Abstract
The tribe Geonomateae is a widely distributed group of 103 species of Neotropical palms which contains six ecologically important understory or subcanopy genera. Although it has been the focus of many studies, our understanding of the evolutionary history of this group, and in particular of the taxonomically complex genus Geonoma, is far from complete due to a lack of molecular data. Specifically, the previous Sanger sequencing-based studies used a few informative characters and partial sampling. To overcome these limitations, we used a recently developed Arecaceae-specific target capture bait set to undertake a phylogenomic analysis of the tribe Geonomateae. We sequenced 3,988 genomic regions for 85% of the species of the tribe, including 84% of the species of the largest genus, Geonoma. Phylogenetic relationships were inferred using both concatenation and coalescent methods. Overall, our phylogenetic tree is highly supported and congruent with taxonomic delimitations although several morphological taxa were revealed to be non-monophyletic. It is the first time that such a large genomic dataset is provided for an entire tribe within the Arecaceae. Our study lays the groundwork not only for detailed macro- and micro-evolutionary studies within the group, but also sets a workflow for understanding other species complexes across the tree of life.
Introduction
Palms (Arecaceae) are an important ecological component (Henderson, 2002; Couvreur et al., 2011) and a useful plant group of tropical ecosystems (Macia et al., 2011; Gruca et al., 2015). The palm family was recently advocated as a model group to understand the evolution of tropical rain forests (Baker and Couvreur, 2013) and numerous studies have investigated their phylogenetic relationships and systematics (Uhl and Dransfield, 1987; Baker et al., 1999; Dransfield et al., 2008; Baker and Dransfield, 2016). However, given the remarkably low rate of molecular evolution observed in palms (Wilson et al., 1990), phylogenetic studies at different taxonomic levels within the Arecaceae based on a few plastid or nuclear genes generally result in poorly resolved phylogenetic trees, especially at the species level (Roncal et al., 2005, 2008; Cuenca et al., 2008; Baker et al., 2011; Bacon et al., 2012a, 2016a,b, 2017; Meerow et al., 2015; Sanín et al., 2016).
The lack of informative genetic markers, combined with insufficient taxonomic sampling, currently limits our understanding of the phylogenetic relationships within the most diverse palm genera in the Neotropics, such as Bactris Jacq. ex Scop., Chamaedorea Willd., and Geonoma Willd. These three genera are mostly small shade-adapted palms and they contain the most abundant palm species in the understory of many Neotropical forests (Vormisto et al., 2004; Balslev et al., 2016, 2017; Ley-lopez and Avalos, 2017; Muscarella et al., 2018). They also often exhibit a high amount of intraspecific phenotypic variation (Roncal, 2006), which is hard to address with a taxonomic classification. This is exemplified by Geonoma, which, with 68 recognized species (Henderson, 2011), is the third most diverse palm genus in the Neotropics. Geonoma belongs to the tribe Geonomateae Luerss., together with five other genera. These five additional genera range in size from two (Welfia H. Wendl.) to 21 species (Calyptrogyne H. Wendl.), and are also small understory palms except for Calyptronoma Griseb. (three species) and Welfia, which can reach up to 15 m and 25 m, respectively. The tribe displays a wide geographical and ecological distribution, occurring from southern Mexico to south-eastern Brazil, including the Caribbean, with species growing from the lowlands up to 3,000 m elevation in the Andes. The tribe has been intensively studied and its main biological aspects, such as taxonomy (Wessels Boer, 1968; Zona, 1995; Stauffer et al., 2003; Henderson, 2005, 2011, 2012; Henderson and Villalba, 2013), ecology (Chazdon, 1992; Knudsen et al., 1998; Sampaio and Scariot, 2008; Pizo and Almeida-Neto, 2009), and phylogenetic relationships (Roncal et al., 2005, 2010, 2011, 2012) have been characterized to some extent. Considerable research has also been dedicated to investigate the phenotypically widely variable species complexes that represent 20% of the species of Geonoma (Borchsenius, 2002; Henderson and Martins, 2002; Roncal, 2006; Roncal et al., 2007; Henderson, 2011; Borchsenius et al., 2016). Despite all these efforts, the evolutionary history of Geonoma and Geonomateae remains only partially understood due to the paucity of DNA sequences, which so far are available only for three nuclear loci and approximately 60% of the species in the tribe.
Obtaining a robust phylogenetic hypothesis for the Geonomateae is therefore crucial to enable a reliable assessment of the systematic relationships of its lineages, but also to provide the foundation to assess the macroevolutionary patterns and the dynamics of diversification in this key palm group. The increasing affordability of next generation sequencing techniques, which offers the possibility to sequence hundreds of loci at a time, has already benefitted many plant phylogenetic studies (e.g., Nicholls et al., 2015; Sass et al., 2016; Mandel et al., 2017; Mitchell et al., 2017). For Arecaceae, while most of genome-scale data initially focused on commercially important species such as the oil palm (Uthaipaisanwong et al., 2012; Singh et al., 2013) and the date palm (Yang et al., 2010; Al-Mssallem et al., 2013), evolutionary biologists have put considerable effort in the last few years to generate genomic data across the whole family and are aiming at a species level phylogenetic tree of all palms (Comer et al., 2015, 2016; Heyduk et al., 2015; Barrett et al., 2016, 2018).
In this context, the recent development of several sequence capture kits for the Arecaceae (Heyduk et al., 2015; de La Harpe et al., 2019) represents an ideal opportunity to fill the gaps in palm phylogenomics. Here, using the bait kit developed by de La Harpe et al. (2019), we sequenced 4,184 genomic regions for 85% of the species of tribe Geonomateae, including 84% of the species of Geonoma and applied both standard and coalescent-based methods to reconstruct the phylogenetic relationships within the tribe. Using substantial intraspecific sampling, we assessed the validity of the species delimitations proposed by Henderson (2011) for the widespread and highly morphologically variable species complexes. We also estimated the phylogenetic informativeness of the DNA regions in the capture kit and proposed a smaller selection of the most useful genomic regions for phylogenetic studies at deep and shallow evolutionary scales within the Arecaceae. Our results show that these new molecular tools increase our understanding of the systematics and evolution in this important group of understory palms and open up new directions of research to test hypotheses about the factors underlying the diversification of species in palms.
Materials and Methods
Taxon Sampling
We gathered a total of 312 samples of either silica-dried leaves or herbarium fragments from specimens stored at the herbarium of Geneva (G) and the Herbario Nacional Colombiano (COL), including 240 samples representing 57 (84%) of the 68 currently recognized species of Geonoma (Supplementary Table S1). Among the 11 missing species of Geonoma, eight are narrow endemics known only from the type collections (G. deneversii A. J. Hend., G. dindoensis A. J. Hend., G. gentryi A. J. Hend. and G. operculata A. J. Hend.) or less than ten herbarium specimens (G. peruviana A. J. Hend., G. sanmartinensis A. J. Hend., G. schizocarpa A. J. Hend. and G. venosa A. J. Hend.). Whenever possible, we sampled several individuals per species and included different subspecies. For widely distributed species, sample selection was designed to cover the greatest possible extant of their geographic distribution. Our sampling also included 65 individuals representing 25 species from the other five genera of the tribe Geonomateae (100% taxon sampling for Asterogyne H. Wendl, 61% for Calyptrogyne, 100% for Calyptronoma, 75% for Pholidostachys H. Wendl. Ex Hook. f., and 50% for Welfia), covering in total 85% of the tribe’s species richness. For the purpose of computing the phylogenetic informativeness of the targeted genomic regions across the whole Arecaceae, we also included seven samples from phylogenetically more distant palm genera, belonging to subfamilies Arecoideae Burnett (Bactris, Cocos L., Socratea H. Karst, and Wettinia Poepp.), Ceroxyloideae Drude (Ceroxylon Bonpl. ex DC.), and Coryphoideae Burnett (Licuala Wurmb).
DNA Extraction, Dual-Indexed Library Preparation, and Target Capture Sequencing
DNA was extracted using the DNeasy® plant mini kit (Qiagen, Venlo, Netherlands) following the supplier’s instructions. DNA quality and degradation were evaluated with agarose gels and a NanodropTM TM spectrophotometer ND-1000 (Thermo Fisher Scientific, Waltham, MA, United States) and DNA was quantified with a Qubit® Fluorometer v 2.2 (Thermo Fisher Scientific, Waltham, MA, United StatesUSA). When possible, a total of 500 ng of DNA were used per sample for library preparation.
DNA samples were fragmented to 400 bp fragments with a bioruptor® ultrasonicator UCD-200TM-EX (Diagenode, Liège, Belgium) with six cycles of 30 s ON, and 90 s OFF. This step was omitted for samples with degraded DNA. Library preparations were performed following de La Harpe et al. (2019). Briefly, sample cleaning, end repair and A-tailing steps were carried out with a KAPA LTP library preparation kit (Roche, Basel, Switzerland), and adaptor ligation and adaptor fill-in reactions steps (Meyer and Kircher, 2010).
A set of 60 dual-index primers were used for amplification, as recommended by Kircher et al. (2011), to avoid inaccuracies in multiplex sequencing. Two sets of 7 bp indexes were generated using the create_index_sequences.py Python program (Meyer and Kircher, 2010): one set of 30 indexes for the P5 Illumina primers, and one set of 30 indexes for the P7 Illumina primers. The index lists were chosen to contain a balanced subset of indexes with an edit distance of 4 to reduce the chance of conversion by sequencing and amplification errors. Adaptor and primer sequences are described in Supplementary Table S2. Eight cycles of PCR were used for most samples, except for 29 low quality and degraded samples for which 12 cycles of PCR were necessary to obtain sufficient DNA amount (Supplementary Table S1). Libraries were quantified with a Qubit® Fluorometer v 2.2. Target capture was performed using the custom kit PopcornPalm developed by de La Harpe et al. (2019) and deposited in Dryad1. This kit targets 4,051 genes and 133 non-genic putatively neutral regions. Target capture was conducted on pooled dual-indexed libraries following myBait® Custom Target Capture Kits protocol v3.0 (Arbor Biosciences, Ann Arbor, MI, United States), with 18 h incubation time at 65°C and 12 cycles of post-capture PCR reactions. Pools of 64 samples were used as template for each target capture hybridization reaction, using an initial amount of 1.2 μg of pooled libraries. The pooled target capture reactions were quantified with a Qubit® Fluorometer v 2.2 before sequencing with an Illumina HiSeq3000 sequencer in paired-end 2 × 150 bp mode.
Read Trimming, Mapping, and SNP Calling
Reads were first trimmed with the program condetri v2.2 (Smeds and Künstner, 2011) using a base quality score of 20 as high-quality threshold parameter before mapping to the Geonoma undata Klotzsch pseudoreference genome described in de La Harpe et al. (2019) with bowtie2 v2.2.5 (Langmead and Salzberg, 2012) and the very-sensitive-local option. Only reads that mapped at a unique location in the genome were kept for analysis.
Before variant calling, PCR duplicates were masked with the software Picard v1.1192, and reads were realigned around indels and base-recalibrated using GATK v3.8 (McKenna et al., 2010). SNPs were then called for targeted genomic regions using UnifiedGenotyper of GATK v3.8 using the EMIT_ALL_SITES option in order to obtain the full sequence of the targets. The main advantage of paired-end 2 × 150 bp read sequencing is the potential recovery of adjacent regions to the exonic targets. For this reason, the entire sequence including UTRs, exons and introns was called for each gene. Sites were filtered with the following parameters using VCFtools v0.1.13 (Danecek et al., 2011): minimum quality >20, no indel allowed, minimum depth of 8× per sample, and maximum of 50% of missing data. For each genomic region the alignment in fasta format was generated using the program vcf-tab-to-fasta3.
Selection of Most Informative Genomic Regions
Because the bait kit developed by de La Harpe et al. (2019) for micro- and macro-evolutionary analyses in palms is large (over 4,000 genomic regions) and contains several fast-evolving DNA regions that are not necessarily useful for phylogenomic studies, we selected a subsample of the most informative genomic regions which we then used to infer the species tree of the Geonomateae. Additionally, we made available a new bait kit for future phylogenomic studies in palms, which combines the subset of genes presented here with the genes from the Heyduk et al.’s kit (2015). Our workflow for gene selection and phylogenomic analyses is summarized in Figure 1. In order to maximize the phylogenetic informativeness of the retained genes for the Arecaceae and not only for tribe Geonomateae, the selection steps were performed on a dataset which contained species from three different Arecaceae subfamilies (Arecoideae, Ceroxyloideae, and Coryphoideae). First, we estimated the phylogenetic informativeness for each gene at different geological time intervals with the program TAPIR (Pond and Muse, 2005; Townsend, 2007; Faircloth et al., 2012). For each alignment, TAPIR estimates the site rates under the best-fitting substitution model and further computes a quantitative measure of the power of the gene to resolve the branching order at different depths of a given phylogenetic tree. To reduce computing time, the analysis was performed on a subset of 20 out of the 312 samples sequenced, which were selected to represent a wide range of evolutionary time scales, from intra-specific variability up to 88 Ma of divergence. The selection included three species of Geonoma (including four samples of G. deversa), two species of Asterogyne, two species of Calyptrogyne, as well as Welfia regia H. Wendl., Bactris gasipaes Kunth, Cocos nucifera L., Socratea exorrhiza (Mart.) H. Wendl, Wettinia maynensis Spruce, Ceroxylon alpinum Bonpl. ex DC., and two species of Licuala. Because TAPIR does not accept missing data, we only considered the genes for which sequence data were available for all 20 samples. Details for this analysis can be found in the Supplementary Material. Then, we selected the most appropriate genes for phylogenomic analyses according to the following criteria: (1) single-copy genes, (2) genes located on one of the 16 chromosomes of the Elaeis guineensis Jacq. reference genome (i.e., no gene on the extra low quality scaffolds), (3) genes absent from the bait kit of Heyduk et al. (2015) to avoid redundancy in the final bait set, (4) genes among the top 500 genes with the highest phylogenetic informativeness measure and/or with the highest mean bootstrap value per gene tree, (5) genes with a minimum mean bootstrap value per gene tree >60, and (6) genes with a minimum of five baits covering their exonic regions. We constrained our selection to a total of 17,091 baits to obtain a maximum of 20,000 baits when combined with the 2,909 baits of the Heyduk’s kit (Heyduk et al., 2015). This option thus allows for coherence among different studies and maximizes the informativeness of the data at the lowest possible cost, since the smallest kit size available at the Arbor Biosciences company (Ann Arbor, MI, United States) is of 20,000 baits.
FIGURE 1
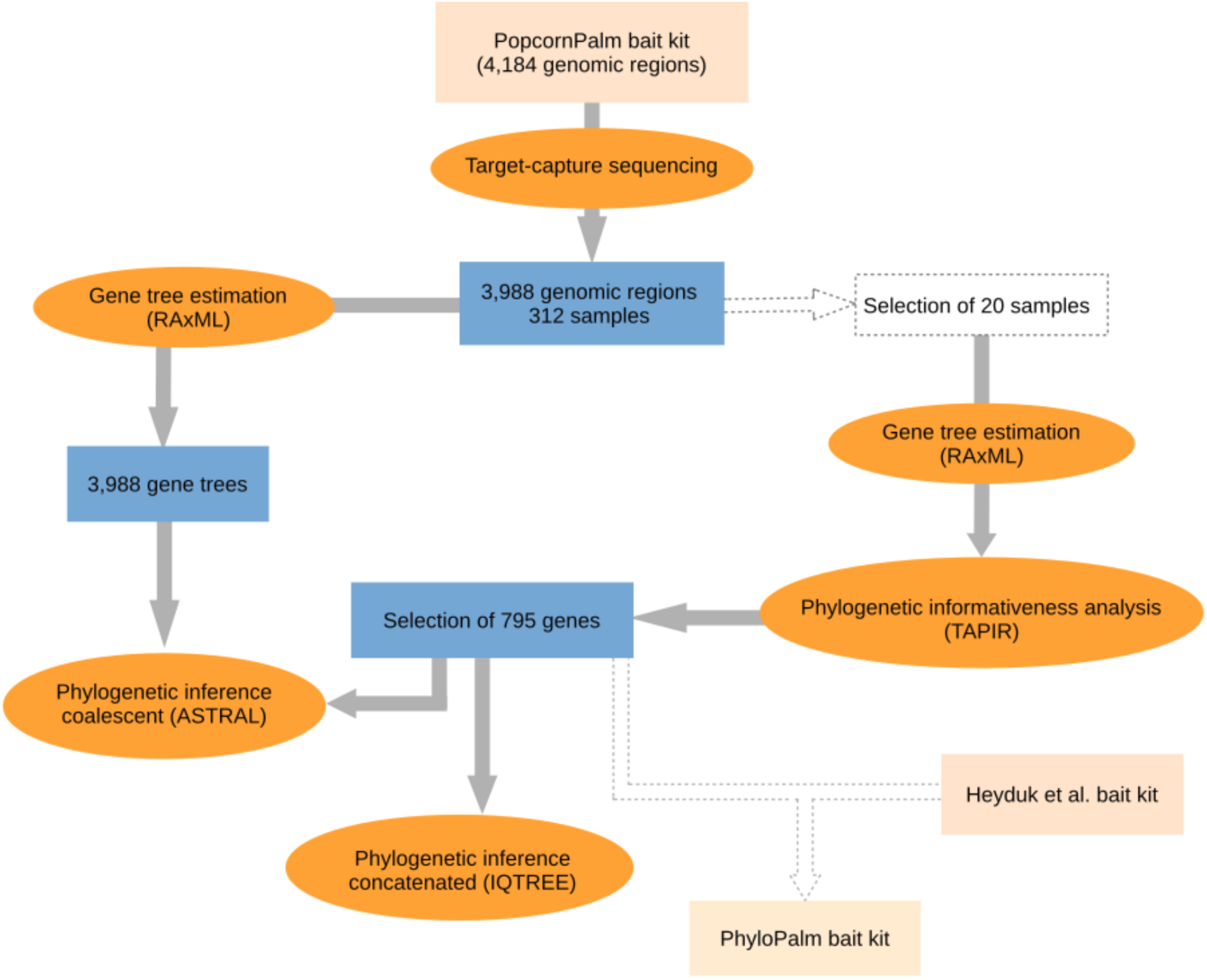
Flowchart illustrating the steps of the phylogenomic analyses. The selection of 795 genes was combined with the bait kit from Heyduk et al. (2015) to provide a new bait kit for future phylogenomic studies.
Phylogenetic Inference
Phylogenetic trees were estimated using both maximum likelihood and coalescent based methods. We used the software IQ-TREE (Nguyen et al., 2015) to estimate, under the maximum likelihood criteria, the topology and branch lengths of the phylogenetic tree for all samples based on the concatenated analysis of the reduced set of genes satisfying the criteria described above. We partitioned the data by gene (Chernomor et al., 2016), using a GTR+GAMMA model of substitutions for each gene, and estimated support using the ultrafast bootstrap option (Hoang et al., 2018). We did not perform model testing because parameter rich models such as GTR+G and GTR+G+I have been shown in simulations to suffice for phylogeny reconstruction (Hoff et al., 2016; Abadi et al., 2019). The consensus tree obtained from this analysis was visualized using Figtree v1.4.3 (Rambaut, 2012). Next, we applied a coalescent approach which takes into account gene tree incongruence due to incomplete lineage sorting (Liu et al., 2009). We used ASTRAL 4.10.12, a two-step coalescent-based method that estimates the species tree given a set of gene trees (Mirarab et al., 2014; Mirarab and Warnow, 2015). All gene trees were first estimated with RaxML 8.2.10 (Stamatakis, 2014), using the GTR+GAMMA model of substitution and support estimated with the -autoMRE option. We then performed ASTRAL on the reduced data set and obtained a measure of branch support by computing local posterior probabilities. The impact of including weakly informative genes in two-step coalescent analyses is debated and while some studies showed that it can help to resolve difficult nodes (Blom et al., 2017) others argued that it reduces the accuracy of species tree estimation (Liu et al., 2015). To test whether including less informative genes would improve our phylogenetic inference we also applied ASTRAL to our full genomic dataset. Additionally, we computed quartet support values to measure the level of gene tree incongruence in our dataset and plotted quartet support for the three possible topologies at each branch using a python script4. All trees were rooted using the two Licuala species as outgroup.
Results
Target Capture Sequencing
In total, we recovered DNA sequences for 3,988 genomic regions out of 4,184. On average, we obtained 2,064,810 reads per sample (Supplementary Table S1). After filtering, a total of 7,438,988 high quality bases including 2,288,308 SNPs were obtained with an average coverage of 30.8× and only 9.3% of missing data. When considering only the samples of Geonoma, 1,102,445 SNPs were recovered.
Phylogenetic Informativeness
Across our data set, phylogenetic informativeness increased with increasing evolutionary divergence times (Figure 2). After applying the selection step, the reduced dataset of 17,091 baits contained 795 genes, ranging from 1,108 to 12,710 bp in length. The corresponding bait kit combining our 795 genes with Heyduk’s baits (Heyduk et al., 2015) is available at the Arbor Biosciences company (Ann Arbor, MI, United States) under the name “PhyloPalm.”
FIGURE 2
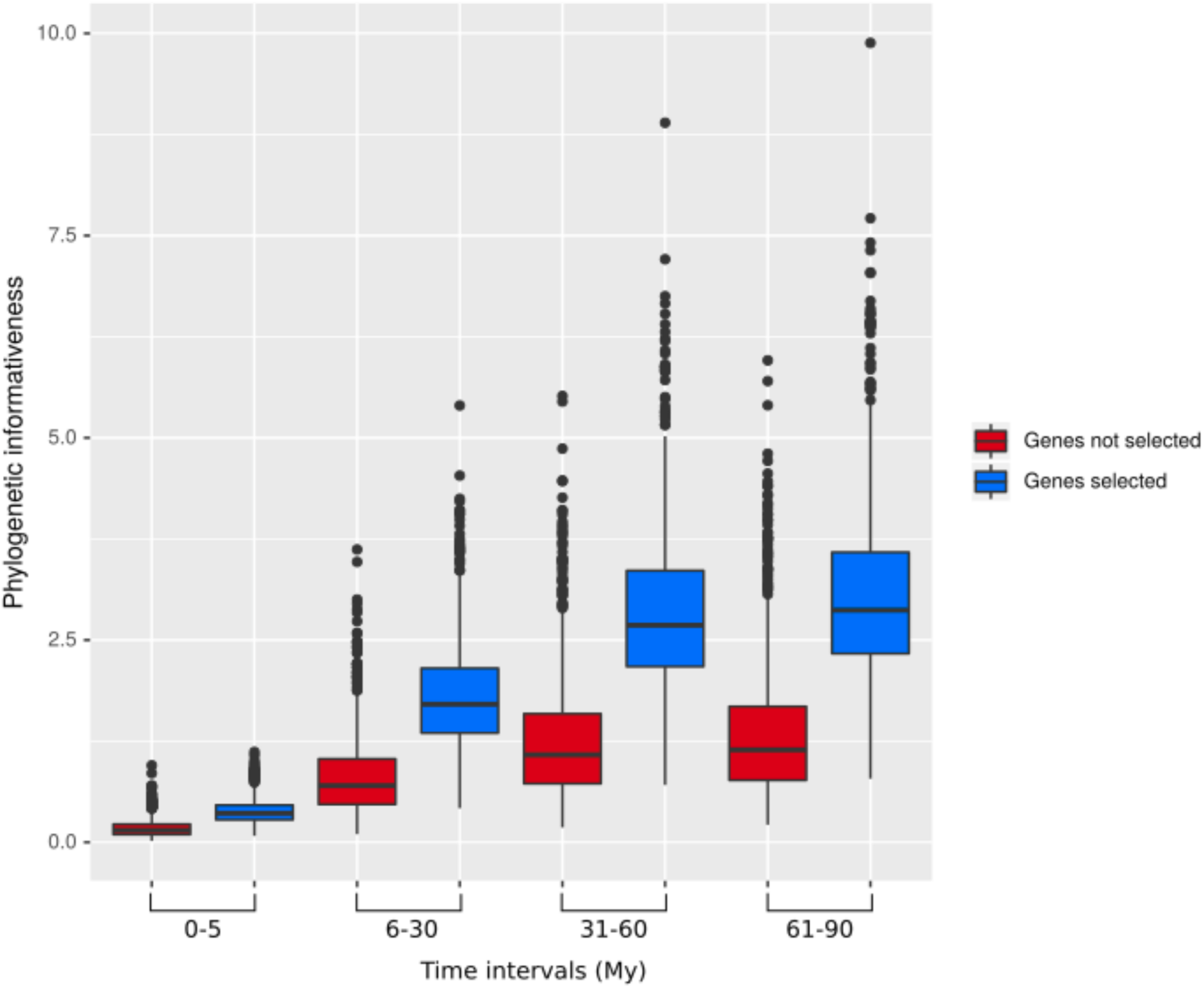
Phylogenetic informativeness of the 795 selected genes (blue) and the remaining genes (red) over different evolutionary time intervals.
Phylogenetic Inference
The total length of the concatenated alignment of the 795 selected genes was 3,064,021 bp. Phylogenetic trees obtained from the different datasets and methods had largely congruent topologies, except for the sister group of Clades XII-XIV (see section “Discussion” for clades numbers). This corresponded to Clade XI in the coalescent analysis of the 795 genes (with local posterior probability [LPP] of 0.59, Figure 3) and to Clades IX-X both in the concatenated analysis (with bootstrap support [BS] of 100%, Figure 4) and the coalescent analysis of the full dataset (with LPP of 0.66). For the 795 genes dataset, the support was slightly higher in the phylogenetic tree obtained with IQ-TREE (96% of nodes with BS >90, Figure 4) than with ASTRAL (89% of LPP >0.9, Figure 3). In the coalescent analyses, support increased with the size of the gene set, with 96% of branches having a LPP >0.9 in the phylogenetic tree obtained from the complete dataset of 3,988 genes. This is expected since the LPP are dependent on the discordance among gene trees but also the number of gene trees analyzed (Sayyari and Mirarab, 2016). Additionally, quartet support values indicated that gene tree incongruence is widespread across the phylogeny (Figure 3). In all analyses Calyptronoma was recovered paraphyletic, with C. plumeriana (Mart.) Lourteig and C. rivalis (O.F. Cook) L.H. Bailey more closely related to Calyptrogyne than to C. occidentalis (Sw.) H.E. Moore. The remaining five genera of tribe Geonomateae were recovered as monophyletic, with BS of 100% in the maximum likelihood phylogenetic tree and posterior probabilities of 1 in the coalescent phylogenetic trees.
FIGURE 3
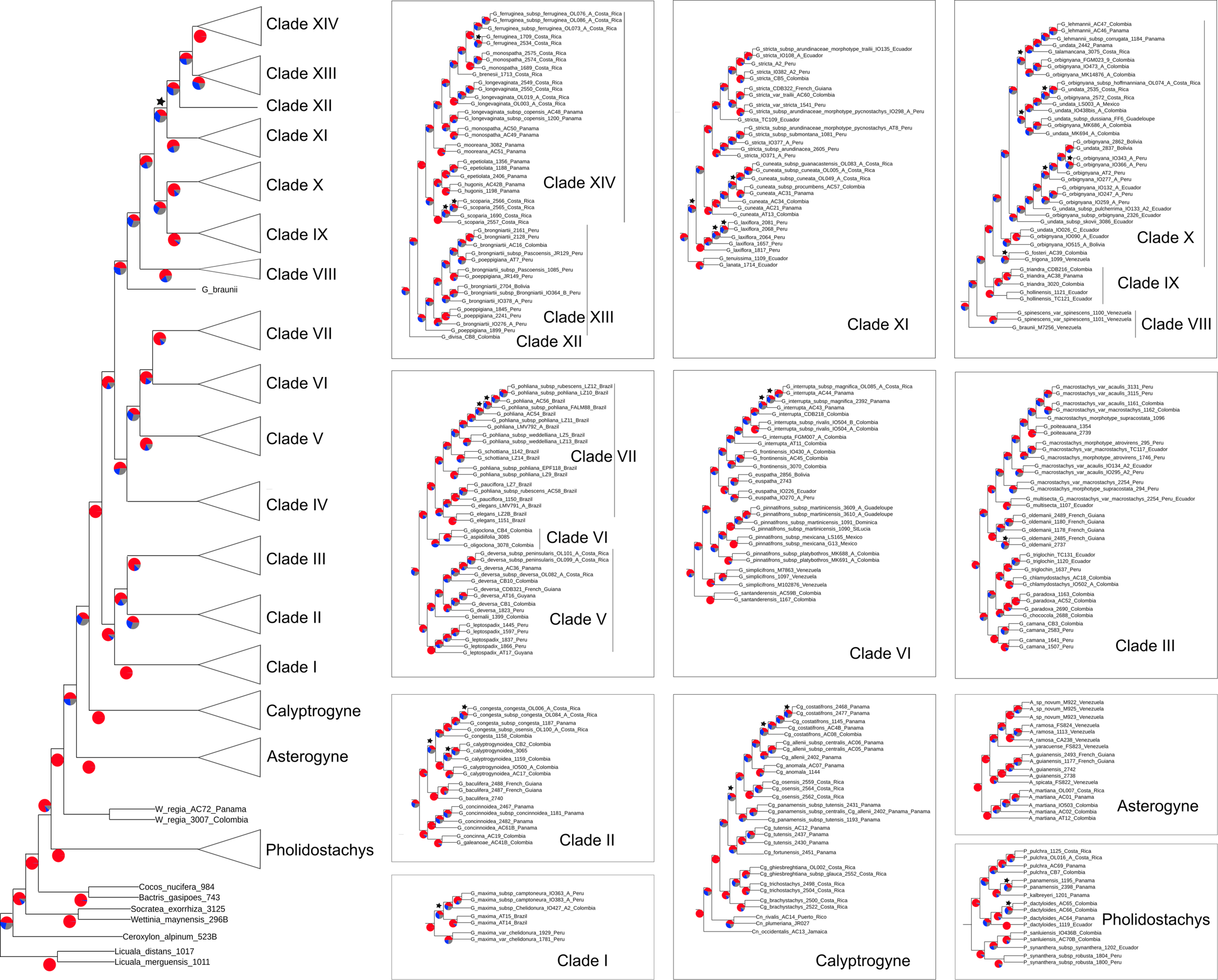
Cladogram inferred with ASTRAL on the set of 795 gene trees. Pie charts indicate for each branch the percentage of gene trees aggreing with the topology of the species tree (red) and the percentage of gene trees supporting the other two alternative topologies (blue and gray). Stars indicate branches with LPP below 0.9.
FIGURE 4
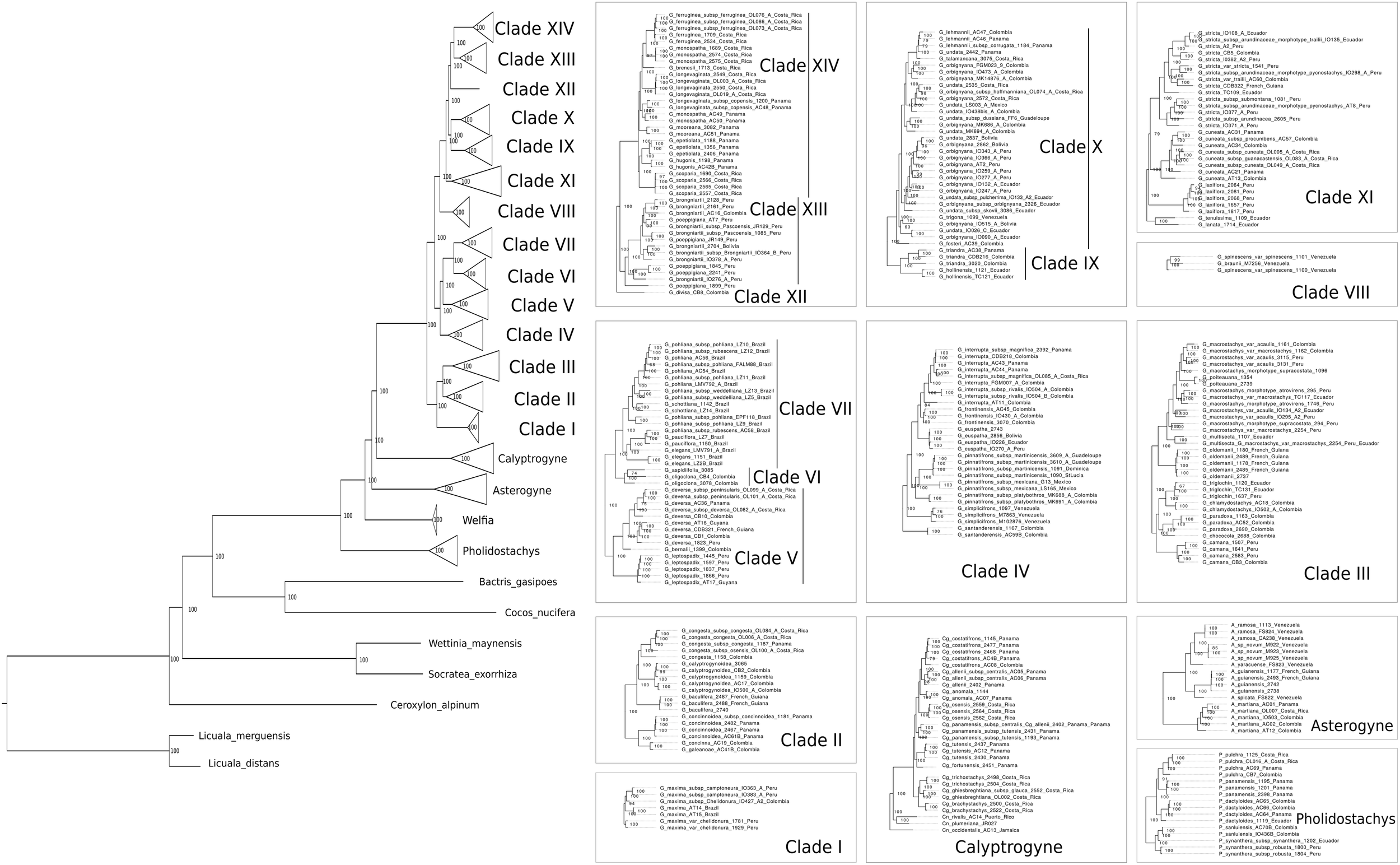
Maximum likelihood phylogeny inferred from the concatenated alignment of the 795 selected genes. Numbers indicate BS.
Discussion
The tribe Geonomateae is an ideal group to study plant evolutionary history in Neotropical rainforests for several reasons. First, it comprises the third largest genus of all Neotropical palms. Second, its species are distributed across all habitat types along the Andean and Central-American mountains as well as the Pacific, Caribbean and Amazonian lowlands, and in many of these areas they represent an important floristic element. Finally, Geonoma includes several species complexes with tremendous morphological variation which renders the taxonomic delimitation of species challenging. Because of these interesting characteristics, the systematics (Henderson et al., 1995; Henderson, 2011), ecology (Chazdon, 1991; Rodriguez-Buritica et al., 2005), and evolution (Roncal et al., 2011, 2012) of Geonoma have received significant attention. However, previous phylogenetic analyses relied on limited taxonomic and molecular sampling, thus preventing a detailed understanding of the phylogenetic relationships within the group. In particular, the phylogenetic trees recovered by Roncal et al. (2005, 2010, 2011, 2012) were not fully resolved and the status of species complexes had not been investigated. In this study, we addressed these shortcomings by applying a target-capture approach, using the baits developed by de La Harpe et al. (2019) to sequence nearly 4,000 genes for 57 species of Geonoma and 25 species of the closely related genera of tribe Geonomateae. We performed concatenation and coalescent-based phylogenetic inferences which resulted in highly similar topologies, despite substantial amount of gene tree incongruence across the phylogeny. We showed that only a fraction of our complete genomic dataset was sufficient to resolve phylogenetic relationships within the Geonomateae (Figures 3, 4).
Implications for the Systematics of Tribe Geonomateae
Phylogenetic relationships between the six genera of Geonomateae were so far poorly understood since various studies recovered different topologies (e.g., Baker et al., 2009; Roncal et al., 2010, 2011, 2012). Here, we were able to fully resolve the intergeneric relationships within the tribe but quartet support values in ASTRAL indicate a high level of gene tree incongruence (Figure 3), which problably explains the contrasted findings of the previous studies. We hypothesize that incomplete lineage sorting caused by the rapid divergence of the genera within tribe Geonomateae, as suggested by the very short branches in the maximum likelihood phylogenetic tree (Figure 4), is responsible for the observed gene tree discordance. Additionally, we confirmed the previously hypothesized paraphyly of the Caribbean endemic genus Calyptronoma and consequently advocate that it should be synonymized under Calyptrogyne, the sister group of Geonoma. The unique sample of P. synanthera (Mart.) H.E. Moore subspecies synanthera did not cluster with our two samples of P. synanthera subspecies robusta (Trail) A.J. Hend., but was instead recovered as sister to P. sanluiensis A. J. Hend. Pholidostachys synanthera subspecies synanthera and P. sanluiensis are Andean taxa which co-occur in the Central Cordillera of Colombia whereas P. synanthera subspecies robusta is a lowland West-Amazonian taxa. Additional sampling would be needed to test whether this placement is the result of hybridization between P. synanthera subspecies synanthera and P. sanluiensis or whether subspecies synanthera and robusta are actually separate species. Finally, we were able to assess the robustness of the current taxonomy for Geonoma, the largest and taxonomically most challenging genus of the tribe, in which the large degree of phenotypic variation has complicated species delimitations for a long time.
Phylogenetic Clades Within Geonoma
Based on our coalescent phylogeny and following the most recent phylogenetic reconstructions of the genus (Henderson, 2011; Roncal et al., 2011), we recognize 14 well-supported clades within Geonoma (Figures 3, 5). We compare our findings with those from the maximum parsimony analysis of 30 morphological traits in the last revision of the group (Henderson, 2011). We use numbers to refer to clades to avoid confusion with the clade names previously used by Roncal et al. (2011) and Henderson (2011).
FIGURE 5
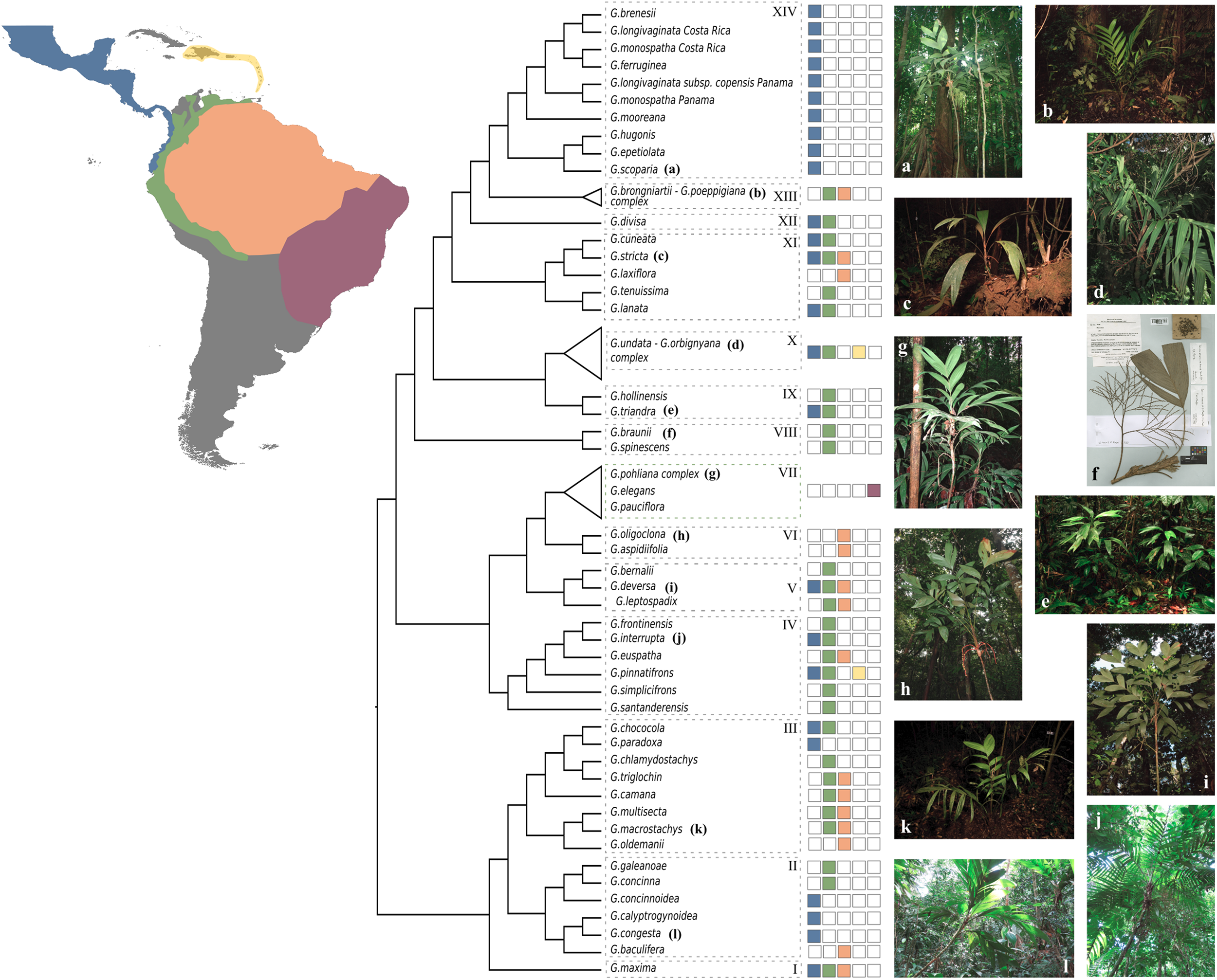
Summary cladogram of the phylogenetic relationships in Geonoma. Dashed boxes with numbers refer to clades mentioned in the discussion. Colored squares show the geographic distribution of species. Photos by Ingrid Olivares (a), FP7-PALMS project archive (b,c,h,i,k), Rodrigo Cámara Leret (e), Fred Stauffer (f), Oriane Loiseau (d,j,l), and Talita Mota Machado (g).
Clade I
This clade comprises a single variable species, G. maxima (Poit.) Kunth. It was included by Henderson (2011) in his G. macrostachys clade, which corresponds to Clade III in our analysis (Figure 5). Geonoma maxima differs, however, from all species of that clade in having a locular epidermis without an operculum, and a higher number of rachillae (4–50 vs. 1–9 rachillae; Henderson, 2011). Henderson (2011) recognized 11 subspecies within this primarily Amazonian lowland species, but the two subspecies with several individuals sampled (G. maxima subsp. camptoneura (Burret) A.J. Hend. and G. maxima subsp. chelidonura (Spruce) A.J. Hend) did not form monophyletic groups (Figures 3, 4).
Clade II
This clade comprises six species (G. baculifera (Poit.) Kunth, G. calyptrogynoidea Burret, G. concinna Burret, G. concinnoidea A.J. Hend, G. congesta H, Wendl. ex Spruce, G. galeanoae A.J. Hend; Figure 5) that mostly grow in the lowlands of the Chocó region from Costa Rica to north-western Ecuador, with only G. baculifera occurring in north-eastern Amazonia and the Guianas. This clade corresponds to Henderson’s (2011)G. congesta clade, which is characterized by the prophyll surfaces with close, equal, parallel, and non-dividing ridges. Henderson (2011) did not include G. galeanoae in this clade despite also sharing this trait, because its position in his morphology-based maximum parsimony tree was unresolved. Our analysis firmly recovers that species as a member of the clade and as sister to G. concinna with strong support (BS of 100, Figure 4 and LPP of 1, Figure 3) emphasizing the taxonomic relevance of this character of the inflorescence bract. Henderson (2011) further recognized two subclades that were also recovered here with good support (BS of 100, Figure 4 and LPP of 1, Figure 3): one including G. concinna and G. concinnoidea (and G. galeanoae in our study), and the other with the remaining species. This latter subclade is characterized by the non-homoplasious character state of staminodial tubes of non-fertilized pistillate flowers projecting and persistent after anthesis (Henderson, 2011).
Clade III
The third clade includes nine species [G. camana Trail, G. chlamydostachys Galeano, G. chococola Wess. Boer, G. macrostachys Mart., G. multisecta (Burret) Burret, G. oldemannii Granv., G. paradoxa Burret, G. poiteauana Kunth, G. triglochin Burret; Figure 5] from the Amazonian lowlands and adjacent regions, except for G. paradoxa from the Pacific coast of Colombia and Ecuador. It largely corresponds to Henderson’s (2011)G. macrostachys clade except for three species (G. deneversi, G. schizocarpa, and G. umbraculiformis) that we did not sequence and G. maxima, which we recovered as an independent clade (see above). We recovered two subclades, one composed of G. macrostachys, G. multisecta, G. poiteauana, and G. oldemannii and the other of G. camana, G. chlamydostachys, G. chococola, G. paradoxa, and G. triglochin (each clade with bootstrap value of 100, Figure 4 and LPP of 1, Figure 3). These clades did not correspond to the two subgroups recognized by Henderson (2011) in his G. macrostachys clade. Geonoma macrostachys is one of the morphologically and taxonomically most complex species in the genus, and Henderson (2011) divided it into several morphotypes, some of which behave as sympatric taxa at the local scale (Roncal, 2006; Roncal et al., 2007; Borchsenius et al., 2016). However, the 12 individuals of G. macrostachys from four morphotypes that we sampled show that, at larger geographic scales, morphotypes do not cluster into monophyletic groups. The species G. poiteauna, which used to be treated as a variety of G. macrostachys (Henderson et al., 1995) and subsequently raised at the species level (Henderson, 2011), is recovered here as nested within G. macrostachys (Figures 3, 4).
Clade IV
This clade includes six species (G. euspatha Burret, G. frontinensis Burret, G. interrupta (Ruiz & Pav.) Mart, G. pinnatifrons Willd, G. santanderensis Galeano & R. Bernal, G. simplicifrons Willd; Figure 5) that largely occur on lower mountain slopes from Costa Rica to Bolivia and northeastern Brazil, as well as in the Antilles. It is essentially identical to Henderson’s (2011)G. interrupta clade, except for G. santanderensis here recovered as sister to the other five species with strong support (BS of 100, Figure 4 and LPP of 1, Figure 3) whereas it was placed within the G. stricta clade in Henderson’s maximum parsimony analysis. Although G. santanderensis shares several specific morphological traits with G. aspidiifolia Spruce and G. oligoclona Trail (such as internodes covered with reddish or brownish scales, rachillae surfaces with spiky, fibrous projections or ridges, staminodial tubes lobed at the apex with the lobes not spreading at anthesis and not acuminate) our phylogenetic analyses reveal that these are homoplasic characters since the species are not closely related. Also, of the two subspecies of G. interrupta we sampled, the monophyletic subspecies rivalis, endemic of the Central Cordillera in Colombia, is nested within the geographically widespread subspecies maxima. The latter is also phenotypically more variable but never includes the rheophytic leaf morphology of subspecies rivalis.
Clade V
This clade includes three species (G. bernalii A. J. Hend, G. deversa (Poit.) Kunth, G. leptospadix Trail; Figure 5) occuring from Costa Rica to Peru and the Guianas. In Henderson’s (2011) maximum parsimony tree, G. bernalii belonged to the G. lanata clade, whereas the other two species were placed in an unresolved polytomy. Geonoma deversa and G. leptospadix are variable and very widespread lowland species which probably hybridize in northeastern Brazil and the Guianas as suggested by the observation of specimens with intermediate morphology (Henderson, 2011) but none of the putative hybrid was sampled here. Geonoma bernalii occurs in northern Colombia and was previously identified as G. leptospadix. In our phylogenetic tree, G. leptospadix appears as sister to a group formed by G. bernalii and G. deversa.
Clade VI
This clade includes two morphologically very similar species (G. aspidiifolia and G. oligoclona; Figure 5) from Amazonia and the Guianan highlands. In fact, out of the two specimens of G. oligoclona, one is recovered more closely related to the single specimen of G. aspidiifolia (with BS of 74, Figure 4 and LPP of 1, Figure 3). In the absence of additional individuals of G. aspidiifolia, it is premature to conclude whether they actually represent a single variable species or two closely related species. In Henderson’s (2011) maximum parsimony tree these two species were placed within the G. stricta clade and were closely related to G. santanderensis.
Clade VII
This clade includes four species (G. elegans Mart., G. pauciflora Mart., G. pohliana Mart., and G. schottiana Mart.; Figure 5) from the Brazilian Atlantic Forest and the Cerrado. It corresponds to Henderson’s (2011)G. schottiana clade and to Roncal et al. (2011) Brazilian Cerrado + Mata Atlantica clade. It appears that this small radiation of south-eastern Brazilian species resulted from a single colonization event that gave rise to four species with considerable inter- and intraspecific variation. In our phylogeny, the two individuals of G. schottiana were recovered as nested within G. pohliana, indicating that similarly to what is observed in other species complexes, morphological taxa are not always underpinned by strong genetic differentiation. Geonoma pohliana is an extremely variable species complex which Henderson (2011) subdivided into 11 subspecies. The three subspecies sampled in our analysis appeared to be randomly mixed. The other two Brazilian species G. elegans and G. pauciflora, were recovered paraphyletic (Figures 3, 4).
Clade VIII
This clade includes two morphologically similar endemic species from Venezuela (G. spinescens H. Wendl. ex Burret and G. braunii (Stauffer) A.J. Hend; Figure 5). Little DNA was obtained from the three herbarium samples and in fact G. braunii was recovered as sister taxa to clades VIII-XVI in the two ASTRAL analyses (Figure 3). However, we believe that this is caused by the lack of DNA sequences for G. braunii. Therefore, despite this uncertainty, we decided to follow the topology of the concatenated analysis and treat the two species as part of a single clade because it is coherent with the fact that G. braunii used to be considered a variety of G. spinescens (Stauffer, 1997). Henderson (2011) treated them as distinct species based on the flower pits alternately arranged in the former and spirally arranged in the latter. The two species were included within Henderson’s (2011)G. lanata clade. In our concatenated analysis, G. spinescens appears paraphyletic because one of the two samples of G. spinescens was found sister to G. braunii (with bootstrap support of 99, Figure 4) but given the low amount of sequence data for G. braunii we can not advocate either of the two possible taxonomic treatment.
Clade IX
This clade includes two species (G. hollinensis A.J. Hend, Borchs & Balslev, and G. triandra (Burret) Wess. Boer; Figure 5) that are distributed from Panama to Ecuador and occur at similar elevations. The geographic distribution of these sister species (G. hollinensis restricted to north-eastern Ecuador and G. triandra found from north-western Ecuador to southern Panama) suggest that vicariance was involved in their divergence. Both species have staminate flowers with three stamens, and were segregated as subgenus Kalbreyera by Wessels Boer (1968). Henderson (2011) placed them together with G. occidentalis in the G. triandra clade but we cannot confirm this placement since G. occidentalis was not included in our study.
Clade X
This clade includes samples of five species (G. lehmannii Dammer ex Burret, G. orbignyana Mart., G. talamancana Grayum, G. trigona (Ruiz & Pav.) A.H. Gentry and G. undata; Figure 5) that occur at high elevations from Mexico to Bolivia, also reaching the Lesser Antilles, plus G. fosteri A.J. Hend. It is largely congruent with the G. undata clade of Henderson (2011) and the Andes + Central American Mountains clade of Roncal et al. (2010, 2011). Except for G. fosteri, the species of this clade share the character state of apiculate and lobed proximal lips of the flower pits. This is one of the taxonomically most complex groups of the genus, and species delimitation has been handled differently over time (Henderson et al., 1995; Henderson, 2011). In our study, the samples assigned to the widespread species G. orbignyana and G. undata were not recovered as phylogenetically independent lineages (Figures 3, 4). Rather than clustering according to taxonomic delimitations, specimens of these two species grouped with strong support by geographic location, forming two main clades (each with BS of 100, Figure 4 and LPP of 1, Figure 3): (a) a central American – north Andean clade composed of specimens from Mexico, Costa-Rica, Panama, the Caribbean, and Colombia; and (b) a north Andean – central Andean clade composed of specimens from Ecuador, Peru, and Bolivia. However, in addition to these two subclades, there was also a separate group at the base of the clade, comprising three specimens from Ecuador and Bolivia in the coalescent analysis (with LPP of 1, Figure 3) or these three specimens plus G. trigona in the concatenated analysis (with BS of 63, Figure 4). The samples of G. lehmannii subsp. corrugata A.J. Hend. and G. talamancana, both occurring in Central America, were nested within the central American – north Andean clade with strong support (BS of 100, Figure 4 and LPP of 1, Figure 3). As these two species have resembling morphologies and similar high elevation habitats to G. undata and G. orbignyana, they may represent locally divergent populations of this broad species complex, in which a novel phenotype has been fixed (e.g., the thick corrugated leaves and the well-developed peduncle in G. lehmannii subsp. corrugata) although it is not underpinned by strong genetic isolation. Another similar case is G. trigona, which, together with G. fosteri is recovered as sister to the rest of Clade X in the coalescent analysis (with LPP of 0.8, Figure 3). The placement of G. fosteri is intriguing because it is morphologically clearly different from the rest of the species in this clade, even though it occurs in the same habitat. Although high-quality DNA was recovered for this sample and the identification of the living specimen from which it was collected was double-checked, we cannot rule out contamination during laboratory work to explain this surprising result.
Clade XI
This clade includes five species (G. cuneata H. Wendl. ex Spruce, G. lanata A.J. Hend, Borchs & Balslev, G. laxiflora Mart., G. stricta (Poit.) Kunth, G. tenuissima H.E. Moore, Figure 5) that Henderson (2011) placed in several distinct clades (G. cuneata, G. lanata, and G. stricta clades). These five species were part of an unresolved clade recovered in Roncal et al. (2012). Geonoma stricta and G. cuneata are two species complexes whereas the three remaining species of this clade have rather low variability (Henderson, 2011). Henderson (2011) recognized nine subspecies in G. stricta, with the most widespread and morphologically variable, subspecies arundinacea, further divided into eight morphotypes. Of the three subspecies included in our analysis, subspecies stricta and montana were nested within arundinacea. For the latter, the two morphotypes sampled (trailii and pycnostachys) were paraphyletic. In G. cuneata, nine subspecies were recognized and the most widespread and morphologically variable, subspecies cuneata was divided into 13 morphotypes (Henderson, 2011). We did not sample enough individuals of the different subspecies of G. cuneata to comment on their recognition using phylogenomics, except that subspecies guanacastensis was nested within subspecies cuneata. Species of this clade are mostly distributed in lowland and lower montane forests with G. cuneata distributed from Nicaragua to Ecuador, G. lanata and G. tenuissima occurring mostly on the western Andean slopes, G. laxiflora in western Amazonia, and the widespread G. stricta overlapping the previous four ranges and reaching central Amazonia and the Guyanas. Although these species are morphologically quite different from each other, which explains why Henderson (2011) recovered them in different clades, they commonly have cane-like stems with yellowish and smooth internodes.
Clade XII
This clade includes a single species, G. divisa H.E. Moore (Figure 5), which is endemic to northwestern Colombia. Henderson (2011) placed this species in his G. stricta clade, alongside G. longivaginata H.Wendl. ex Spruce and G. ferruginea H. Wendl. ex Spruce, but this relationship was not supported at all in our analyses. Morphologically, G. divisa differs from these two species in its tricussately arranged, closely spaced flower pits (Henderson, 2011).
Clade XIII
This clade includes two species (G. brongniartii Mart., G. poeppigiana Mart.; Figure 5) that occur from Colombia to Bolivia. These closely related species are variable and their separation has long been debated (Henderson, 2011). In our phylogenetic tree, these two morphological taxa are mixed together within a single clade rather than forming distinct monophyletic groups (Figures 3, 4). Although we can not rule out sample misidentification, this result could also be explained by alternative hypotheses. Indeed, the geographic distributions of these two species overlap, with G. poeppigiana having a smaller range than G. brongniartii, and Henderson (2011) reported putative hybrids between them in central Peru. All specimens of G. poeppigiana included in our phylogeny come from Peru and so do most specimens of G. brongniartii. If the two species indeed hybridize in the Peruvian area where they co-occur, this may explain why they are mixed in our phylogenetic tree. Alternatively, these samples could represent a single widely variable species (see below).
Clade XIV
This clade includes eight species (G. brenesii Grayum, G. epetiolata H.E. Moore, G. ferruginea, G. hugonis Grayum & de Nevers, G. longivaginata, G. monospatha de Nevers, G. mooreana de Nevers & Grayum, G. scoparia Grayum & de Nevers; Figure 5) from Costa Rica and Panama. It corresponds to Roncal et al.’s (2011) central American clade, whereas in Henderson’s (2011) tree these species were placed in three different clades (namely the G. cuneata, G. lanata, and G. stricta clades). The three samples of G. monospatha from Costa Rica are placed far apart from the two Panamanian samples in our phylogenetic tree (with BS of 100, Figure 4 and LPP of 1, Figure 3). These populations are geographically disjunct, with a gap of several hundred kilometers between them. Furthermore, the Costa Rican specimens have smaller leaves and inflorescences and thrive at higher elevations (mean elevations 1750 m vs. 837 m). All of this suggests that the Costa Rican population may better be treated as a distinct species. Similarly, the two samples of the Panamanian G. longivaginata subspecies copensis A. J. Hend. are more closely related to the two Panamanian samples of G. monospatha (with BS of 100, Figure 4 and LPP of 1, Figure 3) than to the individuals of G. longivaginata from Costa Rica, suggesting that this subspecies may, in fact, also better be treated as a distinct species.
Comparison With Other Phylogenetic Reconstructions
In the latest revision of Geonoma, Henderson (2011) conducted a maximum parsimony phylogenetic analysis of all the species of the genus based on 30 qualitative morphological characters. We used his species-level taxonomy to name our taxa and tried to apply his clade definition to the phylogenetic tree we obtained. We found that many of his clades were supported by our study, although often with the exclusion or inclusion of a few species. For instance, his G. cuneata clade (minus G. cuneata) corresponds to our Clade XIV, his G. macrostachys clade (minus G. maxima) corresponds to our Clade III, his G. schottiana clade corresponds to our Clade VII, his G. undata clade (plus G. fosteri) corresponds to our Clade X, his G. congesta clade (plus G. galeanoae) to our Clade II, and his G. interrupta clade (plus G. santaderensis) to our Clade IV. In contrast, Henderson’s G. lanata and G. stricta clades are not supported at all by our analyses, suggesting that these clades were defined by homoplasic morphological characters with limited phylogenetic information. These characters included rachillae surfaces with spiky, fibrous projections or ridges for the G. stricta clade, and filiform rachillae with extended narrowed sections between the alternately arranged flower pits for the G. lanata clade. Furthermore, the relationships between the clades as recovered by Henderson (2011) are in strong disagreement with the topology obtained from molecular data, indicating that morphology provides little insight on the deep phylogenetic relationships within the genus.
The first molecular phylogeny of Geonoma was based on 20 species and two markers (Roncal et al., 2005). It was later extended to three genes and 43 species (Roncal et al., 2010, 2011, 2012). Using an extended sampling of 57 species and 795 gene regions, our study confirmed many of the findings of these studies for the phylogenetic relationships at intermediate levels of divergence. For instance, our Clades I-III, which together are sister to the other 11 clades, correspond to Roncal’s (2011) Amazon clade, which was also recovered as sister to the remainder of the genus. Furthermore, the internal arrangements of the species in this group are also largely congruent, with G. maxima sister to the remainder of the species in the Amazon clade, and G. calyptrogynoidea and G. congesta (our Clade II) sister to the remainder of the species (our Clade III), although G. baculifera and G. concinna were recovered by Roncal et al. (2010) to be more closely related to species in our Clade III than they were to species in Clade II, where we placed them. Likewise, Roncal et al.’s (2011) Brazilian Cerrado + Mata Atlantica, Andes + Central American Mountains, and Central America clades were also recovered in our phylogenetic tree and the relative arrangements of these clades are overall similar between both studies. In general, previously unresolved phylogenetic relationships were resolved with strong support in our analyses.
Species Delimitation
Henderson (2011) used a statistical approach in which species delimitation was based on the results of a clustering analysis of qualitative morphological characters. The congruence between the results of this method and those of our phylogenetic analysis is striking. Indeed, the majority of species and species complexes for which we included several samples were recovered as monophyletic units in our phylogenetic trees. Furthermore, morphological variation seemed to be correlated to genetic divergence, as indicated by the longer branches of widely variable species in the maximum likelihood tree compared to species with smaller geographic ranges and less variable phenotypes (Figure 4). Although further sampling would be necessary in order to include the full range of variability of some widely distributed species, our study nevertheless supports the validity of the characters used by Henderson (2011) to define species boundaries. Conversely, even though not all our samples had identification at the intraspecific level, our results indicate that the intraspecific divisions as subspecies, varieties or morphotypes generally do not match the genetic clusters. These intraspecific taxa were defined by Henderson (2011) usually based on a few, often variable characters such as the degree of division in the inflorescences. Only in G. pinnatifrons are the subspecies, which were delimited based on geographical distribution, supported by the tree topology (Figures 3, 4). In other species complexes (e.g., in G. cuneata, G. maxima, G. stricta, G. macrostachys, and G. pohliana), intraspecific taxa are not recovered as monophyletic (Figures 3, 4). Furthermore, while our phylogenetic tree revealed a broad North-South differentiation within the mixed G. orbignyana – G. undata group, in general there seems to be no geographic clustering of individuals, especially for widespread Amazonian taxa such as G. macrostachys, G. maxima or G. stricta. Various hypotheses have been proposed to explain this kind of chaotic intraspecific phenotypic variation, such as population contraction and expansion during Pleistocene climatic oscillations (Cronk, 1998), rapid dispersal followed by selection (Cronk, 1998), or niche divergence induced by forest heterogeneity (Henderson, 2011). From a genetic perspective, incomplete lineage sorting or hybridization are commonly invoked to explain the widespread occurrence of plant species complexes similar to those found in Geonoma (Bacon et al., 2012b; Pinheiro et al., 2018). Although our results pointed to high level of gene tree incongruence in the species complexes of Geonoma (Figure 3), likely due to incomplete lineage sorting, it is beyond the scope of our study to test for any of these underlying mechanisms. Further phylogeographic or population genetic studies are needed to understand the origin of the discrepancy between morphological and genetic data in this group.
From a systematic point of view, the remaining issue to be addressed is the taxonomic status of the several non-monophyletic species that were identified by our analysis. First, there are two cases where two species were recovered mixed within a single clade (G. brongniartii with G. poeppigiana and G. orbignyana with G. undata). Second, there are several
instances of geographically restricted species (e.g., G. lehmanii, G. poiteauana, G. talamancana, and G. trigona) which were found to be nested within more widely distributed species, making the latter paraphyletic. For taxonomic classification, there are two fundamentally different approaches to deal with such situations. On one hand, under a lineage species concept, which requires species monophyly, the phylogenetically intermixed “species” of Geonoma would be considered to represent single variable species as was done in other similar cases in plants (Bennett et al., 2008; Barbosa et al., 2012). Applying this approach would entail a reduction in the number of recognized species within Geonoma. On the other hand, some authors have stressed that at the species level paraphyly is not an issue because it is considered to be the natural output of, for example, peripatric speciation or speciation via polyploidy which are common phenomena in plants (Rieseberg and Brouillet, 1994; Crisp and Chandler, 1996). Likewise, the pattern of small-range species nested within large-range paraphyletic species has been suggested to be common in rainforest trees with widespread distribution for which coalescence times are long due to large population size and extensive gene flow (Pennington and Lavin, 2016). Non-monophyletic species can also arise from hybridization, which is widespread in plants (Whitney et al., 2010). Under a genic species concept, species cohesion may be determined by a small number of genes, thus allowing gene flow between species without calling species identity into question (Wu, 2001; Lexer and Widmer, 2008). Therefore, adopting a species concept which places emphasis on the phenotype would result in treating morphologically divergent entities as separate species even if they do no represent evolutionary independent lineages (Freudenstein et al., 2016). From this point of view, the number of species in Geonoma would remain similar to that proposed by Henderson (2011). In the end, how the phylogenetic information presented here is translated into a taxonomic classification is to a certain degree a matter of personal preference, with different researchers favoring different aspects. Thus, some may emphasize morphological or genetic similarities while others would focus on differences, some place more importance on the ability to diagnose taxa while others prioritize evolutionary independence, and so on. We refrain from proposing taxonomic decisions based on our results, since this would require a full assessment of genetic, morphological, and ecological evidence.
Conclusion
By employing a large novel set of molecular markers, we were able to clarify both deep and shallow phylogenetic relationships within the tribe Geonomateae including for Geonoma, one of the largest and taxonomically most challenging Neotropical palm genera. The remaining poorly supported phylogenetic relationships do not reflect a lack of informative genetic data but are rather caused by a high level of gene tree incongruence, as shown by the coalescent analysis. Our phylogenetic analyses revealed two cryptic species of Geonoma in Central America, which will have to be described in further taxonomic work. The intraspecific sampling confirmed in most cases the validity of the taxonomic delimitation of species proposed by Henderson (2011),
even for those with extensive phenotypic variability such as G. cuneata, G. interrupta, G. maxima, G. pinnatifrons, G. macrostachys, or G. stricta. However, we also pointed to several cases where the morphological delimitations do not reflect the genetic clusters, such as the internal delimitations of widely variable species complexes, the clustering of rare endemic species within broader species complexes, or the mixing of two species complexes. These groups that do not show clear genetic boundaries between morphologically recognized taxa remain the main challenge in the systematics of Geonoma. Ultimately, the number of species recognized in Geonoma depends on the species concept one endorses.
Studies at the population level are needed to understand whether the decoupling between morphological and genetic variation in the species complexes is the result of ongoing speciation with gene flow or from secondary contact and hybridization between previously diverged taxa. Although the impossibility of summarizing morphological variation of these groups into a coherent classification scheme may seem frustrating from a taxonomic point of view, we argue that it represents a unique opportunity to better understand the build-up of Neotropical plant diversity. Indeed, species complexes are common in plants and are gaining attention as model groups to study the underlying factors of plant speciation (Pinheiro et al., 2018). In this context, the set of baits recently developed by de La Harpe et al. (2019) will therefore be a useful tool to carry out specific population levels studies.
With this in mind, we provided the baits for a selection including 20% of the most informative genes from the kit developed by de La Harpe et al. (2019) by assessing their phylogenetic informativeness across three Arecaceae subfamilies. We predict that these baits should work over a wide evolutionary timescale in the Arecaceae and will therefore benefit the whole field of palm phylogenomics. Indeed, the smaller size of this kit will make it accordingly more affordable and will reduce the computation time of post-sequencing bioinformatic analyses while maximizing the phylogenetic informativeness at deep and shallow scales across the Arecaceae family. Hence, we believe that it has the potential to be an essential tool in the search toward a complete species-level phylogeny of the Arecaceae family.
Statements
Data availability statement
Targeted sequence reads generated as part of this manuscript are available in NCBI (BioProject PRJNA541164). The list of the 795 selected genes and their corresponding bait sequences in fasta format as well as all gene trees and species trees were deposited in Zenodo (deposit number 2594808).
Author contributions
NS, MK, and CL designed the study. MPa led the sequencing experiment and performed the post-sequencing bioinformatics analyses. MdLH, OL, TM-M, and MPa did the labwork. OL and
DK performed the phylogenetic analyses. CB, HB, FB, AC, TC, MPe, JuR, MS, FS, CL, MK, and NS were part of the Geonoma Consortium set up to perform this study. OL led the writing with significant contributions from all co-authors, in particular MK, IO, and NS. All co-authors commented and agreed on the last version of the manuscript.
Funding
NS, MK, and CL received funding from the Swiss National Science Foundation (CRSII3-147630), NS from the University of Lausanne, JoR from a Banting postdoctoral fellowship (151042) at University of British Columbia, MPe from the Swiss National Science Foundation (Grant No. 31003A_175655/1), TM-M from the CNPq-SWE (205660/2014-2), MS from the Colciencias (Contract No. 173-2016), and HB from the Danish Council for Independent Research – Natural Sciences (4181-00158) and the European Community (FP7 212631).
Acknowledgments
This study would not have been possible without Andrew Henderson’s help. We are deeply grateful to him for identifying many of the samples and for his invaluable comments on an earlier version of the manuscript. We thank Natalia Arcila, Tatiana Boza, Lilian Costa Procopio, Camilo Flórez, María Fernanda González, Rosa Isela Meneses, Vanessa Rojas, Alain Rousteau, Lázaro Santa Cruz, Adrian Tejedor, Johanna Toivonen, and Erickson Urquiaga for their assistance in the field. Gloria Galeano5 and Jean Christophe Pintaud5 for their assistance with fieldwork planning and the identification of some specimens. AAU and COL for providing silica-dried leaves of many samples; G and COL for herbarium fragments; and Rodrigo Bernal and Juan Carlos Copete for providing photos of some vouchers. We also thank the Vital-IT facilities of the Swiss Institute of Bioinformatics for the use of their HPC infrastructure.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00864/full#supplementary-material
Footnotes
1.^https://doi.org/10.5061/dryad.3v9v238
2.^http://broadinstitute.github.io/picard
3.^https://github.com/JinfengChen/vcf-tab-to-fasta
4.^https://github.com/sidonieB/scripts/blob/master/GetQpiechartsFrom ASTRAL.py
5.^Deceased.
References
1
AbadiS.AzouriD.PupkoT.MayroseI. (2019). Model selection may not be a mandatory step for phylogeny reconstruction.Nat. Commun.10:934. 10.1038/s41467-019-08822-w
2
Al-MssallemI. S.HuS.ZhangX.LinQ.LiuW.TanJ.et al (2013). Genome sequence of the date palm Phoenix dactylifera L.Nat. Commun.4:2274. 10.1038/ncomms3274
3
BaconC. D.BakerW. J.SimmonsM. P. (2012a). Miocene dispersal drives island radiations in the palm tribe Trachycarpeae (Arecaceae).Syst. Biol.61426–442. 10.1093/sysbio/syr123
4
BaconC. D.McKennaM. J.SimmonsM. P.WagnerW. L. (2012b). Evaluating multiple criteria for species delimitation: an empirical example using Hawaiian palms (Arecaceae: Pritchardia).BMC Evol. Biol.12:23. 10.1186/1471-2148-12-23
5
BaconC. D.LookS. L.Gutiérrez-PintoN.AntonelliA.TanH. T.KumarP. P.et al (2016a). Species limits, geographical distribution and genetic diversity in Johannesteijsmannia (Arecaceae).Bot. J. Linn. Soc.182318–347. 10.1111/boj.12470
6
BaconC. D.Velásquez-PuentesF.Flórez-RodríguezA.BalslevH.GaleanoG.BernalR.et al (2016b). Phylogenetics of Iriarteeae (Arecaceae), cross-Andean disjunctions and convergence of clustered infructescence morphology in Wettinia.Bot. J. Linn. Soc.182272–286. 10.1111/boj.12421
7
BaconC. D.MoraesM.JaramilloC.AntonelliA. (2017). Endemic palm species shed light on habitat shifts and the assembly of the Cerrado and Restinga floras.Mol. Phylogenet. Evol.110127–133. 10.1016/j.ympev.2017.03.013
8
BakerW. J.AsmussenC. B.BarrowS. C.DransfieldJ.HeddersonT. A. (1999). A phylogenetic study of the palm family (Palmae) based on chloroplast DNA sequences from the trnL - trnF region.Plant Syst. Evol.219111–126. 10.1007/BF01090303
9
BakerW. J.CouvreurT. L. P. (2013). Global biogeography and diversification of palms sheds light on the evolution of tropical lineages. II. Diversification history and origin of regional assemblages.J. Biogeogr.40286–298. 10.1111/j.1365-2699.2012.02794.x
10
BakerW. J.DransfieldJ. (2016). Beyond Genera Palmarum: progress and prospects in palm systematics.Bot. J. Linn. Soc.182207–233. 10.1111/boj.12401
11
BakerW. J.NorupM. V.ClarksonJ. J.CouvreurT. L. P.DoweJ. L.LewisC. E.et al (2011). Phylogenetic relationships among arecoid palms (Arecaceae: Arecoideae).Ann. Bot.1081417–1432. 10.1093/aob/mcr020
12
BakerW. J.SavolainenV.Asmussen-LangeC. B.ChaseM. W.DransfieldJ.ForestF.et al (2009). Complete generic-level phylogenetic analyses of palms (arecaceae) with comparisons of supertree and supermatrix approaches.Syst. Biol.58240–256. 10.1093/sysbio/syp021
13
BalslevH.CopeteJ.PedersenD.BernalR.GaleanoG.DuqueÁ, et al. (2017). “Palm diversity and abundance in the Colombian Amazon,” in Forest structure, function and dynamics in Western Amazonia, ed.MysterR. W. (London: John Wiley & Sons Ltd.).
14
BalslevH.LaumarkP.PedersenD.GrándezC. (2016). Tropical rainforest palm communities in Madre de Dios in Amazonian Peru.Rev. Peru. Biol.233–12.
15
BarbosaA. R.FioriniC. F.Silva-PereiraV.Mello-SilvaR.BorbaE. L. (2012). Geographical genetic structuring and phenotypic variation in the Vellozia hirsuta (Velloziaceae) ochlospecies complex.Am. J. Bot.991477–1488. 10.3732/ajb.1200070
16
BarrettC.SinnB.KingL.MedinaJ.BaconC.LahmeyerS.et al (2018). Phylogenomics, biogeography, and evolution in the American palm genus Brahea.bioRxiv 467779. 10.1101/467779
17
BarrettC. F.BaconC. D.AntonelliA.HofmannT. (2016). An introduction to plant phylogenomics with a focus on palms.Bot. J. Linn. Soc.182234–255. 10.1111/boj.12399
18
BennettJ. R.WoodJ. R. I.ScotlandR. W. (2008). Uncorrelated variation in widespread species: species delimitation in Strobilanthes echinata Nees (Acanthaceae).Bot. J. Linn. Soc.156131–141. 10.1111/j.1095-8339.2007.00756.x
19
BlomM. P. K.BraggJ. G.PotterS.MoritzC. (2017). Accounting for uncertainty in gene tree estimation: summary-coalescent species tree inference in a challenging radiation of Australian lizards.Syst. Biol.66352–366. 10.1093/sysbio/syw089
20
BorchseniusF. (2002). Staggered flowering in four sympatric varieties of Geonoma cuneata (Palmae).Biotropica34603–606. 10.1111/j.1744-7429.2002.tb00580.x
21
BorchseniusF.LozadaT.KnudsenJ. T. (2016). Reproductive isolation of sympatric forms of the understorey palm Geonoma macrostachys in western Amazonia.Bot. J. Linn. Soc.182398–410. 10.1111/boj.12428
22
ChazdonR. L. (1991). Plant size and Form in the understory palm genus Geonoma: are species variations on a theme?Am. J. Bot.78680–694. 10.1002/j.1537-2197.1991.tb12592.x
23
ChazdonR. L. (1992). Patterns of growth and reproduction of Geonoma congesta, a clustered understory palm.Biotropica2443–51.
24
ChernomorO.von HaeselerA.MinhB. Q. (2016). Terrace aware data structure for phylogenomic inference from supermatrices.Syst. Biol.65997–1008. 10.1093/sysbio/syw037
25
ComerJ. R.ZomleferW. B.BarrettC. F.DavisJ. I.StevensonD. W.HeydukK.et al (2015). Resolving relationships within the palm subfamily Arecoideae (Arecaceae) using plastid sequences derived from next-generation sequencing.Am. J. Bot.102888–899. 10.3732/ajb.1500057
26
ComerJ. R.ZomleferW. B.BarrettC. F.StevensonD. W.HeydukK.Leebens-MackJ. H. (2016). Nuclear phylogenomics of the palm subfamily Arecoideae (Arecaceae).Mol. Phylogenet. Evol.9732–42. 10.1016/j.ympev.2015.12.015
27
CouvreurT. L. P.ForestF.BakerW. J. (2011). Origin and global diversification patterns of tropical rain forests: inferences from a complete genus-level phylogeny of palms.BMC Biol.9:44. 10.1186/1741-7007-9-44
28
CrispM. D.ChandlerG. T. (1996). Paraphyletic species.Telopea6813–844. 10.7751/telopea19963037
29
CronkQ. (1998). “The ochlospecies concept,” in Chorology, Taxonomy and Ecology of the Floras of Africa and Madagascar, edsHuxleyC. R.LockJ. M.CutlerD. F. (Kew: Royal Botanic Gardens), 155–170.
30
CuencaA.Asmussen-LangeC. B.BorchseniusF. (2008). A dated phylogeny of the palm tribe Chamaedoreeae supports Eocene dispersal between Africa, North and South America.Mol. Phylogenet. Evol.46760–775. 10.1016/j.ympev.2007.10.010
31
DanecekP.AutonA.AbecasisG.AlbersC. A.BanksE.DePristoM. A.et al (2011). The variant call format and VCFtools.Bioinformatics272156–2158. 10.1093/bioinformatics/btr330
32
de La HarpeM.HessJ.LoiseauO.SalaminN.LexerC.ParisM. (2019). A dedicated target capture approach reveals variable genetic markers across micro-and macro-evolutionary time scales in palms.Mol. Ecol. Resour.19221–234. 10.1111/1755-0998.12945
33
DransfieldJ.UhlN. W.AsmussenC. B.BakerW. J.HarleyM. M.LewisC. E. (2008). Genera Palmarum: The Evolution and Classifi Cation of Palms, 2nd Edn. Kew: Royal Botanical Gardens.
34
FairclothB. C.ChangJ.AlfaroM. E. (2012). TAPIR enables high-throughput estimation and comparison of phylogenetic informativeness using locus-specific substitution models.arXiv preprintarXiv:1202.1215.
35
FreudensteinJ. V.BroeM. B.FolkR. A.SinnB. T. (2016). Biodiversity and the species concept—lineages are not enough.Syst. Biol.66644–656. 10.1093/sysbio/syw098
36
GrucaM.Blach-OvergaardA.BalslevH. (2015). African palm ethno-medicine.J. Ethnopharmacol.165227–237. 10.1016/j.jep.2015.02.050
37
HendersonA. (2002). Evolution and Ecology of Palms.New York, NY: New York Botanical Garden Press.
38
HendersonA. (2005). A multivariate study of Calyptrogyne (Palmae).Syst. Bot.3060–83. 10.1600/0363644053661913
39
HendersonA. (2011). A revision of Geonoma (Arecaceae).Phytotaxa171–271.
40
HendersonA. (2012). A revision of Pholidostachys (Arecaceae).Phytotaxa431–48.
41
HendersonA.GaleanoG.BernalR. (1995). A Field Guide to the Palms of the Americas.Princeton, NJ: Princeton University Press.
42
HendersonA.MartinsR. (2002). Classification of specimens in the Geonoma stricta (Palmae) complex: the problem of leaf size and shape.Brittonia54202–212. 10.1663/0007-196x(2002)054%5B0202:cositg%5D2.0.co;2
43
HendersonA.VillalbaI. (2013). A revision of Welfia (Arecaceae).Phytotaxa11933–44. 10.11646/phytotaxa.119.1.3
44
HeydukK.TrapnellD. W.BarrettC. F.Leebens-mackJ. I. M. (2015). Phylogenomic analyses of species relationships in the genus Sabal (Arecaceae) using targeted sequence capture.Biol. J. Linn. Soc.117106–120. 10.1111/bij.12551
45
HoangD. T.ChernomorO.von HaeselerA.MinhB. Q.VinhL. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation.Mol. Biol. Evol.35518–522. 10.1093/molbev/msx281
46
HoffM.OrfS.RiehmB.DarribaD.StamatakisA. (2016). Does the choice of nucleotide substitution models matter topologically?BMC Bioinformatics17:143. 10.1186/s12859-016-0985-x
47
KircherM.SawyerS.MeyerM. (2011). Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform.Nucleic Acids Res.40:e3. 10.1093/nar/gkr771
48
KnudsenJ. T.AnderssonS.BergmanP.BergmanP.KnudsenJ. T. (1998). Floral scent attraction in Geonoma macrostachys, an understorey palm of the Amazonian rain forest.Oikos85409–418.
49
LangmeadB.SalzbergS. L. (2012). Fast gapped-read alignment with Bowtie 2.Nat. Methods9357–359. 10.1038/nmeth.1923
50
LexerC.WidmerA. (2008). The genic view of plant speciation: recent progress and emerging questions.Philos. Trans. R. Soc. Lond. B Biol. Sci.3633023–3036. 10.1098/rstb.2008.0078
51
Ley-lopezJ. M.AvalosG. (2017). Propagation of the palm flora in a lowland tropical rainforest in Costa Rica: fruit collection and germination patterns.Trop. Conserv. Sci.101–12. 10.1177/1940082917740703
52
LiuL.XiZ.WuS.DavisC. C.EdwardsS. V. (2015). Estimating phylogenetic trees from genome-scale data.Ann. N. Y. Acad. Sci.136036–53. 10.1111/nyas.12747
53
LiuL.YuL.KubatkoL.PearlD. K.EdwardsS. V. (2009). Coalescent methods for estimating phylogenetic trees.Mol. Phylogenet. Evol.53320–328. 10.1016/j.ympev.2009.05.033
54
MaciaM. J.ArmesillaP. J.Cámara-LeretR.Paniagua-ZambranaN.VillalbaS.BalslevH.et al (2011). Palm uses in northwestern South America: a quantitative review.Bot. Rev.77462–570. 10.1007/s12229-011-9086-8
55
MandelJ. R.BarkerM. S.BayerR. J.DikowR. B.GaoT.JonesK. E.et al (2017). The compositae tree of life in the age of phylogenomics.J. Syst. Evol.55405–410. 10.1111/jse.12265
56
McKennaA.HannaM.BanksE.SivachenkoA.CibulskisK.KernytskyA.et al (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data.Genome Res.201297–1303. 10.1101/gr.107524.110
57
MeerowA. W.NoblickL.Salas-LeivaD. E.SanchezV.Francisco-OrtegaJ.JestrowB.et al (2015). Phylogeny and historical biogeography of the cocosoid palms (Arecaceae, Arecoideae, Cocoseae) inferred from sequences of six WRKY gene family loci.Cladistics31509–534. 10.1111/cla.12100
58
MeyerM.KircherM. (2010). Illumina sequencing library preparation for highly multiplexed target capture and sequencing.Cold Spring Harb. Protoc.2010:db.rot5448. 10.1101/pdb.prot5448
59
MirarabS.ReazR.BayzidM. S.ZimmermannT.SwensonM. S.WarnowT. (2014). ASTRAL: genome-scale coalescent-based species tree estimation.Bioinformatics30i541–i548. 10.1093/bioinformatics/btu462
60
MirarabS.WarnowT. (2015). ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes.Bioinformatics31i44–i52. 10.1093/bioinformatics/btv234
61
MitchellN.LewisP. O.Moriarty LemmonE.LemmonA. R.HolsingerK. E. (2017). Anchored phylogenomics improves the resolution of evolutionary relationships in the rapid radiation of Protea L.Am. J. Bot.104102–115. 10.3732/ajb.1600227
62
MuscarellaR.BaconC. D.FaurbyS.AntonelliA.KristiansenS. M.SvenningJ.-C.et al (2018). Soil fertility and flood regime are correlated with phylogenetic structure of Amazonian palm communities.Ann. Bot.123641–655. 10.1093/aob/mcy196
63
NguyenL.-T.SchmidtH. A.von HaeselerA.MinhB. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies.Mol. Biol. Evol.32268–274. 10.1093/molbev/msu300
64
NichollsJ. A.PenningtonR. T.KoenenE. J. M.HughesC. E.HearnJ.BunnefeldL.et al (2015). Using targeted enrichment of nuclear genes to increase phylogenetic resolution in the neotropical rain forest genus Inga (Leguminosae: Mimosoideae).Front. Plant Sci.6:710. 10.3389/fpls.2015.00710
65
PenningtonR. T.LavinM. (2016). The contrasting nature of woody plant species in different neotropical forest biomes reflects differences in ecological stability.New Phytol.21025–37. 10.1111/nph.13724
66
PinheiroF.Dantas-queirozM. V.Palma-silvaC. (2018). Plant species complexes as models to understand speciation and evolution: a review of South American studies.Crit. Rev. Plant Sci.3754–80. 10.1080/07352689.2018.1471565
67
PizoM. A.Almeida-NetoM. (2009). Determinants of fruit removal in Geonoma pauciflora, an understory palm of neotropical forests.Ecol. Res.241179–1186. 10.1007/s11284-009-0599-0
68
PondS. L. K.MuseS. V. (2005). “HyPhy: hypothesis testing using phylogenies,” in Statistical Methods in Molecular Evolution, ed.NielsenR. (New York, NY: Springer New York), 125–181. 10.1007/0-387-27733-1_6
69
RambautA. (2012). FigTree v1. 4. Available at: http://tree.bio.ed.ac.uk/software/figtree/(accessed 28 April 2014).
70
RiesebergL. H.BrouilletL. (1994). Are many plant species paraphyletic?Taxon4321–32. 10.2307/1223457
71
Rodriguez-BuriticaS.OrjuelaM. A.GaleanoG. (2005). Demography and life history of Geonoma orbignyana: an understory palm used as foliage in Colombia.For. Ecol. Manag.211329–340. 10.1016/j.foreco.2005.02.052
72
RoncalJ. (2006). Habitat differentiation of sympatric Geonoma macrostachys (Arecaceae) varieties in Peruvian lowland forests.J. Trop. Ecol.22483–486. 10.1017/s0266467406003270
73
RoncalJ.Blach-OvergaardA.BorchseniusF.BalslevH.SvenningJ. C. (2011). A dated phylogeny complements macroecological analysis to explain the diversity patterns in Geonoma (Arecaceae).Biotropica43324–334. 10.1111/j.1744-7429.2010.00696.x
74
RoncalJ.BorchseniusF.Asmussen-LangeC. B.BalslevH. (2010). “Divergence times in the tribe Geonomateae (Arecaceae) coincide with tertiary geological events,” in Diversity, Phylogeny and Evolution of the Monocotyledons, edsSebergO.PetersenG.BarfodA. S.DavisJ. I. (Aarhus: Aarhus University Press), 245–265.
75
RoncalJ.Francisco-OrtegaJ.AsmussenC. B.LewisC. E. (2005). Molecular phylogenetics of tribe Geonomeae (Arecaceae) using nuclear DNA sequences of Phosphoribulokinase and RNA Polymerase II.Syst. Bot.30275–283. 10.1600/0363644054223620
76
RoncalJ.Francisco-OrtegaJ.LewisC. E. (2007). An evaluation of the taxonomic distinctness of two Geonoma macrostachys (Arecaceae) varieties based on intersimple sequence repeat (ISSR) variation.Bot. J. Linn. Soc.153381–392. 10.1111/j.1095-8339.2007.00619.x
77
RoncalJ.HendersonA.BorchseniusF.CardosoS. R. S.BalslevH. (2012). Can phylogenetic signal, character displacement, or random phenotypic drift explain the morphological variation in the genus Geonoma (Arecaceae)?Biol. J. Linn. Soc.106528–539. 10.1111/j.1095-8312.2012.01879.x
78
RoncalJ.ZonaS.LewisC. E. (2008). Molecular phylogenetic studies of Caribbean palms (Arecaceae) and their relationships to biogeography and conservation.Bot. Rev.7478–102. 10.1007/s12229-008-9005-9
79
SampaioM. B.ScariotA. (2008). Growth and reproduction of the understory palm Geonoma schottiana Mart. in the gallery forest in Central Brazil.Rev. Bras. Bot.31433–442. 10.1590/S0100-84042008000300007
80
SanínM. J.KisslingW. D.BaconC. D.BorchseniusF.GaleanoG.SvenningJ.-C.et al (2016). The neogene rise of the tropical Andes facilitated diversification of wax palms (Ceroxylon: Arecaceae) through geographical colonization and climatic niche separation.Bot. J. Linn. Soc.182303–317. 10.1111/boj.12419
81
SassC.IlesW. J. D.BarrettC. F.SmithS. Y.SpechtC. D. (2016). Revisiting the Zingiberales: using multiplexed exon capture to resolve ancient and recent phylogenetic splits in a charismatic plant lineage.PeerJ4:e1584. 10.7717/peerj.1584
82
SayyariE.MirarabS. (2016). Fast coalescent-based computation of local branch support from quartet frequencies.Mol. Biol. Evol.331654–1668. 10.1093/molbev/msw079
83
SinghR.Ong-AbdullahM.LowE.-T. L.ManafM. A. A.RosliR.NookiahR.et al (2013). Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds.Nature500335–339. 10.1038/nature12309
84
SmedsL.KünstnerA. (2011). ConDeTri-a content dependent read trimmer for Illumina data.PLoS One6:e26314. 10.1371/journal.pone.0026314
85
StamatakisA. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies.Bioinformatics301312–1313. 10.1093/bioinformatics/btu033
86
StaufferF. W. (1997). Estudio morfologico y taxonomico de Geonoma spinescens H. Wendl. ex Burret (Arecaceae) y descripcion de una nueva variedad.Acta Bot. Venezuelica201–10.
87
StaufferF. W.AsmussenC. B.HendersonA.EndressP. K. (2003). A revision of Asterogyne (Arecaceae: Arecoideae: Geonomeae).Brittonia55326–356. 10.1663/0007-196x(2003)055
88
TownsendJ. P. (2007). Profiling phylogenetic informativeness.Syst. Biol.56222–231. 10.1080/10635150701311362
89
UhlN. W.DransfieldJ. (1987). Genera Palmarum. A Classification of Palms Based on the Work of Harold E. Moore, Jr.Lawrence, KS: Allen Press.
90
UthaipaisanwongP.ChanprasertJ.ShearmanJ. R.SangsrakruD.YoochaT.JomchaiN.et al (2012). Characterization of the chloroplast genome sequence of oil palm (Elaeis guineensis Jacq.).Gene500172–180. 10.1016/j.gene.2012.03.061
91
VormistoJ.SvenningJ.HallP.BalslevH. (2004). Diversity and dominance in palm (Arecaceae) communities in terra firme forests in the western Amazon basin.J. Ecol.92577–588. 10.1111/j.0022-0477.2004.00904.x
92
Wessels BoerJ. G. (1968). The geonomoid palms.Meded. Van Het Bot. Mus. En Herb. Van Rijksuniv. Te Utrecht2821–202. 10.3732/ajb.89.11.1772
93
WhitneyK. D.AhernJ. R.CampbellL. G.AlbertL. P.KingM. S. (2010). Patterns of hybridization in plants.Perspect. Plant Ecol. Evol. Syst.12175–182.
94
WilsonM. A.GautB.CleggM. T. (1990). Chloroplast DNA evolves slowly in the palm family (Arecaceae).Mol. Biol. Evol.7303–314.
95
WuC. (2001). The genic view of the process of speciation.J. Evol. Biol.14851–865.
96
YangM.ZhangX.LiuG.YinY.ChenK.YunQ.et al (2010). The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.).PLoS One5:e12762. 10.1371/journal.pone.0012762
97
ZonaS. (1995). A revision of Calyptronoma.Principes39140–151.
Summary
Keywords
Arecaceae, Geonoma, Neotropics, phylogenetic informativeness, phylogenomics, species complexes
Citation
Loiseau O, Olivares I, Paris M, de La Harpe M, Weigand A, Koubínová D, Rolland J, Bacon CD, Balslev H, Borchsenius F, Cano A, Couvreur TLP, Delnatte C, Fardin F, Gayot M, Mejía F, Mota-Machado T, Perret M, Roncal J, Sanin MJ, Stauffer F, Lexer C, Kessler M and Salamin N (2019) Targeted Capture of Hundreds of Nuclear Genes Unravels Phylogenetic Relationships of the Diverse Neotropical Palm Tribe Geonomateae. Front. Plant Sci. 10:864. doi: 10.3389/fpls.2019.00864
Received
14 January 2019
Accepted
17 June 2019
Published
12 July 2019
Volume
10 - 2019
Edited by
Lisa Pokorny, Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, Spain
Reviewed by
Karolina Heyduk, Yale University, United States; Zhen Li, Ghent University, Belgium
Updates
Copyright
© 2019 Loiseau, Olivares, Paris, de La Harpe, Weigand, Koubínová, Rolland, Bacon, Balslev, Borchsenius, Cano, Couvreur, Delnatte, Fardin, Gayot, Mejía, Mota-Machado, Perret, Roncal, Sanin, Stauffer, Lexer, Kessler and Salamin.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolas Salamin, nicolas.salamin@unil.ch
†Share co-last authorship
This article was submitted to Plant Systematics and Evolution, a section of the journal Frontiers in Plant Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.