 Álvaro Calderón-González1
Álvaro Calderón-González1 Nicolas Pouilly2
Nicolas Pouilly2 Stéphane Muños2
Stéphane Muños2 Xavier Grand3Marie Coque3
Xavier Grand3Marie Coque3 Leonardo Velasco1
Leonardo Velasco1 Begoña Pérez-Vich1*
Begoña Pérez-Vich1*- 1Instituto de Agricultura Sostenible (IAS) – Consejo Superior de Investigaciones Científicas (CSIC), Córdoba, Spain
- 2Laboratoire des Interactions Plantes Micro-organismes (LIPM), UMR CNRS-INRA 2594-441, Castanet-Tolosan, France
- 3Biogemma SAS, Paris, France
Sunflower broomrape (Orobanche cumana Wallr.) is a holoparasitic plant that causes major yield losses to sunflower crops in the Old World. Efforts to understand how this parasitic weed recognizes and interacts with sunflowers are important for developing long-term genetic resistance strategies. However, such studies are hampered by the lack of genetic tools for O. cumana. The objectives of this research were to construct a genetic linkage map of this species using SSR and SNP markers, and mapping the Pg locus that is involved in plant pigmentation. The genetic map was developed from the progenies of a cross between the O. cumana inbred lines EK-12 and EK-A1, which originated from populations belonging to two distant and geographically separated gene pools identified in Spain. The inbred lines also differed in plant pigmentation, with EK-A1 lacking anthocyanin pigmentation (pgpg genotype). A genetic map comprising 26 SSR and 701 SNP markers was constructed, which displayed 19 linkage groups (LGs), corresponding to the 19 chromosome pairs of O. cumana. The total length of the map was 1795.7 cM, with an average distance between two adjacent positions of 2.5 cM and a maximum map distance of 41.9 cM. The Pg locus mapped to LG19 between the SNP markers OS02468 and OS01653 at 7.5 and 3.4 cM, respectively. This study constitutes the first linkage map and trait mapping study in Orobanche spp., laying a key foundation for further genome characterization and providing a basis for mapping additional traits such as those having a key role in parasitism.
Introduction
Sunflower broomrape (Orobanche cumana Wallr.) is a holoparasitic plant found in the wild from south-eastern Europe to central Asia parasitizing a few species of the Asteraceae, mainly Artemisia spp. As a parasitic weed, it parasitizes on sunflower and represents one of the most serious production constraints for this crop in many sunflower-producing countries, particularly in Central and Eastern Europe, Spain, Turkey, Israel, Iran, Kazakhstan, and China (Fernández-Martínez et al., 2015). Moreover, the parasite has spread in recent years to new countries where it had not been reported before such as France (Jestin et al., 2014), Tunisia (Amri et al., 2012), and Morocco (Nabloussi et al., 2018), and in countries where the parasite had been traditionally observed in specific areas, it continues spreading to new regions, such as the North of Spain (Fernández-Escobar et al., 2009; Malek et al., 2017).
The genetic interaction between broomrape and sunflower is in most cases governed by the gene-for-gene model for plant-pathogen interactions, in which resistance reactions are governed by the interaction of host genes for resistance and the corresponding pathogen genes for avirulence (Rodríguez-Ojeda et al., 2013b). This kind of interaction has led to the development of resistant sunflower cultivars based on vertical resistance mechanisms, and determines the occurrence of physiological races of broomrape that are controlled by these resistant genes in sunflower (Fernández-Martínez et al., 2015). Vranceanu et al. (1980) described five broomrape races in the early 80 s named as A to E and developed a set of sunflower differential lines to identify them, each carrying a single dominant gene (Or1 through Or5, respectively) conferring resistance to the corresponding race. New races overcoming Or5 resistance appeared from the middle 1990s onward in several countries such as Spain, Romania, Turkey, Bulgaria, Ukraine, and Russia (Fernández-Martínez et al., 2015). Initially, all of them were named as race F though the relationship between the different F races has not been studied. Nowadays, populations overcoming resistance sources to race F, named as races G and H, have been identified in most of these countries (Kaya et al., 2009; Pacureanu-Joita et al., 2009; Shindrova and Penchev, 2012; Antonova et al., 2013; Martín-Sanz et al., 2016). As mentioned for race F, no comparative studies have been conducted between races G and H reported in different countries. For races F and G, monogenic and dominant resistance in sunflower has also been reported (Pacureanu-Joita et al., 2004; Velasco et al., 2012).
As shown by broomrape race evolution, sunflower vertical resistance mechanisms are readily overcome by the parasite. For the development of long-term breeding strategies, it is essential to understand the genetic bases of the host-parasite interaction. However, this is currently hampered by the limited availability of genetic tools in O. cumana, since most of the research has been carried out on the crop host, the sunflower. The few genetic studies in O. cumana have mainly focused on population structure and genetic diversity analyses (Castejón-Muñoz et al., 1991; Gagne et al., 1998; Pineda-Martos et al., 2013, 2014; Guchetl et al., 2014; Molinero-Ruiz et al., 2014; Martín-Sanz et al., 2016). These studies concluded that the populations parasitizing sunflowers were characterized by low intra-population diversity and, in general, low differentiation between populations. Additionally, different mechanisms were postulated for explaining race evolution, such as single-gene mutations within local populations (Pineda-Martos et al., 2013), or genetic recombination of avirulence genes (Martín-Sanz et al., 2016). Classical genetic analyses in O. cumana were started just a few years ago. This in part might be due to the fact that working on genetics of holoparasitic plants signifies additional difficulties to obtain the plant material and segregating generations required for such studies, in such a way that all the plants-segregating generations have to be obtained by artificial inoculation on the corresponding host, which is a labor intensive and time-consuming procedure. Initially, Rodríguez-Ojeda et al. (2010) carried out basic studies for determining the feasibility of the use of inbreeding and hybridisation techniques for carrying out these genetic studies in O. cumana. Later, these authors isolated a line named EK-A1 from a natural mutant lacking anthocyanin pigmentation identified in a population of O. cumana from central Spain, and determined the inheritance of the trait evaluating the phenotype of segregating populations from crosses with plants of a normally pigmented line (Rodríguez-Ojeda et al., 2011). The authors concluded that the trait was controlled by partially dominant alleles at a single locus, which was named Pg. Therefore, this trait showing a monogenic inheritance turned out to be an excellent candidate for conducting trait mapping studies in O. cumana genome. Additionally, the segregating population generated by Rodríguez-Ojeda et al. (2011) probed to be highly polymorphic, since it was constructed from parental lines belonging to the two genetically distant O. cumana gene pools that co-exist in Spain (the Gualdalquivir Valley and the Cuenca gene pools, in southern and central Spain, respectively, Pineda-Martos et al., 2013).
Genetic linkage maps are essential for studying the genome structure and organization. They are also valuable resources in parasitic weeds for locating genes that control traits of interest, such as avirulence or host specificity and ultimately might permit the positional cloning of these genes. In Orobanche spp., there have been no attempts to develop genetic linkage maps, and no trait mapping studies have been carried out to date. Genetic linkage maps in other parasitic weeds are also extremely limited. To our knowledge, the only study detailing a linkage map construction has been reported for Striga hermonthica (Del.) Benth., a parasitic weed of cereals, using amplified fragment length polymorphisms (AFLPs) (Pescott, 2013).
Since O. cumana genetic tools are essential to provide a better knowledge of the sunflower-O. cumana parasitic system for the development of knowledge-based control strategies, and these tools are so far extremely limited for this species and in general for Orobanche spp., the objectives of this research were to (i) develop a genetic linkage map in O. cumana using SSR and SNP markers, and (ii) map as a Mendelian trait the Pg locus involved in plant pigmentation, which was previously studied at the phenotypic level by Rodríguez-Ojeda et al. (2011).
Materials and Methods
Plant Material, Phenotyping and Mapping Population
The mapping population consisted of 91 F2 plants and their corresponding F2:3 families derived from the cross between the O. cumana lines EK-A1 and EK-12, previously reported by Rodríguez-Ojeda et al. (2011). The EK-A1 plants lack pigmentation, having a yellow stem and a white corolla, whereas the EK-12 plants have normal pigmentation showing a bluish-violet stem and bluish to pale-violet corolla (Figure 1). This trait is controlled by partially dominant alleles at a single locus, referred to as Pg. This was demonstrated by Rodríguez-Ojeda et al. (2011) through the independent evaluation of sixteen F2 populations (F2 plant generation) segregating for the plant pigmentation trait and the progenies of 120 F2 plants (F3 plant generation) evaluated in different years. All plants and generations were grown on the sunflower susceptible cultivar DMM as reported by Rodríguez-Ojeda et al. (2011). The growth conditions and phenotypic characterization for plant pigmentation of F2 and F3 plants were previously described by Rodríguez-Ojeda et al. (2011). For the molecular study, a F2 population consisting in 91 F2 plants for which (i) F2:3 families were available and (ii) there was sufficient F3 tissue, was used. Stem and corolla colors were visually evaluated at each 91 F2 plants and their corresponding F2:3 families (F3 plants), considering the phenotypes of the parental lines (unpigmented-yellow stem in EK-A1 plants and a fully pigmented-bluish-violet stem in EK-12 plants, Figure 1), and of the F1-heterozygous plants (greenish-intermediate pigmented, Figure 1; Rodríguez-Ojeda et al., 2011). Plant stems were scored as unpigmented (yellow) and pigmented (bluish violet-fully pigmented and greenish-intermediate pigmented). Pigmented plants (bluish violet and greenish) were included in the same class because of the large number of broomrape plants in some pots which made it difficult to distinguish greenish from bluish-violet plants. A Chi-square test was used to evaluate the proposed segregation ratio for the population used in the molecular study.

Figure 1. Unpigmented O. cumana EK-A1 plants (1, 2), partially pigmented heterozygotes (3, 4), and fully pigmented EK-12 plants (5, 6) showing yellow, greenish, and bluish-violet stems, respectively.
In order to evaluate the potential of the SNPs reported in this study for their use in genotyping other published O. cumana segregating populations and for diversity analyses, three further O. cumana populations used as parental lines in O. cumana crosses described in Rodríguez-Ojeda et al. (2013b) were used. These three populations belonged to the Guadalquivir Valley gene pool and were (i) OC-94 (race E, collected in Sevilla, Spain in 1994), (ii) EK-23 (race F, collected in Córdoba, Spain in 1995), and (iii) SP (race F, collected in Sevilla, Spain in 2004) (Rodríguez-Ojeda et al., 2013b).
Tissue Collection and DNA Extraction
Broomrape shoots were first collected from individual F2 plants (single broomrape shoots), and individual plants from the parental lines EK-A1 and EK-12, and the other three O. cumana populations used for a polymorphism and diversity analysis (OC-94, EK-23, and SP). In order not to affect the F3 seed production, the apical bud of each F2 broomrape shoot was removed after most of the flowers had been formed. Because of the small amount of tissue collected in F2, tissue was also collected from F2:3 families (about 20 young F3 shoots per family) in order to recreate their corresponding F2 genotype. This is an accurate approach useful for species lacking enough tissue in individual F2 plants to yield enough DNA for marker analysis (Kochert, 1994). In this case, 20 young F3 shoots from each F2:3 family were collected before flowering, and bulked by mixing an equal tissue amount of the F3 shoots from each F2:3 family. In all cases, tissue was immediately frozen at -80°C, lyophilized, and ground in a laboratory ball mill. DNA was then extracted following the procedure reported by Pineda-Martos et al. (2013).
Genotyping With SSR Markers
The complete set of 298 O. cumana SSR markers developed by Pineda-Martos et al. (2014) was tested for polymorphism in the parental lines EK-12 and EK-A1, and six individuals from the mapping population. PCR amplification was carried out in 30 μL reaction mixtures, consisting of 50 ng of template DNA, 0.03 U/μL of Taq DNA polymerase (FirePol Taq polymerase, Solis BioDyne, Tartu, Estonia), 1× PCR buffer, 2.5 mM MgCl2, 200 μM dNTPs (Solis BioDyne, Tartu, Estonia), and 0.3 μM of primers. A touchdown PCR program was used on a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA, United States), which consisted of an initial denaturation step of 94°C for 2 min, followed by one cycle of 94°C for 30 s, final annealing temperature (TA) + 10°C for 30 s, and 72°C for 30 s, 9 cycles in which the annealing temperature was decreased by 1 °C, and 32 cycles at 94°C for 30 s, TA for 30 s, and 72°C for 30 s, with a final extension of 10 min at 72°C. Amplified products were separated on 3% MetaPhor (BMA, Rockland, ME, United States) agarose gels in 1× TBE buffer with SafeView Nucleic Acid Stain (NBS Biologicals Ltd., Huntingdon, United Kingdom) incorporated in the gel, in such a way that microsatellite alleles were effectively resolved with size differences between alleles by 2%. A 100 bp DNA ladder (Solis BioDyne, Tartu, Estonia) was used as a standard molecular weight marker to get an approximate size for the DNA fragments. The resultant gel images were scored manually with the aid of Quantity One 1-D Analysis Software (Bio-Rad Laboratories Inc., Hercules, CA, United States). The amplification profile for each microsatellite was scored visually and independently. A set of 28 polymorphic SSR markers showing clearly co-dominant and scorable fragments was selected and genotyped in the mapping population (91 individuals), following the same approach described above.
Genotyping With SNP Markers
A set of 1536 O. cumana SNP markers developed by Coque et al. (2016) through exome capture was used to genotype the parental lines EK-12 and EK-A1 and the mapping population (ninety-one individuals) using competitive allele-specific PCR assays based on KASPTM technology (LGC genomics, Teddington, United Kingdom) on the Limagrain genotyping platform. For this SNP discovery study, a set of 12 broomrape populations representing different level of virulence or aggressiveness and different countries was submitted to transcriptome sequencing. This approach led to the discovery of approximately 368000 bi-allelic SNP among which 1536 were selected for genotyping a Biogemma broomrape collection (around 500 populations). A genetic diversity analysis was conducted and an optimized subset of 198 SNP capturing the maximum of the genetic diversity analysis was selected. Details of the 198 O. cumana SNP markers are reported in Supplementary Table S1. The remainder of the SNP marker information is proprietary. For genetic mapping purpose, monomorphic markers were excluded for the linkage map construction.
In order to evaluate the potential of the SNPs reported in this study for their use in genotyping other O. cumana segregating populations, the number of markers polymorphic between parental genotypes of published mapping populations were determined as follows. The 198 O. cumana SNP marker subset from Supplementary Table S1 was genotyped in 10 individuals from each of the parental genotypes EK-A1, EK-12, EK-23, OC-94, and SP, used to develop the segregating populations EK-12 × EK-23, and OC-94 × SP, segregating for avirulence/virulence (Rodríguez-Ojeda et al., 2013b), and EK-A1 × EK-12, segregating for plant pigmentation (Rodríguez-Ojeda et al., 2011, this study). The individuals were genotyped for these SNP using the KASPTM genotyping assay service provided by LGC Genomics (Herts, United Kingdom). A total of 38 markers and 7 individuals that failed genotyping or had >10% missing data, respectively, were excluded from the analysis. Therefore, polymorphism and genetic diversity analysis was carried out with 160 SNPs markers and the following parameters were calculated: percentage of polymorphic loci (P) and Shannon’s diversity index (I) within each population and percentage of polymorphic loci (P) between populations. In addition, pairwise genetic distances between populations were calculated as the genetic distance coefficient GST using 1000 random permutations to assess significance. The matrix of GST pairwise distances was used as input for a principal coordinates analysis (PCoA). All analyses were carried out using GenAlEx version 6.5 (Peakall and Smouse, 2012).
Genetic Linkage Map Construction and Genetic Mapping of the Pigmentation Gene
Only co-dominant markers were used for the linkage map construction. The segregation of alleles at the SSR or SNP marker loci was checked against the expected ratios for codominant (1:2:1) markers using a chi-square test. The genetic linkage map was constructed with MAPMAKER/EXP (version 3.0b) (Lincoln et al., 1993) using genotyping data from polymorphic and co-dominant markers from the 298 SSR and 1536 SNP marker sets. Map distances in centiMorgan (cM) were converted from recombination fractions using the Kosambi mapping function. Two-point analysis was used to identify linkage groups (LGs) with an LOD score of 6.0 and a maximum distance of 40 cM, except for LGs 13 and 15, in which specific markers were grouped with a LOD value of 3.5. Three-point and multi-point analyses were used to determine the order and interval distances between the markers in each LG. Loci whose position were ambiguous (i.e., those placed automatically at a less-strict LOD of 2.0) were noted. Markers that had the most skewed segregation ratios (P < 0.0001) were excluded from the map. Linkage maps were drawn using MapChart 2.1 software (Voorrips, 2002). The linkage groups were randomly numbered as there are no previously reported O. cumana genetic maps. Simple correlation coefficients (r) between the total number of markers per linkage group and the total linkage group length were calculated. The significance of the correlation coefficients was calculated by the standard testing procedures for r = 0 null hypothesis (Snedecor and Cochran, 1989).
As the “plant pigmentation” trait is controlled by a single gene (Pg, Rodríguez-Ojeda et al., 2011), it was mapped as a Mendelian locus. Accordingly, the genotypes for the Pg gene were inferred from the pigmentation phenotypes in F2 plants and their corresponding F2:3 families. F2 plants were classified as homozygous dominant for the pigmentation gene if they showed a stem similar to EK-12, and showed uniformly pigmented plants in their respective F3 progeny, heterozygous if their F3 segregated for stem color, and homozygous recessive if they were similar to EK-A1 and showed uniformly unpigmented plants in their respective F3 progeny. Linkage analysis for the pigmentation gene was run with MAPMAKER/EXP (version 3.0b) using segregation data for SNP and SSR marker loci and for the Pg locus. Pg mapping was carried out as indicated for SSR and SNP markers, excepting that a LOD threshold of 10 and a maximum distance of 30 cM were used as linkage criteria. Finally, potential candidate genes for the Pg locus were identified using BLAST searches for plant pigment biosynthesis genes at the flavonoid/anthocyanin and carotenoid biosynthesis pathways (MetaCyc, version 20.0; Caspi et al., 2018) against a 622 contigs first draft of the O. cumana sequence genome (Gouzy et al., 2017).
Results
Phenotypic Evaluation of the Mapping Population
Phenotypic evaluation of the mapping population revealed 15 unpigmented F2 plants showing homogeneously unpigmented F3 progenies (a total of 675 unpigmented F3 plants for all the 15 F2:3 families) and 76 pigmented F2 plants. From these, 30 presented homogeneously bluish-violet pigmented F3 progenies (a total of 1146 bluish-violet F3 plants), and 46 showed segregating F3 progenies with both pigmented and unpigmented plants (a total of 1305 pigmented and 452 yellow-unpigmented F3 plants). The number of plants observed in each phenotypic class [15 (unpigmented): 46 (segregating): 30 (fully pigmented)] did not differ significantly from a 1:2:1 genetic proportion expected for one-gene segregation (χ2 = 4.96, P = 0.08).
Genetic Linkage Map Construction
From the 1536 O. cumana SNPs, 1285 were successfully genotyped. Among these, 722 (56.2%) were polymorphic between the parents EK-A1 and EK-12 and segregated accordingly in the mapping population. From the 298 O. cumana SSRs, 168 showed high quality amplification, and 33 (19.6%) were polymorphic (28 co-dominant and 5 dominant). After excluding markers with extremely distorted segregation (P < 0.0001) and dominant markers, 737 markers were used for the linkage analysis. The final genetic linkage map was constructed using 26 SSR and 701 SNP polymorphic and codominant markers. Two additional markers that remained unlinked and 8 markers that remained “unmapped” (grouped in LGs of two or three markers) were not included. The 727 SSR and SNP loci were arranged in 19 linkage groups, which correspond to the 19 chromosome pairs of the O. cumana genome (Schneeweiss et al., 2004; Piednoël et al., 2012; Figure 2 and Supplementary Table S2). The total map length was 1795.7 cM. The average distance between two adjacent positions across the whole map was 2.5 cM, but there were 12 regions on 11 LGs with intervals greater than 20 cM, with the largest interval (41.9 cM) being observed in LG13 (Figure 2 and Supplementary Table S2).
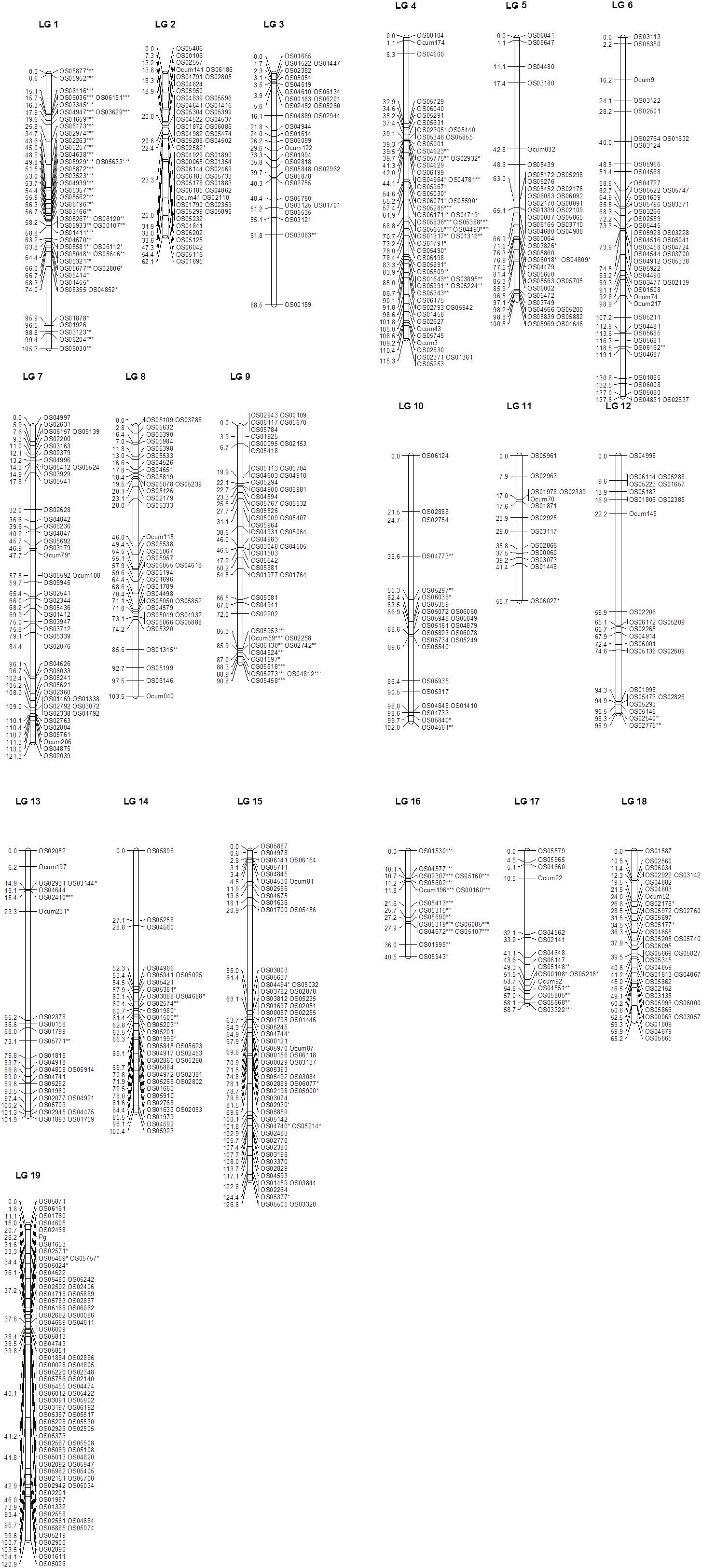
Figure 2. SSR and SNP O. cumana genetic linkage map containing the pigmentation locus Pg. The map is based on segregation of 727 co-dominant SNP and SSR markers, and Pg, in 91 individuals. Linkage groups (LG) follow a randomly selected numeration. Genetic distances are given in centiMorgans (Kosambi) on the left of each LG. The position of the Pg gene associated with plant pigmentation in O. cumana, mapped as a Mendelian trait, is shown at LG19. Prefix Ocum is for SSR marker loci and prefix OS for SNP marker loci. SNP and SSR marker loci labeled as ∗, ∗∗, or ∗∗∗ showed distorted segregation at P < 0.05, P < 0.01, and P < 0.001, respectively.
The number of markers on different LGs ranged from 13 on LG11 to 80 on LG19. The length of different LGs ranged from 40.5 cM on LG16 to 137.6 cM on LG6 (Figure 2 and Supplementary Table S2). The LG length was correlated with the number of markers per linkage group (r = 0.66, p = 0.002, n = 19). LG2 was the most skewed linkage group from the linear fitting, probably due to an excess of markers (a total of 28) clustering in two groups of 0 cM (Figure 2). Excluding this LG resulted in a higher correlation (r = 0.74, p = 0.0004, n = 18). A highly significant (P < 0.001) distorted segregation was observed in LG1, LG4, the bottom of LG9, and LG16 (Supplementary Table S2).
Genetic Mapping of the Pg Pigmentation Gene
Using the genotypic classification of F2 plants (homozygous unpigmented: segregating: homozygous pigmented) based on the F3 evaluation, the locus Pg associated with plant pigmentation mapped as a Mendelian trait was located 28.2 cM downstream from the upper end of LG19, between the SNP markers OS02468 and OS01653, which were 7.5 cM distal and 3.4 cM proximal, respectively, of the Pg locus (Figure 2). Based on the physical position of context sequences of markers flanking Pg on the O. cumana draft genome (OS01653 and OS02571 at positions 4.17 and 4.07 Mbp, respectively, in contig OcIN23s039, and OS02468 and OS04605 at positions 2.51 and 4.41 Mbp, respectively, in contig OcIN23s036), candidate genes for plant pigment biosynthesis at the flavonoid/anthocyanin and carotenoid biosynthesis pathways were searched locating at these contigs. Two contiguous genes for flavonoid biosynthesis and carotenoid biosynthesis were found in contig OcIN23s036. Both genes were coding for carotenoid/flavonoid glucoside glucosyltransferases. The genes were a flavonoid glucoside glucosyltransferase (at position 3.61 Mbp) and a crocetin glucoside glucosyltransferase (at position 3.60 Mbp) whose physical position was between that of OS02468 and OS04605 SNP markers in contig OcIN23s036.
Diversity and Polymorphism SNP Analysis
Evaluation of the potential of SNPs (using the 198 SNP subset from Supplementary Table S1) for genotyping existing segregating populations in O. cumana revealed an extremely low number of polymorphic markers between parental genotypes coming from populations of the Guadalquivir Valley gene pool (OC-94, SP, EK-12, and EK-23), in contrast to a much higher number of polymorphic markers found between EK-A1 from the Cuenca gene pool and the remaining genotypes from the Guadalquivir Valley gene pool, including EK-12 (Table 1). This was coupled with a very low intra-population variation and inter-population diversity for populations from the same gene pool, as shown by diversity parameters (Table 1). Principal Coordinate Analysis revealed a clear separation between parental genotypes from the Guadalquivir Valley gene pool (OC-94, SP, EK-12, and EK-23), and the Cuenca gene pool (EK-A1) (Figure 3), separated along Coordinate 1 that explained 94.14% of the total variation.
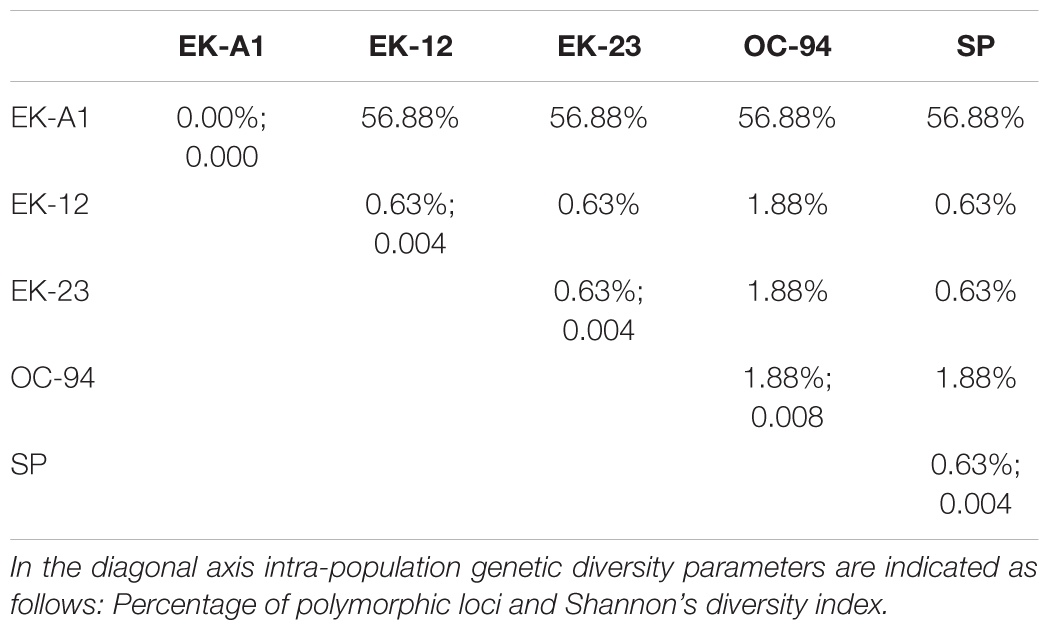
Table 1. Percentage of polymorphic SNP loci between the parental genotypes EK-A1 (Cuenca gene pool), EK-12 (Guadalquivir valley gene pool), EK-23 (Guadalquivir valley gene pool), OC-94 (Guadalquivir valley gene pool), and SP (Guadalquivir valley gene pool) from O. cumana segregating populations.
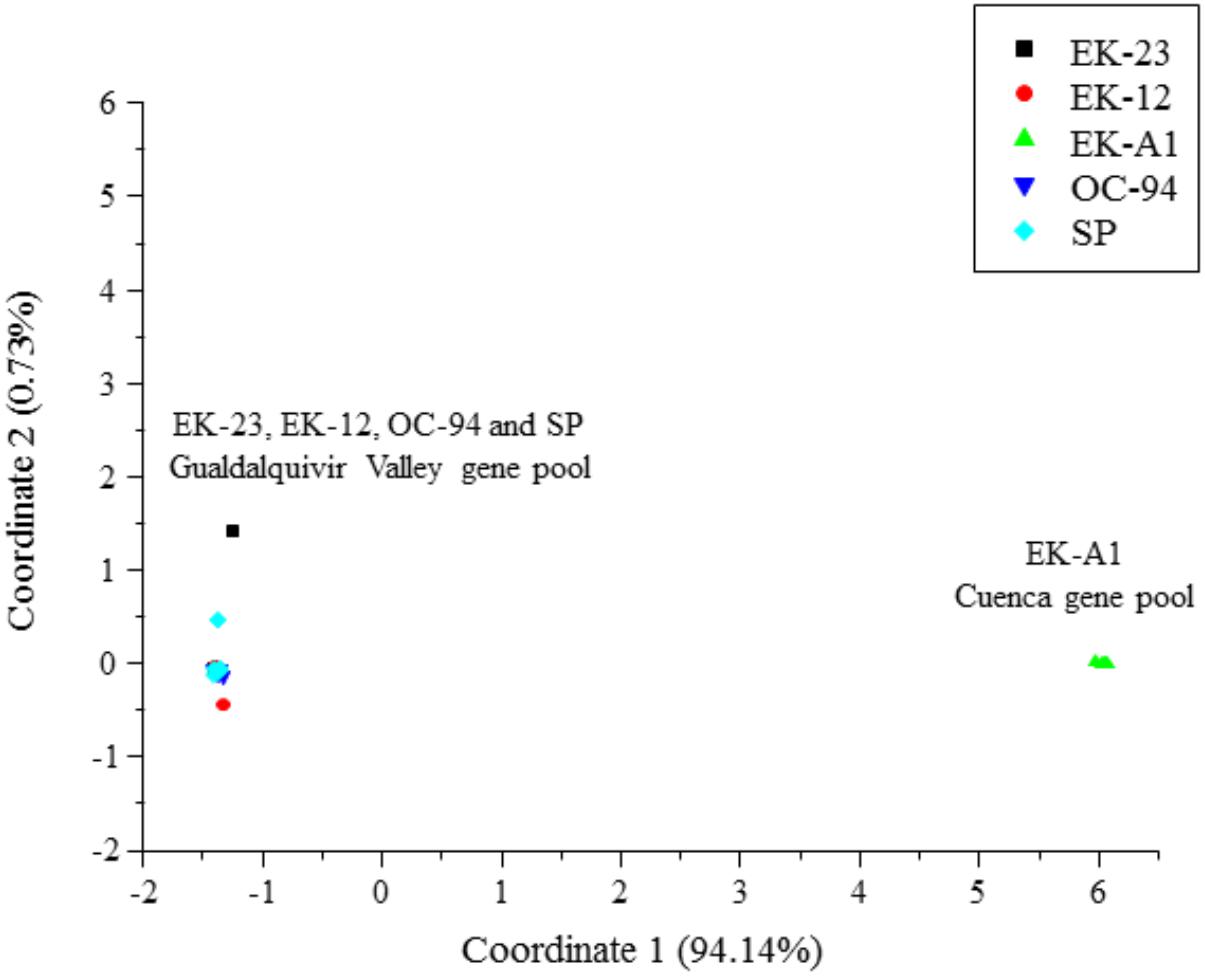
Figure 3. Principal coordinates analysis of the five O. cumana parental genotypes EK-A1, EK-12, EK-23, OC-94, and SP.
Discussion
Genetic studies in O. cumana have been very scarce till date. Rodríguez-Ojeda et al. (2011) studied the inheritance of the unpigmented plant trait in line EK-A1. The trait was found to be controlled by partially dominant alleles at a single locus, named Pg. Another genetic study at the phenotypic level was conducted by Rodríguez-Ojeda et al. (2013b), who evaluated the segregation of virulence in progenies from crosses between race E and race F lines developed from populations collected in southern Spain. Differences in virulence between both types of populations were found to be controlled by a single gene, suggesting that the evolution from race E to race F in that area was produced by a point mutation. Genetic studies in O. cumana, conducted initially at the phenotypic level, can be now be expanded at the molecular level thanks to genetic and genomic advances in this species, such as the development of SSR markers (Pineda-Martos et al., 2014), SNP markers (reported for the first time in this study), or a first draft of the genome sequence (Gouzy et al., 2017). The present study, in which SNP markers have been reported, a genetic linkage map using SSR and SNPs has been constructed and the Pg gene (characterized previously at the phenotypic level by Rodríguez-Ojeda et al., 2011) mapped, is in the same line as these previous studies, providing new genetic tools for understanding genetic mechanisms in this parasitic weed, and ultimately contributing to the development of durable genetic resistance in sunflower.
The development of a genetic linkage map is an important pre-requisite for the identification of genes or QTL underlying important traits. The O. cumana map developed in the present research is the first linkage map reported for any Orobanche/Phelipanche species and represents a significant advance in the study of genome structure and organization, and for mapping genes of importance on the O. cumana genome. This O. cumana genetic map contains 19 LGs, coincident with the basic number of chromosomes in this species (Schneeweiss et al., 2004; Piednoël et al., 2012), a relatively high marker density (a total of 727 SNP and SSR marker loci) and a resolution of 2.5 cM. From 737 polymorphic markers used for linkage analysis, only 10 remained “unmapped,” with 98.6% of the markers falling into one larger LG (with a minimum number of markers of 13, and coverage >40 cM). Therefore, the map developed appears to cover the vast majority of the O. cumana genome. The genetic linkage map information from this study combined with the availability of an O. cumana genome sequence draft and the contig and genetic linkage position of SNP and SSR markers, will be of utility to anchor contigs into scaffolds and ultimately to obtain the chromosome sequences, contributing in this way to the development of new genetic resources for this species.
The total O. cumana map length was 1795.7 cM. Considering the size of its genome to be 1.42 Gb or 1.40 Gb resulting from either densitometry (Weiss-Schneeweiss et al., 2006) or assembly of DNA sequences (Pouilly et al., 2018), respectively, the O. cumana genome-wide recombination rate was estimated as 1.27 cM/Mb. This value is slightly lower than the average value of 1.85 cM/Mb previously reported by Stapley et al. (2017) for plants (averaged from 189 species), and also lower than the average value of 2.6 cM/Mb (ranging from 1.6 to 3.9 cM/Mb) detailed by these authors for five plant species of the Lamiales order, to which the Orobanchaceae family belongs. Interestingly, Stapley et al. (2017) compared the genome-wide recombination rate of parasitic or pathogenic species with free-living species and found that parasitic or pathogenic species had a higher recombination rate compared to their free-living counterparts in animals, but they found no differences in fungi. Unfortunately, no data were reported for parasitic plants as parasitic/pathogenic plant species were not included in their dataset.
The distribution of markers between the 19 linkage groups was fairly uniform with, in general, the largest groups containing the most markers. However, there were markers clustered observed in some regions of the map, especially on LG2, LG5, and LG19. Marker clustering has also been reported in genetic maps in other plant species, and it has been associated with centromeric regions due to the suppression of recombination in the heterochromatic regions surrounding these regions (Haanstra et al., 1999), or to the lack of recombination around genes with evolutionary significance (Jessup et al., 2002; Hao et al., 2004). In addition, despite the short average distance between adjacent markers on the map (2.5 cM), there were 12 gaps larger than 20 cM. These gaps have also been reported in several genetic linkage maps in plant species and may be due to the lack of marker polymorphism and a shortage of marker detection in these regions (Berry et al., 1995) or due to recombination hot spots (Mézard, 2006).
Around 20.5% (149 out of 728) of the mapped markers in the map had significantly distorted segregation ratios. They were mainly clustered on LG1 and LG16 and on specific regions in LG4 and LG9, suggesting that these may be of biological significance. Distorted segregation in specific genomic regions is an inevitable feature observed in many of the marker-based linkage maps in plants. It has been attributed to a range of causes, including deleterious recessive alleles (Berry et al., 1995), self-incompatibility alleles (Barzen et al., 1995), structural rearrangements (Quillet et al., 1995), or differences in DNA content (Jenczewski et al., 1997). It is worth noting that the allele differences that contribute to the relative success of parasitism could also lead to segregation distortion (Thomas et al., 2012). Interestingly, all markers showing segregation distortion clustering at LG1, LG9, and LG16 favored the allele from the pigmented parent EK-12. Although the pigmentation gene Pg did not map in any of those regions, and the lack of pigmentation in O. cumana plants has not been associated to parasitism (González-Torres et al., 1982; Rodríguez-Ojeda et al., 2011), this might suggest a fitness benefit for progenies that inherited specific EK-12 alleles.
Overall SNP polymorphism in the EK-A1 and EK-12 cross was relatively high (more than 50% polymorphic loci). This cross involved parents from two distant gene pools identified in Spain, with the EK-A1 population belonging to the Cuenca gene pool in Central Spain and the EK-12 population being from the Guadalquivir Valley gene pool in Southern Spain (Rodríguez-Ojeda et al., 2011; Pineda-Martos et al., 2013). In addition to these two divergent gene pools in Spain, Pineda-Martos et al. (2013) also described an extremely low inter- and intra- population diversity within each of these two gene pools using SSR markers, which was attributed to a founder effect. This lack of polymorphism within the Guadalquivir Valley gene pool has also been confirmed in this study using SNP markers. Therefore, using parental lines from already described O. cumana distant gene pools to avoid factors decreasing diversity such as founder effects is desirable for genetic mapping and construction of highly saturated genetic maps in O. cumana, since the possibility of detecting polymorphism among parents is increased, resulting in a higher number of segregating loci.
Orobanche spp. are often highly variable in regard to their size, coloration, and pubescence (Rumsey and Jury, 1991). Unpigmented plants (likely lacking anthocyanin pigmentation) have been observed in the populations of several Orobanche spp. (Kreutz, 1995; Rumsey, 2007), including O. cumana (González-Torres et al., 1982). Results previously reported by Rodríguez-Ojeda et al. (2011) showed that the absence of pigmentation in O. cumana was the result of a single-gene mutation, probably involved in anthocyanin biosynthesis. Later, Rodríguez-Ojeda et al. (2013a) proved the unpigmented trait in O. cumana to be very useful for studies on its biology, allowing the determination of cross-fertilization rate in this species. In this study, the pigmentation locus Pg has been mapped to LG19 in the SSR-SNP O. cumana genetic linkage map, between the SNP markers OS01653 and OS02468. This constitutes the first trait mapping study in Orobanche spp. Even though the development of molecular markers for the pigmentation trait may not have drawn interest because the trait is easily distinguishable visually, the location of a gene with a known phenotypic effect in the O. cumana genetic map may be an important reference for future mapping and molecular marker studies in this species. In addition, it might contribute to the identification of causal genes and mutations for stem and flower-color variations in O. cumana. In fact, candidate gene analysis revealed two contiguous genes for carotenoid/flavonoid glucoside glucosyltransferases mapping close to the Pg locus. Glycosylation is often the final step in the biosynthesis of plant secondary metabolites, which enhances their water solubility and chemical stability and alters their biological activity (Yang et al., 2018). The attachment of additional sugar to flavonoid glycosides has also been related to modifications in physiological properties such as color. For instance, anthocyanin color is influenced by glycosylation pattern (Zhang et al., 2014), and its possible role on Orobanche plant pigmentation might be further investigated.
Conclusion
In conclusion, this work represents the first genetic linkage map and trait mapping study for O. cumana, and for any Orobanche/Phelipanche spp., a species and a genus for which there are very limited genetic/genomic information published. Results from this study will contribute to understand the genetic basis of the sunflower- O cumana interaction, which is required for the development of new knowledge-based strategies for broomrape management. In this sense, the reported SNPs and the saturated genetic map constitute valuable genetic resources for different downstream applications such as new SNP-based genetic diversity and population structure analyses, further genome characterization and sequence assembly of the O. cumana genome, and the identification of genes/QTL underlying relevant traits. The mapping of the Pg locus determining plant pigmentation provides one example, and mapping genes/QTL associated to parasitism and virulence using segregating populations generated from parental lines differing for these traits is under way. Locating and eventually cloning genes responsible for these traits will bear direct implications for practical agriculture, since they will represent new targets for rational design of control strategies to this devastating parasitic weed.
Author Contributions
LV and BP-V conceived the study and planned and supervised the research. SM supervised a part of the genotyping. MC conducted the SNP development and genotyping. ÁC-G and NP conducted the SSR genotyping and collaborated in the SNP genotyping. BP-V, XG, and ÁC-G conducted the statistical analyses and map construction. ÁC-G, LV, and BP-V wrote the draft of the manuscript. All authors critically read the manuscript and revised its final version.
Funding
This study has been partially funded by the research project AGL2017-87693-R of the Spanish Ministry of Economy and Innovation (cofunded with EU FEDER Funds), Biogemma and INRA (Institut National de la Recherche Agronomique, Paris, France).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Plácida Nieto and Alberto Merino for the technical support. We also acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00797/full#supplementary-material
TABLE S1 | Details of 198 O. cumana SNP markers, including their context sequence and map positions.
TABLE S2 | Map positions of 727 SNP and SSR marker loci and the pigmentation locus Pg in the O. cumana genetic linkage map. Prefix Ocum is for SSR marker loci and prefix OS for SNP marker loci. Marker loci labelled as ∗, ∗∗, or ∗∗∗ showed distorted segregation at P < 0.05, P < 0.01, or P < 0.001, respectively. Loci whose position were ambiguous (i.e., those placed automatically at a less-strict LOD of 2.0) were labelled in red. Markers highlighted in bold and underlined are SSR or those SNP detailed in Supplementary Table S1.
References
Amri, M., Abbes, Z., Ben Youssef, S., Bouhadida, M., Ben Salah, H., and Kharrat, M. (2012). Detection of the parasitic plant, Orobanche cumana on sunflower (Helianthus annuus L.) in Tunisia. Afr. J. Biotechnol. 11, 4163–4167. doi: 10.5897/AJB11.3031
Antonova, T. S., Araslanova, N. M., Strelnikov, E. A., Ramazanova, S. A., Guchetl, S. Z., and Chelyustnikova, T. A. (2013). Distribution of highly virulent races of sunflower broomrape (Orobanche cumana Wallr.) in the Southern regions of the Russian Federation. Russ. Agric. Sci. 39, 46–50. doi: 10.3103/S1068367413010023
Barzen, E., Mechelke, W., Ritter, E., Schulte-Kappert, E., and Salamini, F. (1995). An extended map of the sugar beet genome containing AFLP and RFLP loci. Theor. Appl. Genet. 90, 189–193. doi: 10.1111/j.1439-0523.1993.tb00641.x
Berry, S. T., Leon, A. J., Hanfrey, C. C., Challis, P., Burkholz, A., Barnes, S. R., et al. (1995). Molecular marker analysis of Helianthus annuus L. 2. Construction of an RFLP linkage map for cultivated sunflower. Theor. Appl. Genet. 91, 195–199. doi: 10.1007/BF00220877
Caspi, R., Billington, R., Fulcher, C. A., Keseler, I. M., Kothari, A., Krummenacker, M., et al. (2018). The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 46, D633–D639. doi: 10.1093/nar/gkx935
Castejón-Muñoz, M., Suso, M. J., Romero-Muñoz, F., and García-Torres, L. (1991). “Isoenzymatic study of broomrape (Orobanche cernua) populations infesting sunflower (Helianthus annuus),” in Proceedings of the 5th International Symposium on Parasitic Weeds, eds J. K. Ransom, L. J. Musselman, A. D. Worsham, and C. Parker (Nairobi: International Maize and Wheat Improvement Center – CIMMYT), 313–319.
Coque, M., André, T., Giménez, R., Archipiano, M., Polovynko, L., Tardin, M. C., et al. (2016). “Study of Orobanche cumana genetic diversity,” in Proceedings of the 19th International Sunflower Conference, eds Y. Kaya and S. Hasancebi (Paris: International Sunflower Association), 734.
Fernández-Escobar, J., Rodríguez-Ojeda, M. I., Fernández-Martínez, J. M., and Alonso, L. C. (2009). Sunflower broomrape (Orobanche cumana Wallr.) in Castilla-León, a traditionally non-broomrape infested area in Northern Spain. Helia 51, 57–64. doi: 10.2298/HEL0951057F
Fernández-Martínez, J. M., Pérez-Vich, B., and Velasco, L. (2015). “Sunflower broomrape (Orobanche cumana Wallr.),” in Sunflower Oilseed. Chemistry, Production, Processing and Utilization, eds E. Martínez-Force, N. T. Dunford, and J. J. Salas (Champaign, IL: AOCS Press), 129–156.
Gagne, G., Roeckel-Drevet, P., Grezes-Besset, B., Shindrova, P., Ivanov, P., Grand-Ravel, C., et al. (1998). Study of the variability and evolution of Orobanche cumana populations infesting sunflower in different European countries. Theor. Appl. Genet. 96, 1216–1222. doi: 10.1007/s001220050859
González-Torres, R., Jiménez-Díaz, R. M., and Melero-Vara, J. M. (1982). Distribution and virulence of Orobanche cernua in sunflower crops in Spain. Phytopathologische Zeitschrift 104, 78–89. doi: 10.1111/j.1439-0434.1982.tb00008.x
Gouzy, J., Pouilly, N., Boniface, M. C., Bouchez, O., Carrère, S., Catrice, O., et al. (2017). “The complete genome sequence of Orobanche cumana (sunflower broomrape),” in Proceedings of the 14th World Congress on Parasitic Plants, (Sheffield: International Plant Parasitic Society).
Guchetl, S. Z., Antonova, T. S., and Tchelustnikova, T. A. (2014). Genetic similarity and differences between the Orobanche cumana wallr. populations from Russia, Kazakhstan, and Romania revealed using the markers of simple sequence repeat. Russ. Agric. Sci. 40, 326–330. doi: 10.3103/S1068367414050103
Haanstra, J. P. W., Wye, C., Verbakel, H., Meijer-Dekens, F., Berg, P. V. D., Odinot, P., et al. (1999). An integrated high-density RFLP-AFLP map of tomato based on two lycopersicon esculentum ×L. pennellii F2 populations. Theor. Appl. Genet. 99, 254–271. doi: 10.1007/s001220051231
Hao, M., Moore, P. H., Liu, Z., Kim, M. S., Yu, Q., Fitch, M. M. M., et al. (2004). High-density linkage mapping revealed suppression of recombination at the sex determination locus in papaya. Genetics 166, 419–436. doi: 10.1534/genetics.166.1.419
Jenczewski, E., Gherardi, M., Bonnin, I., Prosperi, J. M., Olivieri, I., and Huguet, T. (1997). Insight on segregation distortions in two intraspecific crosses between annual species of Medicago(Leguminosae). Theor. Appl. Genet. 94, 682–691.
Jessup, R. W., Burson, B. L., Burow, G. B., Wang, Y.-W., Chang, C., Li, Z., et al. (2002). Disomic inheritance, suppressed recombination, and allelic interaction govern apospory in buffelgrass as revealed by genome mapping. Crop Sci. 42, 1688–1694. doi: 10.2135/cropsci2002.1688
Jestin, C., Lecomte, V., and Duroueix, F. (2014). “Current situation of sunflower broomrape in France,” in Proceedings of the 3rd International Symposium on Broomrape (Orobanche spp.) in Sunflower, (Paris: International Sunflower Association), 28–31.
Kaya, Y., Evci, G., Pekcan, V., Gucer, T., and Yilmaz, M. I. (2009). Evaluation of broomrape resistance in sunflower hybrids. Helia 51, 161–170. doi: 10.2298/hel0951161k
Kochert, G. (1994). “RFLP technology,” in DNA-Based Markers in Plants, eds R. L. Phillips and K. Vasil (Dordrecht: Springer), 8–38.
Kreutz, C. A. J. (1995). Orobanche: The European Broomrape Species. I. Central and Northern Europe. Maastricht: Stichting Natuurpublicaties Limburg.
Lincoln, S., Daley, M., and Lander, E. (1993). Constructing Genetic Linkage Map with Mapmaker/exp. 3.0: A Tutorial and Reference Manual, 3rd Edn. Cambridge: Whitehead Institute for Biometrical Research.
Malek, J., del Moral, L., Fernández-Escobar, J., Pérez-Vich, B., and Velasco, L. (2017). Racial characterization and genetic diversity of sunflower broomrape populations from Northern Spain. Phytopathol. Mediterr. 56, 70–76. doi: 10.14601/Phytopathol_Mediterr-19163
Martín-Sanz, A., Malek, J., Fernández-Martínez, J. M., Pérez-Vich, B., and Velasco, L. (2016). Increased virulence in sunflower broomrape (Orobanche cumana Wallr.) populations from Southern Spain is associated with greater genetic diversity. Front. Plant Sci. 7:589. doi: 10.3389/fpls.2016.00589
Mézard, C. (2006). Meiotic recombination hotspots in plants. Biochem. Soc. Trans. 34, 531–534. doi: 10.1042/BST0340531
Molinero-Ruiz, M. L., García-Carneros, A. B., Collado-Romero, M., Raranciuc, S., Domínguez, J., and Melero-Vara, J. M. (2014). Pathogenic and molecular diversity in highly virulent populations of the parasitic weed Orobanche cumana from Europe. Weed Res. 54, 87–96. doi: 10.1111/wre.12056
Nabloussi, A., Velasco, L., and Assissel, N. (2018). First report of sunflower broomrape, Orobanche cumana Wallr., in Morocco. Plant Dis. 102:457. doi: 10.1094/PDIS-06-17-0858-PDN
Pacureanu-Joita, M., Raranciuc, S., Sava, E., Stanciu, D., and Nastase, D. (2009). Virulence and aggressiveness of sunflower broomrape (Orobanche cumana Wallr.) populations in Romania. Helia 51, 111–118. doi: 10.2298/hel0951111p
Pacureanu-Joita, M., Veronesi, C., Raranciuc, S., and Stanciu, D. (2004). “Parasite-host plant interaction of Orobanche cumana Wallr. (Orobanche cernua Loefl) with Helianthus annuus,” in Proceedings of the 16th International Sunflower Conference, ed. G. J. Seiler (Paris: International Sunflower Association),171–177.
Peakall, R., and Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Pescott, O. (2013). The Genetics of Host Adaptation in the Parasitic Plant Striga Hermonthica. Ph.D. thesis, The University of Sheffield, Sheffield.
Piednoël, M., Aberer, A. J., Schneeweiss, G. M., Macas, J., Novák, P., Gundlach, H., et al. (2012). Next-generation sequencing reveals the impact of repetitive DNA across phylogenetically closely related genomes of Orobanchaceae. Mol. Biol. Evol. 29, 3601–3611. doi: 10.1093/molbev/mss168
Pineda-Martos, R., Velasco, L., Fernández-Escobar, J., Fernández-Martínez, J. M., and Pérez-Vich, B. (2013). Genetic diversity of sunflower broomrape (Orobanche cumana) populations from Spain. Weed Res. 53, 279–289. doi: 10.1111/wre.12022
Pineda-Martos, R., Velasco, L., and Pérez-Vich, B. (2014). Identification, characterization, and discriminatory power of microsatellite markers in the parasitic weed Orobanche cumana. Weed Res. 54, 120–132. doi: 10.1111/wre.12062
Pouilly, N., Gouzy, J., Boniface, M. C., Bouchez, O., Carrère, S., Catrice, O., et al. (2018). “The complete genome sequence of the parasitic weed Orobanche cumana (sunflower broomrape),” in Proceeding of the XXVI Plant and Animal Genome Conference XXVI, (San Diego),
Quillet, M. C., Madjidian, N., Griveau, Y., Serieys, H., Tersac, M., Lorieux, M., et al. (1995). Mapping genetic factors controlling pollen viability in an interspecific cross in Helianthus sect. Helianthus. Theor. Appl. Genet. 91, 1195–1202. doi: 10.1007/BF00220929
Rodríguez-Ojeda, M. I., Pérez-Vich, B., Alonso, L. C., and Fernández-Escobar, J. (2010). The influence of flowering plant isolation on seed production and seed quality in Orobanche Cumana. Weed Res. 50, 145–150. doi: 10.1111/j.1365-3180.2010.00817.x
Rodríguez-Ojeda, M. I., Fernández-Martínez, J. M., Velasco, L., and Pérez-Vich, B. (2013a). Extent of cross-fertilization in Orobanche cumana Wallr. Biol. Plantarum 57, 559–562. doi: 10.1007/s10535-012-0301-1
Rodríguez-Ojeda, M. I., Pineda-Martos, R., Alonso, L. C., Fernández-Escobar, J., Fernández-Martínez, J. M., Pérez-Vich, B., et al. (2013b). A dominant avirulence gene in Orobanche cumana triggers Or5 resistance in sunflower. Weed Res. 53, 322–327. doi: 10.1111/wre.12034
Rodríguez-Ojeda, M. I., Velasco, L., Alonso, L. C., Fernández-Escobar, J., and Pérez-Vich, B. (2011). Inheritance of the unpigmented plant trait in Orobanche cumana. Weed Res. 51, 151–156. doi: 10.1111/j.1365-3180.2010.00830.x
Rumsey, F. J. (2007). A reconsideration of Orobanche maritima Pugsley (Orobanchaceae) and related taxa in southern England and the Channel Islands. Watsonia 26, 473–476.
Rumsey, F. J., and Jury, S. L. (1991). An account of Orobanche L. in Britain and Ireland. Watsonia 18, 257–295.
Schneeweiss, G. M., Palomeque, T., Colwell, A. E., and Weiss-Schneeweiss, H. (2004). Chromosome numbers and karyotype evolution in holoparasitic Orobanche (Orobanchaceae) and related genera. Am. J. Bot. 91, 439–448. doi: 10.3732/ajb.91.3.439
Shindrova, P., and Penchev, E. (2012). Race composition and distribution of broomrape (Orobanche cumana Wallr.) in Bulgaria during 2007-2011. Helia 57, 87–94. doi: 10.2298/hel1257087s
Snedecor, G. W., and Cochran, W. G. (1989). Statistical Methods, 8th Edn. Ames, IA: Iowa State University Press.
Stapley, J., Feulner, P. G. D., Johnston, S. E., Santure, A. W., and Smadja, C. M. (2017). Variation in recombination frequency and distribution across eukaryotes: patterns and processes. Philos. Trans. R. Soc. B 372:20160455. doi: 10.1098/rstb.2016.0455
Thomas, V. P., Fudali, S. L., Schaff, J. E., Liu, Q., Scholl, E. H., Opperman, C. H., et al. (2012). A sequence-anchored linkage map of the plant-parasitic nematode Meloidogyne hapla reveals exceptionally high genome-wide recombination. Genes Genomes Genet. 2, 815–824. doi: 10.1534/g3.112.002261
Velasco, L., Pérez-Vich, B., Yassein, A. A. M., Jan, C. C., and Fernández-Martínez, J. M. (2012). Inheritance of resistance to sunflower broomrape (Orobanche cumana Wallr.) in a interspecific cross between Helianthus annuus and Helianthus debilis subsp. tardiflorus. Plant Breed. 121, 220–221. doi: 10.1111/j.1439-0523.2011.01915.x
Voorrips, R. E. (2002). MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78. doi: 10.1093/jhered/93.1.77
Vranceanu, A. V., Tudor, V. A., Stoenescu, F. M., and Pirvu, N. (1980). “Virulence groups of Orobanche cumana Wallr. differential hosts and resistance sources and genes in sunflower,” in Proceedings of the 9th International Sunflower Conference, (Paris: International Sunflower Association), 74–80.
Weiss-Schneeweiss, H., Greilhuber, J., and Schneeweiss, G. M. (2006). Genome size evolution in holoparasitic Orobanche (Orobanchaceae) and related genera. Am. J. Bot. 93, 148–156. doi: 10.3732/ajb.93.1.148
Yang, B., Liu, H., Yang, J., Gupta, V. K., and Jiang, Y. (2018). New insights on bioactivities and biosynthesis of flavonoid glycosides. Trends Food Sci. Technol. 79, 116–124. doi: 10.1016/j.tifs.2018.07.006
Keywords: linkage map, plant pigmentation, segregating populations, sunflower, sunflower broomrape
Citation: Calderón-González Á, Pouilly N, Muños S, Grand X, Coque M, Velasco L and Pérez-Vich B (2019) An SSR-SNP Linkage Map of the Parasitic Weed Orobanche cumana Wallr. Including a Gene for Plant Pigmentation. Front. Plant Sci. 10:797. doi: 10.3389/fpls.2019.00797
Received: 12 November 2018; Accepted: 03 June 2019;
Published: 19 June 2019.
Edited by:
Agata Gadaleta, University of Bari Aldo Moro, ItalyReviewed by:
Umesh K. Reddy, West Virginia State University, United StatesPedro Martinez-Gomez, Center for Edaphology and Applied Biology of Segura, Spanish National Research Council (CSIC), Spain
Copyright © 2019 Calderón-González, Pouilly, Muños, Grand, Coque, Velasco and Pérez-Vich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Begoña Pérez-Vich, YnBlcmV6QGlhcy5jc2ljLmVz