x B. oleracea), and genome AC (B. napus, originated from B. rapa × B. oleracea). It is known that the characterized resistance genes can confer durable resistance to black rot in genomes A and B (Guo et al., 1991; Ignatov et al., 2000). However, there is limited information on resistance genes from genome C (B. oleracea), and there are no reports on resistance sources against black rot disease for this genome group (Camargo et al., 1995; Saha et al., 2016; Sharma et al., 2016).
Functional genomic and proteomic techniques have been important tools for exploring and understanding plant–pathogen interaction mechanisms. Proteomic studies can provide the link between gene expression and protein abundance and help identify key proteins involved in plant defense and resistance (Kamal et al., 2010a,b; Komatsu and Hossain, 2017). Although mass spectrometer sensitivity and software development have improved protein identification in the last years, there are still some limitations in the detection of low abundant proteins. One alternative to overcome this problem is the analysis of subcellular proteomes. This strategy can reduce sample complexity and provide the identification of a high amount of additional proteins contributing to a better understanding of the metabolic pathways involved (Stekhoven et al., 2014; Wang and Komatsu, 2016). Indeed, analyses of subcellular proteomes have been widely performed and presented a better picture of differential protein abundance under different stress conditions (Peltier et al., 2000; Uberegui et al., 2015). Chloroplasts have an important role in stress response and therefore the study of the chloroplast proteome can bring important contributions for the elucidation of plant defense, especially since this organelle participates actively in plant immune response (Audran et al., 2016).
In a previous study, Ribeiro et al. (2018) analyzed B. oleracea leaves inoculated with Xcc by 2-DE. Although differential protein spots were detected, the 2-DE technique is highly limited, particularly in the detection of low abundant proteins (Corthals et al., 2000; Wang and Hanash, 2003). Therefore, in this study, we performed bottom-up proteomics of inoculated leaves at the same time point (24 h after infiltration), in order to understand protein abundance at an early stage of infection. The total leaf proteome was further complemented with the analysis of chloroplast-enriched samples and the expression levels for 30 genes encoding the identified differential proteins were determined by qRT-PCR. Additionally, one protein was selected for overexpression in Arabidopsis thaliana to verify its involvement in resistance to Xcc.
Materials and Methods
Plant Material and Chloroplast Isolation
In this work, two B. oleracea var. capitata cultivars, one moderately resistant (Astrus Plus – Chile/Seminis®) and one susceptible (Veloce – Brazil/Agristar®), as determined previously by our group (data not published), were used. A schematic figure of the experimental design is presented in the Supplementary Figure S1. The isolate of X. campestris pv. campestris (Xcc) Xcc51, obtained from Embrapa Hortaliças, Brasília, DF, Brazil, was used. Young plants (45 days after sowing) of both cultivars were inoculated with bacterial or saline (0.85% NaCl) solution, according to Santos et al. (2017). Leaves were harvested 24 hours after infiltration (hai), ground in liquid nitrogen and stored at -80°C. Three biological replicates, composed of five plants each, were analyzed. The same samples were used for chloroplast isolation using 5 g of ground material according to Kley et al. (2010), with modification in the percoll gradient. A 40%:80% percoll (Sigma-Aldrich) gradient was used.
Protein Extraction and LC-MS/MS Analysis
Leaves (0.3 g) and isolated chloroplasts (500 μL) were used for protein extraction according to Mot and Vanderleyden (1989), with modifications, as follows: for chloroplast protein extraction, we used a 1:2 extraction buffer (0.7 M sucrose, 0.5 M Tris-HCl, 50 mM EDTA, 0.1 M KCl, and 40 mM DTT):phenol proportion. Precipitated proteins were solubilized in urea/thiourea buffer (7 M urea; 2 M thiourea; 4% CHAPS, and 5 mM DTT) and quantified using the Bradford Reagent (Bio-Rad, Unite States). Approximately 150 μg of proteins from three biological replicates were loaded onto SDS-PAGE and allowed to migrate approximately 1 cm (Supplementary Figure S2) in a 12% resolving gel, as described by Valledor and Weckwerth (2014). Each gel lane containing one biological replicate was cut and submitted to in gel digestion using 5 μg of trypsin (Promega, Madison, WI, Unite States), according to Valledor and Weckwerth (2014). After the digestion procedure, the proteins were quantified using QuibtTM fluorometer (Invitrogen), following the manufacturer’s instructions. Three biological and three technical replicates were analyzed by LC-MS/MS, totalizing nine technical replicates. The peptide samples were desalted according to the protocol described by Rappsilber et al. (2007) and suspended in 50 μL of 4% (v/v) acetonitrile (ACN) and 0.25% (v/v) formic acid.
A total of 2 μg of digested peptides were loaded into a one-dimensional (1D) nano-flow LC-MS/MS system (Thermo Scientific). Peptides were eluted using a monolithic C18 column Acclaim PepMap (Thermo Scientific) of 150 mm in length and 0.075 mm internal diameter. The gradient employed 0.1% formic acid in mobile phase A and 0.1% formic acid and 90% acetonitrile in mobile phase B during 180 min with a controlled flow rate of 400 nL/min from 5 to 35% phase B. The effluent from the nLC column was directly electrosprayed into an Orbitrap Mass Spectrometer (LTQ-Orbitrap XLTM Hybrid Ion Trap, Thermo Scientific), operated in the positive ion mode and set to data-dependent acquisition.
Precursor peptides were detected in the mass range of 400–1,500 m/z and at a resolution of 120 K (at 200 m/z) with a target ion counting of 5 × 105. Tandem MS was performed by the isolation window of 1 atomic mass unit (amu), with CID (collision-induced dissociation) fragmentation in the quadrupole with a normalized collision energy of 35. The automatic gain control (AGC) was defined at 4 × 105 and the max injection time was of 50 ms. Only the 10 most intense precursors in the charge states of 2–6 were subjected to MS2. The dynamic exclusion duration was defined as 15 s with mass error tolerance around 10 ppm. The instrument was operated in max speed mode with cycles of 3 s.
Protein Identification and Quantification
The raw data were processed using the software Progenesis QI (Nonlinear Dynamics, Waters, Durham, NC, United States) and PEAKS® 7 (Bioinformatics Solutions Inc., Waterloo, ON, Canada). A total of four pairwise comparisons (Xcc-inoculated vs. saline solution-inoculated) were performed (Supplementary Figure S3): (1) resistant Xcc-inoculated leaves compared to saline solution-inoculated leaves (LRI:LRC), (2) resistant Xcc-inoculated chloroplast compared to saline solution-inoculated chloroplast (ChRI:ChRC), (3) susceptible Xcc-inoculated leaves compared to saline solution-inoculated leaves (LSI:LSC), (4) susceptible Xcc-inoculated chloroplast compared to saline solution-inoculated chloroplast (ChSI:ChSC). The chromatograms from each comparison were automatically aligned and the alignment was manually revised for inconsistencies. Profile data from the MS scans were used to calculate the relative peptide abundance using the areas under the peaks of extracted ion chromatograms. Quantified features were median normalized and evaluated for statistical significance using ANOVA p ≤ 0.05.
MS/MS files were exported as Mascot generic file (mgf) for peptide identification using PEAKS®7 (Bioinformatics Solutions Inc., Waterloo, ON, Canada) software (Zhang et al., 2012), and searched against the UniProt_Brassica oleracea database (taxonomy ID 3712) on February 20171. The analysis using PEAKS®7 was performed with the following parameters: peptide m/z tolerance – 10 ppm; fragment ion m/z tolerance – 0.5 Da; digestion using trypsin with two missed cleavages allowed; Cys carbamidomethylation as fixed modification and Met oxidation as variable modification. The search results were filtered by FDR < 1%. The SPIDER tool within the software PEAKS®7 was used to find homologous peptides presenting a single amino acid substitution in the database (Han et al., 2005). The data generated was deposited in the MassIVE repository (DOI: 10.25345/C5KG6W).
qRT-PCR Analysis
Total RNA was extracted from the same leaf samples (0.1 g) used for protein analysis by the TRIzolTM Reagent method (InvitrogenTM), following the manufacturer’s instructions. RNA samples were quantified using a NanoDropTM 200 spectrophotometer (Thermo Scientific) and the integrity of the RNA was observed in 1% agarose gel. RNA was treated with TurboTM DNAse (Applied Biosystems/Ambion) and cDNA synthesis was performed using 2 μg of total RNA and the Go ScriptTM Reverse Transcription System (Promega), following the manufacturer’s instructions. A total of 30 genes encoding the differentially abundant proteins identified were selected for qRT-PCR (Supplementary Table S1). SAND (SAND family protein), TBP1 (TATA-box-binding protein 1), TUB6 (Tubulin beta-6), and UBQ1 (Ubiquitin-60S ribosomal protein L40) were used as reference genes. All primers used were designed using Primer3Plus program (Untergasser et al., 2007). qRT-PCR was performed using three biological and three technical replicates, as described by Santos et al. (2017). The analysis was performed in a thermal cycler 7300 Real-Time PCR System (Applied Biosystems). To verify the absence of genomic DNA in the samples, qRT-PCR was performed using RNA as template. For stability evaluation of the reference genes, the geNorm algorithm (Etschmann et al., 2006) was used and the expression analysis was performed with the REST software (Pfaffl et al., 2002).
Overexpression of BoCHI-B4 Like Gene in Arabidopsis thaliana
The gene BoCHI-B4 like (GAQY01039586) encoding the basic endochitinase CHB4-like protein (A0A0D3BPL2) was selected for functional validation. The binary vector pBin61 that carries a transcription cassette with the CaMV 35S promoter and terminator, and the kanamycin resistance gene as selection marker was used (Bendahmane et al., 2002) The BoCHIB4 like gene (BoCHIB4) was synthesized and cloned into the pBIN61 vector by Epoch Life Science Inc. (Missouri City, TX, United States) to generate the construct pBIN61: BoCHIB4, which was used to transform A. thaliana (Col-0), mediated by Agrobacterium tumefaciens (strain GV3130) using the floral dip method (Bent and Clough, 1998). Approximately 0.5 g of seeds of the transformed plants were sterilized and distributed in MS culture medium supplemented with kanamycin (50 mg L-1) resulting in the selection of 15 pBIN61:BoCHIB4 primary transformants. The parental lines and T2-generation were germinated in MS medium containing kanamycin and transferred to substrate and maintained in a growth chamber under a 12 h light:12 h dark photoperiod at 22°C. To confirm transformation, leaves were harvested for DNA extraction, followed by PCR amplification and sequencing using specific primers. DNA of non-transformed plants (wild type Col-0) was used as control.
Molecular and Phenotypical Characterization of Arabidopsis Transgenic Lines
Leaves of T2 homozygous events were harvested for DNA and RNA extraction for Southern blot and qRT-PCR analysis. For Southern blot, DNA extracted from T2-generation plants (10 μg) was digested with XbaI and analyzed using standard procedure (Romano and Vianna, 2015). The RNA preparation and qRT-PCR analysis were performed as described above. The A. thaliana reference genes ACT2 (Actin 2) and EF-1α (elongation factor-1α) were used (Supplementary Table S1). The effect of gene overexpression was confirmed by spraying bacterial solution (Xcc51 OD600 = 0.1) followed by disease development scoring from 1 to 5 days post inoculation (dpi) using a disease index ranging from 0 (no symptom, considered highly resistant) to 4 (full leaf necrosis, classified as highly susceptible), based on (Meyer et al., 2005). A total of five events was evaluated and 15 plants from each event were analyzed, as well as 15 wild type Col-0 plants, used as the control.
Results and Discussion
Proteomic Profile and Gene Expression Analysis of Brassica Leaves and Chloroplast Enriched Samples
In this study, two conditions were compared for the identification of differentially abundant proteins: Xcc-inoculated and saline solution-inoculated (control condition) leaf and chloroplast from resistant and susceptible cultivars (Astrus and Veloce), resulting in four comparisons (Supplementary Figure S3). As observed in our previous studies (data not published), Astrus was moderately resistant to Xcc51 and Veloce was highly susceptible (Figure 1). The LC-MS/MS data analysis resulted in more than 30,000 peptide sequences, corresponding to more than 1,000 protein species (Supplementary Table S2). Since several of the matches corresponded to uncharacterized proteins in the Uniprot database, a second analysis was performed using Blas2GO software to identify the proteins and infer the gene ontology (GO) for biological process. The leaf proteome of the resistant cultivar inoculated with Xcc (LRI) and in the control condition (LRC) revealed a total of 1,424 proteins, while the leaf samples of the susceptible cultivar Veloce inoculated with Xcc (LSI) and in the control condition (LSC) revealed a total of 1,395 proteins (Supplementary Figure S3).

Figure 1. Symptoms of Brassica oleracea cultivars Astrus (moderately resistant) and Veloce (highly susceptible), at different hours after inoculation (hai) with Xanthomonas campestris pv. campestris.
Chloroplast enriched samples from the resistant cultivar inoculated with Xcc (ChlRI) and in the control condition (ChlRC) as well as from the susceptible cultivar Veloce inoculated with Xcc (ChlSI) and in the control condition (ChlSC) were also analyzed (Supplementary Figure S3). The proteins identified in the leaf and chloroplast enriched samples from each cultivar were merged into a single table (Supplementary Table S2) for discussion, totalizing 2,086 proteins in the resistant interaction, referred to as RI:RC and 2037 in the susceptible interaction, referred to as SI:SC (Supplementary Figure S3). Proteins with the same name were aligned for sequence comparison (protein sequence alignment) using ClustalOmega2 and when differences in the sequences were observed they were considered as different protein species. The results showed that the proteome from chloroplast samples contributed with 662 additional proteins in the resistant plants (ChlRI and ChlRC) and 642 in the susceptible (ChlSI and ChlSC) (Supplementary Figure S3), which were not detected in leaf samples. Moreover, a total of 338 differentially abundant proteins were identified in the resistant interaction (RI:RC), 200 of which were obtained from chloroplast enriched samples (Xcc-inoculated compared to the control). The susceptible interaction (SI:SC) revealed 361 differential proteins out of which 175 were identified in chloroplasts (Supplementary Table S3). These results emphasize that the analysis of subproteomes can contribute significantly for the identification of additional proteins (Rolland et al., 2012; Bayer et al., 2015), especially those present in lower abundance (Kim and Kang, 2008).
In this work, we also analyzed the gene expression levels by qRT-PCR of 30 selected genes encoding the differential proteins identified (Figure 2 and Table 1), based on biological process (defense-related), fold-change (≥1.5 increased or decreased in both cultivars) and previous studies (Villeth et al., 2016; Ribeiro et al., 2018). As expected, the expression levels of many mRNAs did not correlate with protein abundance and different clusters could be observed in the heatmap generated to compare these levels (Figure 2). In the resistant cultivar, among the 14 proteins showing increased abundance (statistically validated), 5 corresponding genes showed upregulation (BoAMC4; BoANNA2; BoCHB4; BoRGP1; BoFSD1), and among the 4 proteins showing decreased abundance, 1 corresponding gene showed downregulation (BoPER32) and the others were not statistically significant. Similar results were obtained for the susceptible cultivar: 10 proteins showed increased abundance, out of which 4 corresponding genes were upregulated, while 8 proteins showed decreased abundance and 2 corresponding genes showed downregulation (BoENH1 and BoPRX2F).
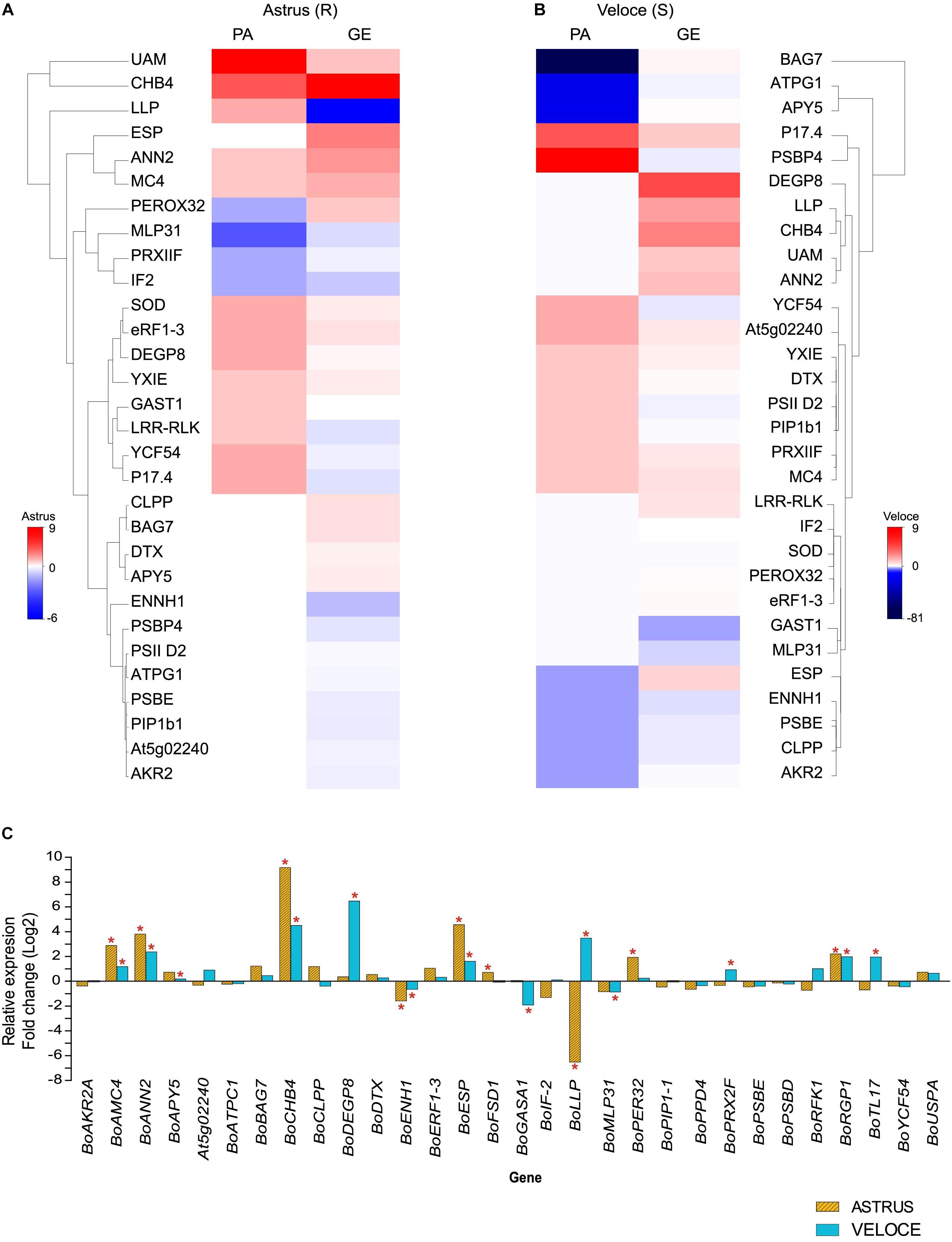
Figure 2. Heatmap showing the correlation between protein abundance (PA) and gene expression (GE) levels in the resistant (A) and in the susceptible interaction (B). (C) Gene expression of 30 genes in leaves of B. oleracea 24 h after inoculation with X. campestris pv. campestris (Xcc) compared to the control condition. The symbol ∗ indicates statistically significant differential expression (p ≤ 0.05). The full information of genes and gene products are presented in Table 1. Bo, Brassica oleracea gene name homologous to A. thaliana.
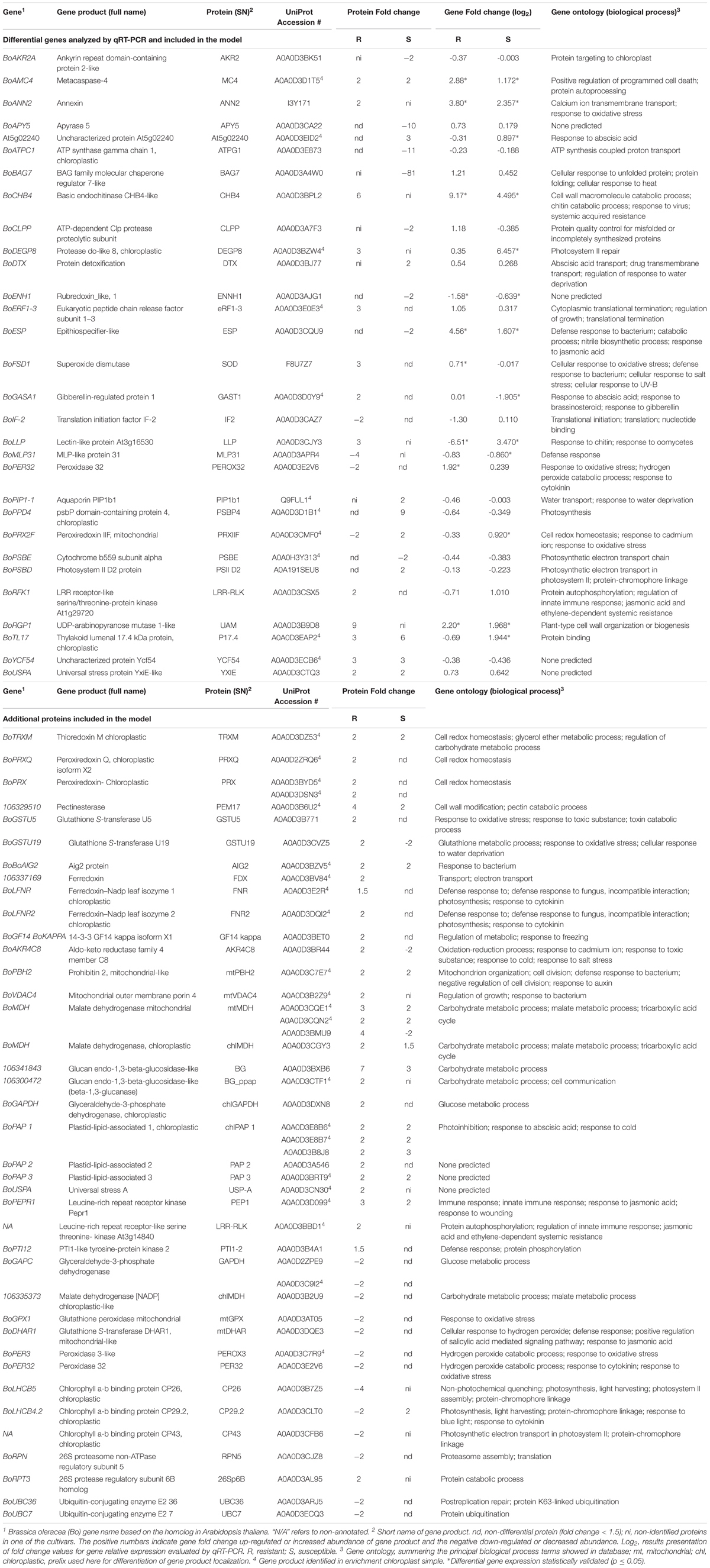
Table 1. Differential proteins and encoding genes analyzed by qRT-PCR analysis (RI:RC and SI:SC interactions) and discussed in the proposed interaction model.
The differences observed between protein abundance and gene expression levels has been widely reported and may be explained by the regulatory processes that can occur after mRNA transcription, including post-transcriptional, translational, post-translational and protein degradation regulation mechanisms, as well as half-life of RNA and of the corresponding proteins (de Sousa Abreu et al., 2009; Lee et al., 2011; Vogel and Marcotte, 2012).
Xcc-Responsive Proteins in the Resistant and Susceptible Interaction
The proteome analysis of the resistant cultivar (RI:RC) revealed 338 differentially abundant proteins (215 increased and 123 decreased) while in the susceptible cultivar comparison (SI:SC) 361 differential proteins (225 increased and 136 decreased) were detected. The GO analysis revealed the same over-represented GO terms in both resistant and susceptible interactions (Supplementary Table S3), including cell metabolism, protein biosynthesis, processing and degradation, photosynthesis, disease/defense response and uncharacterized proteins (proteins with no GO information).
A higher number of energy metabolism proteins (Supplementary Table S3) were identified in the susceptible cultivar, most of which showed decreased abundance. It is known that there is intense activity of the main glycolytic pathways during plant–pathogen interaction and alterations in sugar metabolism in the host plant can be crucial for pathogen control, since both organisms compete for nutrients (reviewed by Kanwar and Jha, 2019). It is important to highlight that most of the differential proteins related to energy metabolism were detected only in chloroplast enriched samples, which reinforces the importance of analyzing organelle enriched samples to get a better picture of the plant–pathogen interaction. It is known that photosynthesis is severely affected during biotic and abiotic stresses since resistance has an energy cost. Although the molecular participation of chloroplast in plant immunity is not clear, it has been shown that chloroplasts can have a crucial role in the plant basal immune system that involves PAMPs signaling, Ca2+ signaling pathways, as well as salicylic and jasmonic acid (JA) production (Grant and Jones, 2009; Padmanabhan and Dinesh-Kumar, 2010; Nomura et al., 2012).
Energy Metabolism Proteins
Three malate dehydrogenase mitochondrial proteins (A0A0D3CQE1; A0A0D3CQN2) showed increased abundance in both cultivars, however, one of them (A0A0D3BMU9) showed increased abundance in the resistant cultivar (4-fold) and decreased abundance in the susceptible one (14-fold), when compared to the control condition. Malate is implicated in many plant metabolic processes, including TCA cycle, Calvin cycle, and in pH regulation and ion transport in roots. Malate dehydrogenase, an important malate metabolizing enzyme, has been associated with plant defense, suggesting that the increased abundance of this enzyme can provide resources for biosynthesis of defense compounds (Rhodes et al., 1968; Walter et al., 1988; Casati et al., 1999). In a previous study, one MDH showed increased abundance in brassica–Xcc resistant interaction and was associated with the activation of photosynthetic metabolism (Villeth et al., 2016). Other metabolism proteins such as fructose-1,6-biphosphate, cytosolic EC 3.1.3.11 (A0A0D3BSL1), basic endochitinase CHI-B4-like, EC 3.2.1.14 (A0A0D3B6J8; A0A0D3BPL2) and UDP-arabinopyranose mutase 1-like (A0A0D3B9D8), were increased in RI (9-, 12-, 9- and 6-fold, respectively) and the first two (A0A0D3BSL1 and A0A0D3B6J8) were decreased in SI (28- and 12-fold, respectively), while UDP-arabinopyranose level was unchanged in SI. In the analysis of gene expression (Figure 2 and Table 1), BoCHI-4 like and BoRGP1 were upregulated, with a higher expression in RI:RC (578 and fivefold, respectively) when compared to SI:SC (23- and 4-fold, respectively), suggesting that these proteins, besides being involved in energy metabolism, can have an important role in plant defense. Based on the proteomic and qRT-PCR results, the basic endochitinase BoCHI-B4-like gene (GAQY01039586.1) was selected for overexpression in the model plant A. thaliana for functional validation.
The metabolic pathways role in defense response process is not well understood, however, our results were consistent with other studies, which suggest that the positive regulation of metabolism can initiate a signaling cascade in the signal transduction pathway, leading to a defense response (Rojas et al., 2014). On the other hand, the pathogen can acquire metabolites from the host cell and the plant can respond to prevent the loss of metabolites by increasing the uptake of monosaccharides, limiting the available extracellular sugar for bacteria. This could be a strategical antimicrobial response, since this competitive reaction can lead to the restriction of the delivery of virulence factors (Vogel and Marcotte, 2012; Couto and Zipfel, 2016; Yamada et al., 2016). Indeed, in Brassica and Arabidopsis it has been demonstrated that sugar transporters, such as SWEET transporters that mediate sugar export are positively regulated upon pathogen infection (Chen et al., 2010, 2012; Jian et al., 2016), which could indicate co-evolution for nutrient competition during plant–pathogen interaction (Chen et al., 2010). Studies have suggested that Xanthomonas effectors can recognize SWEET proteins from the plant and induce sugar export from the cell to be used as carbon source for bacterial growth (Cohn et al., 2014; Huang et al., 2016).
Proteins Involved in Photosynthesis and Protein Biosynthesis, Processing and Degradation
As expected, several photosynthesis-related proteins were differentially abundant in both interactions, such as photosystem II CP43 reaction center protein, chlorophyll a-b binding protein CP29.2 and CP26, Ribulose bisphosphate carboxylase (Supplementary Table S3). Most proteins related to photosynthesis in the resistant interaction showed decreased abundance (18%), which is consistent with previous results obtained by Ribeiro et al. (2018), when analyzing the same cultivar at the same time point by 2-DE. On the other hand, in this study, most photosynthesis-related proteins were increased in the SI:SC interaction. In this study, we used chloroplast enriched samples and observed that several of the proteins with increased abundance were detected in chloroplasts samples (Supplementary Table S3), which once again reinforce the importance of subproteome analysis to better understand the global protein interaction profile.
In this study, a clear imbalance in metabolic and photosynthetic processes in both cultivars could be observed, however, it is possible that the resistant plant may have a higher recovery capacity than the susceptible plant, since homeostasis and repair proteins were more abundant in the resistant interaction than in the susceptible. It is known that the impaired metabolic capacity can directly influence the functioning of the photosynthetic apparatus (Raven et al., 2007), correlating metabolic alterations with response to pathogens.
Another over-represented GO term observed in this study was protein biosynthesis, protein processing and degradation (folding, assembly, fate and degradation). Proteins mainly involved in transcription, translation, post-translational and transduction processes were observed with increased abundance in both interactions, including several ribosomal proteins (30S, 40S, and 50S in RI:RC; 50S and 60S in SI:SC). It is noteworthy that the BAG (Bcl-2 associated athanogene) family molecular chaperone regulator 7-like (A0A0D3A4W0) showed a pronounced decreased abundance (81-fold) in SI compared to the control, and in resistant plants this protein was not detected. qRT-PCR results showed an upregulation trend in RI and downregulation trend in SI (Figure 2). These results suggest post-transcriptional or post-translational regulation events, since mRNA and protein levels were highly distinct. The BAG7 belongs to Class III of BAG family proteins, which is composed by eight proteins encoded by highly conserved genes, widely distributed in living organisms (Weissbach et al., 1994; Takayama et al., 1995). In plants, BAG proteins have been considered multifunctional and known to regulate the cytoprotective process during biotic and abiotic stresses (Doukhanina et al., 2006). Li et al. (2016) identified proteins of this family that confer resistance in Arabidopsis against the fungal pathogen Botrytis cinerea, showing evidence of the participation of BAG proteins in innate immunity processes.
Disease/Defense Response Proteins
Among the disease/defense response proteins identified in our study, most were increased in both cultivars and were involved with oxidative stress (Supplementary Figure S4). However, a higher number of pathogen-related proteins associated with plant responses showed increased abundance in the resistant cultivar (16%), whereas only a few (5%) were increased in the susceptible cultivar. The increased defense proteins identified in both cultivars were annexin (I3Y171), AIG2 (A0A0D3BZV5), ferredoxin (A0A0D3BV84), ferredoxin-NADP leaf isozyme 1 and 2 chloroplastic (A0A0D3E2R1; A0A0D3DQI2) and mitochondrial outer membrane protein 4 (A0A0D3B2Z9).
Several increased proteins in susceptible plants were involved with responses to abscisic acid (ABA), while in resistant plants these proteins showed decreased abundance. ABA is a phytohormone, known as a signaling molecule, responsible for the regulation of abiotic stress response (Taiz et al., 2017). Studies have shown that ABA can suppress the plant immune response, (Kim et al., 2011; Desclos-Theveniau et al., 2012) and in many pathosystems, this phytohormone can act antagonistically to the salicylic acid (SA) pathway. SA is another important phytohormone that can confer plant resistance against pathogens (Audenaert et al., 2002; Jiang et al., 2010). Furthermore, ABA can suppress the MAPK pathway, causing immunosuppression in A. thaliana and possibly in other cruciferous plants (Mine et al., 2017). ABA’s effect during plant–pathogen interaction is considered complex, however, it is possible that the increased abundance of proteins involved with ABA response can be a mechanism, which can favor susceptibility (Kim et al., 2011; Desclos-Theveniau et al., 2012).
The increase of ABA can also lead to the accumulation of other proteins such as aquaporins. In this study, the aquaporins PIP3 (Q9FUC0) and BoPIP1b1 (Q9FUL1) were differentially abundant; the second was evaluated by qRT-PCR and showed a downregulation trend in both cultivars. These aquaporins were identified in chloroplast samples with increased abundance only in the susceptible plants. PIP aquaporins are intramembrane channels important for the transport of water and CO2 in the plant tissues (Luu and Maurel, 2005; Verkman, 2013). The detection of these proteins in chloroplast samples was not expected, however, since we sampled intact and broken chloroplasts, it is possible that some non-chloroplastic proteins were also isolated. Our results suggest that the accumulation of these proteins may be related to ABA, as observed by Aroca et al. (2006), in leaves of Phaseolus vulgaris after ABA treatment. Aquaporins are multifunctional and some isoforms are able to detect pathogen molecular patterns (PAMPs) such as harpins (Zhu et al., 2000; Flexas et al., 2007). The transport of hydrogen peroxide has also been associated to aquaporins (Taiz et al., 2017). It was demonstrated that the loss of function of the gene locus AtPIP1;4 in Arabidopsis cancels the import of apoplastic H2O2 induced by the pathogen and consequently blocks the plant immune response (Tian et al., 2016).
Curiously, another protein named epithiospecifier-like (ESP-like; A0A0D3CQU9) involved in defense response, was decreased in susceptible plants and unchanged in resistant plants. Conversely, the expression of ESP gene was upregulated according to qRT-PCR results in both cultivars (23-fold change in the resistant plant and 3-fold change in the susceptible). In the previous study performed by Ribeiro et al. (2018), another resistant cabbage cultivar (União) was analyzed and ESP protein was exclusively identified in the resistant cultivar infected with Xcc by 2-DE analysis, demonstrating that the regulation of ESP protein can be important for the plant defense against Xcc. The ESP protein is related to the glucosinolate pathway involved in plant protection against herbivory pests. Glucosinolates are secondary metabolites, known as phytoanticipins (preformed antimicrobial compounds), representing one of the first chemical barriers against pathogen attack (Osbourn, 1996). These metabolites can be found extensively in Brassicaceae plants (i.e., broccoli, cabbage, mustard), and are biologically active compounds reported in some processes of plant defense including stress response and antioxidant activities (Bennett and Wallsgrove, 1994; Halkier and Gershenzon, 2006).
Interaction Model of Resistant B. oleracea–Xcc Interaction
Overall, in this study, we observed that the protein profiles of the resistant and susceptible plants were similar, especially regarding the predominant GO terms. However, a higher number of pathogen-related proteins were identified in the resistant plants and therefore we propose a model of this interaction based on protein localization and their role in the cell (Figure 3 and Table 1). This model can help better understand the plant response to Xcc infection and provide candidate genes for the development of more efficient pathogen control strategies.
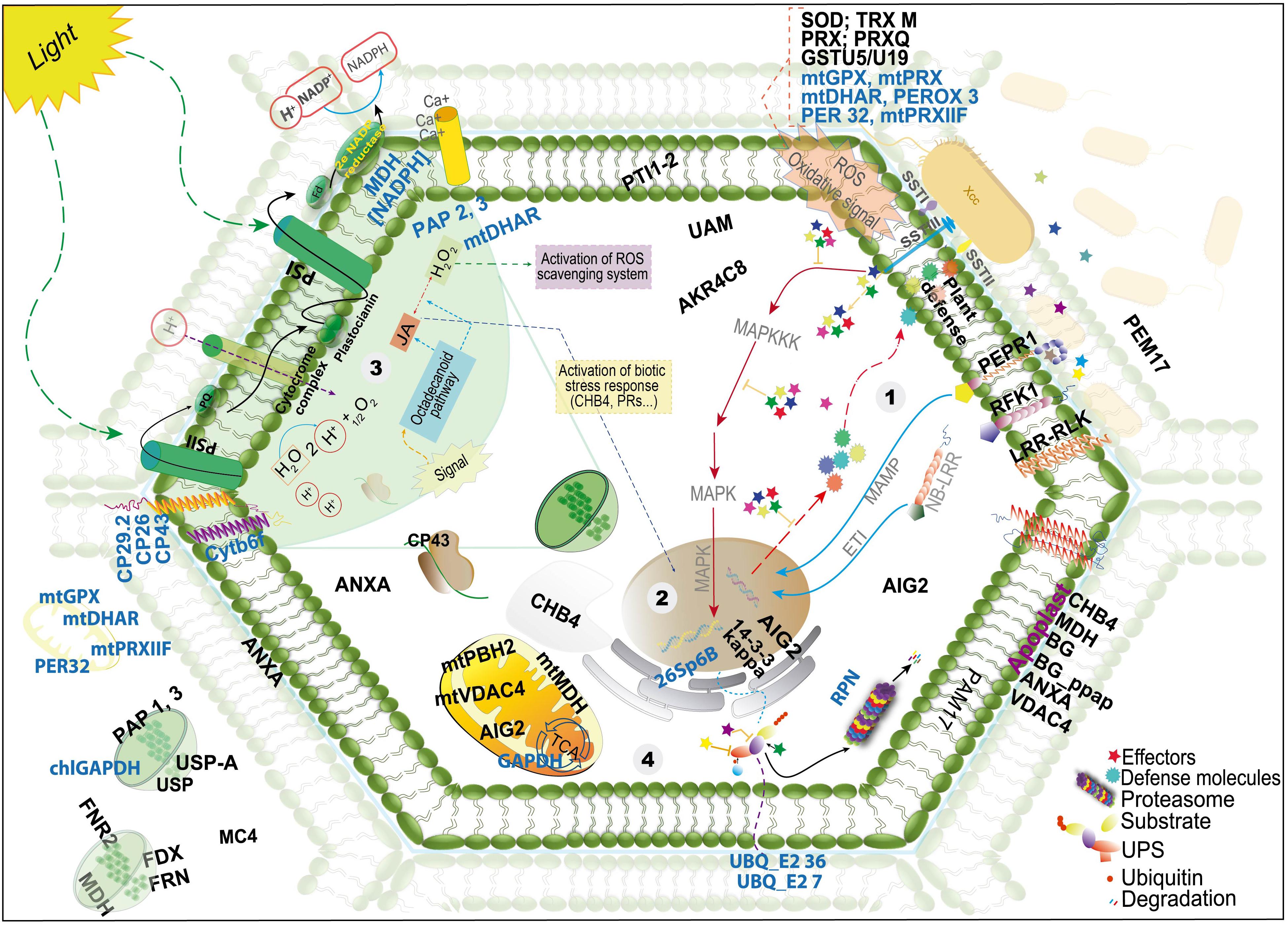
Figure 3. Schematic view of a model proposed with the proteins identified in the resistant Brassica oleracea–Xanthomonas campestris pv. campestris (Xcc) infected leaf and chloroplast-enriched proteomes. The figure shows the localization, as proposed by UniProt database; detailed information on the proteins is presented in Table 1. The names in black and blue indicate proteins with increased and decreased abundance, respectively. The steps begin at the recognition of the pathogen, involving important signaling proteins, activation of molecular defense response pathways and oxidative stress response (steps 1–3), followed by UPS (ubiquitin pathway system) modulation and repair proteins (step 4), as well as alteration of metabolic and photosynthetic pathways (steps 3 and 4).
The classical mechanism of bacterial recognition occurs at the beginning of infection, in an attempt to neutralize the effectors released by the bacterium and repair the damage caused in the cell. In the resistant cultivar Astrus, we found several proteins with increased abundance probably involved in this initial response (see step 1 in the model), such as Lectin-like protein (A0A0D3CJY3), leucine-rich repeat receptor kinase (A0A0D3D099), leucine-rich repeat receptor-like serine threonine- kinase (A0A0D3CSX5). These transmembrane signaling proteins, together with other proteins, such as NB-LRR, can be essential to sense the pathogen and promote systemic immunity (O’Neill and Bowie, 2007; Couto and Zipfel, 2016). Other signaling proteins identified showing increased abundance were the universal stress protein A (A0A0D3CN30) and universal stress PHOS34-like (A0A0D3CTQ3). Although the exact role of PHOS34 in plant defense is not known, studies have reported that this protein can be phosphorylated by MPK3 and MPK6, and after treatment with the flagellin flg22 peptide (Merkouropoulos et al., 2008), suggesting that this protein may be related with cellular signaling in the presence of the bacterium.
Several antioxidant/detoxification proteins were also increased, including superoxide dismutase Fe (F8U7Z7), peroxidase (A0A0D3C7R8), peroxiredoxin (A0A0D3DSN3; A0A0D2ZRQ6; A0A0D3BYD5) and glutathione S-transferase U5 and U19 (A0A0D3B771; A0A0D3CVZ5). The accumulation of ROS can be toxic to the pathogen by inhibiting and/or reducing its survival (Jones and Dangl, 2006; Zhang and Zhou, 2010). However, ROS accumulation can lead to the oxidation of important cell components like lipids and genetic material (Sharma et al., 2016). An intriguing result obtained was the decrease of other extra and intracellular antioxidant proteins including glutathione peroxidase mitochondrial; (A0A0D3AT05; A0A0D3DQE3) peroxidase 3-like (A0A0D3C7R9); peroxiredoxin-mitochondrial (A0A0D3CMF0) and peroxidase 32 (A0A0D3E2V6) in the resistant interaction. This result may indicate that a balance in the abundance of proteins related to oxidative stress, maintaining some proteins with increased abundance and others with decreased abundance may be important for an efficient control of the pathogen without extensive damage to the plant tissue.
Once the pathogen overcomes the first line of defense, other events occur in response to the effector delivery into the cell by the type III secretion system. At this stage, proteins such as NB-LRR proteins interact with the pathogen effectors (Spallek et al., 2009; Marino et al., 2012). In this study, we identified a leucine-rich repeat receptor kinase PEPR1 (A0A0D3D099), which has been reported as the receptor for AtPep1, a peptide elicitor from Arabidopsis that signals activation of innate immune response against pathogens (Yamaguchi et al., 2006) as well as a probable LRR receptor-like serine/threonine-protein kinase At1g29720 RFK1-like (A0A0D3CSX5). Both proteins are integral components of the membrane (model-step 1) and may be involved in triggering a defense response.
Ubiquitination pathway also seems to play an important role in the resistant interaction. The protein 14-3-3-GF14 kappa (A0A0D3BET0), known as a metabolism regulator associated with abiotic stress, was identified and can modulate other proteins by facilitating their degradation by ubiquitins (Fuller et al., 2006; Chang et al., 2009; Liu et al., 2017). The ubiquitin pathway is necessary to tag proteins that should be degraded, however, bacterial effectors may also interact with ubiquitin proteasome system (UPS) as a false system protein (Figure 3). The bacterial effectors can be ubiquitinated and degraded by proteasomes; they can also interfere in the system, act as a ubiquitin ligase or inhibit the specific UPS steps (Collins and Brown, 2010). Proteins related to ubiquitination showed reduced abundance in the present work (A0A0D3CJZ8; A0A0D3ARJ5, A0A0D3ECQ3, A0A0D3BLH4). In a highly resistant plant, ubiquitination proteins also showed reduced abundance at 24 hai (Ribeiro et al., 2018), which may indicate a negative regulation of this pathway, leading to cell death and consequently resulting in limitation of bacterial growth (Spallek et al., 2009; Marino et al., 2012).
Proteins involved with defense against pathogens were also increased including Ferredoxin–NADP leaf isozyme 1 and 2, chloroplastic (A0A0D3E2R1; A0A0D3DQI2) and annexin (I3Y171). Annexins are members of a well-known family of proteins involved in tolerance against environmental stresses and have been studied in tobacco, cotton, Brassica and Arabidopsis plants (Jami et al., 2008; Konopka-Postupolska et al., 2009; Clark et al., 2012; Szalonek et al., 2015). AIG2 (A0A0D3BZV5) is another defense protein, which has not been functionally characterized yet, however, it is known that the corresponding gene is induced in Arabidopsis by the avirulent gene avrRpt2 of Pseudomonas syringae (Reuber and Ausubel, 1996).
Defense response is also highly correlated with the levels of phytohormones, such as JA, ethylene (ET), ABA, and cytokinin. JA is important in the plant defense against various stresses. As seen in the model, the indirect activation of JA by the octadecanoid pathway and H2O2 accumulation can result in the activation of biotic stress response. The JA pathway can activate other pathways such as the signal transduction pathway, inducing the formation of chemical and physical barriers against pathogen or herbivore attacks (Kazan and Manners, 2008). In addition to the lectin proteins identified, plastid lipid-associated protein 2 and 3, chloroplastic (A0A0D3A546; A0A0D3BRT9) were also identified.
Another phytohormone involved in defense signaling is cytokinin, involved in plant development, cellular differentiation and senescence (Hwang et al., 2012). It has been reported that high levels of this hormone increased plant immunity (Swartzberg et al., 2008; Choi et al., 2010; Argueso et al., 2012). In this study, some proteins responsive to cytokinins were increased including 50S Ribosomal Chloroplastic protein (A0A0D3C0C3); binding partner of ACD11 1 isoform X2 (A0A0D3AJE9) and succinate dehydrogenase subunit 5 mitochondrial-like isoform X2 (A0A0D3DK02). Based on these results, it seems that the regulation of proteins responsive to these phytohormones may play an important role in resistance against Xcc.
Secondary metabolites also play an important role in plant defense. Chloroplastic plastid-lipid-associated proteins (1, 2, and 3), were identified with increased abundance and are associated with the storage of carotenoids in plants and sequestration of hydrophobic compounds (Ting et al., 1998; Laizet et al., 2004; Leitner-Dagan et al., 2006).
Finally, when the initial defense mechanisms are not enough to contain the pathogen, cell death can also be activated. In this study, metacaspase 4 (A0A0D3D1T5), an important protein reported as a participant in the cell death mechanism (Kwon and Hwang, 2013), showed increased abundance in both cultivars. qRT-PCR analysis showed that the corresponding gene was upregulated sevenfold in the resistant cultivar and twofold in the susceptible one. Taken together, the model presented here can represent a step-by-step of the defense mechanism in resistant brassica plants, beginning at the recognition of the pathogen, with the activation of important signaling proteins, molecular defense response pathways, and oxidative stress response (steps 1–3 in the model), followed by UPS modulation and repair proteins (step 4), and alteration of metabolic and photosynthetic pathways (steps 3 and 4). The model may also contribute to better understand the molecular responses during the plant–pathogen interaction reflected by the differential abundance of proteins under Xcc infection.
Functional Validation by Overexpression of CHI-B4 Like Protein in A. thaliana
In this study, several candidate proteins were identified, potentially involved in the resistance response to Xcc. One of these proteins, the CHI-B4 like protein, as mentioned above, showed increased abundance in the proteomic analysis and high gene expression levels. Endochitinase-like proteins, are members of the chitinase family that participate in the catabolic process in the cell (Stintzi et al., 1993). The chitinases are also classified as pathogen related proteins (PRs) and endochitinases belong to group 3 of PRs that cleave chitin molecules. In general, studies have related chitinases to plant–fungus interaction (reviewed by Jalil et al., 2015).
A. thaliana plants overexpressing cabbage BoCHB4 gene under the control of CaMV 35S promoter were obtained through transformation. The presence of the transgene was confirmed in five homozygote plants and single copy insertions were observed by Southern blot analysis for each positive event (Figure 4). The transcript level of the transgene was also assessed, and the transgenic lines showed a relative expression level 588-fold higher than the wild type (Col-0). Since CHI-B4 like can also be found in A. thaliana, an alignment of the nucleotide sequences of both genes (endogenous and transgenic) was performed and showed 84% identity with the Arabidopsis gene, and therefore the expression levels detected were probably related to the transgene and not to the endogenous gene.
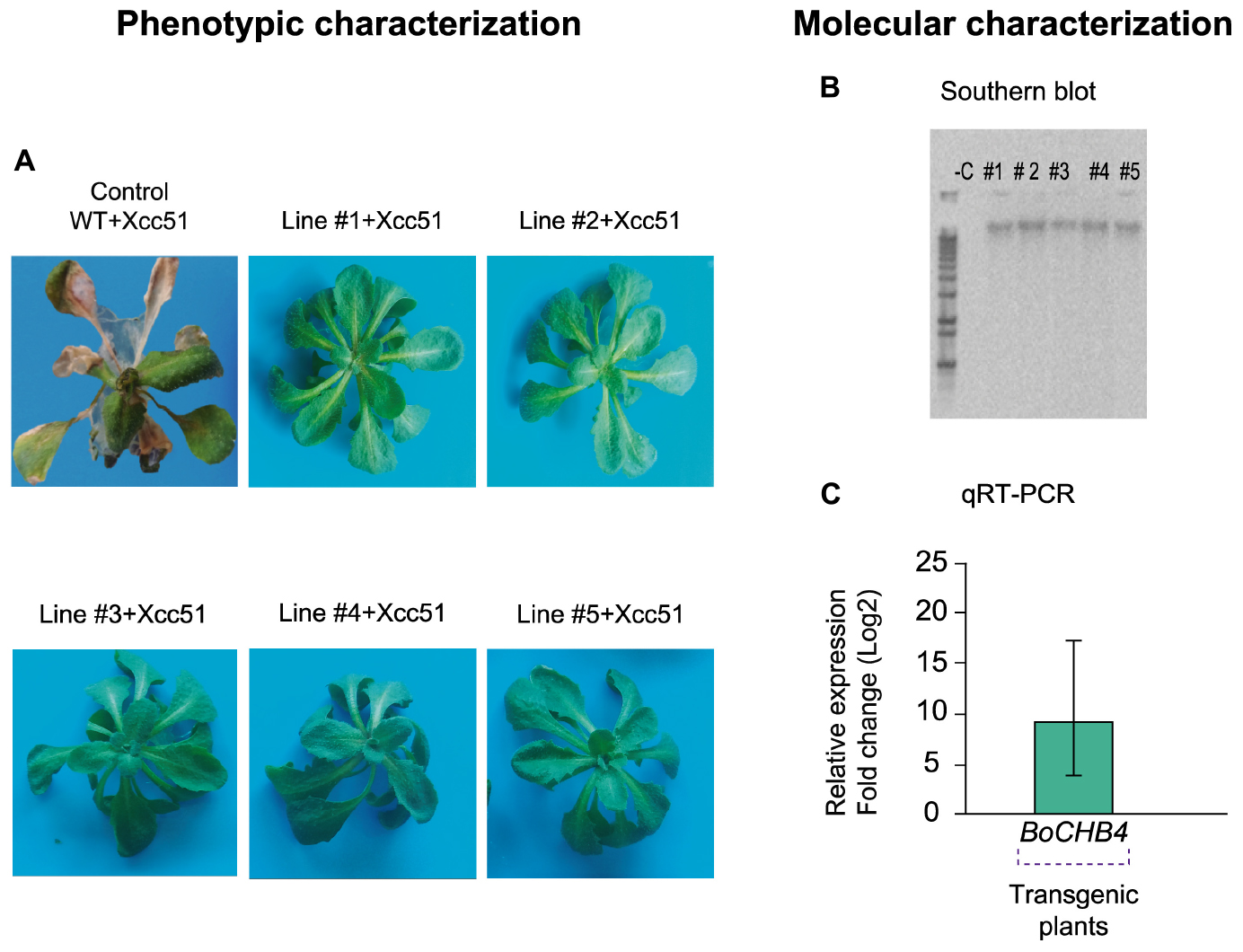
Figure 4. Phenotypic and molecular evaluation of transgenic lines and wild type (WT) Arabidopsis thaliana plants. (A) A. thaliana plants (WT and #1–5 BoCHB4 lines) inoculated with Xanthomonas campestris pv. campestris (Xcc 51) at 5 days after inoculation (dai). (B) Southern blot analysis of A. thaliana transgenic lines (# 1–5) with BoCHB4 radiolabeled probe. (C) Relative expression of BoCHB4 gene in A. thaliana WT and transgenic lines (# 1–5).
A phenotypic evaluation of A. thaliana plants overexpressing BoCHB4 was performed and at 48 hai the WT plants began to show the first symptoms (Figure 4). The phenotypic analysis of the WT and transgenic lines after inoculation of Xcc showed that at 5 days after inoculation (dai), among the surviving plants, the WT replicates were almost totally necrotic and most leaves were dead, while the transgenic plants showed no symptoms even at 15 days after inoculation. According to the disease scoring index based on Meyer et al. (2005), A. thaliana WT Col-0 was highly susceptible, showing severe necrosis and leaf death, while the transformed plants were highly resistant, since no symptoms were observed. Based on these results, we conclude that the overexpression of BoCHB4 can confer resistance against the bacterial pathogen Xcc. This work provides an important contribution regarding the comprehension of resistance mechanisms and offers candidate genes to be used in genetic breeding programs aiming at the development of more efficient strategies for black rot disease control.
Author Contributions
CS performed and designed the experiments, analyzed and interpreted data, and wrote the manuscript. FN, GD, WF, and JJ-N assisted in proteomic data acquisition and analysis. GP, PH, VS, OO-N, and MG-d-S assisted in gene isolation, vector design, and plant transformation. OF analyzed the data and revised the manuscript and AM designed the experiments, wrote the manuscript, and lead the work.
Funding
This research was supported by grants from Embrapa Recursos Genéticos e Biotecnologia, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT), and Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00414/full#supplementary-material
FIGURE S1 | Workflow showing the entire procedure for sample preparation.
FIGURE S2 | SDS-PAGE step prior to LC-MS/MS analysis. The section indicated by the red square was excised and submitted to in gel digestion using trypsin.
FIGURE S3 | Venn diagram showing the total number of proteins identified by the proteomic analysis. (a) Quantitative distribution of proteins from inoculated leaves (LRI) and from the control condition (LRC) as well as from chloroplast-enriched inoculated samples (ChlRI) and from the control condition (ChlRC) in the resistant interaction. (b) Quantitative distribution of proteins from the susceptible inoculated leaves (LSI) and from the control condition (LSC) as well as from chloroplast-enriched inoculated samples (ChlSI) and from the control condition (ChlSC) in the susceptible interaction.
FIGURE S4 | Subclassification of proteins related to disease/defense response, differentially abundant in resistant and susceptible cultivars. (a) Proteins with increased and (b) decreased abundance in the RI:RC comparison. (c) Proteins with increased and (d) decreased abundance in the SI:SC comparison. RI, resistant cultivar inoculated; RC, resistant cultivar control; SI, susceptible cultivar inoculated; SC, susceptible cultivar control.
TABLE S1 | Primers designed for the genes encoding the identified proteins used in qRT-PCR.
TABLE S2 | Brassica oleracea var. capitata protein data obtained by LC-MS/MS. The data in each cultivar are reported in separate spreadsheets.
TABLE S3 | Biological process category of the identified proteins with increased and decreased abundance in both cultivars comparison (RI:RC and SI:SC), as indicated in each spreadsheet.
Footnotes
References
Argueso, C. T., Ferreira, F. J., Epple, P., To, J. P. C., Hutchison, C. E., Schaller, G. E., et al. (2012). Two-component elements mediate interactions between cytokinin and salicylic acid in plant immunity. PLoS Genet. 8:e1002448. doi: 10.1371/journal.pgen.1002448
Aroca, R., Ferrante, A., Vernieri, P., and Chrispeels, M. J. (2006). Drought, abscisic acid and transpiration rate effects on the regulation of PIP aquaporin gene expression and abundance in Phaseolus vulgaris plants. Ann. Bot. 98, 1301–1310. doi: 10.1093/aob/mcl219
Audenaert, K., De Meyer, G. B., and Höfte, M. M. (2002). Abscisic acid determines basal susceptibility of tomato to Botrytis cinerea and suppresses salicylic acid-dependent signaling mechanisms. Plant Physiol. 128, 491–501. doi: 10.1104/pp.010605
Audran, C., Serrano, I., and Rivas, S. (2016). Chloroplasts at work during plant innate immunity. J. Exp. Bot. 67, 3845–3854. doi: 10.1093/jxb/erw088
Bayer, R. G., Stael, S., and Teige, M. (2015). Chloroplast isolation and affinity chromatography for enrichment of low-abundant proteins in complex proteomes. Methods Mol. Biol. 1295, 211–223. doi: 10.1007/978-1-4939-2550-6_16
Bendahmane, A., Farnham, G., Moffett, P., and Baulcombe, D. C. (2002). Constitutive gain-of-function mutants in a nucleotide binding site–leucine rich repeat protein encoded at the Rx locus of potato. Plant J. 32, 195–204. doi: 10.1046/j.1365-313X.2002.01413.x
Bennett, R. N., and Wallsgrove, R. M. (1994). Secondary metabolites in plant defence mechanisms. New Phytol. 127, 617–633. doi: 10.1111/j.1469-8137.1994.tb02968.x
Bent, A. F., and Clough, S. J. (1998). “Agrobacterium germ-line transformation: transformation of Arabidopsis without tissue culture,” in Plant Molecular Biology Manual, eds S. B. Gelvin and R. A. Schilperoort (Dordrecht: Springer), 17–30.
Camargo, L. E. A., Williams, P. H., and Osborn, T. C. (1995). Mapping of quantitative trait loci controlling resistance of Brassica oleracea to Xanthomonas campestris pv. campestris in the field and greenhouse. Phytopathology 85, 1296–1300. doi: 10.1094/Phyto-85-1296
Casati, P., Drincovich, M. F., Edwards, G. E., and Andreo, C. S. (1999). Malate metabolism by NADP-malic enzyme in plant defense. Photosynth. Res. 61, 99–105. doi: 10.1023/A:1006209003096
Chang, I. F., Curran, A., Woolsey, R., Quilici, D., Cushman, J. C., Mittler, R., et al. (2009). Proteomic profiling of tandem affinity purified 14-3-3 protein complexes in Arabidopsis thaliana. Proteomics 9, 2967–2985. doi: 10.1002/pmic.200800445
Chen, L.-Q., Hou, B.-H., Lalonde, S., Takanaga, H., Hartung, M. L., Qu, X.-Q., et al. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532. doi: 10.1038/nature09606
Chen, L.-Q., Qu, X.-Q., Hou, B.-H., Sosso, D., Osorio, S., Fernie, A. R., et al. (2012). Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335, 207–211. doi: 10.1126/science.1213351
Choi, J., Huh, S. U., Kojima, M., Sakakibara, H., Paek, K.-H., and Hwang, I. (2010). The cytokinin-activated transcription factor ARR2 promotes plant immunity via TGA3/NPR1-dependent salicylic acid signaling in Arabidopsis. Dev. Cell 19, 284–295. doi: 10.1016/j.devcel.2010.07.011
Clark, G. B., Morgan, R. O., Fernandez, M. P., and Roux, S. J. (2012). Evolutionary adaptation of plant annexins has diversified their molecular structures, interactions and functional roles. New Phytol. 196, 695–712. doi: 10.1111/j.1469-8137.2012.04308.x
Cohn, M., Bart, R. S., Shybut, M., Dahlbeck, D., Gomez, M., Morbitzer, R., et al. (2014). Xanthomonas axonopodis virulence is promoted by a transcription activator-like effector–mediated induction of a SWEET sugar transporter in cassava. Mol. Plant Microbe Interact. 27, 1186–1198. doi: 10.1094/MPMI-06-14-0161-R
Collins, C. A., and Brown, E. J. (2010). Cytosol as battleground: ubiquitin as a weapon for both host and pathogen. Trends Cell Biol. 20, 205–213. doi: 10.1016/j.tcb.2010.01.002
Corthals, G. L., Wasinger, V. C., Hochstrasser, D. F., and Sanchez, J. C. (2000). The dynamic range of protein expression: a challenge for proteomic research. Electrophoresis 21, 1104–1115. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1104::AID-ELPS1104>3.0.CO;2-C
Couto, D., and Zipfel, C. (2016). Regulation of pattern recognition receptor signalling in plants. Nat. Rev. Immunol. 16, 537–552. doi: 10.1038/nri.2016.77
de Sousa Abreu, R., Penalva, L. O., Marcotte, E. M., and Vogel, C. (2009). Global signatures of protein and mRNA expression levels. Mol. Biosyst. 5, 1512–1526. doi: 10.1039/b908315d
Desclos-Theveniau, M., Arnaud, D., Huang, T.-Y., Lin, G. J.-C., Chen, W.-Y., Lin, Y.-C., et al. (2012). The Arabidopsis lectin receptor kinase LecRK-V.5 represses stomatal immunity induced by Pseudomonas syringae pv. tomato DC3000. PLoS Pathog. 8:e1002513. doi: 10.1371/journal.ppat.1002513
Doukhanina, E. V., Chen, S., Van Der Zalm, E., Godzik, A., Reed, J., and Dickman, M. B. (2006). Identification and functional characterization of the BAG protein family in Arabidopsis thaliana. J. Biol. Chem. 281, 18793–18801. doi: 10.1074/jbc.M511794200
Etschmann, B., Wilcken, B., Stoevesand, K., Von Der, Schulenburg, A., and Sterner-Kock, A. (2006). Selection of reference genes for quantitative real-time PCR analysis in canine mammary tumors using the GeNorm algorithm. Vet. Pathol. 43, 934–942. doi: 10.1354/vp.43-6-934
Flexas, J., Ortuño, M. F., Ribas-Carbo, M., Diaz-Espejo, A., Florez-Sarasa, I. D., and Medrano, H. (2007). Mesophyll conductance to CO2 in Arabidopsis thaliana. New Phytol. 175, 501–511. doi: 10.1111/j.1469-8137.2007.02111.x
Fuller, B., Stevens, S. M. Jr., Sehnke, P. C., and Ferl, R. J. (2006). Proteomic analysis of the 14-3-3 family in Arabidopsis. Proteomics 6, 3050–3059. doi: 10.1002/pmic.200500729
Grant, M. R., and Jones, J. D. G. (2009). Hormone (dis) harmony moulds plant health and disease. Science 324, 750–752. doi: 10.1126/science.1173771
Guo, H., Dickson, M. H., and Hunter, J. E. (1991). Brassica napus sources of resistance to black rot in crucifers and inheritance of resistance. Hortscience 26, 1545–1547. doi: 10.21273/HORTSCI.26.12.1545
Halkier, B. A., and Gershenzon, J. (2006). Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 57, 303–333. doi: 10.1146/annurev.arplant.57.032905.105228
Han, Y., Ma, B. I. N., and Zhang, K. (2005). SPIDER: software for protein identification from sequence tags with de novo sequencing error. J. Bioinform. Comput. Biol. 3, 697–716. doi: 10.1142/S0219720005001247
Huang, S., Antony, G., Li, T., Liu, B., Obasa, K., Yang, B., et al. (2016). The broadly effective recessive resistance gene xa5 of rice is a virulence effector-dependent quantitative trait for bacterial blight. Plant J. 86, 186–194. doi: 10.1111/tpj.13164
Hwang, I., Sheen, J., and Müller, B. (2012). Cytokinin signaling networks. Annu. Rev. Plant Biol. 63, 353–380. doi: 10.1146/annurev-arplant-042811-105503
Ignatov, A., Kuginuki, Y., and Hidam, K. (2000). Distribution and inheritance of race-specific resistance to Xanthomonas campestris pv. campestris in Brassica rapa and B. napus. J. Russ. Phytopathol. Soc. 1, 89–94.
Jalil, S. U., Mishra, M., and Ansari, M. I. (2015). Current view on chitinase for plant defense. Trends Biosci. 8, 6733–6743.
Jami, S. K., Clark, G. B., Turlapati, S. A., Handley, C., Roux, S. J., and Kirti, P. B. (2008). Ectopic expression of an annexin from Brassica juncea confers tolerance to abiotic and biotic stress treatments in transgenic tobacco. Plant Physiol. Biochem. 46, 1019–1030. doi: 10.1016/j.plaphy.2008.07.006
Jian, H., Lu, K., Yang, B., Wang, T., Zhang, L., Zhang, A., et al. (2016). Genome-wide analysis and expression profiling of the SUC and SWEET gene families of sucrose transporters in oilseed rape (Brassica napus L.). Front. Plant Sci. 7:1464. doi: 10.3389/fpls.2016.01464
Jiang, C.-J., Shimono, M., Sugano, S., Kojima, M., Yazawa, K., Yoshida, R., et al. (2010). Abscisic acid interacts antagonistically with salicylic acid signaling pathway in rice–Magnaporthe grisea interaction. Mol. Plant Microbe Interact. 23, 791–798. doi: 10.1094/MPMI-23-6-0791
Jones, J. D. G., and Dangl, J. L. (2006). The plant immune system. Nature 444, 323–329. doi: 10.1038/nature05286
Kamal, A. H. M., Kim, K.-H., Shin, K.-H., Choi, J.-S., Baik, B.-K., Tsujimoto, H., et al. (2010a). Abiotic stress responsive proteins of wheat grain determined using proteomics technique. Aust. J. Crop Sci. 4, 196–208.
Kamal, A. H. M., Kim, K.-H., Shin, K.-H., Kim, D.-E., Oh, M.-W., Choi, J.-S., et al. (2010b). Proteomics-based dissection of biotic stress responsive proteins in bread wheat (Triticum aestivum L.). Afr. J. Biotechnol. 9, 7239–7255.
Kanwar, P., and Jha, G. (2019). Alterations in plant sugar metabolism: signatory of pathogen attack. Planta 249, 305–318. doi: 10.1007/s00425-018-3018-3
Kazan, K., and Manners, J. M. (2008). Jasmonate signaling: toward an integrated view. Plant Physiol. 146, 1459–1468. doi: 10.1104/pp.107.115717
Kim, S. T., and Kang, K. Y. (2008). “Proteomics in plant defense response,” in Plant Proteomics: Technologies, Strategies, and Applications, eds G. K. Agrawal and R. Rakwal (Hoboken, NJ: John Wiley & Sons, Inc.), 585–604. doi: 10.1002/9780470369630.ch40
Kim, T.-H., Hauser, F., Ha, T., Xue, S., Böhmer, M., Nishimura, N., et al. (2011). Chemical genetics reveals negative regulation of abscisic acid signaling by a plant immune response pathway. Curr. Biol. 21, 990–997. doi: 10.1016/j.cub.2011.04.045
Kley, J., Heil, M., Muck, A., Svatoš, A., and Boland, W. (2010). Isolating intact chloroplasts from small Arabidopsis samples for proteomic studies. Anal. Biochem. 398, 198–202. doi: 10.1016/j.ab.2009.11.016
Komatsu, S., and Hossain, Z. (2017). Preface—Plant Proteomic Research. Basel: Multidisciplinary Digital Publishing Institute.
Konopka-Postupolska, D., Clark, G., Goch, G., Debski, J., Floras, K., Cantero, A., et al. (2009). The role of annexin 1 in drought stress in Arabidopsis. Plant Physiol. 150, 1394–1410. doi: 10.1104/pp.109.135228
Kwon, S. I., and Hwang, D. J. (2013). Expression analysis of the metacaspase gene family in Arabidopsis. J. Plant Biol. 56, 391–398. doi: 10.1007/s12374-013-0290-4
Laizet, Y., Pontier, D., Mache, R., and Kuntz, M. (2004). Subfamily organization and phylogenetic origin of genes encoding plastid lipid-associated proteins of the fibrillin type. J. Genome Sci. Technol. 3, 19–28. doi: 10.1166/gl.2004.038
Lee, M. V., Topper, S. E., Hubler, S. L., Hose, J., Wenger, C. D., Coon, J. J., et al. (2011). A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol. Syst. Biol. 7:514. doi: 10.1038/msb.2011.48
Leitner-Dagan, Y., Ovadis, M., Shklarman, E., Elad, Y., David, D. R., and Vainstein, A. (2006). Expression and functional analyses of the plastid lipid-associated protein CHRC suggest its role in chromoplastogenesis and stress. Plant Physiol. 142, 233–244. doi: 10.1104/pp.106.082404
Li, Y., Kabbage, M., Liu, W., and Dickman, M. B. (2016). Aspartyl protease mediated cleavage of BAG6 is necessary for autophagy and fungal resistance in plants. Plant Cell 28, 233–247.
Liu, Z., Jia, Y., Ding, Y., Shi, Y., Li, Z., Guo, Y., et al. (2017). Plasma membrane CRPK1-mediated phosphorylation of 14-3-3 proteins induces their nuclear import to fine-tune CBF signaling during cold response. Mol. Cell 66, 117–128. doi: 10.1016/j.molcel.2017.02.016
Luu, D. T., and Maurel, C. (2005). Aquaporins in a challenging environment: molecular gears for adjusting plant water status. Plant Cell Environ. 28, 85–96. doi: 10.1111/j.1365-3040.2004.01295.x
Marino, D., Peeters, N., and Rivas, S. (2012). Ubiquitination during plant immune signaling. Plant Physiol. 160, 15–27. doi: 10.1104/pp.112.199281
Merkouropoulos, G., Andreasson, E., Hess, D., Boller, T., and Peck, S. C. (2008). An Arabidopsis protein phosphorylated in response to microbial elicitation, AtPHOS32, is a substrate of MAP kinases 3 and 6. J. Biol. Chem. 283, 10493–10499. doi: 10.1074/jbc.M800735200
Meyer, D., Lauber, E., Roby, D., Arlat, M., and Kroj, T. (2005). Optimization of pathogenicity assays to study the Arabidopsis thaliana–Xanthomonas campestris pv. campestris pathosystem. Mol. Plant Pathol. 6, 327–333. doi: 10.1111/j.1364-3703.2005.00287.x
Mine, A., Berens, M. L., Nobori, T., Anver, S., Fukumoto, K., Winkelmüller, T. M., et al. (2017). Pathogen exploitation of an abscisic acid-and jasmonate-inducible MAPK phosphatase and its interception by Arabidopsis immunity. Proc. Natl. Acad. Sci. 114, 7456–7461. doi: 10.1073/pnas.1702613114
Mot, R. D., and Vanderleyden, J. (1989). Application of two-dimensional protein analysis for strain fingerprinting and mutant analysis of Azospirillum species. Can. J. Microbiol. 35, 960–967. doi: 10.1139/m89-158
Nomura, H., Komori, T., Uemura, S., Kanda, Y., Shimotani, K., Nakai, K., et al. (2012). Chloroplast-mediated activation of plant immune signalling in Arabidopsis. Nat. Commun. 3:926. doi: 10.1038/ncomms1926
O’Neill, L. A. J., and Bowie, A. G. (2007). The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364. doi: 10.1038/nri2079
Osbourn, A. E. (1996). Preformed antimicrobial compounds and plant defense against fungal attack. Plant Cell 8, 1821–1831. doi: 10.1105/tpc.8.10.1821
Padmanabhan, M. S., and Dinesh-Kumar, S. P. (2010). All hands on deck—the role of chloroplasts, endoplasmic reticulum, and the nucleus in driving plant innate immunity. Mol. Plant Microbe Interact. 23, 1368–1380. doi: 10.1094/MPMI-05-10-0113
Peltier, J.-B., Friso, G., Kalume, D. E., Roepstorff, P., Nilsson, F., Adamska, I., et al. (2000). Proteomics of the chloroplast: systematic identification and targeting analysis of lumenal and peripheral thylakoid proteins. Plant Cell 12, 319–341. doi: 10.1105/tpc.12.3.319
Pfaffl, M. W., Horgan, G. W., and Dempfle, L. (2002). Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30:e36. doi: 10.1093/nar/30.9.e36
Rappsilber, J., Mann, M., and Ishihama, Y. (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906. doi: 10.1038/nprot.2007.261
Raven, P. H., Evert, R. F., and Eichhorn, S. E. (2007). Biologia Vegetal, 830. Rio de Janeiro: Guanabara.
Reuber, T. L., and Ausubel, F. M. (1996). Isolation of Arabidopsis genes that differentiate between resistance responses mediated by the RPS2 and RPM1 disease resistance genes. Plant Cell 8, 241–249. doi: 10.1105/tpc.8.2.241
Rhodes, M. J. C., Wooltorton, L. S. C., Galliard, T., and Hulme, A. C. (1968). Metabolic changes in excised fruit tissue-I. Factors affecting the development of a malate decarboxylation system during the ageing of disks of pre-climacteric apples. Phytochemistry 7, 1439–1451. doi: 10.1016/S0031-9422(00)88589-1
Ribeiro, D. G., Cunha, G. C. R., Santos, C., Silva, L. P., Oliveira Neto, O. B., Labuto, L. B. D., et al. (2018). Brassica oleracea resistance-related proteins identified at an early stage of black rot disease. Physiol. Mol. Plant Pathol. 104, 9–14. doi: 10.1016/j.pmpp.2018.06.002
Rojas, C. M., Senthil-Kumar, M., Tzin, V., and Mysore, K. S. (2014). Regulation of primary plant metabolism during plant-pathogen interactions and its contribution to plant defense. Front. Plant Sci. 5:17. doi: 10.3389/fpls.2014.00017
Rolland, N., Curien, G., Finazzi, G., Kuntz, M., Maréchal, E., Matringe, M., et al. (2012). The biosynthetic capacities of the plastids and integration between cytoplasmic and chloroplast processes. Annu. Rev. Genet. 46, 233–264. doi: 10.1146/annurev-genet-110410-132544
Romano, E., and Vianna, G. R. (2015). Manual de Transformação Genética de Plantas. Brasília: Embrapa-SPI.
Saha, P., Kalia, P., Sharma, M., and Singh, D. (2016). New source of black rot disease resistance in Brassica oleracea and genetic analysis of resistance. Euphytica 207, 35–48. doi: 10.1007/s10681-015-1524-y
Santos, C., Maximiano, M. R., Ribeiro, D. G., Oliveira-Neto, O. B., Murad, A. M., Franco, O. L., et al. (2017). Differential accumulation of Xanthomonas campestris pv. campestris proteins during the interaction with the host plant: contributions of an in vivo system. Proteomics 17:1700086. doi: 10.1002/pmic.201700086
Sharma, B. B., Kalia, P., Yadava, D. K., Singh, D., and Sharma, T. R. (2016). Genetics and molecular mapping of black rot resistance locus Xca1bc on chromosome B-7 in ethiopian mustard (Brassica carinata A. Braun). PLoS One 11:e0152290. doi: 10.1371/journal.pone.0152290
Spallek, T., Robatzek, S., and Göhre, V. (2009). How microbes utilize host ubiquitination. Cell. Microbiol. 11, 1425–1434. doi: 10.1111/j.1462-5822.2009.01346.x
Stekhoven, D. J., Omasits, U., Quebatte, M., Dehio, C., and Ahrens, C. H. (2014). Proteome-wide identification of predominant subcellular protein localizations in a bacterial model organism. J. Proteomics 99, 123–137. doi: 10.1016/j.jprot.2014.01.015
Stintzi, A., Heitz, T., Prasad, V., Wiedemann-Merdinoglu, S., Kauffmann, S., Geoffroy, P., et al. (1993). Plant ‘pathogenesis-related’ proteins and their role in defense against pathogens. Biochimie 75, 687–706. doi: 10.1016/0300-9084(93)90100-7
Swartzberg, D., Kirshner, B., Rav-David, D., Elad, Y., and Granot, D. (2008). Botrytis cinerea induces senescence and is inhibited by autoregulated expression of the IPT gene. Eur. J. Plant Pathol. 120, 289–297. doi: 10.1007/s10658-007-9217-6
Szalonek, M., Sierpien, B., Rymaszewski, W., Gieczewska, K., Garstka, M., Lichocka, M., et al. (2015). Potato annexin STANN1 promotes drought tolerance and mitigates light stress in transgenic Solanum tuberosum L. plants. PLoS One 10:e0132683. doi: 10.1371/journal.pone.0132683
Taiz, L., Zeiger, E., Møller, I. M., and Murphy, A. (2017). Fisiologia e Desenvolvimento Vegetal. Porto Alegre: Artmed Editora.
Takayama, S., Sato, T., Krajewski, S., Kochel, K., Irie, S., Milian, J. A., et al. (1995). Cloning and functional analysis of BAG-1: a novel Bcl-2-binding protein with anti-cell death activity. Cell 80, 279–284. doi: 10.1016/0092-8674(95)90410-7
Tian, S., Wang, X., Li, P., Wang, H., Ji, H., Xie, J., et al. (2016). Plant aquaporin AtPIP1; 4 links apoplastic H2O2 induction to disease immunity pathways. Plant Physiol. 171, 1635–1650. doi: 10.1104/pp.15.01237
Ting, J. T. L., Wu, S. S. H., Ratnayake, C., and Huang, A. H. C. (1998). Constituents of the tapetosomes and elaioplasts in Brassica campestris tapetum and their degradation and retention during microsporogenesis. Plant J. 16, 541–551. doi: 10.1046/j.1365-313x.1998.00325.x
Uberegui, E., Hall, M., Lorenzo,Ó., Schröder, W. P., and Balsera, M. (2015). An Arabidopsis soluble chloroplast proteomic analysis reveals the participation of the Executer pathway in response to increased light conditions. J. Exp. Bot. 66, 2067–2077. doi: 10.1093/jxb/erv018
Untergasser, A., Nijveen, H., Rao, X., Bisseling, T., Geurts, R., and Leunissen, J. A. M. (2007). Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 35, W71–W74. doi: 10.1093/nar/gkm306
Valledor, L., and Weckwerth, W. (2014). An improved detergent-compatible gel-fractionation LC-LTQ-Orbitrap-MS workflow for plant and microbial proteomics. Methods Mol. Biol. 1072, 347–358. doi: 10.1007/978-1-62703-631-3_25
Villeth, G. R. C., Carmo, L. S. T., Silva, L. P., Santos, M. F., De Oliveira Neto, O. B., Grossi-De-Sá, M. F., et al. (2016). Identification of proteins in susceptible and resistant Brassica oleracea responsive to Xanthomonas campestris pv. campestris infection. J. Proteomics 143, 278–285. doi: 10.1016/j.jprot.2016.01.014
Vogel, C., and Marcotte, E. M. (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232. doi: 10.1038/nrg3185
Walter, M. H., Grima-Pettenati, J., Grand, C., Boudet, A. M., and Lamb, C. J. (1988). Cinnamyl-alcohol dehydrogenase, a molecular marker specific for lignin synthesis: cDNA cloning and mRNA induction by fungal elicitor. Proc. Natl. Acad. Sci. 85, 5546–5550. doi: 10.1073/pnas.85.15.5546
Wang, H., and Hanash, S. (2003). Multi-dimensional liquid phase based separations in proteomics. J. Chromatogr. B 787, 11–18. doi: 10.1016/S1570-0232(02)00335-5
Wang, X., and Komatsu, S. (2016). Plant subcellular proteomics: application for exploring optimal cell function in soybean. J. Proteomics 143, 45–56. doi: 10.1016/j.jprot.2016.01.011
Weissbach, L., Settleman, J., Kalady, M. F., Snijders, A. J., Murthy, A. E., Yan, Y.-X., et al. (1994). Identification of a human rasGAP-related protein containing calmodulin-binding motifs. J. Biol. Chem. 269, 20517–20521.
Yamada, K., Saijo, Y., Nakagami, H., and Takano, Y. (2016). Regulation of sugar transporter activity for antibacterial defense in Arabidopsis. Science 354, 1427–1430. doi: 10.1126/science.aah5692
Yamaguchi, Y., Pearce, G., and Ryan, C. A. (2006). The cell surface leucine-rich repeat receptor for AtPep1, an endogenous peptide elicitor in Arabidopsis, is functional in transgenic tobacco cells. Proc. Natl. Acad. Sci. U.S.A. 103, 10104–10109. doi: 10.1073/pnas.0603729103
Zhang, J., Xin, L., Shan, B., Chen, W., Xie, M., Yuen, D., et al. (2012). PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 11:M111.010587.
Zhang, J., and Zhou, J.-M. (2010). Plant immunity triggered by microbial molecular signatures. Mol. Plant 3, 783–793. doi: 10.1093/mp/ssq035
Keywords: LC-MS/MS, differential protein abundance, qRT-PCR, gene overexpression, plant–pathogen interaction
Citation: Santos C, Nogueira FCS, Domont GB, Fontes W, Prado GS, Habibi P, Santos VO, Oliveira-Neto OB, Grossi-de-Sá MF, Jorrín-Novo JV, Franco OL and Mehta A (2019) Proteomic Analysis and Functional Validation of a Brassica oleracea Endochitinase Involved in Resistance to Xanthomonas campestris. Front. Plant Sci. 10:414. doi: 10.3389/fpls.2019.00414
Received: 27 October 2018; Accepted: 19 March 2019;
Published: 12 April 2019.
Edited by:
Stefanie Wienkoop, University of Vienna, AustriaReviewed by:
Laurence Veronique Bindschedler, Royal Holloway, University of London, United KingdomTong Zhang, Pacific Northwest National Laboratory (DOE), United States
Copyright © 2019 Santos, Nogueira, Domont, Fontes, Prado, Habibi, Santos, Oliveira-Neto, Grossi-de-Sá, Jorrín-Novo, Franco and Mehta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Octavio L. Franco, b2NmcmFuY29AZ21haWwuY29t Angela Mehta, YW5nZWxhLm1laHRhQGVtYnJhcGEuYnI=