 Mohsen Hajheidari
Mohsen Hajheidari Csaba Koncz
Csaba Koncz Marcel Bucher
Marcel Bucher- 1Botanical Institute, Cologne Biocenter, Cluster of Excellence on Plant Sciences, University of Cologne, Cologne, Germany
- 2Department of Plant Developmental Biology, Max Planck Institute for Plant Breeding Research, Cologne, Germany
- 3Biological Research Center, Institute of Plant Biology, Hungarian Academy of Sciences, Szeged, Hungary
The history of life consists of a series of major evolutionary transitions, including emergence and radiation of complex multicellular eukaryotes from unicellular ancestors. The cells of multicellular organisms, with few exceptions, contain the same genome, however, their organs are composed of a variety of cell types that differ in both structure and function. This variation is largely due to the transcriptional activity of different sets of genes in different cell types. This indicates that complex transcriptional regulation played a key role in the evolution of complexity in eukaryotes. In this review, we summarize how gene duplication and subsequent evolutionary innovations, including the structural evolution of nucleosomes and chromatin-related factors, contributed to the complexity of the transcriptional system and provided a basis for morphological diversity.
Introduction
Early organisms on Earth were microscopic, and for the first 2500 million years (Myr), living organisms rarely achieved a complexity higher than two or three cell types (Carroll, 2001). Around 500 Myr ago from the mid-Cambrian to early Ordovician, land plants that are a major focus of this review likely evolved from a lineage of unicellular eukaryotes in charophyte green algae (Stebbins and Hill, 1980; Kenrick and Crane, 1997; Harholt et al., 2016; Del-Bem, 2018; Morris et al., 2018). With the evolution of land plants, these more complex organisms colonized the Earth and transformed the biosphere providing habitable environments for terrestrial organisms by supplying sufficient oxygen and nutrients (Hori et al., 2014). Recent evolutionary analyses indicate that the cell wall, symbiotic signaling pathways, the RPB1 heptapeptide repeats, hormonal biosynthesis or signaling pathways, and desiccation and UV radiation tolerance evolved in charophyte green algae prior to land plants (Stebbins and Hill, 1980; Hajheidari et al., 2013; Hori et al., 2014; Yang and Stiller, 2014; Delaux et al., 2015; Ju et al., 2015; Harholt et al., 2016; Del-Bem, 2018). This demonstrates that charophyte green algae were preadapted to cope with harsh terrestrial environments. The greater complexity of unicellular eukaryotes and the evolution and diversification of land plants could not be possible without the existence of a high level of cellular complexity and elaborate mechanisms for gene regulation in unicellular eukaryotic ancestors (Figure 1).
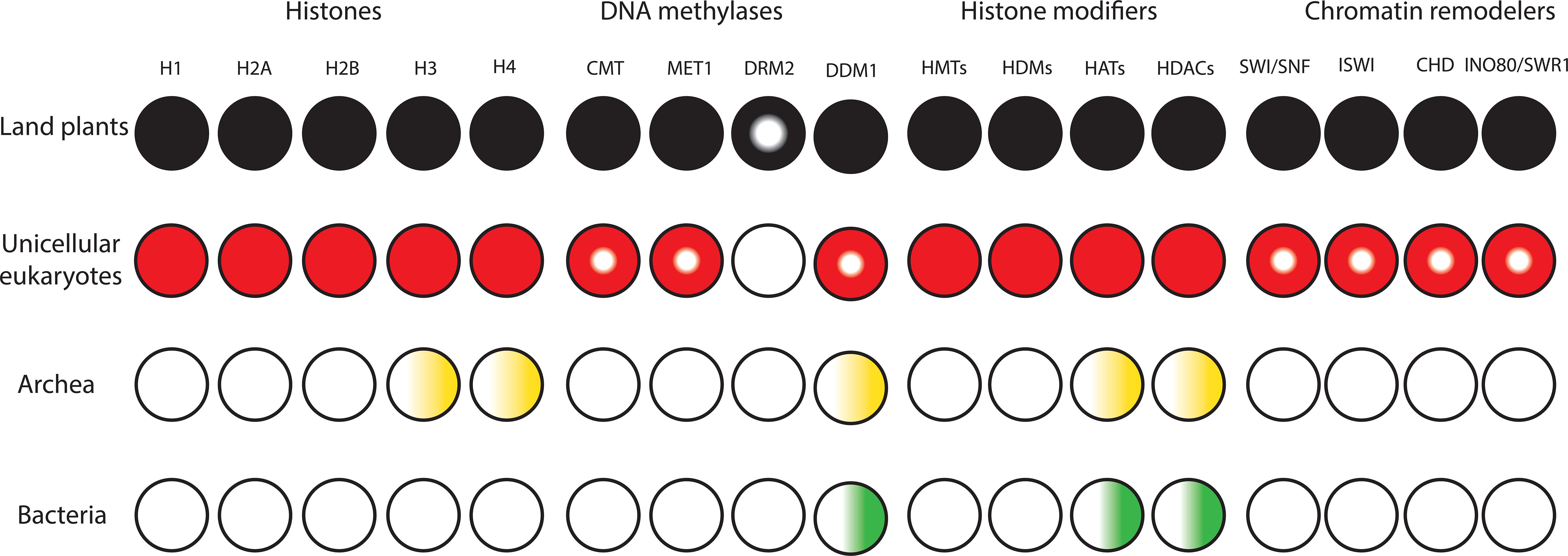
Figure 1. Diversification and expansion of histones, chromatin remodelers and modifiers in the domains of life. Filled circles denote presence of orthologs in all lineages. Semi-filled circles indicate factors present in part of the lineage. Gradients indicate presence of homologs. White circles indicate the lack of homologs.
Eukaryotes have a high degree of cellular complexity. The genomes of most eukaryotes are larger than those of prokaryotes, however, in eukaryotes, in contrast to prokaryotes, genome size does not show a good correlation with gene number (Valentine, 1978; Gregory, 2005). Furthermore, an increase in genome size or gene number is not a good criterion for developmental and morphological complexity. For example, the genome of the bryophyte Physcomitrella patens is about 480 MB and possesses approximately 35,938 genes, while Arabidopsis thaliana, with much higher morphological complexity, has a smaller genome (∼135 MB) containing about 27,235 genes (Rensing et al., 2008). To understand the evolution and diversification of morphological complexity two questions should be addressed. First, which factors were the major genetic resources underpinning morphological complexity? And secondly, how does morphological diversity evolve? We know that all cells of a complex multicellular organism contain the same genome, however, their organs are composed of a variety of cell types that differ dramatically in both structure and function. The distinctiveness of a given cell type is determined by controlled transcriptional activity of distinct sets of genes in a cell lineage. Complexity is a term with different definitions (Carroll, 2001). However, the number of cell types is broadly considered an indicator for morphological/organismal complexity (Carroll, 2001; Levine and Tjian, 2003; Chen et al., 2014). This suggests that complex transcriptional regulation plays a key role in the evolution of complexity in eukaryotes. This is in agreement with a higher proportion of transcription factors in more complex organisms with high evolutionary distances within each kingdom (Table 1). Moreover, the rate of expansion of transcriptional regulators is faster than linearly for every gene added to the genome (Levine and Tjian, 2003; Charoensawan et al., 2010a; Rensing, 2014). However, in many cases evolution is not necessarily accompanied by higher morphological complexity or with an increased number of transcriptional regulators (Wolf and Koonin, 2013).
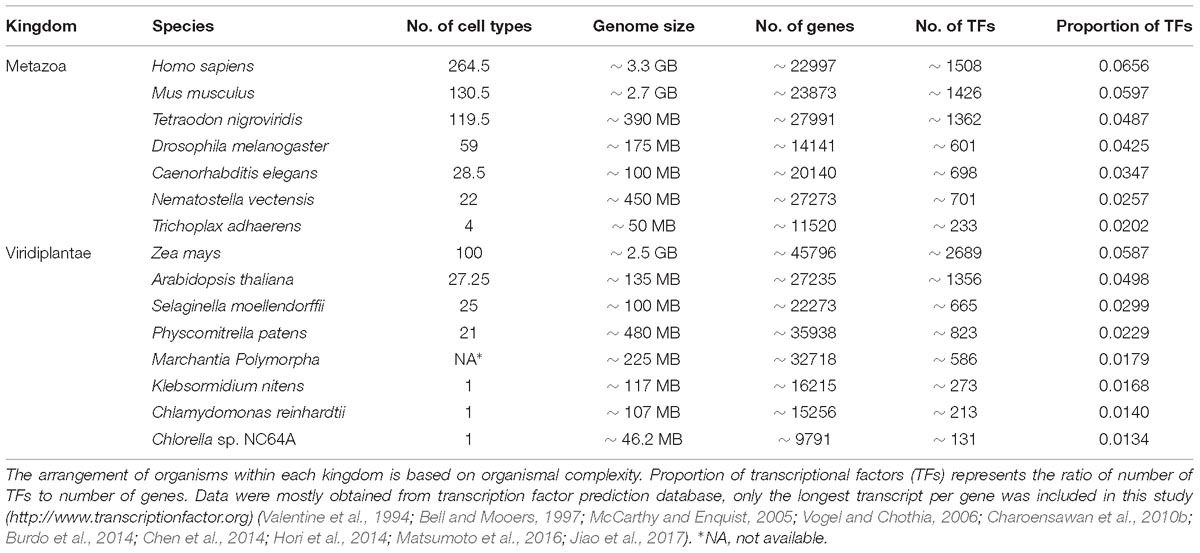
Table 1. Organismal/morphological complexity correlates with the proportion of transcriptional regulators within each kingdom when the evolutionary distance between organisms is high.
Gene Duplication - a Major Driver in the Evolution of Morphological Complexity
Genomic studies have revealed notable increases in the number of genes, intergenic regions, intragenic regions (introns), and transposons from prokaryotes to multicellular eukaryotes. Whole-genome and small-scale duplications are known as essential sources for the evolution of functional novelty and morphological complexity (Ohno, 1970; Lynch and Conery, 2003; Gregory, 2005; Bratlie et al., 2010). Increases in organismal complexity are repeatedly coupled to short-term large-scale increases in gene number in the history of eukaryotes (Ohno, 1970; Gu et al., 2002; McLysaght et al., 2002; Maere et al., 2005; Vanneste et al., 2014). For example, eukaryotic RNA polymerases (PolI, PolII, and PolIII) evolved due to massive gene duplications during the transition from an archaeum to a fully fledged eukaryote (Koonin, 2015). Whole-genome duplications in plants normally lead to genomic instability, alteration of gene expression and cell division abnormalities (Comai, 2005). On the other hand, the genomic plasticity of polyploids is higher than diploids and this may lead to increased tolerance of polyploidy in a broader range of environmental conditions. Recent studies suggest that challenging environmental conditions may positively enhance short-term polyploid establishment and survival (for a detailed review see, Van de Peer et al., 2017). After genome duplication, duplicated genes can have different evolutionary fates. Duplicated genes predominantly become pseudogenes/silent due to non-adaptive accumulation of deleterious mutations (non-functionalization) within a few Myr (Lynch and Conery, 2003; Maere et al., 2005). In an evolutionary study in rodents, it was shown that one copy of duplicates, which is usually the novel daughter copy, experiences a fivefold higher divergence rate within 4–12 Myr after duplication. Subsequently, the divergence rate decreases and after 40.5 Myr returns to preduplication levels (Pegueroles et al., 2013). A subset of duplicates may stay active by different mechanisms. For example, an increase in the expression of duplicates can be beneficial (gene dosage) or both duplicates can be essential to keep the ancestral function (subfunctionalization). In addition, duplicates can be important to maintain the stoichiometric balance (gene balance) or to prevent interference between the products of paralogs (paralog interference). Duplication can also lead to the evolution of novel functions. Neofunctionalization arises after gene duplication resulting in one gene copy keeping the ancestral function and the second copy becoming fixed by positive selection. In addition, functional novelty can also arise due to escape from adaptive conflict (EAC). In this case, the evolution of a novel function in the ancestral copy before duplication has reduced the ability of the gene to carry out the original function and after duplication each copy can freely optimize the ancestral or the novel function (Ohno, 1970; Lynch, 2000; He and Zhang, 2005; Conant and Wolfe, 2008; Des Marais and Rausher, 2008). It has been shown that the decay rates of paralogs derived from small-scale duplications are considerably higher than those derived from large-scale duplications (Maere et al., 2005; Freeling, 2009). Furthermore, after whole genome duplication the retention rate of different genes is not similar. For example, genes that are involved in transcriptional regulation, signal transduction, and development have a higher retention rate than other functional categories (Blanc, 2004; Seoighe and Gehring, 2004; Maere et al., 2005). On the other hand following a large-scale duplication and emergence of polyploid organisms, most of the duplicates are deleted or non-functionalized over time and genome size reduction is accompanied by extensive genome reorganization. This process is called diploidization and leads to the conversion of polyploids to diploids over a period of several Myr and species that emerge by diploidization following polyploidization are called palaeopolyploids. All extant angiosperms are indeed palaeopolyploid (Olsen and Wendel, 2013; Dodsworth et al., 2016). It is also important to consider that whole-genome duplication in animals in general is less common than in plants (Hallinan and Lindberg, 2011; Nossa et al., 2014; Clarke et al., 2015; Schwager et al., 2017; Li et al., 2018).
Alteration of Gene Expression Patterns and Morphological Complexity
Pioneering studies in molecular evolutionary biology revealed that there is relatively little protein divergence among mammalians such as chimps and humans, although their phenotype and behavior are very different (Britten and Davidson, 1971; Wilson et al., 1974; King and Wilson, 1975). These studies led to the proposal that the evolution of complexity occurred more by altering gene regulation than by changing protein sequences. In agreement with this proposal, later studies showed that many homologous proteins, despite long term (∼ 500 Myr) independent evolution in different lineages, are often functionally equivalent (Grens et al., 1995; Halder et al., 1995). Furthermore, vital roles are attributed to conserved protein sequences and their mutations are deleterious or lead to pleiotropic effects and are thus under purifying selection (Grens et al., 1995; Halder et al., 1995; Hoekstra and Coyne, 2007). However, alteration of their expression level or pattern is usually non-deleterious and this is mostly due to the modular nature of cis-regulatory elements (Hoekstra and Coyne, 2007).
The morphological complexity of multicellular organisms relies on spatio-temporal patterns of developmentally important regulatory factors (Spitz and Furlong, 2012). The precise expression patterns of master developmental regulators are mostly governed by enhancers/cis-regulatory modules that integrate signaling and tissue-specific inputs to specify times and locations of gene expression (Shen et al., 2012). Enhancers are short DNA sequences that contain multiple sites for sequence-specific transcription factors (Shlyueva et al., 2014). In prokaryotes, enhancer-dependent gene regulation is less common and the regulatory regions of prokaryotes and unicellular eukaryotes are usually composed of short sequences in the vicinity of the core promoter (Gralla, 1996; Wyrick and Young, 2002). However, enhancers in multicellular eukaryotes are scattered across the genome and found upstream and downstream of genes. The birth of enhancers is mediated by various mechanisms during evolution. Duplication and rapid/subsequent diversification of enhancers is an important source for the genesis of new enhancers (Goode et al., 2011; Vlad et al., 2014). New enhancer sequences can emerge from non-regulatory sequences or older enhancer elements via random genetic drift or adaptive selection (Frankel et al., 2011; Rebeiz et al., 2011; Duque and Sinha, 2015; Villar et al., 2015). Transposable elements (TEs) are also important material for tinkering with eukaryotic transcriptional regulatory systems (Jordan et al., 2003; Cao et al., 2016). Enhancers can control genes that are located far away; therefore, one gene can be regulated by multiple distal and close enhancers with different spatiotemporal activities. Furthermore, one enhancer may regulate the activity of multiple genes. These features facilitate a vast combinatorial complexity of transcriptional regulation with a relatively limited set of genes (Long et al., 2016).
It is important to consider that alteration in heritable gene expression patterns is due either to diversification of cis-regulatory elements or trans-regulatory factors (transcription regulators and non-coding RNAs). Recent studies have quantified the relative contribution of cis- and trans-regulatory factors to the evolution of gene expression, which as shown above is a key player in the evolution of morphological complexity. These studies suggested that trans-regulatory factors have a higher contribution to gene expression alteration than cis-regulatory factors within a given species. However, as sequence divergence or evolutionary distance increase, cis-regulatory differences become the dominant contributor in gene expression alteration. This relative contribution of cis-regulatory elements and trans-regulatory factors in the regulation of gene expression varies amongst taxa (Metzger et al., 2017; Osada et al., 2017).
Many studies have shown evolutionary changes through diversification of regulatory elements or protein-coding sequences (Stern, 1998; Arnaud et al., 2011; Vlad et al., 2014; Kusters et al., 2015; Sicard et al., 2016; Vuolo et al., 2016; Jiang and Rausher, 2018). Reduced complexity (RCO) evolution is an interesting example in plants that shows how gene duplication and subsequent diversification in regulatory elements and coding sequences played a key role in the evolution of morphological diversity within the Brassicaceae family. Vlad et al. (2014) discovered that a tandem duplication of the LATE MERISTEM IDENTITY 1 (LMI1) gene has given rise to two new copies in Cardamine. One of the copies has become a pseudogene owing to accumulation of deleterious mutations, whereas another copy located immediately downstream of the LMI1gene locus is active. LMI1 is expressed in the margins of leaflet, stipules, and flowers. In contrast to LMI1, the novel active copy RCO is essential for the formation of the complex leaves in C. hirsuta. It is expressed at the base of the leaflet and promotes leaflet formation through local growth repression. RCO was lost in the lineage that gave rise to A. thaliana leading to simplification of the leaves in this species. When the RCO promoter drives the expression of the LMI1 gene at the base of leaflets, the LMI1 gene acts similar to RCO and represses the growth at the flank of developing leaflets. This demonstrated that neofunctionalization has occurred due to diversification of regulatory elements. Later studies uncovered that indeed RCO enhancer evolution likely coevolved with a single amino acid change. This change led to the reduction of RCO protein stability, which is required for minimizing the pleiotropic effects of the RCO enhancer (Vuolo et al., 2016). The evolution of domesticated maize (Zea mays ssp. mays) from its wild relative teosinte (Z. mays, ssp. Parviglumis) is also an excellent example of morphological evolution through directional selection during domestication. Since the crop plant maize and teosinte are morphologically very different, taxonomists once placed them in separate genera (Doebley et al., 1997). However, later studies demonstrated that these plants are close relatives and expression alteration of a few transcription factors led to great morphological divergence and played a substantial role in the emergence of cultivated maize from teosinte. Diversification of regulatory elements of teosinte branch1 (tb1) and barren stalk1 (ba1), which encode bHLH transcription factors, had a great impact on positioning of the male inflorescence and conversion of lateral branches of teosinte into the maize ear (Doebley et al., 1997; Gallavotti et al., 2004; Clark et al., 2006). In teosinte, kernels are tightly sealed in a stony casing, while the kernels of crop maize are naked and could readily be consumed by animals or humans. Surprisingly, just a single amino acid change in the SBP-box transcription factor teosinte glume architecture1 (tga1) was the cause of the liberation of kernels from the hardened cupulate fruitcases (Wang et al., 2005, 2015).
The Structural Evolution of the Nucleosome as a Prerequisite Step for Morphological Complexity
To the best of our knowledge, all domains of life rely on DNA to store and inherit genetic information. Factors that alter the conformation of DNA to make it fit inside the cell/nucleus are present in all kingdoms of life and have the potential to influence transcription. Bacteria lack histones and contain nucleoid-associated proteins (NAPs) that are major DNA-binding factors facilitating chromosomal domain formation and organization (Figure 1; Luijsterburg et al., 2008). In bacterial cells, there is no inherent barrier for RNA polymerases to gain access to the DNA (Struhl, 1999; Dillon and Dorman, 2010). Archaeal cells also have circular DNA, as in bacteria. The phylum Crenarchaeota in the archaea domain generally lack histone proteins and their chromosome organization relies on Alba proteins, which are NAPs. However, the phylum Euryarchaeota in archaea mainly contain histone proteins that lack flexible tails at their N-terminus (Williams and Embley, 2014; Peeters et al., 2015). Methanopyrus kandleri and Halobacterium NRC1 in Euryarchaeota contain unusual “doublet histones” that have evolved through an end-to-end duplication of the histone fold. The ancestral gene encoding a doublet histone was split and diverged into H3 and H4 to form H3–H4 tetramers. H2A and H2B likely evolved later through a second specialization of a doublet as well (Ng et al., 2000; Malik and Henikoff, 2003). Eukaryotic histones are derived from a common ancestor shared with Archaea. Archaeal chromatin-like structure is apparently important for DNA protection from thermal denaturation (Reeve, 2003; Sandman and Reeve, 2005). Eukaryotic cells contain very stable, compact, and at the same time very dynamic chromatin. Nucleosomes are the fundamental units of chromatin that consist of ∼147 base pairs of DNA wrapped around a core of eight histone proteins comprising two copies of histone H3, H4, H2A, and H2B. The tails of core histones protrude from the nucleosome core particle and many residues in these tails can be post-translationally modified, influencing all DNA-based processes, including transcription (Venkatesh and Workman, 2015). Chromatin also contains linker DNAs (∼10–90 bp) that connect nucleosomes and interact with histone H1 (Han and Grunstein, 1988; Szerlong and Hansen, 2011; Zhou et al., 2013). In higher eukaryotes, H1 histones have three domains, a highly conserved central globular domain, an unstructured short N-terminal domain, and a long basic C-terminal domain (Ramakrishnan et al., 1993). Linker histone-like proteins are found in eubacteria, which are likely the provenance of H1 histones (Kasinsky et al., 2001). These proteins are similar to the C-terminal domain of H1 histones in higher eukaryotes, however, they have no globular domain. Linker histones are diverse and perform various roles in processes such as chromatin organization, genome stabilization, transcriptional regulation, and embryogenesis (Hergeth and Schneider, 2015; Kotliński et al., 2016; Bayona-Feliu et al., 2017). In contrast to prokaryotes, the compact structure of chromatin in eukaryotes generated an inherent barrier for DNA-based processes. This was one of the key prerequisite steps in the evolutionary trajectory of complex multicellular organisms.
Evolution of Chromatin Remodelers and Modifiers
The compact structure of chromatin in eukaryotes prevents free access of transcription factors to cis-regulatory DNA elements. In addition to transcription factors, proteins involved in replication and repair must be able to access DNA. To tackle this barrier, it was necessary for early eukaryotes to evolve and expand classes of chromatin modifiers and remodelers to facilitate access to DNA (Figure 1). Due to the possession of mitochondria, Eukaryotes had more available energy to encode a higher level of proteins. This together with genome expansion likely generated evolutionary pressure for co-evolution of high density chromatin packaging and chromatin-modifying factors in early eukaryotes (Flaus et al., 2006; Lane and Martin, 2010; Garg and Martin, 2016; Koster et al., 2015; Martin and Sousa, 2016). Chromatin modifiers and remodelers further expanded and diversified in eukaryotes. This led to the establishment of distinct classes of chromatin-modifying factors with unique functional complexes that facilitate binding of transcription factors to cis-regulatory DNA elements in a cell-type-specific manner in higher eukaryotes (Gentry and Hennig, 2014; Sarnowska et al., 2016; Zhou et al., 2016). The major chromatin-modifying factors are DNA methyltransferases (DNMTs), histone deacetylases (HDACs), histone acetyltransferases (HATs), histone methyltransferases (HMTs), histone demethylases (HDMs), and chromatin remodelers.
DNA Methyltransferases
In prokaryotes, as a part of the restriction-modification (RM) systems DNA methylases cooperate with restriction enzymes to protect the genome against foreign DNA. Prokaryotic DNA methylases evolved from ancient RNA-modifying enzymes and are the provenance of eukaryotic DNA methylases. In eukaryotes, multiple independent duplications, losses, and divergences led to the emergence of distinct types of DNA methylases, which are involved in a range of activities, including gene and transposon silencing, imprinting, transcriptional activation, and post-transcriptional regulation (Law and Jacobsen, 2010; Blow et al., 2016; Lyko, 2018). In Arabidopsis, de novo cytosine methylation is catalyzed by DOMAINS REARRANGED METHYLTRANSFERASE2 (DRM2) and the DNA methylation pattern is maintained by METHYLTRANSFERASE 1 (MET1) and CHROMOMETHYLASE 3 (CMT3), as well as DRM2. Interestingly, DNA methylation could create a basis for morphological diversity by regulating DNA binding affinity of transcription factors. For example, epigenetic mutation of the Lcyc gene inhibits its expression and modifies the symmetry of the flowers from bilateral to radial in Linaria vulgaris (Cubas et al., 1999). DNA hyper-methylation in the promoter region of a SBP-box transcription factor, COLORLESS NON-RIPENING (Cnr), leads to colorless and abnormal ripening of fruits in tomato without changes in nucleotide sequence (Manning et al., 2006). DNA methylation in eukaryotes can also be guided by non-coding RNAs. Small RNA-directed DNA methylation (RdDM) pathways play a key role in maintenance of genome stability and developmental regulation (Castel and Martienssen, 2013; Matzke and Mosher, 2014). The canonical RdDM model suggests that the target loci are transcribed by Pol IV and the primary transcripts are converted to dsRNAs by RDR2. These dsRNAs are processed into mature 24nt repeat-associated siRNA (ra-siRNA) by DCL3, methylated by HEN1, and loaded into RISC-like RITS (RNA-induced transcriptional silencing) complexes containing AGO4 and Pol V, which scan the genomic DNA to drive DNA methylation at target loci carrying complementary sequences (Cao et al., 2003; Zilberman et al., 2003; Wierzbicki et al., 2008; Law and Jacobsen, 2010).
The MORC ATPase family is an evolutionary conserved protein family that is prevalent in both prokaryotes and eukaryotes (Iyer et al., 2008). However, in eukaryotes, especially in the plant kingdom it greatly expanded through gene duplication (Dong et al., 2018). Using contextual information, Iyer et al. (2008) suggested that MORC proteins may play a substantial role in the bacterial RM system. MORC proteins are required for meiotic division in animals and pathogen-associated molecular pattern (PAMP)-triggered immunity in plants (Watson et al., 1998; Kang et al., 2012; Liu et al., 2016; Dong et al., 2018). The Arabidopsis genome contains seven MORC genes (AtMORC1-7). It has been demonstrated that MORC1, MORC2, and MORC6 are involved in gene silencing and transposon suppression without changing genome-wide DNA methylation patterns (Moissiard et al., 2012, 2014; Bordiya et al., 2016). However, MORC-mediated transcriptional silencing depends, at least in part, on the interaction with the RdDM components (Lorković et al., 2012; Brabbs et al., 2013; Liu et al., 2016).
Histone Modifiers
Post-translational modification of histones also plays a key role in the regulation of chromatin dynamics. Transcriptionally active chromatins usually contain trimethylated histone H3K4 and highly acetylated histone H3 and H4. In contrast, transcriptionally silent chromatins are enriched in the methylation of lysine 9 and/or 27 of histone H3 (Hebbes and Thorne, 1988; Jenuwein and Allis, 2001; Fischle et al., 2003). Histone methylation is catalyzed by three distinct protein families; the SET domain-containing protein family, the non-SET domain proteins Dot1/Dot1L, and the PRMT1 family. In contrast to histone acetyl/ deacetyltransferases and based on early phylogenetic analysis, it was concluded that the SET domain-containing methyltransferases evolved in the eukaryotic lineage and the bacterial SET domain was the result of horizontal gene transfer from a eukaryotic host (Stephens et al., 1998; Iyer et al., 2003). However, a recent phylogenetic study using an expanded collection of prokaryotic genomes showed that the SET domain is found in free-living bacteria as well as in pathogenic bacteria. Interestingly, these enzymes are involved in the synthesis of secondary metabolites, such as antibiotics in bacteria (Iyer et al., 2011; Alvarez-Venegas, 2014). Thus, the SET domain is also an ancient catalytic domain. The SET-domain proteins are grouped into seven families (Ng et al., 2007) and are members of different complexes with broad functions. For example, polycomb group proteins (PcG) that act as chromatin-based transcriptional repressors, generally form two multimeric complexes, the polycomb repressive complexes 1 (PRC1) and PRC2. The histone methyltransferase Enhancer of Zeste [E(z)], which is the catalytic subunit of PRC2, catalyzes the trimethylation of histone H3 lysine 27 (H3K27me3) via its SET domain (Goodrich et al., 1997; Cao et al., 2002; Czermin et al., 2002). Arabidopsis consists of three H3K27me3 HMTs, CURLY LEAF (CLF), SWINGER (SWN), and MEDEA (MEA). The loss of function mutation of CLF and SWN that act, at least in part, redundantly leads to development of embryo- or callus-like structures in Arabidopsis (Goodrich et al., 1997; Grossniklaus et al., 1998; Chanvivattana et al., 2004). The prior positioning of H3K27me3 by the PRC2 complex is normally required for the recruitment of PRC1 and subsequent monoubiquitylation of histone H2A on lysine 119 (H2AK119ub1). However, PRC2 recruitment through PRC1-dependent H2A119ub1 has also been reported (Landeira et al., 2010; Blackledge et al., 2014). In contrast to PcG, the TRithoraX Group (trxG) proteins activate transcription by catalyzing methylation of histone H3 on lysine 4 (H3K4) via their SET domain. PcG and trxG proteins are essential in establishment and maintenance of cell identity and organ development in higher eukaryotes through permanent/dynamic transcriptional regulation of developmentally important genes (Alvarez-Venegas, 2010; Schuettengruber et al., 2017). Thus, they play a substantial role in morphological complexity. Phylogenetic analysis of the SET-domain proteins suggests that four families of the SET-domain proteins were present before the divergence of plants, metazoans, and fungi and later highly expanded and diverged in each kingdom mostly due to large-scale duplication (Zhang and Ma, 2012).
Histone demethylases are classified into two distinct families, the KDM1/LSD1 and JmjC domain-containing proteins. The catalytic domain of KDM1 genes is the AOD domain. The AOD domain is found in prokaryotes suggesting that prokaryotes are the provenance of eukaryotic KMD1-type HDMs. The eubacterial Cupin genes are likely the ancestor of all JmjC domain-containing proteins. Whole-genome duplication was likely the major driving force for the expansion and diversification of JmjC domain-containing proteins in complex multicellular eukaryotes (Qian et al., 2015). In contrast to eubacterial proteins that contain only the JmjC domain, most of the eukaryotic proteins contain complex architectural domains (Zhou and Ma, 2008; Qian et al., 2015).
Histone acetyltransferases and deacetylases both contain ancient catalytic domains, and members of the GCN5-related N-acetyltransferase (GNAT) superfamily and the histone deacetylase superfamily are found in all kingdoms of life. However, these enzymes were greatly expanded and diversified in multicellular eukaryotes (Leipe and Landsman, 1997; Gregoretti et al., 2004; Boycheva et al., 2014; Marinov and Lynch, 2016). HATs are grouped into two classes according to their intracellular localization, i.e., into A-type and B-type. B-type HATs are localized in the cytoplasm and catalyze acetylation of free histones. However, A-type HATs are localized in the nucleus and catalyze acetylation of the nucleosome core histones. In Arabidopsis, A-type HATs are classified into four groups based on their sequence and structural similarities (Eberharter et al., 1996; Pandey et al., 2002): (1) Gcn5-related N-acetyltransferases (GNATs), (2) The MYST-related HATs, (3) cAMP-responsive element-binding protein (CBP), and (4) TATA-binding protein associated factor (TAFII250). The HDACs are also classified into four groups: (1) Reduced Potassium Dependency 3 (RDP3), (2) Histone DeAcetylase 1 (HDA1), (3) Silent Information Regulator 2 (SIR2), and (4) Histone Deacetylase 2 (HD2) (Shen et al., 2015).
Chromatin Remodelers
Transcription-relevant chromatin remodeling ATPases are classified into four distinct families (SWI/SNF, ISWI/SNF2L, CHD/Mi-2, and INO80/SWR1) that are functionally and genetically non-redundant based on their structure. The catalytic/ATPase domain of remodelers consists of two covalently linked RecA-like lobes. Chromatin remodeling complexes hydrolyze ATP and convert the chemical energy resulting from hydrolysis into mechanical motion, including sliding of the nucleosomes along the DNA, disassembling the nucleosome and exchanging histone variants (Flaus et al., 2006; Bannister and Kouzarides, 2011; Zhou et al., 2016). Phylogenetic studies have suggested that eukaryotic chromatin remodeling ATPases have likely evolved from the ancestral Snf2-like proteins in bacteria after the innovation of chromatin-binding domains in early eukaryotes (Flaus et al., 2006; Koster et al., 2015). The Arabidopsis orthologs of yeast SWI2/SNF2 are BRM, SYD, CHR12/MINU1, and CHR23/MINU2. Structurally, BRM is the closest ortholog to yeast SWI2/SNF2. It contains a helicase/SANT-associated (HAS) domain upstream of ATPase that is a binding platform for nuclear actin-related proteins (Szerlong et al., 2008) and a C-terminal bromodomain, which is capable of binding to acetylated lysine (Dhalluin et al., 1999; Jacobson et al., 2000). In A. thaliana, SWI2/SNF2 proteins assemble into different large complexes and control various activities such as plant growth and development (Sarnowska et al., 2016). The ISWI complexes were initially isolated from D. melanogaster. In A. thaliana, CHROMATIN REMODELING11 (CHR11) and CHR17 are orthologs of ISWI in D. melanogaster. They contain an ATPase domain at their N-terminus and HAND, SANT, and SLIDE domains at their C- terminus. AtISWI proteins, which are functionally redundant, form different complexes with the AtDDT (DNA-binding homeobox and different transcription factors)-domain proteins and control multiple developmental processes (Li et al., 2014). Proteins from the CHD/Mi-2 family contain two tandemly arranged chromodomains at the N-terminus that are able to interact with methylated histones and/or DNA. The CHD/Mi-2 family evolved soon after the onset of the eukaryotic lineage and further expanded in higher eukaryotes (Hargreaves and Crabtree, 2011; Gentry and Hennig, 2014; Koster et al., 2015). Saccharomyces cerevisiae, A. thaliana, and humans consist of one, four, and nine CHD genes, respectively (Koster et al., 2015). CHD remodelers positively or negatively control transcription and are also involved in mRNA processing (Murawska and Brehm, 2011; Hu et al., 2014). The chromatin-remodeling complexes of the INO80 group are INO80 and SWR1 in yeast. A single INO80 and SWR1/PIE1 (PHOTOPERIOD INDEPENDENT EARLY FLOWERING 1) are present in Arabidopsis. The INO80/SWR1 complexes similarly, to other chromatin-remodeling complexes work as transcriptional regulators. In addition, they are implicated in the DNA-repair system and are required for DNA recombination (Noh and Amasino, 2003; Fritsch et al., 2004; Gerhold and Gasser, 2014).
Symbiosis and Multicellularity
It is well documented that mitochondria and chloroplasts of eukaryotic cells, which are descended from α-proteobacteria-like and cyanobacteria-like prokaryotes, respectively, arose through endosymbiosis (Weeden, 1981; Gray et al., 1999). Thus, endosymbiosis played a crucial role in the evolution of cellular complexity. Multicellular organisms harbor a vast diversity of microbes, comprising fungi, bacteria, protists, and viruses, collectively called microbiota (Almario et al., 2017; Durán et al., 2018). Molecular clock estimates of fungal phylogeny suggest that Ascomycota, Basidiomycota, and Glomales, which are major taxonomic groups of terrestrial fungi, were present around 600 myr ago (Redecker et al., 2000) and fossilized spores and fungal hyphae that are very similar to extant arbuscular mycorrhizal fungi (AMF) with the age of 460–480 myr support molecular estimates (Selosse and Le Tacon, 1998; Redecker et al., 2000; Heckman et al., 2001). Considering that early land plants colonized poorly developed soils and did not have true roots, the establishment of AMF symbiosis supplying nutrients, water, and enhancing tolerance to biotic and abiotic stresses was a key event in the terrestrialization process (Redecker et al., 2000; Heckman et al., 2001; Rausch et al., 2001; Kenrick and Strullu-Derrien, 2014; Almario et al., 2017; Xue et al., 2018). In addition to fungi, bacterial micribiota are a substantial part of diverse assemblages of symbiotic microorganisms and are critical for plant survival (Durán et al., 2018). Surprisingly, bacterial symbiosis is required for cell division and morphogenesis in Ulva mutabilis, which is a green macroalgae and an important primary producer in coastal ecosystems (Wichard, 2015). Taken together, these lines of evidences suggest that symbiosis played an important role in the transition from water to land and the evolution of multicellularity. Organism-associated microbes had a great impact on phenotypic extension and host evolution. In evolutionary studies, considering the host and its associated microbiota as a biological entity, the holobiont could be key for a better understanding of the evolution of multicellular organisms (Shropshire and Bordenstein, 2016; Almario et al., 2017; Hassani et al., 2018; Haag, 2018).
Conclusion
In early eukaryotes, due to an increase of genome size, high density packaging of the DNA molecules into the confined space of the nucleus and simultaneous evolution of novel factors controlling the accessibility of DNA was a necessity to ensure all DNA-based processes, including transcriptional regulation. Increased genome size together with higher available energy per gene likely led to the evolution of chromatin structure and chromatin-modifying factors in early eukaryotes (Flaus et al., 2006; Lane and Martin, 2010; Koster et al., 2015; Garg and Martin, 2016; Martin and Sousa, 2016). Although, the origins of catalytic subunits of chromatin remodelers and modifiers can be traced back in prokaryotes, these catalytic subunits and their interacting partners continuously expanded and highly diversified and were finally coopted, while prokaryotes lack chromatin-remodeling and –modifying complexes. The innovation of these complexes was a key prerequisite step in the evolutionary trajectory of complex multicellular eukaryotes. Both symbiotic microbiota and epigenetics are critical for adaptation to environmental conditions, plant survival, and their evolution. However, our knowledge concerning how diversification and expansion of chromatin-related factors and recruitment of symbiotic microbiota led to the complexity of living organisms is low. In addition, the functional links between symbiotic microbiota and epigenetics is largely unknown. In future work, a combination of approaches in ecophysiology, plant-microbe interaction, phylogenomics, molecular biology, systems biology, cell biology, and biochemistry studies on a wide range of unicellular and multicelluar organisms will shed more light on the interrelationship of chromatin-related factors and microbiota community structure and their contribution to the evolution of complex multicellular organisms and the holobiont.
Author Contributions
MH wrote the manuscript. CK and MB critically revised and approved the manuscript for publication.
Funding
Funding of the project was provided by the German Federal Ministry of Education and Research (BMBF, Project No. 031B0200A to MB and MH) within the frame of RECONSTRUCT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Timothy Jobe for the careful correction of the manuscript.
References
Almario, J., Jeena, G., Wunder, J., Langen, G., Zuccaro, A., Coupland, G., et al. (2017). Root-associated fungal microbiota of nonmycorrhizal Arabis alpina and its contribution to plant phosphorus nutrition. Proc. Natl. Acad. Sci. 114:E9403. doi: 10.1073/pnas.1710455114
Alvarez-Venegas, R. (2010). Regulation by polycomb and trithorax group proteins in Arabidopsis. Arab. B 8:e0128. doi: 10.1199/tab.0128
Alvarez-Venegas, R. (2014). Bacterial set domain proteins and their role in eukaryotic chromatin modification. Front. Genet. 5:65. doi: 10.3389/fgene.2014.00065
Arnaud, N., Lawrenson, T., Østergaard, L., and Sablowski, R. (2011). The same regulatory point mutation changed seed-dispersal structures in evolution and domestication. Curr. Biol. 21, 1215–1219. doi: 10.1016/j.cub.2011.06.008
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Bayona-Feliu, A., Casas-Lamesa, A., Reina, O., Bernués, J., and Azorín, F. (2017). Linker histone H1 prevents R-loop accumulation and genome instability in heterochromatin. Nat. Commun. 8:283. doi: 10.1038/s41467-017-00338-5
Bell, G., and Mooers, A. O. (1997). Size and complexity among multicellular organisms. Biol. J. Linn. Soc. 60, 345–363. doi: 10.1006/bijl.1996.0108
Blackledge, N. P., Farcas, A. M., Kondo, T., King, H. W., McGouran, J. F., Hanssen, L. L. P., et al. (2014). Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157, 1445–1459. doi: 10.1016/j.cell.2014.05.004
Blanc, G. (2004). Functional divergence of duplicated genes formed by polyploidy during arabidopsis evolution. Plant Cell 16, 1679–1691. doi: 10.1105/tpc.021410
Blow, M. J., Clark, T. A., Daum, C. G., Deutschbauer, A. M., Fomenkov, A., Fries, R., et al. (2016). The epigenomic landscape of prokaryotes. PLoS Genet. 12:e1005854. doi: 10.1371/journal.pgen.1005854
Bordiya, Y., Zheng, Y., Nam, J.-C., Bonnard, A. C., Choi, H. W., Lee, B.-K., et al. (2016). Pathogen infection and MORC proteins affect chromatin accessibility of transposable elements and expression of their proximal genes in arabidopsis. Mol. Plant Microbe Interact. 29, 674–687. doi: 10.1094/MPMI-01-16-0023-R
Boycheva, I., Vassileva, V., and Iantcheva, A. (2014). Histone acetyltransferases in plant development and plasticity. Curr. Genom. 15, 28–37. doi: 10.2174/138920291501140306112742
Brabbs, T. R., He, Z., Hogg, K., Kamenski, A., Li, Y., Paszkiewicz, K. H., et al. (2013). The stochastic silencing phenotype of Arabidopsis morc6 mutants reveals a role in efficient RNA -directed DNA methylation. Plant J. 75, 836–846. doi: 10.1111/tpj.12246
Bratlie, M. S., Johansen, J., Sherman, B. T., Huang, D. W., Lempicki, R. A., and Drabløs, F. (2010). Gene duplications in prokaryotes can be associated with environmental adaptation. BMC Genomics 11:588. doi: 10.1186/1471-2164-11-588
Britten, R. J., and Davidson, E. H. (1971). Repetitive and non-repetitive DNA sequences and a speculation on the origins of evolutionary novelty. Q. Rev. Biol. 46, 111–138. doi: 10.2307/2822073
Burdo, B., Gray, J., Goetting-Minesky, M. P., Wittler, B., Hunt, M., Li, T., et al. (2014). The Maize TFome - development of a transcription factor open reading frame collection for functional genomics. Plant J. 80, 356–366. doi: 10.1111/tpj.12623
Cao, C., Xu, J., Zheng, G., and Zhu, X. G. (2016). Evidence for the role of transposons in the recruitment of cis-regulatory motifs during the evolution of C4photosynthesis. BMC Genomics 17:201. doi: 10.1186/s12864-016-2519-3
Cao, R., Wang, L., Wang, H., Xia, L., Erdjument-Bromage, H., Tempst, P., et al. (2002). Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043. doi: 10.1126/science.1076997
Cao, X., Aufsatz, W., Zilberman, D., Mette, M. F., Huang, M. S., Matzke, M., et al. (2003). Role of the DRM and CMT3 methyltransferases in RNA-Directed DNA methylation. Curr. Biol. 13, 2212–2217. doi: 10.1016/j.cub.2003.11.052
Carroll, S. B. (2001). Chance and necessity: the evolution of morphological complexity and diversity. Nature 409, 1102–1109. doi: 10.1038/35059227
Castel, S. E., and Martienssen, R. A. (2013). RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet. 14, 100–112. doi: 10.1038/nrg3355
Chanvivattana, Y., Bishopp, A., Schubert, D., Stock, C., Moon, Y. H., and Sung, Z. R. (2004). Interaction of Polycomb-group proteins controlling flowering in Arabidopsis. Development 131, 5263–5276. doi: 10.1242/dev.01400
Charoensawan, V., Wilson, D., and Teichmann, S. A. (2010a). Genomic repertoires of DNA-binding transcription factors across the tree of life. Nucleic Acids Res. 38, 7364–7377. doi: 10.1093/nar/gkq617
Charoensawan, V., Wilson, D., and Teichmann, S. A. (2010b). Lineage-specific expansion of DNA-binding transcription factor families. Trends Genet. 26, 388–393. doi: 10.1016/j.tig.2010.06.004
Chen, L., Bush, S. J., Tovar-Corona, J. M., Castillo-Morales, A., and Urrutia, A. O. (2014). Correcting for differential transcript coverage reveals a strong relationship between alternative splicing and organism complexity. Mol. Biol. Evol. 31, 1402–1413. doi: 10.1093/molbev/msu083
Clark, R. M., Wagler, T. N., Quijada, P., and Doebley, J. (2006). A distant upstream enhancer at the maize domestication gene tb1 has pleiotropic effects on plant and inflorescent architecture. Nat. Genet. 38, 594–597. doi: 10.1038/ng1784
Clarke, T. H., Garb, J. E., Hayashi, C. Y., Arensburger, P., and Ayoub, N. A. (2015). Spider transcriptomes identify ancient large-scale gene duplication event potentially important in silk gland evolution. Genome Biol. Evol. 7, 1856–1870. doi: 10.1093/gbe/evv110
Comai, L. (2005). The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 6, 836–846. doi: 10.1038/nrg1711
Conant, G. C., and Wolfe, K. H. (2008). Turning a hobby into a job: how duplicated genes find new functions. Nat. Rev. Genet. 9, 938–950. doi: 10.1038/nrg2482
Cubas, P., Vincent, C., and Coen, E. (1999). An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401, 157–161. doi: 10.1038/43657
Czermin, B., Melfi, R., McCabe, D., Seitz, V., Imhof, A., and Pirrotta, V. (2002). Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal polycomb sites. Cell 111, 185–196. doi: 10.1016/S0092-8674(02)00975-3
Delaux, P.-M., Radhakrishnan, G. V., Jayaraman, D., Cheema, J., Malbreil, M., Volkening, J. D., et al. (2015). Algal ancestor of land plants was preadapted for symbiosis. Proc. Natl. Acad. Sci. 112, 13390–13395. doi: 10.1073/pnas.1515426112
Del-Bem, L. E. (2018). Xyloglucan evolution and the terrestrialization of green plants. New Phytol. 219, 1150–1153. doi: 10.1111/nph.15191
Des Marais, D. L., and Rausher, M. D. (2008). Escape from adaptive conflict after duplication in an anthocyanin pathway gene. Nature 454, 762–765. doi: 10.1038/nature07092
Dhalluin, C., Carlson, J. E., Zeng, L., He, C., Aggarwal, A. K., and Zhou, M. M. (1999). Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496. doi: 10.1038/20974
Dillon, S. C., and Dorman, C. J. (2010). Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 8, 185–195. doi: 10.1038/nrmicro2261
Dodsworth, S., Chase, M. W., and Leitch, A. R. (2016). Is post-polyploidization diploidization the key to the evolutionary success of angiosperms? Bot. J. Linn. Soc. 180, 1–5. doi: 10.1111/boj.12357
Doebley, J., Stec, A., and Hubbard, L. (1997). The evolution of apical dominance in maize. Nature 386, 485–488. doi: 10.1038/386485a0
Dong, W., Vannozzi, A., Chen, F., Hu, Y., Chen, Z., and Zhang, L. (2018). MORC domain definition and evolutionary analysis of the MORC gene family in green plants. Genome Biol. Evol. 10, 1730–1744. doi: 10.1093/gbe/evy136
Duque, T., and Sinha, S. (2015). What does it take to evolve an enhancer? A simulation-based study of factors influencing the emergence of combinatorial regulation. Genome Biol. Evol. 7, 1415–1431. doi: 10.1093/gbe/evv080
Durán, P., Thiergart, T., Garrido-Oter, R., Agler, M., Kemen, E., Schulze-Lefert, P., et al. (2018). Microbial interkingdom interactions in roots promote arabidopsis survival. Cell 175, 973.e14–983.e14. doi: 10.1016/j.cell.2018.10.020
Eberharter, A., Lechner, T., Goralik-Schramel, M., and Loidl, P. (1996). Purification and characterization of the cytoplasmic histone acetyltransferase B of maize embryos. FEBS Lett. 386, 75–81. doi: 10.1016/0014-5793(96)00401-2
Fischle, W., Wang, Y., and Allis, C. D. (2003). Histone and chromatin cross-talk. Curr. Opin. Cell Biol. 15, 172–183. doi: 10.1016/S0955-0674(03)00013-9
Flaus, A., Martin, D. M. A., Barton, G. J., and Owen-Hughes, T. (2006). Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 34, 2887–2905. doi: 10.1093/nar/gkl295
Frankel, N. S., Erezyilmaz, D. F., McGregor, A. P., Wang, S., Payre, F., and Stern, D. L. (2011). Morphological evolution caused by many subtle-effect substitutions in regulatory DNA. Nature 474, 598–603. doi: 10.1038/nature10200
Freeling, M. (2009). Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453. doi: 10.1146/annurev.arplant.043008.092122
Fritsch, O., Benvenuto, G., Bowler, C., Molinier, J., and Hohn, B. (2004). The INO80 protein controls homologous recombination in Arabidopsis thaliana. Mol. Cell. 16, 479–485. doi: 10.1016/j.molcel.2004.09.034
Gallavotti, A., Zhao, Q., Kyozuka, J., Meeley, R. B., Ritter, M. K., Doebley, J. F., et al. (2004). The role of barren stalk1 in the architecture of maize. Nature 432, 630–635. doi: 10.1038/nature03148
Garg, S. G., and Martin, W. F. (2016). Mitochondria, the cell cycle, and the origin of sex via a syncytial eukaryote common ancestor. Genome Biol. Evol. 8, 1950–1970. doi: 10.1093/gbe/evw136
Gentry, M., and Hennig, L. (2014). Remodelling chromatin to shape development of plants. Exp. Cell Res. 321, 40–46. doi: 10.1016/j.yexcr.2013.11.010
Gerhold, C. B., and Gasser, S. M. (2014). INO80 and SWR complexes: relating structure to function in chromatin remodeling. Trends Cell Biol. 24, 619–631. doi: 10.1016/j.tcb.2014.06.004
Goode, D. K., Callaway, H. A., Cerda, G. A., Lewis, K. E., and Elgar, G. (2011). Minor change, major difference: divergent functions of highly conserved cis-regulatory elements subsequent to whole genome duplication events. Development 138, 879–884. doi: 10.1242/dev.055996
Goodrich, J., Puangsomlee, P., Martin, M., Long, D., Meyerowitz, E. M., and Coupland, G. (1997). A polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nature 386, 44–51. doi: 10.1038/386044a0
Gralla, J. D. (1996). Activation and repression of E. coli promoters. Curr. Opin. Genet. Dev. 6, 526–530. doi: 10.1016/S0959-437X(96)80079-7
Gray, M. W., Burger, G., and Lang, B. F. (1999). Mitochondrial evolution. Science 4:a011403. doi: 10.1126/science.283.5407.1476
Gregoretti, I. V., Lee, Y. M., and Goodson, H. V. (2004). Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 338, 17–31. doi: 10.1016/j.jmb.2004.02.006
Gregory, T. R. (ed.) (2005). “Genome size evolution in animals,” in The Evolution of the Genome, ed. T. R. Gregory (Cambridge: Academic Press), 3–87.
Grens, A., Mason, E., Marsh, J. L., and Bode, H. R. (1995). Evolutionary conservation of a cell fate specification gene: the hydra achaete-scute homolog has proneural activity in Drosophila. Development 121, 4027–4035. doi: 10.1242/dev.00407
Grossniklaus, U., Vielle-Calzada, J. P., Hoeppner, M. A., and Gagliano, W. B. (1998). Maternal control of embryogenesis by MEDEA, a Polycomb group gene in Arabidopsis. Science 280, 446–450. doi: 10.1126/science.280.5362.446
Gu, X., Wang, Y., and Gu, J. (2002). Age distribution of human gene families shows significant roles of both large- and small-scale duplications in vertebrate evolution. Nat. Genet. 31, 205–209. doi: 10.1038/ng902
Haag, K. L. (2018). Holobionts and their hologenomes: evolution with mixed modes of inheritance. Genet. Mol. Biol. 41(1 Suppl. 1), 189–197. doi: 10.1590/1678-4685-gmb-2017-0070
Hajheidari, M., Koncz, C., and Eick, D. (2013). Emerging roles for RNA polymerase II CTD in Arabidopsis. Trends Plant Sci. 18, 633–643. doi: 10.1016/j.tplants.2013.07.001
Halder, G., Callaerts, P., and Gehring, W. J. (1995). New perspectives on eye evolution. Curr. Opin. Genet. Dev. 5, 602–609. doi: 10.1016/0959-437X(95)80029-8
Hallinan, N. M., and Lindberg, D. R. (2011). Comparative analysis of chromosome counts infers three paleopolyploidies in the mollusca. Genome Biol. Evol. 3, 1150–1163. doi: 10.1093/gbe/evr087
Han, M., and Grunstein, M. (1988). Nucleosome loss activates yeast downstream promoters in vivo. Cell 55, 1137–1145. doi: 10.1016/0092-8674(88)90258-9
Hargreaves, D. C., and Crabtree, G. R. (2011). ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420. doi: 10.1038/cr.2011.32
Harholt, J., Moestrup,Ø, and Ulvskov, P. (2016). Why plants were terrestrial from the beginning. Trends Plant Sci. 21, 96–101. doi: 10.1016/j.tplants.2015.11.010
Hassani, M. A., Durán, P., and Hacquard, S. (2018). Microbial interactions within the plant holobiont. Microbiome 6:58. doi: 10.1186/s40168-018-0445-0
He, X., and Zhang, J. (2005). Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics 169, 1157–1164. doi: 10.1534/genetics.104.037051
Hebbes, T. R., and Thorne, A. W. (1988). A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 7, 1395–1402. doi: 10.1007/BF00777468
Heckman, D. S., Geiser, D. M., Eidell, B. R., Stauffer, R. L., Kardos, N. L., and Hedges, S. B. (2001). Molecular evidence for the early colonization of land by fungi and plants. Science 293, 1129–1133. doi: 10.1126/science.1061457
Hergeth, S. P., and Schneider, R. (2015). The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 16, 1439–1453. doi: 10.15252/embr.201540749
Hoekstra, H. E., and Coyne, J. A. (2007). The locus of evolution: evo devo and the genetics of adaptation. Evolution 61, 995–1016. doi: 10.1111/j.1558-5646.2007.00105.x
Hori, K., Maruyama, F., Fujisawa, T., Togashi, T., Yamamoto, N., Seo, M., et al. (2014). Klebsormidium flaccidum genome reveals primary factors for plant terrestrial adaptation. Nat. Commun 5:3978. doi: 10.1038/ncomms4978
Hu, Y., Lai, Y., and Zhu, D. (2014). Transcription regulation by CHD proteins to control plant development. Front. Plant Sci. 5:223. doi: 10.3389/fpls.2014.00223
Iyer, L. M., Abhiman, S., and Aravind, L. (2008). MutL homologs in restriction-modification systems and the origin of eukaryotic MORC ATPases. Biol. Direct. 3:8. doi: 10.1186/1745-6150-3-8
Iyer, L. M., Abhiman, S., and Aravind, L. (2011). Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 101, 25–104. doi: 10.1016/B978-0-12-387685-0.00002-0
Iyer, L. M., Koonin, E. V., and Aravind, L. (2003). Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases. BMC Struct. Biol. 3:1. doi: 10.1186/1472-6807-3-1
Jacobson, R. H., Ladurner, A. G., King, D. S., and Tjian, R. (2000). Structure and function of a human TAF(II)250 double bromodomain module. Science 288, 1422–1425. doi: 10.1126/science.288.5470.1422
Jenuwein, T., and Allis, C. D. (2001). Translating the histone code. Science 293, 1074–1080. doi: 10.1126/science.1063127
Jiang, P., and Rausher, M. (2018). Two genetic changes in cis-regulatory elements caused evolution of petal spot position in Clarkia. Nat. Plants 4, 14–22. doi: 10.1038/s41477-017-0085-6
Jiao, Y., Peluso, P., Shi, J., Liang, T., Stitzer, M. C., Wang, B., et al. (2017). Improved maize reference genome with single-molecule technologies. Nature 546, 524–527. doi: 10.1038/nature22971
Jordan, I. K., Rogozin, I. B., Glazko, G. V., and Koonin, E. V. (2003). Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 19, 68–72. doi: 10.1016/S0168-9525(02)00006-9
Ju, C., Van De Poel, B., Cooper, E. D., Thierer, J. H., Gibbons, T. R., Delwiche, C. F., et al. (2015). Conservation of ethylene as a plant hormone over 450 million years of evolution. Nat. Plants 1:14004. doi: 10.1038/nplants.2014.4
Kang, H. G., Woo Choi, H., Von Einem, S., Manosalva, P., Ehlers, K., Liu, P. P., et al. (2012). CRT1 is a nuclear-translocated MORC endonuclease that participates in multiple levels of plant immunity. Nat. Commun. 3, 1297. doi: 10.1038/ncomms2279
Kasinsky, H. E., Lewis, J. D., Dacks, J. B., and Ausió, J. (2001). Origin of H1 linker histones. FASEB J. 15, 34–42. doi: 10.1096/fj.00-0237rev
Kenrick, P., and Crane, P. R. (1997). The origin and early evolution of plants on land. Nature 389, 33–39. doi: 10.1038/37918
Kenrick, P., and Strullu-Derrien, C. (2014). The origin and early evolution of roots. Plant Physiol. 166, 570–580. doi: 10.1104/pp.114.244517
King, M. C., and Wilson, A. C. (1975). Evolution at two levels in humans and chimpanzees. Science 188, 107–116. doi: 10.1126/science.1090005
Koonin, E. V. (2015). Origin of eukaryotes from within archaea, archaeal eukaryome and bursts of gene gain: eukaryogenesis just made easier? Philos. Trans. R. Soc. B Biol. Sci. 370:20140333. doi: 10.1098/rstb.2014.0333
Koster, M. J. E., Snel, B., and Timmers, H. T. M. (2015). Genesis of chromatin and transcription dynamics in the origin of species. Cell 161, 724–736. doi: 10.1016/j.cell.2015.04.033
Kotliński, M., Rutowicz, K., Kniżewski, Ł, Palusiński, A., Olêdzki, J., Fogtman, A., et al. (2016). Histone H1 variants in Arabidopsis are subject to numerous post-translational modifications, both conserved and previously unknown in histones, suggesting complex functions of H1 in plants. PLoS One 11:e0147908. doi: 10.1371/journal.pone.0147908
Kusters, E., Della Pina, S., Castel, R., Souer, E., and Koes, R. (2015). Changes in cis-regulatory elements of a key floral regulator are associated with divergence of inflorescence architectures. Development 142, 2822–2831. doi: 10.1242/dev.121905
Landeira, D., Sauer, S., Poot, R., Dvorkina, M., Mazzarella, L., Jørgensen, H. F., et al. (2010). Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA Polymerase II to developmental regulators. Nat. Cell Biol. 12, 618–624. doi: 10.1038/ncb2065
Lane, N., and Martin, W. (2010). The energetics of genome complexity. Nature 467, 929–934. doi: 10.1038/nature09486
Law, J. A., and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. doi: 10.1038/nrg2719
Leipe, D. D., and Landsman, D. (1997). Histone deacetylases, acetoin utilization proteins and acetylpolyamine amidohydrolases are members of an ancient protein superfamily. Nucleic Acids Res. 25, 3693–3697. doi: 10.1093/nar/25.18.3693
Levine, M., and Tjian, R. (2003). Transcription regulation and animal diversity. Nature 424, 147–151. doi: 10.1038/nature01763
Li, G., Liu, S., Wang, J., He, J., Huang, H., Zhang, Y., et al. (2014). ISWI proteins participate in the genome-wide nucleosome distribution in Arabidopsis. Plant J 78, 706–714. doi: 10.1111/tpj.12499
Li, Z., Tiley, G. P., Galuska, S. R., Reardon, C. R., Kidder, T. I., Rundell, R. J., et al. (2018). Multiple large-scale gene and genome duplications during the evolution of hexapods. Proc. Natl. Acad. Sci. 115, 4713–4718. doi: 10.1073/pnas.1710791115
Liu, Z. W., Zhou, J. X., Huang, H. W., Li, Y. Q., Shao, C. R., Li, L., et al. (2016). Two components of the RNA-Directed DNA methylation pathway associate with MORC6 and silence loci targeted by MORC6 in Arabidopsis. PLoS Genet. 12:e1006026. doi: 10.1371/journal.pgen.1006026
Long, H. K., Prescott, S. L., and Wysocka, J. (2016). Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167, 1170–1187. doi: 10.1016/j.cell.2016.09.018
Lorković, Z. J., Naumann, U., Matzke, A. J. M., and Matzke, M. (2012). Involvement of a GHKL ATPase in RNA-directed DNA methylation in Arabidopsis thaliana. Curr. Biol. 22, 933–938. doi: 10.1016/j.cub.2012.03.061
Luijsterburg, M. S., White, M. F., Van Driel, R., and Dame, R. T. (2008). The major architects of chromatin: architectural proteins in bacteria, archaea and eukaryotes. Crit. Rev. Biochem. Mol. Biol. 43, 393–418. doi: 10.1080/10409230802528488
Lyko, F. (2018). The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 19, 81–92. doi: 10.1038/nrg.2017.80
Lynch, M. (2000). The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155. doi: 10.1126/science.290.5494.1151
Lynch, M., and Conery, J. S. (2003). The origins of genome complexity. Science 302, 1401–1404. doi: 10.1126/science.1089370
Maere, S., De Bodt, S., Raes, J., Casneuf, T., Van Montagu, M., Kuiper, M., et al. (2005). Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. 102, 5454–5459. doi: 10.1073/pnas.0501102102
Malik, H. S., and Henikoff, S. (2003). Phylogenomics of the nucleosome. Nat. Struct. Biol. 10, 882–891. doi: 10.1038/nsb996
Manning, K., Tör, M., Poole, M., Hong, Y., Thompson, A. J., King, G. J., et al. (2006). A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952. doi: 10.1038/ng1841
Marinov, G. K., and Lynch, M. (2016). Diversity and divergence of dinoflagellate histone proteins. G3 6, 397–422. doi: 10.1534/g3.115.023275
Martin, W. F., and Sousa, F. L. (2016). Early microbial evolution: the age of anaerobes. Cold Spring Harb. Perspect. Biol. 8:a018127. doi: 10.1101/cshperspect.a018127
Matsumoto, T., Wu, J., Itoh, T., Numa, H., Antonio, B., and Sasaki, T. (2016). The Nipponbare genome and the next-generation of rice genomics research in Japan. Rice 9:33. doi: 10.1186/s12284-016-0107-4
Matzke, M. A., and Mosher, R. A. (2014). RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408. doi: 10.1038/nrg3683
McCarthy, M. C., and Enquist, B. J. (2005). Organismal size, metabolism and the evolution of complexity in metazoans. Evol. Ecol. Res. 7, 681–696.
McLysaght, A., Hokamp, K., and Wolfe, K. H. (2002). Extensive genomic duplication during early chordate evolution. Nat. Genet. 31, 200–204. doi: 10.1038/ng884
Metzger, B. P. H., Wittkopp, P. J., and Coolon, J. D. (2017). Evolutionary dynamics of regulatory changes underlying gene expression divergence among Saccharomyces species. Genome Biol. Evol. 9, 843–854. doi: 10.1093/gbe/evx035
Moissiard, G., Bischof, S., Husmann, D., Pastor, W. A., Hale, C. J., Yen, L., et al. (2014). Transcriptional gene silencing by Arabidopsis microrchidia homologues involves the formation of heteromers. Proc. Natl. Acad. Sci. 111, 7474–7479. doi: 10.1073/pnas.1406611111
Moissiard, G., Cokus, S. J., Cary, J., Feng, S., Billi, A. C., Stroud, H., et al. (2012). MORC family ATPases required for heterochromatin condensation and gene silencing. Science 336, 1448–1451. doi: 10.1126/science.1221472
Morris, J. L., Puttick, M. N., Clark, J. W., Edwards, D., Kenrick, P., Pressel, S., et al. (2018). The timescale of early land plant evolution. Proc. Natl. Acad. Sci. 115, E2274–E2283. doi: 10.1073/pnas.1719588115
Murawska, M., and Brehm, A. (2011). CHD chromatin remodelers and the transcription cycle. Transcription 2, 244–253. doi: 10.4161/trns.2.6.17840
Ng, D. W. K., Wang, T., Chandrasekharan, M. B., Aramayo, R., Kertbundit, S., and Hall, T. C. (2007). Plant SET domain-containing proteins: structure, function and regulation. Biochim. Biophys. Acta Gene Struct. Expr. 1769, 316–329. doi: 10.1016/j.bbaexp.2007.04.003
Ng, W. V., Kennedy, S. P., Mahairas, G. G., Berquist, B., Pan, M., Shukla, H. D., et al. (2000). Genome sequence of Halobacterium species NRC-1. Proc. Natl. Acad. Sci. 97, 12176–12181. doi: 10.1073/pnas.190337797
Noh, Y. S., and Amasino, R. M. (2003). PIE1, an ISWI family gene, is required for FLC activation and floral repression in Arabidopsis. Plant Cell 15, 1671–1682. doi: 10.1105/tpc.012161.lelic
Nossa, C. W., Havlak, P., Yue, J. X., Lv, J., Vincent, K. Y., Brockmann, H. J., et al. (2014). Joint assembly and genetic mapping of the Atlantic horseshoe crab genome reveals ancient whole genome duplication. Gigascience 3:9. doi: 10.1186/2047-217X-3-9
Ohno, S. (1970). Evolution by Gene Duplication. London: George Alien & Unwin Ltd.; Berlin: Springer-Verlag. doi: 10.1007/978-3-642-86659-3
Olsen, K. M., and Wendel, J. F. (2013). Crop plants as models for understanding plant adaptation and diversification. Front. Plant Sci. 4:290. doi: 10.3389/fpls.2013.00290
Osada, N., Miyagi, R., and Takahashi, A. (2017). Cis- and trans-regulatory effects on gene expression in a natural population of Drosophila melanogaster. Genetics 206, 2139–2148. doi: 10.1534/genetics.117.201459
Pandey, R., Müller, A., Napoli, C. A., Selinger, D. A., Pikaard, C. S., Richards, E. J., et al. (2002). Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res. 30, 5036–5055. doi: 10.1093/nar/gkf660
Peeters, E., Driessen, R. P. C., Werner, F., and Dame, R. T. (2015). The interplay between nucleoid organization and transcription in archaeal genomes. Nat. Rev. Microbiol. 13, 333–341. doi: 10.1038/nrmicro3467
Pegueroles, C., Laurie, S., and Albà, M. M. (2013). Accelerated evolution after gene duplication: a time-dependent process affecting just one copy. Mol. Biol. Evol. 30, 1830–1842. doi: 10.1093/molbev/mst083
Qian, S., Wang, Y., Ma, H., and Zhang, L. (2015). Expansion and functional divergence of jumonji c-containing histone demethylases: significance of duplications in ancestral angiosperms and vertebrates. Plant Physiol. 168, 1321–1337. doi: 10.1104/pp.15.00520
Ramakrishnan, V., Finch, J. T., Graziano, V., Lee, P. L., and Sweet, R. M. (1993). Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature 362, 219–223. doi: 10.1038/362219a0
Rausch, C., Daram, P., Brunner, S., Jansa, J., Laloi, M., Leggewie, G., et al. (2001). A phosphate transporter expressed in arbuscule-containing cells in potato. Nature 414, 462–470. doi: 10.1038/35106601
Rebeiz, M., Jikomes, N., Kassner, V. A., and Carroll, S. B. (2011). Evolutionary origin of a novel gene expression pattern through co-option of the latent activities of existing regulatory sequences. Proc. Natl. Acad. Sci. 108, 10036–10043. doi: 10.1073/pnas.1105937108
Redecker, D., Kodner, R., and Graham, L. E. (2000). Glomalean fungi from the Ordovician. Science 289, 1920–1921. doi: 10.1126/science.289.5486.1920
Reeve, J. N. (2003). Archaeal chromatin and transcription. Mol. Microbiol. 48, 587–598. doi: 10.1046/j.1365-2958.2003.03439.x
Rensing, S. A. (2014). Gene duplication as a driver of plant morphogenetic evolution. Curr. Opin. Plant Biol. 17, 43–48. doi: 10.1016/j.pbi.2013.11.002
Rensing, S. A., Lang, D., Zimmer, A. D., Terry, A., Salamov, A., Shapiro, H., et al. (2008). The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 319, 64–69. doi: 10.1126/science.1150646
Sandman, K., and Reeve, J. N. (2005). Archaeal chromatin proteins: different structures but common function? Curr. Opin. Microbiol. 8, 656–661. doi: 10.1016/j.mib.2005.10.007
Sarnowska, E., Gratkowska, D. M., Sacharowski, S. P., Cwiek, P., Tohge, T., Fernie, A. R., et al. (2016). The role of SWI/SNF chromatin remodeling complexes in hormone crosstalk. Trends Plant Sci. 21, 594–608. doi: 10.1016/j.tplants.2016.01.017
Schuettengruber, B., Bourbon, H. M., Di Croce, L., and Cavalli, G. (2017). Genome regulation by polycomb and trithorax: 70 years and counting. Cell 171, 34–57. doi: 10.1016/j.cell.2017.08.002
Schwager, E. E., Sharma, P. P., Clarke, T., Leite, D. J., Wierschin, T., Pechmann, M., et al. (2017). The house spider genome reveals an ancient whole-genome duplication during arachnid evolution. BMC Biol. 15:62. doi: 10.1186/s12915-017-0399
Selosse, M. A., and Le Tacon, F. (1998). The land flora: a phototroph-fungus partnership? Trends Ecol. Evol. 13, 15–20. doi: 10.1016/S0169-5347(97)01230-5
Seoighe, C., and Gehring, C. (2004). Genome duplication led to highly selective expansion of the Arabidopsis thaliana proteome. Trends Genet. 20, 461–464. doi: 10.1016/j.tig.2004.07.008
Shen, Y., Wei, W., and Zhou, D. X. (2015). Histone acetylation enzymes coordinate metabolism and gene expression. Trends Plant Sci. 20, 614–621. doi: 10.1016/j.tplants.2015.07.005
Shen, Y., Yue, F., Mc Cleary, D. F., Ye, Z., Edsall, L., Kuan, S., et al. (2012). A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120. doi: 10.1038/nature11243
Shlyueva, D., Stampfel, G., and Stark, A. (2014). Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 15, 272–286. doi: 10.1038/nrg3682
Shropshire, J. D., and Bordenstein, S. R. (2016). Speciation by symbiosis: the microbiome and behavior. mBio 7, e1785-15. doi: 10.1128/mBio.01785-15
Sicard, A., Kappel, C., Lee, Y. W., Woźniak, N. J., Marona, C., Stinchcombe, J. R., et al. (2016). Standing genetic variation in a tissue-specific enhancer underlies selfing-syndrome evolution in Capsella. Proc. Natl. Acad. Sci. 113, 13911–13916. doi: 10.1073/pnas.1613394113
Spitz, F., and Furlong, E. E. M. (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626. doi: 10.1038/nrg3207
Stebbins, G. L., and Hill, G. J. C. (1980). Did multicellular plants invade the land. Am. Nat. 115, 342–353. doi: 10.1086/283565
Stephens, R. S., Kalman, S., Lammel, C., Fan, J., Marathe, R., Aravind, L., et al. (1998). Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282, 754–759. doi: 10.1126/science.282.5389.754
Stern, D. L. (1998). A role of Ultrabithorax in morphological differences between Drosophila species. Nature 396, 463–466. doi: 10.1038/24863
Struhl, K. (1999). Fundamentally different logic of gene regulation in eukaryotes and prokaryotes. Cell 98, 1–4. doi: 10.1016/S0092-8674(00)80599-1
Szerlong, H., Hinata, K., Viswanathan, R., Erdjument-Bromage, H., Tempst, P., and Cairns, B. R. (2008). The HSA domain binds nuclear actin-related proteins to regulate chromatin-remodeling ATPases. Nat. Struct. Mol. Biol. 15, 469–476. doi: 10.1038/nsmb.1403
Szerlong, H. J., and Hansen, J. C. (2011). Nucleosome distribution and linker DNA: connecting nuclear function to dynamic chromatin structure. Biochem Cell Biol. 89, 24–34. doi: 10.1139/O10-139
Valentine, J. W. (1978). The evolution of multicellular plants and animals. Sci. Am. 239, 140–146. doi: 10.1038/scientificamerican0978-140
Valentine, J. W., Collins, A. G., and Meyer, C. P. (1994). Morphological complexity increase in metazoans. Paleobiology 20, 131–142. doi: 10.1017/S0094837300012641
Van de Peer, Y., Mizrachi, E., and Marchal, K. (2017). The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424. doi: 10.1038/nrg.2017.26
Vanneste, K., Baele, G., Maere, S., and Van De Peer, Y. (2014). Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the cretaceous-paleogene boundary. Genome Res. 24, 1334–1347. doi: 10.1101/gr.168997.113
Venkatesh, S., and Workman, J. L. (2015). Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 16, 178–189. doi: 10.1038/nrm3941
Villar, D., Berthelot, C., Aldridge, S., Rayner, T. F., Lukk, M., Pignatelli, M., et al. (2015). Enhancer evolution across 20 mammalian species. Cell 160, 554–566. doi: 10.1016/j.cell.2015.01.006
Vlad, D., Kierzkowski, D., Rast, M. I., Vuolo, F., Dello Ioio, R., Galinha, C., et al. (2014). Leaf shape evolution through duplication, regulatory diversification, and loss of a homeobox gene. Science 343, 780–783. doi: 10.1126/science.1248384
Vogel, C., and Chothia, C. (2006). Protein family expansions and biological complexity. PLoS Comput. Biol. 2:e48. doi: 10.1371/journal.pcbi.0020048
Vuolo, F., Mentink, R. A., Hajheidari, M., Bailey, C. D., Filatov, D. A., and Tsiantis, M. (2016). Coupled enhancer and coding sequence evolution of a homeobox gene shaped leaf diversity. Genes Dev. 30, 2370–2375. doi: 10.1101/gad.290684.116
Wang, H., Nussbaum-Wagler, T., Li, B., Zhao, Q., Vigouroux, Y., Faller, M., et al. (2005). The origin of the naked grains of maize. Nature 436, 714–719. doi: 10.1038/nature03863
Wang, H., Studer, A. J., Zhao, Q., Meeley, R., and Doebley, J. F. (2015). Evidence that the origin of naked kernels during maize domestication was caused by a single amino acid substitution in tga1. Genetics 200, 965–974. doi: 10.1534/genetics.115.175752
Watson, M. L., Zinn, A. R., Inoue, N., Hess, K. D., Cobb, J., Handel, M. A., et al. (1998). Identification of morc (microrchidia), a mutation that results in arrest of spermatogenesis at an early meiotic stage in the mouse. Proc. Natl. Acad. Sci. U.S.A. 95, 14361–14366. doi: 10.1073/pnas.95.24.14361
Weeden, N. F. (1981). Genetic and biochemical implications of the endosymbiotic origin of the chloroplast. J. Mol. Evol. 17, 133–139. doi: 10.1007/BF01733906
Wichard, T. (2015). Exploring bacteria-induced growth and morphogenesis in the green macroalga order Ulvales (Chlorophyta). Front. Plant Sci. 6:86. doi: 10.3389/fpls.2015.00086
Wierzbicki, A. T., Haag, J. R., and Pikaard, C. S. (2008). Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell 135, 635–648. doi: 10.1016/j.cell.2008.09.035
Williams, T. A., and Embley, T. M. (2014). Archaeal “dark matter” and the origin of eukaryotes. Genome Biol. Evol. 6, 474–481. doi: 10.1093/gbe/evu031
Wilson, A. C., Sarich, V. M., and Maxson, L. R. (1974). The importance of gene rearrangement in evolution: evidence from studies on rates of chromosomal, protein, and anatomical evolution. Proc. Natl. Acad. Sci. 71, 3028–3030. doi: 10.1073/pnas.71.8.3028
Wolf, Y. I., and Koonin, E. V. (2013). Genome reduction as the dominant mode of evolution. BioEssays 35, 829–837. doi: 10.1002/bies.201300037
Wyrick, J. J., and Young, R. A. (2002). Deciphering gene expression regulatory networks. Curr. Opin. Genet. Dev. 12, 130–136. doi: 10.1016/S0959-437X(02)00277-0
Xue, L., Klinnawee, L., Zhou, Y., Saridis, G., Vijayakumar, V., Brands, M., et al. (2018). AP2 transcription factor CBX1 with a specific function in symbiotic exchange of nutrients in mycorrhizal Lotus japonicus. Proc. Natl. Acad. Sci. 115, E9239–E9246. doi: 10.1073/pnas.1812275115
Yang, C., and Stiller, J. W. (2014). Evolutionary diversity and taxon-specific modifications of the RNA polymerase II C-terminal domain. Proc. Natl. Acad. Sci. 111, 5920–5925. doi: 10.1073/pnas.1323616111
Zhang, L., and Ma, H. (2012). Complex evolutionary history and diverse domain organization of SET proteins suggest divergent regulatory interactions. New Phytol. 195, 248–263. doi: 10.1111/j.1469-8137.2012.04143
Zhou, B.-R., Feng, H., Kato, H., Dai, L., Yang, Y., Zhou, Y., et al. (2013). Structural insights into the histone H1-nucleosome complex. Proc. Natl. Acad. Sci. 110, 19390–19395. doi: 10.1073/pnas.1314905110
Zhou, C. Y., Johnson, S. L., Gamarra, N. I., and Narlikar, G. J. (2016). Mechanisms of ATP-dependent chromatin remodeling motors. Annu. Rev. Biophys. 45, 153–181. doi: 10.1146/annurev-biophys-051013-022819
Zhou, X., and Ma, H. (2008). Evolutionary history of histone demethylase families: distinct evolutionary patterns suggest functional divergence. BMC Evol. Biol. 8:294. doi: 10.1186/1471-2148-8-294
Keywords: gene duplication, evolution, chromatin, transcriptional regulation, morphological complexity, microbiota, symbiosis
Citation: Hajheidari M, Koncz C and Bucher M (2019) Chromatin Evolution-Key Innovations Underpinning Morphological Complexity. Front. Plant Sci. 10:454. doi: 10.3389/fpls.2019.00454
Received: 10 January 2019; Accepted: 26 March 2019;
Published: 12 April 2019.
Edited by:
Jean-Benoit Charron, McGill University, CanadaReviewed by:
Liangsheng Zhang, Fujian Agriculture and Forestry University, ChinaMatthew David Herron, Georgia Institute of Technology, United States
Copyright © 2019 Hajheidari, Koncz and Bucher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohsen Hajheidari, bS5oYWpoZWlkYXJpQHVuaS1rb2Vsbi5kZQ==