Abstract
Tomato (Solanum lycopersicum) is an important vegetable and fruit crop. Its genome was completely sequenced and there are also a large amount of available expressed sequence tags (ESTs) and short reads generated by RNA sequencing (RNA-seq) technologies. Mapping transcripts including mRNA sequences, ESTs, and RNA-seq reads to the genome allows identifying pre-mRNA alternative splicing (AS), a post-transcriptional process generating two or more RNA isoforms from one pre-mRNA transcript. We comprehensively analyzed the AS landscape in tomato by integrating genome mapping information of all available mRNA and ESTs with mapping information of RNA-seq reads which were collected from 27 published projects. A total of 369,911 AS events were identified from 34,419 genomic loci involving 161,913 transcripts. Within the basic AS events, intron retention is the prevalent type (18.9%), followed by alternative acceptor site (12.9%) and alternative donor site (7.3%), with exon skipping as the least type (6.0%). Complex AS types having two or more basic event accounted for 54.9% of total AS events. Within 35,768 annotated protein-coding gene models, 23,233 gene models were found having pre-mRNAs generating AS isoform transcripts. Thus the estimated AS rate was 65.0% in tomato. The list of identified AS genes with their corresponding transcript isoforms serves as a catalog for further detailed examination of gene functions in tomato biology. The post-transcriptional information is also expected to be useful in improving the predicted gene models in tomato. The sequence and annotation information can be accessed at plant alternative splicing database (http://proteomics.ysu.edu/altsplice).
Introduction
Understanding the transcriptome diversity and gene expression dynamics is critical for developing methods for further improving the quantity and quality of plant products. Tomato (Solanum lycopersicum), one of the important fruit and vegetable crops, has its genome being completely sequenced. The complete genome of the inbred tomato cultivar “Heinz 1706” approximately has a size of 900 megabases (Mb) with a total of 34,727 protein-coding genes predicted (Tomato Genome Consortium, 2012). Recently updated release of the tomato genome assembly (SL3.0, ITAG3.2 release) contains 35,768 gene models1. Since the release of the tomato complete genome sequences, large amounts of RNA-seq data in tomato have been generated in a number of research projects from tissues at variable different developmental stages, growing under different conditions, or challenged with different pathogens, as well as comparative transcriptome analysis with wild tomato plants (Kumar and Khurana, 2014). Mapping RNA-seq data, generated in domesticated and wild tomato plants under various conditions or treatments, to the released complete genome sequences as a reference genome has significantly contributed to our understanding the transcriptome complexity and regulations including differential gene expressions and alternative splicing (AS) in tomato (Wang K. et al., 2016; Dai et al., 2017).
A gene can be transcribed to form two or more RNA transcripts using the process of AS in intron containing eukaryotic organisms (Reddy et al., 2013), thus, significantly increasing the diversities of mRNAs and proteins in the organism. AS commonly occurs in eukaryotes including protists, fungi, plants, and animals (McGuire et al., 2008). Four basic types of AS including exon skipping (ExonS), alternative donor site (AltD), alternative acceptor (AltA) site, and intron retention (IntronR) were commonly found (Wang and Brendel, 2006; Sablok et al., 2011). Various complex types can be formed by combination of basic events (Sablok et al., 2011). While these basic types can be found in all kingdoms of eukaryotes, ExonS is most prevalent event in animals including humans and IntronR is the dominant event in plants (McGuire et al., 2008), suggesting that the splicing mechanisms may be different in animals and plants. Numerous experimental results showed AS plays important roles in many biological processes in plants such as photosynthesis, defense responses, flowering timing, responses to stresses, etc., (Reddy et al., 2013; Staiger and Brown, 2013). AS isoforms may or may not be functional. The functional isoforms may encode distinct functional proteins and the non-functional isoforms are degraded by a process known as nonsense-mediated decay (NMD) (Lewis et al., 2003; Xu et al., 2014).
Alternative splicing has been examined in a number of plant species including the model species, Arabidopsis thaliana, crop plants including rice, maize, sorghum, etc., (Zhang C. et al., 2016). Due to the differences in the amounts of available gene expression data in different plant species, the estimated AS rates vary tremendously from below 10% to ∼70% in intron-containing genes (Zhang C. et al., 2016; Min, 2017). For example, due to relatively large amounts of transcription data available in Arabidopsis, it was estimated that ∼60–70% of multi-exon genes undergoing AS (Filichkin et al., 2010; Marquez et al., 2012; Yu et al., 2016; Zhang R. et al., 2017). Other well analyzed plant species were rice (Oryza sativa) (Wang and Brendel, 2006; Min et al., 2015; Wei et al., 2017; Chae et al., 2017; Zhang et al., 2018), maize (Zea mays) (Thatcher et al., 2014; Min et al., 2015; Thatcher et al., 2016; Mei et al., 2017; Min, 2017), and sorghum (Sorghum bicolor) (Panahi et al., 2014; Min et al., 2015; Abdel-Ghany et al., 2016); fruit plants such as grape (Vitis vinifera) (Vitulo et al., 2014; Sablok et al., 2017), and fiber plants such as cotton (Gossypium raimondii; G. barbadense; G. davidsonii, and G. hirsutum) (Li et al., 2014; Min, 2018; Wang et al., 2018a; Zhu G. et al., 2018). However, the above mentioned species are just few examples of AS analysis in plants, not an exhaustive list of all plants AS work. Further, genome-wide conserved AS events in flowering plant species as well as in monocot species have also been analyzed (Chamala et al., 2015; Mei et al., 2017).
Transcriptome analysis in plants have been carried out intensively using recently developed RNA-sequencing (RNA-seq) technology. A number of well-designed experiments in genome-wide transcriptome analysis for identifying differentially expressed genes and/or AS in tomato, a model plant specifically for fruit development, have been reported in recent few years. These studies include comparative transcriptome analysis of domesticated tomato for identifying differentially expressed genes in different tissues (Lopez-Casado et al., 2012; Koenig et al., 2013; Zouine et al., 2014; Sun and Xiao, 2015; Sundaresan et al., 2016; Wang K. et al., 2016; Zhang Y. et al., 2017), diurnal transcriptome changes (Higashi et al., 2016), global transcriptome profiles of tomato leaf responses to exogenous ABA or cytokinin (Wang et al., 2013; Shi et al., 2013), root transcriptome regulations in response to different plant hormone cytokinin and auxin (Gupta et al., 2013), transcriptome profiles with a focus on fruit development or in different fruit tissues (Ye et al., 2015; Zhang S. et al., 2016; Dai et al., 2017; Ezura et al., 2017) and fruit chilling tolerance (Cruz-Mendívil et al., 2015). RNA-seq data were also collected for analysis of differential gene expressions in response to tomato yellow leaf curl virus (TYLCV) infection in the TYLCV-resistant (R) breeding line and TYLCV-susceptible breeding line (Chen et al., 2013), in response to tobacco rattle virus (TRV) (Zheng et al., 2017), Pseudomonas syringae (Yang et al., 2015; Worley et al., 2016), Xanthomonas perforans (Du et al., 2015), Cladosporium fulvum (Xue et al., 2016), Colletotrichum gloeosporioides (Alkan et al., 2015), Verticillium dahlia (Tan et al., 2015), Meloidogyne incognita (root-knot nematode) (Shukla et al., 2018) and in arbuscular mycorrhiza inoculated and control plants (Zouari et al., 2014). Transcriptome analysis was also carried out with mutants including high pigment mutant (Tang et al., 2013) and SIEIN2-silenced tomato (ethylene insensitive 2) mutant which had a non-ripening phenotype (Wang R.H. et al., 2016).
The aforementioned RNA-seq projects generated large amounts of RNA-seq data in tomato, which provide an unprecedented opportunity for integrating these transcriptome data with publicly available expressed sequence tag (EST) and mRNA sequences for identifying alternatively spiced genes in tomato. However, the integrated study for AS events in tomato has not been reported. Thus, the aim of the current work is to maximize AS identification and generate a comprehensive catalog of alternatively spliced genes and AS events in tomato by integrating ESTs, mRNAs, and RNA-seq data available in public databases. The identified alternatively spliced genes and AS events with detailed annotation information in the work are expected to provide a solid resource for tomato researchers for further detailed functional analysis of these genes in tomato growth and development including fruit production.
Materials and Methods
Genome, EST, and mRNA Sequence Datasets
Tomato genome sequences (version SL3.0) and associated annotation files (ITAG3.20) were downloaded from the International Tomato Genome Sequencing Project (see text footnote 1). Using “Solanum lycopersicum” as “organism” we downloaded 300,665 EST sequences and 53,613 mRNA sequences of tomato from EST and nucleotide database at the National Center for Biotechnology Information (NCBI).
We used a procedure well implemented in our previous analysis for cleaning the data (Min et al., 2015; Min, 2018). The procedure used EMBOSS trimmest tool for trimming the polyA or polyT end (Rice et al., 2000), BLASTN search against UniVec and E. coli database for removal of vector and E. coli contaminants, and BLASTN search against the plant repeat database for removal of the repetitive sequences including transposable elements. A total of 350,141 cleaned EST and mRNA sequences were obtained and combined with 39,095 transcript sequences generated by Scarano et al. (2017) and 250,676 transcript sequences generated by Wang K. et al. (2016). Thus a total of 639,912 sequences were used for assembling using CAP3 (Huang and Madan, 1999). A total of 452,672 putative unique transcripts including 27,791 contigs and 424,881 singlets were obtained for mapping to tomato genome sequences.
Mapping Assembled Transcripts to the Genome
We used ASFinder to map the assembled transcripts tomato genome sequences (Min, 2013). We applied the threshold values as reported previously (Walters et al., 2013). Mapped transcripts having an intron size>100 kb were removed for AS identification in order to avoid chimeric transcripts.
RNA-Seq Data Mapping to the Genome
We downloaded tomato RNA-seq sequence data from the NCBI SRA database2 using SRA Toolkit. The RNA-seq data were retrieved from 27 published papers, which were listed in Supplementary Table S1. In total, 2,543 Gbs RNA-seq data were downloaded. The data from each publication were processed individually. The RNA-seq reads were mapped to tomato genome sequences using TopHat (v2.2.6) with default parameters (Kim et al., 2013). Then the transcript alignment file together with the ITAG3.20 annotation was used as input for Cufflinks (v2.2.1)3 (Trapnell et al., 2010). The GTF (Gene Transfer Format) files generated from each RNA-seq dataset after Cufflinks were merged using Cuffcompare script within the Cufflinks package (Trapnell et al., 2010). The GTF file generated from merged RNA-seq GTF files then was further merged using Cuffcompare script with the GTF file that was generated by the ASFinder for mapping the assembled ESTs and mRNA (transcripts) sequences to the genome to generate a final GTF file for AS analysis. AStalavista was used for AS event classification (Foissac and Sammeth, 2007).
Transcript Functional Annotation
The sequences of the transcripts were retrieved using gtf_to_fasta tool in the tophat package (Kim et al., 2013), based on the GTF file generated by Cuffcompare program after merging the EST and mRNA mapping GTF file and RNA-seq mapping GTF file. They were functionally annotated using a procedure we reported previously (Min, 2018). The annotation information contains protein coding regions (ORF) predciton, assessment of full–length transcript coverage, protein family, and comparison with sequences of predicted gene models. Gene Ontology (GO) information was extracted also using a procedure reported previously (Min, 2018). Transcripts not having BLASTX hit against UniProtKB-SwissProt database were further used for non-coding RNA (ncRNA) identification by using BLASTN search against the non-coding RNA central database (version 10)4 with a cutoff E-value of 1e-5.
Transposable Element Analysis in Introns
Intron sequences were retrieved using an in-house script. Transposable elements in the introns were identified using BLASTN searching against the RepBase (Bao et al., 2015)5 using a cutoff E-value of 1e-5.
Internal Exon and Intron Length Distribution and Exon/Intron Junctions
The lengths of internal exons and introns in all transcripts and sizes of DNA fragments involved in AS were analyzed. The exon and intron junction sequences were extracted from genes not undergoing AS (non-AS genes) and genes undergoing AS (AS-genes). The exon/intron boundary sequence logo was created using the weblogo server6 (Crooks et al., 2004).
Availability of Data
The assembled transcripts and AS events identified in this study along with the predicted gene models, along with the data reported previously in our group including Brachypodium distachyon (Sablok et al., 2011; Walters et al., 2013), pineapple (Wai et al., 2016), and sacred lotus (Nelumbo nucifera) (VanBuren et al., 2013), are available from plant alternative splicing database7 (Walters et al., 2013; VanBuren et al., 2013; Min et al., 2015; Wai et al., 2016, 2018; Sablok et al., 2017; Min, 2017, 2018). BLAST search is also available for searching the transcripts. The datasets for database construction and the supplementary data are publicly available at: http://proteomics.ysu.edu/publication/data/Tomato/.
Results
Features of Assembled Transcripts
In this work we integrated genome mapping data from available ESTs and mRNAs in the public nucleotide database with RNA-seq data downloaded from the NCBI SRA database that were obtained from 27 previous publications (see Supplementary Table S1). A total of 533,707 putative unique transcripts with an average length of 1,350 bp were obtained based on the final GTF file which was generated by merging the mapping GTF file of assembled EST and mRNA sequences and the mapping GTF file of RNA-seq data to tomato genome sequences (Table 1). The basic features of the transcripts in the dataset were summarized (Table 1).
Table 1
| Total unique transcripts | 533707 |
| Average transcript length (bp) | 1350 |
| Total genomic loci with at least one transcript | 260681 |
| Transcripts matching with gene model cDNAs | 260365 |
| Unique gene model cDNAs matching with transcripts | 34522 |
| Transcripts having a BLASTX match against Swiss-Prot database | 226881 |
| Total predicted ORFs from assembled transcripts | 518307 |
| Average length of predicted ORFs (amino acids) | 215 |
| Predicted full-length ORFs | 182325 |
| Predicted ORFs having a PFAM match | 176234 |
Basic features of the assembled unique transcripts in tomato plants.
The mapped transcripts were clustered to a total of 260,681 genomic loci (Table 1), which were significantly higher than the number of protein coding genes (35,768 gene models, 35768 protein sequences and 35,768 cDNAs) annotated in ITAG3.20. Using ungapped BLASTN search with 100% identity and a minimum length of 80 bp in a high score aligned segment, a total of 260,365 transcripts accounting for 48.8% of total transcripts matched with cDNA sequences of gene models. A total of 34,522 (96.5%) unique loci, represented by cDNA sequences, have matched at least one transcript. Assembled transcripts were annotated functionally using BLASTX search against the UniProt-SwissProt database. Among them, a total of 226,881 (42.5%) has a BLASTX hit and 182,325 transcripts were predicted to contain a complete ORF region, i.e., a full-length ORF. Within the assembled transcripts, a total of 518,307 ORFs were predicted with an average length of 215 amino acids and 176,324 ORFs were mapped to a protein family (Pfam) using rpsBLAST (Table 1). Transcripts which were not able to match with a predicted cDNA transcript sequence were likely novel transcripts. Whether the novel transcripts identified in the projects represent the transcription noise, i.e., without any biological significance, or play certain biological roles remain to be examined in future study.
AS Events Identification
Tomato genome has 12 chromosomes (Tomato Genome Consortium, 2012). Chromosome zero (Chr0) is a segment of chromosome that has not been assigned to a specific chromosome (Table 2). There were 369,911 AS events identified from 34,419 genomic loci involving 161,913 transcripts. Although there are variations in the total number of AS events among different chromosomes, the general AS event distribution patterns are consistent across chromosomes (Table 2). Among the four basic AS types, IntronR is the prevalent type of AS event (18.9%), followed by AltA (12.9%) and AltD (7.3%), and ExonS as the least type (6.0%). Various complex types can be formed in transcript isoforms by combination of basic events and 54.9% of AS events were complex types (2).
Table 2
| AltA (%) | AltD (%) | ExonS (%) | IntronR (%) | Others (%) | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr0 | 188 | 11.8 | 108 | 6.8 | 55 | 3.4 | 451 | 28.3 | 794 | 49.7 | 1596 |
| Chr1 | 6410 | 13.5 | 3592 | 7.6 | 2923 | 6.2 | 9269 | 19.5 | 25276 | 53.2 | 47470 |
| Chr2 | 5396 | 10.1 | 3156 | 5.9 | 2379 | 4.5 | 8324 | 15.6 | 34184 | 64.0 | 53439 |
| Chr3 | 5168 | 13.2 | 2856 | 7.3 | 2274 | 5.8 | 7432 | 19.0 | 21476 | 54.8 | 39206 |
| Chr4 | 3988 | 14.0 | 2228 | 7.8 | 1851 | 6.5 | 5567 | 19.5 | 14926 | 52.3 | 28560 |
| Chr5 | 3026 | 13.4 | 1818 | 8.0 | 1434 | 6.3 | 4488 | 19.8 | 11883 | 52.5 | 22649 |
| Chr6 | 4077 | 14.1 | 2173 | 7.5 | 1758 | 6.1 | 5938 | 20.5 | 15071 | 51.9 | 29017 |
| Chr7 | 3379 | 12.3 | 1855 | 6.7 | 1661 | 6.0 | 5106 | 18.6 | 15506 | 56.4 | 27507 |
| Chr8 | 3355 | 13.3 | 1839 | 7.3 | 1537 | 6.1 | 4979 | 19.8 | 13426 | 53.4 | 25136 |
| Chr9 | 3281 | 12.8 | 1995 | 7.8 | 1600 | 6.3 | 4896 | 19.2 | 13786 | 53.9 | 25558 |
| Chr10 | 2883 | 13.6 | 1572 | 7.4 | 1378 | 6.5 | 4509 | 21.3 | 10811 | 51.1 | 21153 |
| Chr11 | 3254 | 13.0 | 1876 | 7.5 | 1630 | 6.5 | 4383 | 17.5 | 13973 | 55.6 | 25116 |
| Chr12 | 3316 | 14.1 | 1905 | 8.1 | 1681 | 7.2 | 4693 | 20.0 | 11909 | 50.7 | 23504 |
| Total | 47721 | 12.9 | 26973 | 7.3 | 22161 | 6.0 | 70035 | 18.9 | 203021 | 54.9 | 369911 |
Summary of alternative splicing events in each chromosome of tomato plants.
Among 369,911 transcripts generated from pre-mRNAs having AS, 150,131 matched to cDNAs of 23,233 unique annotated gene models (See Supplementary Table S2). We noticed that there were some gene models undergoing AS having only one corresponding transcript shown in the table because other transcripts involved in AS did not have a sufficient overlap region with the gene model transcript (Supplementary Table S2). However, the complete information of isoform transcripts can be obtained from the database and the genome browser. Based on the mapping analysis, there were 34,522 unique loci (gene models) having EST/mRNA or RNA-seq mapped, i.e., these genes were supported with transcription data. Thus, using only expressed genes the AS rate in tomato was estimated to be 67.3%. However, when all gene models were used, the estimated AS rate was 65.0%.
Li et al. (2014) reported that transposons were enriched in the retained introns in cotton plants. While only 2.9% of all introns contained transposable elements (TEs), 43% of the retained introns were found to have TEs in the AS transcripts, suggesting TE-insertion may result in IntronR during pre-mRNA splicing in cotton (Li et al., 2014). We retrieved 68,241 retained introns from our datasets with a length >50 bp and found only 812 TEs (1.2%). While in the whole set (138,127) of introns of the predicted gene models, using the same cutoff value, 3,105 TEs (2.3%) were found. Thus, the TE enrichment phenomenon reported by Li et al. (2014) was not found in tomato.
To facilitate identifying project specific AS events which may aid in elucidating the biological significances of AS isoforms, we analyzed AS events in each individual projects (Table 3). Because we used pooled data from different projects with variable data sizes and treatment conditions, it is difficult to directly compare the results from these projects (Supplementary Table S1). However, the overall trend in AS type frequency distribution is consistent with the final combined data (Tables 2, 3), that is, IntronR is the most prevalent type of AS, followed by AltA and AltD, with ExonR as the least frequent type. The RNA-seq mapping information including expression levels, AS event analysis, and information for sequence identifier mapping to the final assembled transcripts are available for downloading at http://proteomics.ysu.edu/publication/data/Tomato/projects/.
Table 3
| Dataa | AltA | AltD | ExonS | IntronR | Others |
|---|---|---|---|---|---|
| ESTs and mRNAs | 13738 | 7779 | 9355 | 17255 | 32448 |
| RNA-seq projects | |||||
| Alkan (2014) | 2664 | 1841 | 1367 | 3966 | 3057 |
| Chen (2013) | 1425 | 1068 | 711 | 2238 | 1528 |
| Cruz-Mendivil (2015) | 122 | 130 | 88 | 329 | 448 |
| Dai (2017) | 2075 | 1440 | 992 | 2574 | 1920 |
| Du (2015) | 2557 | 1873 | 1273 | 3851 | 2878 |
| Ezura (2017) | 254 | 251 | 88 | 3698 | 1658 |
| Gupta (2013) | 2619 | 1909 | 1375 | 4723 | 3437 |
| Higashi (2016) | 1773 | 1256 | 967 | 7274 | 2916 |
| Koenig (2013) | 1791 | 1376 | 1005 | 2774 | 1998 |
| Lopez-Casado (2011) | 565 | 521 | 353 | 995 | 889 |
| Shi (2013) | 1773 | 1431 | 972 | 4806 | 2810 |
| Shukla (2017) | 18581 | 9519 | 7032 | 20625 | 29217 |
| Sun (2015) | 5309 | 3255 | 1876 | 5925 | 4530 |
| Sundaresan (2016) | 1857 | 1286 | 920 | 2461 | 1802 |
| Tan (2015) | 1268 | 954 | 656 | 1863 | 1287 |
| Tang (2013) | 1353 | 1057 | 816 | 1813 | 1401 |
| Wang (2013) | 5324 | 3022 | 1945 | 5945 | 4683 |
| Wang (2016) | 3603 | 2264 | 1857 | 5079 | 4818 |
| Worley (2016) | 1921 | 1308 | 1078 | 3769 | 2402 |
| Xue (2017) | 6455 | 3602 | 3246 | 8886 | 9113 |
| Yang (2015) | 2093 | 1356 | 1257 | 3259 | 2041 |
| Ye (2015) | 1389 | 1127 | 762 | 3678 | 2112 |
| Zhang (2016) | 6109 | 3399 | 2727 | 7825 | 6880 |
| Zhang (2017) | 2493 | 1623 | 1385 | 3121 | 2396 |
| Zheng (2017) | 412 | 376 | 267 | 972 | 683 |
| Zouari (2014) | 2679 | 1820 | 1436 | 3878 | 3091 |
| Zouine (2014) | 2853 | 1935 | 1357 | 3278 | 2453 |
Summary of alternative splicing events identified in EST and mRNA assembly dataset and each RNA-seq dataset in tomato plants.
aThe last name of the first author with year of the publication is used the project ID. The details of the reference for each project can be found in the Supplementary Table S1.
Functional Annotation of Transcripts
All transcripts were functionally annotated including BALSTX search against the UniProtKB-SwissProt database and predicting the ORF regions and completeness of ORFs (see section “Transcript Functional Annotation”). The predicted proteins were further annotated for the protein family (Pfam) analysis. To provide an overview of the protein family distribution in tomato proteome and proteins encoded by genes generating AS isoforms, we used the protein sequences of the gene models for Pfam analysis. Among 35,768 protein sequences of gene models a total of 22,322 entries had PFam matches with a total of 3,319 unique Pfam. Among a total of 23,233 protein sequences generated from AS genes, a total 16,531 had Pfam matches with a total of 3,114 unique Pfam. The top Pfam in the whole tomato proteome and proteins encoded by genes undergoing AS were listed in Table 4. The numbers of proteins in each Pfam varied significantly from 1 member in some families to 631 members in Pkinase (pfam00069). In average 74.1% of Pfam members were alternatively spliced with varying proportions in different protein families (Table 4). In considering the varying Pfam size, number of exons per gene and gene expression levels as well as functional differences of these protein coding genes, such a difference in AS rates in the genes belonging to different protein families is expected. Comparing with our previous Pfam analysis of proteins generated from AS genes in cereal plants and fruit plants, these Pfams found in tomato AS genes were also well conserved in other plant species (Min et al., 2015; Sablok et al., 2017).
Table 4
| Pfam ID | Total | AS genes | % | Pfam abbreviation | Pfam description |
|---|---|---|---|---|---|
| pfam00069 | 631 | 503 | 79.7 | Pkinase | Protein kinase domain |
| pfam07714 | 467 | 388 | 83.1 | Pkinase_Tyr | Protein tyrosine kinase |
| pfam00067 | 337 | 216 | 64.1 | p450 | Cytochrome P450 |
| pfam13041 | 316 | 247 | 78.2 | PPR_2 | PPR repeat family |
| pfam13639 | 223 | 140 | 62.8 | zf-RING_2 | Ring finger domain |
| pfam00931 | 217 | 151 | 69.6 | NB-ARC | NB-ARC domain |
| pfam00076 | 179 | 162 | 90.5 | RRM_1 | RNA recognition motif |
| pfam00249 | 178 | 111 | 62.4 | Myb_DNA-binding | Myb-like DNA-binding domain |
| pfam00201 | 169 | 86 | 50.9 | UDPGT | UDP-glucoronosyl and UDP-glucosyl transferase |
| pfam10536 | 167 | 101 | 60.5 | PMD | Plant mobile domain |
| pfam03171 | 147 | 87 | 59.2 | 2OG-FeII_Oxy | 2OG-Fe(II) oxygenase superfamily |
| pfam00847 | 137 | 62 | 45.3 | AP2 | AP2 domain |
| pfam02519 | 130 | 35 | 26.9 | Auxin_inducible | Auxin responsive protein |
| pfam00141 | 123 | 81 | 65.9 | peroxidase | Peroxidase |
| pfam05699 | 118 | 28 | 23.7 | Dimer_Tnp_hAT | hAT family C-terminal dimerisation |
| pfam00319 | 109 | 34 | 31.2 | SRF-TF | SRF-type transcription factor |
| pfam02458 | 99 | 51 | 51.5 | Transferase | Transferase family |
| pfam14432 | 95 | 68 | 71.6 | DYW_deaminase | DYW family of nucleic acid deaminases |
| pfam00481 | 91 | 79 | 86.8 | PP2C | Protein phosphatase 2C |
| pfam00854 | 86 | 75 | 87.2 | PTR2 | POT family |
| pfam01095 | 85 | 42 | 49.4 | Pectinesterase | Pectinesterase |
| pfam00561 | 84 | 70 | 83.3 | Abhydrolase_1 | alpha/beta hydrolase fold |
| pfam00657 | 83 | 65 | 78.3 | Lipase_GDSL | GDSL-like Lipase/Acylhydrolase |
| pfam00010 | 82 | 70 | 85.4 | HLH | Helix-loop-helix DNA-binding domain |
| pfam03106 | 82 | 54 | 65.9 | WRKY | WRKY DNA -binding domain |
| pfam01554 | 77 | 66 | 85.7 | MatE | MatE |
| pfam13839 | 75 | 63 | 84.0 | PC-Esterase | GDSL/SGNH-like Acyl-Esterase family found |
| pfam00004 | 74 | 64 | 86.5 | AAA | ATPase family associated with various cellular |
| pfam02362 | 74 | 49 | 66.2 | B3 | B3 DNA binding domain |
| pfam00071 | 73 | 58 | 79.5 | Ras | Ras family |
| pfam00083 | 73 | 53 | 72.6 | Sugar_tr | Sugar (and other) transporter |
| pfam05695 | 72 | 13 | 18.1 | DUF825 | Plant protein of unknown function (DUF825) |
| pfam00082 | 71 | 41 | 57.7 | Peptidase_S8 | Subtilase family |
| pfam13499 | 71 | 40 | 56.3 | EF-hand_7 | EF-hand domain pair |
| Others | 17227 | 13078 | 75.9 | ||
| Total | 22322 | 16531 | 74.1 |
Protein families in gene models and alternatively spliced genes in tomato plants∗.
The isoform transcripts generated by alternative pre-mRNA splicing may or may not be functional. Thus the impact of AS on the functionalities of isoforms was assessed using Pfam annotation information of the predicted proteins. Within a total of 369,911 pairs generating AS events, 114,729 (31.0%) pairs did not have Pfam annotation, 164,594 (44.5%) pairs had same Pfam annotation; 64,107 (17.3%) pairs had one isoform with Pfam annotation and one isoform did not have Pfam annotation, suggesting either no protein sequences predicted or a loss of protein functionality; and 26,481 (7.2%) pairs had different Pfam annotation, suggesting a functional domain change in the protein sequences resulting from AS. Thus, the comparative Pfam analysis of the proteins encoded by the isoform transcripts generated by AS revealed that about 24.5% of them may cause functional loss or change in the protein isoforms. Our previous analysis showed that the resulting functional domain loss or change by AS were 19.6% in maize, 20.9% in cotton, and 24.9% in pineapple, respectively (Wai et al., 2016; Min, 2017, 2018). The translation frame changes in AS isoforms is the main reason for protein domain loss or change.
A total of 342,073 transcripts not having a BLASTX hit against the UniProt Swiss-Prot database were further used to search the ncRNA database using BLASTN. The ncRNA database was obtained from the RNAcentral (release 10) with a total of 11,963,117 ncRNA sequences. With a cutoff E-value of 1e-5, we identified a total of 136,643 transcripts sharing similarities with known ncRNAs. The list of ncRNAs can be downloaded at http://proteomics.ysu.edu/publication/data/Tomato/.
Gene Ontology (GO) Analysis
The gene ontology8 is “a community-based bioinformatics resource that supplies information about gene product function using ontologies to represent biological knowledge” (Gene Ontology Consortium, 2014). GO classification has three major categories including the biological processes, molecular functions, and cellular components. We used the protein sequences of gene models to search the UniProtKB/Swiss-Prot database, then retrieved GO IDs based on the UniProt ID mapping table. Among 35,768 protein sequences predicted from gene models, 25,048 of them had a BLASTP hit with UniProtKB/Swiss-Protein dataset. Among them 18,031 were from protein sequences of genes with pre-mRNAs undergoing AS. We then retrieved 155,246 and 114,667 GO IDs for the whole set and AS gene set, respectively. The GO IDs were further mapped to each category using Slim Viewer with plant specific GO terms (McCarthy et al., 2006). The top GO terms in each category were presented in Figure 1. The percentage of each category was calculated based on the total counts in each category.
FIGURE 1
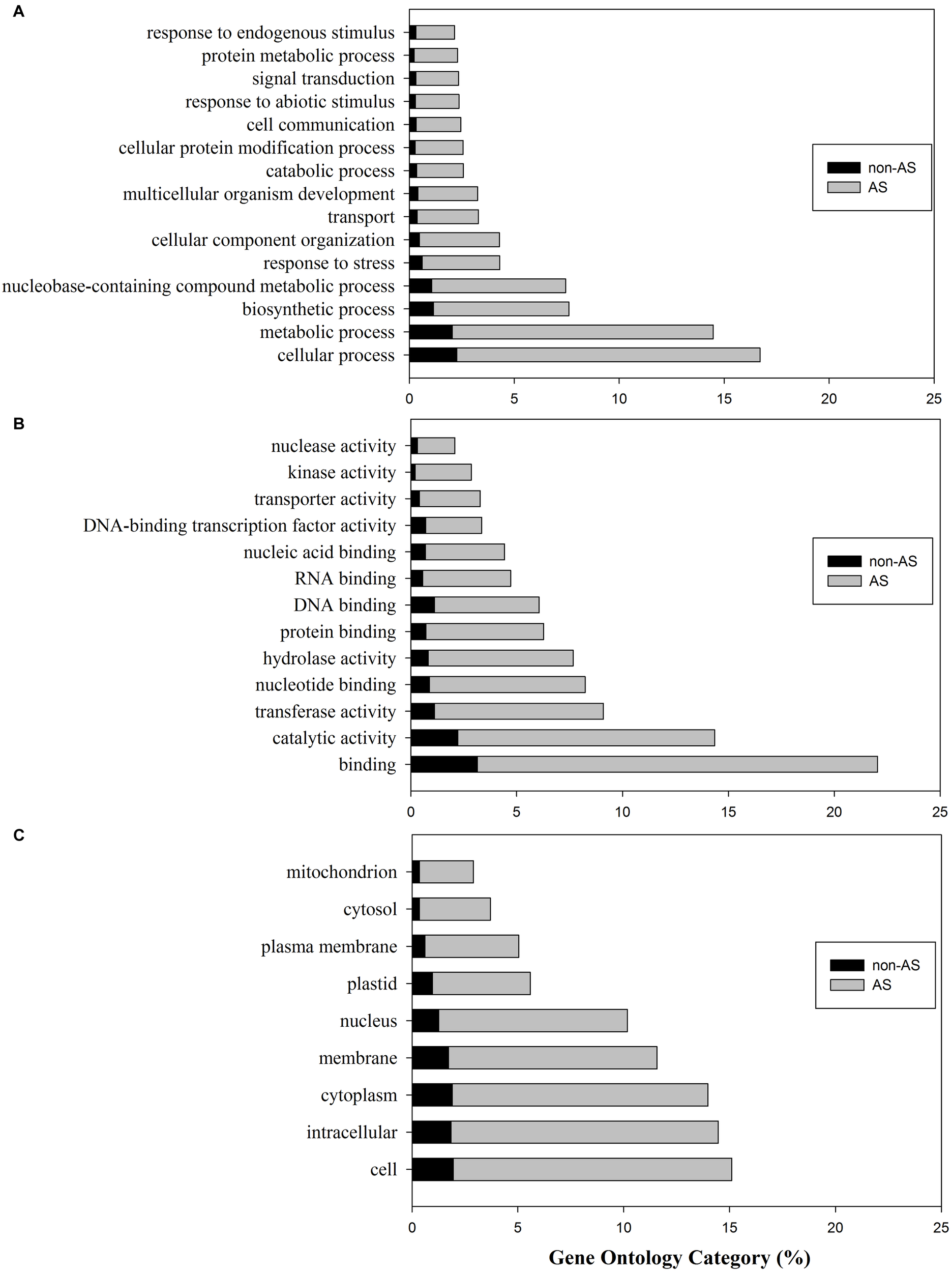
Gene ontology (GO) classification of tomato genes with pre-mRNAs not undergoing alternative splicing (non-AS genes) and genes with pre-mRNA undergoing alternative splicing (AS-genes). (A) Biological process; (B) Molecular function; (C) Cellular components.
In the whole tomato proteome set, the mapped GO term counts were 47,795 in biological processes; 25,989 in molecular functions, and 33517 in cellular components. The comparative analysis showed that 86.5% of genes involved biological processes may undergo alternative splicing (Figure 1A). The top biological processes include cellular process, metabolic process, biosynthetic process, nucleobase-containing compound metabolic process, response to stress, cellular component organization, etc., (Figure 1A). In particular, 153 genes (76.5%) from a total of 200 genes involved in the secondary metablic process (GO:0019748) were found undergoing AS. These genes are involved in the biosynthesis of lycopene, carotenoid, abscisic acid, flavonoid, anthocyanins, etc., (Bovy et al., 2007; Liu et al., 2015). AS analyses involved in secondary metabolism pathways including the flavonoid pathway in tea plant have been carried out, suggesting AS may play important roles in regulation of flavonoids biosynthesis (Zhu J. et al., 2018; Qiao et al., 2019).
Similarly, the majority (86.2%) of tomato gene products in the category of molecular functions were also alternatively spliced (Figure 1B). The top categories of molecular functions include binding, catalytic activity, transferase, nucleotide binding, hydrolase activity, protein binding, etc., (Figure 1B). Cellular components analysis also revealed that ∼86.5% of tomato genes with GO cellular component annotation having pre-mRNAs were alternatively spliced (Figure 1C).
Features of Exons, Introns and Exon-Intron Junctions
The lengths of internal exons and introns, as well as the DNA fragment sizes involved in AS events were calculated based on the RNA to genome mapping information (Table 5 and Figure 2). A total of 215,952 internal exons and 282,296 introns were extracted. The sizes of internal exons varied from 1 to 88,397 bp with a mean value of 282 bp; intron sizes varied from 5 to 313,176 bp with a mean value of 1,352 bp (Table 5). Among internal exons, 84.2% of them had a size of ≤400 bp and 94.6% were ≤1000 bp (Figure 2). In contrast, 55.0% of introns were ≤400 bp and 76.5% were ≤1000 bp. There were 0.01% of internal exons and 1.44% of introns ≥10 kb. In addition, there were 350 introns with a size >100 kb. As we removed alignments in the mapping of EST/mRNA assembled data, these introns were clearly from the RNA-seq mapping. We manually checked some of the transcripts having long introns and found that these extremely long introns were likely due to the mapping of the fused transcripts generated from different genes. The fused transcripts in plants often are ignored as transcription noise. However, it is known that fusion of transcripts in human is related to cancer development (Mertens et al., 2015; Kumar et al., 2016). Similar to what we found in B. distachyon and in fruit plants (Walters et al., 2013; Sablok et al., 2017), DNA fragments involved in AS events were relatively shorter than the average size of the internal exons or introns in tomato (Table 5). Thus, it is reasonable to conclude that small exons tend to be skipped and small introns tend to be retained.
Table 5
| Sample size | Length range (bp) | Mean (bp) | Standard deviation (bp) | |
|---|---|---|---|---|
| Internal exons | 215952 | 1–88397 | 282 | 547 |
| Introns | 282296 | 5–313176 | 1352 | 7609 |
| Retained introns | 70035 | 6–19337 | 366 | 710 |
| Alternative acceptor sites | 47721 | 1–12110 | 145 | 451 |
| Alternative donor sites | 26973 | 1–11238 | 212 | 493 |
| Skipped exons | 22161 | 2–25108 | 214 | 446 |
Summary of internal exon length and intron length of all transcripts, and DNA fragment sizes (bp) involved in alternative splicing events in tomato.
FIGURE 2
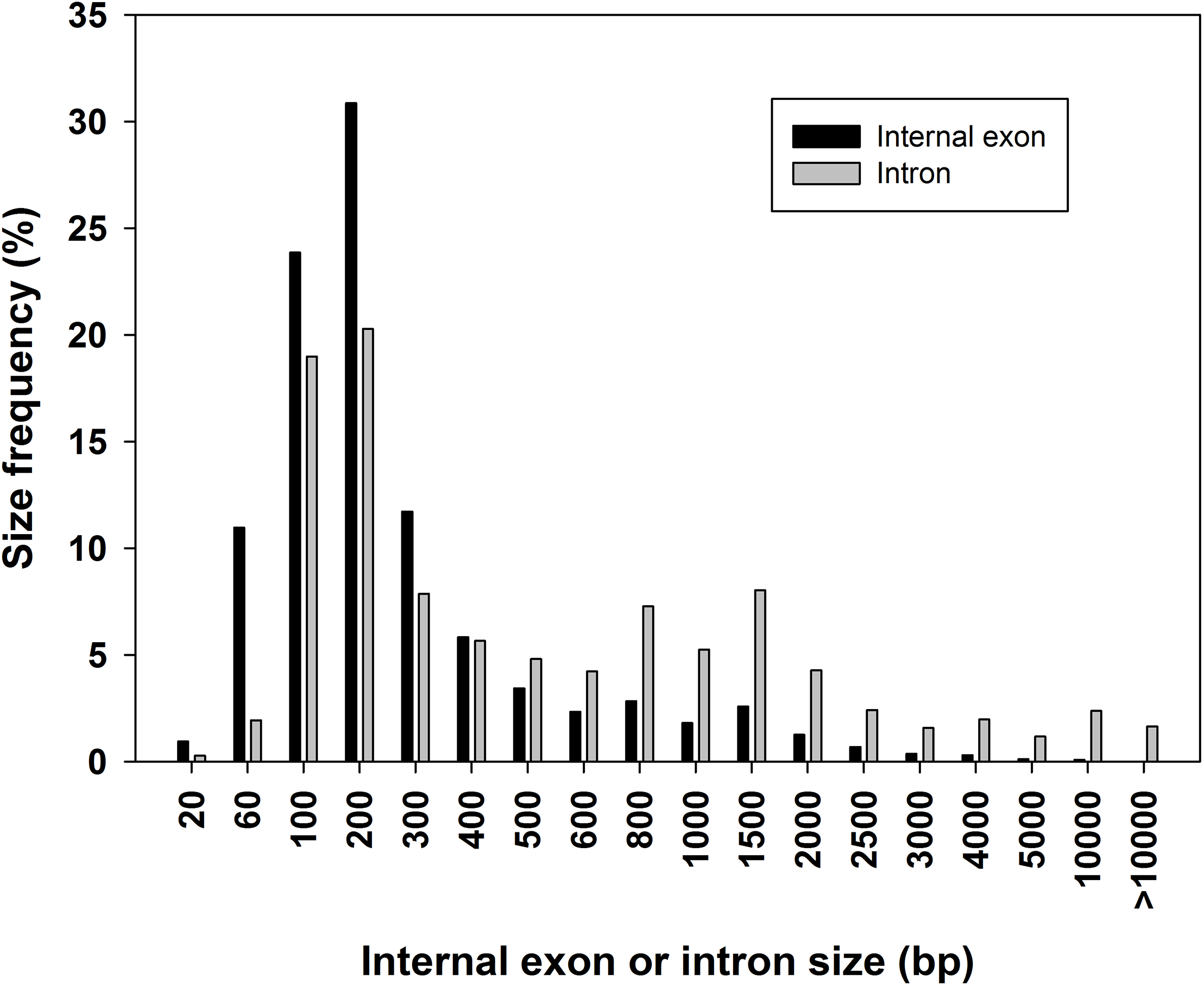
Distribution of internal exon size and intron size in tomato genes. Bin size are right inclusive (e.g., bin 100 comprises sequences of lengths 1–100 bp).
There were a total 46,114 introns extracted from genes not having AS (non-AS genes) and 236,182 introns from genes having AS (AS genes). Two nucleotides from each end of introns were extracted from both non-AS genes and AS genes. The majority of introns (90.6% in average) in both AS genes (90.9%) and non-AS genes (89.0%) had a canonical splicing junction site of 5′-GT..AG-3′ (Table 6). The minor types of splicing sites in introns included 5′-GC..AG-3′, 5′-GC..AT-3′, 5′-AT..AC-3′, and many others types (Table 6). A chi-square test showed there was no significant difference in the frequencies of the types of splicing sites between AS genes and non-AS genes (Table 6). The pictograms showed that the only noticeable difference in nucleotide usage probabilities in the junction sites at the 5′-end of introns between non-AS genes and AS-genes was position 8 (Figure 3, left panels). However, there were noticeable differences in the nucleotide probabilities at the 3′-end of introns but within the 5′-end of the next exonic region of, i.e., at position 14, 16, and 17 (Figure 3, right panels). Whether there is any biological significance remains to be examined.
Table 6
| Types | Total | % | Non-AS gene | % | AS gene | % |
|---|---|---|---|---|---|---|
| 5′-GT..AG-3′ | 255669 | 90.6 | 41053 | 89.0 | 214616 | 90.9 |
| 5′-GC..AG-3′ | 7571 | 2.7 | 1511 | 3.3 | 6060 | 2.6 |
| 5′-GC..AT-3′ | 3060 | 1.1 | 712 | 1.5 | 2348 | 1.0 |
| 5′-AT..AC-3′ | 2751 | 1.0 | 844 | 1.8 | 1907 | 0.8 |
| 5′-CT..AC-3′ | 2067 | 0.7 | 232 | 0.5 | 1835 | 0.8 |
| 5′-GT..AT-3′ | 2036 | 0.7 | 554 | 1.2 | 1482 | 0.6 |
| Others | 9142 | 3.2 | 1208 | 2.6 | 7934 | 3.4 |
| Total | 282296 | 46114 | 236182 |
The usage of different splicing sites at both ends of the introns in tomato.
FIGURE 3
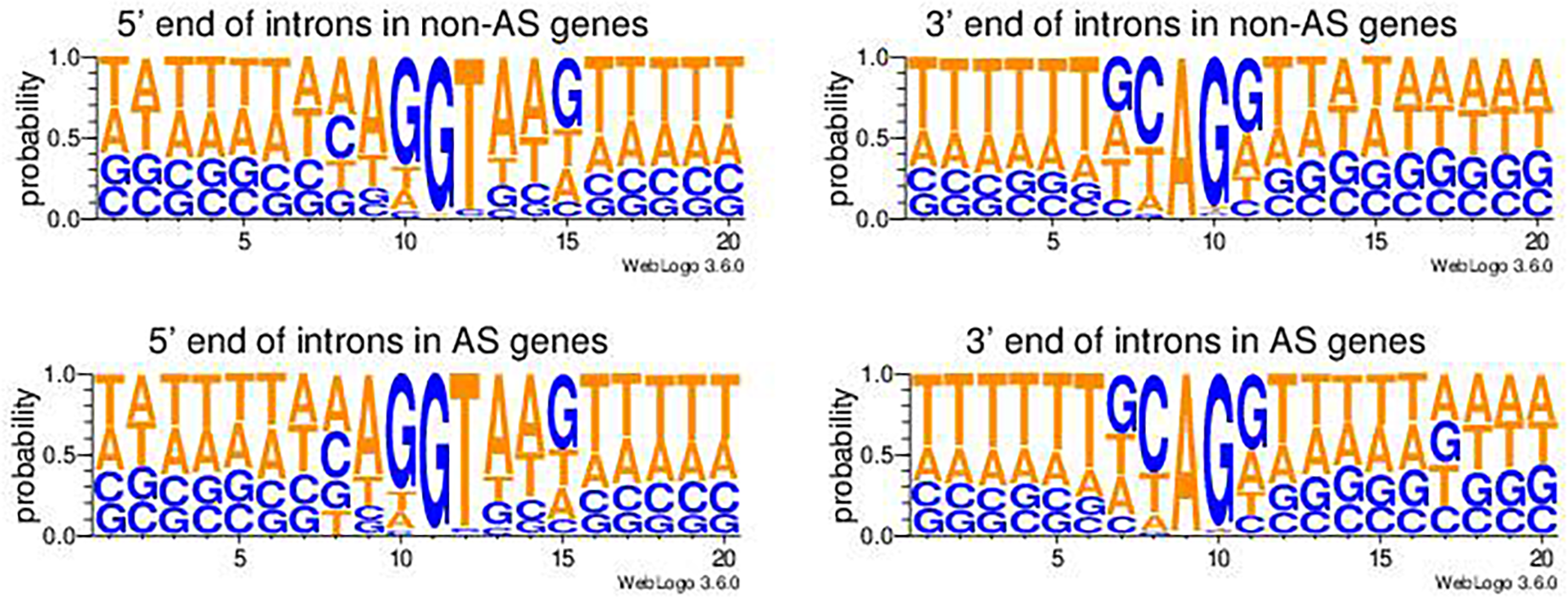
Pictograms of nucleotide probabilities at each position of the exon-intron junctions in genes not undergoing alternative splicing (non-AS genes) and genes undergoing alternative splicing (AS genes). The 5′-end intronic nucleotides are from position 11 to 20 in the left panel pictograms and the 3′-end intronic nucleotides are from position 1 to 10 in the right panel pictorgrams.
Discussion
In this work a much higher number of transcripts, than any previous report, were identified in tomato, as we integrated all currently available EST/mRNA sequences with RNA-seq data generated from 27 RNA-seq projects covering a broad range of biological samples with plants grown under various conditions (see Supplementary Table S1). The human ENCODE project reported the genome is pervasively transcribed (∼76% of the full genome transcribed), and “many novel non-protein-coding transcripts have been identified, with many of these overlapping protein-coding loci and others located in regions of the genome previously thought to be transcriptionally silent” (Encode Project Consortium, 2007; Pennisi, 2012). Thus the current set of transcripts represents the most comprehensive and complete set of transcripts identified in tomato by now.
The basic type distribution patterns of AS events in tomato are consistent with findings in other plant species (Wang and Brendel, 2006; Walters et al., 2013; VanBuren et al., 2013; Thatcher et al., 2014; Min et al., 2015; Sablok et al., 2017). However, the proportion of the complex type is related to the transcriptome sampling size and thus the completeness and the average length of the transcripts (Min et al., 2015; Min, 2017, 2018; Sablok et al., 2017). Long transcripts have more exons covered and thus are able to detect AS isoforms having more than one type of AS event in their sequences. The estimated AS rate in tomato was estimated ∼65.0% in the analysis. This AS rate in tomato is nearly reached to the maximal rate, though such a value may never be able to obtain due to the dynamic nature of transcriptomes. At least this rate is comparable with the rate reported in Arabidopsis (∼60%) and in maize (55%) (Marquez et al., 2012; Mei et al., 2017; Min, 2017). Obtaining such a high rate is clearly due to relatively large number of RNA-seq data were used in current analysis. Using RNA-seq data to detect AS isoforms has been widely accepted as a suitable approach by the research community (Reddy et al., 2012; Sablok et al., 2013), and some of the identified isoforms have been experimentally validated using RT-PCR (Thatcher et al., 2014; Wang K. et al., 2016; Xu et al., 2016). As a meta-analysis in this work, we did not perform any validation on the identified isoforms generated by AS genes. In considering the dynamic nature of AS in responding to the changing environmental conditions and developmental regulations, however, experimental validations are needed in each specific experiment. Pacific BioSciences (PacBio) single-molecule real-time (SMRT) long-read isoform sequencing (Iso-Seq) and Nanopore sequencing from Oxford Nanopore Technologies (ONT) were two tools revolutionizing the way AS are identified (Zhao et al., 2019). The long reads sequencing technologies could avoid the error-prone step during transcripts assembly for RNA-seq reads from Illumina sequencing platform. In future, it will be interesting to validate these AS events based on RNA-seq short reads by using long reads from PacBio or ONT technologies. The list of potential isoforms identified in the work provides a foundation for designing experiments for exploring the biological significances of these AS events.
We also identified a large number of ncRNAs including miRNA and long ncRNAs. The ncRNAs play important regulatory roles in plant biology (Borges and Martienssen, 2015; Chekanova, 2015). A number of studies have reported the biological significances of ncRNAs in tomato plants (Wang et al., 2015, 2018b; Zhu et al., 2015). The list of ncRNAs compiled in the work will aid in further elucidating the roles of ncRNA playing in tomato biology.
Statements
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: http://proteomics.ysu.edu/publication/data/Tomato/.
Author contributions
XM designed the experiments and performed the functional annotation. SC collected and processed the data for genome mapping. XM, FY, and LG analyzed the data. XM and LG wrote the manuscript.
Funding
The work was funded by Youngstown State University (YSU) YSU Research Council grant (Project #13-18) to XM. The College of Graduate Studies of YSU supported the article processing charges. The work was also supported by a Research Professorship award and reassigned time for scholarship from College of Science, Technology, Engineering, and Mathematics and Department of Biological Sciences at YSU to XM.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00689/full#supplementary-material
Footnotes
1.^https://solgenomics.net/organism/Solanum_lycopersicum/genome
2.^https://www.ncbi.nlm.nih.gov/sra/docs/sradownload/
3.^http://cole-trapnell-lab.github.io/cufflinks/
5.^https://www.girinst.org/repbase/
6.^http://weblogo.threeplusone.com/
References
1
Abdel-GhanyS. E.HamiltonM.JacobiJ. L.NgamP.DevittN.SchilkeyF.et al (2016). A survey of the sorghum transcriptome using single-molecule long reads.Nat. Commun.7:11706. 10.1038/ncomms11706
2
AlkanN.FriedlanderG.MentD.PruskyD.FluhrR. (2015). Simultaneous transcriptome analysis of Colletotrichum gloeosporioides and tomato fruit pathosystem reveals novel fungal pathogenicity and fruit defense strategies.New Phytol.205801–815. 10.1111/nph.13087
3
BaoW.KojimaK. K.KohanyO. (2015). Repbase Update, a database of repetitive elements in eukaryotic genomes.Mob. DNA6:11. 10.1186/s13100-015-0041-9
4
BorgesF.MartienssenR. A. (2015). The expanding world of small RNAs in plants.Nat. Rev. Mol. Cell Biol.16:727. 10.1038/nrm4085
5
BovyA.SchijlenE.HallR. D. (2007). Metabolic engineering of flavonoids in tomato (Solanum lycopersicum): the potential for metabolomics.Metabolomics3:399.
6
ChaeS.KimJ. S.JunK. M.LeeS. B.KimM. S.NahmB. H.et al (2017). Analysis of genes with alternatively spliced transcripts in the leaf, root, panicle and seed of rice using a long oligomer microarray and RNA-seq.Mol. Cells40714–730. 10.14348/molcells.2017.2297
7
ChamalaS.FengG.ChavarroC.BarbazukW. B. (2015). Genome-wide identification of evolutionarily conserved alternative splicing events in flowering plants.Front. Bioeng. Biotechnol.3:33. 10.3389/fbioe.2015.00033
8
ChekanovaJ. A. (2015). Long non-coding RNAs and their functions in plants.Curr. Opin. Plant Biol.27207–216. 10.1016/j.pbi.2015.08.003
9
ChenT.LvY.ZhaoT.LiN.YangY.YuW.et al (2013). Comparative transcriptome profiling of a resistant vs. susceptible tomato (Solanum lycopersicum) cultivar in response to infection by tomato yellow leaf curl virus.PLoS One8:e80816. 10.1371/journal.pone.0080816
10
CrooksG. E.HonG.ChandoniaJ. M.BrennerS. E. (2004). WebLogo: a sequence logo generator.Genome Res.141188–1190. 10.1101/gr.849004
11
Cruz-MendívilA.López-ValenzuelaJ. A.Calderón-VázquezC. L.Vega-GarcíaM. O.Reyes-MorenoC.Valdez-OrtizA. (2015). Transcriptional changes associated with chilling tolerance and susceptibility in ‘Micro-Tom’tomato fruit using RNA-Seq.Postharvest Biol. Tec.99141–151. 10.1016/j.postharvbio.2014.08.009
12
DaiQ.GengL.LuM.JinW.NanX.HeP. A.et al (2017). Comparative transcriptome analysis of the different tissues between the cultivated and wild tomato.PLoS One12:e0172411. 10.1371/journal.pone.0172411
13
DuH.WangY.YangJ.YangW. (2015). Comparative transcriptome analysis of resistant and susceptible tomato lines in response to infection by Xanthomonas perforans race T3.Front. Plant Sci.6:1173. 10.3389/fpls.2015.01173
14
Encode Project Consortium (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project.Nature447799–816. 10.1038/nature05874
15
EzuraK.Ji-SeongK.MoriK.SuzukiY.KuharaS.AriizumiT.et al (2017). Genome-wide identification of pistil-specific genes expressed during fruit set initiation in tomato (Solanum lycopersicum).PLoS One12:e0180003. 10.1371/journal.pone.0180003
16
FilichkinS. A.PriestH. D.GivanS. A.ShenR.BryantD. W.FoxS. E.et al (2010). Genome-wide mapping of alternative splicing in Arabidopsis thaliana.Genome Res.2045–58. 10.1101/gr.093302.109
17
FoissacS.SammethM. (2007). ASTALAVISTA: dynamic and flexible analysis of alternative splicing events in custom gene datasets.Nucleic Acids Res.2007W297–W299. 10.1093/nar/gkm311
18
Gene Ontology Consortium (2014). Gene ontology consortium: going forward.Nucleic Acids Res.43D1049–D1056. 10.1093/nar/gku1179
19
GuptaS.ShiX.LindquistI. E.DevittN.MudgeJ.RashotteA. M. (2013). Transcriptome profiling of cytokinin and auxin regulation in tomato root.J. Exp. Bot.64695–704. 10.1093/jxb/ers365
20
HigashiT.TanigakiY.TakayamaK.NaganoA. J.HonjoM. N.FukudaH. (2016). Detection of diurnal variation of tomato transcriptome through the molecular timetable method in a sunlight-type plant factory.Front. Plant Sci.7:87. 10.3389/fpls.2016.00087
21
HuangX.MadanA. (1999). CAP3: A DNA sequence assembly program.Genome Res.9868–877.
22
KimD.PerteaG.TrapnellC.PimentelH.KelleyR.SalzbergS. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions.Genome Biol.14:R36. 10.1186/gb-2013-14-4-r36
23
KoenigD.Jiménez-GómezJ. M.KimuraS.FulopD.ChitwoodD. H.HeadlandL. R.et al (2013). Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato.Proc. Natl. Acad. Sci. U.S.A.110E2655–E2662. 10.1073/pnas.1309606110
24
KumarR.KhuranaA. (2014). Functional genomics of tomato: opportunities and challenges in post-genome NGS era.J. Biosci.39917–929.
25
KumarS.RazzaqS. K.VoA. D.GautamM.LiH. (2016). Identifying fusion transcripts using next generation sequencing.Wiley Interdiscip. Rev. RNA7811–823. 10.1002/wrna.1382
26
LewisB. P.GreenR. E.BrennerS. E. (2003). Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans.Proc. Natl. Acad. Sci. U.S.A.100189–192.
27
LiQ.XiaoG.ZhuY. X. (2014). Single-nucleotide resolution mapping of the Gossypium raimondii transcriptome reveals a new mechanism for alternative splicing of introns.Mol. Plant7829–840. 10.1093/mp/sst175
28
LiuL.ShaoZ.WangQ. (2015). Regulation of carotenoid metabolism in tomato.Mol. Plant.828–39.
29
Lopez-CasadoG.CoveyP. A.BedingerP. A.MuellerL. A.ThannhauserT. W.ZhangS.et al (2012). Enabling proteomic studies with RNA-Seq: the proteome of tomato pollen as a test case.Proteomics12761–774. 10.1002/pmic.201100164
30
MarquezY.BrownJ. W.SimpsonC. G.BartaA.KalynaM. (2012). Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis.Genome Res.221184–1195. 10.1101/gr.134106.111
31
McCarthyF. M.WangN.MageeG. B.NanduriB.LawrenceM. L.CamonE. B.et al (2006). AgBase: a functional genomics resource for agriculture.BMC Genomics7:229. 10.1186/1471-2164-7-229
32
McGuireA. M.PearsonM. D.NeafseyD. E.GalaganJ. E. (2008). Cross-kingdom patterns of alternative splicing and splice recognition.Genome Biol.9:R50. 10.1186/gb-2008-9-3-r50
33
MeiW.BoatwrightL.FengG.SchnableJ. C.BarbazukW. B. (2017). Evolutionarily conserved alternative splicing across monocots.Genetics207465–480. 10.1534/genetics.117.300189
34
MertensF.JohanssonB.FioretosT.MitelmanF. (2015). The emerging complexity of gene fusions in cancer.Nat. Rev. Cancer15371–381. 10.1038/nrc3947
35
MinX. J. (2013). ASFinder: a tool for genome-wide identification of alternatively spliced transcripts from EST-derived sequences.Int. J. Bioinform. Res. Appl.9221–226. 10.1504/IJBRA.2013.053603
36
MinX. J. (2017). Comprehensive cataloging and analysis of alternative splicing in maize.Comp. Mol. Biol.71–11. 10.5376/cmb.2017.07.0001
37
MinX. J. (2018). A survey of alternative splicing in allotetraploid cotton (Gossypium hirsutum L.).Comp. Mol. Biol.81–13. 10.5376/cmb.2018.08.0001
38
MinX. J.PowellB.BraesslerJ.MeinkenJ.YuF.SablokG. (2015). Genome-wide cataloging and analysis of alternatively spliced genes in cereal crops.BMC Genomics16:721. 10.1186/s12864-015-1914-5
39
PanahiB.AbbaszadehB.TaghizadeghanM.EbrahimieE. (2014). Genome-wide survey of alternative splicing in Sorghum bicolor.Physiol. Mol. Biol. Plants20323–329. 10.1007/s12298-014-0245-3
40
PennisiE. (2012). ENCODE project writes eulogy for junk DNA.Science3371159–1161. 10.1126/science.337.6099.1159
41
QiaoD.YangC.ChenJ.GuoY.LiY.NiuS.et al (2019). Comprehensive identification of the full-length transcripts and alternative splicing related to the secondary metabolism pathways in the tea plant (Camellia sinensis).Sci. Rep.9:2709. 10.1038/s41598-019-39286-z
42
ReddyA. S.MarquezY.KalynaM.BartaA. (2013). Complexity of the alternative splicing landscape in plants.Plant Cell253657–3683. 10.1105/tpc.113.117523
43
ReddyA. S.RogersM. F.RichardsonD. N.HamiltonM.Ben-HurA. (2012). Deciphering the plant splicing code: experimental and computational approaches for predicting alternative splicing and splicing regulatory elements.Front. Plant Sci.7:18. 10.3389/fpls.2012.00018
44
RiceP.LongdenI.BleasbyA. (2000). EMBOSS: the european molecular biology open software suite.Trends Genet.16276–277.
45
SablokG.GuptaP. K.BaekJ. M.VazquezF.MinX. J. (2011). Genome-wide survey of alternative splicing in the grass Brachypodium distachyon: an emerging model biosystem for plant functional genomics.Biotechnol. Lett.33629–636. 10.1007/s10529-010-0475-6
46
SablokG.HarikrishnamJ. A.MinX. J. (2013). Next generation sequencing for better understanding alternative splicing: way ahead for model and non-model plants.Transcriptomics1:e103.
47
SablokG.PowellB.BraesslerJ.YuF.MinX. J. (2017). Comparative landscape of alternative splicing in fruit plants.Curr. Plant Biol.929–36. 10.1016/j.cpb.2017.06.001
48
ScaranoD.RaoR.CorradoG. (2017). In Silico identification and annotation of non-coding RNAs by RNA-seq and De Novo assembly of the transcriptome of tomato fruits.PLoS One12:e0171504. 10.1371/journal.pone.0171504
49
ShiX.GuptaS.LindquistI. E.CameronC. T.MudgeJ.RashotteA. M. (2013). Transcriptome analysis of cytokinin response in tomato leaves.PLoS One8:e55090. 10.1371/journal.pone.0055090
50
ShuklaN.YadavR.KaurP.RasmussenS.GoelS.AgarwalM.et al (2018). Transcriptome analysis of root-knot nematode (Meloidogyne incognita)-infected tomato (Solanum lycopersicum) roots reveals complex gene expression profiles and metabolic networks of both host and nematode during susceptible and resistance responses.Mol. Plant Pathol.19615–633. 10.1111/mpp.12547
51
StaigerD.BrownJ. W. (2013). Alternative splicing at the intersection of biological timing, development, and stress responses.Plant Cell253640–3656. 10.1105/tpc.113.113803
52
SunY.XiaoH. (2015). Identification of alternative splicing events by RNA sequencing in early growth tomato fruits.BMC Genomics16:948. 10.1186/s12864-015-2128-6
53
SundaresanS.Philosoph-HadasS.RiovJ.MugasimangalamR.KuravadiN. A.KochanekB.et al (2016). De novo transcriptome sequencing and development of abscission zone-specific microarray as a new molecular tool for analysis of tomato organ abscission.Front. Plant Sci.6:1258. 10.3389/fpls.2015.01258
54
TanG.LiuK.KangJ.XuK.ZhangY.HuL.et al (2015). Transcriptome analysis of the compatible interaction of tomato with Verticillium dahliae using RNA-sequencing.Front. Plant Sci.6:428. 10.3389/fpls.2015.00428
55
TangX.TangZ.HuangS.LiuJ.LiuJ.ShiW.et al (2013). Whole transcriptome sequencing reveals genes involved in plastid/chloroplast division and development are regulated by the HP1/DDB1 at an early stage of tomato fruit development.Planta238923–936. 10.1007/s00425-013-1942-9
56
ThatcherS. R.DanilevskayaO. N.MengX.BeattyM.Zastrow-HayesG.HarrisC.et al (2016). Genome-wide analysis of alternative splicing during development and drought stress in maize.Plant Physiol.170586–599. 10.1104/pp.15.01267
57
ThatcherS. R.ZhouW.LeonardA.WangB. B.BeattyM.Zastrow-HayesG.et al (2014). Genome-wide analysis of alternative splicing in Zea mays: landscape and genetic regulation.Plant Cell263472–3487. 10.1105/tpc.114.130773
58
Tomato Genome Consortium (2012). The tomato genome sequence provides insights into fleshy fruit evolution.Nature485635–641. 10.1038/nature11119
59
TrapnellC.WilliamsB. A.PerteaG.MortazaviA.KwanG.Van BarenM. J.et al (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation.Nat. Biotechnol.28511–515. 10.1038/nbt.1621
60
VanBurenR.WaltersB.MingR.MinX. J. (2013). Analysis of expressed sequence tags and alternative splicing genes in sacred lotus (Nelumbo nucifera Gaertn.).Plant Omics6311–317.
61
VituloN.ForcatoC.CarpinelliE. C.TelatinA.CampagnaD.D’AngeloM.et al (2014). A deep survey of alternative splicing in grape reveals changes in the splicing machinery related to tissue, stress condition and genotype.BMC Plant Boil.14:99. 10.1186/1471-2229-14-99
62
WaiC. M.PowellB.MingR.MinX. J. (2016). Analysis of alternative splicing landscape in pineapple (Ananas comosus).Trop. Plant Biol.9150–160. 10.1007/s12042-016-9168-1
63
WaiC. M.PowellB.MingR.MinX. J. (2018). “Analysis of transcriptome and alternative splicing landscape in pineapple,” in Genetics and Genomics of Pineapple. Plant Genetics and Genomics: Crops and ModelsVol. 22ed.MingR. (Cham: Springer).
64
WaltersB. R.LumG. E.SablokG. A.MinX. J. (2013). Genome-wide landscape of alternative splicing events in Brachypodium distachyon.DNA Res.20163–171. 10.1093/dnares/dss041
65
WangB.BrendelV. (2006). Genome wide comparative analysis of alternative splicing in plants.Proc. Natl. Acad. Sci. U.S.A.1037175–7180. 10.1073/pnas.0602039103
66
WangJ.YuW.YangY.LiX.ChenT.LiuT.et al (2015). Genome-wide analysis of tomato long non-coding RNAs and identification as endogenous target mimic for microRNA in response to TYLCV infection.Sci. Rep.5:16946. 10.1038/srep16946
67
WangK.JiaoZ.XuM.WangY.LiR.CuiX.et al (2016). Landscape and fruit developmental regulation of alternative splicing in tomato by genome-wide analysis.Hortic. Plant J.2338–350. 10.1016/j.hpj.2017.01.007
68
WangR. H.YuanX. Y.MengL. H.ZhuB. Z.ZhuH. L.LuoY. B.et al (2016). Transcriptome analysis provides a preliminary regulation route of the ethylene signal transduction component, SlEIN2, during tomato ripening.PLoS One11:e0168287. 10.1371/journal.pone.0168287
69
WangM.WangP.LiangF.YeZ.LiJ.ShenC.et al (2018a). A global survey of alternative splicing in allopolyploid cotton: landscape, complexity and regulation.New Phytol.217163–178. 10.1111/nph.14762
70
WangM.ZhaoW.GaoL.ZhaoL. (2018b). Genome-wide profiling of long non-coding RNAs from tomato and a comparison with mRNAs associated with the regulation of fruit ripening.BMC Plant Biol.18:75. 10.1186/s12870-018-1300-y
71
WangY.TaoX.TangX. M.XiaoL.SunJ. L.YanX. F.et al (2013). Comparative transcriptome analysis of tomato (Solanum lycopersicum) in response to exogenous abscisic acid.BMC Genomics14:841. 10.1186/1471-2164-14-841
72
WeiH.LouQ.XuK.YanM.XiaH.MaX.et al (2017). Alternative splicing complexity contributes to genetic improvement of drought resistance in the rice maintainer HuHan2B.Sci. Rep.7:11686. 10.1038/s41598-017-12020-3
73
WorleyJ. N.PomboM. A.ZhengY.DunhamD. M.MyersC. R.FeiZ.et al (2016). A novel method of transcriptome interpretation reveals a quantitative suppressive effect on tomato immune signaling by two domains in a single pathogen effector protein.BMC Genomics17:229. 10.1186/s12864-016-2534-4
74
XuP.KongY.SongD.HuangC.LiX.LiL. (2014). Conservation and functional influence of alternative splicing in wood formation of Populus and Eucalyptus.BMC Genomics15:780. 10.1186/1471-2164-15-780
75
XuZ.LuoH.JiA.ZhangX.SongJ.ChenS. (2016). Global identification of the full-length transcripts and alternative splicing related to phenolic acid biosynthetic genes in Salvia miltiorrhiza.Front. Plant Sci.7:100. 10.3389/fpls.2016.00100
76
XueD. Q.ChenX. L.ZhangH.ChaiX. F.JiangJ. B.XuX. Y.et al (2016). Transcriptome analysis of the Cf-12-mediated resistance response to Cladosporium fulvum in tomato.Front. Plant Sci.7:2012. 10.3389/fpls.2016.02012
77
YangY. X.WangM. M.YinY. L.OnacE.ZhouG. F.PengS.et al (2015). RNA-seq analysis reveals the role of red light in resistance against Pseudomonas syringae pv. tomato DC3000 in tomato plants.BMC Genomics16:120. 10.1186/s12864-015-1228-7
78
YeJ.HuT.YangC.LiH.YangM.IjazR.et al (2015). Transcriptome profiling of tomato fruit development reveals transcription factors associated with ascorbic acid, carotenoid and flavonoid biosynthesis.PLoS One10:e0130885. 10.1371/journal.pone.0130885
79
YuH.TianC.YuY.JiaoY. (2016). Transcriptome survey of the contribution of alternative splicing to proteome diversity in Arabidopsis thaliana.Mol. Plant9749–752. 10.1016/j.molp.2015.12.018
80
ZhangC.YangH.YangH. (2016). Evolutionary character of alternative splicing in plants.Bioinform. Biol. Insights9(Suppl. 1), 47–52. 10.4137/BBI.S33716
81
ZhangS.XuM.QiuZ.WangK.DuY.GuL.et al (2016). Spatiotemporal transcriptome provides insights into early fruit development of tomato (Solanum lycopersicum).Sci. Rep.6:23173. 10.1038/srep23173
82
ZhangG.SunM.WangJ.LeiM.LiC.ZhaoD.et al (2018). PacBio full-length cDNA sequencing integrated with RNA-seq reads drastically improves the discovery of splicing transcripts in rice.Plant J.97296–305. 10.1111/tpj.14120
83
ZhangR.CalixtoC. P.MarquezY.VenhuizenP.TzioutziouN. A.GuoW.et al (2017). A high quality Arabidopsis transcriptome for accurate transcript-level analysis of alternative splicing.Nucleic Acids Res.455061–5073. 10.1093/nar/gkx267
84
ZhangY.ZhaoG.LiY.ZhangJ.ShiM.MuhammadT.et al (2017). Transcriptome profiling of tomato uncovers an involvement of cytochrome P450s and peroxidases in stigma color formation.Front. Plant Sci.8:897. 10.3389/fpls.2017.00897
85
ZhaoL.ZhangH.KohnenM.PrasadK.GuL.ReddyA. S. (2019). Analysis of transcriptome and epitranscriptome in plants using PacBio Iso-Seq and Nanopore-based direct RNA sequencing.Front. Genet.10:253. 10.3389/fgene.2019.00253
86
ZhengY.DingB.FeiZ.WangY. (2017). Comprehensive transcriptome analyses reveal tomato plant responses to tobacco rattle virus-based gene silencing vectors.Sci. Rep.7:9771. 10.1038/s41598-017-10143-1
87
ZhuB.YangY.LiR.FuD.WenL.LuoY.et al (2015). RNA sequencing and functional analysis implicate the regulatory role of long non-coding RNAs in tomato fruit ripening.J. Exp. Bot.664483–4495. 10.1093/jxb/erv203
88
ZhuG.LiW.ZhangF.GuoW. (2018). RNA-seq analysis reveals alternative splicing under salt stress in cotton, Gossypium davidsonii.BMC Genomics19:73. 10.1186/s12864-018-4449-8
89
ZhuJ.WangX.XuQ.ZhaoS.TaiY.WeiC. (2018). Global dissection of alternative splicing uncovers transcriptional diversity in tissues and associates with the flavonoid pathway in tea plant (Camellia sinensis).BMC Plant Biol.18:266. 10.1186/s12870-018-1497-9
90
ZouariI.SalvioliA.ChialvaM.NoveroM.MiozziL.TenoreG. C.et al (2014). From root to fruit: RNA-Seq analysis shows that arbuscular mycorrhizal symbiosis may affect tomato fruit metabolism.BMC Genomics15:221. 10.1186/1471-2164-15-221
91
ZouineM.FuY.Chateigner-BoutinA. L.MilaI.FrasseP.WangH.et al (2014). Characterization of the tomato ARF gene family uncovers a multi-levels post-transcriptional regulation including alternative splicing.PLoS One9:e84203. 10.1371/journal.pone.0084203
Summary
Keywords
alternative splicing, gene expression, tomato, mRNA, plant, Solanum lycopersicum, transcriptome
Citation
Clark S, Yu F, Gu L and Min XJ (2019) Expanding Alternative Splicing Identification by Integrating Multiple Sources of Transcription Data in Tomato. Front. Plant Sci. 10:689. doi: 10.3389/fpls.2019.00689
Received
20 February 2019
Accepted
08 May 2019
Published
28 May 2019
Volume
10 - 2019
Edited by
Kranthi Kiran Mandadi, Texas A&M University, United States
Reviewed by
Chaoling Wei, Anhui Agricultural University, China; Daqi Fu, China Agricultural University, China
Updates
Copyright
© 2019 Clark, Yu, Gu and Min.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang Jia Min, xmin@ysu.edu
This article was submitted to Plant Physiology, a section of the journal Frontiers in Plant Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.