 Jorge Vieira1,2Sara Rocha1,2Noé Vázquez3,4Hugo López-Fernández1,2,3,4,5
Jorge Vieira1,2Sara Rocha1,2Noé Vázquez3,4Hugo López-Fernández1,2,3,4,5 Florentino Fdez-Riverola3,4,5Miguel Reboiro-Jato3,4,5
Florentino Fdez-Riverola3,4,5Miguel Reboiro-Jato3,4,5 Cristina P. Vieira1,2*
Cristina P. Vieira1,2*- 1Instituto de Biologia Molecular e Celular (IBMC), Universidade do Porto, Porto, Portugal
- 2Instituto de Investigação e Inovação em Saúde, Universidade do Porto, Porto, Portugal
- 3Escuela Superior de Ingeniería Informática (ESEI), Edificio Politécnico, Universidad de Vigo, Ourense, Spain
- 4Centro de Investigaciones Biomédicas (Centro Singular de Investigación de Galicia), Vigo, Spain
- 5SING Research Group, Instituto de Investigación Sanitaria Galicia Sur (IIS Galicia Sur), SERGAS-UVIGO, Vigo, Spain
Non-self gametophytic self-incompatibility (GSI) recognition system is characterized by the presence of multiple F-box genes tandemly located in the S-locus, that regulate pollen specificity. This reproductive barrier is present in Solanaceae, Plantaginacea and Maleae (Rosaceae), but only in Petunia functional assays have been performed to get insight on how this recognition mechanism works. In this system, each of the encoded S-pollen proteins (called SLFs in Solanaceae and Plantaginaceae /SFBBs in Maleae) recognizes and interacts with a sub-set of non-self S-pistil proteins, called S-RNases, mediating their ubiquitination and degradation. In Petunia there are 17 SLF genes per S-haplotype, making impossible to determine experimentally each SLF specificity. Moreover, domain –swapping experiments are unlikely to be performed in large scale to determine S-pollen and S-pistil specificities. Phylogenetic analyses of the Petunia SLFs and those from two Solanum genomes, suggest that diversification of SLFs predate the two genera separation. Here we first identify putative SLF genes from nine Solanum and 10 Nicotiana genomes to determine how many gene lineages are present in the three genera, and the rate of origin of new SLF gene lineages. The use of multiple genomes per genera precludes the effect of incompleteness of the genome at the S-locus. The similar number of gene lineages in the three genera implies a comparable effective population size for these species, and number of specificities. The rate of origin of new specificities is one per 10 million years. Moreover, here we determine the amino acids positions under positive selection, those involved in SLF specificity recognition, using 10 Petunia S-haplotypes with more than 11 SLF genes. These 16 amino acid positions account for the differences of self-incompatible (SI) behavior described in the literature. When SLF and S-RNase proteins are divided according to the SI behavior, and the positively selected amino acids classified according to hydrophobicity, charge, polarity and size, we identified fixed differences between SI groups. According to the in silico 3D structure of the two proteins these amino acid positions interact. Therefore, this methodology can be used to infer SLF/S-RNase specificity recognition.
Introduction
To avoid self-fertilization and promote out-crossing, many Angiosperms have developed self-incompatible (SI) mechanisms, that allow the pistil to reject self-pollen and thus, only non-genetically related pollen is allowed to effect fertilization (De Nettancourt, 1977). Despite the large diversity of SI mechanisms, the most common pre-zygotic reproductive system in flowering plants is the gametophytic self-incompatibility (GSI) system (Igic et al., 2008). The genes determining pistil (S-pistil) and pollen (S-pollen) GSI specificity have been characterized in self-incompatible Papaveraceae, Solanaceae, Plantaginaceae, and Rosaceae species. In Papaveraceae, the pollen-pistil interaction occurs in the stigma surface during or shortly after germination (Foote et al., 1994). The pistil S-locus is a small protein (∼15 kDa, called PrsS (Papaver rhoeas stigmatic S- protein; Foote et al., 1994) secreted to the pistil surface that binds to the self- pollen S-receptor [called PrpS (Papaver rhoeas pollen S); Wheeler et al., 2009], triggering a Ca (2+) dependent signaling network, that results in pollen inhibition and programmed cell death (Eaves et al., 2014; Wilkins et al., 2014). In Solanaceae, Plantaginaceae, and Rosaceae the rejection of self-pollen occurs during the growth of pollen tubes in the style, and the pistil gene is an extracelular ribonuclease, called S-RNase (Roalson and McCubbin, 2003; McClure, 2009; Nowak et al., 2011), and the pollen gene(s) is(are) F-box protein(s) (Entani et al., 2003; Ushijima et al., 2003; Qiao et al., 2004; Tsukamoto et al., 2005, 2010; Cheng et al., 2006; Nunes et al., 2006; Hua et al., 2007; Sassa et al., 2007; Wheeler and Newbigin, 2007; Kubo et al., 2010, 2015; Minamikawa et al., 2010; Kakui et al., 2011; Okada et al., 2011; Aguiar et al., 2013, 2015; Williams et al., 2014). Therefore, Papaveraceae GSI has evolved independently from that of Solanaceae, Plantaginaceae, and Rosaceae. In Solanaceae, Plantaginaceae, and Rosaceae, both phylogenetic analyses and gene structure analyses (conserved and hypervariable regions, intron number and position) of the S-RNase, shows that the RNase-based system has evolved only once, about 120 million years ago, before the separation of the Asteridae and Rosideae (Igic and Kohn, 2001; Steinbachs and Holsinger, 2002; Vieira et al., 2008; Ramanauskas and Igic, 2017). The shared ancestry, however, does not imply that gene duplications, followed by functional change, could occur and thus, that paralogous genes could be determining S-pistil specificity in different species groups. Indeed, in the Rosaceae family, two gene lineages are determining Prunus (Amygdaleae) and Malus (Maleae) S-pistil and S-pollen specificity (Aguiar et al., 2015). In these species a self- and non-self- recognition mechanisms is present, respectively. So far, only Prunus has an RNase based self-recognition mechanism. In this system, only one S-pollen gene (called SFB), is sufficient to account for the self-S-RNase inhibition (Entani et al., 2003; Ushijima et al., 2003; Ikeda et al., 2004; Tsukamoto et al., 2005, 2010; Nunes et al., 2006; Hua et al., 2007; Aguiar et al., 2015). In this model, a “general inhibitor,” such as the gene products of the F-box like genes in the vicinity of the S-locus region (Matsumoto and Tao, 2016; Chen et al., 2018), inactivate the cytotoxic effect of non-self S-RNases (Luu et al., 2001; Sonneveld et al., 2005).
In the non-self -recognition mechanism multiple S-pollen genes (called SLFs; S-locus F-box in Solanaceae (Wheeler and Newbigin, 2007; Kubo et al., 2010, 2015; Williams et al., 2014, 2015; Sun et al., 2015; Li et al., 2017; Wu et al., 2018), and SFBB; S-locus F-box brothers in Maleae (Sassa et al., 2007; Minamikawa et al., 2010; Kakui et al., 2011; Okada et al., 2011; Aguiar et al., 2013, 2015)) regulate pollen specificity. Each of the encoded S-proteins recognizes and interacts with a sub-set of non-self S-RNases, and mediate their degradation, under a model called the collaborative non-self -recognition model (Kubo et al., 2010, 2015; Aguiar et al., 2013; Williams et al., 2014, 2015; Sun et al., 2015; Pratas et al., 2018).
Petunia inflata and P. axillaris (Solanaceae) are the model species for functional assays on how the non-self -recognition mechanism works. Transgenic P. inflata has been used to show that S-RNases most likely function as a toxin that specifically degrades RNA of incompatible pollen tubes. Indeed, the expression of a mutant form of S3-RNase (where the His residue in the catalytic domain has been replaced with Arg), in a plant with the S1S2 genotype did not confer the ability of the pistils of this transgenic plant to reject S3 pollen (Huang et al., 1994). Nevertheless, transgenic plants carrying pollen from two different S-haplotypes are self-compatible. This “competitive interaction” gain of function observation was used to identify the first Petunia SLF gene (Sijacic et al., 2004). Transgenic assays using the first identified SLF gene in different S-haplotype backgrounds did not produce self-compatible plants, leading to the identification of other SLF genes involved in GSI specificity (Kubo et al., 2010).
The characterization of the behavior of the products of three SLF genes, in the context of four different S-haplotypes revealed that only a sub-set of SLFs interact with a given non-self S-RNase (Kubo et al., 2010). This implies a large number of SLF genes to account for the large number of S-RNases [about 32 in P. inflata and P. hybrida, that are genetically distinct; (Sims and Robbins, 2009)]. Other approaches have been used, such as the use of artificial microRNA to determine which S-RNases are recognized by the product of a particular SLF allele (Sun and Kao, 2013). Co-immunoprecipitation experiments also show that non-self S-pollen proteins and the S-RNases interact during the SI response (Kubo et al., 2010). These experiments show that SLFs are in an SCF complex, as predicted by the protein degradation model in which SLFs are the F-box proteins of the E3 ubiquitin ligase complex that mediates ubiquitination of all non-self S-RNases (Hua and Kao, 2006; Hua et al., 2008).
To get insight into how many SLFs can contribute for the S-pollen specificity determination, Williams et al. (2014) have analyzed the pollen transcriptome of two S-locus homozygous plants. These authors found 17 SLF genes for each S-haplotype, seven of which were already known (Kubo et al., 2010; Williams et al., 2014). For 11 of these genes, segregation analyses support their role in S-pollen specificity determination (Kubo et al., 2010; Williams et al., 2014). Using large-scale next-generation sequencing and genomic PCR techniques, the characterization of SLF genes in eight P. hybrida S-haplotypes revealed that the number of SLF genes per S-haplotype varies from 16 to 20, that have been grouped into 18 types (Kubo et al., 2015). Thus, different related genes are grouped into the same SLF type. The apparent variation in copy number suggests that SLF specificity recognition can either be achieved by a diverged or deleted allele of a SLF gene (Kubo et al., 2015). Indeed, using this observation alone, these authors could predict the SLF specificity of an allele for seven S-RNases.
Phylogenetic analyses based on the annotation of the Solanum lycopersicum and S. tuberosum genomes as well as custom annotations, led to the identification of 13 and 14 genes, respectively. These analyses suggested that diversification of SLFs predated the separation of the Petunia and Solanum genera, although a small number of SLFs are genera specific (Kubo et al., 2015). Since the number of specificities that can be maintained in a population depends on the effective population size of a population/species (Wright, 1939), the difference found could just reflect different effective population sizes of the genera analyzed. Reliable estimates of the rate at which SLF genes emerge is fundamental to understand how the system evolves. This can only be done using a large number of species from different genera. Thus, in this work we used nine genome assemblies from eight Solanum species and 10 genomes from seven Nicotiana species publicly available. Only four of these are self-incompatible species, and thus we searched for both genes and pseudogenes, using Blast DataBase Manager (BDBM1; Vázquez et al., 2019). The high number of genomes analyzed also implies that it is unlikely that a given SLF will be missing in all genomes simply by chance.
Identification of the regions involved in GSI specificity is also fundamental to understand how the system works. Domain-swapping transgenic experiments with similar S-RNase alleles have showed that the two hypervariable regions of S-RNases, are involved in specificity determination (Ide et al., 1991; Matton et al., 1997, 1999; Matsuura et al., 2001). Although there are positively selected amino acid sites in these regions, there are other amino acid sites in other regions of the S-pistil gene, that may be involved in recognition of the S-pollen gene(s) (Vieira et al., 2007; Brisolara-Correa et al., 2015). To address S-pollen specificity, Petunia SLF1 gene has been divided into three functional domains (FD1 from amino acid position 1 to 110; FD2, from amino acid position 110 to 260, and FD3 from amino acid position 260 to 389), and in vivo assays performed. These experiments showed that FD2 is primarily responsible for the strong interaction between an allelic product of SLF1 gene and the S-RNase, and this interaction is negatively modulated by FD1 and FD3 that together determine the specificity of the SLF1 allele (Hua et al., 2007; Fields et al., 2010). Moreover, yeast two hybrid assays show that the C-terminal regions of SLF interact with S-RNases mainly in hypervariable regions, as also obtained in the 3D structural model of interaction of the SLFs and of S-RNase (Li et al., 2017). In the latter study Li and co-authors show, using in vivo domain-swapping transgenic experiments, that the SLF1 C-terminal region (amino acids 265–389) acts as a major specificity domain in vivo. Two amino acid sites (293 and 317) in this region show evidence for positive selection, using 21 sequences from P. inflata, P. hybrida, and P. axillares using PAML (Li et al., 2017), despite the prediction that, under the non-self-recognition model, natural selection favors diversification of SLF genes rather than alleles within an S haplotype (Kakui et al., 2011; Aguiar et al., 2013). Such analyses are here performed for 10 Petunia S-haplotyes for which more than 11 SLF genes are available, and the results deposited at the B+ database2 (Vázquez et al., 2017, 2018). The 16 amino acid positions under positive selection here identified account for the differences described in SI behavior when different alleles of the same SLF gene are analyzed (Kubo et al., 2010, 2015; Williams et al., 2014; Li et al., 2017). Indeed, we classify SLF specificity according to these amino acids sites. We characterized the S-RNase and SLF amino acid properties (hydrophobicity, charge, polarity and size) at positions under positive selection, to infer critical features for GSI specificity determination. The identification of these amino acid sites on the three-dimensional structural model of interaction of the S-pistil and S-pollen proteins shows that the two proteins interact at these amino acid sites.
Materials and Methods
Solanaceae SLFs Dataset
Petunia SLFs were obtained by querying the NCBI nucleotide database3 with the words “Petunia” and “SLF.” We complemented this approach by looking for those Petunia SLF genes listed in Supplementary Table 1 of Kubo et al. (2015) and Williams et al. (2014), that were not retrieved because the word SLF is missing in the NCBI record. The acc. numbers of the 244 CDS used are shown in brackets in Supplementary Figure S1.
To select reference sequences to be used for the identification of SLF CDS in Solanaceae species, we obtained a Bayesian phylogenetic tree [using MrBayes; (Huelsenbeck and Ronquist, 2001)], after using Muscle as implemented in T-coffee (Notredame et al., 2000), to align the sequences, as implemented in ADOPS (Reboiro-Jato et al., 2012). The GTR model of sequence evolution that was used, allowed for among-site rate variation and a proportion of invariable sites. Third codon positions were allowed to have a gamma distribution shape parameter different from that of first and second codon positions. Two independent runs of 1,000,000 generations with four chains each (one cold and three heated chains) were set up. Trees were sampled every 100th generation and the first 2500 samples were discarded (burn-in). The remaining trees were used to compute the Bayesian posterior probabilities of each clade of the consensus tree. The potential scale reduction factor for every parameter was about 1.00 showing that convergence has been achieved. The tree was rooted with a SLF-like gene (a gene (S2-SLF-like1) not involved in GSI specificity determination from P. integrifolia). For each of the identified genes we selected a reference sequence (marked in bold in Supplementary Figure S1).
Nine genomes from eight Solanum species and 10 genomes from seven Nicotiana species (Table 1) were downloaded from NCBI (assembly database). For those genomes having an annotation, the corresponding predicted CDSs were also downloaded (Table 1). Using the Blast DataBase Manager software (BDBM; Vázquez et al., 2019) Reformat FASTA option, sequence headers were reformatted in order to show the species name and accession numbers only. Since SLF genes are always intronless (Wang et al., 2004), then we obtained all open reading frames (ORFs) from these genomes using the Get ORF operation of BDBM (that uses the getorf command of EMBOSS; Vázquez et al., 2019), with a minimum and maximum size of 300 and 10000 bp, respectively. We then prepared a Blast database for each of the resulting FASTA files, using the Make Blast Database option of BDBM (Vázquez et al., 2019). Moreover, using the same software, we created an alias in order to treat all databases of interest as a single one (using BLAST DB Alias operation as implemented in BDBM; Vázquez et al., 2019). A tblastn search was then performed, using as the query the 36 translated Petunia SLF reference sequences without the F- box motif (the first 60 amino acids), and an e-value of 0.05. This approach will retrieve nucleotide sequences showing similarity at the amino acid level with the SLF reference sequences beyond the F-box motif. To the obtained dataset we added the predicted CDSs from the annotated genomes, and removed identical sequences, after merging the headers, using SEDA4 (López-Fernández et al., 2019). As before, sequences were aligned using Muscle and a Bayesian phylogenetic tree was obtained using ADOPS (Reboiro-Jato et al., 2012).
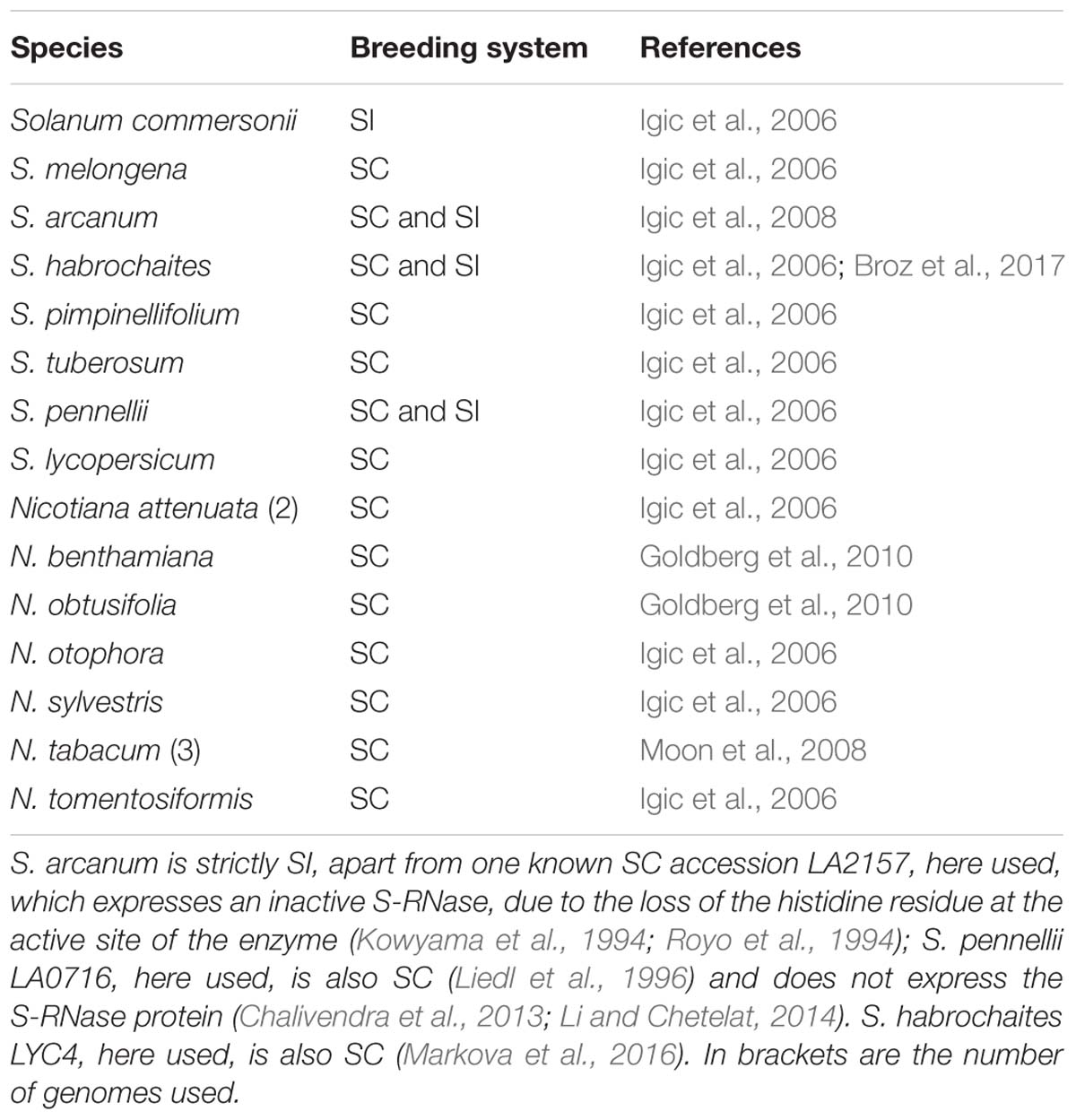
Table 1. Breeding system for the species used in this work.
SLF genes may be missed when using the above mentioned approach, if there are mistakes in the genome sequence, such as insertions and deletions. Moreover, most species here studied are self-compatible (Table 1), and thus the coding region of many SLFs may not be a multiple of three, meaning that they are pseudogenes. Since one of the objectives of this research is to identify all SLF gene lineages present in the Solanaceae genomes, we also used the Splign-Compart and the ProSplign-Compart options (that are insensitive to frameshifts since the annotation is based on similarity at the nucleotide or amino acid level, respectively, and presence of putative splice sites only), as implemented in BDBM (Vázquez et al., 2019). As reference sequences we used for Splign-Compart all sequences shown in Supplementary Figure S2, and for ProSplign-Compart the translation of these sequences. In order to address the hypothesis that there are Solanaceae SLF gene lineages that are not present in Petunia, the P. inflata and P. axillaris genomes were also downloaded from Sol Genomics Network5, and the Splign-Compart as well as the ProSplign-Compart options (implemented in BDBM; Vázquez et al., 2019) used.
Phylogenetic and Positive Selection Analyses
Sequences were aligned using Muscle and a Bayesian phylogenetic tree obtained using MrBayes (Huelsenbeck and Ronquist, 2001), as described above.
Positively selected amino acid sites were inferred using codeML (Yang, 2007) as implemented in ADOPS (Reboiro-Jato et al., 2012), using 10 Petunia S-haplotypes for which, according to (Williams et al., 2014; Kubo et al., 2015), more than 11 SLF genes are available. Phylogenetic trees were obtained as described above. All details can be seen at the B+ database (bpositive.i3s.up.pt; Petunia dataset BP2017000006). Models comparisons were M2a-M1a and M8-M7. We consider as positively selected those amino acid sites that show a probability higher than 95% for both naive empirical Bayes (NEB) or Bayes empirical Bayes (BEB) methods in at least one of the analyses.
Protein Prediction Analyses
The protein sequence of P. integrifolia S3-RNase (M67991), P. hybrida S7-RNase (AB568388), P. axillaris S19-RNase (Kubo et al., 2010), P. integrifolia S2-SFL1 (AY500391), P. hybrida S5-SFL3 (AB568399), and P. hybrida S11-SFL9 (AB933017) were obtained from NCBI. In the N-terminal region of S3-RNase, S7-RNase, and S19-RNase, we removed the first 23 amino acid residues, corresponding to the peptide cleavage site, according to SignalP 4.1 server (Nielsen, 2017). In the N-terminal region of the S5-SLF1, S7-SLF1, S9-SLF1 and S17-SLF1 we removed the first 49 amino acid residues representing the F-box domain. The 3D structure predictions for all proteins (S-RNases without signal peptide and S-SLFs without F– box domains) were modeled by I-Tasser (Yang et al., 2015). For all proteins we used the models with the highest C-score values. The docking of the two proteins was inferred using the HADDOCK server (van Zundert et al., 2016). For both the S-RNase and SLFs, the active residues used in HADDOCK were the amino acids under positive selection (for the S-RNase see (Vieira et al., 2007); for the SLFs see Results) and the two surrounding amino acids. In the case of SLFs we also used the amino acids described in Li et al. (2017) and Wu et al. (2018) as being involved in specificity determination. The passive residues for S-RNases and SLFs were automatically defined around the active residues.
For all HADDOCK clusters showing negative Z-Scores, PISA (Krissinel and Henrick, 2007) was used to calculate the number of hydrogen bonds and salt bridges for the P. hybrida S19-RNase:S11-SFL9. The cluster with the highest number of hydrogen bonds and salt bridges was chosen as being the most likely. This result was used as reference for the inferences regarding the docking of the other S-RNases and SLFs, such as P. integrifolia S3-RNase:S2-SFL1, and P. hybrida S7-RNase:S5-SFL3. This was achieved by selecting from the HADDOCK clusters with negative Z-Scores the one that presented the best structural similarity (according to TM-score) with the chosen model structure of P. hybrida S19-RNase:S11-SFL9, using the TM-align server (Zhang and Skolnick, 2005). All structural images were produced using PyMOL (The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC).
Results
SLF Gene Number in Three Solanaceae Lineages (Petunia, Solanum, and Nicotiana)
In order to estimate how many of the lineages present in Petunia predate the separation of the different Solanaceae species, we need to establish first a set of Petunia nucleotide sequences representative of each gene lineage. The phylogenetic relationship of the 245 SLF Petunia sequences imply a minimum of 36 Petunia SLF genes (Supplementary Figure S1). Of these genes, nine (2, 6, 8, 14, 20, 29, 31, 32, 33 in Supplementary Figure S1) are recent duplications and may be restricted to Petunia species. Five of these genes (9, 11, 13, 17, and 22 in Supplementary Figure S1) represent old lineages and thus were expected to be present in most haplotypes, but have been reported for one S-haplotype only. The Bayesian phylogenetic tree obtained using the 29 Nicotiana and 75 Solanum SLF sequences obtained after using the Get ORF option of BDBM (Vázquez et al., 2019; using the nine available genomes for eight Solanum species, and the 10 available genomes for seven Nicotiana species; Table 1; see Material and Methods), together with one sequence of each Petunia gene, and the nine N. alata SLF sequences (named DDs; Wheeler and Newbigin, 2007), revealed five gene lineages common to the three genera (marked as PNS in Supplementary Figure S2), six present in Petunia and Solanum (PS in Supplementary Figure S2), three in Petunia and Nicotiana (PN in Supplementary Figure S2), and three in Nicotiana and Solanum (NS in Supplementary Figure S2). Therefore, most of the gene lineages seem to be missing at one of the genera. Nevertheless, since most species analyzed are SC, SLFs may be pseudogenes in these species and have frameshifts that precluded their identification using the Get ORF option. Indeed, when using the Splign-Compart option, as implemented in BDBM (Vázquez et al., 2019), 59 SLF sequences are identified (marked in bold in Supplementary Figure S3). The number of gene lineages present in the three genera is then eight (marked as PNS; Figure 1), common to Petunia and Solanum there are three, in Petunia and Nicotiana there are four, and in Nicotiana and Solanum there are four. There are three divergent lineages present in Nicotiana, and two in Solanum, as observed in Petunia. Therefore, the number of SLF genes (17 to 19) is similar in the three genera. This implies that most of the Petunia gene duplications have occurred after the genus split, and are S-haplotype specific. The two Petunia genomes here analyzed revealed only three known SLF genes (SLF2, SLF7, and SLF17; Figure 1). The sequence identified in P. axillaris genome is likely from a novel haplotype, since there are 49 differences between the sequence we retrieved and the P. axillaris S19-SLF2 (AB933033) sequence. For P. inflata the SLF17 sequence is 100% identical to P. integrifolia S2-SLF17 (KJ670458), but the SLF7 sequence is only 99% identical (there are eight nucleotide differences between the two sequences) to the P. integrifolia S2-SLFla (EF614189; an allele of SLF7; Supplementary Figure S1). It is conceivable that the sequence we retrieved comes from a yet to be characterized S-haplotype, but it is also possible that the observed differences are due to sequencing or assembly mistakes. Nevertheless, this approach shows that the Petunia genomes here used either have a low coverage or the S-locus region is difficult to assemble in Petunia.
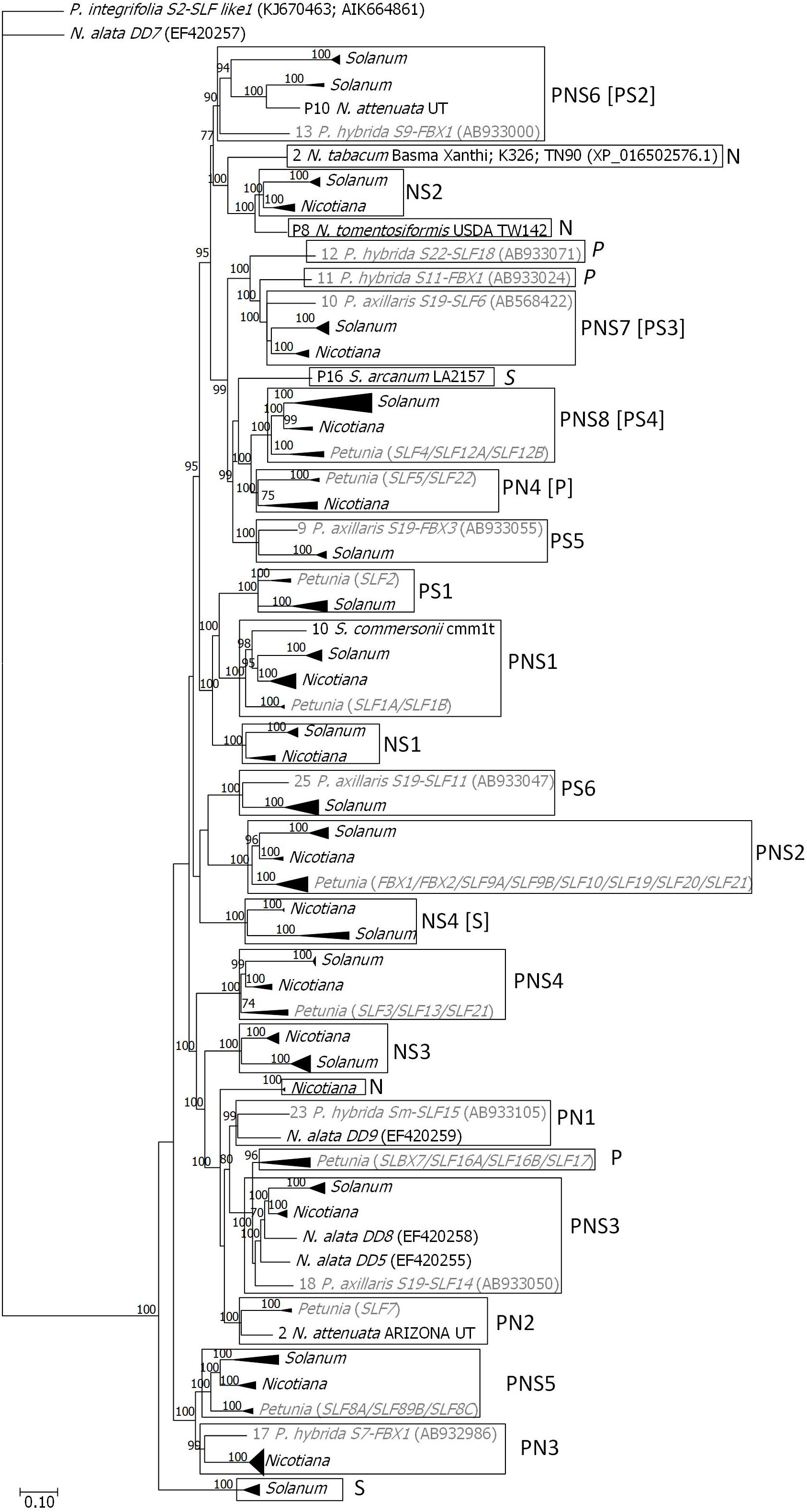
Figure 1. Bayesian phylogenetic tree showing the relationship of the Petunia, Solanum, and Nicotiana SLF sequences, used to infer gene lineages. The tree was rooted with P. integrifolia S2-SLFlike1 (KJ670463; AIK66486). For Petunia (in gray) only one sequence for each SLF gene (1–36; Supplementary Figure S1) was used. Squares represent gene lineages, and P, N, and S are used to identify if the gene lineage is present in Petunia, Nicotiana, and Solanum, respectively. In square brackets is the characterization of each gene lineage when pseudogenes are not considered (Supplementary Figure S2). Numbers above the branches represent posterior credibility values above 70.
For six genomes, including three from SI or SI/SC Solanum species, the number of sequences identified is 16 or more (Table 2), a number similar to that observed in Petunia (Kubo et al., 2015). Only for the SI species S. habrochaites the number of identified SLF sequences is 13. For the SI species, a maximum of three sequences were inferred to be non-functional. The Solanum sequences that belong to the PNS6 and PNS8 lineage (Supplementary Figure S3) are mostly non-functional and thus are unlikely to be inferred to be pseudogenes because of sequencing errors. In the other cases, however, this is a possibility. In N. tabacum at least 20 SLF sequences have been identified, but half are non-functional, which is expected since this is a SC species. In conclusion, the number of SLF genes per S-haplotype in Solanum and Nicotiana seem to be similar to that of Petunia, implying similar effective population sizes for the three species.
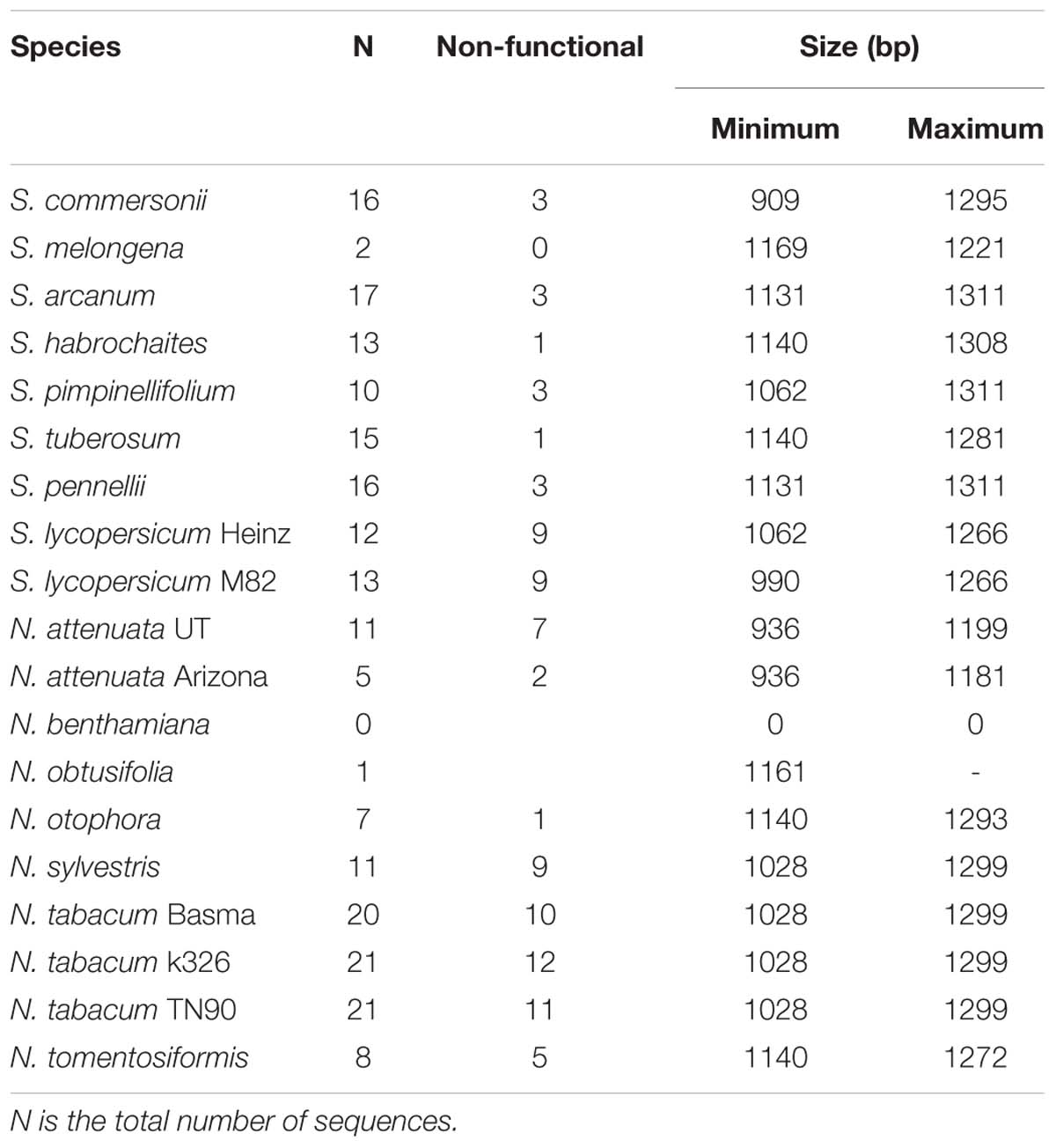
Table 2. Number of SLF sequences (including pseudogenes) and size for the genomes analyzed.
On the Identification of the Amino Acid Sites Determining S-Pollen GSI Specificity
Positively selected amino acid sites have been used to infer the amino acid positions that are, in principle, responsible for GSI specificity (Vieira et al., 2008, 2010; Aguiar et al., 2013, 2015). Under the non-self recognition by multiple factors model, the high intra-haplotypic diversity of S-pollen genes is the result of natural selection favoring diversification within an S-haplotype (Kakui et al., 2011; Aguiar et al., 2013, 2015; Pratas et al., 2018). Therefore, positively selected amino acid sites should be detected when carrying intra-haplotypic analyses (Aguiar et al., 2013, 2015; Pratas et al., 2018). Nevertheless, no strong evidence for positive selection is expected when individual S-pollen genes are considered (Vieira et al., 2009; De Franceschi et al., 2011a,b). A total of 16 amino acids under positive selection are here identified when performing codeML (Yang, 2007) analyses using 10 Petunia S-haplotypes having more than 11 genes characterized (Supplementary Figure S4; B+ database (bpositive.i3s.up.pt; see the Petunia SLF intra haplotype positive selection dataset BP2017000006). It should be noted that two of these amino acid sites are the same that Wu et al. (2018) assigned as being involved in specificity determination between P. integrifolia S2-SLF1 and S3-RNase. None of these amino acid positions are, however, those identified by Li et al. (2017), as determining the pollen S specificity in vivo between S3-RNase and S3l-SLF1 in P. hybrida.
Specificity determination implies physical interactions between the S-pollen and S-pistil proteins at particular amino acid sites. These amino acid sites may present proprieties such as hydrophobicity, size, charge, at a particular S-allele (or group of S-alleles) that allows to distinguish it (them) as non-self. Li et al. (2017) have shown that by swapping one amino acid between two SLFs, located in the C-terminal region (a region that contains a major specificity domain in vivo) that differs in charge and hydrophobicity, is sufficient to modify the specificity of the transgenic P. hybrida S3-SLF1. Therefore, we looked for such amino acid properties at the sites identified as positively selected, by grouping sequences according to specificity recognition described in the literature. According to S2-SLF1 recognition we group the S3-, S7-, S12-, and S13-RNases (group I) as non-self S-RNases, and S2- and S11-RNases (group II) as self (Sun and Kao, 2013; Williams et al., 2014). The two groups of S-RNases differ in two amino acids identified as positively selected (marked in bold and gray in Figure 2A; (Vieira et al., 2007)), at position 76, and 103 (marked with a # and a & in Figure 2A) in what concerns hydrophobicity (group I is hydrophobic), and in the latter position, charge (group I is positively charged), and size (amino acids are small in group II). Dividing the SLF1 sequences into the same groups, there are only two amino acid sites (213, and 262; Figure 2B) identified as positively selected that are different between the groups. The last position is one of the amino acids identified as determining P. integrifolia S2-SLF1/S3-RNase specificity, in Wu et al. (2018). It should be noted that, none of the other amino acid positions identified as putatively involved in specificity determination of P. integrifolia S2-SLF1/S3-RNase (marked with triangles in Figure 2B) are fixed in the groups here used. The amino acid located at position 213 is different in terms of hydrophobicity, size and polarity between the two groups (in group I is hydrophobic, and in group II is tiny and polar; marked with a $ in Figure 2B). In the predicted docking structure of P. integrifolia S2-SLF1/S3-RNase, these amino acids are located in the region of interface between the two proteins (those in red and white; Figures 2C1,C2). Therefore, these amino acid sites could be involved in the recognition of the S3-, S7-, S12-, and S13-RNases as self, and S2- and S11-RNases as non-self by S2- and S11-SLF1. It should be noted that, as expected, in the interface region of the two proteins are also located three of the amino acids identified by Wu et al. (2018), as putatively involved in P. integrifolia S2-SLF1/S3-RNase specificity (marked in orange and white those showing evidence for positive selection). According to Kubo et al. (2015), S5-, S9-, S10-, S11-, S17-, S19-, and S22-SLF3 alleles, that are highly conserved, recognize as non-self the S7-RNase (although only the interaction between S7-RNase and S5- and S11-SLF3 has been confirmed in transformation experiments; group I), and only S11-SLF3B and S7-SLF3, two divergent SLF3 alleles, recognize the S7-RNase as self (both cases are supported by transgenic experiments, group II; Kubo et al., 2015). According to the phylogenetic analyses here performed (Supplementary Figure S1) these two divergent SLF3 sequences are a new gene (SLF21). When these groups of sequences are compared, amino acid 159 is the only site under positive selection that is different between the two groups (underlined in Figure 3A), and thus, could be involved in specificity determination of these sequences. When we compare the S5-, S11-, and S17- RNases (group I) and S7-RNase (group II) at the amino acid positions under positive selection, at amino acid positions 70, 88, 96, 98, and 103, there are differences in charge, hydrophobicity, polarity, size and being aliphatic (group I at amino acid position 70 is hydrophobic, at position 88 is polar and charged, at position 96 is hydrophilic, and in the group II at amino acid position 70 is negatively charged, at position 88 is tiny, at position 96 is hydrophobic, at position 98 is positively charged, and at position 104 is aliphatic; Figure 3B). These positions could be, in principle, involved in specificity recognition of the S7-RNase. When we look at these amino acids in the predicted docking structure of P. hybrida S7-RNase/S5-SLF3 (in red those that are positively selected and show different amino acid proprieties between the two groups; Figures 3C1,C2), these are located in the region of the interface between the two proteins. Therefore, these amino acid sites must be involved in the recognition of the P. hybrida S7-RNase as non-self by S5-SLF3.

Figure 2. Positively selected amino acid sites in the S-RNase (A), SLF1 (B) alignments (highlighted in gray), and in the predicted docking structure of P. integrifolia S3-RNase/S2-SLF1 (as cartoon C1, and surface C2) predicted to be involved in recognition of S3-, S7-, S12-, and S13-RNases (group I) as non-self, and S2-, and S11-RNases (group II) as self by the S2-SLF1 and S11-SLF1. (A) The signal peptide and conserved regions (C1- C5) are marked. The amino acid sites marked in bold are different between the two group of sequences regarding hydrophobicity (#), and charge and size (&). (B) The F-box and FD1-FD3 regions (Hua et al., 2007) are indicated. The amino acid site in bold and marked with a $ is different between the two groups of sequences in terms of hydrophobicity, size and polarity. Triangles indicate the eight putative amino acids at S2-SLF1 determining P. integrifolia S3-RNase specificity, according to Wu et al. (2018). (C1,C2) S3-RNase and S2-SFL1 are shown in green and in cyan, respectively. The amino acids under positive selection are highlighted in blue and red (in A- # and &; in B-$). The amino acids identified by Wu et al. (2018) are in orange and white if they are also here identified as positively selected. ∗Indicate amino acid sites under positive selection that are predicted to form hydrogen bonds and/or salt bridges between P. integrifolia S3-RNase and S2-SFL1.
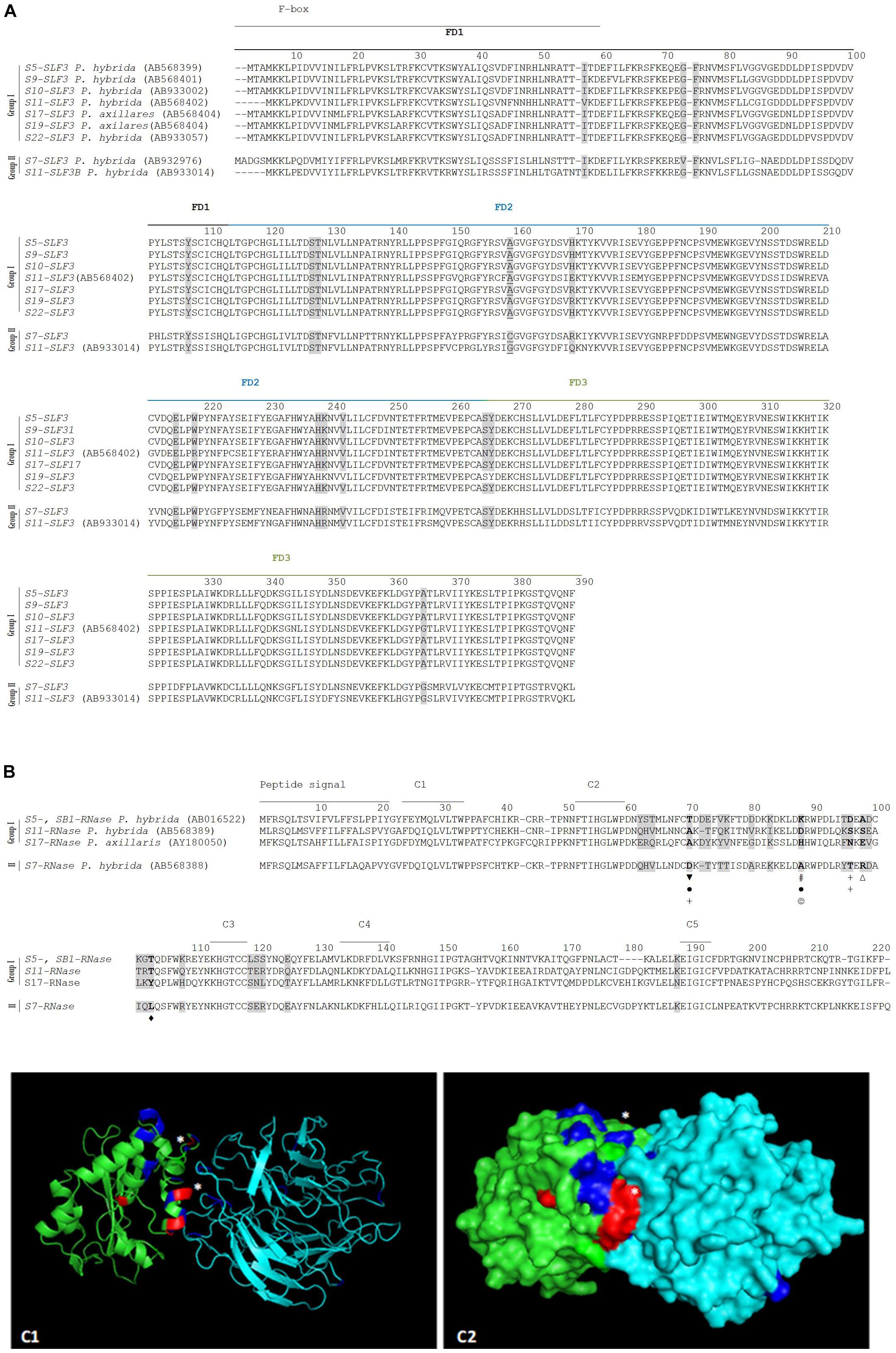
Figure 3. Positively selected amino acid sites at the SLF3 (A), S-RNase (B) alignments (highlighted in gray), and in the predicted docking structure of P. hybrida S7-RNase/S5-SLF3 (as cartoon C1, and surface C2) predicted to be involved in the recognition of S7-RNase as non-self by the S5-, S9-, S10-, S11-, S17-, S19-, and S22-SLF3 (that are highly conserved; Group I), and as self by S11-SLF3B and S7-SLF3 (group II). (A) The F-box and FD1-FD3 regions (Hua et al., 2007) are indicated. The amino acid site underlined is the only amino acid under positive selection different between the two groups of sequences. (B) The S7-RNase is different from S5-, S11-, and S17- RNases (group I) at five (marked in bold) amino acid sites under positive selection in terms of charge (marked with a  , negatively charged
, negatively charged  , positively charged Δ), hydrophobicity (marked with a +), polarity (marked with a #), size (marked with a l’), and being aliphatic (marked with a
, positively charged Δ), hydrophobicity (marked with a +), polarity (marked with a #), size (marked with a l’), and being aliphatic (marked with a  ). (C1,C2) P. hybrida S7-RNase/S5-SLF3 are shown in green and in cyan, respectively. The amino acids under positive selection are highlighted in blue and in red (those that show differences in hydrophobicity, or are aliphatic). ∗Indicate amino acid sites under positive selection that are predicted to form hydrogen bonds and/or salt bridges between P. hybrida S7-RNase/S5-SLF3.
). (C1,C2) P. hybrida S7-RNase/S5-SLF3 are shown in green and in cyan, respectively. The amino acids under positive selection are highlighted in blue and in red (those that show differences in hydrophobicity, or are aliphatic). ∗Indicate amino acid sites under positive selection that are predicted to form hydrogen bonds and/or salt bridges between P. hybrida S7-RNase/S5-SLF3.
S10- and S19-SLF9 alleles are missing, and according to Kubo et al. (2015), all type 9 sequences, that show high sequence conservation, can recognize both S10- and S19-RNases, although functional evidence is only available for S19-RNase and S7-, S11-SLF9. Type 9 sequences are, however, a heterogeneous group and represent four genes (Supplementary Figure S1). We consider all sequences from SLF9A and SLF9B genes as being able to recognize S10- and S19-RNases. When we group the available S-RNases accordingly (S10-, S19- RNases (group I) versus S7-, S11-, and S17-RNases (group II) there are three amino acid positions (14, 45, and 61; Figure 4A) that are different in size (positions 14 and 61) and hydrophobicity (position 45). When we group all available SLF9 alleles there are 12 amino acid positions under positive selection that are conserved, and could be involved in specificity determination of S10- and S19-RNases (Figure 4B). Indeed, when we label them in the predicted docking structure of S11-SLF9/S19-RNase (Figures 4C1,C2), five (those marked with a star in Figure 4B), are located in the region of interface between the two proteins. The amino acid sites under positive selection that show differences in size and hydrophobicity at the S-RNase are also located in this region (Figures 4C1,C2). Therefore, these amino acid sites could be involved in the recognition of the S10-, S19-RNase as non-self by S2-, S3-, S7, S9, S11-, S17-, and S22-SLF9.
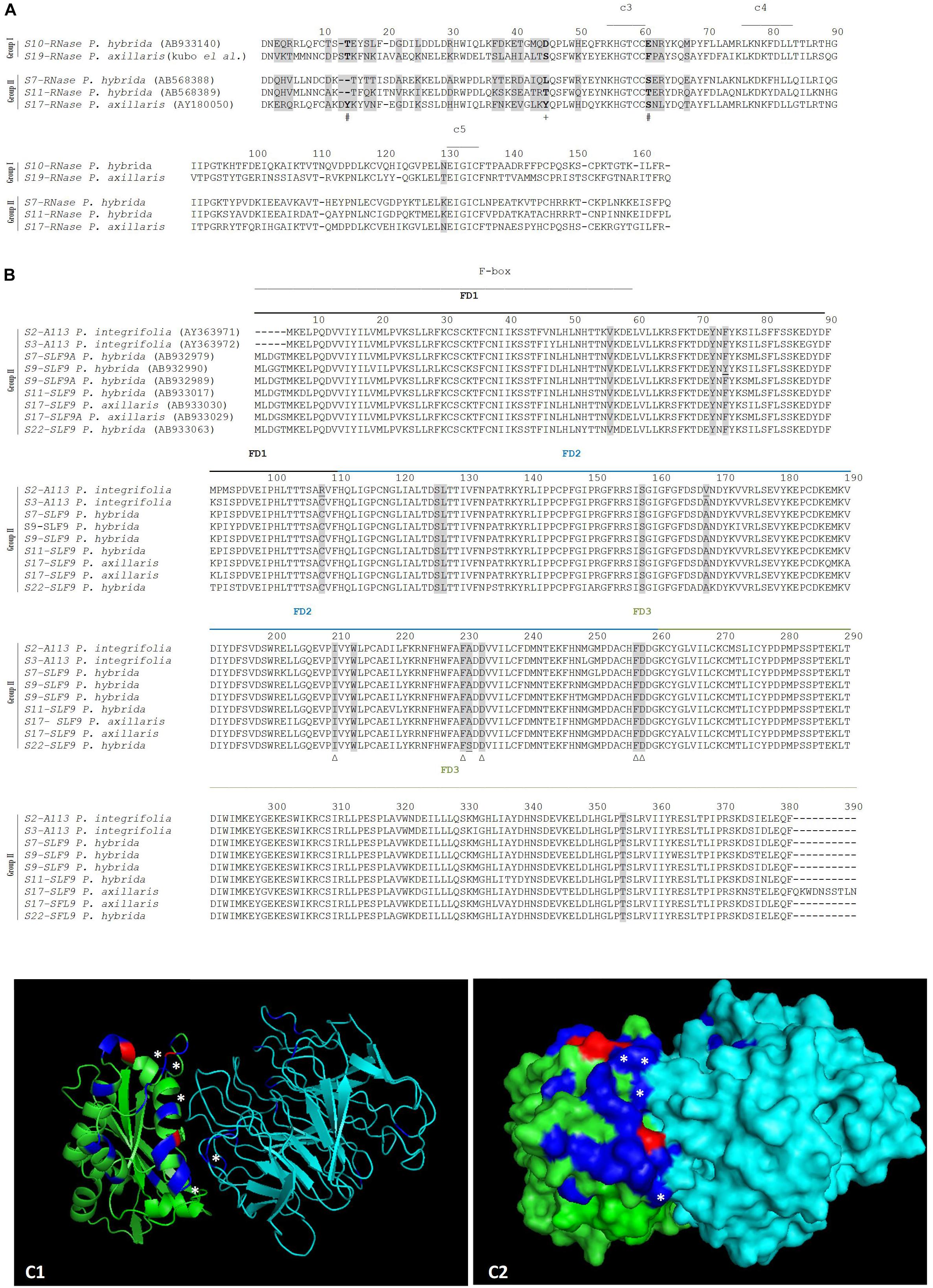
Figure 4. Positively selected amino acid sites at the S-RNase (A), SLF9 (B) alignments (highlighted in gray), and in the predicted docking structure of P. hybrida S11-SLF9/S19-RNase (as cartoon C1, and surface C2) predicted to be involved in the recognition of S10-, and S19-RNase (group I) as non-self by the S2-, S3-S7-, S9-, S11-, S17-, and S22-SLF9A and SLF9B (group II), all showing high sequence conservation. (A) The conserved regions (C3- C5) are marked. The three amino acid positions under positive selection that are different between the two groups are highlighted in bold. Differences in size are marked with a #, and differences in hydrophobicity are marked with a +. (B) The F-box and FD1-FD3 regions (Hua et al., 2007) are indicated. The 12 amino acids under positive selection conserved in SLF9A and SLF9B alleles (those amino acids, highlighted in gray, that are not underlined) could be involved in specificity determination of S10- and S19-RNases. Those located in the region of the interface between the two proteins are marked with a Δ. (C1,C2) P. hybrida S19-RNase/S11-SLF9 are shown in green and in cyan, respectively. The amino acids under positive selection are highlighted in blue and in red (those that show differences in size and hydrophobicity). ∗Indicate amino acid sites under positive selection that are predicted to form hydrogen bonds and/or salt bridges between P. hybrida S19-RNase/S11-SLF9.
Discussion
The number of SLF sequences identified in the different species seems to be more related with difficulties in assembling the genome at the S-locus region (due to the presence of highly repetitive DNA, such as transposable elements, for instance) than with the breeding system of the species analyzed. For instance, for the two SI Petunia species analyzed, although the coverage is as high as 91.3 and 90.2% of their diploid genomes6, only three SLF sequences have been identified. For extant species, SC is usually a recent event, since the average lineage duration of a SC species is on the order of 200 000 years (Goldberg et al., 2010), and thus, SLF sequences are expected to be retrieved in SC species, although many of them as pseudogenes. When this data is analyzed at the genus level, similar number of SLF gene lineages are described in Solanum (17), Nicotiana (19), and Petunia (18), although only eight are present in the three genera. It is possible that these SLF gene lineages have been devoted to the recognition of the same S-RNases in the three genera. The first studies on the characterization of S-RNase alleles from different Solanaceae species, showed evidence of broadly shared ancestral polymorphism (Ioerger et al., 1990; Richman and Kohn, 2000; Igic et al., 2004, 2006; Savage and Miller, 2006). Indeed, at least 83% of the SLF gene lineages were present in the Solanaceae common ancestor. Two (in Nicotiana) to three (in Petrunia and Solanum) SLF lineage genes seem to have appeared after the split of the genera. Thus, assuming one generation per year and the divergence time of 24 MY for the Solanum and Nicotiana genera, and 30 MY for the Solanaceae crown lineages (Sarkinen et al., 2013), the rate of origin of a new SLF gene lineage (and thus a new specificity) is one per 10 MY. This value fits the range for the rate estimates for the origination of new S-alleles [10−6 to 10−9 per gene per generation; (Vekemans and Slatkin, 1994)]. The presence of identical number of SLF gene lineages in the three genera suggests that in the non-self recognition system, 18 SLF genes is the minimum number of S-pollen genes for the system to work when about 30 different specificities (Tsukamoto et al., 2003) are present. This number is compatible with the simulation work performed by Kubo et al. (2015), that found that an average of 16–20 SLFs are enough to recognize the large majority of the S-RNases in Petunia populations. Since the number of S-haplotypes that can be maintained in a population depends of the effective population size, this also suggests no major differences in the effective population size of the species of the three genera. In Solanaceae, a population bottleneck has been suggested only for the common ancestor of Physalis and Witheringia (Paape et al., 2008) and in the genus Lycium (Miller et al., 2008).
Under the non-self recognition model an SLF interacts with a subset of S-RNases, but not with the self-S-RNase (Kubo et al., 2010, 2015; Williams et al., 2014, 2015). It has been proposed that recognition avoidance of the self-S-RNase is achieved by having either a diverged or deleted allele of the SLF type whose product recognizes the S-RNase of that S-haplotype (Kubo et al., 2015). The phylogenetic analyses here performed give support to this hypothesis.
In vivo transgenic assays revealed that different regions of the same allelic form of a SLF protein (S2-SLF1) are involved in specificity determination of the different S-RNases (Sijacic et al., 2004; Kubo et al., 2010; Sun and Kao, 2013; Williams et al., 2014; Li et al., 2017; Wu et al., 2018). For instance, FD3 region of P. inflata S2-SLF1 protein is required for the interaction with the P. inflata S3-RNase, and a maximum of four amino acid sites are involved in specificity determination, but FD2 region contains the amino acids that determine the specificity for P. inflata S7-RNase, and both FD1 and FD2 contain the amino acids that determine the specificity for P. inflata S13-RNase (Wu et al., 2018). Moreover, different allelic pairs of P. hybrida SLF1 (S3-SLF1 and S3L-SLF1 that interacts with P. hybrida S3-RNase), identify the FD3 region as containing the amino acids required for the interaction between S3L-SLF1 and S3-RNase (Li et al., 2017). Furthermore, one amino acid at position 293, when replaced with another with opposite electrostatic potential determines the interaction specificity of P. hybrida S3L-SLF1 and S3-RNase. Interactions between SLF proteins and S-RNases are, thus complex and diverse (Li et al., 2017). Such complexity implies that other methodologies need to be applied to predict those amino acids involved in specificity determination. Amino acid sites determining specificity are expected to be under a different selection regime than the rest of the protein, with selection favoring diversification within a S-haplotype (Kakui et al., 2011; Aguiar et al., 2013, 2015; Pratas et al., 2018). We identified 16 such amino acid positions, and two of these have been identified as involved in specificity determination between S2-SLF1 and S3-RNase in P. integrifolia (Wu et al., 2018). Nevertheless, our analysis was performed using only 10 S-haplotypes for which 11 SLF genes are available, and thus, not all amino acid sites under positive selection have been identified. Therefore, it is not surprising that the amino acid position identified as determining specificity of P. hybrida S3L-SLF1 and S3-RNase, has not been here identified (Li et al., 2017). These putative amino acid sites involved in specificity determination are located at the predicted interaction surface of the two proteins, and those at the S-RNase show distinct amino acid characteristics such as hydrophobicity, size and charge. Therefore, these amino acid positions show all characteristics of being involved in specificity determination. Using this methodology Pratas et al. (2018) identified the putative amino acid sites determining specificity in Malus. Therefore, this methodology can be applied to other species presenting GSI of the non-self recognition type. Although S-RNase and F-box genes belonging to the GSI lineage have been identified in Fabaceae (Aguiar et al., 2015; Ramanauskas and Igic, 2017), Rutaceae, and Malvaceae (Ramanauskas and Igic, 2017) species, further characterization of the S-locus, concerning levels of diversity, expression, segregation analyses and identification of positively selected amino acid sites, is needed to determine if GSI in such species is of the non-self recognition type, before applying the methodology here described.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
JV and CV collected the genome sequence data and performed the analyses. SR performed the analyses on the protein structures. MR-J, FF-R, NV, and HL-F designed and implemented the mentioned operations at BDBM. All authors wrote, read, and approved the final manuscript.
Funding
This work was financed by the project Norte-01-0145-FEDER-000008 -Porto Neurosciences and Neurologic Disease Research Initiative at I3S, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (FEDER). SR is supported by a post-doctoral fellowship under this project. HL-F is supported by a post-doctoral fellowship from Xunta de Galicia (ED481B 2016/068-0). SING group acknowledges Consellería de Educación, Universidades e Formación Profesional (Xunta de Galicia) for the ED431C2018/55-GRC grant and CITI (Centro de Investigación, Transferencia e Innovación) from University of Vigo for hosting its IT infrastructure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00879/full#supplementary-material
Footnotes
- ^ https://www.sing-group.org/BDBM/
- ^ http://bpositive.i3s.up.pt
- ^ https://www.ncbi.nlm.nih.gov/
- ^ https://www.sing-group.org/seda/
- ^ https://solgenomics.net/
- ^ https://www.nature.com/articles/nplants201674
References
Aguiar, B., Vieira, J., Cunha, A. E., Fonseca, N. A., Iezzoni, A., van Nocker, S., et al. (2015). Convergent evolution at the gametophytic self-incompatibility system in Malus and Prunus. PLoS One 10:e0126138. doi: 10.1371/journal.pone.0126138
Aguiar, B., Vieira, J., Cunha, A. E., Fonseca, N. A., Reboiro-Jato, D., Reboiro-Jato, M., et al. (2013). Patterns of evolution at the gametophytic self-incompatibility Sorbus aucuparia (Pyrinae) S pollen genes support the non-self recognition by multiple factors model. J. Exp. Bot. 64, 2423–2434. doi: 10.1093/jxb/ert098
Brisolara-Correa, L., Thompson, C. E., Fernandes, C. L., and de Freitas, L. B. (2015). Diversification and distinctive structural features of S-RNase alleles in the genus Solanum. Mol. Genet. Genomics 290, 987–1002. doi: 10.1007/s00438-014-0969-3
Broz, A. K., Randle, A. M., Sianta, S. A., Tovar-Méndez, A., McClure, B., and Bedinger, P. A. (2017). Mating system transitions in Solanum habrochaites impact interactions between populations and species. New Phytol. 213, 440–454. doi: 10.1111/nph.14130
Chalivendra, S. C., Lopez-Casado, G., Kumar, A., Kassenbrock, A. R., Royer, S., Tovar-Mendez, A., et al. (2013). Developmental onset of reproductive barriers and associated proteome changes in stigma/styles of Solanum pennellii. J. Exp. Bot. 64, 265–279. doi: 10.1093/jxb/ers324
Chen, J., Wang, P., de Graaf, B. H. J., Zhang, H., Jiao, H., Tang, C., et al. (2018). Phosphatidic acid Counteracts S-RNase signaling in pollen by stabilizing the actin cytoskeleton. Plant Cell 30, 1023–1039. doi: 10.1105/tpc.18.00021
Cheng, J., Han, Z., Xu, X., and Li, T. (2006). Isolation and identification of the pollen-expressed polymorphic F-box genes linked to the S-locus in apple (Malus × domestica). Sex Plant Reprod. 19, 175–183. doi: 10.1007/s00497-006-0034-4
De Franceschi, P., Pierantoni, L., Dondini, L., Grandi, M., Sansavini, S., and Sanzol, J. (2011a). Evaluation of candidate F-box genes for the pollen S of gametophytic self-incompatibility in the Pyrinae (Rosaceae) on the basis of their phylogenomic context. Tree Genet. Genomes 7, 663–683. doi: 10.1007/s11295-011-0365-7
De Franceschi, P., Pierantoni, L., Dondini, L., Grandi, M., Sanzol, J., and Sansavini, S. (2011b). Cloning and mapping multiple S-locus F-box genes in European pear (Pyrus communis L.). Tree Genet. Genomes 7, 231–240. doi: 10.1007/s11295-010-0327-5
Eaves, D. J., Flores-Ortiz, C., Haque, T., Lin, Z., Teng, N., and Franklin-Tong, V. E. (2014). Self-incompatibility in Papaver: advances in integrating the signalling network. Biochem. Soc. Trans. 42, 370–376. doi: 10.1042/BST20130248
Entani, T., Iwano, M., Shiba, H., Che, F. S., Isogai, A., and Takayama, S. (2003). Comparative analysis of the self-incompatibility (S-) locus region of Prunus mume: identification of a pollen-expressed F-box gene with allelic diversity. Genes Cells 8, 203–213. doi: 10.1046/j.1365-2443.2003.00626.x
Fields, A. M., Wang, N., Hua, Z., Meng, X., and Kao, T. H. (2010). Functional characterization of two chimeric proteins between a Petunia inflata S-locus F-box protein, PiSLF2, and a PiSLF-like protein, PiSLFLb-S2. Plant Mol. Biol. 74, 279–292. doi: 10.1007/s11103-010-9672-x
Foote, H. C., Ride, J. P., Franklin-Tong, V. E., Walker, E. A., Lawrence, M. J., and Franklin, F. C. (1994). Cloning and expression of a distinctive class of self-incompatibility (S) gene from Papaver rhoeas L. Proc. Natl. Acad. Sci. U.S.A. 91, 2265–2269. doi: 10.1073/pnas.91.6.2265
Goldberg, E. E., Kohn, J. R., Lande, R., Robertson, K. A., Smith, S. A., and Igic, B. (2010). Species selection maintains self-incompatibility. Science 330, 493–495. doi: 10.1126/science.1194513
Hua, Z., and Kao, T. H. (2006). Identification and characterization of components of a putative Petunia S-locus F-box-containing E3 ligase complex involved in S-RNase-based self-incompatibility. Plant Cell 18, 2531–2553. doi: 10.1105/tpc.106.041061
Hua, Z., Meng, X., and Kao, T. H. (2007). Comparison of Petunia inflata S-Locus F-box protein (Pi SLF) with Pi SLF like proteins reveals its unique function in S-RNase based self-incompatibility. Plant Cell 19, 3593–3609. doi: 10.1105/tpc.107.055426
Hua, Z. H., Fields, A., and Kao, T. H. (2008). Biochemical models for S-RNase-based self-incompatibility. Mol. Plant 1, 575–585. doi: 10.1093/mp/ssn032
Huang, S., Lee, H. S., Karunanandaa, B., and Kao, T. H. (1994). Ribonuclease activity of petunia inflata S proteins is essential for rejection of self-pollen. Plant Cell 6, 1021–1028. doi: 10.1105/tpc.6.7.1021
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Ide, H., Kimura, M., Arai, M., and Funatsu, G. (1991). The complete amino-acid-sequence of ribonuclease from the seeds of bitter gourd (Momordica-charantia). FEBS Lett. 284, 161–164. doi: 10.1016/0014-5793(91)80675-S
Igic, B., Bohs, L., and Kohn, J. R. (2004). Historical inferences from the self-incompatibility locus. New Phytol. 161, 97–105. doi: 10.1046/j.1469-8137.2003.00952.x
Igic, B., Bohs, L., and Kohn, J. R. (2006). Ancient polymorphism reveals unidirectional breeding system shifts. Proc. Natl. Acad. Sci. U.S.A. 103, 1359–1363. doi: 10.1073/pnas.0506283103
Igic, B., and Kohn, J. R. (2001). Evolutionary relationships among self-incompatibility RNases. Proc. Natl. Acad. Sci. U.S.A. 98, 13167–13171. doi: 10.1073/pnas.231386798
Igic, B., Lande, R., and Kohn, J. R. (2008). Loss of self-incompatibility and its evolutionary consequences. Int. J. Plant Sci. 169, 93–104. doi: 10.1086/523362
Ikeda, K., Igic, B., Ushijima, K., Yamane, H., Hauck, N. R., Nakano, R., et al. (2004). Primary structural features of the S haplotype-specific F-box protein. SFB, in Prunus. Sex Plant Reprod. 16, 235–243. doi: 10.1007/s00497-003-0200-x
Ioerger, T. R., Clark, A. G., and Kao, T. H. (1990). Polymorphism at the self-incompatibility locus in solanaceae predates speciation. Proc. Natl. Acad. Sci. U.S.A. 87, 9732–9735. doi: 10.1073/pnas.87.24.9732
Kakui, H., Kato, M., Ushijima, K., Kitaguchi, M., Kato, S., and Sassa, H. (2011). Sequence divergence and loss-of-function phenotypes of S locus F-box brothers genes are consistent with non-self recognition by multiple pollen determinants in self-incompatibility of Japanese pear (Pyrus pyrifolia). Plant J. 68, 1028–1038. doi: 10.1111/j.1365-313X.2011.04752.x
Kowyama, Y., Kunz, C., Lewis, I., Newbigin, E., Clarke, A. E., and Anderson, M. A. (1994). Self-compatibility in a Lycopersicon peruvianum variant (LA2157) is associated with a lack of style S-RNase activity. Theor. Appl. Genet. 88, 859–864. doi: 10.1007/BF01253997
Krissinel, E., and Henrick, K. (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. doi: 10.1016/j.jmb.2007.05.022
Kubo, K., Entani, T., Takara, A., Takara, A., Wang, N., Fields, A. M., et al. (2010). Collaborative non-self recognition system in S-RNase-based self-incompatibility. Science 330, 796–799. doi: 10.1126/science.1195243
Kubo, K., Paape, T., Hatakeyama, M., Entani, T., Takara, A., Kajihara, K., et al. (2015). Gene duplication and genetic exchange drive the evolution of S-RNase-based self-incompatibility in Petunia. Nat. Plants 1:14005. doi: 10.1038/nplants.2014.5
Li, J., Zhang, Y., Song, Y., Zhang, H., Fan, J., Li, Q., et al. (2017). Electrostatic potentials of the S-locus F-box proteins contribute to the pollen S specificity in self-incompatibility in Petunia hybrida. Plant J. 89, 45–57. doi: 10.1111/tpj.13318
Li, W., and Chetelat, R. T. (2014). The role of a pollen-expressed cullin1 protein in gametophytic self-incompatibility in Solanum. Genetics 196, 439–442. doi: 10.1534/genetics.113.158279
Liedl, B. E., McCormick, S., and Mutschler, M. A. (1996). Unilateral incongruity in crosses involving Lycopersicon pennellii and L. esculentum is distinct from self-incompatibility in expression, timing and location. Sex Plant Reprod. 9, 299–308.
López-Fernández, H., Duque, P., Henriques, S., Vázquez, N., Fdez-Riverola, F., Vieira, C. P., et al. (2019). Bioinformatics protocols for quickly obtaining large-scale data sets for phylogenetic inferences. Interdiscip. Sci. 11, 1–9. doi: 10.1007/s12539-018-0312-5
Luu, D.-T., Qin, X., Laublin, G., Yang, Q., Morse, D., and Cappadocia, M. (2001). Rejection of S-heteroallelic pollen by a dual-specific S-RNase in Solanum chacoense predicts a multimeric SI pollen component. Genetics 159, 329–335.
Markova, D. N., Petersen, J. J., Qin, X., Short, D. R., Valle, M. J., Tovar-Mendez, A., et al. (2016). Mutations in two pollen self-incompatibility factors in geographically marginal populations of Solanum habrochaites impact mating system transitions and reproductive isolation. Am. J. Bot. 103, 1847–1861. doi: 10.3732/ajb.1600208
Matsumoto, D., and Tao, R. (2016). Recognition of a wide-range of S-RNases by S locus F-box like 2, a general-inhibitor candidate in the Prunus specific S-RNase based self-incompatibility system. Plant Mol. Biol. 91, 459–469. doi: 10.1007/s11103-016-0479-2
Matsuura, T., Sakai, H., Unno, M., Ida, K., Sato, M., Sakiyama, F., et al. (2001). Crystal structure at 1.5-A resolution of Pyrus pyrifolia pistil ribonuclease responsible for gametophytic self-incompatibility. J. Biol. Chem. 276, 45261–45269. doi: 10.1074/jbc.M107617200
Matton, D. P., Luu, D. T., Xike, Q., Laublin, G., O’Brien, M., Maes, O., et al. (1999). Production of an S-RNase with dual specificity suggests a novel hypothesis for the generation of new S alleles. Plant Cell 11, 2087–2097. doi: 10.1105/tpc.9.10.1757
Matton, D. P., Maes, O., Laublin, G., Xike, Q., Bertrand, C., Morse, D., et al. (1997). Hypervariable domains of self-incompatibility RNases mediate allele-specific pollen recognition. Plant Cell 9, 1757–1766. doi: 10.1105/tpc.9.10.1757
McClure, B. (2009). Darwin’s foundation for investigating self-incompatibility and the progress toward a physiological model for S-RNase-based SI. J. Exp. Bot. 60, 1069–1081. doi: 10.1093/jxb/erp024
Miller, J. S., Levin, R. A., and Feliciano, N. M. (2008). A tale of two continents: baker’s rule and the maintenance of self-incompatibility in Lycium (Solanaceae). Evolution 62, 1052–1065. doi: 10.1111/j.1558-5646.2008.00358.x
Minamikawa, M., Kakui, H., Wang, S., Kotoda, N., Kikuchi, S., Koba, T., et al. (2010). Apple S locus region represents a large cluster of related, polymorphic and pollen-specific F-box genes. Plant Mol. Biol. 74, 143–154. doi: 10.1007/s11103-010-9662-z
Moon, H. S., Nicholson, J. S., and Lewis, R. S. (2008). Use of transferable Nicotiana tabacum L. microsatellite markers for investigating genetic diversity in the genus Nicotiana. Genome 51, 547–559. doi: 10.1139/G08-039
Nielsen, H. (2017). Predicting secretory proteins with signalp. Methods Mol. Biol. 1611, 59–73. doi: 10.1007/978-1-4939-7015-5_6
Notredame, C., Higgins, D. G., and Heringa, J. (2000). T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217. doi: 10.1006/jmbi.2000.4042
Nowak, M. D., Davis, A. P., Anthony, F., and Yoder, A. D. (2011). Expression and trans-specific polymorphism of self-incompatibility RNases in coffea (Rubiaceae). PLoS One 6:e21019. doi: 10.1371/journal.pone.0021019
Nunes, M. D., Santos, R. A., Ferreira, S. M., Vieira, J., and Vieira, C. P. (2006). Variability patterns and positively selected sites at the gametophytic self-incompatibility pollen SFB gene in a wild self-incompatible Prunus spinosa (Rosaceae) population. New Phytol. 172, 577–587. doi: 10.1111/j.1469-8137.2006.01838.x
Okada, K., Tonaka, N., Taguchi, T., Ichikawa, T., Sawamura, Y., Nakanishi, T., et al. (2011). Related polymorphic F-box protein genes between haplotypes clustering in the BAC contig sequences around the S-RNase of Japanese pear. J. Exp. Bot. 62, 1887–1902. doi: 10.1093/jxb/erq381
Paape, T., Igic, B., Smith, S. D., Olmstead, R., Bohs, L., and Kohn, J. R. (2008). A 15-Myr-old genetic bottleneck. Mol. Biol. Evol. 25, 655–663. doi: 10.1093/molbev/msn016
Pratas, M. I., Aguiar, B., Vieira, J., Nunes, V., Teixeira, V., Fonseca, N. A., et al. (2018). Inferences on specificity recognition at the malusxdomestica gametophytic self-incompatibility system. Sci. Rep. 8:1717. doi: 10.1038/s41598-018-19820-1
Qiao, H., Wang, F., Zhao, L., Zhou, J., Lai, Z., Zhang, Y., et al. (2004). The F-box protein AhSLF-S2 controls the pollen function of S-RNase based self-incompatibility. Plant Cell 16, 2307–2322. doi: 10.1105/tpc.104.024919
Ramanauskas, K., and Igic, B. (2017). The evolutionary history of plant T2/S-type ribonucleases. PeerJ 5:e3790. doi: 10.7717/peerj.3790
Reboiro-Jato, D., Reboiro-Jato, M., Fdez-Riverola, F., Vieira, C. P., Fonseca, N. A., and Vieira, J. (2012). ADOPS-automatic detection of positively selected sites. J. Integr. Bioinform. 9:200. doi: 10.2390/biecoll-jib-2012-200
Richman, A. D., and Kohn, J. R. (2000). Evolutionary genetics of self-incompatibility in the Solanaceae. Plant Mol. Biol. 42, 169–179. doi: 10.1007/978-94-011-4221-2_8
Roalson, E. H., and McCubbin, A. G. (2003). S-RNases and sexual incompatibility: structure, functions, and evolutionary perspectives. Mol. Phylogenet. Evol. 29, 490–506. doi: 10.1016/s1055-7903(03)00195-7
Royo, J., Kunz, C., Kowyama, Y., Anderson, M., Clarke, A. E., and Newbigin, E. (1994). Loss of a histidine residue at the active site of S-locus ribonuclease is associated with self-compatibility in Lycopersicon peruvianum. Proc. Natl. Acad. Sci. U.S.A. 91, 6511–6514. doi: 10.1073/pnas.91.14.6511
Sarkinen, T., Bohs, L., Olmstead, R. G., and Knapp, S. (2013). A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): a dated 1000-tip tree. BMC Evol. Biol. 13:214. doi: 10.1186/1471-2148-13-214
Sassa, H., Kakui, H., Miyamoto, M., Suzuki, Y., Hanada, T., Ushijima, K., et al. (2007). S locus F-box brothers: multiple and pollen-specific F-box genes with S haplotype-specific polymorphisms in apple and Japanese pear. Genetics 175, 1869–1881. doi: 10.1534/genetics.106.068858
Savage, A. E., and Miller, J. S. (2006). Gametophytic self-incompatibility in Lycium parishii (Solanaceae): allelic diversity, genealogical structure, and patterns of molecular evolution at the S-RNase locus. Heredity 96, 434–444. doi: 10.1038/sj.hdy.6800818
Sijacic, P., Wang, X., Skirpan, A. L., Wang, Y., Dowd, P. E., McCubbin, A. G., et al. (2004). Identification of the pollen determinant of S-RNase mediated self-incompatibility. Nature 429, 302–305. doi: 10.1038/nature02523
Sims, T. L., and Robbins, T. P. (2009). Gametophytic Self-Incompatibility in Petunia. New York, NY: Springer.
Sonneveld, T., Tobutt, K. R., Vaughan, S. P., and Robbins, T. P. (2005). Loss of pollen S function in two self-compatible selections of Prunus avium is associated with deletion/mutation of an S haplotype-specific F-box gene. Plant Cell 17, 37–51. doi: 10.1105/tpc.104.026963
Steinbachs, J. E., and Holsinger, K. E. (2002). S-RNase mediated gametophytic self-incompatibility is ancestral in eudicots. Mol. Biol. Evol. 19, 825–829. doi: 10.1093/oxfordjournals.molbev.a004139
Sun, P., and Kao, T. H. (2013). Self-incompatibility in Petunia inflata: the relationship between a self-incompatibility locus F-box protein and its non-self S-RNases. Plant Cell 25, 470–485. doi: 10.1105/tpc.112.106294
Sun, P., Li, S., Lu, D., Williams, J. S., and Kao, T. H. (2015). Pollen S-locus F-box proteins of Petunia involved in S-RNase based self-incompatibility are themselves subject to ubiquitin-mediated degradation. Plant J. 83, 213–223. doi: 10.1111/tpj.12880
Tsukamoto, T., Ando, T., Takahashi, K., Omori, T., Watanabe, H., Kokubun, H., et al. (2003). Breakdown of self-incompatibility in a natural population of Petunia axillaris caused by loss of pollen function. Plant Physiol. 131, 1903–1912. doi: 10.1104/pp.102.018069
Tsukamoto, T., Ando, T., Watanabe, H., Marchesi, E., and Kao, T. H. (2005). Duplication of the S-locus F-box gene is associated with breakdown of pollen function in an S-haplotype identified in a natural population of self-incompatible Petunia axillaris. Plant Mol. Biol. 57, 141–153. doi: 10.1007/s11103-004-6852-6
Tsukamoto, T., Hauck, N. R., Tao, R., Jiang, N., and Iezzoni, A. F. (2010). Molecular and genetic analyses of four nonfunctional S- haplotype variants derived from a common ancestral S- haplotype identified in sour cherry L. Genetics 184, 411–427. doi: 10.1534/genetics.109.109728
Ushijima, K., Sassa, H., Dandekar, A. M., Gradziel, T. M., Tao, R., and Hirano, H. (2003). Structural and transcriptional analysis of the self-incompatibility locus of almond: identification of a pollen-expressed F-box gene with haplotype-specific polymorphism. Plant Cell 15, 771–781. doi: 10.1105/tpc.009290
van Zundert, G. C. P., Rodrigues, J., Trellet, M., Schmitz, C., Kastritis, P. L., Karaca, E., et al. (2016). The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428, 720–725. doi: 10.1016/j.jmb.2015.09.014
Vázquez, N., López-Fernández, H., Vieira, C. P., Fdez-Riverola, F., Vieira, J., and Reboiro-Jato, M. (2019). BDBM 1.0: a desktop application for efficient retrieval and processing of high qualitity sequence data and application to the identification of the putative coffea S-locus. Interdiscip. Sci. 11, 57–67. doi: 10.1007/s12539-019-00320-3
Vázquez, N., Vieira, C. P., Amorim, B. S., Torres, A., López-Fernández, H., Fdez-Riverola, F., et al. (2017). “Automated collection and sharing of adaptive amino acid changes data,” in Proceedings of the 11th International Conference on Practical Applications of Computational Biology and Bioinformatics. PACBB 2017, eds F. Fdez-Riverola, M. Mohamad, M. Rocha, J. De Paz, and cpsfnmPintocpefnm T. (Berlin: Springer).
Vázquez, N., Vieira, C. P., Amorim, B. S. R., Torres, A., López-Fernández, H., Fdez-Riverola, F., et al. (2018). Large scale analyses and visualization of adaptive amino acid changes projects. Interdiscip. Sci. 10, 24–32. doi: 10.1007/s12539-018-0282-7
Vekemans, X., and Slatkin, M. (1994). Gene and allelic genealogies at a gametophytic self-incompatibility locus. Genetics 137, 1157–1165.
Vieira, J., Ferreira, P. G., Aguiar, B., Fonseca, N. A., and Vieira, C. P. (2010). Evolutionary patterns at the RNase based gametophytic self - incompatibility system in two divergent Rosaceae groups (Maloideae and Prunus). BMC Evol. Biol. 10:200. doi: 10.1186/1471-2148-10-200
Vieira, J., Fonseca, N. A., and Vieira, C. P. (2008). An S-RNase-based gametophytic self-incompatibility system evolved only once in eudicots. J. Mol. Evol. 67, 179–190. doi: 10.1007/s00239-008-9137-x
Vieira, J., Fonseca, N. A., and Vieira, C. P. (2009). RNase-based gametophytic self-incompatibility evolution: questioning the hypothesis of multiple independent recruitments of the S-pollen gene. J. Mol. Evol. 69, 32–41. doi: 10.1007/s00239-009-9249-y
Vieira, J., Morales-Hojas, R., Santos, R. A., and Vieira, C. P. (2007). Different positively selected sites at the gametophytic self-incompatibility pistil S-RNase gene in the Solanaceae and Rosaceae (Prunus, Pyrus, and Malus). J. Mol. Evol. 65, 175–185. doi: 10.1007/s00239-006-0285-6
Wang, L., Dong, L., Zhang, Y., Zhang, Y., Wu, W., Deng, X., et al. (2004). Genome-wide analysis of S-Locus F-box-like genes in Arabidopsis thaliana. Plant Mol. Biol. 56, 929–945. doi: 10.1007/s11103-004-6236-y
Wheeler, D., and Newbigin, E. (2007). Expression of 10 S-class SLF-like genes in Nicotiana alata pollen and its implications for understanding the pollen factor of the S locus. Genetics 177, 2171–2180. doi: 10.1534/genetics.107.076885
Wheeler, M. J., de Graaf, B. H., Hadjiosif, N., Perry, R. M., Poulter, N. S., Osman, K., et al. (2009). Identification of the pollen self-incompatibility determinant in Papaver rhoeas. Nature 459, 992–995. doi: 10.1038/nature08027
Wilkins, K. A., Poulter, N. S., and Franklin-Tong, V. E. (2014). Taking one for the team: self-recognition and cell suicide in pollen. J. Exp. Bot. 65, 1331–1342. doi: 10.1093/jxb/ert468
Williams, J. S., Der, J. P., dePamphilis, C. W., and Kao, T. H. (2014). Transcriptome analysis reveals the same 17 S-locus F-box genes in two haplotypes of the self-incompatibility locus of Petunia inflata. Plant Cell 26, 2873–2888. doi: 10.1105/tpc.114.126920
Williams, J. S., Wu, L., Li, S., Sun, P., and Kao, T. H. (2015). Insight into S-RNase-based self-incompatibility in Petunia: recent findings and future directions. Front. Plant Sci. 5:41. doi: 10.3389/fpls.2015.00041
Wu, L., Williams, J. S., Wang, N., Khatri, W. A., San Roman, D., and Kao, T. H. (2018). Use of domain-swapping to identify candidate amino acids involved in differential interactions between two allelic variants of Type-1 S-Locus F-Box protein and S3-RNase in Petunia inflata. Plant Cell Physiol. 59, 234–247. doi: 10.1093/pcp/pcx176
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang, Y. (2015). The I-TASSER suite: protein structure and function prediction. Nat. Methods 12, 7–8. doi: 10.1038/nmeth.3213
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Keywords: Solanaceae, SLFs, S-RNase, self-incompatibility, specificity recognition, positive selection, BDBM
Citation: Vieira J, Rocha S, Vázquez N, López-Fernández H, Fdez-Riverola F, Reboiro-Jato M and Vieira CP (2019) Predicting Specificities Under the Non-self Gametophytic Self-Incompatibility Recognition Model. Front. Plant Sci. 10:879. doi: 10.3389/fpls.2019.00879
Received: 10 April 2019; Accepted: 20 June 2019;
Published: 04 July 2019.
Edited by:
Tzung-Fu Hsieh, North Carolina State University, United StatesReviewed by:
Gustavo C. MacIntosh, Iowa State University, United StatesJose A. Traverso, University of Granada, Spain
Copyright © 2019 Vieira, Rocha, Vázquez, López-Fernández, Fdez-Riverola, Reboiro-Jato and Vieira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina P. Vieira, Y2d2aWVpcmFAaWJtYy51cC5wdA== orcid.org/0000-0002-7139-2107