 Siwen Liu
Siwen Liu Bo Wu
Bo Wu Shuxia Lv
Shuxia Lv Zongzhuan Shen
Zongzhuan Shen Rong Li
Rong Li Ganjun Yi
Ganjun Yi Chunyu Li
Chunyu Li Xiuwu Guo
Xiuwu Guo- 1College of Horticulture, Shenyang Agricultural University, Shenyang, China
- 2Key Laboratory of South Subtropical Fruit Biology and Genetic Resource Utilization, Ministry of Agriculture, Key Laboratory of Tropical and Subtropical Fruit Tree Research of Guangdong Province, Institution of Fruit Tree Research, Guangdong Academy of Agricultural Sciences, Guangzhou, China
- 3College of Bioscience and Biotechnology, Shenyang Agricultural University, Shenyang, China
- 4Jiangsu Key Lab for Solid Organic Waste Utilization, Key Laboratory of Plant Nutrition and Fertilization in Low-Middle Reaches of the Yangtze River, Ministry of Agriculture, College of Resources and Environmental Sciences, Nanjing Agricultural University, Nanjing, China
Fusaric acid (FA) is an important secondary metabolite of many Fusarium species and involved in the wilt symptoms caused in banana by Fusarium oxysporum f. sp. cubense (Foc). To investigate the evolution characteristics of the 12 Foc FA biosynthetic genes (FUB), coding sequences of the 12 FUB genes and three housekeeping genes, EF-1α/RPB1/RPB2 (translation elongation factor-1α/RNA polymerase II subunit I/RNA polymerase II subunit II), were subjected to genetic diversity analysis, phylogenetic analysis, recombination detection, and selective pressure analysis. The results of selective pressure analysis showed that the 15 genes were mainly subjected to negative selection. However, a significantly higher number of silent mutations, which could not be simply explained by selective pressure difference, were observed in the 12 FUB genes in Foc than in the three housekeeping genes. Infraspecies phylogeny and recombination detection analysis showed that significantly more horizontal gene transfer (HGT) events (normalized) had occurred in the FUB genes than in the three housekeeping genes. In addition, many of these events involved outgroup isolates and significantly increased the genetic diversity of FUB genes in Foc. The infraspecies phylogenetic analysis suggested that the polyphyletic phylogeny proposed for Foc requires further discussion, and the divergence of race 1, race 4, and the common ancestor of several F. oxysporum (Fo) isolates pathogenic to nonbanana plants should have diverged over a short period. Finally, our results suggest that the FUB genes in Fo should have benefited from HGT to gain a relatively high genetic diversity to respond to different host plants and environments despite mainly being subject to negative selection.
Introduction
Fusarium oxysporum (Fo) f. sp. cubense (Foc), the causal agent of Fusarium wilt of banana (Musa spp.), which is also known as Panama disease, is the most important soil-borne pathogen limiting banana production in the world (Ploetz, 2015). Like other Fusarium f. sp., Foc produces many toxic secondary metabolites; among these, fusaric acid (FA) is one of the most studied mycotoxins (Desjardins and Proctor, 2007; Bohni et al., 2016; Singh et al., 2017; López-Díaz et al., 2018) and a host-nonspecific toxin. FA is involved in the toxicity of Fusarium spp. towards plants, animals, and human beings (Ghazi et al., 2017; Singh et al., 2017; López-Díaz et al., 2018) and in interactions with environmental microorganisms (Brown et al., 2015; Bohni et al., 2016; Simonetti et al., 2018). FA has been reported to be phytotoxic to different plants involving multiple biological processes. For instance, it has been reported to chelate metal ions inside tomato (López-Díaz et al., 2018), disturb the water balance in cucumber (Wang et al., 2015), and cause programmed cell death in tobacco suspension cells (Jiao et al., 2013). In Panama disease, FA production by Foc is involved in disturbing the water balance and causing the wilt symptom (Dong et al., 2012; Li et al., 2013; Dong et al., 2014) and has been shown to be essential for its virulence on banana plantlets (Ding et al., 2018).
Due to the important function of FA in many Fusarium spp., much attention has been paid to the genes involved in its biosynthetic and related regulatory processes (Brown et al., 2012; Niehaus et al., 2014; Brown et al., 2015; Studt et al., 2016; Ding et al., 2018). Like many other secondary metabolite biosynthetic genes, which are generally located in gene clusters (Hoogendoorn et al., 2018), a FUB gene cluster consisting of 12 genes (FUB1 to FUB12) was identified in different Fusarium species (Brown et al., 2015; Studt et al., 2016). The functions of the FUB genes in Fusarium were predicted through the functions of their annotated homologs, including a polyketide synthase (PKS, FUB1), an unknown protein (FUB2), an aspartate kinase (FUB3), a serine hydrolase (FUB4), a homoserine O-acetyltransferase (FUB5), an NAD(P)-dependent dehydrogenase (FUB6), an O-acetylhomoserine (thiol-)lyase (FUB7), a nonribosomal peptide synthetase (NPRS)-like enzyme (FUB8), an FMN-dependent dehydrogenase (FUB9), two C6 transcription factors (FUB10 and FUB12), and a major facilitator superfamily transporter (FUB11) (Studt et al., 2016). FA production is differentially altered in deletion mutants of each FUB gene (Brown et al., 2015; Studt et al., 2016), suggesting that the importance of the 12 genes is different. FUB1 is the key gene in the FUB gene cluster, and FA production was abolished or reduced by 95% in the Fo FUB1 deletion mutant (Brown et al., 2015; López-Díaz et al., 2018). FA production was abolished in deletion mutants of FUB10 in Fusarium verticillioides (Fv), Fusarium fujikuroi (Ff), and Fo, and FUB10 has been proven to be a transcription factor positively regulating the transcription of other FUB genes (Brown et al., 2015; Studt et al., 2016). Deletion mutants of the other transcription factor-encoding gene, FUB12, exhibited incomplete loss of FA production in Fv and Ff (Brown et al., 2015; Studt et al., 2016). Fo deletion mutants of FUB3, FUB6, or FUB8 also abolished FA production, but in corresponding Fv mutants, FA production was reduced to only a certain extent (Brown et al., 2015). A FUB4 deletion mutant of a Foc race 4 strain lost its capacity for FA production and was significantly weakened in its virulence towards banana (Ding et al., 2018).
The infraspecies phylogeny of Foc has been suggested to be complex. Three races of Foc that are pathogenic to different host banana types have been reported: race 1, affecting Gros Michel (AAA) and some AAB or ABB bananas; race 2, pathogenic to ABB cooking bananas; race 4, affecting Cavendish bananas; and race 1 and race 2 suspects (Mostert et al., 2017). Race 4 isolates were further divided into two groups according to their different requirements of disease-predisposing conditions: tropical race 4 (TR4) and subtropical race 4 (STR4) (Ploetz, 2015; Ploetz et al., 2015). According to vegetative compatibility, Foc isolates were assigned to 24 vegetative compatibility groups (VCGs) (Fourie et al., 2009). Foc was suggested to have multiple evolutionary origins based on different nuclear and mitochondrial genes (O’Donnell et al., 1998; Fourie et al., 2009; Czislowski et al., 2018). In the study of Fourie et al. (2009), polyphyletic phylogeny was supported for not only Foc but also race 1 and race 4, and the same sets of VCGs were generally clustered together. An infraspecies phylogeny analysis based on three housekeeping genes (EF-1α/RPB1/RPB2) supported monophyletic phylogeny of race 1 VCGs and race 4 VCGs (Czislowski et al., 2018). However, significant discordance was observed between some of the phylogenetic trees based on the SIX (Secreted In Xylem) genes and the infraspecies tree, suggesting horizontal gene transfer (HGT) events in the SIX genes in Foc (Czislowski et al., 2018). The study of Maryani et al. (2019) based on EF-1α/RPB1/RPB2 also suggested that the Foc TR4 group should be monophyletic, but Foc race 1 isolates were clustered into multiple lineages. Horizontal gene transfer rather than convergent evolution was suggested to explain the polyphyletic phylogeny of race 1 and race 4 observed (Czislowski et al., 2018), and it was also suggested to have contributed to the high diversity level observed in race 1 compared with TR4 (Maryani et al., 2019). According to the infraspecies analysis in the study of Czislowski et al. (2018), some Fo f. sp. isolates and nonpathogenic Fo isolates should have originated from Foc race 1. Foc has no known sexual cycle and is supposed to undergo sexual production only rarely, if at all (Kerényi et al., 2004; Ploetz, 2015), and the mechanism of HGT in Foc remains unknown.
As stated above, FA production plays an important role in pathogen plant/environmental microorganism interactions in many Fusarium (sub)species including Foc. Considering its specific function, we hypothesize that the conservation of protein function (negative selection) should be the main selective force on Foc FUB genes when the host and environment are relatively stable, but selection of new advantageous mutations (positive selection) should also be possible if the fungus is adapting to a new host or environment. Based on coding sequences (CDSs) of the 12 FUB genes and three housekeeping genes from 33 Foc isolates, this study aimed to reveal the genetic diversity and evolutionary characteristics including nonsynonymous/synonymous substitution ratio and HGT in FUB genes which are largely determined by the selective forces. And in the meantime, an effort was also made to infer the infraspecies phylogeny of Foc.
Materials and Methods
Isolates and Group Assignments
Whole-genome sequences of 33 Foc isolates (Table 1) collected from multiple countries in South Asia, South Africa, and America were obtained from the Foc genome sequencing project (data unpublished). Published genomes of five Fo isolates, II5 (NRRL#54006, GCA_000260195.2, belonging to Foc race 4), Fo25433 (NRRL#54006, GCA_000260175.2), Fo47 (NRRL#54002, GCA_000271705.2), Fo4287 (NRRL#34936, GCA_000149955.2), and Fo26406 (NRRL#26406, GCA_002318975.1), and an outgroup Fv isolate, Fv7600 (GCA_000149555.1), were downloaded from the National Center for Biotechnology Information (NCBI) assembly database. The Northern Regional Research Laboratory (NRRL) codes were assigned by the Agricultural Research Service Culture Collection, US Department of Agriculture, and the IDs following the NRRL codes in the brackets are GenBank accession numbers. A total of 16 race 1 isolates from the Foc genome sequencing project were assigned to R1, including two nonpathogenic Fo isolates JB255 and JB553, which should belong to R1 according to phylogenetic analysis; and 18 race 4 isolates, including 17 from the Foc genome sequencing project and II5, were assigned to R4. Fo4287, Fo47, and Fo26406, which formed a monophyletic group in the phylogenetic analysis in this study (Figures 2A, B), were assigned to the 3Fo group. Fo25433 was not assigned to the R1 group considering its special identity, although it should have originated from R1 according to phylogeny analysis.
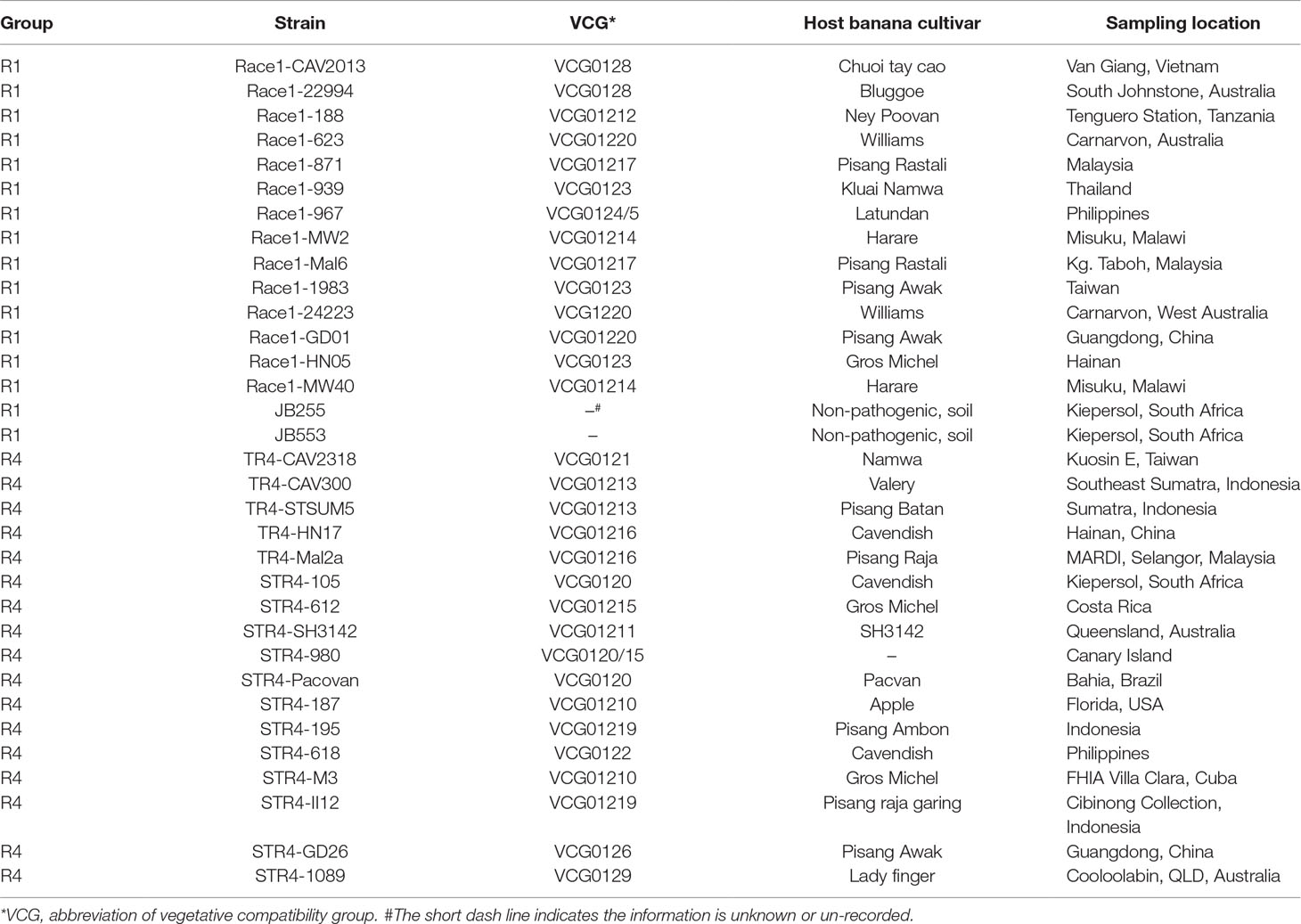
Table 1 Information of sequenced Fo isolates.
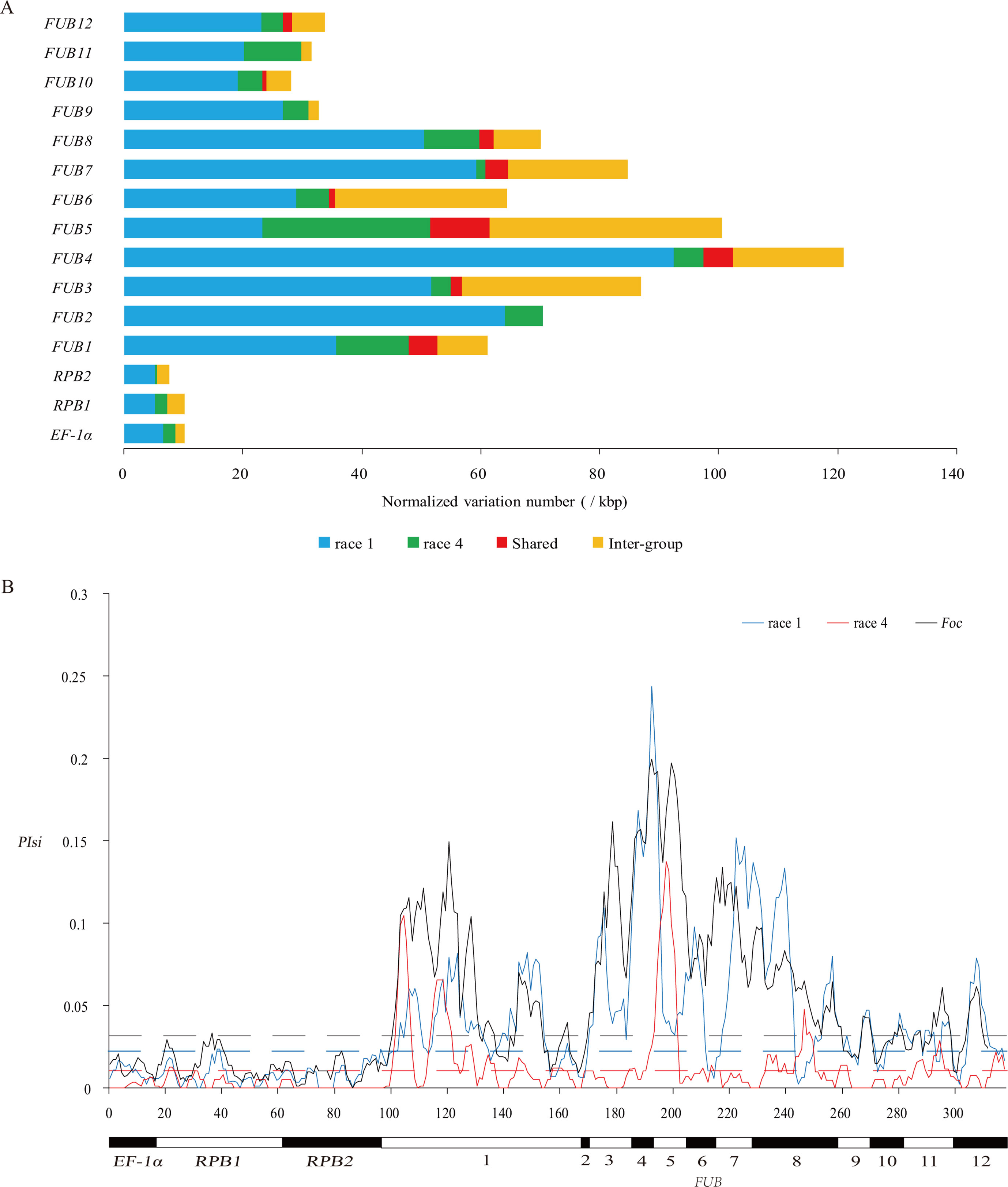
Figure 1 Genetic diversity of 34 Foc isolates based on 15 genes. (A) Distribution of intragroup and intergroup variations in Foc isolates based on 15 genes. Blue and green bars indicate the number of intragroup variations specific in R1 and R4, respectively. The red bar indicates the amount of intragroup variation shared by R1 and R4 isolates. Yellow bar indicates the number of intergroup variations bet ween R1 and R4. (B) Distribution of PIsi on windows of 100 silent sites across the 15 genes. The horizontal dashed lines indicate the threshold PIsi value, which was significantly (p < 0.01) higher than the PIsi level of the three housekeeping genes in R1, R4, and Foc.
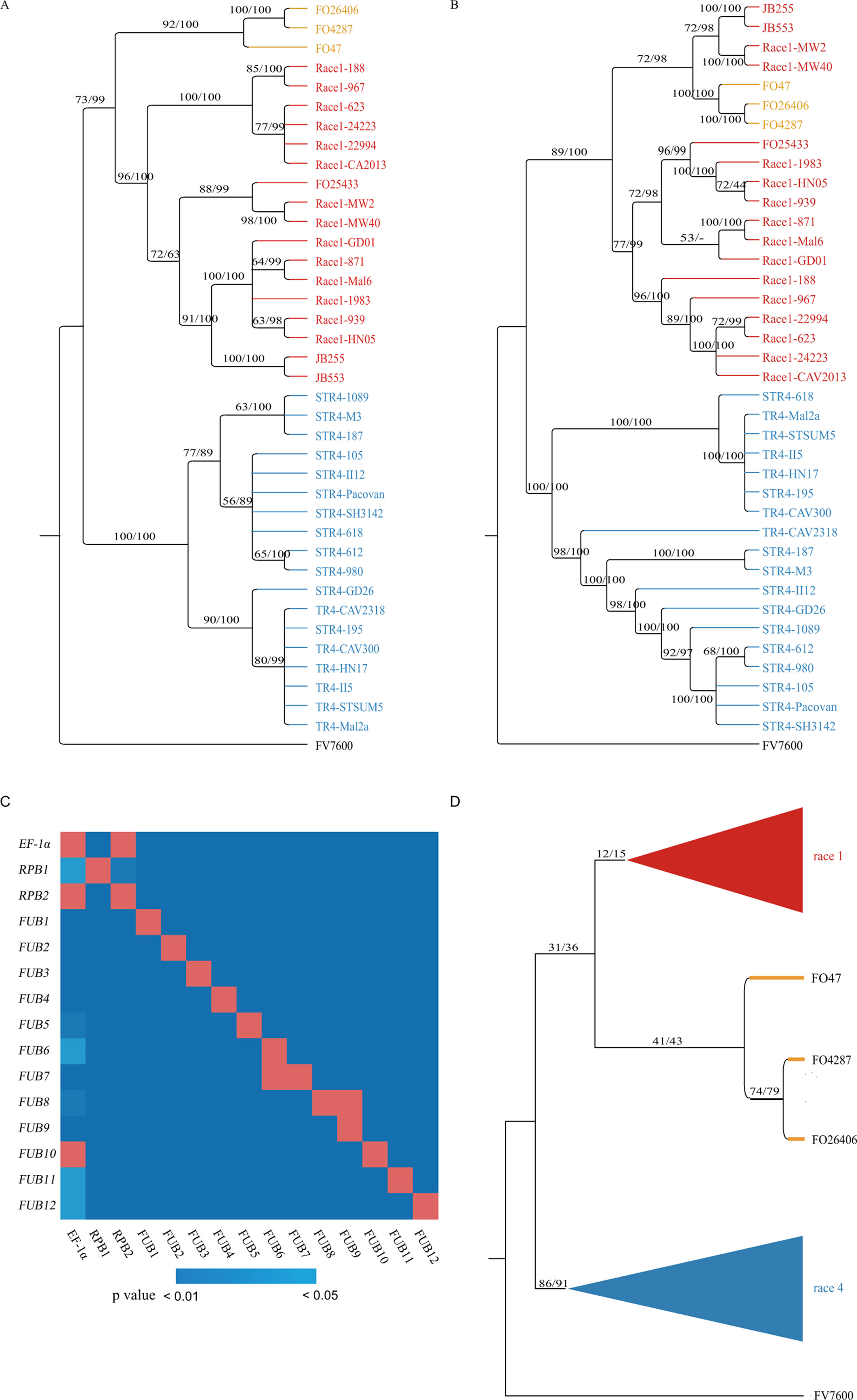
Figure 2 Foc infraspecies phylogeny inference and discordance between phylogenetic trees based on different genes. (A and B) Phylogenetic trees constructed based on concatenated sequences of EF-1α/RPB1/RPB2 and the 12 FUB genes, respectively. The support values on the branches are ML bootstrap proportions/BI posterior probabilities. R1, R4 and 3FO isolates were denoted by red, blue, and orange colors respectively. (C) SH test of the tree concordance between different genes. The best ML tree based on genes listed on the vertical axis was tested using the best tree and sequence of the gene listed on the horizontal axis. The color of the squares was determined by the p value of the SH test: blue indicated significance, and red indicated nonsignificance (p > 0.05). (D) Infraspecies tree inferred using the BSRRSS method. The numbers on branches are the support rates of the branch in 1,000 randomly sampled 1-kbp/2-kbp gene segments.
Sequence Alignment and Genetic Diversity Analysis
CDSs of EF-1α, RPB1, RPB2, and 12 FUB (FUB1–12) genes were obtained from the reference genome sequence of Foc race 4 isolate II5 (GenBank accession no. from MH972571 to 973155) (Guo et al., 2014). Then, the CDSs of the 15 genes were pulled out from all the genomes according to the results of local BLASTN alignment using II5 CDSs as queries, and no highly similar (>95% nucleotide similarity) paralog was identified for any gene. Multiple sequence alignment for each gene and concatenated sequences of EF-1α/RPB1/RPB2, the FUB gene cluster and the 15 genes was carried out using Clustal X version 2 (Larkin et al., 2007). Some of the CDSs were slightly different (probably due to different gene models applied in genome annotations or mutations causing transcript alteration that could not be distinguished and should not be a problem in this study) and were adjusted using their genome sequences to accommodate the CDS of the Foc II5 isolate.
Parameters, including Pi, the number of intragroup/intergroup variations, the number of synonymous/nonsynonymous mutations, the number of haplotypes, Dxy (the average number of nucleotide substitutions per site between populations), and the haplotype diversity of R1, R4 and Foc were calculated for each gene using DnaSP v6.12.01 (Rozas et al., 2017). Some of the mutations occurred on the same codons and could be assigned to be either non-synonymous or synonymous, depending on their occurrence orders. A total 9 such mutations were not assigned to either class. Silent loci in CDSs are generally subjected to neutral evolution in the genome, and the synonymous substitution rates among different genes have been shown to match a uniform point mutation rate across genes in Escherichia coli (Maddamsetti et al., 2015). A high level of synonymous mutations could be utilized as a potential signal of HGT (Proctor et al., 2013; Chen et al., 2018). Because the three housekeeping genes were mainly subjected to negative selection, which would not induce loss of PIsi, the PIsi level in these three genes was utilized as the control level to detect regions with significantly increased PIsi. PIsi was calculated in 100 silent site windows with a step size of 25 across the concatenated sequences of the 15 genes in R1, R4 and Foc using DnaSP v6.12.01. The first 97 windows were located within the three housekeeping genes and used as controls. The 99% cumulative probability threshold PIsi value was calculated for R1, R4 and Foc based on the normal distribution parameters calculated from the 97 windows in EXCEL, and PIsi values higher than this threshold are supposed to be significantly (p < 0.01) higher than those for the three housekeeping genes.
Infraspecies Phylogenetic Analysis
With the aligned sequences, jModelTest-2.1.10 (Darriba et al., 2012) was applied to choose the best models for phylogenetic tree construction based on ML and BI. Then, PhyML 3.1 (Guindon et al., 2010) was applied to produce ML trees using the best-fit model for concatenated sequences of EF-1α/RPB1/RPB2 and the FUB gene cluster and individual genes with 1,000 bootstrap replicates, and MrBayes v3.2 was used for tree construction by BI (Ronquist et al., 2012). Partitioned analysis unlinking models and parameters among different gene loci was applied on concatenated sequences by MrBayes v3.2. The SH test (Shimodaira and Hasegawa, 2001) was implemented in RAxML v8.2.12 (Stamatakis, 2014) to determine whether there was a significant difference in the tree topologies supported by different genes.
When concatenated gene sequences were used in phylogenetic inference, large regions of weak phylogenetic signals (produced when three or more species diverged in a short time) that support the true species tree topology could be obscured by short regions of strong phylogenetic signals caused by recent HGT events, and in this case, the standard nonparametric bootstrap could have lost its power (Heled and Drummond, 2010). In theory, the introgressed genetic material should account for a smaller proportion of the genome than the original species/infraspecies. Accordingly, the BSRRSS method was carried out by our Python script by calculating the branch support rate of branches in the best trees (obtained from 500 independent ML tree searches using RAxML v8.2.12) constructed for 1,000 randomly sampled 1 or 2 kbp gene segments from the concatenated sequence. A majority rule extended consensus tree was obtained from the 1000 best trees using RAxML v8.2.12, which should be a more reliable species tree. The low support rate of inner-group branches could have derived from either HGT or low sequence difference; thus, our main attention would be focused on the splitting of Fo groups. In theory, it would be more reasonable to sample an equal number of gene segments from each ‘unlinked’ locus (at most four in this study, EF-1α, RPB1, RPB2 and the FUB gene cluster), but considering that the gene sequences of the three housekeeping genes were short, it was not applied in this study.
Recombination Detection
Recombination detection was carried out using 7 different algorithms implemented in RDP v.4.95 (Martin et al., 2015), including RDP, Chimaera (Posada and Crandall, 2001), BootScan (Salminen et al., 1995), 3Seq (Boni et al., 2007), GENECONV (Padidam et al., 1999), MaxChi (Smith, 1992) and SiScan (Gibbs et al., 2000). The detected recombination events were required to have a Bonferroni corrected P-value < 0.05 and to be supported by topological evidence and at least two different methods. Two recombination events detected only by MaxChi that could explain the significant tree differences between EF-1α, RPB2, and RPB1 were also accepted because they were well supported by the infraspecies phylogenetic analysis. Manual inspection and correction were carried out by checking whether the recombined region supported a tree topology (using the ML and BI methods implemented in the software) different from that based on the ML/BI tree based on the three housekeeping genes. To analyze if there were significant differences in the number of recombination events in the three housekeeping genes and the FUB gene cluster, a chi-square test was carried out against the null hypothesis that the number of recombination events should be proportional to the length of the analyzed regions.
Ka/Ks and Selective Pressure Analysis
Ka and Ks were calculated between each pair of isolates from Foc and 3Fo using DNAsp v6.12.01, and the means of the Ka/Ks values of all of the genes were compared by one-way ANOVA implemented in SPSS v22 (IBM, New York).
Selection analysis of branches and amino acid sites was carried out using algorithms implemented in the web server Datamonkey 2.0 (Weaver et al., 2018). Isolates with introgressed outgroup genetic material were excluded from the analysis of the corresponding recombinant genes. Branch-site model-based methods aBSREL (Smith et al., 2015) and BUSTED (Murrell et al., 2015) were carried out to test whether the branches leading to R1, R4 and 3Fo had been subjected to positive selection. ML-based methods FEL, SLAC (Kosakovsky Pond and Frost, 2005) and MEME (Murrell et al., 2012) were performed for selective pressure analysis of individual sites.
Results
Genetic Diversity of Foc Based on 12 FUB Genes and Three Housekeeping Genes
CDSs of three housekeeping genes (EF-1α, RPB1, and RPB2) and 12 FUB genes with a total length of 33,168 bp were subtracted from the assembled genome sequences of 16 race 1 group (R1) isolates and 18 race 4 group (R4) isolates (Table 1). A total of 1,505 single-nucleotide variations were detected in the CDSs (Table S1), including 1,464 biallelic and 41 triallelic loci. Of the 1,505 variations (1,546 mutations), 1,228 mutations were synonymous, 309 were nonsynonymous, and the remaining nine mutations were not assigned (explained in the Materials and Methods section). No highly deleterious mutation that caused an open reading frame shift or protein truncation was detected in any of the 15 genes in any isolate.
A higher level of genetic diversity was identified in R1 than in R4. Based on the 15 genes, 12 different genotypes were identified from the 16 R1 isolates, and nine different genotypes were identified from the 18 R4 isolates (Figure S1). More group-specific intragroup variations were detected in R1 than in R4 on all genes except FUB5 (Figure 1A and Table S1). A total of 71 intragroup variations in FUB genes were shared between the two groups, which is most likely a sign of horizontal transfer between the two groups rather than convergent evolution. No shared intragroup variation was discovered in the three housekeeping genes. In total, 298 variations (51 nonsynonymous and 247 synonymous) were identified as intergroup variations in the 15 genes. The nucleotide diversity (Pi) of the R1 was generally higher (>2-fold and on average 6.43-fold) than that of the R4 in all genes except FUB5, on which R1 had lower (0.82-fold) Pi than R4. The Pi in the FUB5 gene of R4 was significantly higher (on average 5.35-fold, p < 0.001) than the Pi in any other gene of the same group. The haplotype diversity of R1 is higher than that of R4 for all genes, including FUB5.
As shown in Table S1, the Pi in the FUB genes of both R1 and R4 is significantly (on average 7.5- and 11.1-fold, respectively, p < 0.01) higher than that in the three housekeeping genes. Differences in Pi could be the result of the combined effect of different selective pressures and different amounts of HGT, but differences in the number of synonymous variations were supposed to be affected by only HGT. According to the results (Table S1), the number of synonymous substitutions per synonymous site (Ks) between R1 and R4 was significantly (p < 0.01) higher for FUB genes than for the three housekeeping genes. As shown in Figure 1B, significantly higher nucleotide diversity of silent variations (PIsi) (p < 0.01) was observed in 77.3%, 45.7%, and 76.0% of the regions of the FUB gene cluster than in the three housekeeping genes in R1, R4, and Foc, respectively. The observed high Ks and PIsi in FUB genes should indicate introgression of outgroup genetic material into the FUB genes in either or both group(s).
Infraspecies Phylogeny Analysis
Molecular data on the 15 genes were used to infer the infraspecific phylogeny of Fo. To reveal the phylogenetic relationships of the sequenced Fo isolates and four published Fo isolates, Fo4287, Fo26406, Fo47, and Fo25433, maximum likelihood (ML) and Bayesian inference (BI)-based phylogenetic methods were applied on concatenated nucleotide sequences of EF-1α/RPB1/RPB2 and 12 FUB genes with Fv7600 as the outgroup. As shown in Figure 2A and B, phylogenetic trees obtained by different phylogenetic methods on the same gene set were highly consistent, and in phylogenetic trees based on both concatenated sequences, isolates of the same VCGs were clustered into the same clades. However, the phylogenetic trees based on the EF-1α/RPB1/RPB2 gene set were poorly supported [significantly worse by the Shimodaira–Hasegawa (SH) test at the 1% level] by the 12-FUB-gene set and vice versa. In the trees based on the three housekeeping genes, four isolates (JB255, JB553, Race1-MW2, and Race1-MW40) were clustered in the R1, but in the 12-FUB-gene cluster, they clustered out of R1 and formed a monophyletic group with Fo4287, Fo26406, and Fo47, which are three nonbanana Fo isolates (referred to as the 3Fo group). The phylogenetic relationship of R1 isolates and R4 isolates based on EF-1α/RPB1/RPB2 should be robust, since any recombination signal was seldom detected in R1 and R4 isolates on these three genes (see the following section).
To check the difference in the phylogeny of the 15 genes, ML and BI-based methods were applied to each of the 15 genes for all of the above accessions. In trees based on both concatenated sequences and most genes, the nonbanana isolate Fo25433 was highly supported as belonging to R1. As shown in Figure S2, contradictory branches were widely observed in the 15 gene trees. The results of SH tests showed that the molecular trees based on most genes were significantly different (p < 0.05, Figure 2C), and only EF-1α and RPB2 did not reject each other (p > 0.05 by SH test), but both rejected the tree based on RPB1, showing that at least one HGT event occurred in the three genes. For the trees based on EF-1α and RPB2, the tree topology supports the idea that the 3FO diverged from the common ancestor of R1 and R4, while the tree based on RPB1 supported that 3Fo and R1 shared a more recent common ancestor than with R4, which is also supported by phylogenetic trees based on both concatenated gene sets (Figures 2A, B).
Considering the widely present topological differences among gene trees, a method named branch support rate in randomly sampled segments (BSRRSS) was used to infer a more reliable infraspecies tree and assess the support rate of controversial branches. Randomly sampled 1- and 2-kbp segments from the CDSs of the 15 genes were both applied in this study and produced similar results. As shown in Figure 2D, in the branches best supported by randomly sampled 1-kbp (2-kbp) gene segments, the monophyly of R1 and the monophyly of R4 were supported by 12% (15%) and 86% (91%) of sampled segments, respectively. The monophyletic clade compromising R1 and 3Fo was supported by 31% (36%) of sampled 1-kbp (2-kbp) segments, and the ratios of sampled 1-kbp (2-kbp) segments supporting the monophyly of R1 and R4 and the monophyly of 3Fo and R4 were 15% (15%) and 4% (2%), respectively. When most recombinant R1 and R4 isolates identified in the following section were excluded from the analysis, the support for the monophyly of R1 and 3Fo was only slightly increased to 35% (38%), while the support for monophyly of R1 increased to 77% (81%). Considering that at most four loci with different lengths were sampled, the support for the monophyletic clade including R1 and 3Fo is not robust based on the 15 genes according to the BSRRSS test method. In the BSRRSS analysis, most 1- and 2-kbp gene segments did not contain phylogenetic signals supporting either monophyletic phylogeny or polyphyletic phylogeny of Foc, indicating that the differentiation of R1, R4, and 3Fo should have occurred in a short period, leaving only a weak phylogenetic signal in the genes.
Enrichment of HGT Events for FUB Genes
According to the above results, HGT should have played a role in producing some of the Foc genotypes observed in this study, such as the four isolates JB255, JB553, Race1-MW2, and Race1-MW40. To reveal the distribution of recombination events that led to the 15 analyzed genes in the Fo isolates, seven different methods implemented in RDP4 were applied, and the topology in Figure 2D was assumed to be the true infraspecies tree in the process. Three recombination events were detected to have occurred in the three housekeeping genes. Two of the events were supported by only one method and were HGTs of the outgroup (which diverged earlier than the divergence of R1 and R4) EF-1α and RPB2 genes into the 3Fo isolates, which could explain the conflict between the phylogenetic trees based on EF-1α, RPB2, and RPB1. The third recombination was identified within the R4 with support from two methods and involved HGT of partial RPB1 of the TR4-STSUM5-like isolate into STR4-GD26. As shown in Figure 3, a significantly greater number of well-supported (detected by at least two methods and passing manual inspection) recombination events (14.7-fold, p < 0.001) were detected in the FUB gene cluster (1.41 recombination/kbp) than in the three housekeeping genes (0.096 recombination/kbp) in the Fo isolates. The recombination detection methods were supposed to detect only relatively large recombinant regions (Martin et al., 2015), and there should be an undetected recombination because the PIsi values were significantly higher in the FUB genes than in the three housekeeping genes in more regions (Figure 1B).
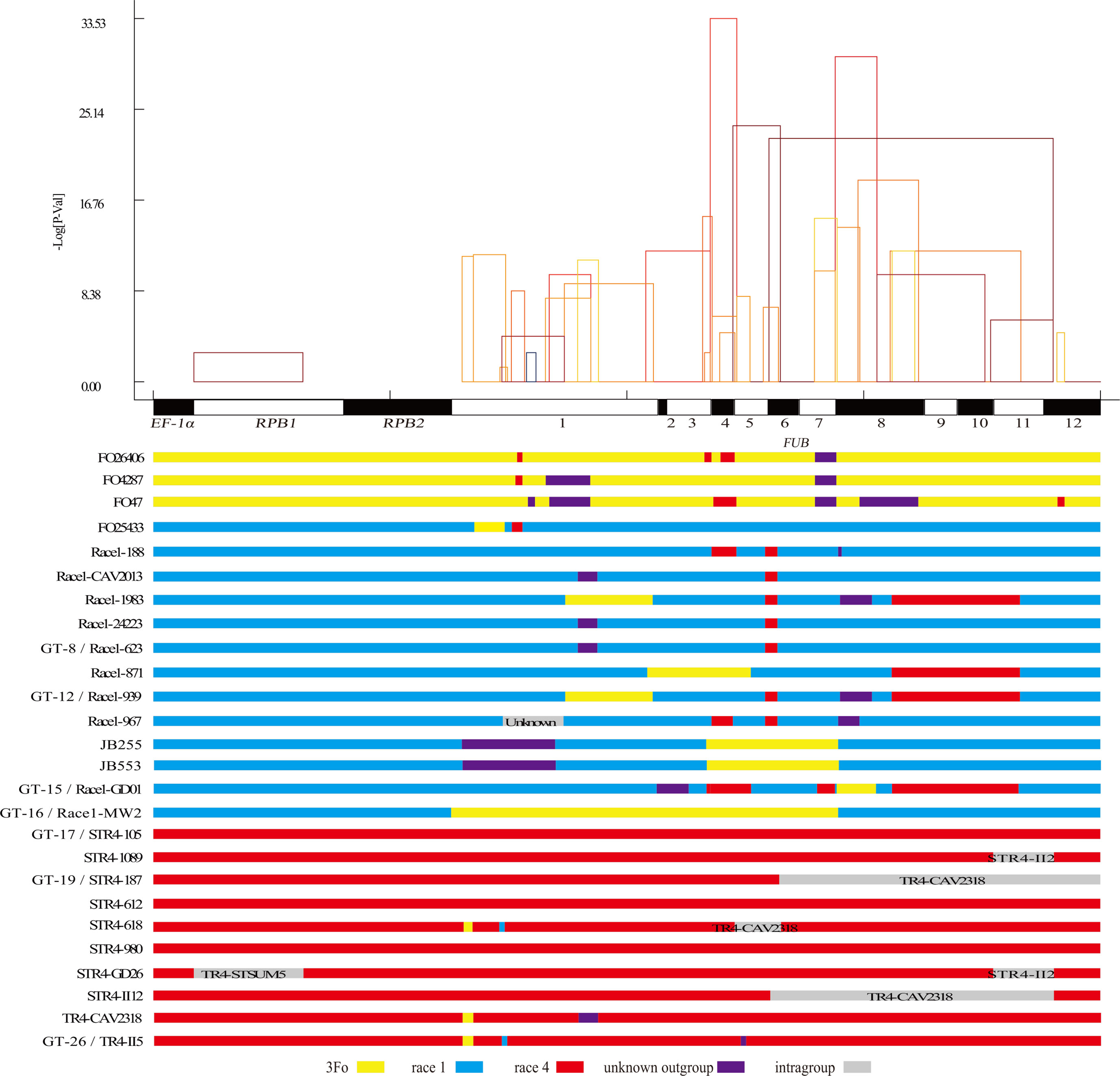
Figure 3 Distribution of inferred recombination events in the 15 genes. Top panel: each rectangle represents an inferred recombination event, and the horizontal coordinates of the two vertical edges of the rectangles indicate the inferred most likely recombination points of the recombination event by RDP4. The height (vertical axis) of each rectangle is the lg (p value) of the recombination event inferred by the algorithm implemented in RDP4. Bottom panel: introgressed gene segments identified in the 26 different Fo genotypes. Gray bands indicate intragroup recombination events in R1, R4, or 3Fo, and the name of putative source isolate (most likely the isolate which is the most similar to the source isolate) was shown on the graph.
As shown in Figure 3, bottom panel, many more outgroup genetic materials were transferred into R1 than into R4, which partially explained why R1 had a much higher genetic diversity than R4. Large regions of horizontal gene-transferred outgroup genetic materials were observed in the FUB genes in Race1-MW2, Race1-MW40, JB553, and JB255. A recombination region that involved the horizontal transfer of ancient outgroup genetic material in FUB5 was detected in II5 and five other R4 isolates, which should explain why the R4 had higher genetic diversity in only FUB5. In two R1 isolates, JB255 and JB553, which have lost their pathogenicity towards banana, FUB1 was identified to be recombined with an outgroup isolate.
Selective Pressure on Fo Genes
The average Ka/Ks values for the 15 genes over different Fo isolates suggested that most coding loci on the 15 genes should have been subjected to negative selection (Table S1), because all of these values were much smaller than 1. The FUB genes other than FUB11 had a significantly higher mean Ka/Ks values than each of the three housekeeping genes (Figure 4). The largest average Ka/Ks value was observed for FUB2 (0.166 ± 0.122), and the Ka/Ks values between some isolates from 3Fo and R4 were as high as 0.84, suggesting that many loci of this gene could have been subjected to neutral evolution in some branches.
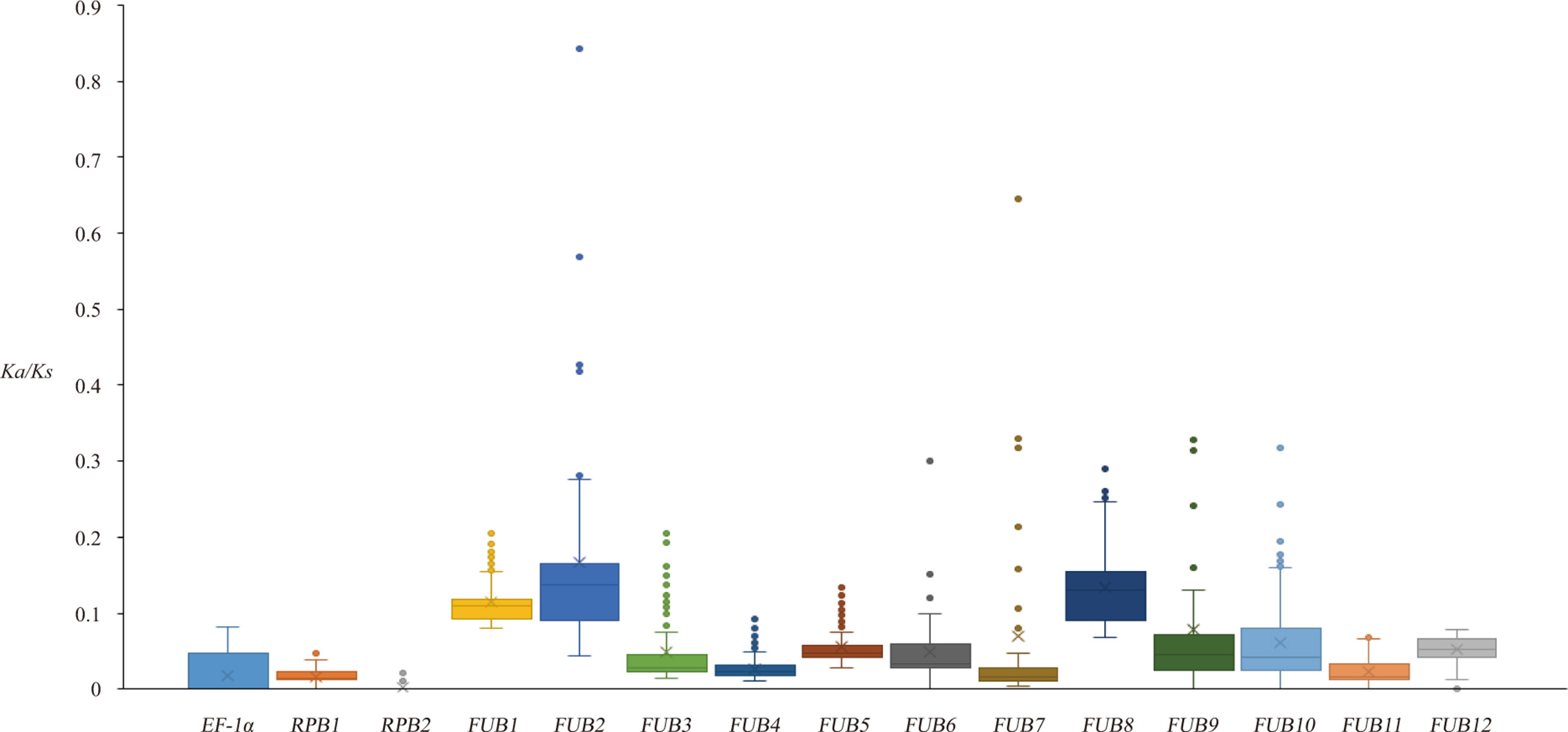
Figure 4 Boxplot of Ka/Ks distribution between Fo isolates based on the 15 genes. The mean Ka/Ks value is indicated by the small crosses on the graph.
Branch-site models were applied to test for positive selection on the branches leading to R1, R4, and 3Fo groups. Positive selection was not identified in any of the tested branches for the three housekeeping genes and 12 FUB genes. Site analysis was carried out to test whether there were any codons subjected to positive or negative selection on the FUB genes. As shown in Table S2, 173 negatively selected amino acid sites but not a single positively selected site was detected (p < 0.05 by at least two methods) on the 12 FUB genes, showing that the translations of analyzed genes were mainly subjected to negative selection or neutral selection.
Discussion
FA is an important virulence factor in Fo and other Fusarium species that are associated with the wilt symptom in banana and other plants (Li et al., 2013; Dong et al., 2014; Singh et al., 2017; Ding et al., 2018). Considering the function of FA involved in interactions with the host and environmental microorganisms (Brown et al., 2015), revealing the evolution characteristics of the FUB genes could help us understand how Fo isolates have evolved to accommodate different host types and environments. Indeed, in this study, we demonstrated that the genetic diversity and phylogeny of Foc isolates did have some specific characteristics in the 12 FUB genes compared with the housekeeping genes.
Our results showed that all 12 FUB genes in Foc were mainly subjected to negative selection according to selection pressure analysis (Figure 4) and that not a single highly deleterious mutation was identified on any of the 12 FUB genes in any isolate. The conservation of the FUB genes observed in this study suggested most of the FUB genes were functionally important, consistent with the report that FA is essential for the virulence of Foc against banana (Ding et al., 2018). Consistent with the report that the functions and importance of the 12 FUB genes are different (Brown et al., 2015), significant differences in selective pressure were found among the 12 FUB genes according to Ka/Ks analysis. The function of FUB2 is unknown and seemed to be unimportant in deletion analysis (Brown et al., 2015); however, the Ka/Ks analysis suggested that FUB2 was conserved in some phylogenetic branches, though it has the highest average Ka/Ks value and had probably been subjected to neutral evolution during the divergence of 3Fo and R4. Additionally, no single fixed variation was observed in FUB2 between R1 and R4 (Table S1), which suggested that it could also have a function in some unknown circumstances. FUB11 (FA transporter) and FUB7 [O-acetylhomoserine (thiol-)lyase] are important in transporting FA from the intracellular space to the extracellular space and in FA production, respectively (Brown et al., 2015), and in this study, FUB11 and FUB7 were shown to be the most conserved of the 12 FUB genes by the Ka/Ks analysis. FUB1 has been known as the key gene in the gene cluster and encodes a PKS, and a recombinant FUB1 gene including a partial gene from an unknown outgroup isolate was observed in both JB255 and JB553, which have lost their pathogenicity towards banana. The correlation between the two facts deserves further experimental verification.
Significantly higher genetic diversity was observed in the FUB genes than in the three housekeeping genes, which is not explained by the difference in selective pressure. According to our analysis, the high genetic diversity of the FUB genes was mainly derived from significantly enriched introgression of outgroup genetic materials into the Foc groups, as shown in Figures 1B and 3. In other words, recombination at FUB genes could have been positively selected, significantly enlarging the gene pool in Foc and other formae speciales of Fo. Because mutations in FUB genes have been mainly reported to affect the regulation and efficiency of FA production in Fusarium isolates (Brown et al., 2015), some recombination events should have the ability to change the pattern of FA production in Fo isolates. Considering that FA is involved in the interaction of Fusarium isolates with both plant hosts and environmental microorganisms (Brown et al., 2015; Bohni et al., 2016; López-Díaz et al., 2018), when the plant host and living environment are relatively stable, the Foc FUB genes should be mainly subjected to negative selection. However, when Foc isolates were brought into a new environment, recombination of the FUB genes with local Fo lineages or even other Fusarium species (ancient outgroup genetic materials were identified in some Foc isolates) could have a positive effect on their adaption, and this process is supposed to save much more time than developing new beneficial mutations. Several recombination events were also supposed to have occurred at the SIX genes, which encode the only identified family of effectors in Fo, as reported by Czislowski et al. (2018). These results suggested that recombination enrichment might be a common phenomenon for genes involved in pathogen–host interactions or interactions with environmental microorganisms in Fo; however, this hypothesis still needs further verification.
The previously inferred polyphyletic phylogeny (Fourie et al., 2009; Czislowski et al., 2018) of race 1, race 4, and Foc is not sufficiently robust according to our analysis. Polyphyletic phylogeny of Foc was inferred based on the concatenated sequence of EF-1α/RPB1/RPB2 in the study of Czislowski et al. (2018), and the inferred infraspecies tree also supported that race 1 and race 4 should be two monophyletic groups. However, as shown in our results based on individual genes, only the gene tree based on RPB1 supported the polyphyletic phylogeny of analyzed Foc isolates, while EF-1α and RPB2 supported the opposite case, which indicated that the phylogenetic inference based on the concatenated sequence could have resulted in a well-supported (by bootstrap or posterior probability) but incorrect tree (Heled and Drummond, 2010). Moreover, recombination analysis suggested that a recombination event had occurred at RPB1 or that two separate recombination events had occurred at EF-1α and RPB2, respectively, in the 3Fo group, depending on which infraspecies tree is accurate. In addition, in the study of Czislowski et al. (2018) and our study, introgression of outgroup genetic materials was observed at both the SIX genes and the FUB genes in both R1 and R4 isolates, which could easily render gene trees supporting polyphyletic phylogeny of R1 and R4 similar to that discovered in a previous study (Fourie et al., 2009), although the difference in the results could also be caused by the difference in Foc isolates used. The BSRRSS analysis in this study showed that the polyphyletic phylogeny of Foc was supported by <40% of the regions of the 15 analyzed genes and that R1, R4, and 3Fo should have diverged within a short time period, which made the phylogeny signal representing the true intraspecific phylogeny relatively weak and easily covered by HGT events. This study also supported that Fo25433, which is a pathogen detected on cotton, should have originated from Foc race 1, and the infraspecies tree in the study of Czislowski et al. (2018) also suggested that many formae speciales of Fo should have originated from the Foc race 1. Considering the limited number of sequenced genes and the HGT commonly observed in all the studies, to achieve a highly reliable infraspecific phylogeny tree for Fo, it is necessary to sequence more genome regions of more isolates and resolve the complex recombination events in sequenced isolates first.
In conclusion, this study showed that negative selection on a majority of amino acids and enriched HGT events are the main evolutionary characteristics of the 12 FUB genes. Though no positive selection signal on any gene or amino acid has been detected in this study, the genetic diversity on the 12 FUB genes was greatly increased due to significantly enriched HGT events, suggesting some of the natural mutations could have an adaptive effect and positively selected. But such mutations were not detected in this study, either because they are too limited in number compared with synonymous mutations or because they do not exist in the coding regions. Infraspecies phylogeny analysis in this study suggested that Foc race 1, Foc race 4, and some other Fo f. sp. should have diverged in a short time, and the previously suggested polyphyly of Foc still needs more evidence.
Author Contributions
SL, BW, and SXL analyzed the data and wrote the manuscript; ZS, RL, GY, CL and XG conceived and designed the experiments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2018YFD1000300), Guangdong Science and Technology Project (2016B020202006, 2016B020233002 and 2014B050502007), National Natural Science Fund (31471740), 948 Project of the Department of Agriculture (2011-G16) and Project of Science and Technology from the Guangzhou Science and Information Bureau (201707010154), Key Collaborative Project of Industry-University-Research of Guangzhou Committee on Scientific and Technological Innovation (201704020031), International Collaborative Project from Ministry of Science and Technology of China (2013DFB30400), National Banana Industry and Technology System Project (nycytx-33), Central Public-interest Scientific Institution Basal Research Fund (1630052017020), Guangdong Academy of Agricultural Sciences Foundation (201815) and Science and Technology Plan Project of Guangdong Province (2016B020201007).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01069/full#supplementary-material
References
Bohni, N., Hofstetter, V., Gindro, K., Buyck, B., Schumpp, O., Bertrand, S., et al. (2016). Production of fusaric acid by Fusarium spp. in pure culture and in solid medium co-cultures. Molecules 21, 370. doi: 10.3390/molecules21030370
Boni, M. F., Posada, D., Feldman, M. W. (2007). An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 176, 1035–1047. doi: 10.1534/genetics.106.068874
Brown, D. W., Butchko, R. A. E., Busman, M., Proctor, R. H. (2012). Identification of gene clusters associated with fusaric acid, fusarin, and perithecial pigment production in Fusarium verticillioides. Fungal Genet. Biol. 49, 521–532. doi: 10.1016/j.fgb.2012.05.010
Brown, D. W., Lee, S.-H., Kim, L.-H., Ryu, J.-G., Lee, S., Seo, Y., et al. (2015). Identification of a 12-gene fusaric acid biosynthetic gene cluster in Fusarium species through comparative and functional genomics. Mol. Plant Microbe Interact. 28, 319–332. doi: 10.1094/MPMI-09-14-0264-R
Chen, N. W. G., Serres-Giardi, L., Ruh, M., Briand, M., Bonneau, S., Darrasse, A., et al. (2018). Horizontal gene transfer plays a major role in the pathological convergence of Xanthomonas lineages on common bean. BMC Genomics 19, 606. doi: 10.1186/s12864-018-4975-4
Czislowski, E., Fraser-Smith, S., Zander, M., O’Neill, W. T., Meldrum, R. A., Tran-Nguyen, L. T. T., et al. (2018). Investigation of the diversity of effector genes in the banana pathogen, Fusarium oxysporum f. sp. cubense, reveals evidence of HGT. Mol. Plant Pathol. 19, 1155–1171. doi: 10.1111/mpp.12594
Darriba, D., Taboada, G. L., Doallo, R., Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. doi: 10.1038/nmeth.2109
Desjardins, A. E., Proctor, R. H. (2007). Molecular biology of Fusarium mycotoxins. Int. J. Food Microbiol. 119, 47–50. doi: 10.1016/j.ijfoodmicro.2007.07.024
Ding, Z., Yang, L., Wang, G., Guo, L., Liu, L., Wang, J., et al. (2018). Fusaric acid is a virulence factor of Fusarium oxysporum f. sp. cubense on banana plantlets. Trop. Plant Pathol. 43, 297–305. doi: 10.1007/s40858-018-0230-4
Dong, X., Ling, N., Wang, M., Shen, Q., Guo, S. (2012). Fusaric acid is a crucial factor in the disturbance of leaf water imbalance in Fusarium-infected banana plants. Plant Physiol. Biochem. 60, 171–179. doi: 10.1016/jplaphy.2012.08.004
Dong, X., Xiong, Y., Ling, N., Shen, Q., Guo, S. (2014). Fusaric acid accelerates the senescence of leaf in banana when infected by Fusarium. World J. Microbiol. Biotechnol. 30, 1399–1408. doi: 10.1007/s11274-013-1564-1
Fourie, G., Steenkamp, E. T., Gordon, T. R., Viljoen, A. (2009). Evolutionary relationships among the Fusarium oxysporum f. sp. cubense vegetative compatibility groups. Appl. Environ. Microbiol. 75, 4770–4781. doi: 10.1128/AEM.00370-09
Ghazi, T., Nagiah, S., Tiloke, C., Sheik Abdul, N., Chuturgoon, A. A. (2017). Fusaric acid induces DNA damage and post-translational modifications of p53 in human hepatocellular carcinoma (HepG2) cells. J. Cell Biochem. 118, 3866–3874. doi: 10.1002/jcb.26037
Gibbs, M. J., Armstrong, J. S., Gibbs, A. J. (2000). Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16, 573–582. doi: 10.1093/bioinformatics/16.7.573
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Guo, L., Han, L., Yang, L., Zeng, H., Fan, D., Zhu, Y., et al. (2014). Genome and transcriptome analysis of the fungal pathogen Fusarium oxysporum f. sp. cubense causing banana vascular wilt disease. PloS One 9, e95543. doi: 10.1371/journal.pone.0095543
Heled, J., Drummond, A. J. (2010). Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 27, 570–580. doi: 10.1093/molbev/msp274
Hoogendoorn, K., Barra, L., Waalwijk, C., Dickschat, J. S., van der Lee, T. A. J., Medema, M. H. (2018). Evolution and diversity of biosynthetic gene clusters in Fusarium. Front. Microbiol. 9, 1158. doi: 10.3389/fmicb.2018.01158
Jiao, J., Zhou, B., Zhu, X., Gao, Z., Liang, Y. (2013). Fusaric acid induction of programmed cell death modulated through nitric oxide signalling in tobacco suspension cells. Planta 238, 727–737. doi: 10.1007/s00425-013-1928-7
Kerényi, Z., Moretti, A., Waalwijk, C., Oláh, B., Hornok, L. (2004). Mating type sequences in asexually reproducing Fusarium species. Appl. Environ. Microbiol. 70, 4419–4423. doi: 10.1128/AEM.70.8.4419-4423.2004
Kosakovsky Pond, S. L., Frost, S. D. W. (2005). Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22, 1208–1222. doi: 10.1093/molbev/msi105
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Li, C., Zuo, C., Deng, G., Kuang, R., Yang, Q., Hu, C., et al. (2013). Contamination of bananas with beauvericin and fusaric acid produced by Fusarium oxysporum f. sp. cubense. PloS One 8, e70226. doi: 10.1371/journal.pone.0070226
López-Díaz, C., Rahjoo, V., Sulyok, M., Ghionna, V., Martín-Vicente, A., Capilla, J., et al. (2018). Fusaric acid contributes to virulence of Fusarium oxysporum on plant and mammalian hosts. Mol. Plant Pathol. 19, 440–453. doi: 10.1111/mpp.12536
Maddamsetti, R., Hatcher, P. J., Cruveiller, S., Médigue, C., Barrick, J. E., Lenski, R. E. (2015). Synonymous genetic variation in natural isolates of Escherichia coli does not predict where synonymous substitutions occur in a long-term experiment. Mol. Biol. Evol. 32, 2897–2904. doi: 10.1093/molbev/msv161
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., Muhire, B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1, vev003. doi: 10.1093/ve/vev003
Maryani, N., Lombard, L., Poerba, Y. S., Subandiyah, S., Crous, P. W., Kema, G. H. J. (2019). Phylogeny and genetic diversity of the banana Fusarium wilt pathogen Fusarium oxysporum f. sp. cubense in the Indonesian centre of origin. Stud. Mycol. 92, 155–194. doi: 10.1016/j.simyco.2018.06.003
Mostert, D., Molina, A. B., Daniells, J., Fourie, G., Hermanto, C., Chao, C.-P., et al. (2017). The distribution and host range of the banana Fusarium wilt fungus, Fusarium oxysporum f. sp. cubense, in Asia. PloS One 12, e0181630. doi: 10.1371/journal.pone.0181630
Murrell, B., Weaver, S., Smith, M. D., Wertheim, J. O., Murrell, S., Aylward, A., et al. (2015). Gene-wide identification of episodic selection. Mol. Biol. Evol. 32, 1365–1371. doi: 10.1093/molbev/msv035
Murrell, B., Wertheim, J. O., Moola, S., Weighill, T., Scheffler, K., Kosakovsky Pond, S. L. (2012). Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8, e1002764. doi: 10.1371/journal.pgen.1002764
Niehaus, E.-M., Bargen, K. W., von Espino, J. J., Pfannmüller, A., Humpf, H.-U., Tudzynski, B. (2014). Characterization of the fusaric acid gene cluster in Fusarium fujikuroi. Appl. Microbiol. Biotechnol. 98, 1749–1762. doi: 10.1007/s00253-013-5453-1
O’Donnell, K., Kistler, H. C., Cigelnik, E., Ploetz, R. C. (1998). Multiple evolutionary origins of the fungus causing Panama disease of banana: concordant evidence from nuclear and mitochondrial gene genealogies. Proc. Natl. Acad. Sci. 95, 2044–2049. doi: 10.1073/pnas.95.5.2044
Padidam, M., Sawyer, S., Fauquet, C. M. (1999). Possible emergence of new geminiviruses by frequent recombination. Virology 265, 218–225. doi: 10.1006/viro.1999.0056
Ploetz, R., Freeman, S., Konkol, J., Al-Abed, A., Naser, Z., Shalan, K., et al. (2015). Tropical race 4 of Panama disease in the Middle East. Phytoparasitica 43, 283–293. doi: 10.1007/s12600-015-0470-5
Ploetz, R. C. (2015). Fusarium wilt of banana. Phytopathology 105, 1512–1521. doi: 10.1094/PHYTO-04-15-0101-RVW
Posada, D., Crandall, K. A. (2001). Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc. Natl. Acad. Sci. 98, 13757–13762. doi: 10.1073/pnas.241370698
Proctor, R. H., van Hove, F., Susca, A., Stea, G., Busman, M., van der Lee, T., et al. (2013). Birth, death and horizontal transfer of the fumonisin biosynthetic gene cluster during the evolutionary diversification of Fusarium. Mol. Microbiol. 90, 290–306. doi: 10.1111/mmi.12362
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61 (3), 539–542. doi: 10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Salminen, M. O., Carr, J. K., Burke, D. S., McCutchan, F. E. (1995). Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res. Hum. Retroviruses 11, 1423–1425. doi: 10.1089/aid.1995.11.1423
Shimodaira, H., Hasegawa, M. (2001). CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17, 1246–1247. doi: 10.1093/bioinformatics/17.12.1246
Simonetti, E., Roberts, I. N., Montecchia, M. S., Gutierrez-Boem, F. H., Gomez, F. M., Ruiz, J. A. (2018). A novel Burkholderia ambifaria strain able to degrade the mycotoxin fusaric acid and to inhibit Fusarium spp. growth. Microbiol. Res. 206, 50–59. doi: 10.1016/j.micres.2017.09.008
Singh, V. K., Singh, H. B., Upadhyay, R. S. (2017). Role of fusaric acid in the development of ‘Fusarium wilt’ symptoms in tomato: physiological, biochemical and proteomic perspectives. Plant Physiol. Biochem. 118, 320–332. doi: 10.1016/j.plaphy.2017.06.028
Smith, J. M. (1992). Analyzing the mosaic structure of genes. J. Mol. Evol. 34, 126–129. doi: 10.1007/BF00182389
Smith, M. D., Wertheim, J. O., Weaver, S., Murrell, B., Scheffler, K., Kosakovsky Pond, S. L. (2015). Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 32, 1342–1353. doi: 10.1093/molbev/msv022
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Studt, L., Janevska, S., Niehaus, E.-M., Burkhardt, I., Arndt, B., Sieber, C. M. K., et al. (2016). Two separate key enzymes and two pathway-specific transcription factors are involved in fusaric acid biosynthesis in Fusarium fujikuroi. Environ. Microbiol. 18, 936–956. doi: 10.1111/1462-2920.13150
Wang, M., Sun, Y., Sun, G., Liu, X., Zhai, L., Shen, Q., et al. (2015). Water balance altered in cucumber plants infected with Fusarium oxysporum f. sp. cucumerinum. Sci. Rep. 5, 7722. doi: 10.1038/srep07722
Keywords: Panama disease, Fusarium oxysporum f. sp. cubense, fusaric acid, horizontal gene transfer, phylogeny
Citation: Liu S, Wu B, Lv S, Shen Z, Li R, Yi G, Li C and Guo X (2019) Genetic Diversity in FUB Genes of Fusarium oxysporum f. sp. cubense Suggests Horizontal Gene Transfer. Front. Plant Sci. 10:1069. doi: 10.3389/fpls.2019.01069
Received: 27 September 2018; Accepted: 07 August 2019;
Published: 04 September 2019.
Edited by:
Miguel Dita, Brazilian Agricultural Research Corporation (EMBRAPA), BrazilReviewed by:
Harold Meijer, Wageningen University & Research, NetherlandsGerda Fourie, University of Pretoria, South Africa
Copyright © 2019 Liu, Wu, Lv, Shen, Li, Yi, Li and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunyu Li, bGljaHVueXU4ODFAMTYzLmNvbQ==; Xiuwu Guo, Z3VveHcxOTU5QDE2My5jb20=