 Deng-Feng Xie1
Deng-Feng Xie1 Huan-Xi Yu1
Huan-Xi Yu1 Megan Price2
Megan Price2 Chuan Xie1
Chuan Xie1 Yi-Qi Deng1
Yi-Qi Deng1 Jun-Pei Chen1
Jun-Pei Chen1 Yan Yu1Song-Dong Zhou1
Yan Yu1Song-Dong Zhou1 Xing-Jin He1*
Xing-Jin He1*- 1Key Laboratory of Bio-Resources and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, China
- 2Sichuan Key Laboratory of Conservation Biology on Endangered Wildlife, College of Life Sciences, Sichuan University, Chengdu, China
The genus Allium (Amaryllidaceae, Allioideae) is one of the largest monocotyledonous genera and it includes many economically important crops that are cultivated for consumption or medicinal uses. Recent advances in molecular phylogenetics have revolutionized our understanding of Allium taxonomy and evolution. However, the phylogenetic relationships in some Allium sections (such as the Allium section Daghestanica) and the genetic bases of adaptative evolution, remain poorly understood. Here, we newly assembled six chloroplast genomes from Chinese endemic species in Allium section Daghestanica and by combining these genomes with another 35 allied species, we performed a series of analyses including genome structure, GC content, species pairwise Ka/Ks ratios, and the SSR component, nucleotide diversity and codon usage. Positively selected genes (PSGs) were detected in the Allium lineage using the branch-site model. Comparison analysis of Bayesian and ML phylogeny on CCG (complete chloroplast genome), SCG (single copy genes) and CDS (coding DNA sequences) produced a well-resolved phylogeny of Allioideae plastid lineages, which illustrated several novel relationships with the section Daghestanica. In addition, six species in section Daghestanica showed highly conserved structures. The GC content and the GC3s content in Allioideae species exhibited lower values than studied non-Allioideae species, along with elevated pairwise Ka/Ks ratios. The rps2 gene was lost in all examined Allioideae species, and 10 genes with significant posterior probabilities for codon sites were identified in the positive selection analysis, seven of them are associated with photosynthesis. Our study uncovered a new species relationship in section Daghestanica and suggested that the selective pressure has played an important role in Allium adaptation and evolution, these results will facilitate our further understanding of evolution and adaptation of species in the genus Allium.
Introduction
Allium L. is the single genus of Allieae and belongs to the subfamily Allioideae (Amaryllidaceae) as per update APG IV (Chase et al., 2016). It is one of the largest genera of the monocotyledons (∼ 920 species) and includes many economically important crops (Herden et al., 2016; Zhu et al., 2017). The new classification of Allium was made by Friesen et al. (2006), who first suggested that the genus Allium is monophyletic. Despite extensive work on the genus, taxonomical and phylogenetic uncertainties remain in some subgenera or sections. For example, Allium section Daghestanica (Tscholok.) N. Friesen. has recently been proposed to be a small group (Friesen et al., 2006) containing more than 10 species globally, with six being endemic to China according to Li et al. (2010). The six Chinese endemics are primarily distributed in the southeast fringe of the Qinghai-Tibet Plateau (QTP): A. chrysanthum Regel, A. chrysocephalum Regel, A. herderianum Regel, A. rude J.M.Xu, A. xichuanense J.M.Xu, and A. maowenense J.M.Xu (Figure 1). Early studies placed A. rude, A. xichuanense, A. chrysocephalum and A. herderianum into sect. Rhiziridium G. Don, A. chrysanthum was placed into sect. Schoenoprasum G. Don, and the A. maowenense was classified into sect. Haplostemon Boiss (Xu et al., 1994; Chen et al., 2000). Li et al. (2010) then reclassified the species into sect. Daghestanica according to molecular phylogenetic analyses, and was confirmed by morphological evidences produced by Yu et al. (2018). These previous studies have significantly advanced the phylogeny and taxonomy of the six species, yet a consensus of the six species’ exact relationships have not been reached. In particular, uncertainties remain because A. herderianum was not sampled in previous phylogenetic studies (Li et al., 2010).

Figure 1. The flower morphological characters of Section Daghestanica species. (A) A. chrysanthum, (B) A. rude, (C) A. xichuanense, (D) A. chrysocephalum, (E) A. maowenense, and (F) A. herderianum.
Many nuclear genes and chloroplast genomes were recently employed in Allium studies (Friesen et al., 2006; Nguyen et al., 2008; Kim and Yoon, 2010; Li et al., 2010; Lee et al., 2017; Jin et al., 2018), which provide valuable information for the phylogenetic study of section Daghestanica. In particular, the whole chloroplast genomes that possess highly conserved gene structure and gene content, and lower substitution rate than nuclear DNA (especially in inverted repeat regions), offer promising solutions to phylogeny uncertainties (Wolfe et al., 1987; Raubeson et al., 2005; Parks et al., 2009). The chloroplast genome sequence of A. cepa is such a single circular molecule of 153,440 bp length with a quadripartite structure (containing 132 genes) that includes two copies: LSC (large single-copy) and SSC (small single-copy), which are separated by an IR (inverted repeats) region (Kim et al., 2015).
In addition to their applicability for phylogenetic studies, whole chloroplast genomes can provide insights into other evolutionary processes, such as chloroplast inheritance, domestication studies and adaptive evolution. Adaptive evolution, defined as the adaptability improvement of species during their evolutionary processes, and it is always driven by evolutionary processes such as natural selection, which act on genetic variations produced by mutations, genetic recombination and gene flow (Scottphillips et al., 2014) and resulted in biodiversity at every level of biological organization (Hall et al., 2008). Therefore, selection pressures that species experienced in their evolutionary processes constitute another interesting aspect in chloroplast genomes analyses. Recent studies detected many positively selected chloroplast genes [genes with Ka (non-synonymous substitution) greater than Ks (synonymous substitutions)]. For example, rbcL and nuclear rbcS genes in Flaveria (Kapralov et al., 2011), as well as clpP1 exon in three distantly related taxa of Oenothera (Erixon and Oxelman, 2008).
Species of Allium are all perennial herbs, are distributed widely from the dry subtropics to the boreal zones, and are characterized by diversified morphological features (e.g., bulbs, leaves, and flowers). Furthermore, the habitat of Allium species varies from dry and well-drained soils to moist and organic soils, that can be in swamps or water (Block, 2010). Therefore, Allium is considered as a successful taxon due to its wide distribution and diversification (Li et al., 2010). Generally, substitution rates of angiosperm cp genomes are slow and are minimally affected by adaptive evolution (Erixon and Oxelman, 2008), excluding several genes that may evolve very rapidly due to the effects of positive selection (Ivanova et al., 2017). Previous studies have found that the positive selection is expected to accelerate the Ka value yet it does not affect the Ks value (Fan et al., 2018). However, little is known about the positive selection and adaptation of Allium species.
In this study, the whole cp genomes of Chinese species in section Daghestanica were sequenced using the next-generation sequencing platform. Combined with another 35 cp genomes previously published (including species from Asparagaceae, Allioideae, Agapanthoideae, Asphodeloideae, Asphodelaceae, and Iridaceae), here we provide the comprehensive analysis of cp genomes for Allium and species in allied families based on present cp genome data. We aimed to generate a robust phylogeny of extant Allium cp genome data, and used this phylogeny to: (1) reconstruct the phylogeny of Chinese species in Allium section Daghestanica based on the cp genome data and analyze the species relationships at the plastid level. (2) compare the cp genome structure of species within section Daghestanica, in genus Allium (Allioideae) and other allied families; and (3) investigate selective or adaptive evolution in the cp genomes of Allium species.
Materials and Methods
Plant Materials and DNA Extraction
We collected the fresh leaves from each field site (Supplementary Table S1) and were immediately used for DNA extraction. The total genomic DNA was extracted from leaf tissues with a modified Cetyl Trimethyl Ammonium Bromide (CTAB) method (Doyle, 1987).
Plastome Genome Sequencing and Assembling
All genome data were sequenced using an Illumina Hiseq 2500 platform by Biomarker Technologies, Inc (Beijing, China). High-quality reads were obtained using the CLC Genomics Workbench v7.5 (CLC Bio, Aarhus, Denmark). Reference-guided assembly was then performed to reconstruct the chloroplast genomes using the program MITObim v1.7 (Christoph et al., 2013). In order to obtain accurate sequences, each species was assembled four times with the reference genomes A. cepa (KM088014), A. sativum (KY085913), A. victorialis (NC_037240), and A. obliquum (LT699701). Gaps that appeared in the assembled cp genomes were corrected by Sanger sequencing and the primers were designed using Lasergene 7.1 (DNASTAR, Madison, WI, United States). The primers and amplifications were shown in Supplementary Table S2. The program DOGMA (Wyman et al., 2004) was used to annotate the whole cp genome, and subsequently corrected within GENEIOUS R11 (Biomatters, Ltd., Auckland, New Zealand). Final plastid genome maps were drawn using OGDRAW (Lohse et al., 2013).
GC Content and Species Pairwise Ka/Ks Ratios
GC content of the complete chloroplast genome (CCG) and the third position GC content of codons in each species were calculated using PAML v4.8 (Yang, 2007). Each CDS sequence of all species was extracted and aligned with MAFFT v. 7 (Katoh and Standley, 2013). Pairwise Ka/Ks ratios of all species were calculated using the concatenated single-CDS alignments with KaKs Calculator version 2.0 (Wang et al., 2010).
SSRs Characterization and Chloroplast Genome Nucleotide Diversity
Perl script MISA (Thiel et al., 2003) was used to search microsatellites loci in the cp genomes with parameters being set as 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. The DnaSP version 5.1 (Librado and Rozas, 2009) was used to calculate the nucleotide diversity of genes in LSC, SSC, and IR regions.
Indices of Codon Usage
Codon usage in these genes was assessed using the program codon W 1.4.4 (J. Peden)1. Five values were used to estimate the extent of bias toward codons: the codon adaptation index (CAI), codon bias index (CBI), frequency of optimal codons (Fop), GC content of synonymous third codons positions (GC3s), and the effective number of codons (ENC).
Phylogenetic Analyses
In order to investigate the relationships of the six Allium species, all available complete genome sequences in allied families were downloaded from NCBI, including 20 species from Asparagaceae, 10 species in Allioideae (all Allium species), one species in Agapanthoideae, 2 species in Asphodeloideae, one species in Asphodelaceae and one species in Iridaceae (Supplementary Table S3). Firstly, all single-copy genes were extracted from all 41 taxa, and alignments of each gene were generated and trimmed. These alignments were then concatenated to produce an alignment of all single copy genes, which were used for phylogenetic analysis. Maximum likelihood (ML) analyses were performed using RAxML 8.2.8 (Stamatakis, 2014) with GTR + G model and 1,000 bootstrap replicates. Bayesian analyses were performed with MrBayes v. 3.2.5 (Ronquist and Huelsenbeck, 2003) under the GTR + I + Γ substitution model. The Markov chain Monte Carlo (MCMC) algorithm was run for 1 × 108 generations, with one tree sampled every 1000 generations. The first 20% of trees were discarded as burn-in, and the remaining trees were used to build a 50% majority-rule consensus tree. The stationarity was considered to be reached when the average standard deviation of split frequencies remained below 0.001. In view of the utility of different cp regions, phylogenetic analyses were performed for the CCG data and the CDS sequences respectively.
Positive Selected Analyses
An optimized branch-site model (Yang and Dos, 2011) and Bayesian Empirical Bayes (BEB) methods (Yang et al., 2005) were used to identify genes under positive selection in Allium species (Allioideae) compared to species in non-Allioideae families. The single-copy CDS sequences of all 41 taxa were extracted and the software MUSCLE v3.6 (Edgar, 2004) was used in sequence alignment according to their amino acid sequences. The alignments of the DNA codon sequences were further trimmed by TRIMAL v1.2 (Capellagutiérrez et al., 2009), and the final alignments were used to perform the positive selection analyses. The branch-site model was implemented to assess potential positive selection in specifically designated Allioideae lineage in the PAML v4.8 package (Yang, 2007). The ratio (ω) of the non-synonymous substitution rate (Ka) to the synonymous substitutions rate (Ks) was used to measure the selective pressure. The ratio ω > 1, ω = 1, and ω < 1 suggest positive selection, neutral selection and negative selection, respectively (Yang and Nielsen, 2002). The log-likelihood values were calculated and tested according to Lan et al. (2017). The BEB method was applied to compute the posterior probabilities of amino acid sites to identify whether these specific sites were under positive selection (codon sites with a high posterior probability) (Yang et al., 2005). A gene with a test p-value < 0.05 and with positively selected sites was considered as a positively selected gene (PSG). The Jalview v2.4 (Clamp et al., 2004) was used to view the amino acid sequences of PSGs.
Results
Chloroplast Features of Allium Species
The complete cp genomes of six Allium species ranged from 153,605 bp (A. herderianum) to 153,710 bp (A. chrysocephalum) in length, with the minimum and maximum differences being 3 and 105 bp, respectively (Table 1 and Figure 2). All six cp genomes showed a typical quadripartite structure that was similar to those of most land plants. The cp genome consisted of a pair of IR regions (26,446–26,512 bp) separated by the LSC (82,658–82,815 bp) and SSC (17,950–18,000 bp) regions. The GC content ranged from 37.7–37.8%, indicating nearly identical levels among the six Allium cp genomes. In addition, the six Allium cp genomes encoded 132 functional genes, with 86 protein-coding genes, 38 tRNA genes, and eight ribosomal RNA genes (Table 1 and Supplementary Table S4). The length and GC contents of the non-coding regions in the six Allium species were lower than the whole cp genome and the coding regions (Table 1). The length, GC content and gene components of the 41 species were included in the Supplementary Table S5.
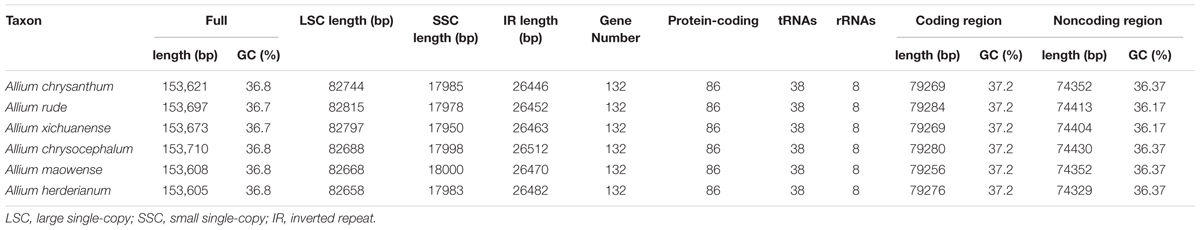
Table 1. Summary of complete chloroplast genomes of Section Daghestanica species.
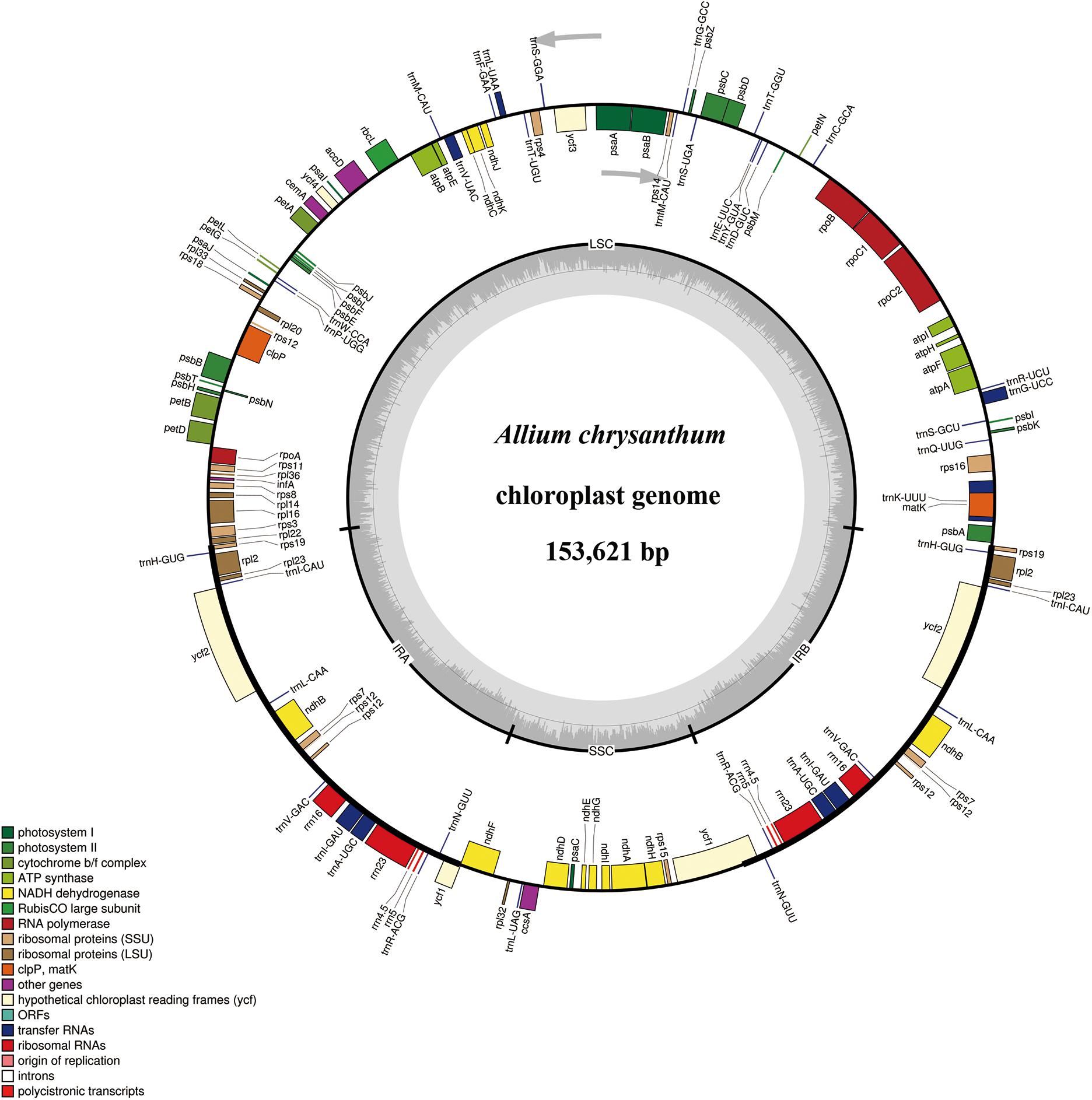
Figure 2. Gene maps of the Section Daghestanica species chloroplast (cp) genomes. Genes shown inside the circle are transcribed clockwise, and those outside are transcribed counterclockwise. Genes belonging to different functional groups are color-coded. The darker gray color in the inner circle corresponds to the GC content, and the lighter gray color corresponds to the AT content. SSU, small subunit; LSU, large subunit; ORF, open reading frame.
GC Content Distribution and the Ka/Ks Ratios of Species Pairwise
The total and the third position GC content were compared between 41 species (belonging to Asparagaceae, Allioideae, Agapanthoideae, Asphodeloideae, Asphodelaceae, and Iridaceae). Lower GC contents were observed at the total nucleotides level (<38.5%) and the third codon positions (<36.0%) in most of Allium (Allioideae) species compared to species in non-Allioideae families (Figure 3 and Supplementary Table S6).
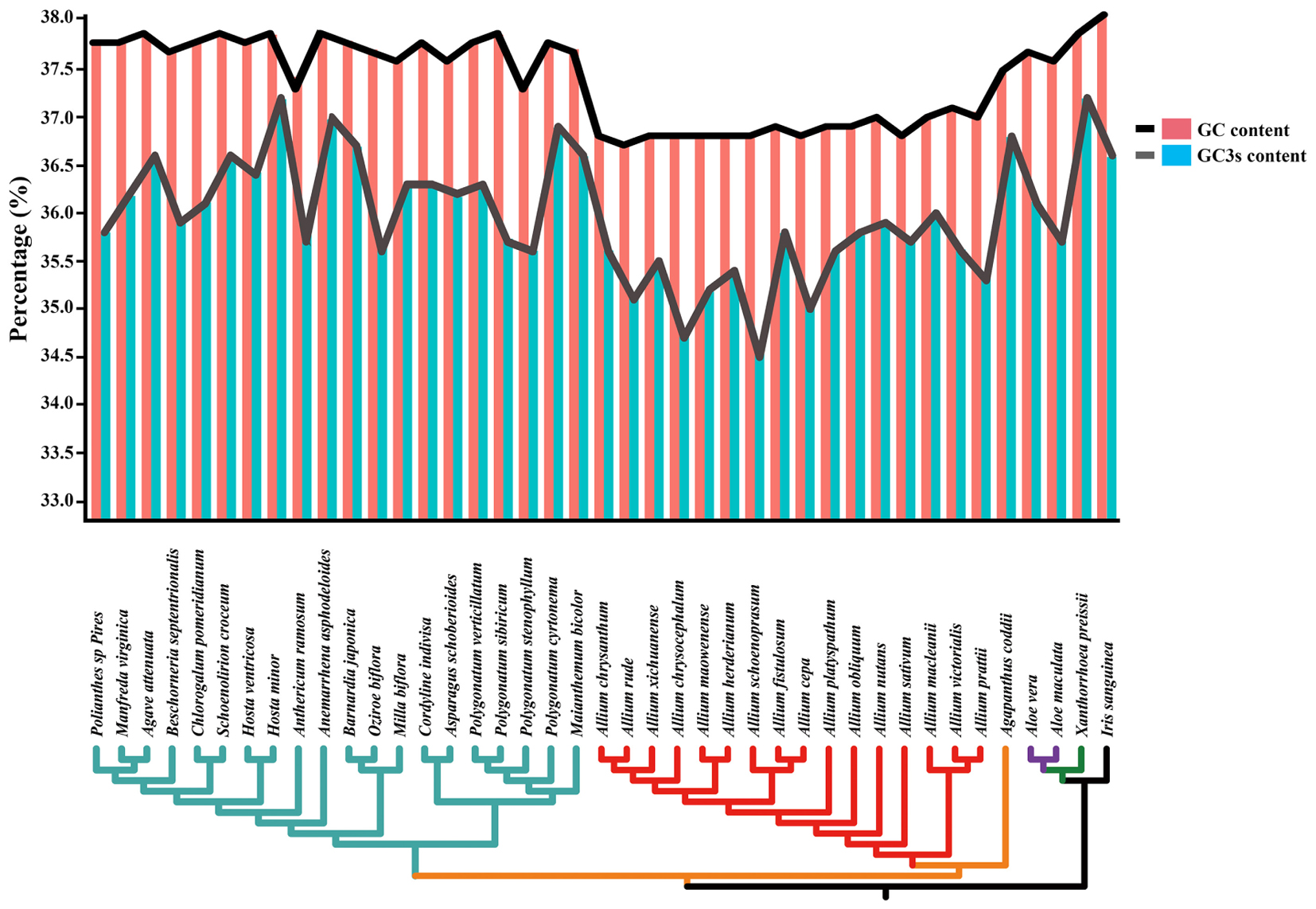
Figure 3. Changes in plastid GC content of all 41 taxa. This graph shows the total GC content (red bar and black line) and the third codon position GC content (blue bar and gray line) of each species.
The pairwise Ka/Ks ratios of each species pair were calculated (Figure 4), which provided information of selective pressure that acted on individual sequences. Much higher pairwise Ka/Ks ratios were observed in Allium (Allioideae) species pairs than non-Allioiseae species pairs (Figure 4 and Supplementary Table S7). In addition, high Ka/Ks ratios were also detected in other species (e.g., species in Hosta and Cordyline) (Figure 4 and Supplementary Table S7).
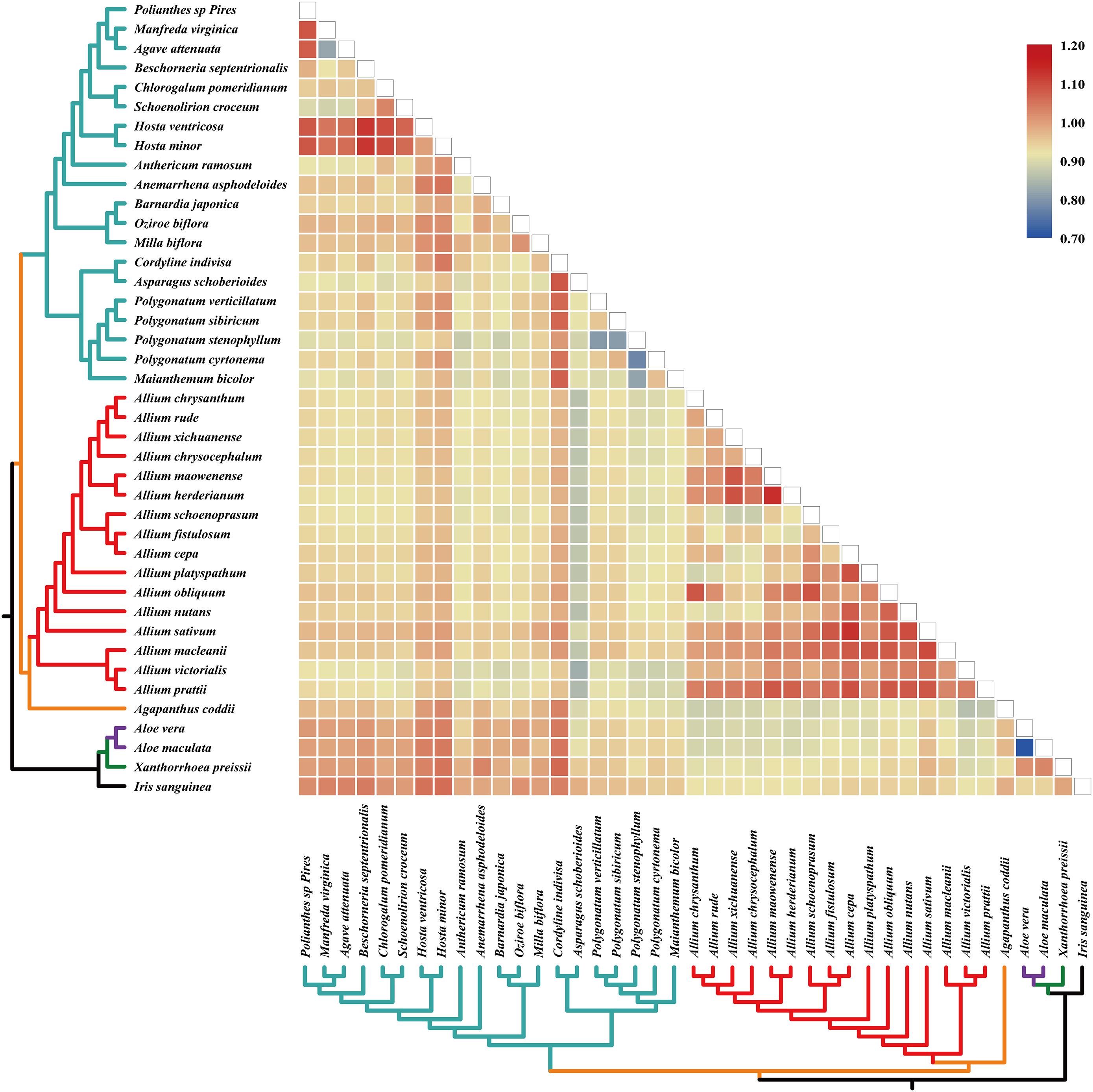
Figure 4. Pairwise Ka/Ks ratios in Allium (Allioideae) and other families. This heatmap shows pairwise Ka/Ks ratios between every sequence in the multigene nucleotide alignment. Allium (Allioideae) are shown on red branches. The scale factors associated with each value are shown on the right-hand side of the figure.
Repeat Sequences Variations, the Nucleotide Diversity, Codon Usage and Gene Loss
We detected numerous microsatellites (SSRs) in the six Allium cp genomes, ranging from 179 (A. maowenense) to 193 (A. chrysocephalum) (Supplementary Figure S1). The most abundant were mono-nucleotide repeats, where the number varied from 63 in A. chrysocephalum to 74 in A. rude, followed by tetra-nucleotides and di-nucleotide repeats, while the penta-nucleotide repeats were the least abundant (Supplementary Figure S1 and Supplementary Table S8). The overall length of the five categories of perfect SSRs ranged from 9 to 25 bp in the six Allium species (Supplementary Table S8).
The nucleotide diversity values in the LSC regions ranged from 0.0006 to 0.07823 with a mean value of 0.0310 (from 0.0035 to 0.0722 with the average value was 0.0465 in SSC regions), while the value was from 0.0000 to 0.0311 with a mean value of 0.0084 in the IRs regions (Supplementary Figure S2). Six genes with high nucleotide diversity were detected (>0.0700), these were trnK-UUU, matK, trnG-UCC, trnG-GCC, ndhF and rps15. Five genes (i.e., accD, clpP, rpl16, ccsA and ndhA) with nucleotide diversity more than 0.05500 were also detected.
The pattern of codon usage bias in the Allium (Allioideae) and non-Allioideae were investigated. We found that five parameters involved in codon usage bias were lower in Allium (Allioideae) species than non-Allioideae species, except the CAI that was lower in the family Asparagaceae (Figure 5).
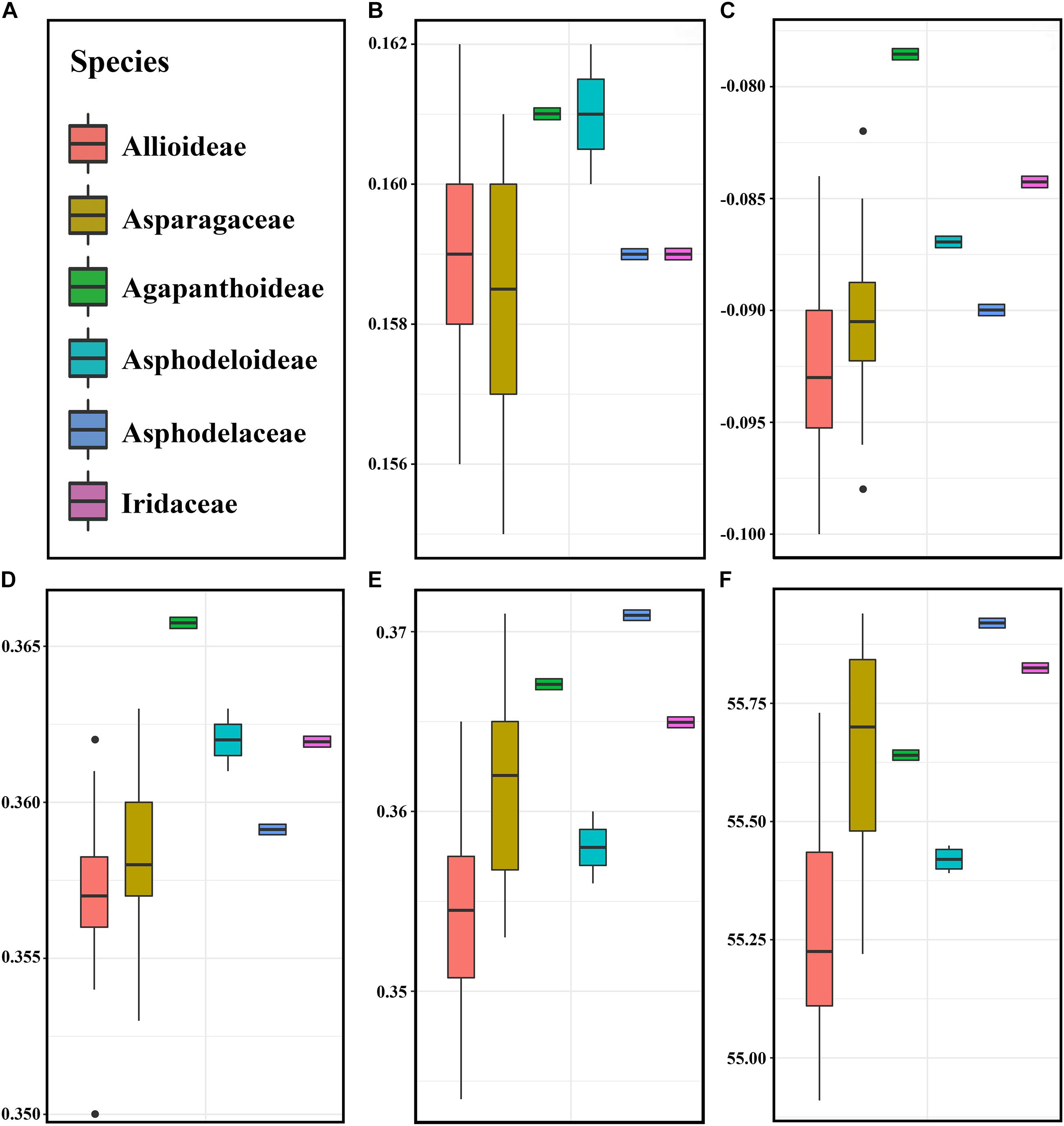
Figure 5. The comparative analysis of codon usage bias in Allium (Allioideae) and other family species. (A) Labels representing each of families. (B) CAI (Codon adaptation index), (C) CBI (Codon bias index), (D) FOP (Frequency of optimal codons index), (E) GC3s (GC of synonymous codons in 3rd position), (F) ENC (Effective number of codons).
As shown in Figure 6, we found that the gene rps2 was lost in all Allium (Allioideae) species. In addition, four genes infA, rps16, psbZ, and ndhD were lost in four Allium species with different degree (A. sativum, A. macleanii, A. platyspathum, and A. victorialis). Gene cemA, infA, rps19, and ycf1 were lost in some species of Asparagaceae, and gene rpl32 and infA were missing in Aloe vera and A. maculate of Asphodeloideae.
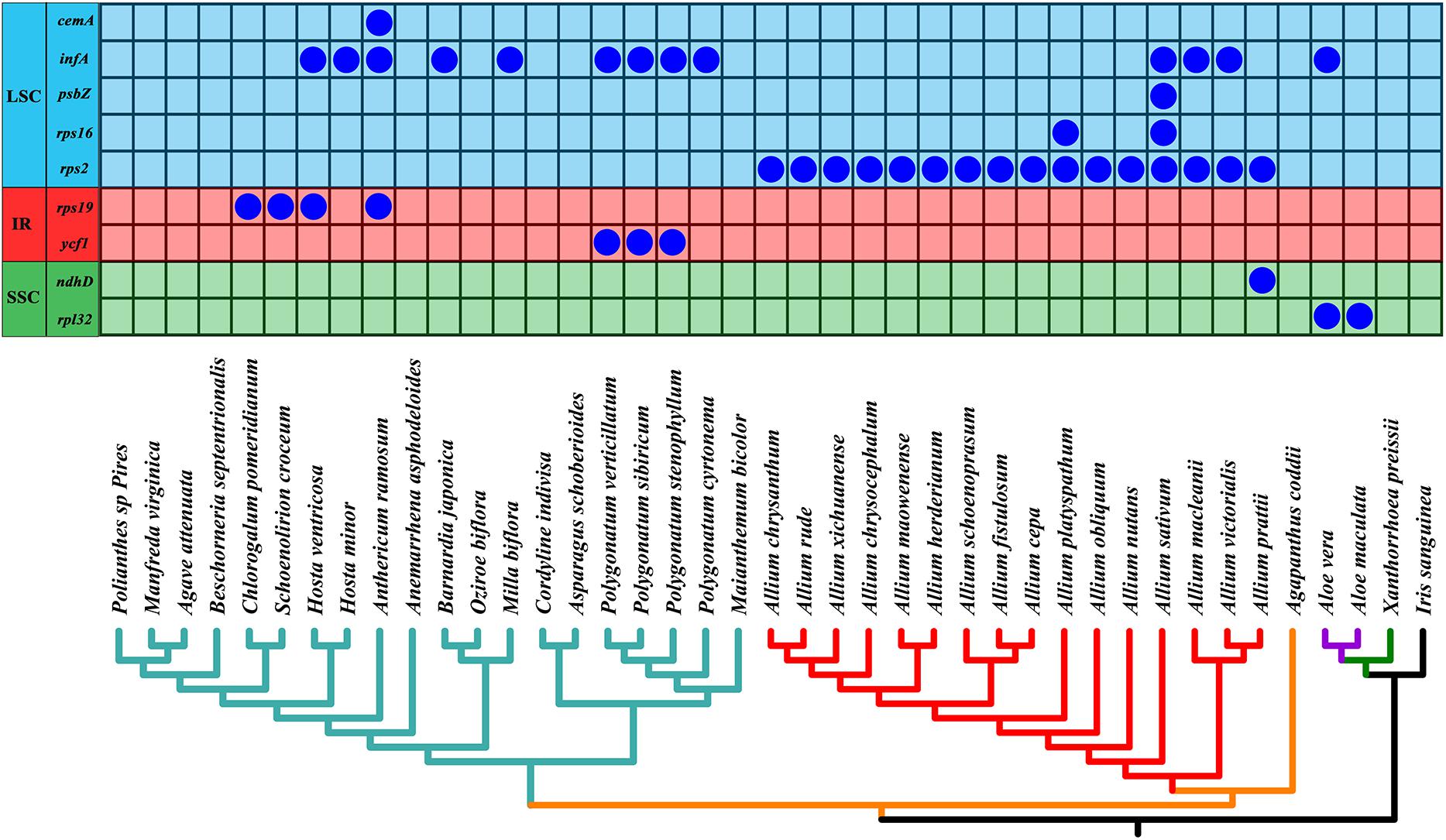
Figure 6. Loss of chloroplast protein-coding genes in the phylogeny of all 41 taxa. Below is the phylogeny of all 41 species based on chloroplast genomes as shown in Figure 7. Different chloroplast regions were indicated at the left side. Allioideae are shown on red branches. IR, inverted repeat; LSC, large single-copy region; SSC, small single-copy region.
Characteristics of cp Genome and Phylogenetic Analysis
Complete chloroplast genome of the six species in section Daghestanica were newly sequenced in this study, and were deposited in GenBank (Supplementary Table S3). These plastid genomes are similar to previously published Allium plastomes in size, structure and gene content (Filyushin et al., 2016, 2018; Lee et al., 2017; Jin et al., 2018). The CCG data set had an aligned length of 192056 bp, within which 24727 parsimony-informative sites (PICs, 12.87 %) were detected. The SCG (single copy genes) possessed 54401 bp aligned nucleotides with 6755 PICs (12.41 %). CDS (coding DNA sequences) data set had an aligned length of 45954 bp nucleotides with 5464 PICs (11.89 %). Comparing these data sets with CCG, the percentages of PICs, SCG and CDS were reduced.
We reconstructed separate phylogenetic trees based on different methods: Bayesian and ML analyses on CCG. Bayesian and ML analyses recovered almost identical trees from each data set. There was strong support for the monophyly of each family were revealed based on CCG data (Figure 7). The topological structures from the SCG and CDS are similar to that from CCG, and all lineages possess high bootstrap values (Supplementary Figure S3). Agapanthus coddii from Agapanthoideae had strong support to be a sister to the Allioideae, and Asparagaceae was supported to be the sister of Agapanthoideae and Allioideae. For Chinese species in section Daghestanica, A. maowenense was closely clustered with A. herderianum, and A. chrysanthum is sister to the A. rude. All six species were closely clustered in one lineage (Figure 7 and Supplementary Figure S3).
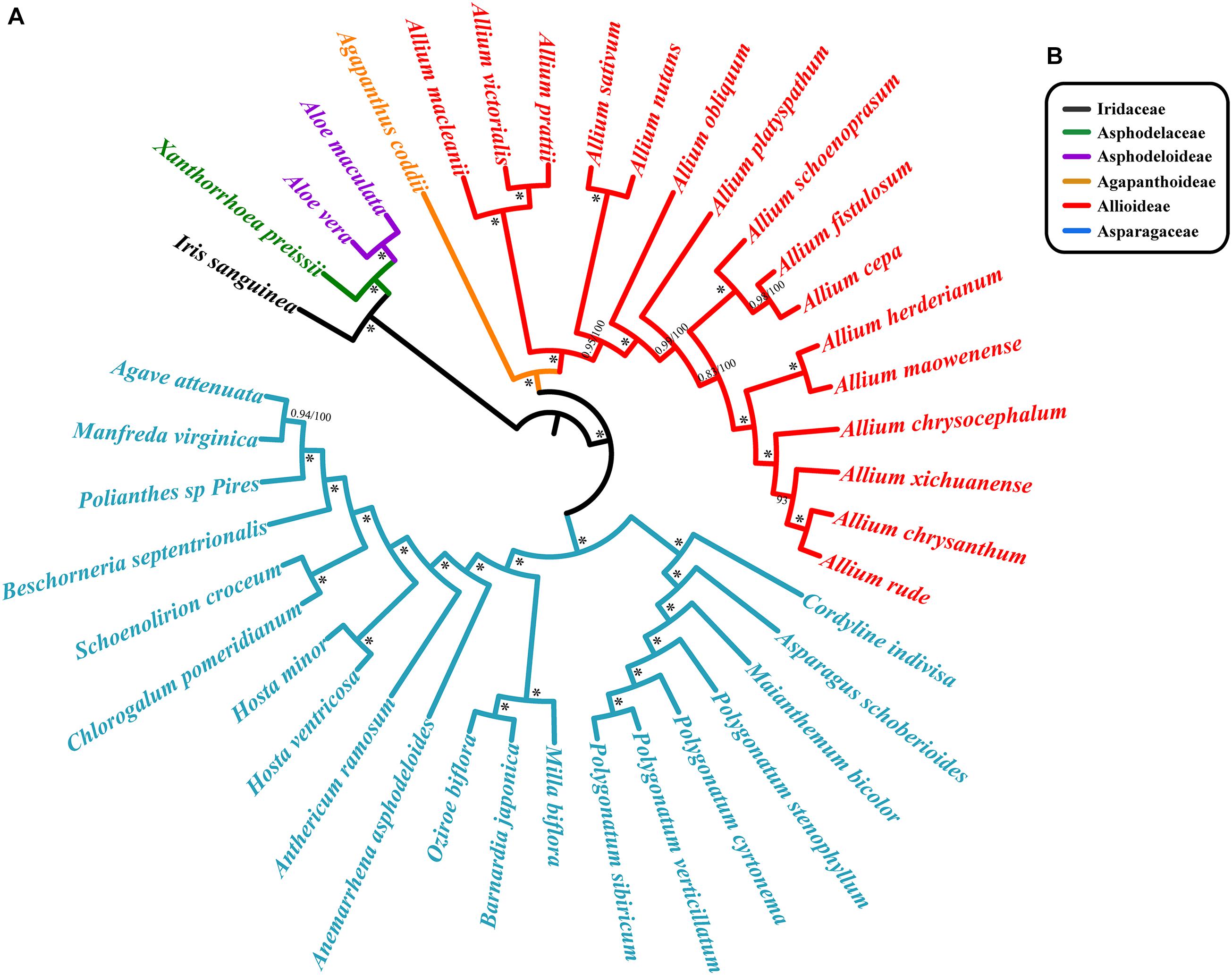
Figure 7. Phylogenetic relationships of section Daghestanica species with related 35 species based on the whole cp genomes. (A) Tree constructed by Bayesian inference (BI) and maximum likelihood (ML) with the posterior probabilities of BI and the bootstrap values of ML above the branches, respectively. ∗ Represent maximum support in all two analyses. (B) Families of each species belong to, color of bar is consistent with the species’ color.
Positive Selection Analyses
There were 50 single-copy CDS genes initially considered for the positive selection analysis (Supplementary Table S9), but 44 were eventually selected after filtering (Table 2). All p-values were not significant in each gene range, however, ten protein coding genes (petA, psbD, psbE, ycf3, psaI, rps4, psbM, ndhE, ndhG, and rpoC1) were found with significant posterior probabilities suggesting sites with positive selection in the BEB test. Among them, most genes only had one positive selective site, whereas rpoC1 gene possessed six positive selective sites, followed by petA and ndhE that had five and three positive selective sites, respectively (Figure 8, Supplementary Figure S4, and Table 2).
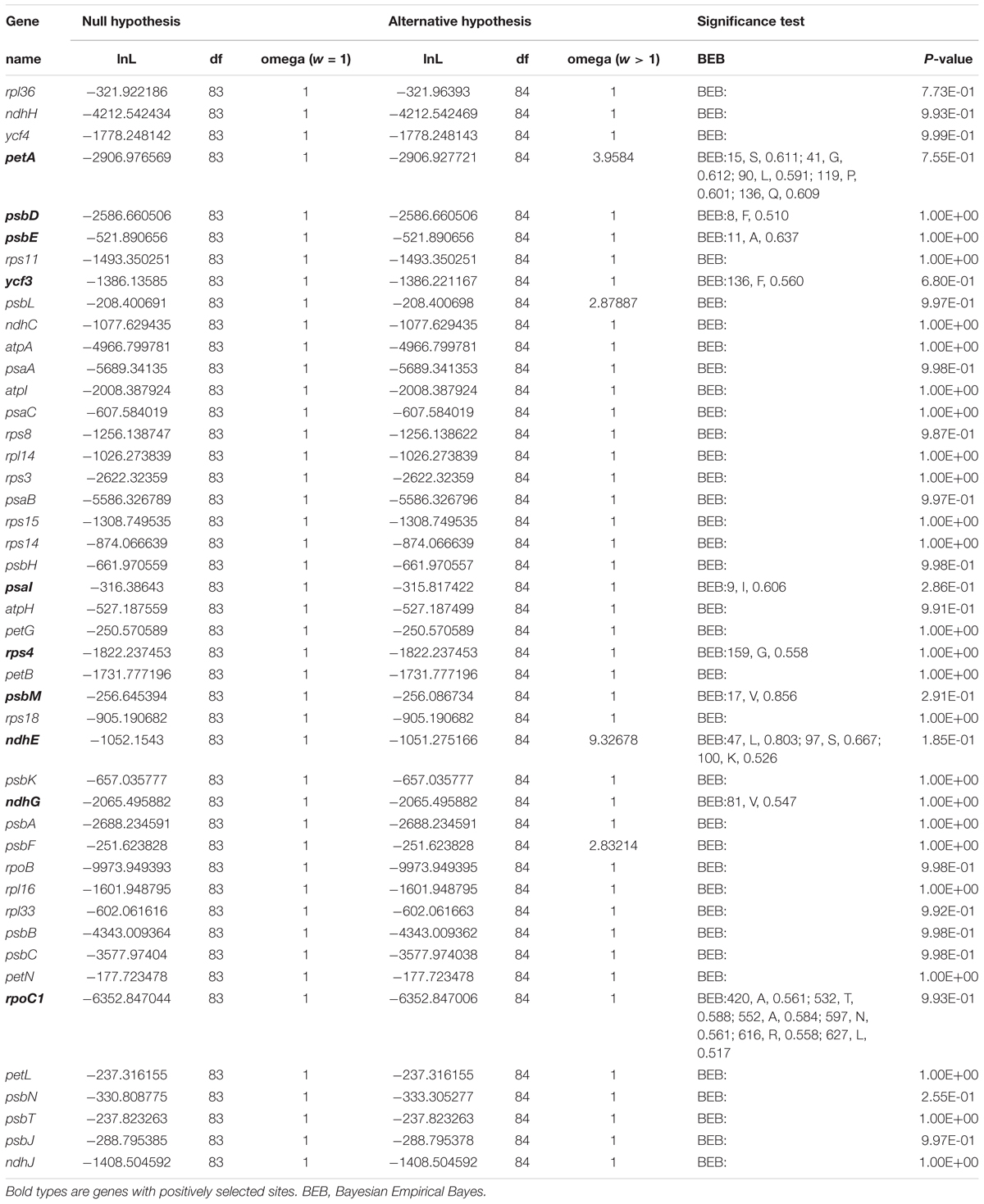
Table 2. The potential positive selection test based on the branch-site model.

Figure 8. Partial alignment of two out of 10 positively selected genes. (A) Partial aligned amino acids sequences of the petA gene; (B) partial aligned amino acids sequences of the rpoC1 gene. The red blocks stand for the amino acids in Allium (Allioideae) with a high BEB posterior probability.
Discussion
Sequence Differentiation of Chinese Species in Allium Section Daghestanica
Recently, chloroplast genomes have been used to evaluate the genetic divergence among related species (Bellusci et al., 2008; Song et al., 2015; Xie et al., 2018). Comparative genome analysis of the six Chinese Allium section Daghestanica species showed highly conserved structures, which can be inferred from the similar gene number, gene component, genome size and the types of simple sequence repeats (SSRs) (Table 1, Supplementary Tables S4, S8, and Supplementary Figure S1). Due to the high polymorphic rate, SSRs have been recognized as one of the main sources of molecular markers and have been extensively researched in phylogenetic and biogeographic studies of populations (Powell et al., 1995; Provan et al., 1997; Pauwels et al., 2012). Six genes (trnK-UUU, matK, trnG-UCC, trnG-GCC, ndhF and rps15) with nucleotide diversity more than 0.0700 and five genes (accD, clpP, rpl16, ccsA, and ndhA) with nucleotide diversity more than 0.05500 were detected (Supplementary Figure S2). Among these loci, clpP, accD, ndhF, rps15, and ccsA have been previously detected as highly variable regions in different plants (Kim and Lee, 2005; Dong et al., 2012; Qian et al., 2013; Hu et al., 2016). We believe that these SSRs and genes with high nucleotide diversity are good sources for interspecies phylogenetic analysis in the future.
The Phylogenetic Analysis of Chinese Species in Section Daghestanica
The results of our phylogenetic analysis strongly support that Allium is monophyletic, which is in accordance with previous studies (Friesen et al., 2006; Nguyen et al., 2008; Li et al., 2010). We also found that Agapanthus coddii was closely related to Allium (Allioideae) (Figure 7 and Supplementary Figure S3). The relationships of Chinese species in section Daghestanica were resolved: the position of A. herderianum was confirmed, which showed a close relationship with A. maowenense, and differentiated early. A. chrysanthum was tightly clustered with A. rude, which is inconsistent with Li et al. (2010), who showed that A. chrysanthum was closely related to A. xichuanense, and A. rude was clustered with A. chrysocephalum. Our results may be more reliable, since we used the whole chloroplast genome (CCG) (Figure 7), SCG and CDS data (Supplementary Figure S3) respectively in phylogeny reconstruction compared to the rps16 fragment in study of Li et al. (2010), and our results were also supported by the morphological characteristics in Yu et al. (2018), which showed similar testa cells of A. chrysanthum and A. rude that are not parallel and irregular in long axis, and their filaments are longer than perianth segments (Figure 1). Although the A. herderianum has a close relationship with A. maowenense, the morphological characteristics of them are distinct different, and A. maowenense is most differentiated in morphology among the six species in terms of its perianth color, green midrib in perianth (Figures 1, 7). In addition, Yu et al. (2018) found that the testa cells of A. maowenense are parallel and irregular, which are obviously different from the other five species. The outer perianth segments of A. chrysocephalum and A. herderianum are boat-shaped, and their style are longer than perianths, these characteristics are distinct from the other four species (Figure 1). The leaves of A. chrysocephalum are flat falcate and in A. herderianum are semiterete and fistulose, easily differentiating the two species. However, we did not find a close relationship between A. chrysocephalum and A. herderianum (Figure 7). Therefore, our study uncovered a new relationship of Chinese species in Allium section Daghestanica using the whole cp genome data.
The Adaptation Evolution of Allium (Allioideae) Plastome
A previous study using 150 species cp genomes showed that the GC content of these species ranged from 19.5 to 42.1% (Smith, 2009). We found that Allioideae species typically exhibited lower GC content than other non-Allioideae families’ species (Figure 3). There are two reasons that may result in lower GC content of plastid DNA, firstly, a neutral mutation process such as AT-mutation pressure or AT-biased gene conversion, which will reduce the GC content (Howe et al., 2003; Kusumi and Tachida, 2005; Khakhlova and Bock, 2010), and secondly, that selection for translational efficiency may lead to the lack of G and C observed in plastid genomes (Morton, 1993, 1998; And and Voelker, 2003). We suggest that mutation pressure from the evolution process may be a crucial factor resulting in the low GC content of Allioideae. The Ka/Ks ratios of Allioideae species are exceptionally high compared with those observed within non-Allioideae families’ species and this may be an indication of an elevated mutation rate in Allioideae plastid DNA (Figure 4). Mutations during evolutionary processes leading to reduced GC content has also been found in mitochondrial DNA, nucleomorph DNA, and in the genomes of symbionts, parasites, and pathogenic bacteria (Ogata et al., 2001; And and Voelker, 2003; Lane et al., 2007; Smith and Lee, 2008).
Elevated pairwise Ka/Ks ratios were observed in Allioideae species pairs compared to non-Allioideae species pairs (Figure 4 and Supplementary Table S7). The elevated Ka/Ks ratios are unlikely to be explained by changes in codon preference since we did not obtain obvious codon usage bias in Allioideae species (Figure 5). It is possible that other factors (e.g., habitat environment and adaptation evolution) may have contributed to the elevated Ka/Ks ratios. Allioideae is a variable group that is spread widely across the Holarctic region from the dry subtropics to the boreal zone (Friesen et al., 2006; Li et al., 2010). Furthermore, species in Allioideae grow in various conditions from dry and well-drained soils to moist and organic soils, with most growing in sunny locations, and a number of species also grow in forests, or even in swamps or water (Block, 2010); The environment imposes stressful living conditions on Allioideae species and results in species divergence. The Ka/Ks ratios have been widely used to infer the evolutionary dynamics and identify adaptive signatures among species (Yang and Bielawski, 2000; Fay and Wu, 2003; Ai et al., 2015), and elevated Ka/Ks ratios indicate species may have undergone more selective forces (Hurst, 2002). Thus, the elevated Ka/Ks ratios observed throughout the Allioideae may suggest species in Allioideae undergo some selection pressure that is unknown.
Gene losses and gains are considered as important adaptive processes that greatly contribute to trait evolution (Hahn et al., 2007; Ding et al., 2012). In this study, we found that the gene rps2 was lost in all Allium (Allioideae) species, with infA, rps16, psbZ, ndhD, cemA, rps19, ycf1, and rpl32 were lost in some species of Allioideae, Asparagaceae and Asphodeloideae (Figure 6). Gene infA, which codes for translation initiation factor 1, was lost in an early ancestor of Fabales and Cucurbitales (Millen et al., 2001), and it was found as pseudogene in many genus (e.g., Albuca, Behnia, Camassia, and Echeandia) in study of McKain et al. (2016). The study of Steele et al. (2012) identified that gene rps16, rpl32, and rps19 were missing from various taxa throughout Asparagales, and these shared losses were suggested as the result of common ancestry. The gene rps2 was also identified as a pseudogene in Chlorophytum rhizopendulum (McKain et al., 2016). However, the mechanism underlying the loss of the rps2 gene in Allioideae was poorly understood. A previous study indicated that the product of the rps2 gene plays an important role in defense signal transduction (Bent et al., 1994). Although we detected the loss of the rps2 gene in Allioideae, we did not find a cause. Therefore, further studies are needed to examine whether specific factors were associated with the loss of the rps2 genes in the Allioideae.
Positive Selection of Allium (Allioideae) Plastome
We investigated PSGs to detect genes in the Allioideae lineage that may have evolved to adapt to environmental conditions. Ten genes with significant posterior probabilities for codon sites were identified in the BEB test, although the positive selection was not significant in all genes (p-value > 0.05) (Figure 8, Supplementary Figure S4, and Table 2). Yang et al. (2005) suggested that codon sites with higher posterior probability can be regarded as positively selected sites, and genes that possessing the positively selected sites may be evolving under divergent selective pressures, which indicate that these ten genes may be under positive selection pressure. Notably, we found that seven of these ten genes are associated with photosystem I and II subunits (psbD, psbE, psbM, and psaI), NADH-dehydrogenase subunits (ndhE and ndhG) and subunits of cytochrome b/f complex (petA) (Table 2). Photosystem I and II are sites of the photosynthetic light reactions of plants (Golbeck, 1987), and are integral membrane protein complexes that use light energy to produce the high energy carriers ATP and NADPH (Weiss et al., 1991; Yamori and Shikanai, 2016). NADH-dehydrogenase subunits and cytochrome b/f complex are essential in the electron transport chain for generation of ATP (Weiss et al., 1991; Cramer et al., 2011; Xiao et al., 2012), and are all important components for photosynthesis of plants. Therefore, all these genes are indispensable components for photosynthesis, which is the most important process for plant growth and development (Bryant and Frigaard, 2006). Among all PSGs, we found that the rpoC1 gene possessed the maximum number of sites under positive selection in Allioideae species (Figure 8, Table 2, and Supplementary Figure S4). This suggests that the rpoC1 gene may play a pivotal role in the adaptive evolution of Allioideae species. We also observed site-specific selection in rps4 gene that has important role in the chloroplast ribosome (Rogalski et al., 2006; Tiller and Bock, 2014). Most of genes mentioned above have been reported under positive selection in previous studies (Dong et al., 2018; Fan et al., 2018; Wu et al., 2018). Species in Allioideae are mostly characterized by tunicated bulbs and narrow basal leaves (Li et al., 2010), and these are key traits that likely contributed to their adaptation to diverse harsh environments, and generated and maintained high levels of plant diversity. The results of high Ka/Ks ratios also suggested positive selection existed in Allioideae species (Figure 4 and Supplementary Table S7). Consequently, most PSGs may have played key roles in the adaptation of species in the Allioideae during the evolution process.
Author Contributions
D-FX, YY, X-JH, and Y-QD conceived and designed the experiments. D-FX, Y-QD, YY, and H-XY analyzed the sequence data and drafted the manuscript. D-FX, S-DZ, H-XY, CX, and J-PC participated in data analysis and manuscript drafting. D-FX, MP, X-JH, and YY revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31872647, 31570198, and 31500188) and the Chinese Ministry of Science and Technology through the National Science and Technology Infrastructure Platform Project (Grant No. 2005DKA21403-JK).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge Hao Li, Fu-Min Xie, and Xin Yang for their help in materials collection. We would like to thank Jun Wen, Juan Li, and Jiao Huang for their help in software use.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00460/full#supplementary-material
FIGURE S1 | Analysis of simple sequence repeats (SSRs) in chloroplast genomes of Section Daghestanica species. Number of different SSR types detected in each species.
FIGURE S2 | The nucleotide diversity of the whole chloroplast genomes of Section Daghestanica species. LSC, large single-copy region; IRs, inverted repeats region; SSC, small single-copy region.
FIGURE S3 | Phylogenetic tree reconstruction of 41 taxa based on (A) the single copy gene sequences and (B) the CDS sequences. Numbers above the branches are the posterior probabilities of BI and the bootstrap values of ML, respectively.
FIGURE S4 | Partial alignment of amino acids sequences in another eight positively selected genes. (A–H): psaI, psbD, psbE, psbM, ndhE, and ndhG, ycf3 and rps4. The red blocks stand for the amino acids in Allioideae with a high BEB posterior probability.
TABLE S1 | Information for sample collection.
TABLE S2 | Primers used for gap closure in this study.
TABLE S3 | The Genbank accessions of all 41 taxa cp genome sequences used this study.
TABLE S4 | Gene content in Section Daghestanica species cp genomes. ∗Genes with two copies.
TABLE S5 | Summary of complete chloroplast genomes of all 41 taxa used in this study.
TABLE S6 | The statistics of codon usage bisa in all 41 taxa used in this study.
TABLE S7 | Summary of Pairwise Ka/Ks ratios in Allium (Allioideae) and other families.
TABLE S8 | Statistics of simple sequence repeats in each species of Section Daghestanica.
TABLE S9 | List of single-copy genes extracted from cp genome.
Footnotes
References
Ai, B., Gao, Y., Zhang, X. L., Tao, J. J., Kang, M., and Huang, H. W. (2015). Comparative transcriptome resources of eleven Primulina species, a group of “stone plants” from a biodiversity hotspot. Mol. Ecol. Res. 15, 619–632. doi: 10.1111/1755-0998.12333
And, K. D., and Voelker, L. R. L. (2003). Molecular biology of mycoplasmas. Annu. Rev. Microbiol. 50, 25–57.
Bellusci, F., Pellegrino, G., Palermo, A. M., and Musacchio, A. (2008). Phylogenetic relationships in the orchid genus Serapias L. based on noncoding regions of the chloroplast genome. Mol. Phylogenet. Evol. 47, 986–991. doi: 10.1016/j.ympev.2008.03.019
Bent, A. F., Kunkel, B. N., Dahlbeck, D., Brown, K. L., Schmidt, R., Giraudat, J., et al. (1994). RPS2 of Arabidopsis thaliana: a Leucine-rich repeat class of plant disease resistance genes. Science 265, 1856–1860. doi: 10.1126/science.8091210
Block, E. (2010). Garlic and Other Alliums: The Lore and the Science. Cambridge: Royal Society of Chemistry.
Bryant, D. A., and Frigaard, N. U. (2006). Prokaryotic photosynthesis and phototrophy illuminated. Trends Microbiol. 14, 488–496. doi: 10.1016/j.tim.2006.09.001
Capellagutiérrez, S., Sillamartínez, J. M., and Gabaldón, T. (2009). Trimal: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chase, M. W., Christenhusz, M. J. M., Fay, M. F., Byng, J. W., Judd, W. S., Soltis, D. E., et al. (2016). An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1016/j.jep.2015.05.035
Chen, X. Q., Liang, S. Y., Xu, J. M., and Minoru, N. T. (2000). “Liliaceae,” in Flora of China, Vol. 24, eds Z. Y. Wu and P. H. Raven (Beijing: Science Press), 73–263.
Christoph, H., Lutz, B., and Bastien, C. (2013). Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129. doi: 10.1093/nar/gkt371
Clamp, M., Cuff, J., Searle, S. M., and Barton, G. J. (2004). The Jalview Java alignment editor. Bioinformatics 20, 426–427. doi: 10.1093/bioinformatics/btg430
Cramer, W. A., Yamashita, E., Baniulis, D., and Hasan, S. S. (2011). The Cytochrome b6f Complex of Oxygenic Photosynthesis. Hoboken, NJ: John Wiley Sons, Ltd.
Ding, Y., Zhou, Q., and Wang, W. (2012). Origins of new genes and evolution of their novel functions. Annu. Rev. Ecol. Evol. Syst. 43, 345–363. doi: 10.1146/annurev-ecolsys-110411-160513
Dong, W., Liu, J., Yu, J., Wang, L., and Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One 7:e35071. doi: 10.1371/journal.pone.0035071
Dong, W. L., Wang, R. N., Zhang, N. Y., Fan, W. B., Fang, M. F., and Li, Z. H. (2018). Molecular evolution of chloroplast genomes of Orchid species: insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 19:716. doi: 10.3390/ijms19030716
Doyle, J. J. (1987). A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 19:113. doi: 10.1186/1471-2105-5-113
Erixon, P., and Oxelman, B. (2008). Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the chloroplast clpP1 gene. PLoS One 3:e1386. doi: 10.1371/journal.pone.0001386
Fan, W. B., Wu, Y., Yang, J., Shahzad, K., and Li, Z. H. (2018). Comparative chloroplast genomics of Dipsacales species: insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front. Plant Sci. 9:689. doi: 10.3389/fpls.2018.00689
Fay, J. C., and Wu, C. (2003). Sequence divergence, functional constraint, and selection in protein evolution. Annu. Rev. Genomics Hum. Genet. 4, 213–235. doi: 10.1146/annurev.genom.4.020303.162528
Filyushin, M. A., Beletsky, A. V., Mazur, A. M., and Kochieva, E. Z. (2016). The complete plastid genome sequence of garlic Allium sativum L. Mitochondrial DNA B Resour. 1, 831–832. doi: 10.1080/23802359.2016.1247669
Filyushin, M. A., Beletsky, A. V., Mazur, A. M., and Kochieva, E. Z. (2018). Characterization of the complete plastid genome of lop-sided onion Allium obliquum L. (Amaryllidaceae). Mitochondrial DNA B Resour. 3, 393–394. doi: 10.1080/23802359.2018.1456369
Friesen, N., Fritsch, R. M., and Blattner, F. R. (2006). Phylogeny and intrageneric classification of Allium (Alliaceae) based on nuclear ribosomal DNA ITS sequences. Aliso 22, 372–395. doi: 10.5642/aliso.20062201.31
Golbeck, J. H. (1987). Structure, function and organization of the photosystem I reaction center complex. Biochim. Biophys. Acta 895, 167–204. doi: 10.1016/S0304-4173(87)80002-2
Hahn, M. W., Demuth, J. P., and Han, S. G. (2007). Accelerated rate of gene gain and loss in Primates. Genetics 177, 1941–1949. doi: 10.1534/genetics.107.080077
Hall, B. K., Hallgrímsson, B., and Strickberger, M. W. (2008). Strickberger’s Evolution: the Integration of Genes, Organisms and Populations. Burlington, MA: Jones & Bartlett Learning.
Herden, T., Hanelt, P., and Friesen, N. (2016). Phylogeny of Allium L. subgenus Anguinum (G. Don. ex W.D.J. Koch) N. Friesen (Amaryllidaceae). Mol. Phylogenet. Evol. 95, 79–93. doi: 10.1016/j.ympev.2015.11.004
Howe, C. J., Barbrook, A. C., Koumandou, V. L., Nisbet, R. E., Symington, H. A., and Wightman, T. F. (2003). Evolution of the chloroplast genome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 99–107. doi: 10.1098/rstb.2002.1176
Hu, Y., Woeste, K. E., and Zhao, P. (2016). Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 7:1955. doi: 10.3389/fpls.2016.01955
Hurst, L. D. (2002). The Ka/ Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18, 486–487. doi: 10.1016/S0168-9525(02)02722-1
Ivanova, Z., Sablok, G., Daskalova, E., Zahmanova, G., Apostolova, E., Yahubyan, G., et al. (2017). Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Front. Plant Sci. 8:204. doi: 10.3389/fpls.2017.00204
Jin, F. Y., Xie, D. F., Zhou, S. D., and He, X. J. (2018). Characterization of the complete chloroplast genome of Allium prattii. Mitochondrial DNA B Resour. 3, 153–154. doi: 10.1080/23802359.2018.1436994
Kapralov, M. V., Kubien, D. S., Andersson, I., and Filatov, D. A. (2011). Changes in Rubisco kinetics during the evolution of C4 photosynthesis in Flaveria (Asteraceae) are associated with positive selection on genes encoding the enzyme. Mol. Biol. Evol. 28, 1491–1503. doi: 10.1093/molbev/msq335
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Khakhlova, O., and Bock, R. (2010). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j.1365-313X.2006.02673.x
Kim, K. J., and Lee, H. L. (2005). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kim, S., Park, J. Y., and Yang, T. (2015). Comparative analysis of the complete chloroplast genome sequences of a normal male-fertile cytoplasm and two different cytoplasms conferring cytoplasmic male sterility in onion (Allium cepa L.). J. Hortic. Sci. Biotechnol. 90, 459–468. doi: 10.1080/14620316.2015.11513210
Kim, S., and Yoon, M. K. (2010). Comparison of mitochondrial and chloroplast genome segments from three onion (Allium cepa L.) cytoplasm types and identification of a trans-splicing intron of cox2. Curr. Genet. 56, 177–188. doi: 10.1007/s00294-010-0290-6
Kusumi, J., and Tachida, H. (2005). Compositional properties of green-plant plastid genomes. J. Mol. Evol. 60, 417–425. doi: 10.1007/s00239-004-0086-8
Lan, Y., Sun, J., Tian, R., Bartlett, D. H., Li, R., Wong, Y. H., et al. (2017). Molecular adaptation in the world’s deepest-living animal: insights from transcriptome sequencing of the hadal amphipod Hirondellea gigas. Mol. Ecol. 26, 3732–3743. doi: 10.1111/mec.14149
Lane, C. E., Van den Heuvel, K., Kozera, C., Curtis, B. A., Parsons, B. J., Bowman, S., et al. (2007). Nucleomorph genome of Hemiselmis andersenii reveals complete intron loss and compaction as a driver of protein structure and function. Proc. Natl. Acad. Sci. U.S.A. 104, 19908–19913. doi: 10.1073/pnas.0707419104
Lee, J., Chon, J. K., Lim, J. S., Kim, E. K., and Nah, G. (2017). Characterization of complete chloroplast genome of Allium victorialis and its application for barcode markers. Plant Breed. Biotechnol. 5, 221–227. doi: 10.9787/PBB.2017.5.3.221
Li, Q. Q., Zhou, S. D., He, X. J., Yu, Y., Zhang, Y. C., and Wei, X. Q. (2010). Phylogeny and biogeography of Allium (Amaryllidaceae: Allieae) based on nuclear ribosomal internal transcribed spacer and chloroplast rps16 sequences, focusing on the inclusion of species endemic to China. Ann. Bot. 106, 709–733. doi: 10.1093/aob/mcq177
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). Organellar genome DRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
McKain, M. R., Mcneal, J. R., Kellar, P. R., Eguiarte, L. E., Pires, J. C., and Leebens-Mack, J. (2016). Timing of rapid diversification and convergent origins of active pollination within Agavoideae (Asparagaceae). Am. J. Bot. 103, 1717–1729. doi: 10.3732/ajb.1600198
Millen, R. S., Olmstead, R. G., Adams, K. L., Palmer, J. D., Lao, N. T., Heggie, L., et al. (2001). Many parallel losses of infA from chloroplast DNA during Angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13, 645–658. doi: 10.1105/tpc.13.3.645
Morton, B. R. (1993). Chloroplast DNA codon use: evidence for selection at the psbA locus based on tRNA availability. J. Mol. Evol. 37, 273–280. doi: 10.1007/BF00175504
Morton, B. R. (1998). Selection on the codon bias of chloroplast and cyanelle genes in different plant and algal lineages. J. Mol. Evol. 46, 449–459. doi: 10.1007/PL00006325
Nguyen, N. H., Driscoll, H. E., and Specht, C. D. (2008). A molecular phylogeny of the wild onions (Allium; Alliaceae) with a focus on the western North American center of diversity. Mol. Phylogenet. Evol. 47, 1157–1172. doi: 10.1016/j.ympev.2007.12.006
Ogata, H., Audic, S., Renestoaudiffren, P., Fournier, P. E., Barbe, V., Samson, D., et al. (2001). Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science 293, 2093–2098.
Parks, M., Cronn, R., and Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84. doi: 10.1186/1741-7007-7-84
Pauwels, M., Vekemans, X., Godé, C., Frérot, H., Castric, V., and Saumitoulaprade, P. (2012). Nuclear and chloroplast DNA phylogeography reveals vicariance among European populations of the model species for the study of metal tolerance, Arabidopsis halleri (Brassicaceae). New Phytol. 193, 916–928. doi: 10.1111/j.1469-8137.2011.04003.x
Powell, W., Morgante, M., Mcdevitt, R., Vendramin, G. G., and Rafalski, J. A. (1995). Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proc. Natl. Acad. Sci. U.S.A. 92, 7759–7763. doi: 10.1073/pnas.92.17.7759
Provan, J., Corbett, G., Mcnicol, J. W., and Powell, W. (1997). Chloroplast DNA variability in wild and cultivated rice (Oryza spp.) revealed by polymorphic chloroplast simple sequence repeats. Genome 40, 104–110. doi: 10.1139/g97-014
Qian, J., Song, J., Gao, H., Zhu, Y., Xu, J., Pang, X., et al. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS One 8:e57607. doi: 10.1371/journal.pone.0057607
Raubeson, L. A., Jansen, R. K., and Henry, R. J. (2005). “Chloroplast genomes of plants,” in Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants, ed. R. J. Henry (Cambridge, MA: CABI Press), 45–68. doi: 10.1079/9780851999043.0045
Rogalski, M., Ruf, S., and Bock, R. (2006). Tobacco plastid ribosomal protein S18 is essential for cell survival. Nucleic Acids Res. 34, 4537–4545. doi: 10.1093/nar/gkl634
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Scottphillips, T. C., Laland, K. N., Shuker, D. M., Dickins, T. E., and West, S. A. (2014). The niche construction perspective: a critical appraisal. Evolution 68, 1231–1243. doi: 10.1111/evo.12332
Smith, D. R. (2009). Unparalleled GC content in the plastid DNA of Selaginella. Plant Mol. Biol. 71, 627–639. doi: 10.1007/s11103-009-9545-3
Smith, D. R., and Lee, R. W. (2008). Mitochondrial genome of the colorless green alga Polytomella capuana: a linear molecule with an unprecedented GC content. Mol. Biol. Evol. 25, 487–496. doi: 10.1093/molbev/msm245
Song, Y., Dong, W., Liu, B., Xu, C., Yao, X., Gao, J., et al. (2015). Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machilus balansae in the family Lauraceae. Front. Plant Sci. 6:662. doi: 10.3389/fpls.2015.00662
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Steele, P. R., Hertweck, K. L., Mayfield, D., Mckain, M. R., Leebens-Mack, J., and Pires, J. C. (2012). Quality and quantity of data recovered from massively parallel sequencing: examples in Asparagales and Poaceae. Am. J. Bot. 99, 330–348. doi: 10.3732/ajb.1100491
Thiel, T., Michalek, W., Varshney, R. K., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Tiller, N., and Bock, R. (2014). The Translational apparatus of plastids and its role in plant development. Mol. Plant 7, 1105–1120. doi: 10.1093/mp/ssu022
Wang, D., Zhang, Y., Zhang, Z., Jiang, Z., and Yu, J. (2010). KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8, 77–80. doi: 10.1016/S1672-0229(10)60008-3
Weiss, H., Friedrich, T., Hofhaus, G., and Preis, D. (1991). The respiratory-chain NADH dehydrogenase (complex I) of mitochondria. Eur. J. Biochem. 3, 563–576. doi: 10.1111/j.1432-1033.1991.tb15945.x
Wolfe, K. H., Li, W. H., and Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. U.S.A. 84, 9054–9058. doi: 10.1073/pnas.84.24.9054
Wu, Y., Liu, F., Yang, D. G., Li, W., Zhou, X. J., Pei, X. Y., et al. (2018). Comparative chloroplast genomics of Gossypium species: insights into repeat sequence variations and phylogeny. Front. Plant Sci. 9:376. doi: 10.3389/fpls.2018.00376
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xiao, J. W., Li, J., Ouyang, M., Yun, T., He, B. Y., Ji, D. L., et al. (2012). DAC is involved in the accumulation of the cytochrome b6/f complex in Arabidopsis. Plant Physiol. 160, 1911–1922. doi: 10.1104/pp.112.204891
Xie, D. F., Yu, Y., Deng, Y. Q., Li, J., Liu, H. Y., Zhou, S. D., et al. (2018). Comparative analysis of the chloroplast genomes of the Chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. Int. J. Mol. Sci. 19:E1847. doi: 10.3390/ijms19071847
Xu, J., Xue, P., Zhu, S., and Jing, W. (1994). A new species of Allium L. from Sichuan and its karyotype. Acta Phytotaxa Sin. 32, 356–358.
Yamori, W., and Shikanai, T. (2016). Physiological functions of cyclic electron transport around Photosystem I in sustaining photosynthesis and plant growth. Annu. Rev. Plant Biol. 67, 81–106. doi: 10.1146/annurev-arplant-043015-112002
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., and Bielawski, J. P. (2000). Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 15, 496–503. doi: 10.1016/S0169-5347(00)01994-7
Yang, Z., and Dos, R. M. (2011). Statistical properties of the branch-site test of positive selection. Mol. Biol. Evol. 28, 1217–1228. doi: 10.1093/molbev/msq303
Yang, Z., and Nielsen, R. (2002). Codon-Substitution Models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 19, 908–917. doi: 10.1093/oxfordjournals.molbev.a004148
Yang, Z., Wong, W. S. W., and Nielsen, R. (2005). Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. doi: 10.1093/molbev/msi097
Yu, H. X., Guo, X. L., Zhou, S. D., and He, X. J. (2018). Pollen and seed micro-morphology comparison and taxonomic significance of the Chinese Allium Sect. Daghestanica. Acta Bot. Boreal Occident Sin. 38, 0061–0067.
Keywords: Allium, Allioideae, chloroplast genome, phylogeny analyses, adaptive evolution, positive selection
Citation: Xie D-F, Yu H-X, Price M, Xie C, Deng Y-Q, Chen J-P, Yu Y, Zhou S-D and He X-J (2019) Phylogeny of Chinese Allium Species in Section Daghestanica and Adaptive Evolution of Allium (Amaryllidaceae, Allioideae) Species Revealed by the Chloroplast Complete Genome. Front. Plant Sci. 10:460. doi: 10.3389/fpls.2019.00460
Received: 23 December 2018; Accepted: 27 March 2019;
Published: 30 April 2019.
Edited by:
Michael R. McKain, The University of Alabama, United StatesReviewed by:
Nikolai Friesen, University of Osnabrück, GermanyCarolina Carrizo García, Instituto Multidisciplinario de Biologia Vegetal (IMBIV), Argentina
Copyright © 2019 Xie, Yu, Price, Xie, Deng, Chen, Yu, Zhou and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xing-Jin He, eGpoZUBzY3UuZWR1LmNu