 Jillian M. Lang1,2†
Jillian M. Lang1,2† Alvaro L. Pérez-Quintero1,2†
Alvaro L. Pérez-Quintero1,2† Ralf Koebnik2
Ralf Koebnik2 Elysa DuCharme1
Elysa DuCharme1 Soungalo Sarra3
Soungalo Sarra3 Hinda Doucoure4
Hinda Doucoure4 Ibrahim Keita4Janet Ziegle5
Ibrahim Keita4Janet Ziegle5 Jonathan M. Jacobs1,2,6
Jonathan M. Jacobs1,2,6 Ricardo Oliva7Ousmane Koita4
Ricardo Oliva7Ousmane Koita4 Boris Szurek2
Boris Szurek2 Valérie Verdier1,2*
Valérie Verdier1,2* Jan E. Leach1*
Jan E. Leach1*- 1Department of Bioagricultural Sciences and Pest Management, Colorado State University, Fort Collins, CO, United States
- 2IRD, Cirad, Univ. Montpellier, IPME, Montpellier, France
- 3Centre Régional de Recherche Agronomique de Niono, Institut d’Economie Rural, Bamako, Mali
- 4Laboratoire de Biologie Moléculaire Appliquée, Université des Sciences Techniques et Technologiques de Bamako, Bamako, Mali
- 5Pacific Biosciences, Menlo Park, CA, United States
- 6Department of Plant Pathology, Infectious Disease Institute, Ohio State University, Columbus, OH, United States
- 7International Rice Research Institute, Los Baños, Philippines
Xanthomonas oryzae (Xo) are globally important rice pathogens. Virulent lineages from Africa and Asia and less virulent strains from the United States have been well characterized. Xanthomonas campestris pv. leersiae (Xcl), first described in 1957, causes bacterial streak on the perennial grass, Leersia hexandra, and is a close relative of Xo. L. hexandra, a member of the Poaceae, is highly similar to rice phylogenetically, is globally ubiquitous around rice paddies, and is a reservoir of pathogenic Xo. We used long read, single molecule real time (SMRT) genome sequences of five strains of Xcl from Burkina Faso, China, Mali, and Uganda to determine the genetic relatedness of this organism with Xo. Novel transcription activator-like effectors (TALEs) were discovered in all five strains of Xcl. Predicted TALE target sequences were identified in the Leersia perrieri genome and compared to rice susceptibility gene homologs. Pathogenicity screening on L. hexandra and diverse rice cultivars confirmed that Xcl are able to colonize rice and produce weak but not progressive symptoms. Overall, based on average nucleotide identity (ANI), type III (T3) effector repertoires, and disease phenotype, we propose to rename Xcl to X. oryzae pv. leersiae (Xol) and use this parallel system to improve understanding of the evolution of bacterial pathogenicity in rice agroecosystems.
Introduction
Rice is a staple crop for more than half the world. Severe rice diseases, such as bacterial leaf streak (BLS) caused by Xanthomonas oryzae pv. oryzicola (Xoc) and bacterial blight (BB), caused by X. o. pv. oryzae (Xoo), are increasing in prevalence in parts of Asia and sub-Saharan Africa and cause significant yield losses. In Asia, perennial weeds are considered an important source of primary pathogen inoculum for these two diseases (Ou, 1985; Mew, 1987).
Southern cutgrass (Leersia hexandra Swartz) is a common grass found in the southern United States, South America, Africa, and Asia. It is a member of the Poaceae family and is closely related to rice, but diverged from Oryza approximately 14 mya (Guo and Ge, 2005). L. hexandra is an invasive species that frequently grows along rivers and canals surrounding rice paddies. Because of its close relationship to Oryza spp., Leersia spp. are included as outgroups in phylogenetic studies. Recent genome investigations of Leersia perrieri, a cutgrass found in Madagascar, were done to compare repetitive elements and transposable elements among Oryza sp. and to uncover orthologs of the important submergence tolerance gene, SUB1 (Copetti, 2013; Copetti et al., 2015; dos Santos et al., 2017). One-third of the L. perrieri genome was found to consist of repeats. The high amount of newly discovered repeats (35%) indicates that the L. perrieri genome is evolving rapidly relative to the Oryza genus (Copetti, 2013).
Early reports of phytoremediation by L. hexandra showed this grass’ capacity to sequester Cr, Cu, and Ni, and it has now been proposed as a tool in wastewater treatment (Liu et al., 2011; Wang et al., 2012; You et al., 2013). Interestingly, this and other grasses in this genus, such as Leersia sayanuka, Leersia oryzoides, and Leersia japonica, are susceptible to Xoo (Noda and Yamamoto, 2008) and can serve as reservoirs for inoculum. Xoo strains isolated from symptomless L. hexandra cause BB symptoms in rice, and, in artificially inoculated weed plants, Xoo multiplied without evidence of disease (Gonzalez et al., 1991) implicating this grass as an alternative host for the pathogen. These and other findings reinforce that effective integrated management of crop diseases must incorporate knowledge of pathogen interactions with weedy species.
The species Xo is highly diverse, and is represented by distinct lineages of Xoo from Asia and Africa, Xoc from Asia and Africa, and strains not assigned as a pathovar from the United States, Xo (Gonzalez et al., 2007; Triplett et al., 2011; Hajri et al., 2012; Poulin et al., 2015; Triplett and Leach, 2016). The term pathovar is used to refer to a strain or set of strains with the same or similar characteristics, differentiated at infrasubspecific level from other strains of the same species or subspecies on the basis of distinctive pathogenicity to one or more plant hosts1. Poulin et al. (2015) used multi-locus variable-number tandem-repeat analysis (MLVA) to investigate genetic structures of microbial populations (Zhao et al., 2012; Poulin et al., 2015), and suggested that Xoo and Xoc from Africa had a common Asian ancestor; this conclusion was based on the fact that the allelic richness, or number of alleles, was significantly less in these populations. However, further analyses on an extensively sampled set of isolates are needed to confirm this ancestral hypothesis.
Xanthomonas spp. inject effector proteins into plant host cells to elicit disease via a type-III (T3) secretion system (White et al., 2009). These proteins can confer pathogenicity and/or dictate host specificity (Jacques et al., 2016). Xanthomonas spp. are most notable for production of transcription activator-like effectors (TALEs). TALEs influence host gene expression by directly binding to specific sequences [effector binding elements (EBEs)] in the target promoter as dictated by repeat-variable di-residues (RVDs) at the 12 and 13 amino acid position in the central repeat region (CRR) (Boch et al., 2009; Moscou and Bogdanove, 2009; White et al., 2009; Bogdanove and Voytas, 2011). The CRR contains different numbers of repeats, each with 33–35 amino acids. TALEs may enhance diseases by targeting susceptibility (S) genes, or may trigger a resistance response through activation of an “executor” resistance gene (R) expression (Boch et al., 2014; Hutin et al., 2015).
The presence of TALE effectors is variable in the genus (Jacques et al., 2016). Xoo contain nine to 20 TALEs while Xoc can contain up to 29. US Xo do not contain any TALEs, and due to this absence, have been employed as a tool to study TALE effector biology in rice (Ryba-White et al., 1995; Verdier et al., 2012). New sequencing technologies and predictive algorithms have accelerated the characterization of TALEs and their host gene targets. In particular, long read, single molecule real time (SMRT) sequencing (Pacific Biosciences, Menlo Park, CA, United States) has enabled the rapid assembly of TALE sequences that are otherwise laborious to capture due to their highly repetitive structure (Eid et al., 2009; Booher et al., 2015; Wilkins et al., 2015; Grau et al., 2016; Peng et al., 2016; Quibod et al., 2016; Tran et al., 2018). Collectively, TALE repertoires (TALomes) encoding polymorphic groups that have contrasted abilities to induce susceptibility target genes potentially underlie host adaptation at a small evolutionary scale (Doucouré et al., 2018). The Xo group has clearly undergone significant evolution influenced by geography, environment, and host, and TALomes can provide critical insight into how these events occur.
Xanthomonas campestris pv. leersiae (Xcl), a pathogen of L. hexandra, was previously shown to group distinctly from Xo by host range and phylogenetic analysis (Fang et al., 1957; Parkinson et al., 2009). However, using a multi-locus sequence alignment (MLSA) analysis Triplett et al. (2011) showed that Xcl strain NCPPB4346, which was isolated from southern cutgrass in China, groups within the Xo cluster, yet it could not be assigned to any described pathovar. A more recently isolated strain, BAI23 from weeds in Burkina Faso, showed high sequence similarity with Xcl NCPPB4346 based on a MLSA analysis as well as the presence of TALEs (Wonni et al., 2014). Together, the two strains form a distinct genetic cluster within X. oryzae.
Prediction algorithms based on the TALE’s specific RVD pattern and their corresponding degenerate DNA code has facilitated identification of plant target genes whose promoters contain EBEs for TALE binding (Doyle et al., 2012; Grau et al., 2013, 2016; Pérez-Quintero et al., 2013; Booher and Bogdanove, 2014; Yang et al., 2014). Many S genes are transporters (sugar or sulfate) or transcription factors and upon induction facilitate bacterial colonization and symptom development (Hutin et al., 2015; Tran et al., 2018). Although a large body of work is available on TALEs from Xoo and Xoc, no information has been reported on the TALEs from Xcl, how they compare to those in other X. oryzae, and the nature of their predicted targets within Leersia spp. In this study, we used comparative genomics, identification of T3 effectors, TALomes, and disease phenotyping to characterize Xcl. We used gene target prediction algorithms to identify potential Xcl TALE gene targets in draft Leersia genome sequences. Finally, we provide evidence to support renaming Xcl to X. oryzae pv. leersiae (Fang et al., 1957) and will refer to this organism as Xol throughout this work.
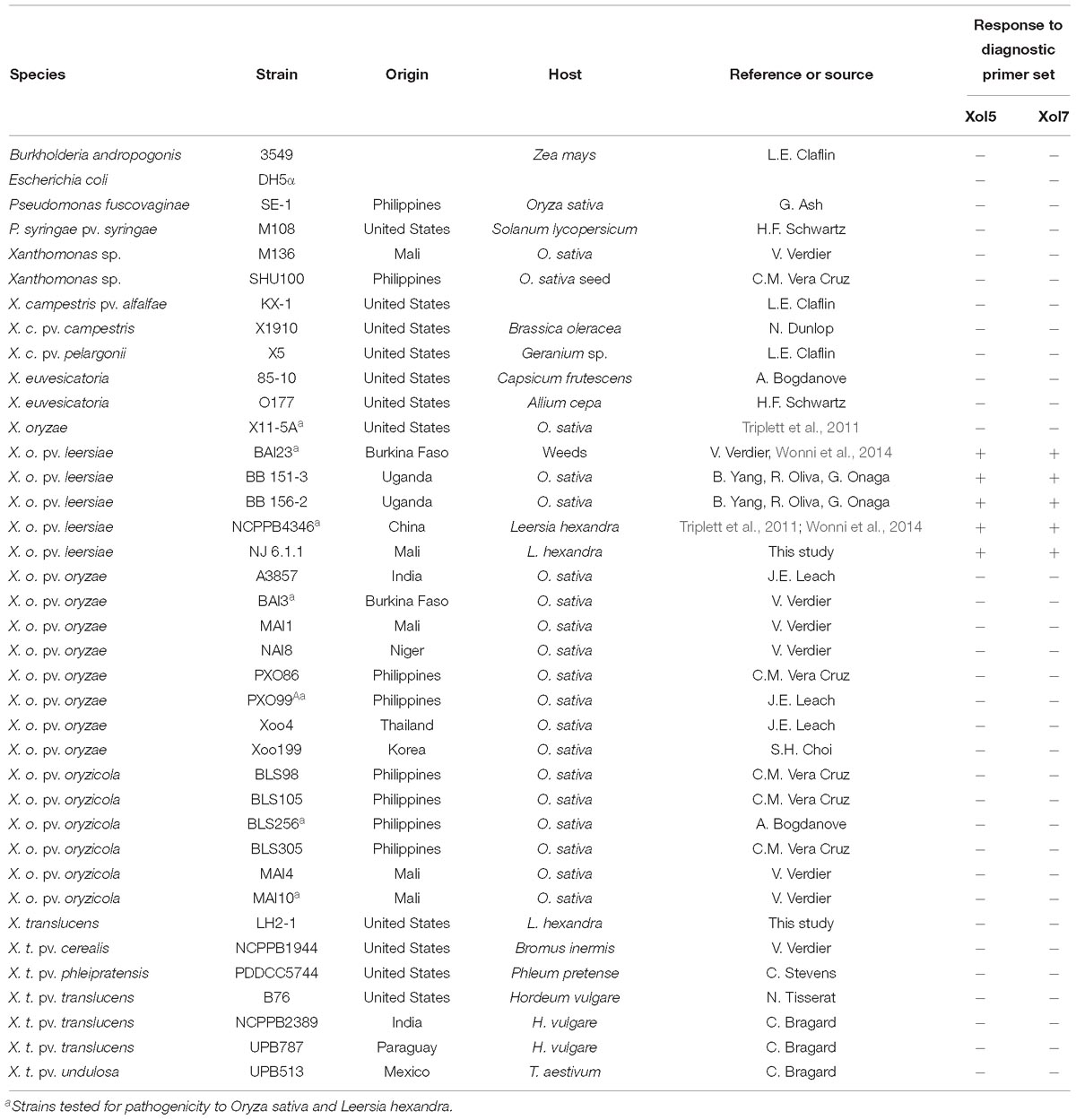
Table 1. Bacterial strains used in phenotyping and molecular diagnostics.
Materials and Methods
Bacterial Strains and Plant Varieties
Bacterial strains included in this study are listed in Table 1. Bacteria were cultured on peptone sucrose agar (PSA) at 28°C for plant inoculations. Genomic DNA for sequencing was isolated from Xol strains BAI23, BB 151-3, BB 156-2, NCPPB4346, and NJ 6.1.1 grown for 48 h on nutrient agar at 28°C (Lang et al., 2014; Wonni et al., 2014). Barley (Hordeum vulgare L. cultivar Morex), wheat (Triticum aestivum cv. Chinese Spring) and tobacco (Nicotiana tabacum) were grown in a growth chamber at 22°C, 50% relative humidity, and 16 h of light. Rice (Oryza sativa) varieties included in pathogenicity assays were Azucena, Carolina Gold, Cypress, IR64, and Nipponbare.
Southern cutgrass (L. hexandra) was collected in Texas, United States, and seed was propagated at Colorado State University. The seed was scarified then germinated in porous ceramic silica (Greens Grade, Profile Products, LLC, Buffalo Grove, IL, United States) and 0.5x Hoagland’s solution with the following modifications: 2.5 mM KNO3, 1 mM MgSO4, 3 mM KH2PO4, 2.5 Ca(NO3)2, 0.05 mM FeSO4, and 0.1 mM (Na)2EDTA (Hoagland and Arnon, 1950). Seed was incubated in a petri dish in the dark for 8 days, then in the light for 30 days at 28°C. Germinated seeds were transplanted into 1-gallon pots with equal parts Greens Grade and ProMix BX (ProMix, Quakertown, PA, United States), and grown in a greenhouse (27 ± 1°C, 16 h day length, and 80–85% relative humidity). Additional plants were obtained from the two mother plants via rhizome propagation. Propagation began 30 days after planting with subsequent propagation every week to allow time for new growth. To promote root growth, plants were placed in the dark for 24 h.
Pathogenicity Assays
Rice varieties were inoculated at 4–5 week-old with suspensions (108 CFU mL−1) of bacterial strains listed in Table 1. Bacterial suspensions were both infiltrated into the intercellular spaces of rice leaves on either side of the main vein with a needleless syringe and inoculated by leaf clipping as described (Kauffman et al., 1973; Reimers and Leach, 1991). Two leaves were inoculated on each of three to six separate plants; water was included as a negative control. The entire experiment was conducted twice. Lesions were measured at 12 days post inoculation (dpi), and bacterial numbers in planta were quantified as previously described (Verdier et al., 2012).
Molecular Diagnostics
To test relationships of Xol to Xo (Notomi et al., 2000; Triplett et al., 2011; Wonni et al., 2014), a diagnostic multiplex and loop mediated isothermal amplification (LAMP) PCR were used (Lang et al., 2010, 2014; Wonni et al., 2014). Previously described universal US Xo primers were also tested to differentiate Xol from this novel US clade within the species (Triplett et al., 2011). UniqPrimer was employed to compare draft Xol genomes (BAI23 and NCPPB4346) and generate primers specific to Xol as previously described (Ash et al., 2014; Lang et al., 2014, 2017; Juanillas et al., 2018). Specificity was validated by screening Xol primers against diverse pools of bacterial genomic DNA (Table 1). Primers, expected product size, and optimal annealing temperatures are listed in Supplementary Table S1.
Genome Sequencing and Assembly
Long read, SMRT sequencing (PacBio, Menlo Park, CA, United States) data were generated for five Xol strains and for the Xo strain X11-5A, to be used as an outgroup. DNA for SMRT sequencing was isolated by culturing strains on nutrient agar for 48 h then using the Genomic DNA buffer set and Genomic-tips according manufacturer instructions (Qiagen, Valencia, CA, United States). SMRT sequence was assembled using HGAP v4 (PacBio, Menlo Park, CA, United States). Genomes were circularized using circulator (Hunt et al., 2015). Assemblies and raw data have been deposited in NCBI (BioProject IDs PRJNA522807 and PRJNA522811; BioSample accessions SAMN03862116, SAMN02469650, SAMN10956066-68, SAMN10956070; raw sequencing files SRX5417793-98; Assembly accessions CP036251-56). Assembly CP036251 (X11-5A) replaces draft assembly GCF000212755.1 and assembly CP036253 (NCPPB4346) replaces draft assembly GCF001276975.1 (also in Bioproject PRJNA257008). Accessions for all genomes used in this study are listed in Supplementary Table S2.
Phylogenomics and Bioinformatic Analyses
In addition to the five Xol sequenced strains, all completely sequenced X. oryzae genomes were obtained from the NCBI to be used for comparisons, as well as representative genomes from other Xanthomonas species. All genomes and accessions can be found in Supplementary Table S2.
Average nucleotide identity (ANI) values were obtained using the ANI-matrix script from the enveomics collection (v1.3) (Rodriguez-R and Konstantinidis, 2016). Parsimony trees based on pan-genome SNPs were obtained using KSNP (v3.0) (Gardner et al., 2015). Multi-locus sequence analysis was made by identifying 33 housekeeping genes in all genome using Amphora2 (Wu and Scott, 2012). Concatenated amino acid sequences of these genes were then aligned using MUSCLE (v3.8.31) (Edgar, 2004), and neighbor-joining bootstrapped trees were generated using functions (dist.ml; model = “Blosum62”, NJ, bootstrap.pml) of the R package phangorn (v2.4.0) (Schliep, 2011).
Automated annotation of proteins used in this manuscript for all genomes was made using Prokka (v1.14-dev) (Seemann, 2014), annotation for public versions available in the NCBI was made with the NCBI Prokaryotic Genome Annotation Pipeline (PGAP). Groups of orthologs and trees based on presence/absence of ortholog genes were generated using OrthoFinder (v 2.2.6) (Emms and Kelly, 2015). Dotplots of whole genome alignments were obtained using Gepard (v. 1.4, using word length 100) (Krumsiek et al., 2007). Genome duplications were then quantified using minimap (v.1) (Li, 2016) as implemented in minidot2 (parameters = −g 1000 −k 50 −w 5 −L 100). Colinear gene regions were identified using DAGchainer (Haas et al., 2004) (parameters = −E 1e-20 −s −g 2000 −x 200 −A 4) using the blast results from orthofinder as inputs. Insertion sequences (ISs) were identified using ISEScan (v1.6) (Xie and Tang, 2017).
Non-TALE T3 effectors were identified by BLASTP (v. 2.6.0+, results were filtered keeping hits with −evalue < 0.0001, > 0% identity in >40% the query length) (Boratyn et al., 2013) of consensus effectors sequences obtained from http://xanthomonas.org/ against the protein sequences obtained using Prokka. TALE sequences were extracted from each genome using in-house Perl scripts3. Neighbor joining trees were generated from concatenated nucleotide TALE N and C terminal sequences, alignments were made using MUSCLE (Edgar, 2004) and trees were generated using functions (dist.ml; model = “JC”, NJ, bootstrap.pml) of the phangorn (v2.4.0) (Schliep, 2011).
Transcription activator-like effector repeat sequences were aligned using DisTAL v1.2 (Pérez-Quintero et al., 2015) and neighbor joining trees were generated based on DisTAL genetic distances using the R package ape (Paradis et al., 2004). TALE groups were defined using the function cutree in R (height = 4.8) on the DisTAL tree. Predictions for TALE binding sites were made using Talvez on the promoters (−1 kb upstream of the translation start site) of annotated genes in the L. perrieri (v1.4) and O. sativa cv. Nipponbare (v. MSU7) as previously described (Pérez-Quintero et al., 2013).
Results
Xol Is Pathogenic to Rice and Southern Cutgrass
To better understand the biology of Xol, we established a host range by screening for pathogenicity on rice and southern cutgrass (L. hexandra) using different inoculation techniques. Relative to the virulent Philippine Xoc strain BLS256, the Xol strains were less aggressive to rice, and caused less expansive water-soaked leaf streaking on several rice varieties. Xol BAI23 was more aggressive than NCPPB4346 on rice varieties Cypress and IR64, and caused more disease than the US Xo strain X11-5A on Azucena. Rice variety Carolina Gold was resistant to Xol, exhibiting a hypersensitive response and no lesion expansion after infiltration. Both Xol strains caused longer lesions on southern cutgrass than Xo, but were not as aggressive as Xoc BLS256 on rice. Xo produced water-soaked spots at the point of infiltration that did not expand on southern cutgrass (Figure 1).
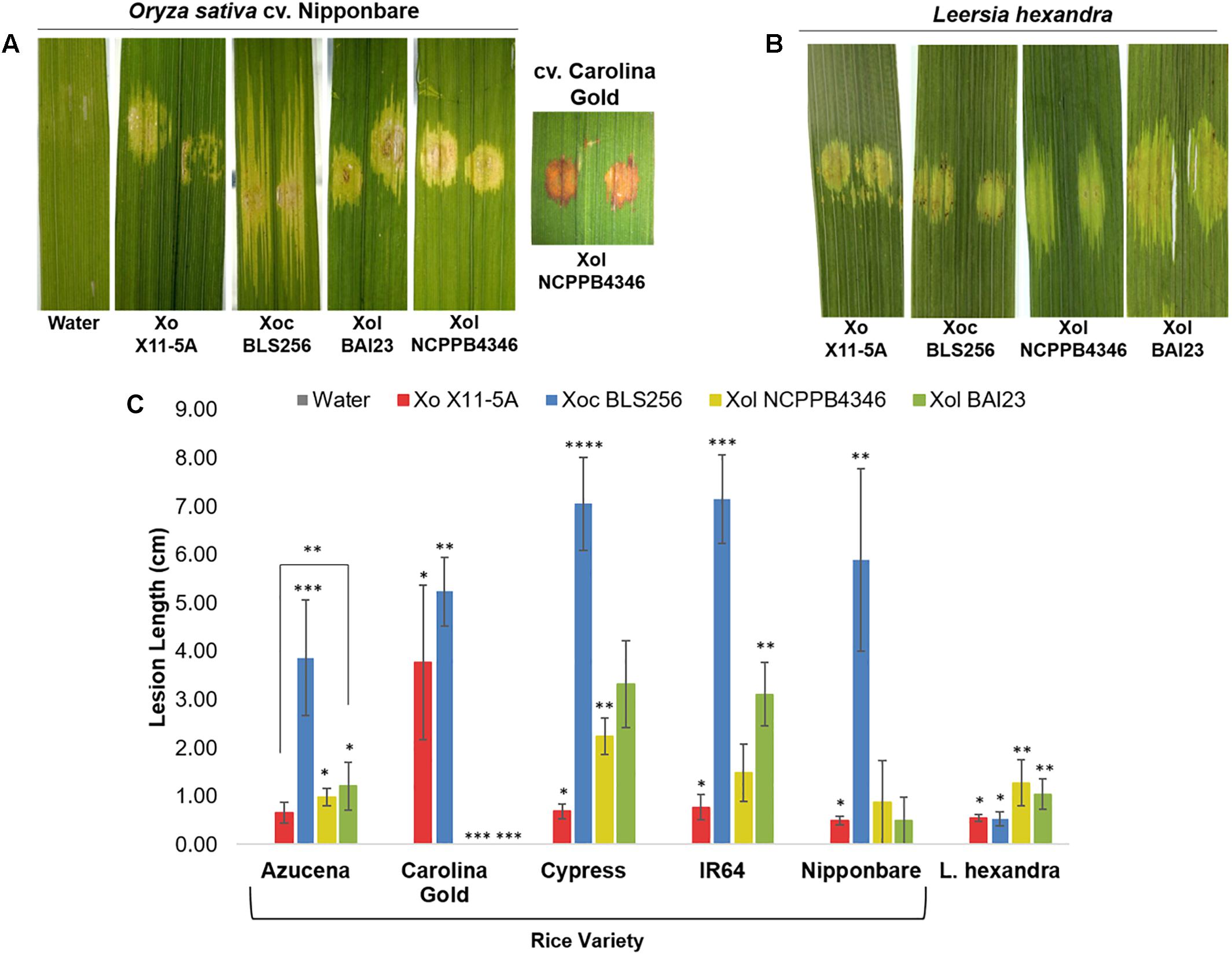
Figure 1. X. oryzae (campestris) pv. leersiae (Xcl) and X. oryzae cause water soaking on reciprocal hosts, but virulence is highest on original host. Phenotypes and quantitative lesion lengths caused by Xanthomonas oryzae (Xo) X11-5A, X. o. pv. oryzicola (Xoc) BLS256 and X. o. pv. leersiae NCPPB4346 on (A,C) diverse rice varieties and (B,C) wild Leersia hexandra were measured 12 days post infiltration inoculation. An asterisk denotes a significant difference between strains on each variety (p ≤ 0.05). Error bars represent ± SD.
After clip inoculations, lesions caused by Xol did not expand on rice or southern cutgrass, unlike those caused by the vascular pathogen Xoo (Supplementary Figure S1). When infiltrated into leaves, populations of Xol were equivalent to Xoc BLS256 and Xo X11-5A on rice cvs. Nipponbare or Azucena after 72 hpi (Figure 2). On southern cutgrass, their native host, Xol grew to a significantly higher population than Xo X11-5A. Xol did not cause disease on wheat or barley (Supplementary Figure S2). Phenotyping Nicotiana species can serve as a screen for the ability of microbes, particularly Xanthomonas spp., to elicit a non-host resistance response (Gonzalez et al., 2007). Xol caused minor chlorosis, but not water soaking nor a hypersensitive response, when infiltrated into N. tabacum, similar to the phenotype caused by Xoo PXO99A and Xoc BLS256 while Xoo BAI3 and Xo US 11-5A caused a strong hypersensitive response at the site of infiltration on N. tabacum similar to prior reports (Gonzalez et al., 2007) (Supplementary Figure S2).
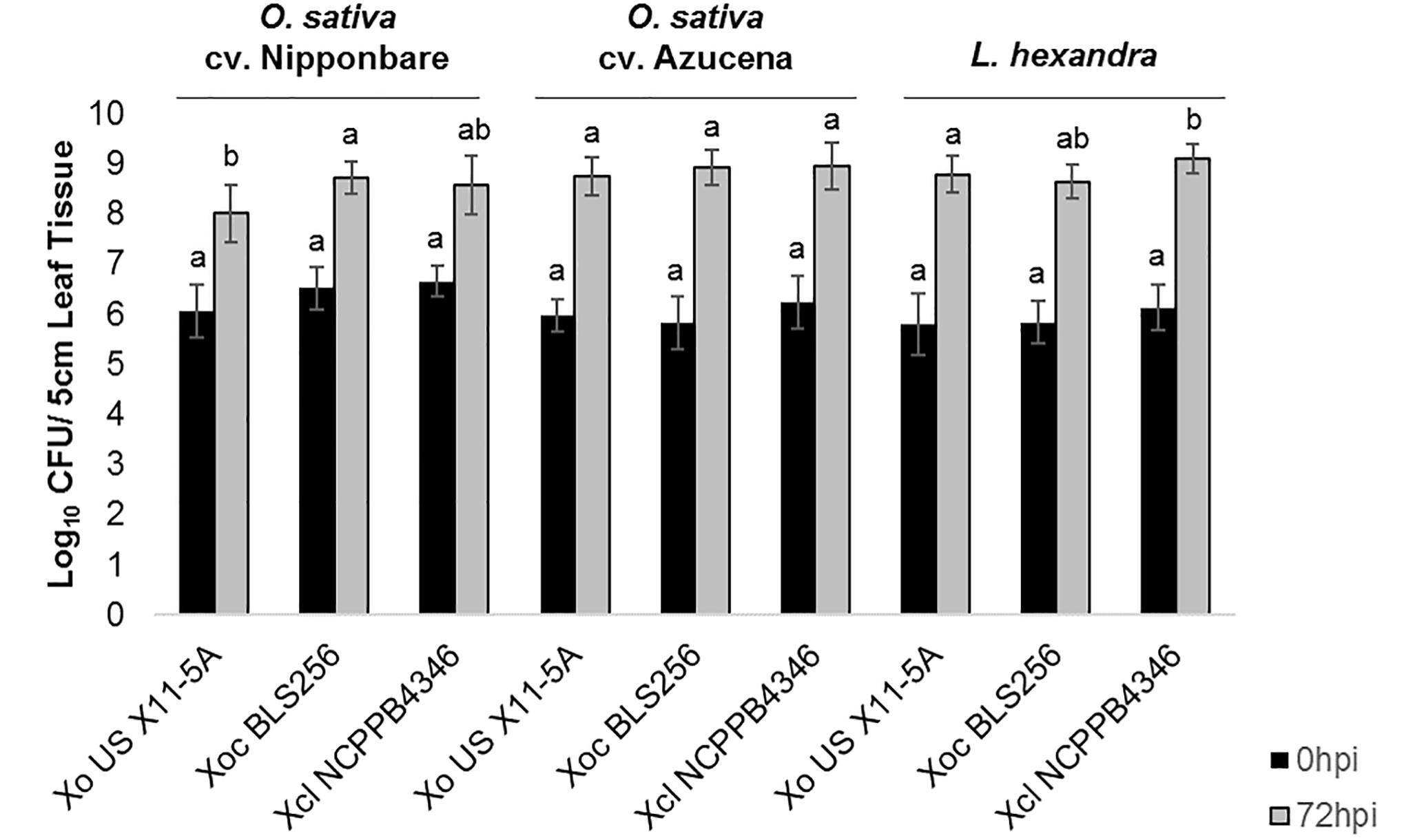
Figure 2. X. oryzae pv. leersiae can grow and colonize rice as effectively as rice pathogens and rice pathogens can colonize Leersia hexandra as effectively as X. oryzae pv. leersiae. Bacterial population growth in leaves of rice varieties Nipponbare and Azucena and L. hexandra inoculated with X. oryzae X11-5A, X. oryzae pv. oryzicola BLS256, and X. oryzae pv. leersiae NCPPB4346 were quantified at 0 and 72 h post inoculation (hpi). Population sizes were measured in a 5 cm leaf segment infiltrated with each strain. Error bars represent ± SD of six independent leaves, and letters denote treatments significantly different from one another on each variety (p ≤ 0.05).
Our studies show that Xol is pathogenic to southern cutgrass and mildly pathogenic on diverse varieties of rice, but does not cause disease on barley or wheat, and that some rice varieties exhibit resistance to Xol. After inoculation, Xol causes symptoms on rice that are most similar to Xoc, i.e., expanding lesions when introduced into the intercellular spaces (leaf infiltration) and no spreading lesions when introduced into the xylem vessels (clipping).
X. campestris pv. leersiae Belongs to the X. oryzae Species and Is Phylogenetically Close to Xoc
Amplification of Xol DNA with primers specific for Xo but not Xoc, Xoo, or US Xo suggested that Xol were related to X. oryzae, but distinct from other Xo pathovars (Lang et al., 2010; Triplett et al., 2011) (Supplementary Figure S3). We then used SMRT sequencing technology to derive complete genomes of five available Xol strains from China (NCPPB4346), Burkina Faso (BAI23), Mali (NJ611), and Uganda (BB151-3 and BB156-2). We calculated pairwise ANI among these and all fully sequenced X. oryzae genomes (Rodriguez-R and Konstantinidis, 2016). This analysis showed that Xol strains were 99–100% identical to one another, and were 97–99% identical to US Xo, Xoc, and Xoo. Xol were most similar to Xoc (∼98.5%). They were 76–91% similar to other Xanthomonas species. X. vasicola was the next most similar Xanthomonas species to oryzae, sharing 91% ANI with Xol and all Xo (Figure 3).We generated parsimony phylogenetic trees based on pan-genome SNPs using kSNP3 (Gardner et al., 2015), which showed again that Xol strains were closely related to Xoc (Figure 3). Neighbor-joining trees generated based on MLSA and on presence/absence of ortholog families showed similar groupings, although the placement of African Xoo in the tree was variable (Figure 3 and Supplementary Figure S4).
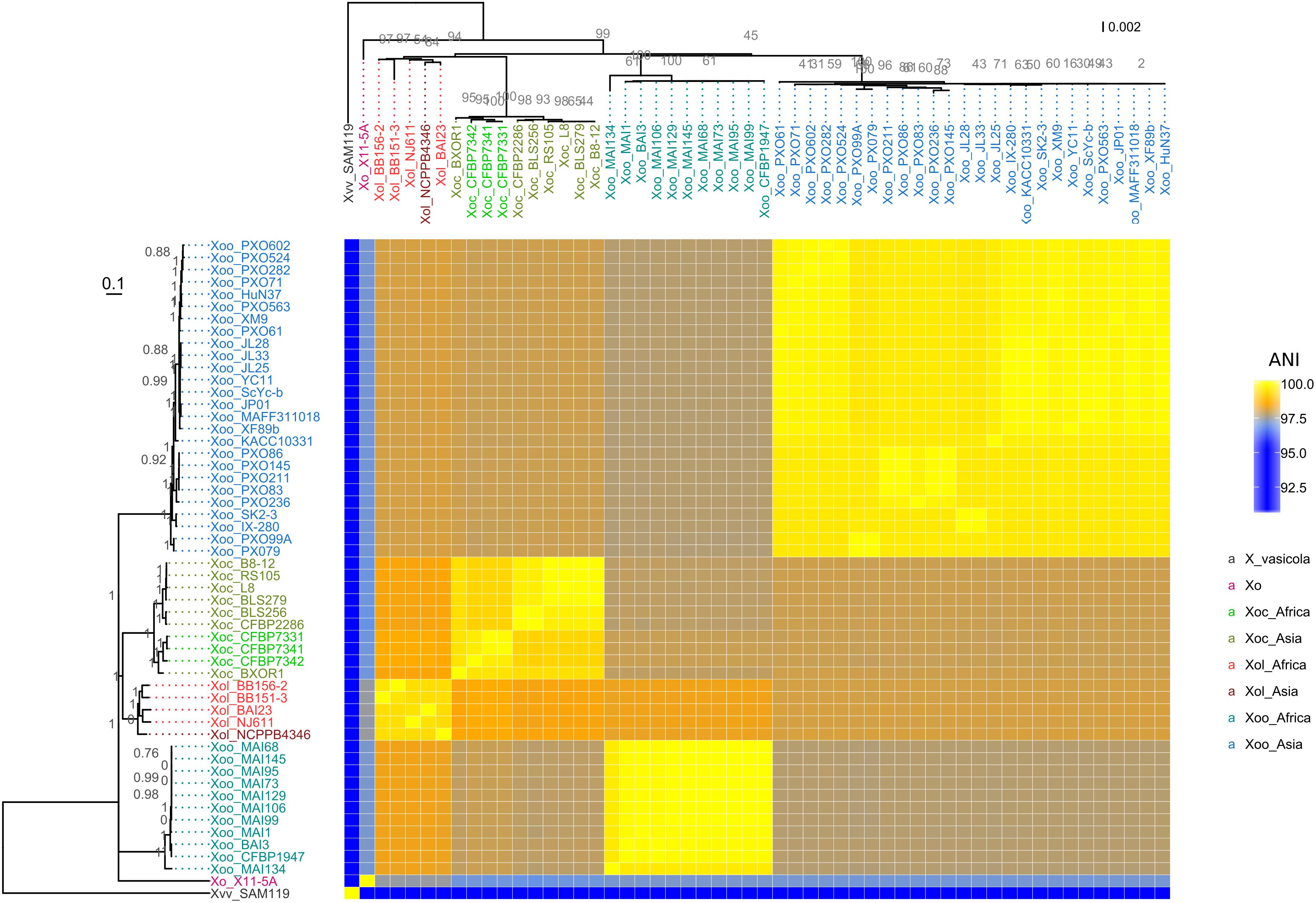
Figure 3. X. oryzae pv. leersiae (Xol) is a member of X. oryzae (Xo) and is closely related to X. oryzae pv. oryzicola (Xoc). The heatmap shows pairwise average nucleotide identity (ANI) values between fully sequenced X. oryzae genomes. (Left) Consensus parsimony tree generated with based on shared pangenome SNPs, numbers in gray indicate node support as outputed by kSNP3, heatmap rows are ordered according to this tree. (Top) MLSA neighbor-joining tree based on concatenated alignments of 33 housekeeping genes, numbers in gray indicate bootstrap support for branches, and heatmap columns are ordered according to this tree. X. oryzae pv. oryzae is abbreviated as Xoo and X. vasicola pv. vasculorum as Xvv. All species abbreviations are followed by strain name.
These combined data indicate that our sequenced strains are more closely related to Xo than other Xanthomonas species and therefore, combined with previously reported MLSA data (Triplett et al., 2011; Wonni et al., 2014), we recommend that the formal taxonomic placement of Xcl be included in the species “oryzae.” Further biological support for this shift from host range and effector repertoires is described below.
To avoid misidentification of Xol as a Xoc or Xoo in future studies, genomes were compared to identify regions of specificity to base diagnostic primer design with UniqPrimer (Juanillas et al., 2018). Two primer sets were validated for specificity against over 30 closely and distantly related bacteria (Supplementary Table S1). Both primer sets consistently amplified only control Xol strains and did not amplify any other bacterial strain tested (Table 1).
Rice Associated X. oryzae Show High Genome Plasticity Compared to X. oryzae pv. leersiae
We generated dotplots to visualize pairwise whole genome alignments to further compare the genomes of X. oryzae strains. These alignments showed several genome rearrangements between the different Xol strains with respect to each other and to other X. oryzae groups. Curiously, we noticed that self-alignments of genomes of pathovars oryzae and oryzicola overall showed more genomic duplication than those of Xol (Figure 4A). To quantify this, we identified colinear regions within each genome that showed these possible genomic duplication events are present in different frequencies in each X. oryzae lineage; and that they are more frequent in Xoc and Asian Xoo, and less frequent in Xol and African Xoo (Figure 4B). Notably, all X. oryzae examined exhibited overall more genomic duplications than other Xanthomonas species (Figure 4B).
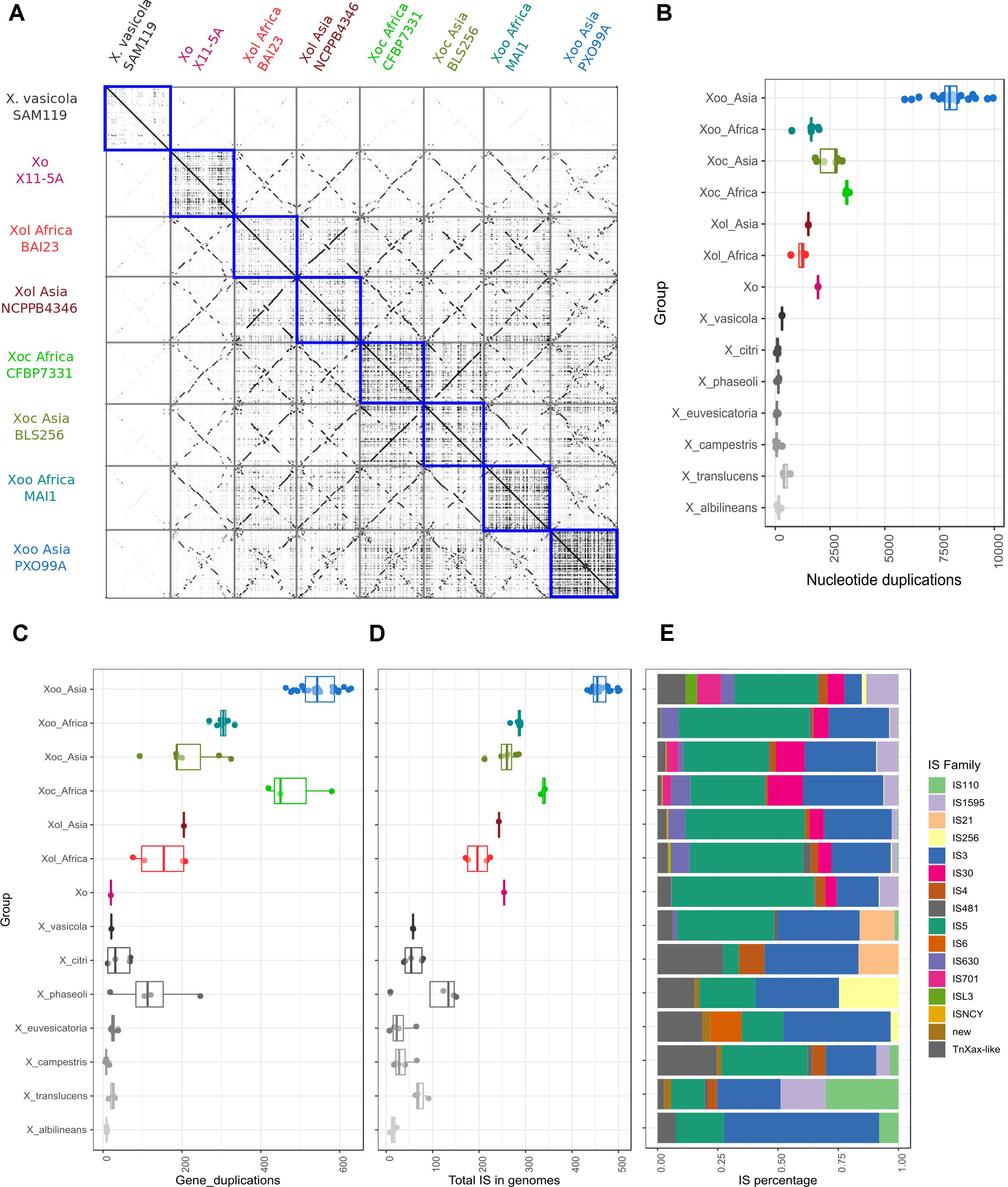
Figure 4. X. oryzae pv. leersiae genomes show generally less plasticity than other X. oryzae genomes but more than other Xanthomonas. (A) Dotplots showing pairwise whole genome alignments between representative genomes of each X. oryzae group, blue squares highlight self-alignments showing high amounts of duplication and rearrangements in X. oryzae pv. oryzicola and pv. oryzae when compared to X. oryzae pv. leersiae. (B) Boxplot showing total duplicated nucleotide sequences (at least 100 bp) in self-alignments of whole genomes for each group, each dot represents the total regions detected for one strain aligned to itself. (C) Gene duplication involving at least four co-linear genes were identified based on alignments of annotated proteins in a genome against itself, boxplot shows total duplication events detected for each strain. (D) Total number of insertion sequences (ISs) identified in genomes of each group. (E) Distribution of IS families identified within each group, percentages are calculated based on the average of each family in all strains analyzed for each group.
We also identified colinear arrangements of homologous genes within each genome, meaning genome duplications that involve multiple genes, and again, these were more frequent in Xoc and Xoo than in Xol (Figure 4C). Since some of these duplications may be mediated by duplicative transposition we annotated and quantified insertion sequences (ISs) in the different X. oryzae genomes, which indeed revealed a higher frequency of IS in the groups with higher amounts of duplicated regions (Figure 4). The distribution of IS families was overall similar among X. oryzae, with Xol’s most resembling the distribution of African Xoo (Figure 4E).
Non-TAL T3E Repertoires of Xol Are Similar to Xoc
Computational prediction of T3E among annotated proteins in each genome revealed that the Xol T3E repertoire resembles more closely that of Xoc. Nonetheless, some features are unique to Xol, specifically all Xol strains possess a xopD gene that is absent in all other Xo genomes. XopAH is also present in all Xol strains and is shared only by two Xoc strains. On the other hand, Xol strains lack xopO which is present in all Xoc strains, and some Xol strains seem to lack the otherwise universal effectors xopW and xopK (Supplementary Figure S5).
Xol Strains Have Distinct TALE Repertoires With Some Similarities to African Xoo
Previous Southern blot analyses using conserved TALE probes predicted that Xol BAI23 and NCPPB4346 had five and four TALEs, respectively (Wonni et al., 2014). However, Southern blot analysis cannot resolve TALEs that are close in size. We found in our whole genome sequences that Xol strains contained 12 or 13 TALEs each, which is more than the nine TALEs per genome found in African Xoo, and less than what is commonly found in Xoc (22–29 TALEs) and Asian Xoo (13–20 TALEs). No truncated TALEs were identified in any Xol genome. As previously reported, no TAL effectors were found in the Xo strain X11-5A (Ryba-White et al., 1995; Triplett et al., 2011).
Phylogenetic trees based on the N and C termini of X. oryzae TALEs showed that Xol TALEs form a distinct group, but seem to be close to African Xoo TALEs, despite the overall genomic similarities with Xoc (Supplementary Figure S6). We also constructed trees based on similarities in the CRR of using DisTAL, which grouped Xol and African Xoo together in a subgroup that also includes Xoc (Figure 5A). We used the DisTAL tree to define TALE groups based on repeat region similarities, Xol TALEs were classified in 12 groups (Figure 5B). One of the groups contained TALEs from Xoc and Xol strains, while all the others were exclusive to Xol. Of these, seven groups were present in all five Xol strains. One of these groups, present in the four African Xol strains, contains the RVD combination “TI” which has not been previously reported in other species.
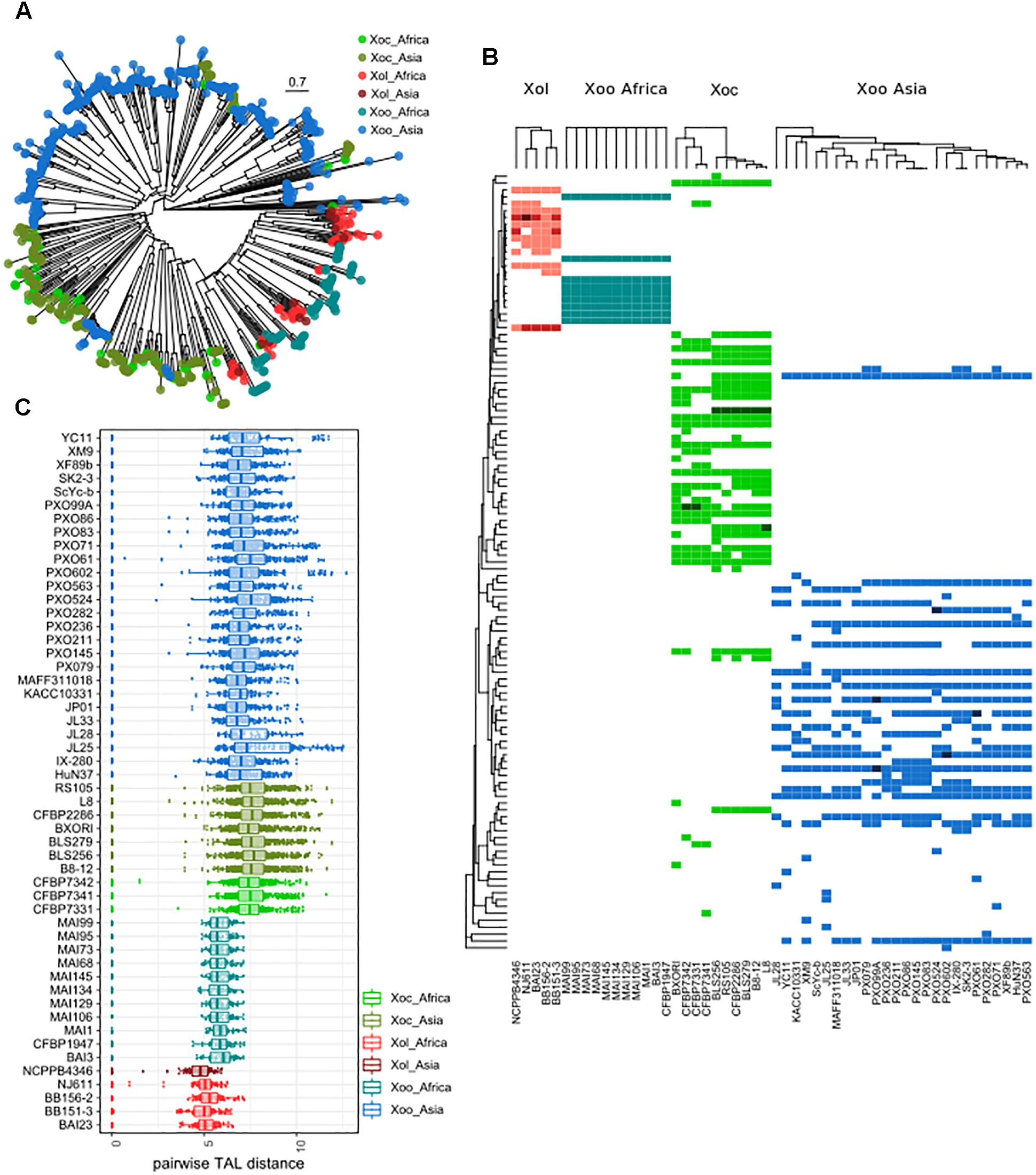
Figure 5. TALEs from X. oryzae pv. leersiae are unique and closely related to African X. oryzae pv. oryzae TALEs. (A) neighbor-joining tree based on TALE repeat arrangement alignments obtained using DisTAL. Each point represents a single TALE. (B) TALEs were classified in group according to the DisTAL tree, the heatmap shows presence/absence of each TALE group in X. oryzae strains, darker colors indicate multiple TALs from one group found in one strain. Tree to the left indicates average DisTAL distance between repeat arrangements for each TALE group. Top trees show hierarchical clustering of each X. oryzae group based on the presence/absence pattern of TALE groups. (C) Dotplots shows pairwise distance between all TALE repeat arrangements from each strain as calculated with DisTAL, each dot represents a pair of TALEs within one genome.
All Xol strains contained at least two TALEs that were classified within the same group, that is, within each Xol genome there are at least two TALEs with nearly identical repeat arrangements (Figure 5B). By examining pairwise genetic distances between the repeat regions of TALEs, we saw that TALEs from Xol are on average more similar to each other than TALEs within other X. oryzae groups, indicating that they are possibly less diversified and/or more redundant (Figure 5C).
Xol TALE Targets in Cutgrass and Rice Are Different From Xoc and Xoo Targets
Talvez (Pérez-Quintero et al., 2013) was used to predict host targets for each TALE in our dataset in the promoters of annotated genes in the L. perrieri (v1.4) and O. sativa (vMSU7) genomes. We identified ortholog pairs between both genomes using reciprocal BLAST. Comparisons of the predictions between both genomes for Xol TALEs revealed very few cases where an ortholog pair was predicted to be a target in both genomes (Supplementary Figure S7), and highlighted that the promoters (and thus the targeted genes) in these two hosts are very different.
We then compared predictions for all Xol lineages in both genomes and calculated the overlap between predictions for each strain. As a result, we saw that each X. oryzae group has a distinct group of predicted targets with relatively little overlap with other groups. In the case of Xol, the highest overlap was found with predictions for Xoc TALEs (10–11% shared predicted targets) (Figure 6). Given the differences in the genomes of their hosts and that Xol TALEs have unique repeat sequences, we expect their targets to be likewise unique. Additionally, we looked for orthologs of known susceptibility (S) genes targeted by Xoc or Xoo TALEs (e.g., SWEETs, OsSULTR3;6) and queried whether they were among the top predictions for Xol TALEs. No known Xoc or Xoo target homolog was found. It is, however, possible, that while not targeting the direct orthologs of these S genes, Xol TALEs may be inducing similar functions, since the predicted targets contain genes annotated with similar functions to known S genes including sulfate transporters, nodulins and various families of transcription factors (Supplementary Table S3). Expression data and further experiments are necessary to effectively identify the biological mechanisms of these TALEs.
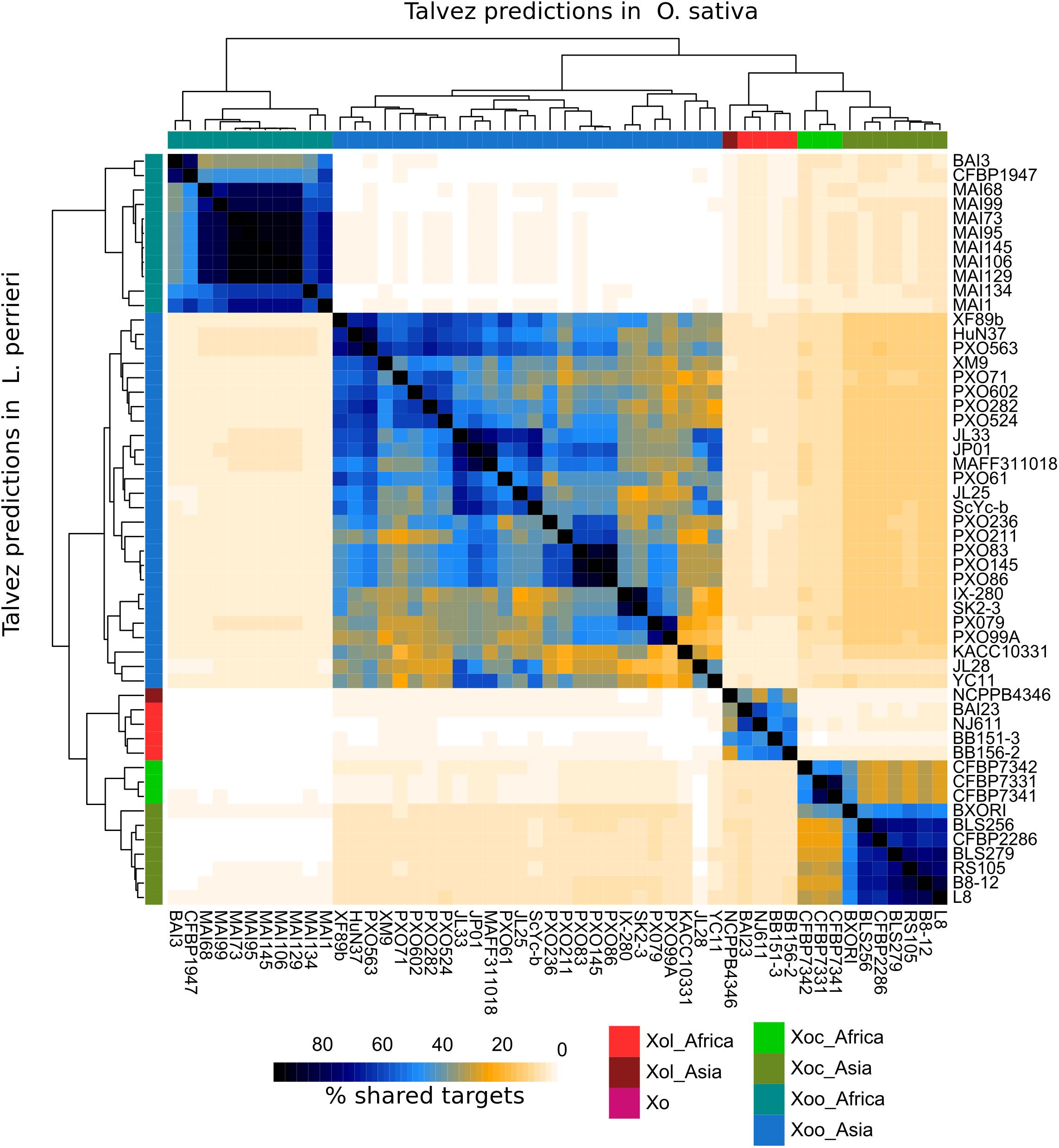
Figure 6. TALEs from X. oryzae pv. leersiae (Xol) are predicted to target distinct genes from other X. oryzae [Xo, X. oryzae pv. oryzae (Xoo), X. oryzae pv. oryzicola (Xoc)] strains. Predictions for binding sites were made for each TALE in the promoters (–1 kb) of annotated genes in the L. perrieri and O. sativa genomes, top 200 predicted targets for each TALE were kept, and predictions were aggregated for each strain. For each pair of strains, the overlap of predictions was calculated as the number of common genes predicted as TALE targets in both strains divided by the total genes predicted as targets in either strain, times 200 (intersect × 2 × 100/union). The lower left triangle part of the heatmap shows percentage of shared targets in the L. perrieri genome for each pair of strains, and the upper right shows shared targets in the O. sativa genome. Trees at the top and left show hierarchical clustering based on stared targets in each genome. Accession numbers for genomes used in this analysis are available in Supplementary Table S2.
Discussion
Xanthomonas oryzae pv. leersiae, which has been isolated from the pervasive weed species L. hexandra surrounding rice paddies (Fang et al., 1957; Wonni et al., 2014), was historically grouped as a distinct species and pathovar (Vauterin et al., 1995; Triplett et al., 2011; Wonni et al., 2014). We compared pathogenicity of multiple strains on diverse cereal hosts and complete genomes of five Xol strains from Burkina Faso, China, Mali, and Uganda. Similar to Xoc, Xol strains caused water soaked lesions on rice and L. hexandra, but were not virulent to wheat and barley. ANI is a widely accepted baseline beyond DNA–DNA hybridization for taxonomic placement of prokaryotes into a species, not a pathovar, at a threshold of >95–96% (Konstantinidis and Tiedje, 2005; Goris et al., 2007; Richter and Rossello-Mora, 2009; Kim et al., 2014; Bull and Koike, 2015). In phylogenetic analyses, these five Xol strains, representing geographic and temporal diversity, grouped more closely with Xo pathovars than other members of this genus, and were above a species delineation threshold in ANI analyses. Therefore, we propose re-naming these strains from Xcl to Xol (Fang et al., 1957) comb. nov.
Xanthomonas oryzae pv. leersiae colonize and cause water-soaking on rice leaves, but the southern cutgrass isolates are not as aggressive as Xo isolated from rice. The lesions caused by Xol were phenotypically similar to rice BLS caused by Xoc and these strains did not cause disease when introduced into rice or southern cutgrass by leaf-clipping. Thus, we suggest that Xol are not systemic pathogens, and are more like Xoc than the systemic relative Xoo.
T3Es, as important contributors to bacterial pathogenicity, may define host range, and can also inform lineages and evolutionary relationships among different populations of related bacteria (Arlat et al., 1991; Hajri et al., 2012; Wonni et al., 2014; Schwartz et al., 2015). Studies in X. oryzae have shown that different lineages are shaped by T3E repertoires and reflect phenotypic adaptation to their agroecosystems (Hajri et al., 2012; Quibod et al., 2016). Likewise, this study sought to uncover similarities between Xo and Xol effector repertoires that could further inform their evolutionary lineage. Out of a set of previously defined core effectors for X. oryzae [avrBs2, avrBs3 (TALEs), xopL, xopN, xopP, xopQ, xopV, xopW, xopY, xopAA, xopAB, xopAE, and xopF1) (Hajri et al., 2012), Xol strains contain all except xopW, which is absent in three of the five strains. Although present in strains BAI23 and NJ611, xopW contains a large IS. This IS most likely prevented its amplification in previous studies (Wonni et al., 2014). Interestingly, this same IS was identified in other African Xoc strains, consistent with a shared evolutionary origin with Xol (Hajri et al., 2012). On the other hand, xopD is present in Xol but absent in other X. oryzae. XopD is a SUMO protease mimic that suppresses host defense responses during Xanthomonas euvesicatoria infection (Kim et al., 2008, 2013). In addition to Xol, xopD is predicted to be present in X. campestris pv. campestris, X. euvesicatoria, and Acidovorax citrulli4. Absence in other Xo suggests an independent acquisition by Xol. Conversely, loss of this effector at some time by Xoo or Xoc could have occurred, but further validation of these hypotheses is necessary. Since it is not known what level of virulence, and/or host specificity any T3E conveys for Xol, future work should include functional validations.
We also assessed TALE diversity in Xol strains, using genomic assemblies based on long reads generated using SMRT sequencing. While according to phenotyping, phylogenomics, and non-TALE Type III effector repertoires, Xol most closely resembles Xoc, the Xol TALE sequences more closely resemble African Xoo. Xol and African Xoo also possess, on average, smaller TALomes (TALEs per genome) than Xoc or Asian Xoo. Furthermore, their TALEs also have less diverse repeat arrangements as evidenced by shorter genetic distances in pairwise TALE repeat alignments. A possible explanation for this feature is that TALEs from Xol and African Xoo more closely resemble the ancestral TALE repertoire for the group and have not undergone the extensive diversification found in Xoc and Asian Xoo. A hypovirulent strain of Xoc was isolated from rice in the Yunnan province of China that also contains nine TALEs. This strain was used in heterologous expression assays to determine targets and function of Tal7 (Cai et al., 2017). Unfortunately, the TALome of this strain is unavailable at this time and it is unclear if this strain is related to the Xol strains characterized in this study.
The expansion of Xoc and Xoo TALomes is curious since many of the TALEs they carry seem not to be required for virulence and some may even have redundant functions (Pérez-Quintero et al., 2013; Cernadas et al., 2014). Given their repetitive nature and a general tendency toward homogenization of repeats, TALEs have been proposed as being selected for evolvability, that is, selected for their ability to quickly recombine (Schandry et al., 2016). Having an expanded TALome would further allow frequent recombination and generation of new TALE variants, as reflected in bigger evolutionary distances between repeat arrangements. This selection of a bigger TALome has been proposed to be driven by extensive breeding for resistance in the host plant (Schandry et al., 2018). When exposed to a resistant host population, a pathogen population can benefit from carrying a heterogeneous and redundant set of effectors, since preexisting isolates harboring a set advantageous for the new conditions would then be selected, in what has been proposed as a type of evolutionary “bet-hedging” strategy (Win et al., 2012). It is then possible that Xoc and Asian Xoo have historically encountered more resistance, possibly related to domestication of its primary host, than Xol and African Xoo, leading to a selection for expanded TALomes.
At least two resistance genes (Xa1 and Xo1) in O. sativa specifically recognize TALEs (Ji et al., 2016; Triplett et al., 2016), and Xoc and Xoo seem to have benefited from their expanded TALomes by selecting TALE variants (iTALEs or truncTALEs) that can specifically suppress resistance mediated by these genes (Ji et al., 2016; Read et al., 2016). Meanwhile Xol infecting L. hexandra, as African Xoo originally infecting Oryza glaberrima (Gonzalez et al., 2007), may have never encountered similar resistance in its host, and thus lacks these TALE variants and cannot cause disease on varieties carrying Xo1 (Figure 1).
TALome expansion may have been accompanied by, or be a consequence of, higher genome plasticity in the X. oryzae clade as evidenced by a high amount of genome and gene duplication and a high frequency of IS elements, these measures being generally lower in Xol. Within the clade, differences may be once more related to resistance in the host, with the more plastic genomes (Asian Xoo) matching more variable host populations. The requirement of plastic genomes in the clade as a whole has been hypothesized to be a consequence of rice cultivation through millennia (Salzberg et al., 2008; Bogdanove and Voytas, 2011), and suggest the ancestor of the clade faced an already variable population. Which then raises the question of where Xol falls within this scenario of adaptation to a cultivated crop?
Rice and southern cutgrass are closely related members of the Poaceae. Leersia species are often used as an outgroup in phylogenetic and, most recently, genomic investigations (Copetti et al., 2015). Evidence of genome duplication events and defense response genes shared by rice and Leersia species has been reported (Jacquemin et al., 2009; Xiao et al., 2009). It is plausible that based on their genetic and evolutionary relatedness that respective pathogens of Oryza and Leersia evolved independently. The US strains of Xo, which do not contain intact TALEs, could have been progenitors of other Xo pathovars, having acquired TALEs over time to enhance virulence on rice or Leersia spp. (Triplett et al., 2011). Significant trade between the United States, Africa, and Asia could allow for movement of strains across continents. However, it is not clear if Xol is currently present in the United States, despite early reports of L. hexandra being an alternate host for Xoo (Gonzalez et al., 1991). Alternatively, this system may represent a sympatric scenario where Xoo, Xoc, Xo, and Xol all lived in the same habitat, providing the opportunity to exchange genetic material and to adapt to their respective host while maintaining basic homology (Jacques et al., 2016). In this scenario, given its similar infection biology and overall similarities to Xoc, Xol may be a specialized subgroup originating from a Xoc-like population able to colonize southern cutgrass, or vice versa.
How specifically adapted Xol strains are to either L. hexandra or rice remains a fascinating question, since Xol could potentially represent an emerging pathogen for rice. Here we have shown that Xol can, to some extent, infect rice, and two of the isolates sequenced in this work were originally recovered from symptomatic rice leaves in a field (BB 151-3 and BB 156-2). To get insights into host adaptation, we attempted to predict targets for Xol TALEs in L. hexandra using L. perrieri, the only available genome from the genus, as proxy and O. sativa. Overall, the predictions indicate that Xol TALEs induce different sets of genes than other X. oryzae, and that different genes are induced in rice and Leersia sp. given that relatively few orthologues are predicted as targets in both genomes. Of particular interest, given the phenotypic similarities with Xoc, is the predicted targeting of genes annotated as sulfate transporters. Four genes in the L. perrieri genome corresponding to orthologs of sulfate transporters from rice (LOC_Os03g09940, LOC_Os03g09970, LOC_Os03g09980, and LOC_Os09g06499) were predicted to be targeted by TALEs from at least one Xol strain with high prediction scores (Supplementary Table S3). The primary virulence target of Tal2g from Xoc is OsSULTR3;6 which is a member of the sulfate transporter family 3 (Takahashi et al., 2011; Cernadas et al., 2014). It is feasible that the TALEs targeting sulfate transporters in Leersia mirror the virulence function of Tal2g. However, transcriptomic data and further biological validation are required since it is still possible that the few-shared genes in the predictions are true targets and similar functions are required for virulence in both hosts.
Conclusion
In summary, we propose Xol, which was isolated from rice and southern cutgrass (L. hexandra), a weedy grass closely related to rice, as a new member of the X. oryzae species. Genomic analysis and disease phenotyping on various hosts demonstrated the close relationship of Xol to the rice pathogens Xoo and Xoc. T3E and TALE content of the Xol indicated that this group of organisms uses similar virulence mechanisms to the rice pathogens. While weeds such as southern cutgrass are not agronomic crops, they are competitors for resources and potential reservoirs of pathogen inoculum, they are important in management considerations for rice growers. Interfering in any agroecosystem requires comprehensive consideration. Certain Leersia sp. are used as banker plants for the critically important rice brown plant hopper (Zheng et al., 2017), therefore integrated management of weeds surrounding rice paddies will require prospecting and balance of all possible pests. The fact that they harbor a pathogen group that can also impact rice emphasizes that, in general, more attention should be focused to the surrounding ecosystem in rice production and more broadly in any crop rotation as a general management strategy. Research contributing toward understanding the Xol/rice/southern cutgrass pathosystem will be significant for all rice-producing countries.
Author Contributions
JML, AP-Q, RK, BS, VV, and JEL conceived and designed experiments. JML, AP-Q, RK, ED, and JJ performed the experiments. SS, HD, IK, RO, OK, and VV collected and provided new Xol strains. JML, AP-Q, RK, and JZ analyzed the data. RK, RO, OK, BS, VV, and JEL provided resources and supervision. JML, AP-Q, BS, VV, and JEL developed the manuscript.
Funding
This research was supported by a Marie Curie IOF Fellowship (EU Grant PIOF-GA-2009-235457 to VV); the Embassy of France in the United States, Office of Science and Technology – STEM Chateaubriand Fellowship Program (to JML); USDA NIFA Postdoctoral Fellowship Award No. 2016-04706 (to JJ); USDA’s National Institute of Food and Agriculture, award # 2018-67013-28490 (to JJ, JML, and JEL); and by IRD JEAI Coana (to SS and OK).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Geoffrey Onaga for strain collection in Uganda; Dr. Bing Yang for Xol BB 151-3 and BB 156-2 genome sequence; and Emily Luna for technical support. We thank Drs. Mathilde Hutin, Céline Pesce, Sébastian Cunnac, and Tuan T. Tran for seed, constructive discussions, and technical support and Dr. Young-Ki Jo for providing Leersia hexandra. This article is based upon work from COST Action CA16107 EuroXanth, supported by COST (European Cooperation in Science and Technology).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00507/full#supplementary-material
Footnotes
- ^ www.isppweb.org/about_tppb_naming.asp
- ^ https://github.com/thackl/minidot
- ^ https://github.com/alperezq/experimenTAL
- ^ www.xanthomonas.org
References
Arlat, M., Gough, C., Barber, C., Boucher, C., and Daniels, M. (1991). Xanthomonas campestris contains a cluster of hrp genes related to larger hrp cluster of Pseudomonas solancearum. Mol. Plant Microbe. Interact. 4, 593–601. doi: 10.1094/MPMI-4-593
Ash, G. J., Lang, J. M., Triplett, L. R., Stodart, B. J., Verdier, V., Cruz, C. V., et al. (2014). Development of a genomics-based LAMP (Loop-mediated isothermal amplification) assay for detection of Pseudomonas fuscovaginae from rice. Plant Dis. 98, 909–915. doi: 10.1094/PDIS-09-13-0957-RE
Boch, J., Bonas, U., and Lahaye, T. (2014). TAL effectors - pathogen strategies and plant resistance engineering. New Phytol. 204, 823–832. doi: 10.1111/nph.13015
Boch, J., Scholze, H., Schornack, S., Landgraf, A., Hahn, S., Kay, S., et al. (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512. doi: 10.1126/science.1178811
Bogdanove, A. J., and Voytas, D. F. (2011). TAL effectors: customizable proteins for DNA targeting. Science 333, 1843–1846. doi: 10.1126/science.1204094
Booher, N. J., and Bogdanove, A. J. (2014). Tools for TAL effector design and target prediction. Methods 69, 121–127. doi: 10.1016/j.ymeth.2014.06.006
Booher, N. J., Carpenter, S. C. D., Sebra, R. P., Wang, L., Salzberg, S. L., Leach, J. E., et al. (2015). Single molecule real-time sequencing of Xanthomonas oryzae genomes reveals a dynamic structure and complex TAL (transcription activator-like) effector gene relationships. Microb. Genomics 1:e000032. doi: 10.1099/mgen.0.000032
Boratyn, G. M., Camacho, C., Cooper, P. S., Coulouris, G., Fong, A., Ma, N., et al. (2013). BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 41, W29–W33. doi: 10.1093/nar/gkt282
Bull, C. T., and Koike, S. T. (2015). Practical benefits of knowing the enemy: modern molecular tools for diagnosing the etiology of bacterial diseases and understanding the taxonomy and diversity of plant-pathogenic bacteria. Annu. Rev. Phytopathol. 53, 157–180. doi: 10.1146/annurev-phyto-080614-120122
Cai, L., Cao, Y., Xu, Z., Ma, W., Zakria, M., Zou, L., et al. (2017). A transcription activator-like effector Tal7 of Xanthomonas oryzae pv. oryzicola activates rice gene Os09g29100 to suppress rice immunity. Sci. Rep. 7:5089. doi: 10.1038/s41598-017-04800-8
Cernadas, R. A., Doyle, E. L., Niño-Liu, D. O., Wilkins, K. E., Bancroft, T., Wang, L., et al. (2014). Code-assisted discovery of TAL effector targets in bacterial leaf streak of rice reveals contrast with bacterial blight and a novel susceptibility gene. PLoS Pathog. 10:e1003972. doi: 10.1371/journal.ppat.1003972
Copetti, D. (2013). “Genomic resources for Leersia perrieri: an outgroup species for the genus Oryza,” in Proceedings of the Plant and Animal Genome VIII (Plant and Animal Genome), (San Diego, CA).
Copetti, D., Zhang, J., El Baidouri, M., Gao, D., Wang, J., Barghini, E., et al. (2015). RiTE database: a resource database for genus-wide rice genomics and evolutionary biology. BMC Genomics 16:538. doi: 10.1186/s12864-015-1762-3
dos Santos, R. S., Farias, D., da, R., Pegoraro, C., Rombaldi, C. V., Fukao, T., et al. (2017). Evolutionary analysis of the SUB1 locus across the Oryza genomes. Rice 10:4. doi: 10.1186/s12284-016-0140-3
Doucouré, H., Pérez-Quintero, A. L., Reshetnyak, G., Tekete, C., Auguy, F., Thomas, E., et al. (2018). Functional and genome sequence-driven characterization of TAL effector gene repertoires reveals novel variants with altered specificities in closely related Malian Xanthomonas oryzae pv. oryzae strains. Front. Microbiol. 9:1657. doi: 10.3389/fmicb.2018.01657
Doyle, E. L., Booher, N. J., Standage, D. S., Voytas, D. F., Brendel, V. P., VanDyk, J. K., et al. (2012). TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 40, W117–W122. doi: 10.1093/nar/gks608
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Fang, C. T., Ren, H. C., Chen, T. K., Chu, Y. K., Faan, H. C., and Wu, S. C. (1957). A comparison of rice bacterial leaf blight organism with the bacterial leaf streak organism of rice and Leersia hexandra Swartz. Acta Phytopathol. Sin. 3, 99–124.
Gardner, S. N., Slezak, T., and Hall, B. G. (2015). kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878. doi: 10.1093/bioinformatics/btv271
Gonzalez, C., Szurek, B., Manceau, C., Mathieu, T., Sere, Y., and Verdier, V. (2007). Molecular and pathotypic characterization of new Xanthomonas oryzae strains from West Africa. Mol. Plant Microbe. Interact. 20, 534–546. doi: 10.1094/MPMI-20-5-0534
Gonzalez, C., Xu, G., Li, H., and Cosper, J. W. (1991). Leersia hexandra, an alternate host for Xanthomonas campestris pv. oryzae in Texas. Plant Dis. 75, 159–162. doi: 10.1094/PD-75-0159
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Grau, J., Reschke, M., Erkes, A., Streubel, J., Morgan, R. D., Wilson, G. G., et al. (2016). AnnoTALE: bioinformatics tools for identification, annotation, and nomenclature of TALEs from Xanthomonas genomic sequences. Sci. Rep. 6:21077. doi: 10.1038/srep21077
Grau, J., Wolf, A., Reschke, M., Bonas, U., Posch, S., and Boch, J. (2013). Computational predictions provide insights into the biology of TAL effector target sites. PLoS Comput. Biol. 9:e1002962. doi: 10.1371/journal.pcbi.1002962
Guo, Y.-L., and Ge, S. (2005). Molecular phylogeny of Oryzeae (Poaceae) based on DNA sequences from chloroplast, mitochondrial, and nuclear genomes. Am. J. Bot. 92, 1548–1558. doi: 10.3732/ajb.92.9.1548
Haas, B. J., Delcher, A. L., Wortman, J. R., and Salzberg, S. L. (2004). DAGchainer: a tool for mining segmental genome duplications and synteny. Bioinformatics 20, 3643–3646. doi: 10.1093/bioinformatics/bth397
Hajri, A., Brin, C., Zhao, S., David, P., Feng, J., Koebnik, R., et al. (2012). Multilocus sequence analysis and type III effector repertoire mining provide new insights into the evolutionary history and virulence of Xanthomonas oryzae. Mol. Plant Pathol. 13, 288–302. doi: 10.1111/j.1364-3703.2011.00745.x
Hoagland, D. R., and Arnon, D. I. (1950). The Water-Culture Method for Growing Plants Without Soil. Berkeley, CA: University of California.
Hunt, M., Silva, N., De Otto, T. D., Parkhill, J., Keane, J. A., and Harris, S. R. (2015). Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 16:294. doi: 10.1186/s13059-015-0849-0
Hutin, M., Pérez-Quintero, A. L., Lopez, C., and Szurek, B. (2015). MorTAL Kombat: the story of defense against TAL effectors through loss-of-susceptibility. Front. Plant Sci. 6:535. doi: 10.3389/fpls.2015.00535
Jacquemin, J., Laudié, M., and Cooke, R. (2009). A recent duplication revisited: phylogenetic analysis reveals an ancestral duplication highly-conserved throughout the Oryza genus and beyond. BMC Plant Biol. 9:146. doi: 10.1186/1471-2229-9-146
Jacques, M.-A., Arlat, M., Boulanger, A., Boureau, T., Carrère, S., Cesbron, S., et al. (2016). Using ecology, physiology, and genomics to understand host specificity in Xanthomonas. Annu. Rev. Phytopathol. 54, 163–187. doi: 10.1146/annurev-phyto-080615-100147
Ji, Z., Ji, C., Liu, B., Zou, L., Chen, G., Yang, B., et al. (2016). Interfering TAL effectors of Xanthomonas oryzae neutralize R-gene-mediated plant disease resistance. Nat. Commun. 7:13435. doi: 10.1038/ncomms13435
Juanillas, V. M. J., Dereeper, A., Beaume, N., Droc, G., Dizon, J., Mendoza, J. R., et al. (2018). Rice galaxy: an open resource for plant science. bioRxiv [Preprint]. doi: 10.1101/358754
Kauffman, H. E., Reddy, A. P. K., Hsieh, S. P. Y., and Merca, S. D. (1973). Improved technique for evaluating resistance of rice varieties to Xanthomonas oryzae. Plant Dis. Rep. 57, 537–541.
Kim, J.-G., Stork, W., and Mudgett, M. B. (2013). Xanthomonas type III effector XopD desumoylates tomato transcription factor SlERF4 to suppress ethylene responses and promote pathogen growth. Cell Host Microbe 13, 143–154. doi: 10.1016/j.chom.2013.01.006
Kim, J.-G., Taylor, K. W., Hotson, A., Keegan, M., Schmelz, E. A., and Mudgett, M. B. (2008). XopD SUMO protease affects host transcription, promotes pathogen growth, and delays symptom development in Xanthomonas-infected tomato leaves. Plant Cell 20, 1915–1929. doi: 10.1105/tpc.108.058529
Kim, M., Oh, H.-S., Park, S.-C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Krumsiek, J., Arnold, R., and Rattei, T. (2007). Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23, 1026–1028. doi: 10.1093/bioinformatics/btm039
Lang, J. M., DuCharme, E., Ibarra Caballero, J., Luna, E., Hartman, T., Ortiz-Castro, M., et al. (2017). Detection and characterization of Xanthomonas vasicola pv. vasculorum (Cobb 1894) comb. nov. causing bacterial leaf streak of corn in the United States. Phytopathology 107, 1312–1321. doi: 10.1094/PHYTO-05-17-0168-R
Lang, J. M., Hamilton, J. P., Diaz, M. G. Q., Van Sluys, M. A., Burgos, M. R. G., Vera Cruz, C. M., et al. (2010). Genomics-based diagnostic marker development for Xanthomonas oryzae pv. oryzae and X. oryzae pv. oryzicola. Plant Dis. 94, 311–319. doi: 10.1094/pdis-94-3-0311
Lang, J. M., Langlois, P., Nguyen, M. H. R., Triplett, L. R., Purdie, L., Holton, T. A., et al. (2014). Sensitive detection of Xanthomonas oryzae pathovars oryzae and oryzicola by loop-mediated isothermal amplification. Appl. Environ. Microbiol. 80, 4519–4530. doi: 10.1128/AEM.00274-14
Li, H. (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110. doi: 10.1093/bioinformatics/btw152
Liu, J., Duan, C., Zhang, X., Zhu, Y., and Lu, X. (2011). Potential of Leersia hexandra Swartz for phytoextraction of Cr from soil. J. Hazard. Mater. 188, 85–91. doi: 10.1016/j.jhazmat.2011.01.066
Mew, T. W. (1987). Current status and future prospects of research on bacterial blight of rice. Annu. Rev. Phytopathol. 25, 359–382. doi: 10.1146/annurev.py.25.090187.002043
Moscou, M. J., and Bogdanove, A. J. (2009). A simple cipher governs DNA recognition by TAL effectors. Science 326:1501. doi: 10.1126/science.1178817
Noda, T., and Yamamoto, T. (2008). Reaction of Leersia grasses to Xanthomonas oryzae pv. oryzae collected from Japan and Asian countries. J. Gen. Plant Pathol. 74, 395–401. doi: 10.1007/s10327-008-0118-0
Notomi, T., Okayama, H., Masubuchi, H., Yonekawa, T., Watanabe, K., Amino, N., et al. (2000). Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28:e63. doi: 10.1093/nar/28.12.e63
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Parkinson, N., Cowie, C., Heeney, J., and Stead, D. (2009). Phylogenetic structure of Xanthomonas determined by comparison of gyrB sequences. Int. J. Syst. Evol. Microbiol. 59, 264–274. doi: 10.1099/ijs.0.65825-0
Peng, Z., Hu, Y., Xie, J., Potnis, N., Akhunova, A., Jones, J., et al. (2016). Long read and single molecule DNA sequencing simplifies genome assembly and TAL effector gene analysis of Xanthomonas translucens. BMC Genomics 17:21. doi: 10.1186/s12864-015-2348-9
Pérez-Quintero, A. L., Lamy, L., Gordon, J., Escalon, A., Cunnac, S., Szurek, B., et al. (2015). QueTAL: a suite of tools to classify and compare TAL effectors functionally and phylogenetically. Front. Plant Sci. 6:545. doi: 10.3389/fpls.2015.00545
Pérez-Quintero, A. L., Rodriguez-R, L. M., Dereeper, A., López, C., Koebnik, R., Szurek, B., et al. (2013). An improved method for TAL effectors DNA-binding sites prediction reveals functional convergence in TAL repertoires of Xanthomonas oryzae strains. PLoS One 8:e68464. doi: 10.1371/journal.pone.0068464
Poulin, L., Grygiel, P., Magne, M., Gagnevin, L., Rodriguez-R, L. M., Forero Serna, N., et al. (2015). New multilocus variable-number tandem-repeat analysis tool for surveillance and local epidemiology of bacterial leaf blight and bacterial leaf streak of rice caused by Xanthomonas oryzae. Appl. Environ. Microbiol. 81, 688–698. doi: 10.1128/AEM.02768-14
Quibod, I. L., Perez-Quintero, A., Booher, N. J., Dossa, G. S., Grande, G., Szurek, B., et al. (2016). Effector diversification contributes to Xanthomonas oryzae pv. oryzae phenotypic adaptation in a semi-isolated environment. Sci. Rep. 6:34137. doi: 10.1038/srep34137
Read, A. C., Rinaldi, F. C., Hutin, M., He, Y., Triplett, L. R., and Bogdanove, A. J. (2016). Suppression of Xo1-mediated disease resistance in rice by a truncated, non-DNA-binding TAL effector of Xanthomonas oryzae. Front. Plant Sci. 7:1516. doi: 10.3389/fpls.2016.01516
Reimers, P. J., and Leach, J. E. (1991). Race-specific resistance to Xanthomonas oryzae pv. oryzae conferred by bacterial blight resistance gene Xa-10 in rice Oryza sativa involves accumulation of a lignin-like substance in host tissues. Physiol. Mol. Plant Pathol. 38, 39–55. doi: 10.1016/S0885-5765(05)80141-9
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. Peer J. Prepr. 4:e1900v1. doi: 10.7287/peerj.preprints.1900v1
Ryba-White, M., Notteghem, J. L., and Leach, J. E. (1995). Comparison of Xanthomonas oryzae pv. oryzae strains from Africa, North America, and Asia by restriction fragment length polymorphism analysis. Int. Rice Res. Newsl. 20, 25–26.
Salzberg, S. L., Sommer, D. D., Schatz, M. C., Phillippy, A. M., Rabinowicz, P. D., Tsuge, S., et al. (2008). Genome sequence and rapid evolution of the rice pathogen Xanthomonas oryzae pv. oryzae PXO99A. BMC Genomics 9:204. doi: 10.1186/1471-2164-9-204
Schandry, N., de Lange, O., Prior, P., and Lahaye, T. (2016). TALE-like effectors are an ancestral feature of the Ralstonia solanacearum species complex and converge in DNA Targeting Specificity. Front. Plant Sci. 7:1225. doi: 10.3389/fpls.2016.01225
Schandry, N., Jacobs, J. M., Szurek, B., and Perez-Quintero, A. L. (2018). A cautionary TALE: how plant breeding may have favoured expanded TALE repertoires in Xanthomonas. Mol. Plant Pathol. 19, 1297–1301. doi: 10.1111/mpp.12670
Schliep, K. P. (2011). phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593. doi: 10.1093/bioinformatics/btq706
Schwartz, A. R., Potnis, N., Timilsina, S., Wilson, M., Patané, J., Martins, J., et al. (2015). Phylogenomics of Xanthomonas field strains infecting pepper and tomato reveals diversity in effector repertoires and identifies determinants of host specificity. Front. Microbiol. 6:535. doi: 10.3389/fmicb.2015.00535
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Takahashi, H., Buchner, P., Yoshimoto, N., Hawkesford, M. J., and Shiu, S.-H. (2011). Evolutionary relationships and functional diversity of plant sulfate transporters. Front. Plant Sci. 2:119. doi: 10.3389/fpls.2011.00119
Tran, T. T., Pérez-Quintero, A. L., Wonni, I., Carpenter, S. C. D., Yu, Y., Wang, L., et al. (2018). Functional analysis of African Xanthomonas oryzae pv. oryzae TALomes reveals a new susceptibility gene in bacterial leaf blight of rice. PLoS Pathog. 14:e1007092. doi: 10.1371/journal.ppat.1007092
Triplett, L., and Leach, J. (2016). Host mechanisms for resistance to TAL effectors: thinking outside the UPT box. Physiol. Mol. Plant Pathol. 95, 66–69. doi: 10.1016/j.pmpp.2016.02.002
Triplett, L. R., Cohen, S. P., Heffelfinger, C., Schmidt, C. L., Huerta, A. I., Tekete, C., et al. (2016). A resistance locus in the American heirloom rice variety Carolina Gold Select is triggered by TAL effectors with diverse predicted targets and is effective against African strains of Xanthomonas oryzae pv. oryzicola. Plant J. 87, 472–483. doi: 10.1111/tpj.13212
Triplett, L. R., Hamilton, J. P., Buell, C. R., Tisserat, N. A., Verdier, V., Zink, F., et al. (2011). Genomic analysis of Xanthomonas oryzae isolates from rice grown in the United States reveals substantial divergence from known X. oryzae pathovars. Appl. Environ. Microbiol. 77, 3930–3937. doi: 10.1128/AEM.00028-11
Vauterin, L., Hoste, B., Kersters, K., and Swings, J. (1995). Reclassification of Xanthomonas. Int. J. Syst. Bacteriol. 45, 472–489. doi: 10.1099/00207713-45-3-472
Verdier, V., Triplett, L. R., Hummel, A. W., Corral, R., Cernadas, R. A., Schmidt, C. L., et al. (2012). Transcription activator-like (TAL) effectors targeting OsSWEET genes enhance virulence on diverse rice (Oryza sativa) varieties when expressed individually in a TAL effector-deficient strain of Xanthomonas oryzae. New Phytol. 196, 1197–1207. doi: 10.1111/j.1469-8137.2012.04367.x
Wang, D., Zhang, X., Liu, J., Zhu, Y., Zhang, H., Zhang, A., et al. (2012). Oxalic acid enhances Cr tolerance in the accumulating plant Leersia hexandra Swartz. Int. J. Phytoremediation 14, 966–977. doi: 10.1080/15226514.2011.636406
White, F. F., Potnis, N., Jones, J. B., and Koebnik, R. (2009). The type III effectors of Xanthomonas. Mol. Plant Pathol. 10, 749–766. doi: 10.1111/j.1364-3703.2009.00590.x
Wilkins, K. E., Booher, N. J., Wang, L., and Bogdanove, A. J. (2015). TAL effectors and activation of predicted host targets distinguish Asian from African strains of the rice pathogen Xanthomonas oryzae pv. oryzicola while strict conservation suggests universal importance of five TAL effectors. Front. Plant Sci. 6:536. doi: 10.3389/fpls.2015.00536
Win, J., Chaparro-Garcia, A., Belhaj, K., Saunders, D. G. O., Yoshida, K., Dong, S., et al. (2012). Effector biology of plant-associated organisms: concepts and perspectives. Cold Spring Harb. Symp. Quant. Biol. 77, 235–247. doi: 10.1101/sqb.2012.77.015933
Wonni, I., Cottyn, B., Detemmerman, L., Dao, S., Ouedraogo, L., Sarra, S., et al. (2014). Analysis of Xanthomonas oryzae pv. oryzicola population in Mali and Burkina Faso reveals a high level of genetic and pathogenic diversity. Phytopathology 104, 520–531. doi: 10.1094/PHYTO-07-13-0213-R
Wu, M., and Scott, A. J. (2012). Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28, 1033–1034. doi: 10.1093/bioinformatics/bts079
Xiao, W., Liu, H., Li, Y., Li, X., Xu, C., Long, M., et al. (2009). A rice gene of de novo origin negatively regulates pathogen-induced defense response. PLoS One 4:e4603. doi: 10.1371/journal.pone.0004603
Xie, Z., and Tang, H. (2017). ISEScan: automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 33, 3340–3347. doi: 10.1093/bioinformatics/btx433
Yang, J., Zhang, Y., Yuan, P., Zhou, Y., Cai, C., Ren, Q., et al. (2014). Complete decoding of TAL effectors for DNA recognition. Cell Res. 24, 628–631. doi: 10.1038/cr.2014.19
You, S.-H., Zhang, X.-H., Liu, J., Zhu, Y.-N., and Gu, C. (2013). Feasibility of constructed wetland planted with Leersia hexandra Swartz for removing Cr, Cu and Ni from electroplating wastewater. Environ. Technol. 35, 187–194. doi: 10.1080/09593330.2013.822006
Zhao, S., Poulin, L., Rodriguez, R. L., Serna, N. F., Liu, S. Y., Wonni, I., et al. (2012). Development of a variable number of tandem repeats typing scheme for the bacterial rice pathogen Xanthomonas oryzae pv. oryzicola. Phytopathology 102, 948–956. doi: 10.1094/PHYTO-04-12-0078-R
Keywords: Xanthomonas oryzae, transcription activator-like effectors (TALEs), agroecosystem, cutgrass, rice
Citation: Lang JM, Pérez-Quintero AL, Koebnik R, DuCharme E, Sarra S, Doucoure H, Keita I, Ziegle J, Jacobs JM, Oliva R, Koita O, Szurek B, Verdier V and Leach JE (2019) A Pathovar of Xanthomonas oryzae Infecting Wild Grasses Provides Insight Into the Evolution of Pathogenicity in Rice Agroecosystems. Front. Plant Sci. 10:507. doi: 10.3389/fpls.2019.00507
Received: 21 December 2018; Accepted: 02 April 2019;
Published: 30 April 2019.
Edited by:
Dawn Arnold, University of the West of England, United KingdomReviewed by:
Gongyou Chen, Shanghai Jiao Tong University, ChinaDavid J. Studholme, University of Exeter, United Kingdom
Copyright © 2019 Lang, Pérez-Quintero, Koebnik, DuCharme, Sarra, Doucoure, Keita, Ziegle, Jacobs, Oliva, Koita, Szurek, Verdier and Leach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valérie Verdier, dmFsZXJpZS52ZXJkaWVyQGlyZC5mcg== Jan E. Leach, SmFuLkxlYWNoQGNvbG9zdGF0ZS5lZHU=
†These authors have contributed equally to this work