 Fengrui Ren
Fengrui Ren Chong Ren
Chong Ren Zhan Zhang
Zhan Zhang Wei Duan
Wei Duan David Lecourieux
David Lecourieux Shaohua Li
Shaohua Li Zhenchang Liang
Zhenchang Liang- 1Beijing Key Laboratory of Grape Sciences and Enology, Laboratory of Plant Resources, Institute of Botany, Chinese Academy of Sciences, Beijing, China
- 2College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
- 3EGFV, Bordeaux Sciences Agro, INRA, Université de Bordeaux, Villenave d’Ornon, France
- 4Sino-Africa Joint Research Center, Chinese Academy of Sciences, Wuhan, China
Clustered regularly interspersed short palindromic repeats (CRISPR)/Cas system is an efficient targeted genome editing method. Although CRISPR/Cas9-mediated mutagenesis has been applied successfully in grape, few studies have examined the technique’s efficiency. To optimize CRISPR/Cas9 editing efficiency in Vitis vinifera, we surveyed three key parameters: GC content of single guide RNA (sgRNA), variety of transformant cells used, and SpCas9 expression levels in transgenic cell mass. Four sgRNAs with differing GC content were designed to target exon sites of the V. vinifera phytoene desaturase gene. Suspension cells of ‘Chardonnay’ and ‘41B’ varieties were used as the transgenic cell mass. Both T7EI and PCR/RE assays showed that CRISPR/Cas9 editing efficiency increases proportionally with sgRNA GC content with 65% GC content yielding highest editing efficiency in both varieties. Additionally, gene editing was more efficient in ‘41B’ than in ‘Chardonnay.’ CRISPR/Cas9 systems with different editing efficiency showed different SpCas9 expression level, but compared with GC content of sgRNA, SpCas9 expression level has less influence on editing efficiency. Taken together, these results help optimize of CRISPR/Cas9 performance in grape.
Introduction
Targeted genome editing (TGE) using site-specific nucleases (SSNs) is a popular technique for studying gene function and new traits (Lee et al., 2016). These powerful SSN tools introduce targeted DNA double-strand breaks to trigger DNA repair pathways involving either non-homologous end-joining (NHEJ) or homologous recombination (HR) (Symington and Gautier, 2011). Gene editing is performed with zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), or clustered regulatory interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system (CRISPR/Cas9). However, designing appropriate constructs for the first two techniques is complex and costly, leading to a preference for the CRISPR/Cas (Lozano-Juste and Cutler, 2014). Of the three types of CRISPR/Cas systems (I, II, and III), type II (Cas9 protein) is the most popular because it is highly effective in facilitating RNA-guided SSNs (Cong et al., 2013; Wu et al., 2014). CRISPR/Cas9 has been used for TGE in a wide variety of animals and plants. Examples of the former include mammalian cells, zebrafish embryos (Hwang et al., 2013), and mice (Yang et al., 2013). Examples of the latter include Arabidopsis (Feng et al., 2013; Li et al., 2013), tobacco (Shan et al., 2014), wheat (Shan et al., 2014), grape (Ren et al., 2016), sorghum (Jiang et al., 2013), maize (Chen and Gao, 2014), soybean (Jacobs et al., 2015), and tomato (Brooks et al., 2014; Pan et al., 2016). Despite this popularity, the CRISPR/Cas system still has unresolved limitations, such as off-target mutagenesis, low editing efficiency, variation in specificity, the presence of protospacer adjacent motif (PAM) sequences and recalcitrant sgRNA/targets.
Grapes (Vitis vinifera) are one of the most important and economically valuable fruit crops worldwide. Due to ongoing climate change, there is also an increasingly urgent need for breeding improvement. Although whole-genome sequence is available for grapevine (V. vinifera ‘Pinot Noir’) (Jaillon et al., 2007), functional genomics has been hampered by the lack of stable and efficient genetic transformation protocols. Fortunately, a recent study found success in using CRISPR/Cas9 for targeted mutagenesis in grapevine (Ren et al., 2016). Subsequently, the CRISPR/Cas9 system was also successfully used to target the grapevine genes VvWRKY52 (a transcription factor) (Wang et al., 2018) and VvPDS (phytoene desaturase) (Nakajima et al., 2017). Nevertheless, editing efficiency remains an area that requires further improvement.
The development of a robust transformation system suitable for a wide range of cultivars is necessary for improving CRISPR/Cas9 efficiency in grape. Currently, some grape cultivars are recalcitrant to Agrobacterium-mediated transformation, though this method is effective in several others (Iocco et al., 2001). Furthermore, agrotransformation is unreliable and inefficient in grape compared with model plants, such as Arabidopsis thaliana and Nicotiana benthamiana. Desired transgenic grape lines thus take much longer to develop than in traditional model plants. These between-species differences are due to numerous parameters, including Agrobacterium strain, culture medium, antibiotic concentration, and temperature. Clearly, there is a pressing need to understand the exact factors that influence efficiency of CRISPR/Cas9 genome editing in grapevine.
In this study, we investigated three parameters that can potentially be optimized to improve TGE in grape. The first factor is the GC content of sgRNA used to target our gene of interest (phytoene desaturase, VvPDS). Phytoene desaturase is part of the carotenoid biosynthetic pathway, and its reduction or loss results in a photobleaching phenotype due to chlorophyll photooxidation (Qin et al., 2007). Therefore, plants carrying functionally disrupted PDS have distinct albino and dwarf morphologies that are easily identifiable from wild-type. The second is cultivar used for transformation, while the last is SpCas9 gene expression in transgenic cell masses (CMs). By analyzing the relationship between these key parameters and determining CRISPR/Cas9 efficiency under different conditions, we provided the basis for a high efficiency CRISPR/Cas9-mediated genome editing protocol. Our findings should help to promote the development of functional genomics and breeding improvement in grape.
Materials and Methods
Plant Materials
‘Chardonnay’ (V. vinifera L.) and ‘41B’ (V. vinifera ‘Chasselas’ × V. berlandieri) were derived from induced embryogenic calli. The former was cultured in 100 mL flasks filled with 25 mL of liquid CSM medium [MS basal medium supplemented with 0.5 g/L glutamic acid, 1 mg/L 2-naphthoxyacetic acid (NOA), 5.0 mL/L glycerol, 20 g/L maltose, pH 5.8]. The latter was cultivated in liquid GM medium [MS medium 1/2 Macro, with 1 g/L N-Z-Amine A, 1 mg/L 2-naphthoxyacetic acid (NOA), 4.6 g/L glycerol, Maltose 18 g/L, 1 mL/L Vitamins GMox1000, pH 5.8]. Suspension cells were shaken at 117 rpm and 27°C in the dark. Cells were sub-cultured every 7 days.
Extraction of Genomic DNA
Genomic DNA was extracted from wild-type (WT) and resistant CM using CTAB (Zhang et al., 2016). First, 700 μL pre-heated CTAB buffer was added into 100 mg CM ground in liquid nitrogen. The mixture was then incubated at 65°C for 20 min before 700 μL chloroform was added to each sample. After centrifugation at 12,000 rpm for 5 min, the supernatant was transferred to a new tube, followed by the addition 500 μL isopropanol and incubation at 4°C for 30 min. The solution was centrifuged again at 12,000 rpm for 10 min to separate out genomic DNA. The DNA pellet was washed with 500 μL of 70% ethanol, and dissolved in 100 μL ddH2O to measure via spectrophotometer.
Cloning of VvPDS Exon
The VvPDS exon was amplified from genomic DNA of both cultivars using High-Fidelity DNA polymerase KOD-plus Neo (TOYOBO, Japan). Specific primers (Supplementary Table S1) were designed based on the homologous gene VIT_09s0002g00100 from the EnsemblPlants1. Thermocycling conditions were as follows: 95°C for 5 min; 45 cycles of 95°C for 10 s, 57°C for 30 s, and 68°C for 30 s; followed by a final extension at 68°C for 5 min. The PCR product was cloned into the pClone007 Simple Vector (TSINGKE), and around five clones were sequenced.
Design of sgRNA and Assembly of CRISPR/Cas9 Construct
CRISPR/Cas9 target sites were designed from verified sequences with the online tools Grape-CRISPR Database2. Four target sites (Table 1) were selected for designing target sgRNAs based on their GC content, location in the gene, and off-target possibilities. The pP1C.4 vector is the backbone of the CRISPR/Cas9 vector carrying plant-optimized Streptococcus pyogenes Cas9 protein-coding gene (Genloci, China). To obtain the AtU6-sgRNA cassette, reverse primers (Supplementary Table S1) comprising of 20 bp sgRNA sequences and an adaptor were used. To amplify the AtU6 promoter fragment, PCR was used to combine the AtU6 promoter and sgRNA. Amplified AtU6-sgRNA fragments containing adaptors could be inserted into the homologous sites of the linearized vector by the homologous recombination method.

Table 1. Summary of the six selected sgRNA.
Growth, Transformation of Grape Suspension Cells
Grape cells were transformed using an Agrobacterium tumefaciens cocultivation method (Martinelli and Mandolino, 1994). Final plasmids were introduced into A. tumefaciens using the freeze-thaw method described by manufacturer protocols. The prepared A. tumefaciens suspension was inoculated with suspension cell cultures collected though a brief centrifugation. After that, for rapid selection, we transferred them into selective liquid CSM medium and liquid GM medium with 5 mg/L hygromycin until resistant CMs were generated. The transformation events with the same Cas9/sgRNA construction were repeated three times at the same time to keep the activity of grape suspension cells and A. tumefaciens consistent. The hygromycin and liquid medium needed to be replaced every 7 days.
Identification of PCR-Generated Exogenous T-DNA Insertion
Specific primers for the hygromycin-resistance gene (Supplementary Table S1) were used for PCR with High-Fidelity DNA polymerase KOD-plus Neo (TOYOBO, Japan). The thermocycler was set at 95°C for 5 min; 34 cycles of 95°C for 10 s, 60°C for 30 s, and 68°C for 30 s; followed by a final extension at 68°C for 5 min. Amplicons were separated on an EtBr-stained agarose gel (1.0%), then cloned into the pclone007 vector for Sanger sequencing.
Detection and Sequencing of Mutations in Transgenic CMs
Prepared genomic DNA of transgenic CMs was used as a template to amplify genomic fragments containing target sites. Specific primers (Supplementary Table S1) were designed to PCR-amplify a 300–600 bp product containing the target site. The PCR reaction was performed with High-Fidelity DNA polymerase KOD-plus Neo in a total volume of 50 μL at 95°C for 5 min; 45 cycles of 98°C for 10 s, 57°C for 30 s and 68°C for 30 s; followed by a final extension at 68°C for 5 min. Amplicons were cloned into pclone007. Approximately 10 clones were sequenced to search for the chimerism in CM.
Identification of CRISPR/Cas9 Efficiency
Two strategies were selected to determine CRISPR/Cas9 system efficiency (Shan et al., 2014). For both assays, PCR was first performed following procedures described above. Amplicons of sgRNACr1-CM and sgRNACr4-CM lines were then digested with the appropriate, assay-specific enzymes, following manufacturer protocol. Bands were visualized with gel electrophoresis. The WT-CM line was used as a positive control. Band intensity (used for estimating indel frequency) was quantified in ImageJ. Three biological replicates were performed for each transgenic and WT line.
PCR/Restriction Enzyme (RE) Assay
This assay requires a RE site that can be disrupted by CRISPR/Cas9-induced mutations. Digestion with the restriction enzyme would then yield un-cleaved bands in PCR. The RE used on sgRNACr1-CM and sgRNACr4-CM amplicons were SspI and EcoRI (NEB, United States), respectively. An EtBr-stained 2.5% agarose gel was used to separate bands.
Indel frequency was calculated with the following formula:
where a is intensity of the undigested PCR product, while b and c are intensities of the two digested products. Mean indel efficiency was calculated from the three replicates.
T7EI Assay
When the target gene does not include an appropriate RE site, the T7EI assay is a better option for assessing CRISPR/Cas9 efficiency. The T7EI nuclease can digest CRISPR/Cas9-induced mismatched dsDNA, leaving behind WT and mutant homoduplexes. Amplicons of sgRNACr3-CM and sgRNACrP1-CM were digested with T7EI enzyme (NEB, United States). An EtBr-stained 3.0% agarose gel was used for separating bands.
The indel frequency was calculated from the following formula:
where a is intensity of undigested PCR product, while b and c are intensities of the two digested products. Mean indel efficiency was calculated from the three independent replicates.
Quantitative RT-PCR Analysis
Total RNA was extracted from ‘Chardonnay’ and ‘41B’ CMs using TRIzol reagent (Invitrogen, United States). Complementary DNA was synthesized from 1 μg of RNA using the HiScript Q RT SuperMix for qPCR (+ gDNA wiper) kit (Vazyme, China) following the manufacturer-provided protocol. Quantitative RT-PCR was performed in a final volume of 20 μL on a CFX96 Real-Time System (Bio-Rad, United States), with SpCas9 specific primers (Supplementary Table S1). Grape Actin1 (AY680701) was used as an internal control. Relative expression level was calculated using the 2-ΔΔCT method. All experiments were performed with three biological replicates and three technical replicates.
Results
Selection of sgRNA for Constructing VvPDS and CRISPR/Cas9 Expression Vectors
Four DNA fragments from VvPDS exons were cloned and sequenced. We observed that ‘Chardonnay’ and ‘41B’ cultivars had nearly identical VvPDS exons to their homologs from the ‘Pinot Noir’ reference genome (PN40024, Supplementary Figure S1). Four 20 bp target sequences with the NGG PAM in VvPDS were designed as sgRNA complementary sites. These sgRNA sites were located in the fourth, second, sixth, and seventh exons, capturing 25, 30, 50, 65% GC content, respectively (Table 1). Arabidopsis U6 promoter (AtU6) was used to drive expression of the four targets, while CaMV 35S promoter drove SpCas9 expression. Expression cassettes were inserted into the pCACRISPR/Cas9 binary vector using the homologous recombination method (Ren et al., 2016) (Figure 1).
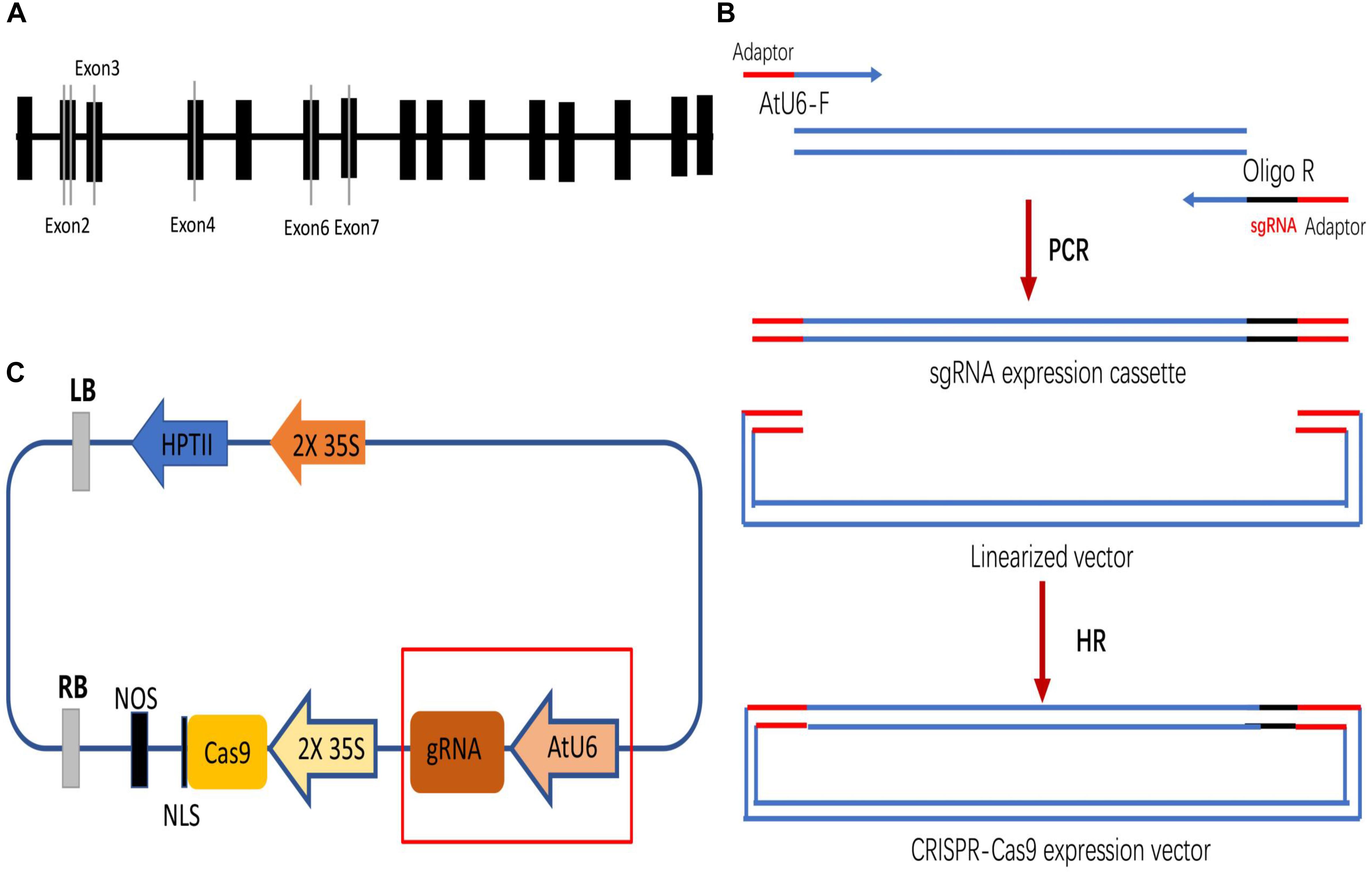
Figure 1. Selecting target sites in the VvPDS gene and constructing Cas9/sgRNA expression vectors. (A) Four sgRNAs (gray lines) were selected in the VvPDS exon sequence (black boxes). (B) Schematic diagram of the protocol for expression vector construction. (C) Schematic diagram of the assembled Cas9/sgRNAs expression vector (pCACRISPR/Cas9) for Agrobacterium-mediated suspension-cell transformation.
Grape Transformation and Identification of Positive Transgenic CMs
‘Chardonnay’ and ‘41B’ cells were transformed with A. tumefaciens EHA105 containing the CRISPR/Cas9 vector. After co-culturing for 2 days, inoculated CMs were transferred to media containing antibiotics hygromycin and cefotaxime for 8–10 weeks for screening. Transgenic CM appeared yellow (Figure 2). Successful transformation was validated through PCR with specific primers for hygromycin-resistant gene; eight tested CMs contained the expected exogenous T-DNA insertions (Figure 2 and Supplementary Table S1).

Figure 2. Grape transformation and T-DNA identification. (A) Selection of ‘Chardonnay’ cells in liquid medium containing antibiotics. (B) Selection of ‘41B’ cells in liquid medium containing antibiotics. Yellowish, resistant transgenic cell masses in red boxes and brown, non-resistant cell masses in blue boxes. (C) Identification of exogenous T-DNA insertion in sgRNA-CMs. The PCR template was genomic DNA of sgRNA-CMs, containing specific primers for the hygromycin-resistance gene. Plasmid of the constructed vector (P4) and wild-type DNA (WT) were used as a positive control and a negative control, respectively. T-DNA insertion was detected in sgRNACr1-41B (B1), sgRNACr1-char (C1), sgRNACr3-41B (B3), sgRNACr3-char (C3), sgRNACr4-41B (B4), sgRNACr4-char (C4), sgRNACrP1-41B (BP1), and sgRNACrP1-char (CP1).
Detection and Sequencing of Mutations in Transgenic CMs
Every 4 weeks post-agrotransformation, we sequenced DNA fragments containing target sites from eight transgenic sgRNA-PDS CMs (sgRNACr1-char, sgRNACr3-char, sgRNACr4-char, sgRNACrP1-char, sgRNACr1-41B, sgRNACr3-41B, sgRNACr4-41B, sgRNACrP1-41B). After 12 weeks, indels were detected in sgRNACr3-41B and sgRNACr3-char; after 16 weeks, indels were observed in sgRNACr4-char sgRNACr4-41B. Indels for sgRNACr1 and sgRNACrP1 were detected after 20 and 24 weeks in ‘41B’ and in ‘Chardonnay,’ respectively.
Sanger sequencing showed that fragments from eight sgRNA-CMs (sgRNACr1-char, sgRNACr3-char, sgRNACr4-char, sgRNACrP1-char, sgRNACr1-41B, sgRNACr3-41B, sgRNACr4-41B, sgRNACrP1-41B) contained indels at the target sites (Figure 3). Thus, CRISPR/Cas9 successfully edited four out of six target sites, using four different sgRNAs with 25, 30, 50, and 65% GC content.
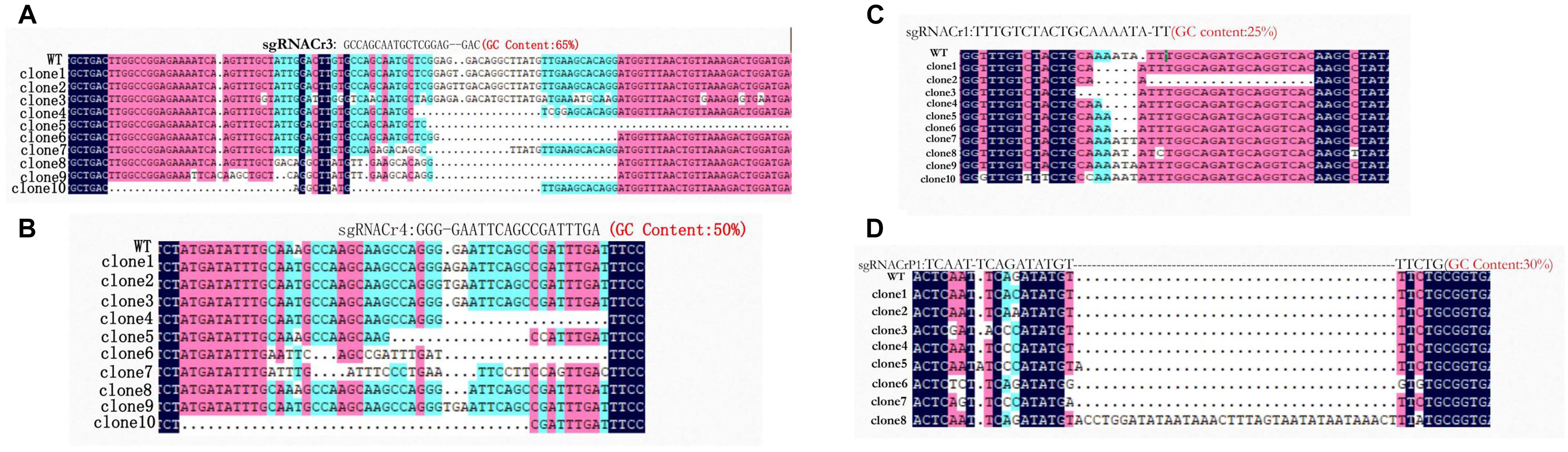
Figure 3. Detection and sequencing of targeted VvPDS mutations in transgenic CMs. (A) DNA sequences of mutations at target site sgRNACr3 in ‘41B’ (clone 1 to clone 6) and in ‘Chardonnay’ (clone 7 to clone 10). (B) Sequence alignment of mutations at target site sgRNACr4 in ‘41B’ (clone 1 to clone 7) and in ‘Chardonnay’ (clone 8 to clone 10). (C) Targeted mutagenesis of VvPDS at target site sgRNACr1 in ‘41B’ (clone 1 to clone 5) and in ‘Chardonnay’ (clone 5 to clone 10). (D) Mutated DNA sequences at target site sgRNACrP1 in ‘41B’ (clone 1 to clone 6) and in ‘Chardonnay’ (clone 7 to clone 8). Homologous nucleotides are shaded, and different colors indicate different homology levels: 100% homology, black; ≥75%, red; ≥50%, blue.
CRISPR/Cas9 Efficiency Using sgRNA Differing in GC Content
We hypothesized that using sgRNA with higher GC content increased CRISPR/Cas9 efficiency, because mutations were detected far more quickly in sgRNACr3-CMs and sgRNACr4-CMs than in sgRNACr1-CMs and sgRNACrP1-CMs. We tested this hypothesis using PCR/RE and T7EI assays.
Results from the digestion of PCR products with SspI revealed a band indicative of indels at the target site in sgRNACr1-41B and sgRNACr1-char. Contrary to these results, when PCR products obtained with templates coming from WT-CMs were digested with SspI, there was no additional band, as would be expected when indels are absent. Band intensity suggested that mean indel frequencies of sgRNACr1-41B and sgRNACr1-char were 22.2 and 30.3%, respectively (Figure 4A).
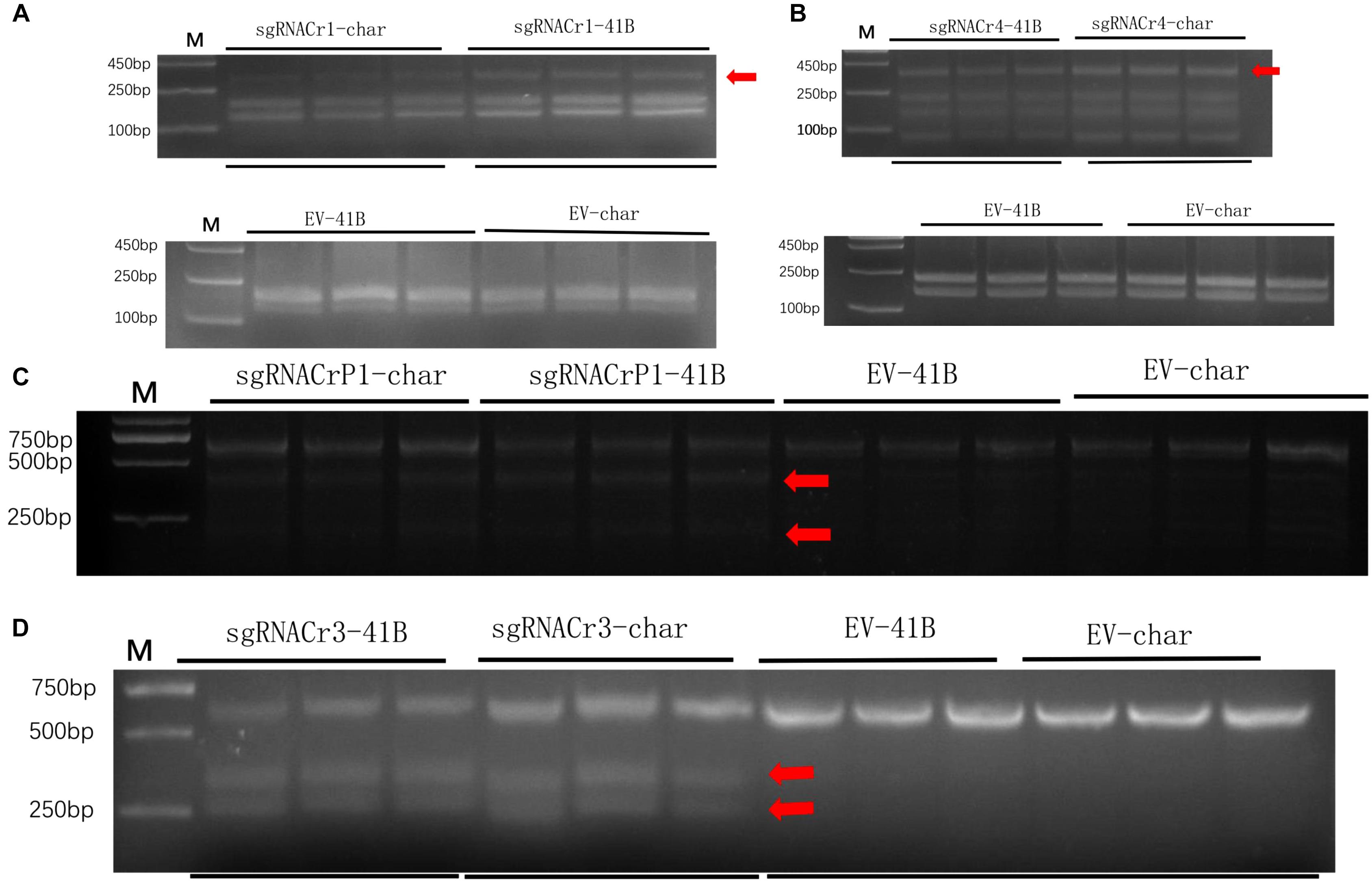
Figure 4. Efficiency of CRISPR/Cas9 using guide RNAs with differing GC content in 41B and “Chardonnay.’ Agarose gels illustrating indel frequency in sgRNACr1-CM lines (A) and sgRNACr4-CM lines (B) detected using PCR/RE assays of transformed cell masses (red arrowheads indicate mutated bands). Agarose gels showing mutagenesis frequency in sgRNACrP1-CM lines (C) and sgRNACr3-CM lines (D) detected using T7EI assays of transformed cell masses (red arrowheads indicate cleaved mutated bands). ‘-41B’ labels indel frequency at each target site in ‘41B,’ while ‘-char’ labels indel frequency in ‘Chardonnay.’ WT-41B and WT-char refer to wild-type, untransformed cell masses. Mean indel efficiency was calculated from three independent replicates.
Digestion with EcoRI similarly identified indels in the target sites of sgRNACr4-41B and sgRNACr4-char, whereas no targets in either of WT-CMs had indels (bands) (Figure 4B). Band intensity revealed an average indel frequency of 82.9 and 55.1% for sgRNACr4-41B and sgRNACr4-char, respectively.
Digestion with T7E1 did not yield any additional fragments in WT-CMs, confirming the lack of mutations. However, both sgRNACrP1-41B and sgRNACrP1-char contained indel mutations, as represented by appearance of new bands. Average indel frequency was around 35.4 and 25.6% for the two sgRNACrP1-CMs, respectively (Figure 4C).
Digestion with T7EI also showed CRISPR/Cas9-mutated target sites in sgRNACr3-41B and sgRNACr3-char CMs, but not in WT-CMs (Figure 4D). Average indel frequency for sgRNACr3-41B and sgRNACr3-char was 86.6 and 59.9%, respectively.
Regardless of cultivar, CRISPR/Cas9 editing efficiency dropped significantly when using sgRNA with low GC content (25 and 30%) than those with high GC content (50 and 65%) (Table 2). The highest editing efficiency in both cultivars occurred when we used sgRNA with 65% GC content, suggesting that CRISPR/Cas9 maybe more efficient in the system with higher GC content sgRNA (>50%) than in those with lower GC content sgRNA (<30%). In addition, for sgRNACr1, there is no statistically significant difference between two cultivars. However, under the identical GC contents, sgRNA-41B had a higher mutation rate than sgRNA-char.
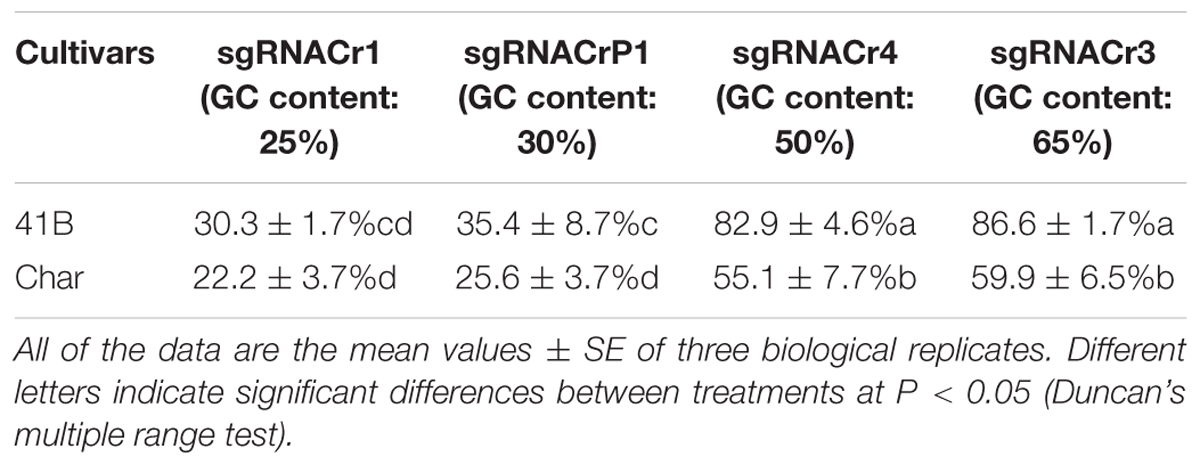
Table 2. Summary of CRISPR/Cas9-mediated targeted editing efficiency in ‘41B’ and ‘Chardonnay’ grape cultivars.
SpCas9 Expression Analysis in ‘41B’ and ‘Chardonnay’ Transgenic Cells
The results of qRT-PCR revealed that all transgenic sgRNA-CM lines exhibited 35S-promoter-driven SpCas9 expression (Figure 5). In ‘Chardonnay’ transgenic cells, SpCas9 expression was detected with different CRISPR/Cas9-induced editing efficiency but no statistically significant difference was found between the four transgenic samples. To make sure the correlation existed between editing efficiency, SpCas9 expression, and GC content of sgRNA, Pearson’s test analysis was used. In ‘Chardonnay,’ the correlation coefficient (R2) between editing efficiency and SpCas9 expression was 0.887 (P-value = 0.113); the correlation coefficient (R2) between editing efficiency and GC content was 0.971 (P-value = 0.029). In ‘41B’ transgenic cells, for SpCas9 expression and GC content, the R2 coefficient was 0.89 (P-value = 0.11) and 0.958 (P-value = 0.042), respectively. These results suggested that the GC content of sgRNA, but not SpCas9 expression level, might be the limiting factor for genome editing.
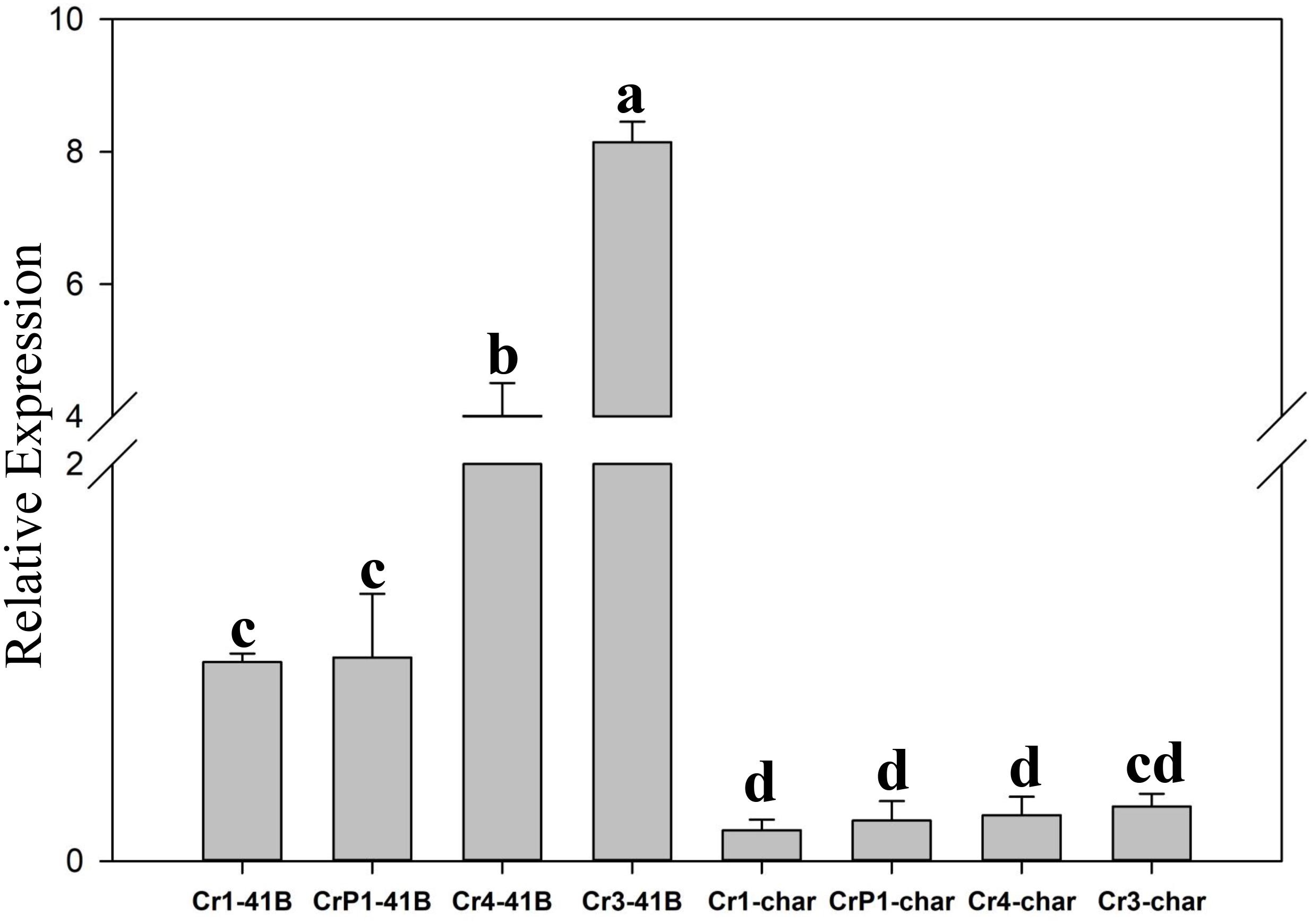
Figure 5. Expression analysis of Cas9 gene using qRT-PCR. Cas9 expression in sgRNACr1-char (Cr1-char), sgRNACr3-char (Cr3-char), sgRNACr4-char (Cr4-char), sgRNACrP1-char (P1-char), sgRNACr1-41B (Cr1-41B), sgRNACr3-41B (Cr3-41B), sgRNACr4-41B (Cr4-41B), and sgRNACrP1-41B (CrP1-41B). The control was Cr1-41B. Grape Actin1 was the internal control. Mean expression was calculated from three independent replicates. Vertical bars indicate standard errors of the mean. Different letters indicate significant differences between treatments at P < 0.05 (Duncan’s multiple range test).
Discussion
In the present study, we successfully generated transgenic grapevine in two cultivars through knocking out VvPDS with the CRISPR/Cas9 system. As PDS gene was widely used in verification of the feasibility of CRISPR/Cas9 system in many species, such as poplar (Fan et al., 2015), potato (Gao et al., 2015), tobacco (Zhang et al., 2016) and watermelon (Tian et al., 2017), we chose VvPDS as an effective “tool” gene to study the CRISPR/Cas9 system efficiency in grape. We will use other functional genes to verify the results we found about CRISPR/Cas9 system efficiency in further study. Although the same VvPDS-knockout transgenic plants had been generated previously using V. vinifera ‘Neo Muscat’ (Nakajima et al., 2017), the authors did not investigate CRISPR/Cas9 efficiency in detail. They reported that the rate of desired mutations remained low even 4 months post-transformation with two sgRNAs that they had designed. We calculated the GC content of their sgRNAs to be 25 and 45%. In this study, we used their sgRNA (PDS-t2) with 25% GC content, along with five other sgRNAs that we designed with the Grape-CRISPR Database2 to vary in GC. Four sgRNAs were effective in both ‘Chardonnay’ and ‘41B’ after 24 weeks of transgenic CM selection. Sanger sequencing showed that these four had GC contents of 25, 30, 50, and 65%. Together, our results and previous findings clearly support the hypothesis that CRISPR/Cas9 efficiency is related to sgRNA GC content.
Repair pathways involving NHEJ or HDR introduce small indels at target sites after TGE (Araki and Ishii, 2014). Therefore, indel rates can be used to predict genome editing efficiency (Shan et al., 2014; Tian et al., 2017; LeBlanc et al., 2018). In our study, we calculated mutation rates and CRISPR/Cas9 efficiency based on band intensity observed from gel electrophoresis. Our data led to the conclusion that high GC content in sgRNA increases mutation rate in both ‘Chardonnay’ and ‘41B.’ Our findings corroborate previous research using CRISPR/Cas9 to target IdnDH in ‘Chardonnay’ (Ren et al., 2016), where the sgRNA with 65% GC content yielded greater efficiency (higher indel rates) than the sgRNA with 35% GC. Beside our work, in another study it was reported that the mutation efficiency in the T1 (GC content 55%) and T4 (GC content 65%) site targeting VvWKY58 was 53.63 and 64.91%, respectively (Wang et al., 2018). Additional research suggested that GC content of the target sites also influenced mutation efficiency in tomato: the high editing efficiency (84.00–100.00%) was detected in sgRNAs with GC content above 50%, whereas the sgRNA with GC content containing a relatively low GC content (40%) exhibited lower editing efficiency (72.70%) (Pan et al., 2016). However, even for genes from “41B” cultivar edited with CRISPR/Cas9 system with high GC content sgRNA, it took 12 weeks to detect mutations. We plan to further confirm editing efficiency through investigating whether the ratio of albino (VvPDS knockout) plantlets correlates with indel rate and optimize the CRISPR/Cas9 system according to the data we got in a future study.
Grape transgenesis studies typically employ ‘Chardonnay’ and ‘41B’ (Lecourieux et al., 2010; Nicolas et al., 2013, 2014), whereas studies of CRISPR/Cas9 application in grape used ‘Chardonnay’ and ‘Thompson Seedless’ (Wang et al., 2018). In our study, we compared CRISPR/Cas9 efficiency in ‘Chardonnay’ and in ‘41B’ to determine whether grape genotype was an influential factor. Independent of sgRNA GC content, CRISPR/Cas9 efficiency was higher in ‘41B’ than in ‘Chardonnay.’ The results of SpCas9 expression analysis suggested that SpCas9 showed different expression level in CRISPR/Cas9 system with different editing efficiency. However, further data analysis showed that statistically significant correlation was found between GC content and CRISPR/Cas9 efficiency in ‘Chardonnay’ and in ‘41B.’ In ‘41B,’ two transgenic samples with high GC content of sgRNA, sgRNACr3-41B and sgRNACr4-41B, showed significantly higher SpCa9 expression level as well as editing efficiency than others. This result suggested that SpCas9 expression levels may influence CRISPR/Cas9-induced editing efficiency, but further correlation analysis showed that editing efficiency was more closely related with GC content than SpCas9 expression levels. Taken together, our data suggest that sgRNA GC content and CM genotype (i.e., cultivar) used for transformation are major limiting parameters governing the efficiency of CRISPR/Cas9-mediated targeted mutagenesis.
Conclusion
We successfully used CRISPR/Cas9 to knock out VvPDS in both ‘Chardonnay’ and ‘41B’ grape cultivars. Our findings supported the hypothesis that sgRNAs with high GC content improved editing efficiency in grapevine. Moreover, we showed that editing efficiency also depends on selecting the appropriate cultivar. Altogether, our study provides valuable data for efforts to optimize the use of CRISPR/Cas9 gene editing in grape.
Author Contributions
FR performed the experiments and wrote the manuscript. CR and ZZ helped with experiments and data analysis. ZL designed the experiments with the assistance of WD, DL, and SL. DL and ZL reviewed the manuscript. All authors have read and approved the final manuscript.
Funding
This work was supported by National Key Research and Development Program of China, grants from STS project of Chinese Academy of Sciences (KFJ-STS-ZDTP-025), Major Science and Technology Program of Ningxia Hui Autonomous Region (2016BZ06), Agricultural Breeding Project of Ningxia Hui Autonomous Region (NXNYYZ20150203), and National Natural Science Foundation of China (31772266).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Fatma Lecourieux for grape transformation, Chengcheng for revising the manuscript, and Renkun Tang for help with the SigmaPlot.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00612/full#supplementary-material
Footnotes
References
Araki, M., and Ishii, T. (2014). International regulatory landscape and integration of corrective genome editing into in vitro fertilization. Reprod. Biol. Endocrinol. 12:108. doi: 10.1186/1477-7827-12-108
Brooks, C., Nekrasov, V., Lippman, Z. B., and Van Eck, J. (2014). Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-associated9 system. Plant Physiol. 166, 1292–1297. doi: 10.1104/pp.114.247577
Chen, K., and Gao, C. (2014). Targeted genome modification technologies and their applications in crop improvements. Plant Cell Rep. 33, 575–583. doi: 10.1007/s00299-013-1539-6
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi: 10.1126/science.1231143
Fan, D., Liu, T., Li, C., Jiao, B., Li, S., Hou, Y., et al. (2015). Efficient CRISPR/Cas9-mediated targeted mutagenesis in Populus in the first generation. Sci. Rep. 5:12217. doi: 10.1038/srep12217
Feng, Z., Zhang, B., Ding, W., Liu, X., Yang, D. L., Wei, P., et al. (2013). Efficient genome editing in plants using a CRISPR/Cas system. Cell Res. 23, 1229–1232. doi: 10.1038/cr.2013.114
Gao, J., Wang, G., Ma, S., Xie, X., Wu, X., Zhang, X., et al. (2015). CRISPR/Cas9-mediated targeted mutagenesis in Nicotiana tabacum. Plant Mol. Biol. 87, 99–110. doi: 10.1007/s11103-014-0263-0
Hwang, W. Y., Fu, Y., Reyon, D., Maeder, M. L., Tsai, S. Q., Sander, J. D., et al. (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229. doi: 10.1038/nbt.2501
Iocco, P., Franks, T., and Thomas, M. R. (2001). Genetic transformation of major wine grape cultivars of Vitis vinifera L. Transgen. Res. 10, 105–112. doi: 10.1023/A:1008989610340
Jacobs, T. B., Lafayette, P. R., Schmitz, R. J., and Parrott, W. A. (2015). Targeted genome modifications in soybean with CRISPR/Cas9. BMC Biotechnol. 15:16. doi: 10.1186/s12896-015-0131-2
Jaillon, O., Aury, J. M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467. doi: 10.1038/nature06148
Jiang, W., Zhou, H., Bi, H., Fromm, M., Yang, B., and Weeks, D. P. (2013). Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 41:e188. doi: 10.1093/nar/gkt780
LeBlanc, C., Zhang, F., Mendez, J., Lozano, Y., Chatpar, K., Irish, V. F., et al. (2018). Increased efficiency of targeted mutagenesis by CRISPR/Cas9 in plants using heat stress. Plant J. 93, 377–386. doi: 10.1093/nar/gkt780
Lecourieux, F., Lecourieux, D., Vignault, C., and Delrot, S. (2010). A sugar-inducible protein kinase, VvSK1, regulates hexose transport and sugar accumulation in grapevine cells. Plant Physiol. 152, 1096–1106. doi: 10.1104/pp.109.149
Lee, J., Chung, J. H., Kim, H. M., Kim, D. W., and Kim, H. (2016). Designed nucleases for targeted genome editing. Plant Biotechnol. J. 14, 448–462. doi: 10.1111/pbi.12465
Li, J. F., Norville, J. E., Aach, J., Mccormack, M., Zhang, D., Bush, J., et al. (2013). Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 31, 688–691. doi: 10.1111/pbi.12465
Lozano-Juste, J., and Cutler, S. R. (2014). Plant genome engineering in full bloom. Trends Plant Sci. 19, 284–287. doi: 10.1016/j.tplants.2014.02.014
Martinelli, L., and Mandolino, G. (1994). Genetic transformation and regeneration of transgenic plants in grapevine (Vitis rupestris S.). Theor. Appl. Genet. 88, 621–628. doi: 10.1007/BF01253963
Nakajima, I., Ban, Y., Azuma, A., Onoue, N., Moriguchi, T., Yamamoto, T., et al. (2017). CRISPR/Cas9-mediated targeted mutagenesis in grape. PLoS One 12:e0177966. doi: 10.1371/journal.pone.0177966
Nicolas, P., Lecourieux, D., Gomes, E., Delrot, S., and Lecourieux, F. (2013). The grape berry-specific basic helix-loop-helix transcription factor VvCEB1 affects cell size. J. Exp. Bot. 64, 991–1003. doi: 10.1093/jxb/ers374
Nicolas, P., Lecourieux, D., Kappel, C., Cluzet, S., Cramer, G., Delrot, S., et al. (2014). The basic leucine zipper transcription factor abscisic acid response element-binding factor2 is an important transcriptional regulator of abscisic acid-dependent grape berry ripening processes. Plant Physiol. 164, 365–383. doi: 10.1104/pp.113.231977
Pan, C., Ye, L., Qin, L., Liu, X., He, Y., Wang, J., et al. (2016). CRISPR/Cas9-mediated efficient and heritable targeted mutagenesis in tomato plants in the first and later generations. Sci. Rep. 6:24765. doi: 10.1038/srep24765
Qin, G., Gu, H., Ma, L., Peng, Y., Deng, X. W., Chen, Z., et al. (2007). Disruption of phytoene desaturase gene results in albino and dwarf phenotypes in Arabidopsis by impairing chlorophyll, carotenoid, and gibberellin biosynthesis. Cell Res. 17, 471–482. doi: 10.1038/cr.2007.40
Ren, C., Liu, X., Zhang, Z., Wang, Y., Duan, W., Li, S., et al. (2016). CRISPR/Cas9-mediated efficient targeted mutagenesis in Chardonnay (Vitis vinifera L.). Sci. Rep. 6:32289. doi: 10.1038/srep32289
Shan, Q., Wang, Y., Li, J., and Gao, C. (2014). Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 9, 2395–2410. doi: 10.1038/nprot.2014.157
Symington, L. S., and Gautier, J. (2011). Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271. doi: 10.1146/annurev-genet-110410-132435
Tian, S., Jiang, L., Gao, Q., Zhang, J., Zong, M., Zhang, H., et al. (2017). Efficient CRISPR/Cas9-based gene knockout in watermelon. Plant Cell Rep. 36, 399–406. doi: 10.1007/s00299-016-2089-5
Wang, X., Tu, M., Wang, D., Liu, J., Li, Y., Li, Z., et al. (2018). CRISPR/Cas9-mediated efficient targeted mutagenesis in grape in the first generation. Plant Biotechnol. J. 16, 844–855. doi: 10.1111/pbi.12832
Wu, X., Scott, D. A., Kriz, A. J., Chiu, A. C., Hsu, P. D., Dadon, D. B., et al. (2014). Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 32, 670–676. doi: 10.1038/nbt.2889
Yang, H., Wang, H., Shivalila, C. S., Cheng, A. W., Shi, L., and Jaenisch, R. (2013). One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379. doi: 10.1016/j.cell.2013.08.022
Keywords: CRISPR/Cas9, optimization, GC content, grape, gene expression, gene editing efficiency
Citation: Ren F, Ren C, Zhang Z, Duan W, Lecourieux D, Li S and Liang Z (2019) Efficiency Optimization of CRISPR/Cas9-Mediated Targeted Mutagenesis in Grape. Front. Plant Sci. 10:612. doi: 10.3389/fpls.2019.00612
Received: 31 October 2018; Accepted: 25 April 2019;
Published: 16 May 2019.
Edited by:
Vladimir Orbovic, University of Florida, United StatesReviewed by:
Yosvanis Acanda Artiga, University of Florida, United StatesJorge Lozano-Juste, Instituto de Biología Molecular y Celular de Plantas (IBMCP), Spain
Copyright © 2019 Ren, Ren, Zhang, Duan, Lecourieux, Li and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenchang Liang, WkwyNDlAaWJjYXMuYWMuY24=