 Hongryul Ahn
Hongryul Ahn Kyuri Jo
Kyuri Jo Dabin Jeong
Dabin Jeong Minwoo Pak
Minwoo Pak Jihye Hur
Jihye Hur Woosuk Jung
Woosuk Jung Sun Kim
Sun Kim- 1Bioinformatics Institute, Seoul National University, Seoul, South Korea
- 2Interdisciplinary Program in Bioinformatics, Seoul National University, Seoul, South Korea
- 3Department of Computer Science and Engineering, Seoul National University, Seoul, South Korea
- 4Department of Crop Science, Konkuk University, Seoul, South Korea
Transcription factor (TF) has a significant influence on the state of a cell by regulating multiple down-stream genes. Thus, experimental and computational biologists have made great efforts to construct TF gene networks for regulatory interactions between TFs and their target genes. Now, an important research question is how to utilize TF networks to investigate the response of a plant to stress at the transcription control level using time-series transcriptome data. In this article, we present a new computational network, PropaNet, to investigate dynamics of TF networks from time-series transcriptome data using two state-of-the-art network analysis techniques, influence maximization and network propagation. PropaNet uses the influence maximization technique to produce a ranked list of TFs, in the order of TF that explains differentially expressed genes (DEGs) better at each time point. Then, a network propagation technique is used to select a group of TFs that explains DEGs best as a whole. For the analysis of Arabidopsis time series datasets from AtGenExpress, we used PlantRegMap as a template TF network and performed PropaNet analysis to investigate transcriptional dynamics of Arabidopsis under cold and heat stress. The time varying TF networks showed that Arabidopsis responded to cold and heat stress quite differently. For cold stress, bHLH and bZIP type TFs were the first responding TFs and the cold signal influenced histone variants, various genes involved in cell architecture, osmosis and restructuring of cells. However, the consequences of plants under heat stress were up-regulation of genes related to accelerating differentiation and starting re-differentiation. In terms of energy metabolism, plants under heat stress show elevated metabolic process and resulting in an exhausted status. We believe that PropaNet will be useful for the construction of condition-specific time-varying TF network for time-series data analysis in response to stress. PropaNet is available at http://biohealth.snu.ac.kr/software/PropaNet.
1. Introduction
A transcription factor (TF) is a protein that regulates expression levels of a target gene (TG) by binding to a specific DNA sequence on the promoter regions of target genes (Latchman, 1997). TFs activate or repress target genes, contributing to expression of genes at the right time and in the right amount throughout the life of the cell and the organism. Groups of TFs function in a coordinated fashion to direct various biological mechanisms such as development (Lobe, 1992), signal transduction (Pawson, 1993), response to environmental change (Chen et al., 2002), and regulation of cell cycle (Meyyappan et al., 1996).
When a plant is exposed to stress, TFs that are activated in response to stress rapidly propagate stress signals to other genes in the cell by regulating multiple downstream genes (Zhu, 2016). In recent genetic engineering experiments of plants, over-expression of specific transcription factors enhanced resistance to stress. For example, over-expression of TFs such as OsAP37 (Oh et al., 2009), OsNAC9 (Redillas et al., 2012), OsNAC10 (Jeong et al., 2010), MYB96 (Seo et al., 2011), OsbZIP12 (Joo et al., 2014), and OsbZIP23 (Karaba et al., 2007) induced drought resistance phenotype in rice and Arabidopsis. Thus, the TF gene regulatory network (or TF network) should be considered as an essential source of information when detecting stress response signaling genes.
However, many computational methods for investigating the stress response genes of plants do not utilize TF networks. Algorithms such as EDISA (Supper et al., 2007), a two-step 3D clustering algorithm (Supper et al., 2007), OPTricluster (Tchagang et al., 2012), and TimesVector (Jung et al., 2017) perform 3D clustering which takes advantage of gene, time and phenotype information, yet, they do not take account of TF network. The primary goal of the methods is to detect a group of genes that show same expression patterns in all phenotypes (coherent response) or show same patterns except in one phenotype (single response) or show different patterns for different phenotypes (independent response). All of these methods successfully detected important gene clusters, but it was not sufficient to detect regulatory relationship between clustered genes without prior knowledge of TF-TG network.
Construction of transcriptional networks has been extensively investigated in both computational biology and experimental biology. In this paper, the TF gene regulatory network (or TF network) refers to a graph-based representation that contains regulatory relationships between TFs and their target genes. Constructing a TF network is a very difficult task since the number of possible TF-TG relationships to be considered for the construction of a TF network is over 30 million (1,500 TF × 20,000 TG); There are 20,418, 22,619 and 27,665 protein-coding genes (i.e., TGs) in human, mouse, and Arabidopsis species, respectively, according to the ENSEMBL (Zerbino et al., 2017). However, a few number of TF network databases have been produced, thanks to the large scale efforts of research communities, e.g., PlantTFDB (Jin et al., 2016), HumanTFDB and AnimalTFDB (Hu et al., 2018).
There are three different strategies for building TF networks. First, the most fundamental approach would be to collect TF-TG relationships from literatures. Biologists conducted various types of biological experiments such as over-expression, knock-down or knock-out of genes, in-depth expression level profiling, for elucidating TF-TG relationships. The number of TF-TG relations in the literature-based databases is relatively small, but the information is reliable. TRRUST (Han et al., 2017) contains 8,444 and 6,552 TF-TG interactions for 800 and 828 TFs for human and mouse species, respectively. In ATRM (Jin et al., 2015), 1,431 TF-TG interactions for 324 TFs are curated for Arabidopsis. HTRIdb (Bovolenta et al., 2012), PAZAR (Portales-Casamar et al., 2007), TFactS (Essaghir et al., 2010), TRED (Zhao et al., 2005) and TFe (Yusuf et al., 2012) are examples of the literature-based TF network construction.
The second approach is based on computational predictions from a large collection of gene expression datasets that are measured using high-throughput technologies such as microarray (Schena et al., 1995) and RNA sequencing (Mortazavi et al., 2008). ARACNe (Margolin et al., 2006) uses an information theoretic framework based on the data processing inequality theorem. It was successfully used for the reconstruction of context-specific transcriptional networks in multiple tissue types (Lefebvre et al., 2010). Then, many methods were developed for the construction of TF networks: GENIE3 (Huynh-Thu et al., 2010), NARROMI (Zhang et al., 2012), Ennet (Sławek and Arodź, 2013), and Wisdom of crowds (Marbach et al., 2012). Following these successes, the DREAM (Dialogue for Reverse Engineering Assessments and Methods) challenge competition was initiated. Yip's method (Yip et al., 2010) (for DREAM3) and GENIE3 (Huynh-Thu et al., 2010) (for DREAM4 and DREAM5) won the competitions. These approaches were further developed to investigate dynamics of TF networks. Various gene regulatory network (GRN) inference methods using time-course data have been developed with model-based and model-free approaches. Model-based methods formulate the expression of a target gene as a function of its regulators. The representative model-based methods, such as ScanBMA (Young et al., 2014), Inferelator (Bonneau et al., 2006), BGRMI (Iglesias-Martinez et al., 2016), Fused-Lasso (Omranian et al., 2016). BGRMI (Iglesias-Martinez et al., 2016), use ridge regression, LASSO and Bayesian Model Averaging (BMA) techniques. Model-free methods compute the degree of regulation based on model-free and time-lag regression. TD-ARACNE (Zoppoli et al., 2010) used mutual information to measure time-delayed dependency between two genes. DyeGENIE3 (Huynh-Thu and Geurts, 2018) and BTNET (Park et al., 2018b) used tree-based ensemble regression methods.
The third approach uses experimental data that measure affinity of TFs. The discovery that formaldehyde can crosslink histones to DNA (Brutlag et al., 1969) initiated Chromatin Immunoprecipitation (ChIP) assay that utilized crosslinked complexes and analysis of the associated DNA (Solomon and Varshavsky, 1985). Formaldehyde produces both protein-nucleic acid and protein-protein crosslinks in vivo by a reaction with amino and imino groups of amino acids and of DNA (Orlando, 2000). ChIP assays performed with crosslinking have made it possible to identify interactions that would not withstand the isolation procedure without crosslinking (Hoffman et al., 2015). ChIP assay has been ubiquitous in a multiple variations, one of which is ChIP-on-chip that combines ChIP with DNA microarray (Ren et al., 2000). Several studies identified binding sites for TFs by ChIP-on-chip in plants including Arabidopsis (Thibaud-Nissen et al., 2006). ChIP sequencing (ChIP-seq) technology was developed independently by three research groups in 2007 (Barski et al., 2007; Johnson et al., 2007; Mikkelsen et al., 2007) and it has been used to identify genomic regions that TF binds to, also known as, transcription factor binding sites (TFBSs). It crosslinks DNA and associated TFs, shears DNA-TF complexes into 500 bp DNA fragments by sonication or nuclease digestion, immunoprecipitates the targeted TF complexes using an appropriate protein-specific antibody, and then determines the sequence of the DNA fragments. With ChIP-seq and several other variants of immunoprecipitation assay such as ChIP-chip (Ren et al., 2000), ChIP-exo (Rhee and Pugh, 2011), ChIA-PET (Fullwood and Ruan, 2009), a number of ChIP-seq-like datasets for different species, tissues and cell lines have been generated and are freely available in databases such as Gene Expression Omnibus (GEO) (Barrett et al., 2013), Sequence Read Archive (SRA) (Kodama et al., 2011) and ENCODE (Landt et al., 2012). We can locate a binding motif sequence of a TF by processing ChIP-seq dataset and predict the target genes by searching the binding motif sequence on the promoter region of target genes. Now, some of the databases are providing TF-TG relationships by predicting binding sites for the collective TFs: TRANSFAC (Matys et al., 2006) a well-known commercial database; ENCODE (Landt et al., 2012), JASPAR (Khan et al., 2017) and ChIP-Atlas (Oki et al., 2018) for model organisms; GTRD (Yevshin et al., 2016), ChIPBase (Yang et al., 2012), Cistrome (Zheng et al., 2018b) and Factorbook (Wang et al., 2012) for human and mouse species; PlantRegMap (Jin et al., 2016) for plant species.
2. Motivation
Investigating time-varying dynamics of TF network upon abiotic stress is the main research question. We can use a template network from existing TF networks that are surveyed in the previous section. A biological experiment can be designed to investigate how a plant responds to stress over time by measuring transcriptome data at different time points under stress. Then, cell's response at the transcriptome level can be easily detected by measuring differentially expressed genes (DEGs), control vs. under stress, at each time point. By constructing TF networks that include DEGs and TFs, we can gain insight into how the responses of the TFs differ in gene expression levels under each stress, i.e., DEGs. There are two major issues with this approach.
1. Contributions of TF to DEGs differ for different TFs. It is not enough to consider only TFs that show significant expression changes during stress. It is known that TFs that show little change in expression levels function in response to stress as much as those that show large change in expression levels. In other words, the amount of change in expression level of a TF is not necessarily proportional to the significance of its role in the response to the stress (Ehlting et al., 2008; Larkindale and Vierling, 2008). In addition, determining major regulators is not trivial since a TF can target other TFs, forming multi-layered relationships to reach DEGs. How do we know which TFs are the major regulators?
2. Since there are many TFs and DEGs at each time point, TF networks are huge in size, which are too big to be interpreted. How can we construct small but informative TF networks at each time point so that we can interpret dynamics of TF networks?
Unfortunately, there is few computational methods to achieve this goal of investigating the dynamics of TF networks from time-series gene expression data. There are some interesting prior works, but they are not designed for answering this research question. DREM (Schulz et al., 2012) is a pioneering method for the identification of regulatory TFs from the time-series gene expression data analysis. It first partitions genes into gene clusters, then defines branch time points where gene expression patterns are diverging, and then investigates the effect of TFs to the downstream genes utilizing known TF-TG relationships to detect regulatory TFs for gene clusters. However, DREM uses only direct TF-TG information. Therefore, it cannot consider the effects of interactions between TFs that might cause indirect influence on differential expression of genes. TimeTP (Jo et al., 2016) is another method for detecting major TFs using Influence Maximization (IM), one of the widely used analysis techniques for social network analysis. TimeTP produces a list of TFs having influence on pathway genes, but it is not designed to investigate time-varying dynamics of TF networks. There are network construction methods that utilized the literature information. For example, Wanke et al. (2010b) constructed a network of MAPK (mitogen-activated protein kinase) signaling pathway including receptors, second messengers and TFs from known interactions in literature. In general, literature based network construction requires quite efforts involving manual curation. Thus, these methods are not suitable for large scale network construction involving multiple pathways.
In this article, we propose PropaNet to investigate dynamics of TF networks from time-series transcriptome data through network simulation analysis that mimics the regulatory mechanism of TF. PropaNet uses state-of-the-art network analysis technologies, influence maximization and network propagation techniques, to perform a two-stage simulation analysis as below.
1. An influence maximization technique is used to produce a ranked list of TFs at each time point, in the order of TFs that have influence on a larger number of DEGs.
2. A network propagation technique is used to select a group of TFs that explains DEGs best as a whole. The process is done by iterating a network propagation simulation by adding a TF at a time, going down the list of TFs determined by the influence maximization technique.
We used PropaNet to interpret time-series analysis transcriptome data under cold and heat stress in Arabidopsis. From the analysis, we successfully identified major TFs and constructed TF networks at each time point. “A major TF” indicates a TF that has large direct or indirect influence on DEGs (estimated by influence maximization) and propagates more influence on more significant DEGs (estimated by network propagation). Performance of PropaNet was compared with simple correlation-based methods and other methods for time series analysis using clustering and network information. PropaNet showed better performance in finding cold- and heat-specific transcription factors and their target genes by incorporating network information and its novel strategy for selecting major regulatory TFs.
3. Methods
3.1. Problem Definition of PropaNet
The PropaNet analysis takes three types of input data: time-series gene expression data EX that are measured at multiple time-points, a template TF network G and a set of target genes TGset. PropaNet detects major TFs that particularly target TGset defined by users. TGset can range from a small set of pathway genes to whole DEGs of the gene expression data. The goal of the PropaNet analysis is to elucidate time-varying networks of major TFs and their target genes at each time point by the network-based analysis on the template TF network. Terminologies used in this paper are defined as follows:
Definition 3.1. Let EX, G and TGset be time-series gene expression data, TF network and a set of targeted genes, respectively. EX is a set of gene expression values ei,j,k of gene i measured at time point j = 0, …, T from replicate k = 1, …, K. A set of differentially expressed genes for time point j = 1, …, T compared to the initial time point j = 0 is defined as DEGsetj. From the template network G, a time-specific network Gj is generated using a gene set Vj = (TGset ∩ DEGsetj) ∪ TFset. A time-specific network Gj is defined as Gj = (Vj, Ej) with nodes Vj (TFs and TGs) and edges Ej (TF-TF and TF-TG pairs). For each node and edge, a weight of a node p : V ↦ ℜ (a map of node to weight) and a weight of an edge w : E ↦ ℜ (a map of edge to weight) exist. Differential expression levels di, j and correlation coefficient that are defined below is assigned to p and w, respectively.
• Dj is a set of differential expression levels at time j, the element of which di,j is the differential expression level of a gene i at the time point j = 1, …, T with respect to the initial time point j = 0. That is, di,j is calculated from comparing two sets of expression values {ei,j,1, …, ei,j,k} and {ei,0,1, …, ei,0,k} by an existing DEG detection algorithm such as limma (Ritchie et al., 2015) or DESeq2 (Anders and Huber, 2010). di,j can be Z-scores from limma or log2 fold change from DESeq2.
• is a Pearson's correlation coefficient (PCC) between gene i and i′ computed using two sets of expression values {ei,j,k} and where j = 1, …, T, k = 1, …, K.
• DEGsetj is a set of differentially expressed genes (DEGs) at time point j where a DEG is defined as a gene with p-value(di,j) < 0.05.
• MTFsetj is a minimal set of major TFs that explains the change of expression levels of DEGsetj at time point j.
• MTFnetj is a time-varying TF network at time point j that shows the regulatory system explaining how MTFsetj controls DEGsetj.
PropaNet outputs a set of major regulatory TFs MTFsetj and their time-varying network including target genes MTFnetj for each time point j = 1, …, T. PropaNet operates in three steps as below and the process is visualized in Figure 1.
• Step 1. Instantiation of time-specific TF networks from a template network. A time-specific network consists of DEG and TFs at each time point.
• Step 2. Time-specific measurement of the influence of each TF by influence maximization. TFs in the network are ranked along with their influence on DEGs via the network topology (including non-direct targets).
• Step 3. Identification of time-specific major regulatory TFs by network propagation. The TF set is constructed by adding a TF at a time, following down the ranked list of TFs.
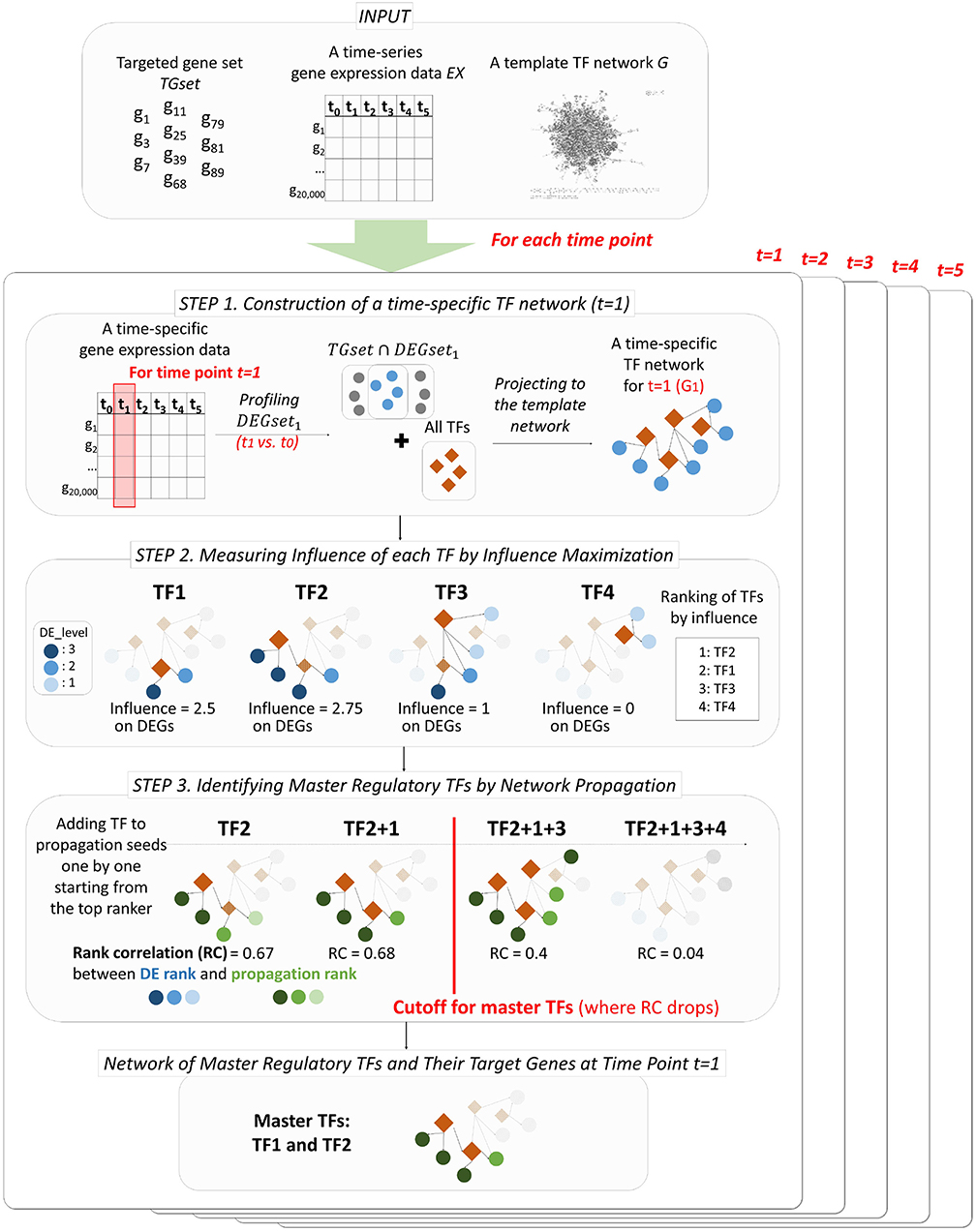
Figure 1. Workflow of PropaNet. The PropaNet analysis takes three types of input data: time-series gene expression data that are measured at multiple time-points, a template TF network and a set of targeted genes. The goal of the PropaNet analysis is to elucidate time-varying networks at each time point. It uses the influence maximization technique to produce a ranked list of TFs at each time point, in the order of TF that explains DEGs better. Then, a network propagation technique is used to select a group of TFs that explains DEGs best as a whole. The process is done by iterating a network propagation simulation by adding a TF at a time, going down the list of TFs determined by the influence maximization technique.
3.2. Step 1. Instantiation of Time-Specific TF Networks
The first step of PropaNet is to construct a time-specific networks Gj for each time point j = 1, …, T by mapping TFs and a intersected gene set of user-defined target genes and DEGs to a template network (i.e., a gene set Vj = (TGset ∩ DEGsetj) ∪ TFset for time j) (Figure 1). DEGs are determined by comparing gene expressions at each time point j = 1, …, T with time j = 0 (i.e., 0 h after stress). The inputs of PropaNet (a template TF network and DEG profiles) are designed to be user-determined.
3.3. Step 2. Time-Specific Measurement of the Influence of Each TFs by Influence Maximization
The goal of this step is to rank TFs in the order of influence to TGset ∩ DEGsetj at each time point j. Influence maximization (IM) is an algorithmic technique used in network influence analysis to select a set of seed nodes to maximize the spread of influence (the expected number of influenced nodes) from a given network (Li et al., 2018). Labeled influence maximization (Li et al., 2011) is a modified version of IM, applied only to a set of pre-selected nodes. TimeTP (Jo et al., 2016) used the labeled influence maximization algorithm to determine TFs that regulates the perturbed sub-pathways. We used a modified version of the labeled influence maximization algorithm to rank TFs in terms of their influence to TGset ∩ DEGsetj at time point j.
We provide more detailed explanation of the labeled influence maximization algorithm of PropaNet (Algorithm 1). Input of the algorithm for time j is (Gj = (Vj, Ej), TFset, Dj) to measure the influence of each TF (∈ TFset) to the targeted genes (∈ Vj\TFset) on the time-specific TF network Gj. IM algorithm first initializes the weights of node, DE(s), as the absolute differential expression level ds,j for all s ∈ V and the influence of TF, IL(t), as 0 for t ∈ TFset. Then, it generates sub-graph G′ from G by selecting edges with a probability of 1 − p for each edge (line 4), where p is the weight of the edge in the original graph G. Then, the influence IL(t) increases by the for where is nodes that the TF can reach in the generated sub-graph G′. After repeating the above procedure for Round times, the algorithm produces the final output IL of all TFs at the time point j.

Algorithm 1: Influence Maximization on TF-TG network (G, TFset,D)
3.4. Step 3. Time-Specific Identification of Major Regulatory TFs by Network Propagation
Network propagation is a graph-based analytic paradigm that propagates information of a node to nearby nodes through the edges at each iteration step. This process is repeated for a fixed number of steps or until convergence. Since the value of a node influences not only the values of its direct network neighbors but also those of its distant neighbors, network propagation is known to perform better than direct neighbor search methods and shortest path search methods for a problem of prioritizing genes that are associated with seed genes (Cowen et al., 2017).
Network propagation is mathematically equivalent to random walks on a graph. We can think of p0(v) as an amount of information of a node v at the beginning of iteration 0. At each iteration k, the amount of information at each node v is influenced by the sum of the information at the neighboring (adjacent) nodes N(v) at iteration k − 1, in proportion to the weights on the corresponding edges, according to the following equation:
where w(u, v) is the weight of the edges (u, v) in the input network. The propagation process described in Equation 1 can be written in matrix notation as follows:
where W is a normalized version of the adjacency matrix of the input network. Another version of the propagation process is the random walk with restart (RWR). RWR performs the random walk and restarts at a rate of α:
where the parameter α is thought of the trade-off between prior information (restart) and network smoothing (random walk). After k iterations, the values in the resulting vector pk(v) give us a ranking of each node v that diffused from the initial value of seed nodes.
PropaNet simulates the TF-centered regulation process based on the network propagation. Each step of network propagation outputs ranked list of nodes in the network. The objective function, or the stopping criteria, is to find a ranked list of genes that are most similar to the ranked list of DEGs in terms of their p-values. This can be easily determined by computing Spearman's rank correlation coefficient (SCC) between the two ranked lists. More formally, the simulation is evaluated by the comparison of the ranking between the differential expression of observation DE(v) and the inferred expression of network propagation IP(v). This simulation is independently processed for each time point j. It first initializes the information of nodes, IP(v) as 0 for v ∈ V. At the time point j, we now have a list of TFs and their influence score, IL(t), that are measured in the previous step. It, then, initialize the most influencing TF (i.e., argmaxt IL(t)) as a set of seed S and conducts network propagation on the TF network G to update IP(v). Then, it measures the similarity of ranking, SCC, between IP(v) and DE(v). It adds the next most influencing TF into the set of seed S, performs network propagation, computes SCC, and decides whether accepting the TF or not accepts; the TF is accepted if SCC increases or declined otherwise. It continues this process for the list of TFs until the coverage of target genes exceeds the half number of DEG at the time point. Finally, it produces S as a set of major regulatory TFs.
3.5. Data Description
The experiments for the evaluation of PropaNet were conducted using two time-series gene expression datasets measured under thermal stress, AtGenExpress (Kilian et al., 2007) and E-MTAB-375 (Caldana et al., 2011), and a TF network, PlantRegMap (Jin et al., 2016).
• AtGenExpress dataset. AtGenExpress (Kilian et al., 2007) dataset was a well curated dataset that measured time-series gene expression data for multiple treatments from multiple tissues of Arabidopsis, making it four-dimensional dataset (gene, time, condition, and tissue). In this analysis, the AtGenExpress data of thermal (cold and heat) stress from shoot tissues were used. The raw data, derived by AtGenExpress experiments, were downloaded from GEO (Barrett et al., 2013) for cold (GSE5621) and heat (GSE5628) stresses. Also, zero time point data were downloaded from control time-series samples (GSE5620). Among the dataset of heat stress in AtGenExpress, recovery phase data were excluded to focus on the response of stress. Then, the data were processed using justRMA function of affy R library (Wagner, 2016) with default options (background and RMA normalization). To handle multiple probes in a single gene, custom CDF (Dai et al., 2005) was also used as input of justRMA to summarize expression levels for gene IDs. DEGs of each time point were determined by limma (Ritchie et al., 2015) package using replicates. In this way, the gene expression dataset of 14 samples (7 time points including 0 time point × 2 replicates) for cold stress and 10 samples (5 time points including 0 time point × 2 replicates) for heat stress and the corresponding DEG profiles were generated. The detailed information of the AtGenExpress samples was summarized in Supplementary Table 1.
• E-MTAB-375 dataset. A dataset E-MTAB-375 (Caldana et al., 2011) of light condition with 22 time points under cold and heat stress was used. Since E-MTAB-375 provided no replicates at each time point, we used the first and the second time point data (i.e., T = 0 and 5 min) of non-stress time-series for the replicate data at zero time point (i.e., T = 0 min). Accordingly, the 5-min time point data of stress (cold/heat) time-series were discarded for consistency of time point. The raw data of E-MTAB-375 were downloaded from ArrayExpress (Kolesnikov et al., 2014) and processed in the same way as the AtGenExpress data were processed. The detailed information of the E-MTAB-375 samples was summarized in Supplementary Table 2.
• PlantRegMap TF network. PlantRegMap (Jin et al., 2016) (http://plantregmap.cbi.pku.edu.cn) was used as a template network in our analysis. PlantRegMap was a TF network that was constructed mainly from TF ChIP-seq data. Other existing TF networks, such as ATTED-II (Obayashi et al., 2017), AraNet (Lee et al., 2014), and ATRM (Jin et al., 2015), were constructed by integrating multiple information such as TF ChIP-seq data, literature, and co-expression information. Since we were not able to evaluate how accurate each of the networks is, we decided to use PlantRegMap since we thought that PlantRegMap was the most unbiased network mainly form TF ChIP-seq data. After the PlantRegMap network including 688 TFs and 192,385 regulatory relations was downloaded, we computed PCC between the TF-TG pairs from the experimental gene expression data and assigned PCC values to the weights of edges of the template network.
3.6. Experiment Procedures for Evaluation of PropaNet
To evaluate PropaNet, we performed four types of experiments as below.
• Investigation on biological implications of time varying networks in response to thermal stress in Arabidopsis. We demonstrated PropaNet by applying it to the AtGenExpress microarray dataset to investigate the response mechanism of thermal (cold and heat) stress in Arabidopsis. The temperature-related stress has been investigated extensively in scientific, agricultural, and industrial fields because of recent climate and weather extremes, derived by global warming. Moreover, climate and weather extremes would be worse as global warming continues; a special report of Intergovernmental Panel on Climate Change (IPCC) in 2018 predicts with high confidence that global warming is likely to reach 1.5°C between 2,030 and 2,052 if it continues to increase at the current rate (IPCC, 2018).
Using the AtGenExpress dataset and the PlantRegMap (Jin et al., 2016) template network, PropaNet generated time varying networks in response to thermal stress. Then, enrichment analysis was conducted for the Gene Ontology (GO) (Consortium, 2018) terms and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) pathways to investigate the biological function of the time varying networks by performing Fisher's exact test with Benjamini-Hochberg correction (Benjamini and Hochberg, 1995) using statsmodels (Seabold and Perktold, 2010) Python library. In addition, PropaNet TF networks were compared to a literature-based network, ATRM (Jin et al., 2016), a literature-based network including 1,432 TF-TG edges, to investigate how many TF-TG relationships in the predicted network were reported in the literature.
• Performance comparison with existing tools. This experiment quantitatively compared PropaNet with existing methods at TF and gene levels. For the TF level comparison, the PCC, SCC, entropy and DREM (Schulz et al., 2012) methods predicted the stress-related TFs by analyzing the AtGenExpress dataset, and the accuracy of prediction of each tool was measured in terms of F1 score by comparing the predicted TFs to ground truth TFs. For the gene level comparison, existing methods to determine stress-responsive genes from time-series data, EDISA (Supper et al., 2007), OPTricluster (Tchagang et al., 2012), and DREM (Schulz et al., 2012), were compared with PropaNet by running them on the AtGenExpress dataset and comparing the resulting genes to the ground truth genes in the same way as TF level comparison analysis did.
To define the ground truth genes, a list of genes that were annotated with cold/heat response-related GO terms, such as “response to cold,” “cold acclimation,” and “cellular response to cold” for cold stress, and “response to heat,” “heat acclimation,” “cellular response to heat,” and “cellular heat acclimation” for heat stress was collected from the TAIR database (Lamesch et al., 2012). Then, 33 and 20 TFs and 330 and 158 genes were collected as ground truth genes for cold and heat stress, respectively.
The precision, recall, and F1 scores were used as accuracy measures to investigate how many ground truth genes in the predicted networks, which were defined as follows:
where TP, FP, and FN were the number of correctly positive-predicted genes, falsely positive-predicted genes, and falsely negative-predicted genes, respectively.
• The effect of utilizing non-stress time-series sample as control. Many biological experiments were designed to compare treated vs. control samples. Use of control samples can be used to eliminate the effects of background biological mechanisms (e.g., circadian rhythm) and help focus on mechanisms related to treated samples. To investigate the effect of utilizing control sample data, we developed a modified version of PropaNet, “PropaNetC,” to utilize non-stress time-series control sample data for generating time-varying networks.
After processing stress and non-stress time-series data, PropaNetC identified DEGs at each time point compared to the initial time point (0 h) for each of control and treated samples separately. PropaNetC then selected seed DEGs at each time point, t, by subtracting the DEGs of control samples from DEGs of stress samples (i.e., , where “−” was set difference operation). Using the seed DEGs, PropaNetC conducted further steps, influence maximization and network propagation, in the same way as PropaNet did. We performed analysis with and without using control samples, i.e., PropaNet and PropaNetC, on the AtGenExpress dataset and compared the resulting TF networks.
• Analysis of the effect of the number of time points. In this experiment, we investigated how PropaNet handled the dataset with many time points. The goal of the experiment was to compare two datasets with different time points as below:
– Cold stress data
- AtGenExpress, 7 time points between 0 and 24 h from shoots
- E-MTAB-375, 22 time points between 0 and 21 h 20 min under low-light condition
– Heat stress data
- AtGenExpress, 5 time points between 0 and 3 h from shoots
- E-MTAB-375, 22 time points between 0 and 21 h 20 min under normal-light condition
To investigate the effects of the number of time points and the length of time intervals, we compared how many genes were overlapped between two adjacent time points. The reason for this criterion was to investigate on the effect of the densely measured transcriptome data in the time domain for the construction of time varying TF networks. The overlap of two adjacent networks was defined as the number of gene sets common between two adjacent time points. Quantitatively, the overlap was defined as , where A and B gene sets at two adjacent time points.
4. Results
4.1. Investigation on Biological Implications of Time Varying Networks in Response to Thermal Stress in Arabidopsis
PropaNet investigated time-varying TF networks using time-series gene expression data measured at seven (j = 0, …, 6) and five (j = 0, …, 4) time points for cold and heat stresses, respectively. DEGs were detected at each of the six time points (j = 1, …, 6) and four time points (j = 1, …, 4) for cold and heat stresses, DEGs were detected at each time point with respect to zero time point (j = 0). The number of DEGs were 41, 23, 524, 2,262, 6,129, 7,656 for cold stress, and 1,177, 522, 1,915 and 7,424 for heat stress. Then, PropaNet produced six-time-point-networks (j = 1, …, 6) for cold stress (Supplementary Table 3) and four-time-point-networks (j = 1, …, 4) for heat stress (Supplementary Table 4). Figures 2, 3 show (1) the visualization of time varying TF networks for each time point, (2) interesting TFs that have relatively many downstream genes in the network, and (3) the enriched GO terms and pathways of the target genes, for the analyses of cold and heat stress data networks.
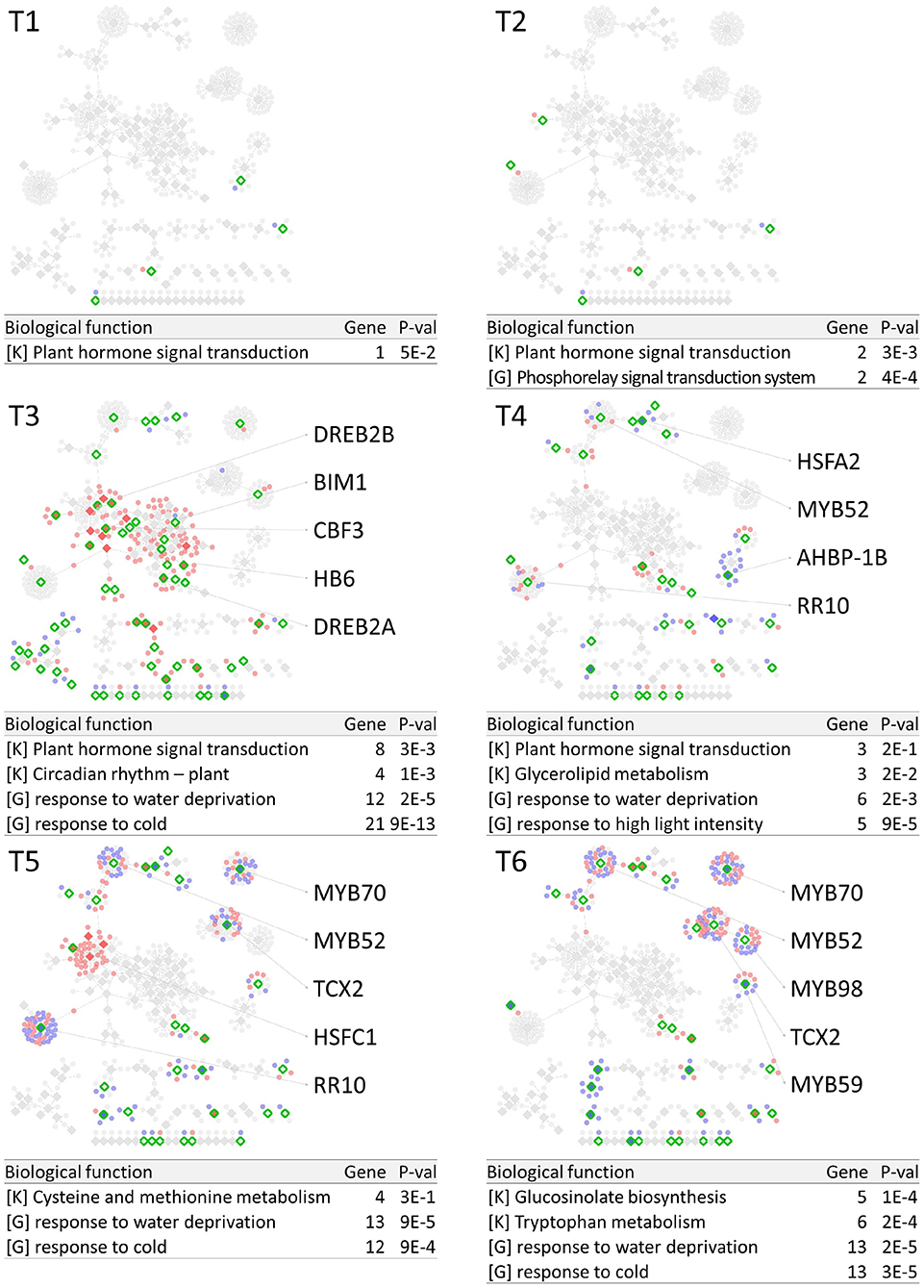
Figure 2. Time-varying network of six time points (T1 ~ T6) for cold stress experiment data in Arabidopsis. Red and blue nodes represent up/down DEGs, and green border rhombuses represent the identified regulatory TFs. Tables show top-2 enriched terms for each of KEGG pathways [K] and GO terms [G] with adjusted p-values by Benjamini-Hochberg correction. Some interesting TFs are named in the figure.
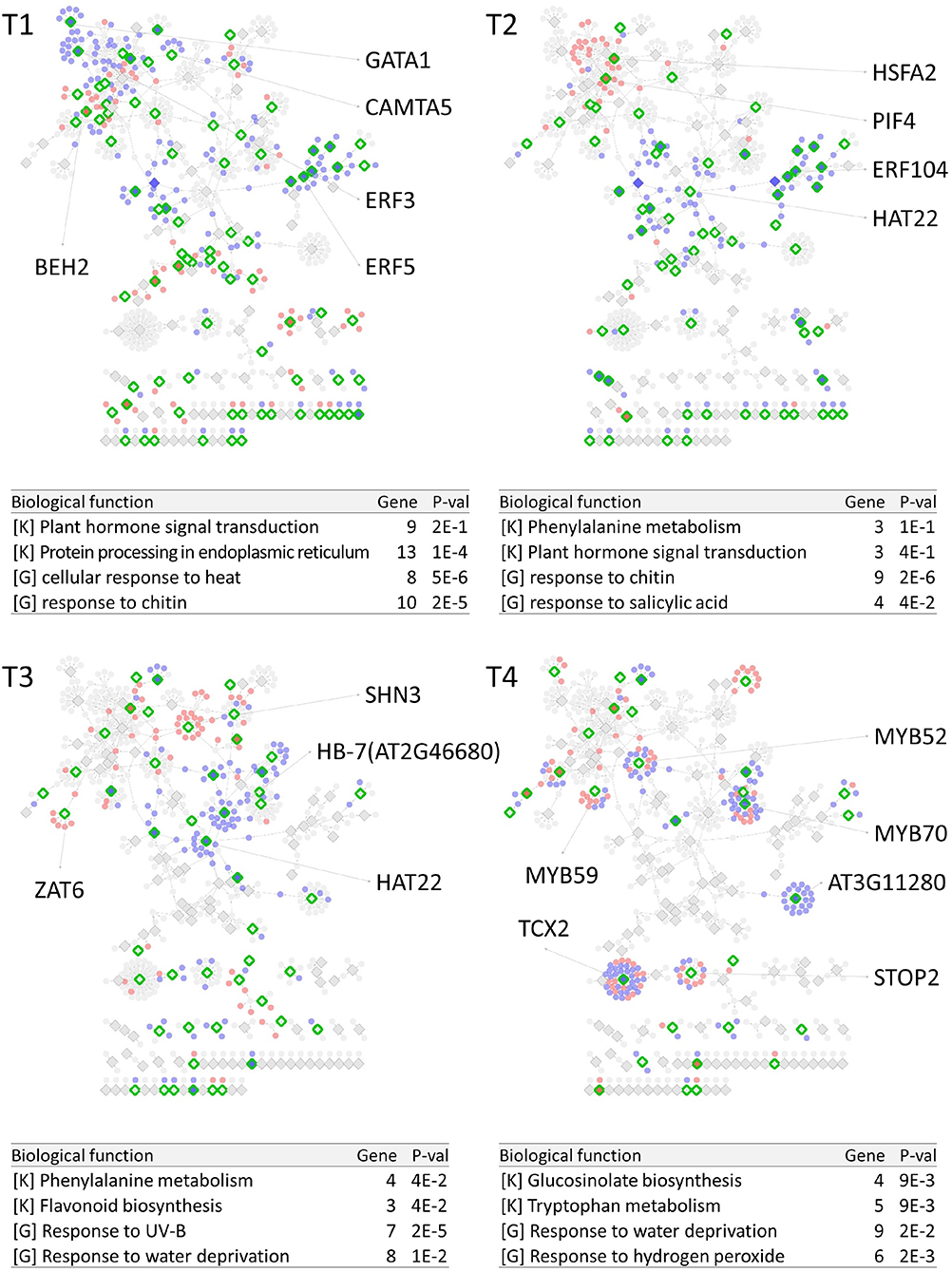
Figure 3. Time-varying network of four time points (T1 ~ T4) for heat stress experiment data in Arabidopsis. Red and blue nodes represent up/down DEGs, and green border rhombuses represent the identified regulatory TFs. Tables show top-2 enriched terms for each of KEGG pathways [K] and GO terms [G] with adjusted p-values by Benjamini-Hochberg correction. Some interesting TFs are named in the figure.
The time-varying networks had a modular structure of genes, i.e., clusters of genes were spread out throughout the network. In addition, neighboring modules had causal relationship where a module activated at the previous time point cause the activation of neighboring modules. This trend showed propagation of TFs to other genes over time under cold and heat stress. An interesting observation is that the cold stress network showed more delayed response than other types of stress in terms of the number of DEGs on the time domain, which was also reported in an earlier study on AtGenExpress dataset (Wanke et al., 2010a). Most genes in cold stress network show little response in the early time point (T1 and T2), but stress response starts from T3 time point in cold stress network while the heat stress network showed response at the very first time point, T1. In addition, major regulatory TFs that have many target genes in the network were well-known TFs related to each stress. For example, CBF3 was found in the T3 cold stress network, which is the most well-known TF for the response to cold stress that initiates global gene expression change to the cold stress (Medina et al., 2011). In addition, cold stress is known to be closely related to drought and heat stresses. Cold, and osmotic stresses are known to induce expression of many of the same genes and downstream genes in Arabidopsis (Xiong et al., 2002). In our result, the drought stress-responsive TFs such as DREB2A and DREB2B appeared in the early response stage (T3) cold stress network. Heat shock factors such as HSFA2 and HSFC1 appeared in T3 and T4 cold stress networks, and they are documented to be induced in cold stress to regulate downstream heat-shock-related proteins (Swindell et al., 2007). In heat stress network, HSFA2 (Schramm et al., 2006), ERF family TFs (Mizoi et al., 2012) that are known to regulate the expression level of downstream genes in heat stress appeared in the heat stress network.
The GO and KEGG enrichment analysis of cold stress networks showed the “response to cold” and “response to water” GO terms and the “plant hormone signal transduction” KEGG pathway were enriched, which are known to be related to cold stress in the literature (Eremina et al., 2016). For heat stress analysis, the “response to chitin” GO term and the “hormone signal transduction” KEGG pathway were enriched, and these terms are known to be related to heat stress in the literature (Eremina et al., 2016).
To investigate how many TF-TG relationships in the PropaNet network were reported through the previous experiments, the PropaNet network was compared to a literature-based network, ATRM (Jin et al., 2016). Among 580 and 687 edges of the PropaNet networks for cold and heat stress, 80 and 64 edges were literature-supported. The overlap between the edges of PropaNet network and the edges of ATRM network was statistically significant (p < 10−7 and p < 0.0098 by Fisher's exact test for cold and heat stress networks). Among the literature-supported edges, 7 edges were supported by the literatures of cold-specific experiments, such as DREB2A→LTI78 (Xiong et al., 2001), DREB2A→LTI30 (Chung and Parish, 2008), CBF3→ERD10 (Seki et al., 2002), HOS10→LTI78, HOS10→COR15A, HOS10→NATA1, and HOS10→ADH1 (Zhu et al., 2005). Also, 10 edges of the PropaNet heat stress network were supported by the literatures of heat-specific, such as HSFA9→HSP101 (Kotak et al., 2007), WRKY39→MBF1C (Li et al., 2010), DREB2A→HSP70 (Sakuma et al., 2006), HSFA2→APX2 (Charng et al., 2007), HSF1→HSP17.6A (Nishizawa et al., 2006), HSF1→GolS1, HSF1→GolS2, HSF1→MIPS2, HSFA2→GolS1, and HSFA2→GolS2 (Busch et al., 2005).
4.2. Performance Comparison With Existing Tools
The performance comparison was done at TF and gene level using ground truth genes. For the TF level comparison, PropaNet was compared with the PCC, SCC, entropy and DREM (Schulz et al., 2012) methods, and PropaNet performed best for both cold and heat AtGenExpress datasets (Figure 4A). The precision, recall, and F1 scores of the TF level comparison were summarized in Supplementary Table 5.
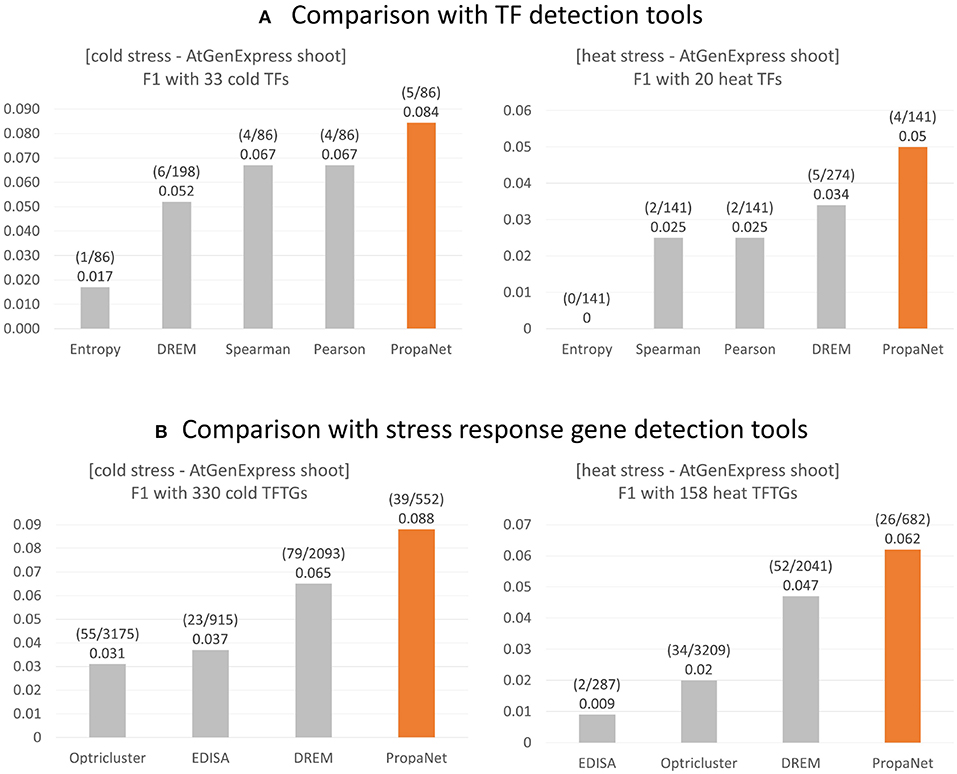
Figure 4. Performance comparison of stress response gene detection tools. (A) For major TF selection, PropaNet was compared with PCC, SCC, entropy, and DREM in terms of detecting how many ground truth TFs, TFs that were annotated by cold/heat stress GO terms. (B) For regulatory network construction, PropaNet was compared with EDISA, OPTricluster, DREM in terms of detecting how many ground truth stress response genes that were annotated by cold/heat stress GO terms. F1 score was used to measure the accuracy of tools.
For the gene level comparison, existing methods to determine stress-responsive genes from time-series data, EDISA (Supper et al., 2007), OPTricluster (Tchagang et al., 2012), and DREM (Schulz et al., 2012), were compared with PropaNet. PropaNet showed the best performances for both cold and heat stress datasets (Figure 4B). The precision, recall, and F1 scores of the gene level comparison were summarized in Supplementary Table 6.
4.3. Effects of Utilizing Non-stress Time-Series Sample as Control
PropaNetC is a modified version of PropaNet to utilize non-stress time-series control sample data. We performed analysis using PropaNet and PropaNetC on the AtGenExpress datasets and investigated how much the resulting networks were overlapped. About 60% of analysis results in cold stress and over 98% in heat stress were overlapped (Figures 5A,B). The GO enrichment analysis for cold stress showed that the cold stress-related GO term, “response to cold,” was enriched in analysis results from PropaNet and PropaNetC overlapping genes(p < 10−8). The GO term, “circadian rhythm,” was enriched only in the results from PropaNet-specific genes (p < 0.0012) (Figure 5C). In detail, circadian rhythm-related genes, such as LNK3, ERD7, WNK1, CCR2, and GI, were not enriched in the analysis result from PropaNetC. This observation suggests that the use of non-stress time-series control sample data could eliminate the effects of background biological mechanisms, e.g., circadian rhythm from the network analysis result.
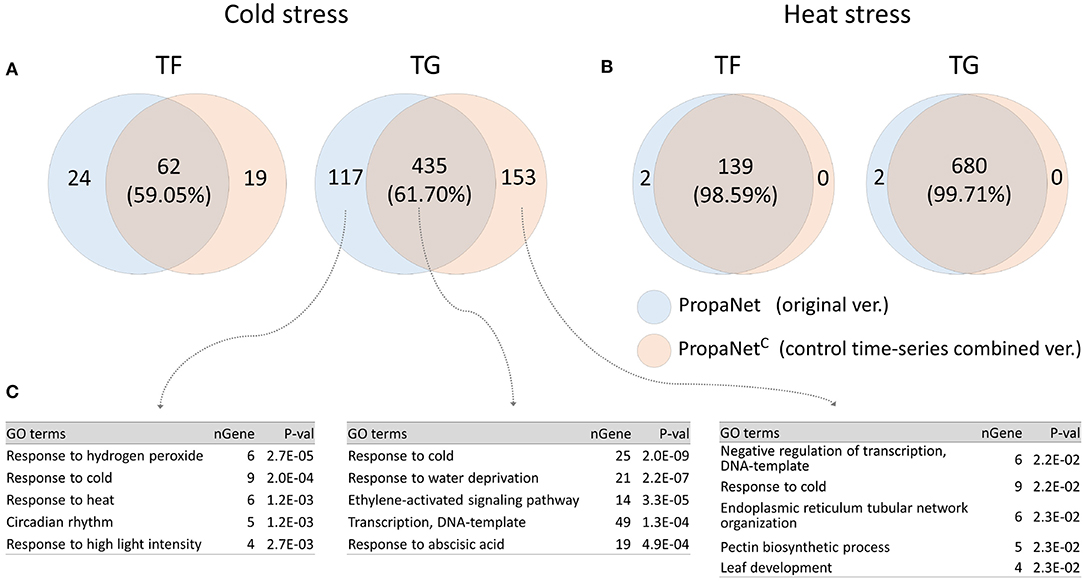
Figure 5. Effects of using non-stress time-series control samples. PropaNetC was a modified version that utilized non-stress time-series control data. The Venn diagram showed that PropaNet and PropaNetC produced about 60% overlapped results in cold stress (A) over 98% overlapped results in heat stress (B). (C) Enriched GO terms in target genes for cold stress showed that a list of circadian rhythm genes were excluded in the analysis result by PropaNetC (p < 10−8).
4.4. Effects of the Number of Time-Points
We performed PropaNet analysis on two datasets with different time points, AtGenExpress (7 and 5 time points for cold and heat stress) and E-MTAB-375 (22 time points). We then investigated the overlap between the resulting networks of adjacent time points. Genes in the networks of many (densely sampled) time points (E-MTAB-375 dataset) were overlapped more, 69% and 38% in average, between adjacent time points than the resulting genes of small (loosely sampled) time points (AtGenExpress dataset), 25% and 20% in average, for cold and heat stress, respectively (Figure 6). This result is reasonable and shows the possibility of estimating gene expression values at unobserved time points as shown in our previous work (Kang et al., 2019).
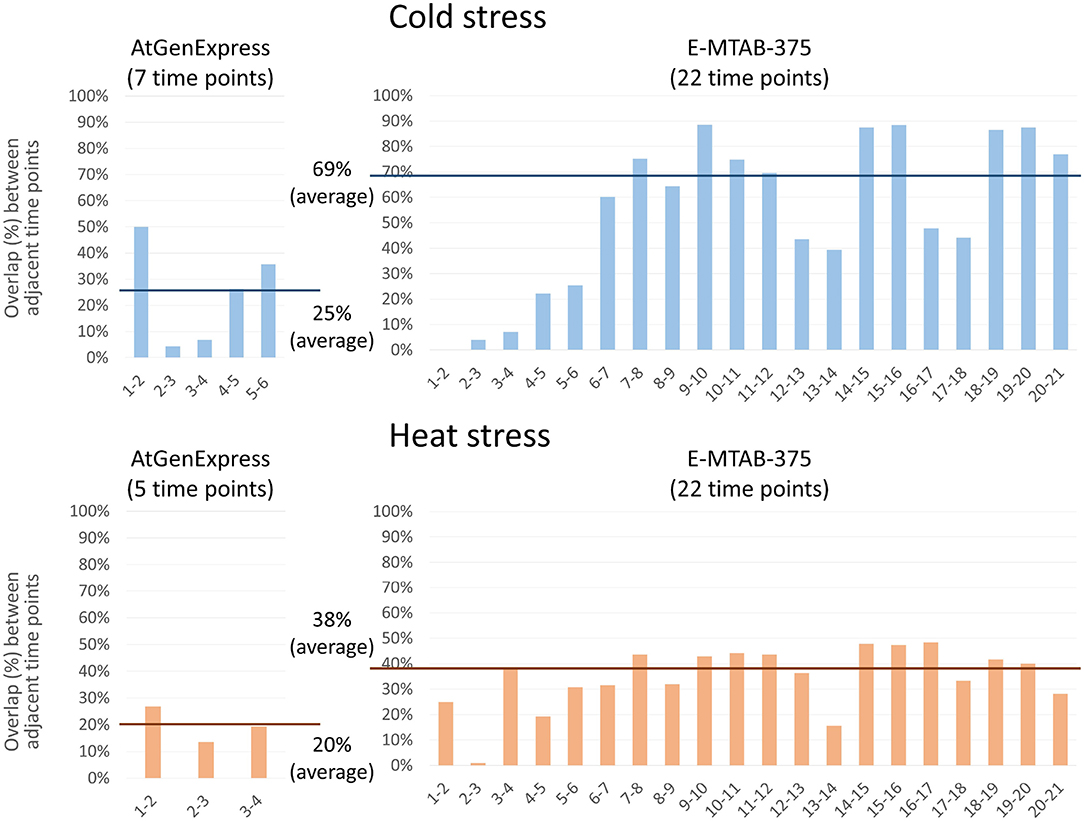
Figure 6. Effect of the number of time-points. PropaNet performed network construction analysis using AtGenExpress (nTimePoints = 7 and 5, for cold and heat stress) and E-MTAB-375 datasets (nTimePoints = 22, for both cold and heat stress). Then overlap between the results of adjacent time points were measured. The results of many time points (E-MTAB-375) showed more overlap between adjacent time points, 69% and 38% in average, than the results of small time points (AtGenExpress), 25% and 20% in average, for cold and heat stress, respectively.
5. Discussions
5.1. PropaNet Results and Stress Response Genes
The effects of temperature on plants are broad. The characteristics of effects of temperature on plant growth could be classified by imposed severity, duration, the ramp rates of changes, recovering condition and the developmental stages of the plant. Usually, the ambient temperature is not a stressful treatment. However, it may depend on the duration of exposure. The physiological consequences of sudden temperature treatment have been extensively studied. However, most of the experimental designs focused on experimentally available conditions and tissues such as leaves, roots, and fruits. Scrutinizing overall relationship of genes provides many of the unrevealed contexts of signal transfers and hierarchies among transcription factors.
In samples curated in AtGenExpress dataset (Kilian et al., 2007), the growth conditions of the pre-treated are different in terms of the treated periods. The plants grew under long-day conditions such as 16 h light and 8 h dark at a light intensity of 150 umol photons flux per square meter per second during the pre-treated stage. However, the photon flux was changed in the cold room to 60 umol in the same unit at steady state up to 24 h. It is our understanding that the circadian rhythm which maintained during the pre-treated stage might be interfered when the cold treatment started, and plants were exposed to the lower intensity light along with the cold temperature. Therefore, it is reasonable to accept that genes were affected by changes in light condition and circadian rhythm as well as cold stress. To remove the effect of diurnal cycling, light signaling and light dependent development, we compared the results of PropaNet with and without using non-stressed control samples. The major TFs described in the following sections are TFs that were detected even after removing the effect of diurnal cycle and light signaling.
5.1.1. Biological Implications of Major TFs Detected From Cold Stress Data
A first glance at the results of cold stress at T1 stage, the appearance of AMS (ABORTED MICROSPORES) gene is matching with the previous reviews of temperature sensing mechanism of plants (Ma et al., 2012). AMS like TF has edges to genes like CYP81D11, UGT78D2, and AT4G00040. CYP81D11 might function as a monooxygenase, and its expression pattern suggests that it is involved in plant detoxification processes (Köster et al., 2012). UGT78D2 glycosylates the hydroxyl group at C3 to form cyanidin3-O-glucosides and plays a role in quercetin and kaempferol glycosylation in Arabidopsis (Li et al., 2017). AT4G00040 encodes chalcone and stilbene synthase family protein and interacts with AMS that binds to the promoter region of the putative CHS gene (Xu et al., 2010). It is noteworthy that bZIP proteins such as bZIP60, AHBP-1B and an HDZip (homeodomain-leucine zipper) protein HB5, are detected in early stages of cold stress T1 and T2. These bZIPs are known to be related to the response to stress as follows. The expression of bZIP60 is upregulated by ER stress inducers (Zhang et al., 2017), and AHBP-1B is responsible to a pathogen (Sun et al., 2018). A HDzip protein HB5 is a positive regulator of ABA pathway (Perotti et al., 2017). In addition, RGA1 that is a GRAS family TF regulates one of the histone deacetylase complex subunits. It may be involved in the nucleosome stability and possibly has a function as a transcription regulator (Zheng et al., 2018a). Those genes are relatively short-lived but have functioned as the early regulators although we could not identify the successive link to the signal cascade.
At T3 stage, major TFs of the networks are bHLH proteins (PIF4, MYC2), AP2/ERF proteins (RAP2.12, DREB1A, DREB2A, DREB2B, ERF9), heat shock protein (HSFA2), HOS10 and ZFHD1. It is remarkable that PIF4 which is known as a thermosensing TF is detected in our analysis. The expression of PIF4 is known to be gradually decreased at night under the light/dark transition (Nusinow et al., 2011). However, PIF4 gene expression seems to maintain stable status without fluctuation up until 6h after cold stress started under the low light intensity and then tend to decrease slowly in AtGenExpress data. PIFs are known to interact with other bHLH proteins including MYC2 and light and thermosensing genes such as Cry1 and PhyB (Park et al., 2018a; Xu et al., 2018). Our analysis, however, detected other potential targets genes such as RLP31, CYCD1, TPPH and AT1G19000 and these relationships remained even after the varying diurnal expression trajectories of the light signaling components were subtracted. This result suggests other unknown functions of PIF4 and their target genes in stress responses besides light and thermosensing. APETALA2/Ethylene Responsive Factor (AP2/ERF) family TFs are related to biotic and abiotic stress regulation in plants (Zhang et al., 2018). Among the AP2/ERF proteins found by PropaNet, DREB1A was also detected as a cold stress-specific marker gene in terms of the transcript abundance and fold change (Wanke et al., 2010a). DREB2A is known as a key transcriptional activator that induces many heat- and drought-responsive genes in Arabidopsis (Mizoi et al., 2019). HSFA2 is a well-studied thermo-responsive gene. In our analysis, HSFA2 interacted with heat shock factors like GolS1, GolS2, HSP101, MBF1C, and ELIP1. When the control samples are considered, however, the network of HSFA2 and their target genes was detected only at the last time point, which suggests that these genes may react to light changes in control samples during the middle stages of cold stress, but the effect may be removed from the last time point network. HOS10 negatively regulates COR gene that acts downstream of the CBF proteins and might control ABA-mediated cold acclimation (Janmohammadi and Mahfoozi, 2013). ZFHD1 belongs to a family that binds to the promoter region of the early responsive to dehydration stress 1 (ERD1) gene and upregulates several stress-inducible genes (Elfving et al., 2011).
In the late stage of cold stress, MYB genes (MYB52, MYB70, MYB98, MYB59) act as central TFs of signaling cascades. MYB52 is known to be involved in ABA, drought, salt, and cold responses (Yu et al., 2016). MYB59 is shown to be involved in cell cycle regulation and presumed to control K-specific negative regulation of primary root elongation in Arabidopsis (Nishida et al., 2017). MYB59 targets several kinases and phosphatases, therefore it is reasonable to guess it could regulate such activities. MYB70 is reported that it negatively regulates genes related to developmental, hormonal and stress signaling pathways (Barah et al., 2015). In our analysis, MYB70 targets many of histone-related genes including histone H2A that takes the position of the variant H2A.Z. MYB98 is a specific transcription factor in synergid and regulates the expression of the female attractant LURE1 (Li et al., 2015). MYB98 targets TCX2 (Tesmin/ TSO1 like CXC 2) which is a metal binding protein. TCX2 is a member of seven homologs of TSO1 and shows highly similar expression patterns compared to TSO1 (Sijacic et al., 2011) which forms a TSO1-MYB3R1 module that regulates cell proliferation with differentiation and is involved in floral organ differentiation, meristem regulation, and gametophyte development. TSO1/LIN54 to MYB3R1/ B-MYB regulatory module in plants and animals, which plays a critical role in coordinating the cell cycle with the cell fate commitment (Sijacic et al., 2011; Wang et al., 2018).
As shown in the list of major TFs, a number of TFs belong to a few protein families such as bZIP, AP2/ERF, heat shock proteins and MYB families. The major TFs of the same family can be detected together because they have common target genes or they are interaction partners for each other composing a dimmers or multimers. The interactions between TFs in the latter case have also been detected in an existing network analysis of AtGenExpress dataset (Wanke et al., 2010b).
5.1.2. Biological Implications of Major TFs Detected From Heat Stress Data
In the first stage, our analysis revealed that many of AP2/ERF and hormone-related genes such as ERF2, ERF3, ERF38, RAP2.10, GATA1 are responsible to heat stress. ERFs form a representative TF family, AP2/ERF, that plays a role in development and stress response process of plants (Vogel et al., 2014). GATA is a transcription regulator, subdivided into four classes, and they are known to be involved in the control of greening, plant development, GA metabolism, and a lot of biological processes (Behringer and Schwechheimer, 2015). Contrast with cold stress, during the high-temperature treatment, WRKY57 is the TF at the initial response even though AMS like TF also is detected in our analysis at the first stage of temperature response. Unlike with cold stress, most plants grow well under higher ambient temperature. Higher ambient temperature is accelerating growth and differentiation. In some case, some organs in G0 stage of cell cycle turn on cell division process, resulting in the additional growth and re-differentiation.
In the following stages, HSFA2, MYB28 and ZAT6 are detected as major TFs. HSFA2 is detected as a major TF in both cold and heat stress data analysis. However, the target genes of HSFA2 are different under cold and heat stress. During the heat treatment, HSFA2 shows a relationship with partly common (HSP101, MBF1C, ELIP1, GolS1, and GolS2) and different sets (SIP2 and APX2) of gene comparing with cold treatment. SIP2 is presumed to be involved in phloem unloading of raffinose in sink leaves. Raffinose and other members of the raffinose family oligosaccharides are involved in stress tolerance and act as antioxidants (Clauw et al., 2016). APX2 encodes cytosolic ROS scavenging enzyme and is shown to be activated in response to heat stress in an HSFA3-dependent manner (Katano et al., 2018). In addition, an early study of AtGenExpress dataset found HSFA2 as a heat stress-specific marker gene by its transcript abundance and fold change (Wanke et al., 2010a). MYB28 is a remarkable major TF which interacts with CYP83A1, CYP83B1, MTO1, and SUR1. The CYP family is made up of a wide variety of monooxygenases containing a prosthetic heme group, and CYP83A1 is required for aliphatic glucosinolate biosynthesis (Nagano et al., 2014). CYP83B1 catalyzes the first committed step in the indole glucosinolate sub-pathway, converting indole-3-acetaldoxime to an S-alkyl-thiohydroximate adduct (Maharjan et al., 2014). MTO1 and CGS1 construct the carbon/amino skeleton derived from Asp with the sulfur moiety donated by Cys (Hacham et al., 2017). SUR1 encodes the C-S lyase that functions in indole glucosinolates biosynthesis. SUR1 is important to IAOx-dependent IAA synthesis (Kong et al., 2014). ZAT6, zinc finger of Arabidopsis thaliana 6, was also found as one of the plant core environmental stress response genes of Arabidopsis responsive to diverse stress conditions, by its dynamic gene expression trajectory (Hahn et al., 2013).
5.1.3. Dynamic Changes of Major TFs and Their Functions in Cold and Heat Stress
We observed dynamic changes of TF-TG grouping and their biological functions in the time course analysis of cold and heat stress data as shown in Figures 2, 3, respectively. Under the cold condition, we can see only a few TF genes that had a signal at the initial T1 and T2 stages. Major grouping occurs in T3 stage, which suggests the defense system related stress may be turned on at this stage, showing AP2/ERF proteins (RAP2.12, DREB2A, DREB2B, ERF9) as major TFs in our analysis. The TF-TG group of T3 stage propagates to the outside of the network in the following T4, T5 and T6 stages to regulate cell and organ restructuring against hazardous effects of cold temperature. About 5–6 TF-TG groups in the later time points have such major TFs like MYB52, 59, 70, 98, and TCX2. It seems that TF-TG network changes in order to prepare for whole-plant-wide harmful cold stress. These changes are focusing on osmotic defense mechanism and structural reforming. However, under the higher temperature condition, the initial responses of the plant are merged into hormone metabolism. We could not observe that critical changes during T1 and T2 stages. In our analysis, as strong growth control compounds, such as gibberellin, ethylene and auxin-related TFs like AP2/ERF and GATA, were detected in early stages. This growth and reforming stages turn to defensive stages in T3 and T4. Many of the stress-related TFs like MYB, TCX2, and ZAT6 were observed as major TFs in T3 and T4 stages under heat stress.
5.2. Advantages and Limitations of PropaNet
Comparative analysis results showed that PropaNet detected known stress-responsive genes more accurately than other time series analysis methods (see section 4.2). The advanced performance of PropaNet can be explained with its three characteristics distinguished from other methods. First, PropaNet considers indirect regulatory power of a TF though consideration of multiple steps of transcription regulation (by influence maximization) while existing tools consider direct targets only. Second, PropaNet takes into account of the regulatory power of multiple TFs simultaneously (by network propagation). Third, PropaNet uses “differential expression” values (such as z-scores or log2 fold changes) to prioritize TFs that target more significant DEGs.
We conjecture that the consideration of multiple steps of regulation of a TF and simultaneous regulations of multiple TFs is the main reason why PropaNet performed better than existing tools in terms of F1 scores. Additionally, the use of PropaNetC that considers non-treated control samples in PropaNet analysis showed to reduce the effect of background biological mechanisms (e.g., circadian rhythm) on time-varying networks (see section 4.3). Therefore, it is recommended to use PropaNetC to eliminate the effects of background biological mechanisms when both treated case and non-treated control samples are available.
On the other hand, there are a few limitations of PropaNet that have to be considered before analysis. PropaNet assumes the reliability of a template TF network that is given by the user as input. In our experiment, we used PlantRegMap as a template network, which was generated by identifying TF binding sites using TF ChIP-seq and searching the DNA sequence motif on the promoter regions of the target genes. Then, it appended TF-target interactions that were found in the literature. Thus, it has the possibility to include false TF-target interactions; (1) The antibodies that are used in TF ChIP experiments are known to have the range of affinity to bind the un-targeted but similar-structured TFs. (2) The quality of ChIP-seq data can vary depending on the experimenters and the year of data generation. (3) TF ChIP-seq experiments are conducted in a particular condition, so it is possible that the TF interactions change in a different condition. Use of extended versions of PlantRegMap or protein-protein interaction networks may provide more detailed information on the plant response to cold and heat stress, which has to be studied in the future. In addition, PropaNet detects TFs of which differential expressions are positively correlated with DEGs, which cannot capture inhibitory relationship between TF and target genes. This is due to the limitation of random walk process that might not converge with negative weights. Thus, incorporating negative weights to detect inhibitory TFs can be a future work for PropaNet as well.
Data Availability
All datasets for this study are included in the manuscript and the Supplementary Files.
Author Contributions
SK and WJ designed the project. SK, KJ, and HA directed the development of the algorithm. SK and KJ developed a modified version of influence maximization algorithm. HA collected and processed the data. KJ, DJ, MP, and HA implemented the program of the algorithm and conducted experiments of network construction. WJ and JH biologically interpreted the network analysis results. HA, KJ, SK, and WJ wrote the paper.
Funding
This work was supported by National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT (No. NRF-2017M3C4A7065887) and the Collaborative Genome Program for Fostering New Post-Genome Industry of the National Research Foundation (NRF) funded by the Ministry of Science and ICT (MSIT) (No. NRF-2014M3C9A3063541). This work was supported for WJ by the Agenda program (No. PJ0143072019), Rural Development of Administration of Republic of Korea.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00698/full#supplementary-material
References
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Barah, P., B N, M. N., Jayavelu, N. D., Sowdhamini, R., Shameer, K., and Bones, A. M. (2015). Transcriptional regulatory networks in Arabidopsis thaliana during single and combined stresses. Nucl. Acids Res. 44, 3147–3164. doi: 10.1093/nar/gkv1463
Barrett, T., Wilhite, S. E., Ledoux, P., Evangelista, C., Kim, I. F., Tomashevsky, M., et al. (2013). NCBI GEO: archive for functional genomics data sets–update. Nucl. Acids Res. 41, D991–D995. doi: 10.1093/nar/gks1193
Barski, A., Cuddapah, S., Cui, K., Roh, T.-Y., Schones, D. E., Wang, Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. doi: 10.1016/j.cell.2007.05.009
Behringer, C., and Schwechheimer, C. (2015). B-GATA transcription factors–insights into their structure, regulation, and role in plant development. Front. Plant Sci. 6:90. doi: 10.3389/fpls.2015.00090
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.). 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bonneau, R., Reiss, D. J., Shannon, P., Facciotti, M., Hood, L., Baliga, N. S., et al. (2006). The Inferelator: an algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 7:R36. doi: 10.1186/gb-2006-7-5-r36
Bovolenta, L. A., Acencio, M. L., and Lemke, N. (2012). HTRIdb: an open-access database for experimentally verified human transcriptional regulation interactions. BMC Genomics 13:405. doi: 10.1186/1471-2164-13-405
Brutlag, D., Schlehuber, C., and Bonner, J. (1969). Properties of formaldehyde-treated nucleohistone. Biochemistry 8, 3214–3218. doi: 10.1021/bi00836a013
Busch, W., Wunderlich, M., and Schöffl, F. (2005). Identification of novel heat shock factor-dependent genes and biochemical pathways in Arabidopsis thaliana. Plant J. 41, 1–14. doi: 10.1111/j.1365-313X.2004.02272.x
Caldana, C., Degenkolbe, T., Cuadros-Inostroza, A., Klie, S., Sulpice, R., Leisse, A., et al. (2011). High-density kinetic analysis of the metabolomic and transcriptomic response of Arabidopsis to eight environmental conditions. Plant J. 67, 869–884. doi: 10.1111/j.1365-313X.2011.04640.x
Charng, Y. Y., Liu, H. C., Liu, N. Y., Chi, W. T., Wang, C. N., Chang, S. H., et al. (2007). A heat-inducible transcription factor, HsfA2, is required for extension of acquired thermotolerance in Arabidopsis. Plant Physiol. 143, 251–262. doi: 10.1104/pp.106.091322
Chen, W., Provart, N. J., Glazebrook, J., Katagiri, F., Chang, H.-S., Eulgem, T., et al. (2002). Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell 14, 559–574. doi: 10.1105/tpc.010410
Chung, S., and Parish, R. W. (2008). Combinatorial interactions of multiple cis-elements regulating the induction of the Arabidopsis XERO2 dehydrin gene by abscisic acid and cold. Plant J. 54, 15–29. doi: 10.1111/j.1365-313X.2007.03399.x
Clauw, P., Coppens, F., Korte, A., Herman, D., Slabbinck, B., Dhondt, S., et al. (2016). Leaf growth response to mild drought: natural variation in Arabidopsis sheds light on trait architecture. Plant Cell 28, 2417–2434. doi: 10.1105/tpc.16.00483
Consortium, G. O. (2018). The gene ontology resource: 20 years and still GOing strong. Nucl. Acids Res. 47, D330–D338. doi: 10.1093/nar/gky1055
Cowen, L., Ideker, T., Raphael, B. J., and Sharan, R. (2017). Network propagation: a universal amplifier of genetic associations. Nat. Rev. Genet. 18:551. doi: 10.1038/nrg.2017.38
Dai, M., Wang, P., Boyd, A. D., Kostov, G., Athey, B., Jones, E. G., et al. (2005). Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucl. Acids Res. 33:e175. doi: 10.1093/nar/gni179
Ehlting, J., Chowrira, S. G., Mattheus, N., Aeschliman, D. S., Arimura, G.-I., and Bohlmann, J. (2008). Comparative transcriptome analysis of arabidopsis thaliana infested by diamond back moth (plutella xylostella) larvae reveals signatures of stress response, secondary metabolism, and signalling. BMC Genomics 9:154. doi: 10.1186/1471-2164-9-154
Elfving, N., Davoine, C., Benlloch, R., Blomberg, J., Brännström, K., Müller, D., et al. (2011). The Arabidopsis thaliana Med25 mediator subunit integrates environmental cues to control plant development. Proc. Natl. Acad. Sci. U.S.A. 108, 8245–8250. doi: 10.1073/pnas.1002981108
Eremina, M., Rozhon, W., and Poppenberger, B. (2016). Hormonal control of cold stress responses in plants. Cell. Mol. Life Sci. 73, 797–810. doi: 10.1007/s00018-015-2089-6
Essaghir, A., Toffalini, F., Knoops, L., Kallin, A., van Helden, J., and Demoulin, J.-B. (2010). Transcription factor regulation can be accurately predicted from the presence of target gene signatures in microarray gene expression data. Nucl. Acids Res. 38:e120. doi: 10.1093/nar/gkq149
Fullwood, M. J., and Ruan, Y. (2009). ChIP-based methods for the identification of long-range chromatin interactions. J. Cell. Biochem. 107, 30–39. doi: 10.1002/jcb.22116
Hacham, Y., Matityahu, I., and Amir, R. (2017). Transgenic tobacco plants having a higher level of methionine are more sensitive to oxidative stress. Physiol. Plant. 160, 242–252. doi: 10.1111/ppl.12557
Hahn, A., Kilian, J., Mohrholz, A., Ladwig, F., Peschke, F., Dautel, R., et al. (2013). Plant core environmental stress response genes are systemically coordinated during abiotic stresses. Int. J. Mol. Sci. 14, 7617–7641. doi: 10.3390/ijms14047617
Han, H., Cho, J.-W., Lee, S., Yun, A., Kim, H., Bae, D., et al. (2017). TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucl. Acids Res. 46, D380–D386. doi: 10.1093/nar/gkx1013
Hoffman, E. A., Frey, B. L., Smith, L. M., and Auble, D. T. (2015). Formaldehyde crosslinking: a tool for the study of chromatin complexes. J. Biol. Chem. 290, 26404–26411. doi: 10.1074/jbc.R115.651679
Hu, H., Miao, Y. R., Jia, L. H., Yu, Q. Y., Zhang, Q., and Guo, A. Y. (2018). AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucl. Acids Res. 47, D33–D38. doi: 10.1093/nar/gky822
Huynh-Thu, V. A., and Geurts, P. (2018). dynGENIE3: dynamical GENIE3 for the inference of gene networks from time series expression data. Sci. Rep. 8:3384. doi: 10.1038/s41598-018-21715-0
Huynh-Thu, V. A., Irrthum, A., Wehenkel, L., and Geurts, P. (2010). Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 5:e12776. doi: 10.1371/journal.pone.0012776
Iglesias-Martinez, L. F., Kolch, W., and Santra, T. (2016). BGRMI: a method for inferring gene regulatory networks from time-course gene expression data and its application in breast cancer research. Sci. Rep. 6:37140. doi: 10.1038/srep37140
IPCC (2018). “2018: summary for policymakers,” in Global Warming of 1.5°C. An IPCC Special Report on the impacts of global warming of 1.5°C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty, eds M.-D. Valérie, P. Zhai, H.-O. Pörtner, D. C. Roberts, J. Skea, P. R. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan, R. Pidcock, S. Connors, J. R. Matthews, Y. Chen, X. Zhou, M. Gomis, E. Lonnoy, M. Maycock, and T. Waterfield (Geneva: World Meteorological Organization), 32.
Janmohammadi, M., and Mahfoozi, S. (2013). Regulatory network of gene expression during the development of frost tolerance in plants. Curr. Opin. Agricult. 2, 11–19. Retrieved from: http://cuopag.com
Jeong, J. S., Kim, Y. S., Baek, K. H., Jung, H., Ha, S. H., Do Choi, Y., et al. (2010). Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice under field drought conditions. Plant Physiol. 153, 185–197. doi: 10.1104/pp.110.154773
Jin, J., He, K., Tang, X., Li, Z., Lv, L., Zhao, Y., et al. (2015). An Arabidopsis transcriptional regulatory map reveals distinct functional and evolutionary features of novel transcription factors. Mol. Biol. Evol. 32, 1767–1773. doi: 10.1093/molbev/msv058
Jin, J., Tian, F., Yang, D. C., Meng, Y. Q., Kong, L., Luo, J., et al. (2016). PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucl. Acids Res. 45, D1040–D1045. doi: 10.1093/nar/gkw982
Jo, K., Jung, I., Moon, J. H., and Kim, S. (2016). Influence maximization in time bounded network identifies transcription factors regulating perturbed pathways. Bioinformatics 32, i128–i136. doi: 10.1093/bioinformatics/btw275
Johnson, D. S., Mortazavi, A., Myers, R. M., and Wold, B. (2007). Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502. doi: 10.1126/science.1141319
Joo, J., Lee, Y. H., and Song, S. I. (2014). Overexpression of the rice basic leucine zipper transcription factor OsbZIP12 confers drought tolerance to rice and makes seedlings hypersensitive to ABA. Plant Biotechnol. Rep. 8, 431–441. doi: 10.1007/s11816-014-0335-2
Jung, I., Jo, K., Kang, H., Ahn, H., Yu, Y., and Kim, S. (2017). TimesVector: a vectorized clustering approach to the analysis of time series transcriptome data from multiple phenotypes. Bioinformatics 33, 3827–3835. doi: 10.1093/bioinformatics/btw780
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kang, H., Ahn, H., Jo, K., Oh, M., and Kim, S. (2019). mirTime: identifying condition-specific targets of MicroRNA in time-series transcript data using Gaussian process model and spherical vector clustering. Bioinformatics. doi: 10.1093/bioinformatics/btz306. [Epub ahead of print].
Karaba, A., Dixit, S., Greco, R., Aharoni, A., Trijatmiko, K. R., Marsch-Martinez, N., et al. (2007). Improvement of water use efficiency in rice by expression of HARDY, an Arabidopsis drought and salt tolerance gene. Proc. Natl. Acad. Sci. U.S.A. 104, 15270–15275. doi: 10.1073/pnas.0707294104
Katano, K., Kataoka, R., Fujii, M., and Suzuki, N. (2018). Differences between seedlings and flowers in anti-ROS based heat responses of Arabidopsis plants deficient in cyclic nucleotide gated channel 2. Plant Physiol. Biochem. 123, 288–296. doi: 10.1016/j.plaphy.2017.12.021
Khan, A., Fornes, O., Stigliani, A., Gheorghe, M., Castro-Mondragon, J. A., van der Lee, R., et al. (2017). JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucl. Acids Res. 46, D260–D266. doi: 10.1093/nar/gkx1126
Kilian, J., Whitehead, D., Horak, J., Wanke, D., Weinl, S., Batistic, O., et al. (2007). The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 50, 347–363. doi: 10.1111/j.1365-313X.2007.03052.x
Kodama, Y., Shumway, M., and Leinonen, R. (2011). The sequence read archive: explosive growth of sequencing data. Nucl. Acids Res. 40, D54–D56. doi: 10.1093/nar/gkr854
Kolesnikov, N., Hastings, E., Keays, M., Melnichuk, O., Tang, Y. A., Williams, E., et al. (2014). ArrayExpress update-simplifying data submissions. Nucl. Acids Res. 43, D1113–D1116. doi: 10.1093/nar/gku1057
Kong, W., Li, Y., Zhang, M., Jin, F., and Li, J. (2014). A novel Arabidopsis microRNA promotes IAA biosynthesis via the indole-3-acetaldoxime pathway by suppressing SUPERROOT1. Plant Cell Physiol. 56, 715–726. doi: 10.1093/pcp/pcu216
Köster, J., Thurow, C., Kruse, K., Meier, A., Iven, T., Feussner, I., et al. (2012). Xenobiotic- and jasmonic acid-inducible signal transduction pathways have become interdependent at the Arabidopsis CYP81D11 promoter. Plant Physiol. 159, 391–402. doi: 10.1104/pp.112.194274
Kotak, S., Vierling, E., Bäumlein, H., and von Koskull-Döring, P. (2007). A novel transcriptional cascade regulating expression of heat stress proteins during seed development of Arabidopsis. Plant Cell 19, 182–195. doi: 10.1105/tpc.106.048165
Lamesch, P., Berardini, T. Z., Li, D., Swarbreck, D., Wilks, C., Sasidharan, R., et al. (2012). The Arabidopsis information resource (TAIR): improved gene annotation and new tools. Nucl. Acids Res. 40, D1202–D1210. doi: 10.1093/nar/gkr1090
Landt, S. G., Marinov, G. K., Kundaje, A., Kheradpour, P., Pauli, F., Batzoglou, S., et al. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831. doi: 10.1101/gr.136184.111
Larkindale, J., and Vierling, E. (2008). Core genome responses involved in acclimation to high temperature. Plant Physiol. 146, 748–761. doi: 10.1104/pp.107.112060
Latchman, D. S. (1997). Transcription factors: an overview. Int. J. Biochem. Cell Biol. 29, 1305–1312. doi: 10.1016/S1357-2725(97)00085-X
Lee, T., Yang, S., Kim, E., Ko, Y., Hwang, S., Shin, J., et al. (2014). AraNet v2: an improved database of co-functional gene networks for the study of Arabidopsis thaliana and 27 other nonmodel plant species. Nucl. Acids Res. 43, D996–D1002. doi: 10.1093/nar/gku1053
Lefebvre, C., Rajbhandari, P., Alvarez, M. J., Bandaru, P., Lim, W. K., Sato, M., et al. (2010). A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol. Syst. Biol. 6:377. doi: 10.1038/msb.2010.31
Li, F. H., Li, C. T., and Shan, M. K. (2011). “Labeled influence maximization in social networks for target marketing,” in 2011 IEEE Third International Conference on Privacy, Security, Risk and Trust and 2011 IEEE Third International Conference on Social Computing (Boston, MA: IEEE), 560–563.
Li, H. J., Zhu, S. S., Zhang, M. X., Wang, T., Liang, L., Xue, Y., et al. (2015). Arabidopsis CBP1 is a novel regulator of transcription initiation in central cell-mediated pollen tube guidance. Plant Cell 27, 2880–2893. doi: 10.1105/tpc.15.00370
Li, P., Li, Y.-J., Zhang, F.-J., Zhang, G.-Z., Jiang, X.-Y., Yu, H.-M., et al. (2017). The Arabidopsis UDP-glycosyltransferases UGT79B2 and UGT79B3, contribute to cold, salt and drought stress tolerance via modulating anthocyanin accumulation. Plant J. 89, 85–103. doi: 10.1111/tpj.13324
Li, S., Zhou, X., Chen, L., Huang, W., and Yu, D. (2010). Functional characterization of Arabidopsis thaliana WRKY39 in heat stress. Mol. Cells 29, 475–483. doi: 10.1007/s10059-010-0059-2
Li, Y., Fan, J., Wang, Y., and Tan, K.-L. (2018). Influence maximization on social graphs: a survey. IEEE Trans. Knowl. Data Eng. 30, 1852–1872. doi: 10.1109/TKDE.2018.2807843
Lobe, C. G. (1992). “9 transcription factors and mammalian development,” in Current Topics in Developmental Biology, Vol. 27, ed R. A. Pedersen (New York, NY: Elsevier), 351–383. doi: 10.1016/S0070-2153(08)60539-6
Ma, X., Feng, B., and Ma, H. (2012). AMS-dependent and independent regulation of anther transcriptome and comparison with those affected by other Arabidopsis anther genes. BMC Plant Biol. 12:23. doi: 10.1186/1471-2229-12-23
Maharjan, P. M., Dilkes, B. P., Fujioka, S., Pěnčík, A., Ljung, K., Burow, M., et al. (2014). Arabidopsis gulliver1/superroot2-7 identifies a metabolic basis for auxin and brassinosteroid synergy. Plant J. 80, 797–808. doi: 10.1111/tpj.12678
Marbach, D., Costello, J. C., Küffner, R., Vega, N. M., Prill, R. J., Camacho, D. M., et al. (2012). Wisdom of crowds for robust gene network inference. Nat. Methods 9:796. doi: 10.1038/nmeth.2016
Margolin, A. A., Nemenman, I., Basso, K., Wiggins, C., Stolovitzky, G., Dalla Favera, R., et al. (2006). ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7:S7. doi: 10.1186/1471-2105-7-S1-S7
Matys, V., Kel-Margoulis, O. V., Fricke, E., Liebich, I., Land, S., Barre-Dirrie, A., et al. (2006). TRANSFAC® and its module TRANSCompel®: transcriptional gene regulation in eukaryotes. Nucl. Acids Res. 34(Suppl. 1):D108–D110. doi: 10.1093/nar/gkj143
Medina, J., Catalá, R., and Salinas, J. (2011). The CBFs: three Arabidopsis transcription factors to cold acclimate. Plant Sci. 180, 3–11. doi: 10.1016/j.plantsci.2010.06.019
Meyyappan, M., Atadja, P. W., and Riabowol, K. T. (1996). Regulation of gene expression and transcription factor binding activity during cellular aging. Neurosignals 5, 130–138. doi: 10.1159/000109183
Mikkelsen, T. S., Ku, M., Jaffe, D. B., Issac, B., Lieberman, E., Giannoukos, G., et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553. doi: 10.1038/nature06008
Mizoi, J., Kanazawa, N., Kidokoro, S., Takahashi, F., Qin, F., Morimoto, K., et al. (2019). Heat-induced inhibition of phosphorylation of the stress-protective transcription factor DREB2A promotes thermotolerance of Arabidopsis thaliana. J. Biol. Chem. 294, 902–917. doi: 10.1074/jbc.RA118.002662
Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2012). AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 1819, 86–96. doi: 10.1016/j.bbagrm.2011.08.004
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621. doi: 10.1038/nmeth.1226
Nagano, M., Ishikawa, T., Ogawa, Y., Iwabuchi, M., Nakasone, A., Shimamoto, K., et al. (2014). Arabidopsis Bax inhibitor-1 promotes sphingolipid synthesis during cold stress by interacting with ceramide-modifying enzymes. Planta 240, 77–89. doi: 10.1007/s00425-014-2065-7
Nishida, S., Kakei, Y., Shimada, Y., and Fujiwara, T. (2017). Genome-wide analysis of specific alterations in transcript structure and accumulation caused by nutrient deficiencies in Arabidopsis thaliana. Plant J. 91, 741–753. doi: 10.1111/tpj.13606
Nishizawa, A., Yabuta, Y., Yoshida, E., Maruta, T., Yoshimura, K., and Shigeoka, S. (2006). Arabidopsis heat shock transcription factor A2 as a key regulator in response to several types of environmental stress. Plant J. 48, 535–547. doi: 10.1111/j.1365-313X.2006.02889.x
Nusinow, D. A., Helfer, A., Hamilton, E. E., King, J. J., Imaizumi, T., Schultz, T. F., et al. (2011). The ELF4–ELF3–LUX complex links the circadian clock to diurnal control of hypocotyl growth. Nature 475:398. doi: 10.1038/nature10182
Obayashi, T., Aoki, Y., Tadaka, S., Kagaya, Y., and Kinoshita, K. (2017). ATTED-II in 2018: a plant coexpression database based on investigation of the statistical property of the mutual rank index. Plant Cell Physiol. 59:e3. doi: 10.1093/pcp/pcx191
Oh, S. J., Kim, Y. S., Kwon, C. W., Park, H. K., Jeong, J. S., and Kim, J. K. (2009). Overexpression of the transcription factor AP37 in rice improves grain yield under drought conditions. Plant Physiol. 150, 1368–1379. doi: 10.1104/pp.109.137554
Oki, S., Ohta, T., Shioi, G., Hatanaka, H., Ogasawara, O., Okuda, Y., et al. (2018). ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 19:e46255. doi: 10.15252/embr.201846255
Omranian, N., Eloundou-Mbebi, J. M., Mueller-Roeber, B., and Nikoloski, Z. (2016). Gene regulatory network inference using fused LASSO on multiple data sets. Sci. Rep. 6:20533. doi: 10.1038/srep20533
Orlando, V. (2000). Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem. Sci. 25, 99–104. doi: 10.1016/S0968-0004(99)01535-2
Park, E., Kim, Y., and Choi, G. (2018a). Phytochrome b requires pif degradation and sequestration to induce light responses across a wide range of light conditions. Plant Cell 30, 1277–1292. doi: 10.1105/tpc.17.00913
Park, S., Kim, J. M., Shin, W., Han, S. W., Jeon, M., Jang, H. J., et al. (2018b). BTNET: boosted tree based gene regulatory network inference algorithm using time-course measurement data. BMC Syst. Biol. 12:20. doi: 10.1186/s12918-018-0547-0
Pawson, T. (1993). Signal transduction-a conserved pathway from the membrane to the nucleus. Dev. Genet. 14, 333–338. doi: 10.1002/dvg.1020140502
Perotti, M. F., Ribone, P. A., and Chan, R. L. (2017). Plant transcription factors from the homeodomain-leucine zipper family I. Role in development and stress responses. IUBMB Life 69, 280–289. doi: 10.1002/iub.1619
Portales-Casamar, E., Kirov, S., Lim, J., Lithwick, S., Swanson, M. I., Ticoll, A., et al. (2007). PAZAR: a framework for collection and dissemination of cis-regulatory sequence annotation. Genome Biol. 8:R207. doi: 10.1186/gb-2007-8-10-r207
Redillas, M. C., Jeong, J. S., Kim, Y. S., Jung, H., Bang, S. W., Choi, Y. D., et al. (2012). The overexpression of OsNAC9 alters the root architecture of rice plants enhancing drought resistance and grain yield under field conditions. Plant Biotechnol. J. 10, 792–805. doi: 10.1111/j.1467-7652.2012.00697.x
Ren, B., Robert, F., Wyrick, J. J., Aparicio, O., Jennings, E. G., Simon, I., et al. (2000). Genome-wide location and function of DNA binding proteins. Science 290, 2306–2309. doi: 10.1126/science.290.5500.2306
Rhee, H. S., and Pugh, B. F. (2011). Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147, 1408–1419. doi: 10.1016/j.cell.2011.11.013
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl. Acids Res. 43:e47. doi: 10.1093/nar/gkv007
Sakuma, Y., Maruyama, K., Qin, F., Osakabe, Y., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2006). Dual function of an Arabidopsis transcription factor DREB2A in water-stress-responsive and heat-stress-responsive gene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 18822–18827. doi: 10.1073/pnas.0605639103
Schena, M., Shalon, D., Davis, R. W., and Brown, P. O. (1995). Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270, 467–470. doi: 10.1126/science.270.5235.467
Schramm, F., Ganguli, A., Kiehlmann, E., Englich, G., Walch, D., and von Koskull-Döring, P. (2006). The heat stress transcription factor HsfA2 serves as a regulatory amplifier of a subset of genes in the heat stress response in Arabidopsis. Plant Mol. Biol. 60, 759–772. doi: 10.1007/s11103-005-5750-x
Schulz, M. H., Devanny, W. E., Gitter, A., Zhong, S., Ernst, J., and Bar-Joseph, Z. (2012). DREM 2.0: improved reconstruction of dynamic regulatory networks from time-series expression data. BMC Syst. Biol. 6:104. doi: 10.1186/1752-0509-6-104
Seabold, S., and Perktold, J. (2010). “Statsmodels: econometric and statistical modeling with python,” in 9th Python in Science Conference (Austin), 57–61.
Seki, M., Narusaka, M., Ishida, J., Nanjo, T., Fujita, M., Oono, Y., et al. (2002). Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 31, 279–292. doi: 10.1046/j.1365-313X.2002.01359.x
Seo, P. J., Lee, S. B., Suh, M. C., Park, M. J., Go, Y. S., and Park, C. M. (2011). The MYB96 transcription factor regulates cuticular wax biosynthesis under drought conditions in Arabidopsis. Plant Cell 23, 1138–1152. doi: 10.1105/tpc.111.083485
Sijacic, P., Wang, W., and Liu, Z. (2011). Recessive antimorphic alleles overcome functionally redundant loci to reveal TSO1 function in Arabidopsis flowers and meristems. PLoS Genet. 7:e1002352. doi: 10.1371/journal.pgen.1002352
Sławek, J., and Arodź, T. (2013). ENNET: inferring large gene regulatory networks from expression data using gradient boosting. BMC Syst. Biol. 7:106. doi: 10.1186/1752-0509-7-106
Solomon, M. J., and Varshavsky, A. (1985). Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures. Proc. Natl. Acad. Sci. U.S.A. 82:6470. doi: 10.1073/pnas.82.19.6470
Sun, T., Busta, L., Zhang, Q., Ding, P., Jetter, R., and Zhang, Y. (2018). TGACG-BINDING FACTOR 1 (TGA1) and TGA4 regulate salicylic acid and pipecolic acid biosynthesis by modulating the expression of SYSTEMIC ACQUIRED RESISTANCE DEFICIENT 1 (SARD1) and CALMODULIN-BINDING PROTEIN 60g (CBP60g). New Phytol. 217, 344–354. doi: 10.1111/nph.14780
Supper, J., Strauch, M., Wanke, D., Harter, K., and Zell, A. (2007). EDISA: extracting biclusters from multiple time-series of gene expression profiles. BMC Bioinformatics 8:334. doi: 10.1186/1471-2105-8-334
Swindell, W. R., Huebner, M., and Weber, A. P. (2007). Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genomics 8:125. doi: 10.1186/1471-2164-8-125
Tchagang, A. B., Phan, S., Famili, F., Shearer, H., Fobert, P., Huang, Y., et al. (2012). Mining biological information from 3D short time-series gene expression data: the OPTricluster algorithm. BMC Bioinformatics 13:54. doi: 10.1186/1471-2105-13-54
Thibaud-Nissen, F., Wu, H., Richmond, T., Redman, J. C., Johnson, C., Green, R., et al. (2006). Development of Arabidopsis whole-genome microarrays and their application to the discovery of binding sites for the TGA2 transcription factor in salicylic acid-treated plants. Plant J. 47, 152–162. doi: 10.1111/j.1365-313X.2006.02770.x
Vogel, M. O., Moore, M., König, K., Pecher, P., Alsharafa, K., Lee, J., et al. (2014). Fast retrograde signaling in response to high light involves metabolite export, MITOGEN-ACTIVATED PROTEIN KINASE6, and AP2/ERF transcription factors in Arabidopsis. Plant Cell 26, 1151–1165. doi: 10.1105/tpc.113.121061
Wagner, F. (2016). pyAffy: an efficient Python/Cython implementation of the RMA method for processing raw data from Affymetrix expression microarrays. PeerJ Preprints 4:e1790v1. doi: 10.7287/peerj.preprints.1790v1
Wang, J., Zhuang, J., Iyer, S., Lin, X., Whitfield, T. W., Greven, M. C., et al. (2012). Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22, 1798–1812. doi: 10.1101/gr.139105.112
Wang, W., Sijacic, P., Xu, P., Lian, H., and Liu, Z. (2018). Arabidopsis TSO1 and MYB3R1 form a regulatory module to coordinate cell proliferation with differentiation in shoot and root. Proc. Natl. Acad. Sci. U.S.A. 115, E3045–E3054. doi: 10.1073/pnas.1715903115
Wanke, D., Berendzen, K. W., Kilian, J., and Harter, K. (2010a). Insights Into the Arabidopsis Abiotic Stress Response From the AtGenExpress Expression Profile Dataset, Ch. 10. Weinheim: John Wiley & Sons, Ltd.
Wanke, D., Hahn, A., Kilian, J., Harter, K., and Berendzen, K. W. (2010b). “Regulatory networks: inferring functional relationships through Co-expression,” in Quantum Bio-Informatics III, eds L. Accardi, W. Freudenberg, and O. Masanori (Singapore: World Scientific), 405–424.
Xiong, L., Ishitani, M., Lee, H., and Zhu, J.-K. (2001). The Arabidopsis LOS5/ABA3 locus encodes a molybdenum cofactor sulfurase and modulates cold stress–and osmotic stress–responsive gene expression. Plant Cell 13, 2063–2083. doi: 10.1105/tpc.13.9.2063
Xiong, L., Schumaker, K. S., and Zhu, J.-K. (2002). Cell signaling during cold, drought, and salt stress. Plant Cell 14(Suppl. 1):S165–S183. doi: 10.1105/tpc.000596
Xu, F., He, S., Zhang, J., Mao, Z., Wang, W., Li, T., et al. (2018). Photoactivated CRY1 and phyB interact directly with AUX/IAA proteins to inhibit auxin signaling in Arabidopsis. Mol. Plant 11, 523–541. doi: 10.1016/j.molp.2017.12.003
Xu, J., Yang, C., Yuan, Z., Zhang, D., Gondwe, M. Y., Ding, Z., et al. (2010). The ABORTED MICROSPORES regulatory network is required for postmeiotic male reproductive development in Arabidopsis thaliana. Plant Cell 22, 91–107. doi: 10.1105/tpc.109.071803
Yang, J.-H., Li, J.-H., Jiang, S., Zhou, H., and Qu, L.-H. (2012). ChIPBase: a database for decoding the transcriptional regulation of long non-coding RNA and microRNA genes from ChIP-Seq data. Nucl. Acids Res. 41, D177–D187. doi: 10.1093/nar/gks1060
Yevshin, I., Sharipov, R., Valeev, T., Kel, A., and Kolpakov, F. (2016). GTRD: a database of transcription factor binding sites identified by ChIP-seq experiments. Nucl. Acids Res. 45, D61–D67. doi: 10.1093/nar/gkw951
Yip, K. Y., Alexander, R. P., Yan, K.-K., and Gerstein, M. (2010). Improved reconstruction of in silico gene regulatory networks by integrating knockout and perturbation data. PLoS ONE 5:e8121. doi: 10.1371/journal.pone.0008121
Young, W. C., Raftery, A. E., and Yeung, K. Y. (2014). Fast Bayesian inference for gene regulatory networks using ScanBMA. BMC Syst. Biol. 8:47. doi: 10.1186/1752-0509-8-47
Yu, Y. T., Wu, Z., Lu, K., Bi, C., Liang, S., Wang, X. F., et al. (2016). Overexpression of the MYB37 transcription factor enhances abscisic acid sensitivity, and improves both drought tolerance and seed productivity in Arabidopsis thaliana. Plant Mol. Biol. 90, 267–279. doi: 10.1007/s11103-015-0411-1
Yusuf, D., Butland, S. L., Swanson, M. I., Bolotin, E., Ticoll, A., Cheung, W. A., et al. (2012). The transcription factor encyclopedia. Genome Biol. 13:R24.
Zerbino, D. R., Achuthan, P., Akanni, W., Amode, M. R., Barrell, D., Bhai, J., et al. (2017). Ensembl 2018. Nucl. Acids Res. 46, D754–D761. doi: 10.1093/nar/gkx1098
Zhang, B., Su, L., Hu, B., and Li, L. (2018). Expression of AhDREB1, an AP2/ERF transcription factor gene from peanut, is affected by histone acetylation and increases abscisic acid sensitivity and tolerance to osmotic stress in Arabidopsis. Int. J. Mol. Sci. 19:1441. doi: 10.3390/ijms19051441
Zhang, S.-S., Yang, H., Ding, L., Song, Z.-T., Ma, H., Chang, F., et al. (2017). Tissue-specific transcriptomics reveals an important role of the unfolded protein response in maintaining fertility upon heat stress in Arabidopsis. Plant Cell 29, 1007–1023. doi: 10.1105/tpc.16.00916
Zhang, X., Liu, K., Liu, Z.-P., Duval, B., Richer, J.-M., Zhao, X.-M., et al. (2012). NARROMI: a noise and redundancy reduction technique improves accuracy of gene regulatory network inference. Bioinformatics 29, 106–113. doi: 10.1093/bioinformatics/bts619
Zhao, F., Xuan, Z., Liu, L., and Zhang, M. Q. (2005). TRED: a transcriptional regulatory element database and a platform for in silico gene regulation studies. Nucl. Acids Res. 33(Suppl. 1):D103–D107. doi: 10.1093/nar/gki004
Zheng, H., Zhang, F., Wang, S., Su, Y., Ji, X., Jiang, P., et al. (2018a). MLK1 and MLK2 coordinate RGA and CCA1 activity to regulate hypocotyl elongation in Arabidopsis thaliana. Plant Cell 30, 67–82. doi: 10.1105/tpc.17.00830
Zheng, R., Wan, C., Mei, S., Qin, Q., Wu, Q., Sun, H., et al. (2018b). Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucl. Acids Res. 47, D729–D735. doi: 10.1093/nar/gky1094
Zhu, J., Verslues, P. E., Zheng, X., Lee, B. H., Zhan, X., Manabe, Y., et al. (2005). HOS10 encodes an R2R3-type MYB transcription factor essential for cold acclimation in plants. Proc. Natl. Acad. Sci. U.S.A. 102, 9966–9971. doi: 10.1073/pnas.0503960102
Zhu, J.-K. (2016). Abiotic stress signaling and responses in plants. Cell 167, 313–324. doi: 10.1016/j.cell.2016.08.029
Keywords: transcription factor, gene regulatory network inference, time-varying, plant stress, influence maximization, network propagation
Citation: Ahn H, Jo K, Jeong D, Pak M, Hur J, Jung W and Kim S (2019) PropaNet: Time-Varying Condition-Specific Transcriptional Network Construction by Network Propagation. Front. Plant Sci. 10:698. doi: 10.3389/fpls.2019.00698
Received: 04 January 2019; Accepted: 09 May 2019;
Published: 14 June 2019.
Edited by:
Henrik Aronsson, University of Gothenburg, SwedenCopyright © 2019 Ahn, Jo, Jeong, Pak, Hur, Jung and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Woosuk Jung, anVuZ3dAa29ua3VrLmFjLmty; Sun Kim, c3Vua2ltLmJpb2luZm9Ac251LmFjLmty
†These authors have contributed equally to this work